

Abstract
T cells engineered to express chimeric antigen receptors (CARs) have revolutionised the field of cellular therapy for cancer. Despite its success, this strategy has some recognised limitations and toxicities. Hence, there is growing interest in developing novel cellular therapies based on non‐αβ T‐cell immune effector cells, including NK cells that offer clear advantages in cancer immunotherapy. As a result, NK cells are being explored as an alternative platform for CAR engineering and are becoming recognised as important players in the next generation of cellular therapies targeting cancer. In this review, we highlight preclinical and clinical studies of CAR‐NK cells derived from different sources and discuss strategies under investigation to enhance the antitumor activity of these engineered innate immune cells.
Keywords: allogeneic, cancer immunotherapy, CAR, cellular therapy, NK cells, off‐the‐shelf product
NK cells can be derived from various sources (PB, UCB, HSC, iPSC, NK cell lines) and can be engineered to express a chimeric antigen receptor (CAR) to target various surface antigens on cancer cells. These CAR‐NK cells can be used as off‐the‐shelf adoptive cellular therapy to treat patients with various malignancies.

Introduction
This past decade has seen impressive advances in the cellular therapy field. Autologous T cells genetically engineered to express a chimeric antigen receptor (CAR) have induced durable clinical responses in patients with B‐cell acute lymphoblastic leukaemia (B‐ALL) or B‐cell lymphomas as two striking examples. 1 Despite this clinical success, manufacturing complexities and severe toxicities are persistent limitations of CAR T‐cell therapy. 2 These challenges highlight the need to investigate alternative immune effector cells as potential vehicles for CAR engineering with an improved safety profile and the potential to be used as allogeneic off‐the‐shelf products.
CARs are engineered receptor molecules that direct immune effector cell functions through specific antigen recognition by its immunoglobulin‐like extracellular domain, the single‐chain variable fragment (scFv). 3 Once equipped with a CAR, T cells are redirected to recognise and kill tumor cells expressing the target antigen. 4 Thus far, four cellular therapies, all CD19‐targeted T‐cell products, have been approved by the Food and Drug Administration (FDA): tisagenlecleucel (KYMRIAH, Novartis), axicabtagene ciloleucel (Yescarta, Kite Pharma) and brexucabtagene autoleucel (KTE‐X19, Tercartus). Tisagenlecleucel was shown to be effective in children and young adults with B‐ALL and adult patients with aggressive B‐cell lymphomas. 5 , 6 Axicabtagene ciloleucel is approved for the treatment of patients with relapsed or refractory aggressive B‐cell lymphomas. 7 Recently, brexucabtagene autoleucel was shown to have impressive activity in patients with relapsed or refractory mantle cell lymphoma. 8 Additionally, a fourth CD19‐targeted product, lisocabtagene maraleucel (Juno Therapeutics), was just granted FDA approval for the treatment of relapsed or refractory large B‐cell lymphomas. 9 CAR T‐cell therapies targeting various other antigens are also under investigation for haematologic malignancies, including T‐cell leukaemias and lymphomas, myeloid malignancies and multiple myeloma.
Large‐scale use of CAR T‐cell therapy (Table 1) faces a number of challenges. First of all, treatment with these modified cells can cause serious adverse events, such as cytokine release syndrome (CRS) and immune effector cell‐associated neurotoxicity syndrome (ICANS), which may increase the length of hospitalisation and raise the cost of therapy. 10 Second, manufacturing of autologous CAR T cells can be a cumbersome, expensive and lengthy procedure, 11 as it involves the collection of T cells from heavily pretreated patients, their transduction and expansion, and finally infusion of the CAR T products. 11 Given these shortcomings, there is growing interest in other subsets of immune effectors, such as natural killer (NK) cells. NK cells represent an essential component of the innate immune system and play an important role in the first line of defence against pathogens and cancer. 12 As their name indicates, NK cells are specialised killers with the natural ability to eliminate abnormal cells without the need for prior sensitisation, as they rely on the balance between activating and inhibitory signals from germline‐encoded receptors. 12 Unlike allogeneic T cells, allogeneic NK cells do not cause graft‐versus‐host disease and thus pave the way for a potentially off‐the‐shelf product for cell therapy.
Table 1.
Clinical trials for CD19‐directed CAR T cell therapy
| Clinical trial | ELIANA trial (NCT02435849) | JULIET trial (NCT02445248) | ZUMA‐1 trial (NCT02348216) | TRANSCEND NHL 001 trial (NCT 02631044) | ZUMA‐2 trial (NCT02601313) |
|---|---|---|---|---|---|
| Drug | Tisagenlecleucel (Kymriah, Novartis) | Tisagenlecleucel (Kymriah, Novartis) | Axicabtagene ciloleucel (Yescarta, Kite) | Lisocabtagene maraleucel (Juno Therapeutics) | Axicabtagene ciloleucel KTE‐X19 (Yescarta, Kite) |
| Target | CD19 | CD19 | CD19 | CD19 | CD19 |
| Stimulatory and co‐stimulatory domains | CD3 ζ and 1‐4BB | CD3 ζ and 1‐4BB | CD3 ζ and CD28 | CD3 ζ and 1‐4BB | CD3 ζ and CD28 |
