

Abstract
Introduction:
Osimertinib is an effective third-generation tyrosine kinase inhibitor (TKI) for EGFR-mutant lung cancers. However, treatment for patients with acquired resistance to osimertinib remains challenging. We characterized a novel EGFR mutation in exon 20 that was acquired while on osimertinib.
Methods:
A 79-year-old woman had disease progression during third-line treatment with osimertinib for an EGFR L858R/T790M-mutant lung cancer. Sequencing of circulating cell-free DNA showed EGFR L858R, an acquired novel EGFR M766Q mutation in exon 20, and no evidence of EGFR T790M. Homology modeling was done to investigate the effects of M776Q on binding to osimertinib. L858R and L858R/M766Q mutations were retrovirally introduced into Ba/F3 and NIH/3T3 cells and evaluated for sensitivity to first-generation (erlotinib), second-generation (afatinib, neratinib, and poziotinib), and third-generation TKIs (osimertinib) by MTS and colony formation assays. EGFR-mediated signaling pathways were interrogated by western blotting.
Results:
Modeling suggested that EGFR M766Q could disrupt osimertinib binding. L858R/M766Q double-mutant cells were 12-fold more resistant to osimertinib, and more than 250-fold more resistant to erlotinib and afatinib, as compared to L858R-mutant cells. In contrast, double-mutant cells remained sensitive to neratinib and poziotinib at clinically relevant doses (IC50, 4.3 and 1.3 nM, respectively). This was corroborated by the effect of the TKIs on colony formation and EGFR signaling.
Conclusions:
Acquisition of EGFR M766Q exon 20 mutation is a novel mechanism of acquired resistance to osimertinib. EGFR-mutant lung cancers with an acquired EGFR M766Q mutation in the setting of osimertinib-resistance may be sensitive to neratinib and poziotinib.
Keywords: lung adenocarcinoma, EGFR, osimertinib-resistance, neratinib, poziotinib
Introduction
Osimertinib is a third-generation EGFR-tyrosine kinase inhibitor (TKI) with high efficacy in NSCLC patients harboring the EGFR T790M resistance mutation1. Most EGFR T790M-mutant patients ultimately progress on osimertinib treatment, and up to half of those patients show loss of T790M and acquisition of other EGFR alterations2, 3. Treatment for these patients remains challenging. Because osimertinib is increasingly being used as frontline therapy for EGFR-mutant lung cancers, we expect exon 20 resistance mutations will become more common. We describe a novel EGFR M766Q exon 20 mutation with retention of L858R, but loss of T790M, in a patient with lung adenocarcinoma who progressed on third-line osimertinib therapy. Our findings suggest that neratinib or poziotinib may be a beneficial therapeutic option for patients whose recurrence after osimertinib therapy lacks T790M, but harbors rare EGFR mutations at or near M766.
Materials and Methods
Patient and Clinical Sample Collection
Sequencing data and clinical history were obtained through an IRB-approved protocol at the Rutgers Cancer Institute of New Jersey. As part of routine care on progression of disease, plasma was obtained and sent for sequencing (Guardant Health).
Homology Modeling
We used the Swiss-Model automated homology modeling server4 to extract the structural information from the template structure (PDB ID:4zau), model mutated sidechains using a backbone-dependent sidechain rotamer library and resolve unfavorable interactions or steric clashes. We did not model EGFR S306L because there was no structural precedent, and based on its location in EGFR, we predicted that it is unlikely to affect the kinase catalytic domain.
Experimental Assays
NIH/3T3 (ATCC) and IL3-dependent Ba/F3 (DSMZ) cells were obtained in 2018 and maintained, infected and selected as described5. Cells were confirmed negative for mycoplasma6. Inhibitors were purchased from Selleck Chemicals. Plasmids were a gift from Matthew Meyerson (Addgene #11012/#32072). Mutations were introduced using the QuikChange XL Site-Directed Mutagenesis Kit (Agilent). MTS assays, soft agar colony-forming assays and western blotting were performed following standard methods. IC50 values were calculated using GraphPad Prism 5.0. Significance was determined by a two-tailed t-test. Anti-EGFR (#2232), P-EGFR (#3777), ERK1/2 (#4695), P-ERK1/2 (#9101), and P-AKT (#4060) antibodies were from Cell Signaling Technology. Anti-AKT (#5298) and vinculin (#73614) antibodies were from Santa Cruz Biotechnology.