| Study design | Single‐arm, multicentered, unblinded phase 2 | Single‐arm, multicenter, open labeled, phase 2 | Single‐arm, multicenter, unblinded, phase 1–2 | Single‐arm, multicenter, multicohort, unblinded, phase 1–2 | Multicenter, phase 2 trial |
| Population | Patients within 3–23 years old with R/R B‐ALL | Patients 18 years or older with R/R DLBCL who received at least 2 prior lines of therapy (de novo DLBCL or from transformed FL, and high‐grade B‐cell lymphoma with MYC rearrangement plus rearrangement of Bcl2, Bcl6 or both) | Patients 18 years or older with refractory large B cell lymphoma (DLBCL, transformed follicular lymphoma, primary mediastinal B cell lymphoma) | Patients 18 years or older with R/R large B‐cell lymphoma (transformed or de novo DLBCL, primary mediastinal B cell lymphoma and high‐grade B‐cell lymphoma with MYC rearrangement plus rearrangement of Bcl2, Bcl6 or both), FL grade 3B or relapsed MCL | Patients 18 years or older with R/R mantle cell lymphoma |
| Previous lines of treatment, range (median) | 1–8 (3) | 1–8 (not reported) | 2–4 (3) | 2–4 (3) | 1–5 (3) |
| Previous autologous stem cell transplantation, n (%) | 46 (61) | 18 (33) | Not reported | 90 (33) | 29 (43) |
| Lymphodepletion chemotherapy | Fludarabine 30 mg/m2 IV (daily doses for 4 doses) and cyclophosphamide 500 mg/m2 IV (daily doses for 2 doses) | Fludarabine 25 mg/m2 IV (daily doses for 3 days) and cyclophosphamide 250 mg/m2 IV (daily doses for 3 doses) or bendamustine 90 mg/m2 IV (daily doses for 2 days) | Fludarabine 30 mg/m2 IV (daily doses for 3 days) and cyclophosphamide 500 mg/m2 IV (daily doses for 3 doses | Fludarabine 30 mg/m2 IV (daily doses for 3 doses) and cyclophosphamide 300 mg/m2 IV (daily doses for 3 doses) | Fludarabine 30 mg/m2 IV (daily doses for 3 days) and cyclophosphamide 500 mg/m2 IV (daily doses for 3 doses |
| T cell dose | Single dose of 0.2–5 × 106 cells/kg (patients < or = 50 kg) or 0.1–2.5 × 106 cells/kg (patients > 50 kg) | Single dose of 0.1–6 × 108 cells (median 3 × 108 cells) | Single dose of 2 × 106 cells/kg (minimum 1 × 106 cells/kg) |
Sequential infusions
(Median 91 × 106 T cells; range 44–156 × 106 T cells) |
Single dose of 2 × 106 cells/kg (minimum 1 × 106 cells/kg) |
| Primary endpoints | Overall remission rate (CR/CRi) by month 3 | Best overall response rate (CR + PR) | Objective response (CR + PR) | Best overall response rate (CR + PR), incidence of adverse events and probability of dose‐limiting toxicities | Objective response (CR+ PR) |
| Response rates | ORR 81% (CR 60% and CRi 21%) | Best ORR 52% (CR 40% and PR 12%) | Objective response 83% (CR 58% and PR 25%) |
Objective response
|
Objective response 93% (CR 67% and PR 27%) |
| Adverse events | |||||
| B‐cell aplasia | All patients with response to treatment | Only one patient had normal B‐cell count before infusion | 25% had persistent B cell aplasia at 24 months post infusion | 92% had B cell aplasia at baseline | None |
| CRS, n (%) | 58 (77) | 64 (58) | 100 (92) | 113 (42) | 62 (91) |
| Neurologic events, n (%) | 30 (40) | 23 (21) | 72 (67) | 80 (30) | 43 (63) |
| Pyrexia, n (%) | 30 (40) | 39 (35) | 94 (87) | 45 (17) | 62 (91) |
| Febrile neutropenia, n (%) | 27 (36) | 18 (16) | 39 (36) | 25 (9) | Not reported |
| Hypotension, n (%) | 22 (29) | 29 (26) | 63 (58) | 60 (22) | 35 (51) |
| Elevated AST, n (%) | 20 (27) | Not reported | 19 (18) | Not reported | 21 (31) |
In this review, we focus on recent advances in the development of CAR‐engineered NK cells as an alternative immune cell effector for the treatment of different malignancies, highlighting advantages over CAR T‐cell therapy and the preclinical and clinical milestones that have been achieved so far.
Advantages of NK cell over T cells for CAR engineering
CD8+ T cells are effector lymphocytes whose function depends on the activation of the T‐cell receptor (TCR) after it recognises a specific antigen bound to a major histocompatibility complex (MHC) class I on the surface of target cells. 13 , 14 Thus, CD8+ T‐cell activation depends on a first specific signal generated by the TCR, leading to secretion of granzyme and perforin, which then kill malignant or infected cells. 13 Although NK cells share certain cytotoxic functions with CD8+ T cells, they do not require prior antigen priming before they are able to kill their targets. Instead, NK cells rely on the balance between activating and inhibitory inputs generated by several germline‐encoded receptors for their cytotoxic functions towards pathogens and transformed cells. 15 , 16 Once equipped with CARs, they maintain their ability to be triggered by these innate receptors, while antigen recognition is redirected towards CAR‐specific targets. This feature, absent in T cells, preserves NK cell‐mediated cytotoxicity towards malignant cells, even in the event of target antigen loss or downregulation.
Thus far, the approved CAR T‐cell products have been autologous because of the risk of GVHD with the use of allogeneic T cells. 17 However, harvesting autologous T cells from cancer patients can be difficult as a result of lymphopenia induced by multiple lines of therapy and/or high disease burden in patients with leukaemia. The autologous T cells must be transduced and expanded before infusion, a manufacturing process that can take precious days away from patients in need of urgent treatment. 18 In contrast to T cells, allogeneic NK cells are not associated with GVHD 19 , 20 , 21 and readily available as an allogeneic off‐the‐shelf product, making them an attractive option for cellular therapy.