Results
Case Report
A 79-year-old never-smoker female presented with a right upper lobe mass and underwent a right upper lobectomy in November 2007. Pathology revealed a 5-cm well-differentiated adenocarcinoma with areas of bronchoalveolar growth pattern, no pleural or lymphovascular invasion, and 0/7 involved lymph nodes (T2N0M0). In August 2008, she had multiple thyroid nodules and underwent a right partial thyroidectomy, which revealed well-differentiated adenocarcinoma, similar to her lung adenocarcinoma, confirming metastatic disease. Subsequently, genomic testing of the lung mass revealed an EGFR L858R mutation, and she was started on erlotinib in May 2009. Her dose fluctuated between 150 mg and 100 mg daily due to side effects including diarrhea, sores on scalp, alopecia, and curling of her eyelashes and toenails. A repeat lung biopsy in May 2014 after radiographic disease progression again demonstrated adenocarcinoma. Genomic testing identified EGFR L858R and T790M mutations. After seeking consultation elsewhere, she entered a clinical trial with rociletinib, but experienced adverse events including hyperglycemia and thrombocytopenia, and displayed evidence of disease progression in September 2016. She was started on osimertinib, 40 mg daily in September 2016, and she had stable disease and clinical benefit.
In August 2017, imaging studies showed disease progression in the lung and possible new osseous metastases. She then presented to RCINJ. Our review confirmed the progressive disease, but the patient refused a biopsy. A blood sample was sent for circulating cell-free tumor-DNA sequencing, which revealed mutant allele frequencies: EGFR L858R (0.6%), TP53 V203M (0.2%), and “Variants of uncertain clinical significance” including: EGFR S306L (3.3%), M766Q (0.2%). EGFR S306L was reported in one other lung adenocarcinoma in cBioportal (www.cbioportal.com), although the clinical implications and biological effects of this mutation are unknown. Mutations in EGFR at M766 have not been reported in cBioportal; however, an EGFR M677T mutation was reported to be activating7. Based on the report we recommended restarting erlotinib. Her preference was to continue osimertinib and not to pursue other treatments. She died in January 2019 due to complications of progressive disease.
Homology Modeling of EGFR Double-Mutant Structure
In the osimertinib co-crystal structure with wild-type EGFR (PDB ID:4zau), residue M766 stabilizes position T790 with favorable non-covalent contact of 4.4 Angstrom (Fig. 1). In the double mutant model, Q766 comes within 3.7 Angstrom of T790, too close for legitimate van der Waals interaction (Fig. 1B). Q766 appears to push T790 forward into the inhibitor binding site, thereby weakening osimertinib binding.
Figure 1.
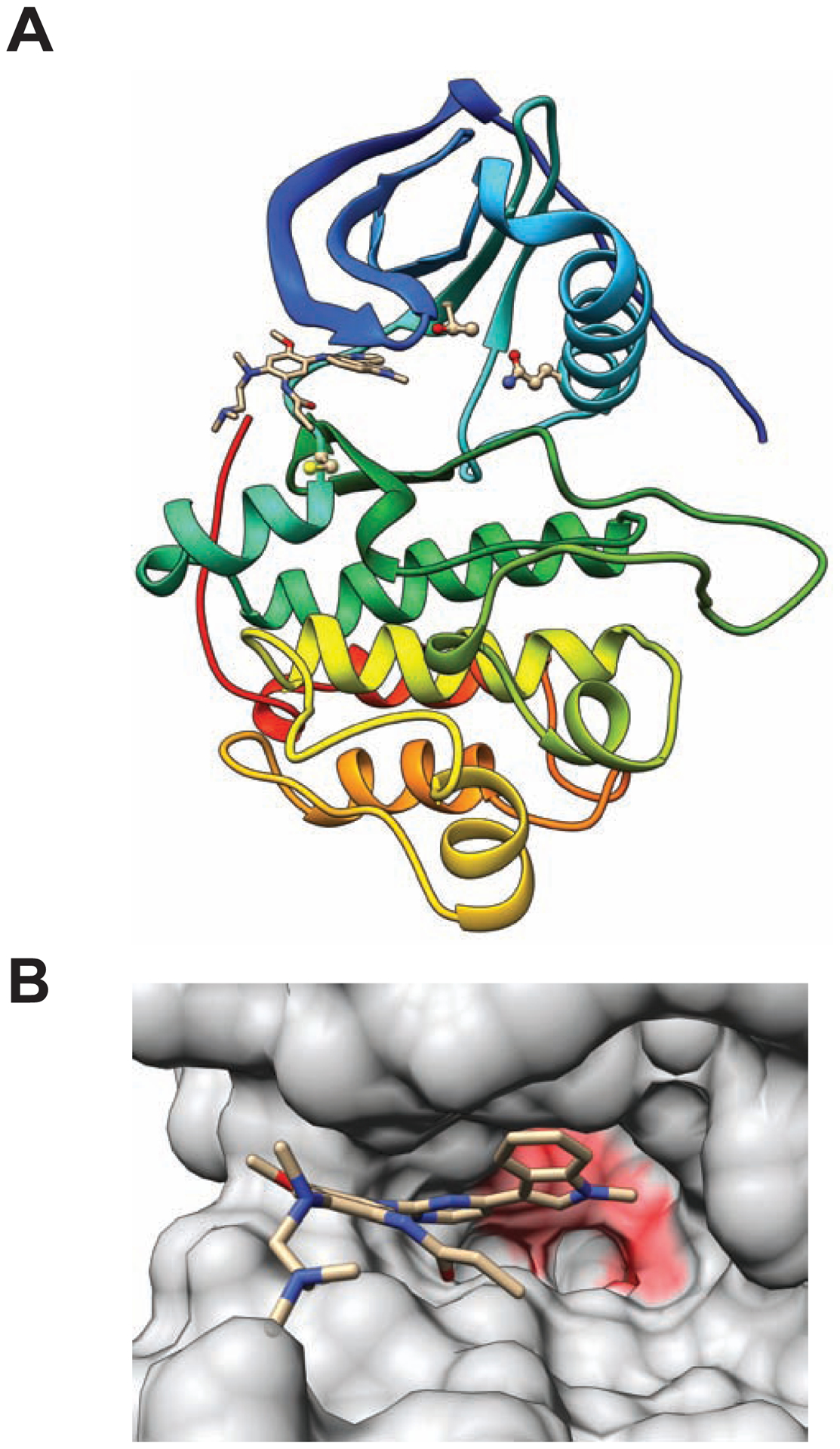
Homology models of EGFR L858R/M766Q double mutant with bound osimertinib. (A) Protein-ligand complex structure. Protein chain shown with cartoon representation; inhibitor shown with stick figure representation; and residues Q766, T790, and C797 shown with ball and stick representations. (B) Inhibitor binding site. Solvent-accessible protein surface is shown with semi-transparent representation. Osimertinib is shown with stick representation. Surface features overlying residues with predicted unfavorable steric clashes are colored red.
Response of Cells Expressing EGFR L858R/M766Q to EGFR-TKIs
We generated Ba/F3 cells stably expressing EGFR L858R alone or in cis with M766Q, and EGFR L858R/T790M/C797S-expressing Ba/F3 cells to serve as a control for osimertinib resistance. All mutations induced IL-3-independence (Supplement 1), and mutant proteins were expressed at equivalent levels (Supplement 2). Ba/F3 cells expressing EGFR L858R/M766Q were more than 10-fold more resistant to osimertinib compared to cells expressing EGFR L858R (IC50 50.62 nM vs. 4.20 nM) (Fig. 2A; Table 1), consistent with other reported osimertinib-resistance mutations, including EGFR L858R/L718V and hotspot mutations near C7978, 9. Patients who lose T790M upon osimertinib resistance may display sensitivity to earlier generation TKIs such as erlotinib or afatinib10. However, Ba/F3 cells expressing L858R/M766Q were resistant to both erlotinib (IC50 4,450 nM vs. 16.5 nM) and afatinib (IC50 46.4 nM vs. 0.16 nM), compared to the single EGFR L858R mutation (Fig. 2B, C; Table 1).
Figure 2.