Finally, CAR T‐cell therapy has been associated with serious clinical adverse events such as CRS and ICANS. 10 , 22 In fact, in the phase I and phase II clinical trials that led to the approval of CD19‐targeted CAR T‐cell therapy, CRS of any grade was experienced by 58–92% of patients, while neurologic events of any grade were seen in 21–67% of patients who received an infusion. 5 , 6 , 7 Recently, our group has shown that CAR‐NK cells lack measurable toxicity in patients with non‐Hodgkin lymphoma and chronic lymphocytic leukaemia. 23
Potential sources of NK cells for CAR engineering
NK cells can be derived from multiple sources for use in CAR engineering, as discussed below and shown in Table 2 and Figure 1.
Table 2.
Pre‐clinical studies on CAR NK therapy
| Study | Target | Malignancy | Stimulatory and co‐stimulatory domains | Source of NK cells | Culture and expansion | Engineering method |
|---|---|---|---|---|---|---|
| Liu et al.23 | CD19 | B‐cell malignancies | CD3ζ and CD28 | Cord blood | Genetically modified K562 feeder cells co‐expressing IL‐21 and CD137, ectopic IL‐15 and exogenous hrIL‐2 a | Retrovirus |
| Herrera et al. 66 | CD19 | B‐cell malignancies | CD3ζ and 4‐1BB | Peripheral and cord blood | Exogenous hrIL‐2 and IL‐15 | Lentivirus |
| Muller et al. 65 | CD19 | B‐cell malignancies | CD3ζ and CD28 | Peripheral blood | Exogenous IL‐15 | Lentivirus or alpharetrovirus |
| Quintarelli et al. 64 | CD19 | B‐cell malignancies | CD3ζ and 4‐1BB | Peripheral blood | NK Cell Expansion kit containing antibodies against NKp46 and CD2 and exogenous hrIL‐2 or IL‐15 | Retrovirus |
| Goodridge et al. 67 | CD19 | B‐cell malignancies | CD3ζ and 2B4 | iPSC | Autonomous IL‐15 receptor | Unknown |
| Romanski et al. 60 | CD19 | B‐ALL | CD3ζ | NK‐92 | Exogenous hrIL‐2 | Retrovirus |
| Oelsner et al. 62 | Flt3 | B‐ALL | CD3ζ and CD28 | NK‐92 | Exogenous hrIL‐2 | Lentivirus |
| Muller et al. 59 | CD20 | B‐cell malignancies | CD3ζ | NK‐92 | Exogenous hrIL‐2 | Retrovirus |
| Tassev et al. 63 | EBNA | EBV+ B‐lymphoblastic cell lines | CD3ζ and 4‐1BB | NK‐92MI b | No cytokines, feeder cells or stimulation beads | Retrovirus |
| Boissel et al. 61 | CD19 | CLL | CD3ζ | NK‐92 | Exogenous hrIL‐2 | Electroporation |
| Jiang et al. 71 | CD138 | Multiple myeloma | CD3ζ | NK‐92MI b | No cytokines, feeder cells or stimulation beads | Lentivirus |
| Chu et al. 73 | CS1 | Multiple myeloma | CD3ζ and CD28 | NK‐92 | Exogenous hrIL‐2 | Lentivirus |
| Chen et al. 76 | CD3 | Peripheral T‐cell lymphoma | CD3ζ, CD28 and 4‐1BB | NK‐92 | Exogenous hrIL‐2 | Lentivirus |
| Chen et al. 75 | CD5 | Peripheral T‐cell lymphoma | CD3ζ, CD28 and 4‐1BB | NK‐92 | Exogenous hrIL‐2 | Lentivirus |
| You et al. 77 | CD7 | T‐ALL | CD3ζ, CD28 and 4‐1BB | NK‐92MI b | No cytokines, feeder cells or stimulation beads | Electroporation |
| Han et al. 48 | EGFR and EGFRvIII | Glioblastoma | CD3ζ and CD28 | NK‐92 and NKL c | Exogenous hrIL‐2 | Lentivirus |
| Murakami et al. 82 | EGFRvIII | Glioblastoma | CD3ζ, CD28 and 4‐1BB | KHYG‐1 | Exogenous hrIL‐2 | Lentivirus |
| Genssler et al. 81 |
EGFR EGFRvIII d |
Glioblastoma | CD3ζ and CD28 | NK‐92 | Exogenous hrIL‐2 | Lentivirus |
| Zhang et al. 85 | HER2 | Glioblastoma | CD3ζ and CD28 | NK‐92 | Exogenous hrIL‐2 | Lentivirus |
| Muller et al. 82 | EGFRvIII | Glioblastoma | DAP12 | NK cell line YTS | No cytokines | Lentivirus |
| Sahm et al. 85 | EpCAM | Breast cancer | CD3ζ and CD28 | NK‐92 | Exogenous hrIL‐2 and ectopic IL‐15 | Lentivirus |
| Liu et al. 88 | HER2 | Breast cancer | CD3ζ and CD28 | NK‐92 | Exogenous hrIL‐2 | Electroporation |
| Chen et al. 89 | EGFR and EGFRvIII | Breast cancer brain metastases | CD3ζ and CD28 | NK‐92 | Exogenous hrIL‐2 | Lentivirus |
| Liu et al. 90 | EGFR | Triple negative breast cancer | CD3ζ, 4‐1BB and CD28 | Peripheral blood | Exogenous hrIL‐2 | Lentivirus |
| Hu et al. 91 | Tissue Factor | Triple negative breast cancer | CD3ζ, 4‐1BB and CD28 | NK‐92MI b | No cytokines, feeder cells or stimulation beads | Lentivirus |
| Li et al. 54 | Mesothelin | Ovarian cancer | CD3ζ and multiple co‐stimulatory domains including DAP10, 2B4 and 4‐1BB |
NK‐92 iPSC |
Exogenous hrIL‐2 After NK cell differentiation, feeder cells expressing membrane bound IL‐21 and exogenous hrIL‐2 |
Transposon plasmids |
| Uherek et al. 92 | HER2 | Ovarian and breast cancer | CD3ζ | NK‐92 | Exogenous hrIL‐2 | Retrovirus |
B‐ALL, B‐cell acute lymphoblastic leukemia; CLL, chronic lymphocytic leukemia; iPSC, induced pluripotent stem cell; T‐ALL, T‐cell acute lymphoblastic leukemia.
hrIL‐2: human recombinant interleukin‐2.
NK‐92MI: genetically modified NK‐92MI cells produce high levels of IL‐2 and possess a high‐affinity CD16 variant.
NKL is cell line derived from an adult with NK cell leukemia.
Dual specific NK cells targeting wildtype EGFR and mutated EGFRvIII.
Figure 1.
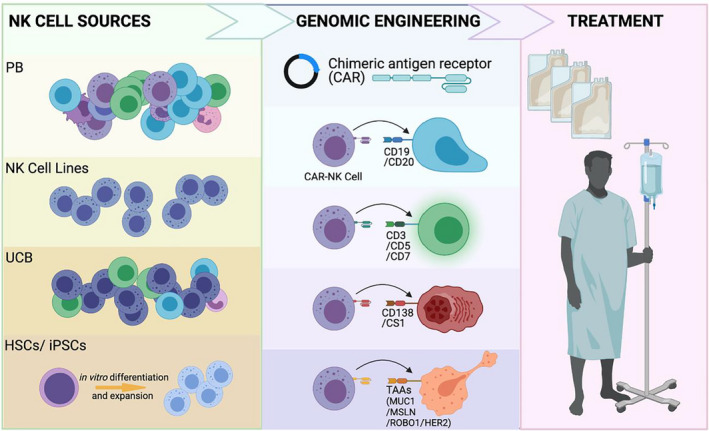
CAR‐NK cells for antitumor therapy. HSCs, haematopoietic stem cells; iPSCs, induced pluripotent stem cells; MSLN, mesothelin; PB, peripheral blood; TAA, tumor‐associated antigen; UCB, umbilical cord blood.