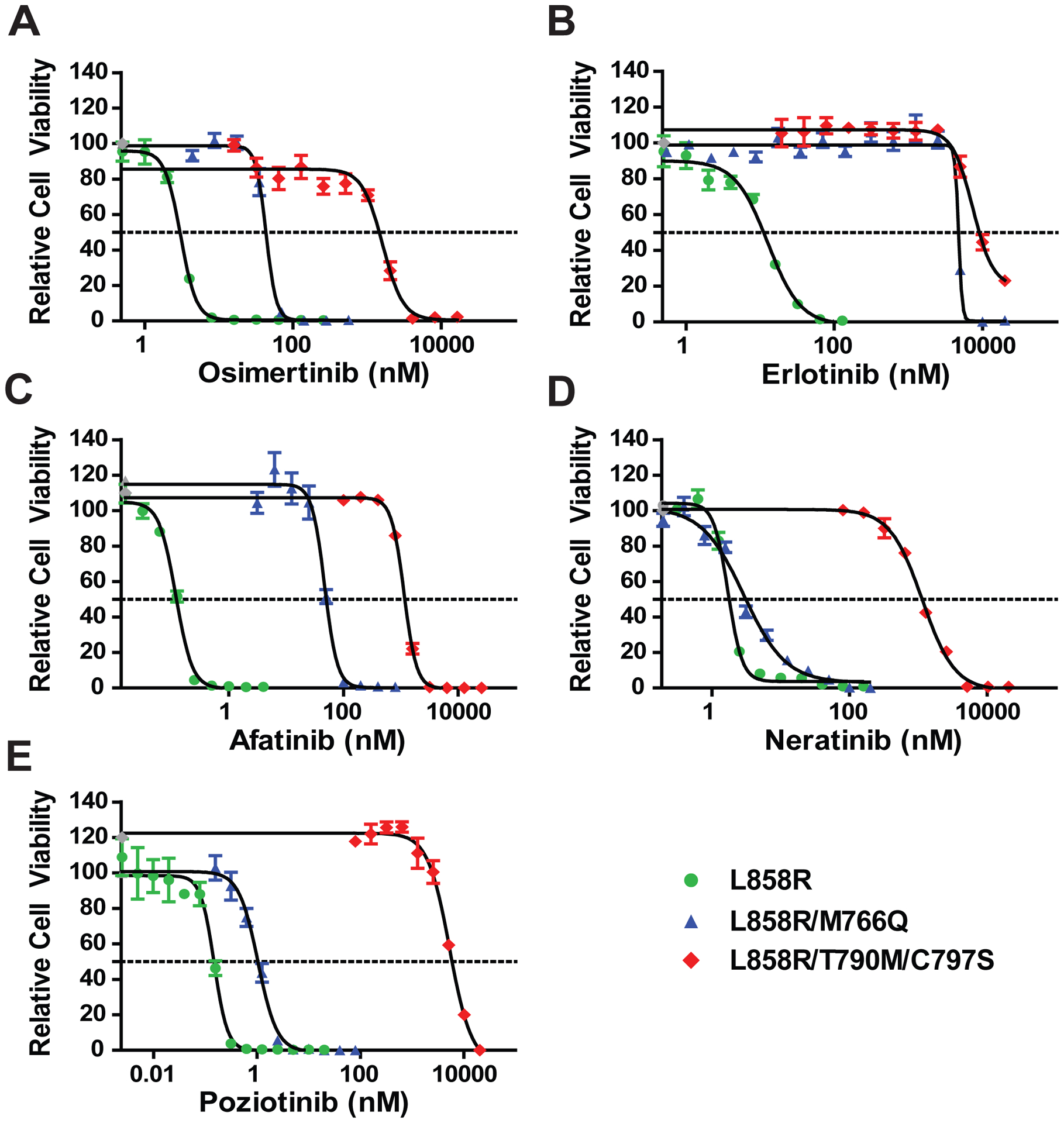
Ba/F3 cells expressing EGFR L858R/M766Q are sensitive to the second-generation TKIs, neratinib and poziotinib.
Cells were treated with specified TKI for 72 hours and assessed for relative viability as compared to the untreated control via MTS assay. (A) Cells expressing EGFR L858R/M766Q were resistant to osimertinib treatment. (B, C) Despite the loss of T790M, EGFR L858R/M766Q did not respond to first-generation erlotinib (B) or second-generation afatinib (C). (D, E) Cells expressing EGFR L858R/M766Q were sensitive to both neratinib (D) and poziotinib (E), which are dual HER2/EGFR TKIs. EGFR L858R/T790M/C797S-expressing cells were tested as controls. Graphs and analyses were from at least two independent experiments. Gray points represent extrapolated values, and error bars show standard error of the mean (SEM).
Table 1.
IC50 values of Ba/F3 cells expressing mutant EGFR constructs for various generations of EGFR-TKIs.
| Osimertinib | Erlotinib | Afatinib | Neratinib | Poziotinib | |
|---|---|---|---|---|---|
| EGFR L858R | 4.20 nM | 16.50 nM | 0.16 nM | 3.42 nM | 0.15 nM |
| EGFR L858R/M766Q | 50.62 nM | 4,450 nM | 46.40 nM | 4.32 nM | 1.33 nM |
| EGFR L858R/T790M/C797S | 1,630 nM | 7,400 nM | 1,500 nM | 1,830 nM | 5,300 nM |
Neratinib is a dual HER2/EGFR irreversible inhibitor with generally low activity in clinical trials potentially because of dose-limiting diarrhea11. However responses were seen in NSCLC patients with the EGFR G719X and other exon 18 mutations11, 12. EGFR L858R/M766Q and EGFR L858R displayed equivalent sensitivity to neratinib at low concentrations (IC50, 4.32 nM and 3.42 nM respectively) (Fig. 2D, Table 1).
Poziotinib is an irreversible pan-EGFR TKI that successfully inhibits exon 20 EGFR/HER2 insertion mutations13, and is currently in Phase II clinical trials for EGFR exon 20 insertion mutations in NSCLC. Exon 20 insertions sterically hinder the binding of osimertinib, but models have shown that the smaller size and unique shape of poziotinib enables better binding of the compound to the drug-binding pocket, allowing it to overcome structural changes induced by exon 20 insertions13. Poziotinib successfully inhibited the growth of EGFR L858R/M766Q expressing cells at clinically achievable concentrations (IC50, 1.33 nM) (Fig. 2E, Table 1).
Similar to the MTS assays, NIH/3T3 cells expressing EGFR L858R/M766Q were significantly less sensitive to osimertinib, erlotinib and afatinib in soft agar colony-forming assays than cells expressing EGFR L858R (Fig. 3A, B), whereas the single and double mutant-expressing cells were similarly sensitive to neratinib and poziotinib (Fig. 3A, B).
Figure 3.