Peripheral blood NK cells
NK cells (classically defined by flow cytometry as CD56‐positive, CD3‐negative, CD19‐negative, CD14‐negative and CD45‐positive 24 ) are short‐lived innate lymphoid cells with a turnover time of about 2 weeks in the circulation at steady state. 25 , 26 They typically represent 10–15% of peripheral blood (PB) lymphocytes, 27 , 28 but their frequency can vary widely among healthy individuals, from 0% to 60% PB‐NK cells (CD3−CD56+ cells) in one study. 27 NK cells can be divided into two major subsets according to their phenotype: CD56brightCD16dim or CD56dimCD16bright cells, the latter being the most frequent in PB. 27 Unlike T cells, NK cells do not display rearranged receptors such as TCRs. Hence, their diversity reflects the combination of germline‐encoded activating (e.g. NKG2C, NKG2D and natural cytotoxic receptors such as NKp30, NKp44 and NKp46) and inhibitory (e.g. killer immunoglobulin‐like receptors [so‐called KIRs] and NKG2A) receptors on their surface. 29 Human PB‐NK cells are typically characterised by a mature cytotoxic CD56dimCD16bright subset 30 with little variation among individuals. 25 Overall, however, PB‐NK cell phenotypic diversity seems to be donor‐specific and highly variable among individuals. 25 Furthermore, while inhibitory receptors are mainly genetically regulated to retain self‐tolerance, NK cell‐activating receptors adapt to diverse environmental stimuli in response to pathogens and malignancies. 29 Finally, PB‐NK cell functionality and diversity can be modulated by exposure to cytokines such as interleukin (IL)‐2, IL‐15, IL‐18, IL‐12 and IL‐21. 25 , 31
In summary, as a candidate source of an off‐the‐shelf product, PB‐NK cells possess the advantages of easy accessibility and mature phenotypic signature. However, donor variability complicates dose standardisation as PB‐NK cells lack a single and uniform renewable source. 32
Umbilical cord blood NK cells
NK cells can also be derived from umbilical cord blood (CB). In fact, their absolute number in CB by volume and fraction is higher than found in PB, representing 20–30% of the CB lymphocytic pool. 28 On the other hand, CB‐NK cells show lower diversity and display a more immature phenotype than do PB‐NK cells. 30 , 33 The CB‐NK cell phenotypic signature features a higher expression of CD56bright, NKG2A, CD94, c‐kit (CD117), Trail, CD62L and CD27, together with a lower expression of CD16, KIRs, NKG2D, NKG2C, TIGIT and DNAM‐1. 30 , 33 The transcriptome of CB‐NK cells also shows lower levels of maturation markers such as T‐bet, eomesodermin, perforin and granzymes. 30 , 33 Consistent with these data, when freshly isolated without any ex vivo expansion, CB‐NK cells function poorly in co‐cultures with tumor cells, as evidenced by their lower cytokine production and diminished degranulation together with decreased cytotoxicity against tumor targets. 30 , 33
Despite their initial immature profile, CB‐NK cells can be successfully expanded and induced to develop more mature characteristics by exposure to cytokines. For example, when cultured in lower concentrations of IL‐2 (200 IU), CB‐NK cells fail to proliferate, probably because of their lower CD25 expression. 33 However, when exposed to higher concentrations of IL‐2 (1000 IU), their proliferation rates improve significantly together with their degranulation and cytotoxic capacity. 33 CB‐NK cell expansion can be further improved by co‐culture with ‘feeder cells’ such as genetically modified K562 cells (erythroleukaemia cell line) that express membrane‐bound IL‐21, 4‐1BB ligand and CD48, which increases the proliferation rate by more than 1000‐fold. 34 Finally, CB‐NK cells can be successfully transduced after expansion with ‘feeder cells’ and/or cytokines. After transduction with a CD19‐directed CAR, the expanded CB‐NK cells show enhanced metabolic fitness and greater cytotoxicity against CD19‐positive tumor than in non‐transduced NK cells. 34 , 35
Thus, CB units offer a rich source of NK cells for immunotherapy. They are readily available in global CB banks, can be expanded using feeder cells and cytokines, and genetically manipulated to express CARs. Nonetheless, similar to PB‐NK cells, their phenotype and yields differ significantly among donors, and they lack a single renewable source. 32
NK cells from umbilical cord CD34+ progenitors
NK cells can also be generated from CD34+ progenitor cells isolated from umbilical CB. This allows the production of a more homogenous and well‐defined NK cell product than does the use of PB‐NK or CB‐NK cells. 36 CD34+ haematopoietic progenitors can be successfully differentiated into CD56+ NK cells and then expanded with cytokines in a stepwise fashion. 37 It should be stressed, however, that cells derived by this method do not seem to be as mature as PB‐NK cells, as they express high levels of CD56, NKG2A and CD94, and display variable KIR levels. 38 Moreover, despite their cytotoxic competency against leukaemic cells, 39 these CD56+ cells show low expression of CD16 (FcγRIII), resulting in poor antibody‐dependent cellular cytotoxicity (ADCC). 40 To the best of our knowledge, only one study has successfully introduced a CD19‐targeted CAR into CD34+ progenitors followed by NK differentiation. 41
Hence, although CD34+ cells from CB can be used as a source for large‐scale production of a homogenous population of CD56+ NK cells, they are immature and display little ADCC capacity even after expansion, thus requiring additional steps to enhance potency for clinical application.
NK‐92 cell line
To avoid some of the difficulties in handling primary NK cells, investigators have turned their efforts to developing a safe immortalised NK cell line for cellular therapy. NK‐92 cells, for example, were derived from a 50‐year‐old patient who had a large granular lymphocyte non‐Hodgkin lymphoma characterised by a CD56+, CD3− and CD16− population. 42 These cells, which are IL‐2‐dependent, have cytolytic activity against K562 (erythroleukaemia) and Daudi (Burkitt lymphoma) cell lines. 42 Infusing cells derived from a cancer patient might appear to be a perilous alternative; however, when irradiated, these cells have no malignant potential, neither in mice nor in humans, and they maintain their cytotoxic capacity. 43 Administration of irradiated NK‐92 cells seems safe with no significant adverse effects, 44 and these cells can be effectively engineered to improve their function. For example, NK‐92 cells do not express CD16 (FcγRIIIa), the main receptor involved in ADCC and a powerful activating signalling pathway that has been implicated in NK cell cytotoxic capacity. However, these cells can be genetically modified to express a high‐affinity CD16 receptor variant that improves their antitumor performance in the presence of diverse monoclonal antibodies. 45 In addition, NK‐92 cells can be modified to express CARs, a strategy that is being tested in a phase I/phase II clinical trials for diverse types of malignancies (NCT02892695 and NCT02742727). Unfortunately, the lifespan of these cells is quite short because of their pre‐infusion irradiation, which hinders their in vivo proliferation, an essential factor in tumor control. 32 Less frequently, other NK cell lines derived from individuals with NK cell leukaemia, such as NKL, 46 KHYG‐1 47 and YTS, 48 have been genetically manipulated to express CARs.
Thus, NK‐92 cells are a renewable, homogenous and genetically modifiable population of NK cells that constitute a potential source for off‐the‐shelf cellular therapy, keeping in mind that the clinical application of this product could be limited by its diminished antitumor potency (because of irradiation) when compared to NK cells from other sources.
NK cells from induced pluripotent stem cells
NK cells can be successfully generated from human‐induced pluripotent stem cells (iPSCs) through a stepwise approach: first by co‐culturing iPSCs with bone marrow‐derived stromal cells followed by exposure to a specific cytokine cocktail to drive NK differentiation. 49 , 50 , 51 Moreover, iPSC‐derived NK cells develop activating and inhibitory receptors (including KIRs, natural cytotoxic receptors and CD16) that are typical of their mature counterparts, and they demonstrate in vitro and in vivo antitumor activity. 49 , 50 , 52 After differentiation of iPSCs into NK cells, they can be expanded by co‐culture with feeder cells, such as genetically modified artificial antigen‐presenting cells (aAPCs). 51
Not surprisingly, iPSC‐derived NK cells have been genetically modified to express CARs. The first experiences with CAR introduction into iPSC‐derived NK cells were in the HIV infection domain, in which a CD4‐targeting CAR possessing a CD3ζ intracellular domain controlled viral replication in vitro and in mouse xenograft models. 53 Chimeric antigen receptors directed to tumor antigens have also been transduced into iPSC‐derived NK cells to direct their activity and improve antitumor cytotoxicity. 54
In summary, iPSC‐derived NK cells remain a compelling homogenous source of cellular therapy. These cells can be genetically manipulated and produced in large scale, showing mature phenotypic profile together with antitumor cytotoxicity. 52 , 54 , 55 Nonetheless, they lack important activation markers that might limit their antitumor cytotoxicity. 56 Hence, their efficacy in in vivo models and in the clinical setting remains to be shown.
To conclude this section, NK cells can be isolated, expanded and engineered from various sources using different methodologies, and each source has specific advantages and disadvantages as depicted in Figure 2.
Figure 2.
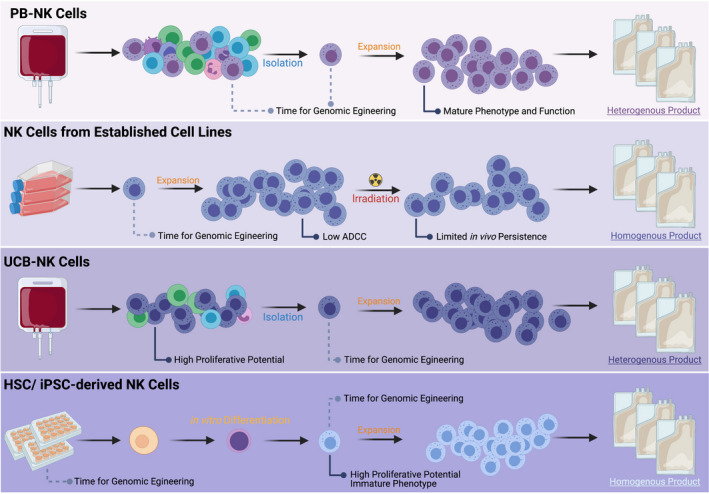
Different sources of allogeneic NK cells for engineering. ADCC, antibody‐dependent cellular cytotoxicity; HSC, haematopoietic stem cell; iPSC, induced pluripotent stem cell; PB, peripheral blood; UCB, umbilical cord blood.
Preclinical studies of CAR‐engineered NK cells
The concept of improving NK cell function by modifying the cells to express a CAR molecule has great appeal, leading several research groups to test this strategy over the past decade, both in vitro and in vivo in mouse xenograft models of cancer. These studies have explored different sources of NK cells, alternative methods for NK cell culture, expansion and transduction, and different plasmid constructs and vectors (Table 2).
B‐cell malignancies
NK cells from diverse sources have been genetically modified to express CARs to redirect their activity against B‐cell malignancies. So far, CD19 has been the most commonly surveyed antigen because of its enriched expression in malignant B cells and lack of expression in most normal cells. 57 , 58 Innovative approaches have been employed in the investigation of CAR‐NK cells for the treatment of B‐cell malignancies.
First‐generation CARs carrying a single CD3ζ costimulatory moiety have been introduced into NK‐92 cells using retroviral transduction. 59 , 60 Two studies have explored this approach with both adopting CD8 as the CAR hinge and transmembrane domain. 59 , 60 The difference between these CARs was their single‐chain variable fragment (scFV) specificity, as one targeted CD19 60 and the other CD20. 59 Boissel et al. 61 also produced first‐generation CAR‐armed NK‐92 cells targeting CD19 to test against CLL cells. Their construct displayed a CD8 hinge and transmembrane domain, and the plasmid delivery was performed using electroporation. 61 NK‐92 cells have also been engineered with second‐generation CARs. Oelsner et al. 62 worked on a second‐generation construct carried by a lentiviral vector and possessing CD3ζ and CD28 stimulatory domains together with a CD28 transmembrane element followed by a modified CD8 hinge. To direct cytotoxicity against Fms‐like tyrosine kinase (Flt3)‐positive B‐ALL cells, they built a CAR immunoglobulin‐like domain specific for Flt3, which led to enhanced antitumor activity. These CAR‐engineered NK‐92 cells required consecutive administration of IL‐2 while in culture.
Another group redirected the antitumor activity of NK‐92MI cells, which are genetically modified NK‐92 cells that independently produce IL‐2 and express a high‐affinity CD16 variant. 63 They did so by introducing into these cells a TCR‐like CAR targeting an HLA‐restricted Epstein–Barr virus (EBV)‐encoded nuclear antigen (EBNA). Their CAR scFv specifically recognised EBNA3C peptide bound to major histocompatibility complexes (MHCs) in EBV‐positive B‐ALL cells. In this study, the CAR hinge and transmembrane domain were CD8‐derived, and its costimulatory moieties were 4‐1BB together with CD3ζ. Retroviral vectors were used for the transduction of NK‐92MI cells. It should be noted that the strategies using NK‐92 subsets need pre‐infusion irradiation, which might decrease cell proliferation and persistence in vivo.
Moreover, three different studies have investigated the introduction of CD19‐targeted CARs into PB‐NK cells, all second‐generation CARs containing a CD3ζ moiety in addition to either 4‐1BB or CD28 intracellular domains. 64 , 65 , 66 To link the extracellular immunoglobulin‐like element to the costimulatory domains, a CD8 hinge combined with either CD28 or CD8 transmembrane structures was used. 64 , 65 Lentivirus or retrovirus vectors were used to transduce these PB‐NK cells, 64 , 65 , 66 which were successfully expanded by cytokine exposure including human recombinant IL‐2 and/or IL‐15. 65 , 66 Quintarelli et al. 64 also added antibodies against NKp46 and CD2 to enhance NK cell stimulation. All these CD19‐targeted PB‐NK cells showed in vitro activity against B‐cell malignancies. 64 , 65 , 66
iPSC‐derived NK cells have also been genetically modified to express CAR. Goodridge et al. 67 introduced a CD19‐targeting CAR combined with an NKG2D transmembrane region together with a CD3ζ and a 2B4 stimulatory domains (NCT04245722). These iPSC‐derived NK cells also possessed a cytokine‐autonomous IL‐15 signalling through a recombinant fusion protein of IL‐15 and IL‐15 receptor alpha to enhance proliferation and persistence. This approach confers the advantage of cytokine independence to these modified cells. These CAR‐armed iPSC‐derived NK cells also display a high‐affinity non‐cleavable CD16 variant to improve ADCC and, in combination with rituximab, they showed enhanced in vitro antitumor cytotoxicity.
Our group has investigated fourth‐generation CB‐derived CAR‐transduced NK cells. After isolation and selection from CB, NK cells were co‐cultured with genetically modified K562‐based feeder cells expressing membrane‐bound IL‐21, 4‐1BB and SLAMF4 in IL‐2‐supplemented growth medium. 35 This strategy promotes CB‐NK cell proliferation and expansion, providing a source for a large‐scale clinical product. Four to six days after isolation, the cells are transduced with a retroviral vector encoding a CD19‐targeting scFv, CD3ζ signalling domain along with CD28 costimulatory domain, IL‐15 to improve NK cell proliferation and persistence together with an inducible caspase‐9 (iCas9) to be used as a safety switch in the event of toxicity. These CB‐derived CAR‐NK cells showed improved proliferation and persistence together with enhanced in vitro and in vivo antitumor activity against B‐cell malignancies. We have also genetically manipulated NK cell checkpoints to enhance CAR‐NK cell fitness by deleting the cytokine‐inducible SH2‐containing protein (CIS) gene with CRISPR‐Cas9 technique. This strategy enhanced CAR‐NK cell aerobic glycolysis because of increased Akt/mTOR together with c‐MYC signalling resulting in better antitumor activity. 68 These promising preclinical data were pivotal in the design of subsequent clinical trials testing the feasibility, safety and efficacy of CB‐derived CAR‐NK cells as cellular therapy for patients with B‐cell malignancies.