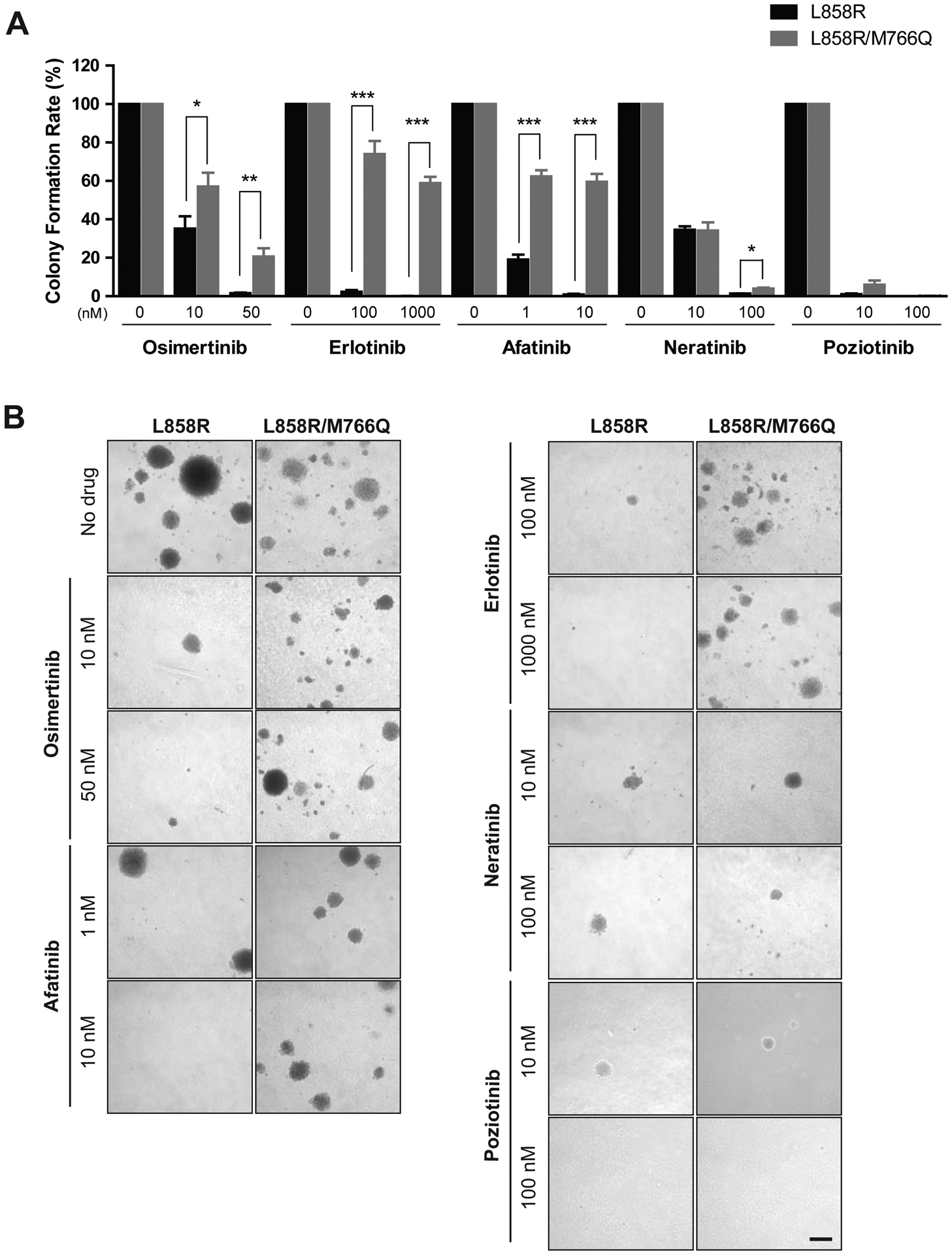
EGFR L858R/M766Q reduces the inhibition of osimertinib, erlotinib and afatinib, but not neratinib or poziotinib, on colony formation.
1 × 105 NIH/3T3 cells expressing EGFR L858R or L858R/M766Q were seeded in agar with or without TKI. (A) Quantification of colony formation rate, relative to the rate of formation without drug (4X objective), from 3 replicates each and two independent experiments; Asterisks indicate significant difference between cells expressing EGFR L858R vs. L858R/M766Q (*p < 0.05, **p < 0.01, and ***p < 0.00001). (B) Representative images (10X) of colonies with or without TKI after 3 weeks; Scale bar indicates 0.5 mm.
EGFR L858R-expressing Ba/F3 cells showed inhibited EGFR and downstream pathways (AKT and ERK1/2) in a dose-dependent manner caused by osimertinib, erlotinib and afatinib, but the EGFR L858R/M766Q double-mutant cells displayed decreased inhibition (Fig. 4A–C). In contrast, neratinib and poziotinib inhibited phosphorylation of EGFR, AKT and ERK1/2 at similar concentrations between double- and single-mutant cells (Fig. 2D, E).
Figure 4.
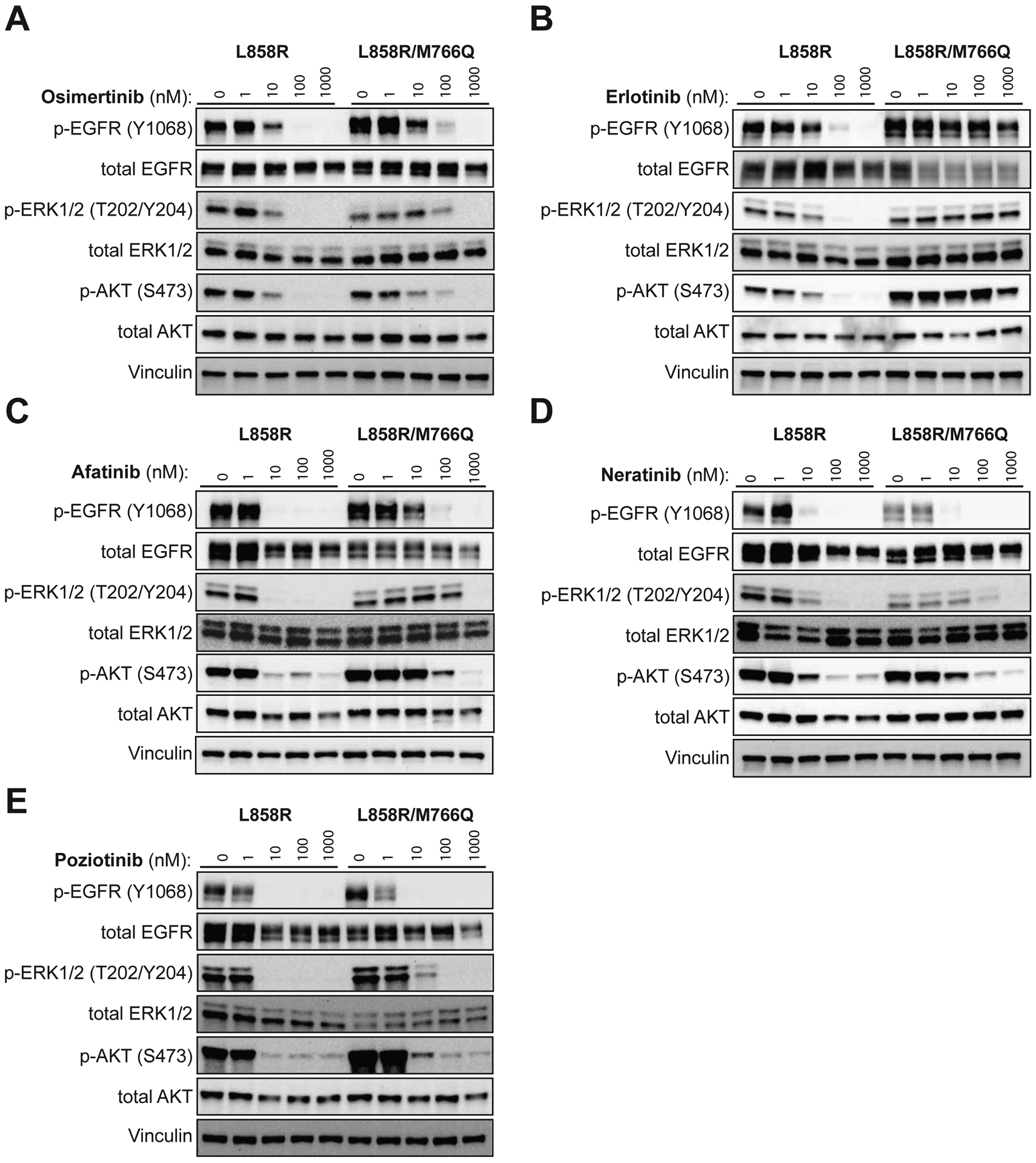
Addition of M766Q reduces the inhibition of phosphorylation of EGFR and downstream pro-growth proteins by osimertinib, erlotinib and afatinib.
Transduced Ba/F3 cells were plated at 1 × 106 cells/mL at hour 0. At hour 2, the drug or DMSO matching the concentration of the highest drug plate was added to the media for 4 hours. (A, B, C) EGFR L858R/M766Q exhibits increased levels of phosphorylated pro-growth proteins, including P-EGFR, P-AKT and P-ERK1/2, as compared to EGFR L858R, when challenged with osimertinib (A), erlotinib (B) and afatinib (C). (D, E) The levels of inhibited phosphorylation of EGFR L858R, EGFR L858R/M766Q and downstream proteins are equivalent when challenged with neratinib (D) or poziotinib (E).