Multiple myeloma
Despite the impressive advances in the treatment of multiple myeloma (MM), it still remains an incurable disease. 69 As a result, the cellular therapy domain has been investigated and attempts have been made to develop CAR‐NK cells against MM.
CD138 (syndecan‐1) is a classical marker of plasma cells and highly expressed in MM. Its expression in MM cells is associated with enhanced proliferation, cell survival pathways and decreased apoptosis. 70 NK‐92MI cells have been modified by lentiviral transduction to express a first‐generation CD138‐targeting CAR, carrying a CD3ζ intracellular domain and a flexible CD8 hinge portion. These CD138‐targeting CAR‐NK cells showed enhanced anti‐MM activity in vitro and in xenograft mouse models. 71
Another candidate antigen for the treatment of MM is CS1 (SLAMF7 or CRACC), a surface glycoprotein highly expressed in plasma cells and MM. 72 Chu et al. have worked on the introduction of a CS1‐specific CAR into NK‐92 cells. Using a lentiviral gene delivery system, they have created a second‐generation CS1‐targeting CAR‐NK‐92 comprising CD3ζ together with a CD28 intracellular stimulatory domain. These armed NK‐92 cells showed directed cytotoxicity against CS1‐positive MM cells and inhibited tumor growth in a xenograft MM model. 73 However, one pitfall to this approach is that normal lymphocytes and NK cells also express CS1, which can result in fratricide not only in vitro but also in vivo, limiting NK cell numbers and their anti‐myeloma activity.
T‐cell malignancies
T‐cell malignancies, including peripheral T‐cell lymphoma and T‐cell acute lymphoblastic leukaemia (T‐ALL), are haematologic malignancies with limited therapeutic options. 74 Adoptive immune cell therapy with NK‐CAR engineering is a potential option in the therapeutic arsenal of these patients. Normal and malignant T cells express very specific antigens, such as CD3 and CD5, which are not expressed on NK cells and hence can be targeted by CAR‐NK cells.
So far, three third‐generation CAR NK cells have been investigated for the treatment of T‐cell malignancies. Chen et al. developed a CD5‐directed CAR NK‐92 cell possessing a CD8 hinge and transmembrane portions. Its intracellular element was composed of CD3ζ together with a 4‐1BB and a CD28 costimulatory domains. 75 Using this same construct backbone, this group created a third‐generation CD3‐targeting CAR NK‐92 cells. 76 These cells were transduced using a lentiviral system. 75 , 76 Lastly, You et al. 77 have developed a CD7‐targeting CAR NK‐92 cells also bearing a CD3ζ intracellular domain together with a 4‐1BB and a CD28 costimulatory moieties. As opposed to the two previously described CAR NK‐92 cells, these CD7‐specific NK‐92 cells possess a Fc hinge and NK‐92 that were transfected by electroporation. 77 The CD5‐, CD3‐ and CD7‐specific NK‐92 cells showed in vitro cytotoxicity against T‐ALL and T‐cell lymphoma cells and outperformed non‐transduced NK‐92 cells in a T‐cell leukaemia mouse xenograft model. 75 , 76 , 77 CAR‐NK cells might be more suitable than CAR T cells for therapy of T‐cell malignancies because of their lack of expression of T‐cell antigens and consequently no fratricide.
Solid tumors
Unfortunately, despite the remarkable success of CAR T cells in the treatment of haematologic malignancies, similar outcomes have not been observed in the therapy of solid tumors with modified T cells. These poor results are most likely because of the lack of adequate homing capacity together with the adverse immunosuppressive solid tumor microenvironment. In this context, several investigators have been researching the potential of CAR‐NK cell therapy against solid malignancies such as glioblastoma, melanoma, breast, ovarian and prostate cancers (Table 2).
Glioblastoma (GBM), a highly lethal primary brain tumor, expresses epidermal growth factor (EGFR), which is implicated in tumor proliferation and migration. 78 This receptor can be detected at very low levels in normal brain tissue, and its gene amplification has been well characterised in the setting of GBM. 79 , 80 However, 30–40% of patients with GBM carry a mutant self‐active variant of EGFR, named EGFRvIII, which increases tumor aggressiveness. 80 Both isoforms, EGFR and EGFRvIII, have been explored as antigen targets for CAR‐directed therapy. Han et al. developed a second‐generation CAR bearing a CD3ζ signalling and CD28 costimulatory domains together with an scFv region targeting both EGFR and EGFRvIII. 48 These EGFR‐directed NK cells showed enhanced in vitro cytotoxicity, and their intracranial injection in an orthotopic mouse model resulted in improved tumor control and increased survival. 48 Since both EGFR and its mutant form EGFRvIII have variable expression in GBM, Gressler et al. tested a novel dual‐targeting CAR construct targeted both EGFR and EGFRvIII and showed that NK‐92 cells expressing this CAR have superior antitumor cytotoxicity compared to their single targeting counterpart. 81 Murakami et al. introduced a third‐generation EGFRvIII‐CAR into the KHYG‐1 NK cell line. Their EGFRvIII‐CAR KHYG‐1 NK cells carried a CD3ζ, CD28 and 4‐1BB intracellular domains and induced apoptosis in EGFRvIII‐positive GBM cells. 82 Using a different NK cell line (YTS NK cell line), Müller et al. generated an EGFRvIII‐targeting NK cells with a DAP12 intracellular stimulatory domain. In addition, they also induced a constitutive expression of CXCR4 in these EGRFvIII‐CAR NK cells to improve their homing to the tumor. With this approach, they achieved tumor‐specific chemotaxis and directed tumor cytotoxicity. 83 Lastly, the growth factor receptor tyrosine kinase Erb2 (HER2) is overexpressed in GBM tissue 84 and has been explored as an alternative target for the treatment of patients with GBM. Zhang et al. introduced a second‐generation HER2‐directed CAR carrying CD3ζ in addition to a CD28 intracellular moiety. Their HER2‐CAR NK‐92 cells displayed enhanced cytotoxicity not only in vitro but also in xenograft mouse models. 85 In all these preclinical GBM studies, lentivirus vectors were used for NK cell transduction. Hence, intracranially injected antigen‐directed CAR‐NK cells seem to be a promising option for the treatment of GBM.