Discussion
EGFR mutation testing is standard-of-practice for advanced-stage NSCLC patients with evidence of adenocarcinoma histology. Osimertinib is commonly used to treat patients who develop the EGFR T790M resistance mutation, and is also increasingly used in the first-line setting. The best therapeutic course of action in patients whose tumors progress after an initial response to osimertinib is not clear. This is the first report of a patient whose lung cancer recurred after osimertinib therapy with an acquired EGFR L858R/M766Q mutation.
Collectively, our data confirmed that EGFR L858R/M766Q is capable of causing clinical resistance to osimertinib. This novel exon 20 mutation also mediated resistance against first- and second-generation TKIs commonly given to patients who have lost a detectable EGFR T790M mutation after treatment with osimertinib. EGFR activity is regulated by repositioning of the αC-helix, which rotates into the ATP-site in the active state, and rotates outwards in the inactive state. Exon 20 insertional mutations are believed to cause conformational transitions which decrease the binding affinity of ATP-site TKIs14. Although we did not rule out some effect of EGFR S306L, the modeling and cell-based data support that EGFR M766Q was likely a driving factor behind the osimertinib resistance observed in our patient. We also cannot rule out that the cells harboring EGFR L858R and M766Q were initially present, possibly subclonally, at diagnosis, and then were selected for and expanded during treatment with osimertinib. The presence of pre-existing resistant subclones of other TKI resistance mutations, such as T790M has been demonstrated, and may be a significant source of acquired resistance to targeted therapy.
EGFR L858R/M766Q-expressing cells responded to neratinib and poziotinib at clinically achievable concentrations and, similar to the classical L858R mutation. Although neratinib was approved by the FDA for certain HER2+ breast cancer patients, it is not approved for NSCLC due to dose-limiting toxicities11. Use of prophylactic loperamide has been reported to reduce incidence and severity of diarrhea and improve tolerance to neratinib15. Poziotinib is another potential treatment candidate for patients who progress on osimertinib with exon 20 mutations, or insertions at M766 or in the αC-helix, although it also has significant side effects13. Additional analysis of large-scale clinical data can provide insight into how frequently this mutation is encountered. The work presented here suggest that neratinib and poziotinib should be evaluated in the clinic for this novel mechanism of acquired resistance to osimertinib in EGFR-mutant lung cancer.
Supplementary Material
Acknowledgements
We thank Dr. Hua Zhong and Dr. Michele Patrizii for their technical assistance.
Funding:
This work was supported by the National Institutes of Health [NCI R01CA190578; NCI P30CA072720]; AHEPA Charitable Fund; and jointly supported by the National Science Foundation, the National Institute of General Medical Sciences, the National Cancer Institute, and the Department of Energy [NSF-DBI 1338415].
Financial support:
Sharon R. Pine (NCI R01CA190578); Shridar Ganesan (AHEPA Charitable Fund). The RCSB PDB is jointly funded by the National Science Foundation, the National Institute of General Medical Sciences, the National Cancer Institute, and the Department of Energy (NSF-DBI 1338415).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest disclosure statement: Shridar Ganesan has received support from Roche, Foghorn Therapeutics, Foundation Medicine, Merck and Inspirata. Helena Yu has consulted for AstraZeneca and has received travel compensation from Lilly. Her institution has received financial support for clinical research she is involved in from AstraZeneca, Lilly, Novartis, Daiichi, Astellas, and Pfizer. The other authors declare no potential conflicts of interest.
References
- 1.Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med 2017;376:629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piotrowska Z, Isozaki H, Lennerz JK, et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov 2018;8:1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le X, Puri S, Negrao MV, et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin Cancer Res 2018;24:6195–6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018;46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saxon JA, Sholl LM, Janne PA. EGFR L858M/L861Q cis Mutations Confer Selective Sensitivity to Afatinib. J Thorac Oncol 2017;12:884–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan KM, Fischer BS, Lee FY, et al. Gamma Secretase Inhibition by BMS-906024 Enhances Efficacy of Paclitaxel in Lung Adenocarcinoma. Mol Cancer Ther 2017;16:2759–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruan Z, Katiyar S, Kannan N. Computational and Experimental Characterization of Patient Derived Mutations Reveal an Unusual Mode of Regulatory Spine Assembly and Drug Sensitivity in EGFR Kinase. Biochemistry 2017;56:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Li Y, Ou Q, et al. Acquired EGFR L718V mutation mediates resistance to osimertinib in non-small cell lung cancer but retains sensitivity to afatinib. Lung Cancer 2018;118:1–5. [DOI] [PubMed] [Google Scholar]
- 9.Yang Z, Yang N, Ou Q, et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin Cancer Res 2018;24:3097–3107. [DOI] [PubMed] [Google Scholar]
- 10.Minari R, Bordi P, Tiseo M. Third-generation epidermal growth factor receptor-tyrosine kinase inhibitors in T790M-positive non-small cell lung cancer: review on emerged mechanisms of resistance. Transl Lung Cancer Res 2016;5:695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sequist LV, Besse B, Lynch TJ, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J Clin Oncol 2010;28:3076–3083. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi Y, Togashi Y, Yatabe Y, et al. EGFR Exon 18 Mutations in Lung Cancer: Molecular Predictors of Augmented Sensitivity to Afatinib or Neratinib as Compared with First- or Third-Generation TKIs. Clin Cancer Res 2015;21:5305–5313. [DOI] [PubMed] [Google Scholar]
- 13.Robichaux JP, Elamin YY, Tan Z, et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med 2018;24:638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yasuda H, Park E, Yun CH, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med 2013;5:216ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hurvitz S, Chan A, Iannotti N, et al. Abstract P3-14-01: Effects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients with HER2+ early-stage breast cancer: The CONTROL trial. Cancer Research 2018;78:P3-14-01–P13-14-01. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.