Breast cancer is the most common malignancy among females in the United States. Not surprisingly, CAR‐NK cells have been investigated as an alternative therapeutic approach for this disease. Targeting diverse antigens, three breast cancer‐directed CARs carrying a CD3ζ intracellular domain together with a CD28 costimulatory moiety were introduced into NK‐92 cells. In this setting, the epithelial cell adhesion molecule (EpCAM) has been explored as a target antigen being overexpressed in carcinomas. 86 Sahm et al. generated EpCAM‐targeting NK‐92 cells, and the svFc element of their CAR was linked to a CD8 hinge followed by a CD28 transmembrane domain. Moreover, their NK‐92 cells were armed with a membrane‐bound IL‐15 molecule capable of trans‐binding to the IL‐15 receptor on the cell surface to improve NK cell proliferation and persistence. These genetically modified NK cells showed enhanced tumor killing in vitro. 87 On the other hand, Liu H et al. developed a second‐generation HER2‐directed CAR for the treatment of breast cancer. Their modified NK‐92 cells demonstrated specific in vitro cytotoxicity and, when injected into a xenograft breast cancer mouse model, these animals showed prolonged survival. 88 Chen et al. created a second‐generation CAR with an scFv portion specific to both EGFR and EGFRvIII. Using a xenograft mouse model of breast cancer brain metastasis, they combined the intracranial injection of both their armed NK‐92 cells and an oncolytic virus to enhance tumor immunogenicity and consequently NK cell activation. Delayed tumor growth was observed with the combination of these two treatments. 89 Different from the previous groups, Liu et al. created a third‐generation EGFR‐directed CAR carrying a CD3ζ portion together with CD28 and 4‐1BB costimulatory domains for the treatment of triple‐negative breast cancer. Their CAR had a CD8 hinge linked to a CD28 transmembrane element. These EGFR‐targeting CAR NK cells were directly injected into the tumor in both a cell line‐derived and a patient's tumor‐derived breast cancer mouse model, and they were able to better control tumor growth than in controls. 90 Finally, Hu et al. developed an innovative CAR‐NK cell approach for the treatment of triple‐negative breast cancer, which consisted of an scFv portion targeting tissue factor (TF) on cancer cells. This TF‐specific CAR carried a CD28 transmembrane domain linked either to an IgG hinge or directly to the scFv element. Intracellular stimulatory domains comprised CD3ζ, CD28 and 4‐1BB moieties. These TF‐directed CAR‐NK‐92MI cells hampered tumor growth in a breast cancer xenograft mouse model. 91
Additionally, some investigators have turned their attention to the investigation of CAR‐NK‐based therapies for the treatment of ovarian cancer. Uherek et al. developed first‐generation CAR NK‐92 cells directed to HER2, carrying a CD8 hinge and a CD3ζ intracellular stimulatory domain. These cells showed cytotoxicity specific to HER2‐positive cells and controlled tumor growth in mouse xenograft models. 92 Li et al created human mesothelin‐targeting NK cells for the treatment of ovarian cancer. They initially screened different combinations of this scFv domain with diverse transmembrane and intracellular domains by introducing the CARs into NK‐92 cells. The transmembrane elements investigated were NKG2D, NKp44, NKp46, CD16 and CD28, while the stimulatory domains studied were CD3ζ, 4‐1BB, 2B4, DAP10 and DAP12. The best CAR‐NK cell performers had NKG2D transmembrane domain combined with CD3ζ and 2B4 stimulatory moieties, and two of them carried either an additional 4‐1BB or DAP10. Further investigations introduced the three most cytotoxic CARs into iPSC‐derived NK cells. iPSC‐derived NK cells carrying the combination of NKG2D transmembrane domain in addition to CD3ζ and 2B4 showed better performance in vitro and, when tested in animal models, showed enhanced antitumor activity. 54
In summary, innovative CAR‐NK cell strategies have been investigated for the treatment of solid malignancies. Hopefully, preclinical data will be translated to the clinic to improve the therapeutic arsenal of patients with non‐haematologic cancers.
CAR‐NK cells in the clinic
Given the impressive outcomes of CD19‐targeted T‐cell therapy in patients with B‐cell malignancies, 6 , 7 , 8 seven phase I/phase II trials are currently investigating CAR NK cell treatment in this patient population. Three of these studies, not yet recruiting, were designed to target CD19 or CD22 (or both) in patients with relapsed or refractory B‐cell lymphoma (trials NCT03692767, NCT03690310, NCT03824964 and NCT04639739). The source of NK cells, signalling domains and vectors used for these studies are not disclosed. Two other trials, one completed (NCT00995137) and the second suspended for interim review (NCT01974479), are investigating the infusion of haploidentical second‐generation CAR NK cells targeting CD19 and possessing CD3ζ and 4‐1BB signalling domains in patients with relapsed or refractory B‐ALL. In the latter, electroporation was used to introduce the plasmid into expanded NK cells. Finally, one study using an iPSC‐derived CAR NK cell product targeting CD19, bearing 2B4‐CD3ζ intracellular domains and expressing a high‐affinity non‐cleavable CD16 to enhance ADCC potential along with a recombinant fusion of IL‐15 and IL‐15 receptor alpha to improve persistence, 67 is now recruiting patients (NCT04245722).
There are also clinical trials testing CAR NK cells for patients with solid tumors. In this setting, a dose‐escalation phase I study is recruiting patients with metastatic solid tumors to investigate the safety of autologous or allogeneic NKG2D ligand‐targeted CAR‐NK cells transfected via mRNA electroporation (NCT03415100). Additional trials are exploring the infusion of CAR NK cells targeting distinct tumor antigens such as Muc1 for multiple solid cancers (NCT02839954), PSMA for castrate‐resistant prostate cancer (NCT03692663), mesothelin for epithelial ovarian cancer (NCT03692637) and ROBO1 for various solid tumors (NCT03940820 and NCT03941457). The vectors used to transduce NK cells in these studies were not disclosed.
The first‐in‐human clinical trial of CB‐derived CAR‐NK cells to treat patients with relapsed or refractory B‐cell haematologic malignancies (NCT03056339) was led by our group. 23 In this dose‐escalation phase I/phase II clinical study, we are evaluating the safety and efficacy of CB‐NK cells modified to express anti‐CD19CAR, IL‐15 and inducible caspase‐9 (anti‐CD19CAR.IL15.iCas9). Eleven heavily pretreated patients with CD19‐positive cancers, including non‐Hodgkin lymphomas (NHL) and chronic lymphocytic leukaemia (CLL), have been treated with a single infusion of CAR NK cells following lymphodepletion chemotherapy. The overall response rate was 73% (8 of 11 patients) including 64% (7 of 11 patients) with complete responses and one case with resolution of Richter's transformation but persistence of CLL. Response durations could not be assessed owing to the study design as patients were allowed to receive consolidation such as a transplant once they had achieved a response. In contrast to large trials of CD19‐directed CAR T‐cell therapy (Table 1), none of our patients showed evidence of neurotoxicity, CRS, haemophagocytic lymphohistiocytosis or GVHD, nor were any admitted to the intensive care unit because of toxicities related to CAR‐NK cell treatment. A quantitative real‐time polymerase chain reaction assay, used to measure the expansion and persistence of CAR‐NK cells, detected these cells for at least 12 months in some patients. 23 These encouraging results ratify the clinical feasibility, short‐term efficacy and safety of CAR‐engineered CB‐NK cell therapy.
Some clinical trials are currently testing CAR‐modified NK‐92 cells for the treatment of both haematologic and solid cancers. In the haematologic cancers, CAR NK‐92 cells targeting BCMA, CD7, CD19 and CD33 are being investigated as a therapeutic option for patients with multiple myeloma (NCT03940833), CD7‐positive haematologic malignancies (NCT02742727), CD19‐positive lymphoid malignancies (NCT02892695) and CD33‐positive acute myeloid leukaemias (NCT02944162). All are single‐armed unblinded phase 1 and phase 2 trials for patients with relapsed or refractory diseases. Both the CD7‐ and the CD33‐targeting CAR‐NK cells have CD28, 4‐1BB and CD3ζ as their signalling domains. One phase 2 open‐labelled clinical trial in the solid tumor domain is recruiting patients with HER2‐positive neuroblastoma to receive intracranial injections of HER2‐directed CAR NK‐92 cells carrying CD3ζ and CD28 signalling components (NCT03383978).
In summary, the preclinical and early clinical data described above are promising and support the application of NK cells as a useful platform for CAR engineering to target a range of malignancies.
Conclusions and future directions
Genetically engineered immune effector cells equipped with CARs are potent additions to the immunotherapeutic arsenal against cancer. CAR T cells have produced remarkable clinical results in patients with relapsed or refractory B‐cell malignancies and multiple myeloma and are beginning to make inroads against solid tumors. Nonetheless, this therapeutic strategy has a number of limitations that underscore the need to identify alternative sources of immune cells, not only to overcome the safety issues, but also to lower the cost of such therapies and increase their accessibility to patients. Among the top candidates in the search, NK cells are especially promising. They have proved safe, technically feasible and potent in patients with lymphoid malignancies although a number of questions remain to be elucidated in future studies, including (1) the durability of the response, (2) the susceptibility of these cells to different suppressive elements in the tumor microenvironment, (3) their ability to overcome immune checkpoints and (4) their long‐term memory following immune encounters with tumor. Nevertheless, advances in engineering and gene‐editing techniques and new insights from the field of immunometabolism promise to address some of these issues and put CAR‐NK cells on the map as a clinical option for the treatment of haematologic and solid malignancies.
Conflict of interest
KR, MD and The University of Texas MD Anderson Cancer Center (MDACC) have an institutional financial conflict of interest with Takeda Pharmaceutical for the licensing of the technology related to CAR‐NK cell research reported here. MD Anderson has implemented an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MDACC's conduct of any other ongoing or future research related to this relationship. KR and The University of Texas MD Anderson Cancer Center have an institutional financial conflict of interest with Affimed GmbH. Because MD Anderson is committed to the protection of human subjects and the effective management of its financial conflicts of interest in relation to its research activities, MD Anderson is implementing an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MD Anderson's conduct of any other ongoing or future research related to this relationship. KR participates on Scientific Advisory Board for GEMoaB, AvengeBio, Kiadis, GSK and Bayer.
Author contributions
May Daher: Conceptualization; Writing‐original draft; Writing‐review & editing. Luciana Melo Garcia: Conceptualization; Writing‐original draft; Writing‐review & editing. Ye Li: Conceptualization; Writing‐original draft; Writing‐review & editing. Katayoun Rezvani: Conceptualization; Funding acquisition; Writing‐original draft; Writing‐review & editing.
Acknowledgments
This work was supported in part by the generous philanthropic contributions to The University of Texas MD Anderson Cancer Center Moon Shots Program, by grants from Stand Up To Cancer (SU2C‐AACR‐DT29‐19), CPRIT (RP160693), the National Institute of Health, National Cancer Institute (1 R01 CA211044‐01, 5 P01CA148600‐03, P50CA100632‐16 and 5P50CA127001‐12) and the Cancer Center Support (CORE) Grant (CA016672) that support the Flow Cytometry and Cellular Imaging Facility at MD Anderson Cancer Center.
References
- 1. Kalos M, Levine BL, Porter DL et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3: 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cerrano M, Ruella M, Perales M‐A et al. The advent of CAR T‐cell therapy for lymphoproliferative neoplasms: integrating research into clinical practice. Front Immunol 2020; 11: 888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med 2014; 65: 333–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med 2018; 379: 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schuster SJ, Bishop MR, Tam CS et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B‐cell lymphoma. N Engl J Med 2019; 380: 45–56. [DOI] [PubMed] [Google Scholar]
- 6. Maude SL, Laetsch TW, Buechner J et al. Tisagenlecleucel in children and young adults with B‐cell lymphoblastic leukemia. N Engl J Med 2018; 378: 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Locke FL, Ghobadi A, Jacobson CA et al. Long‐term safety and activity of axicabtagene ciloleucel in refractory large B‐cell lymphoma (ZUMA‐1): a single‐arm, multicentre, phase 1–2 trial. Lancet Oncol 2019; 20: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang M, Munoz J, Goy A et al. KTE‐X19 CAR T‐cell therapy in relapsed or refractory Mantle‐cell lymphoma. N Engl J Med 2020; 382: 1331–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abramson JS, Palomba ML, Gordon LI et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B‐cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 2020; 396: 839–852. [DOI] [PubMed] [Google Scholar]
- 10. Chou CK, Turtle CJ. Assessment and management of cytokine release syndrome and neurotoxicity following CD19 CAR‐T cell therapy. Expert Opin Biol Ther 2020; 20: 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tyagarajan S, Spencer T, Smith J. Optimizing CAR‐T cell manufacturing processes during pivotal clinical trials. Mol Ther Methods Clin Dev 2020; 16: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rezvani K. Adoptive cell therapy using engineered natural killer cells. Bone Marrow Transplant 2019; 54(Suppl 2): 785–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nat Rev Immunol 2018; 18: 485–497. [DOI] [PubMed] [Google Scholar]
- 14. Samelson LE, Klausner RD. The T‐cell antigen receptor. Structure and mechanism of activation. Ann NY Acad Sci 1988; 540: 1–3. [DOI] [PubMed] [Google Scholar]
- 15. Pazina T, Shemesh A, Brusilovsky M, Porgador A, Campbell KS. Regulation of the functions of natural cytotoxicity receptors by interactions with diverse ligands and alterations in splice variant expression. Front Immunol 2017; 8: 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prager I, Watzl C. Mechanisms of natural killer cell‐mediated cellular cytotoxicity. J Leukoc Biol 2019; 105: 1319–1329. [DOI] [PubMed] [Google Scholar]
- 17. Anwer F, Shaukat A‐A, Zahid U et al. Donor origin CAR T cells: graft versus malignancy effect without GVHD, a systematic review. Immunotherapy 2017; 9: 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mohty M, Dulery R, Gauthier J et al. CAR T‐cell therapy for the management of refractory/relapsed high‐grade B‐cell lymphoma: a practical overview. Bone Marrow Transplant 2020; 55: 1525–1532. [DOI] [PubMed] [Google Scholar]
- 19. Ruggeri L, Capanni M, Urbani E et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002; 295: 2097–2100. [DOI] [PubMed] [Google Scholar]
- 20. Olson JA, Leveson‐Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010; 115: 4293–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shah N, Li LI, McCarty J et al. Phase I study of cord blood‐derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol 2017; 177: 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brudno JN, Kochenderfer JN. Recent advances in CAR T‐cell toxicity: mechanisms, manifestations and management. Blood Rev 2019; 34: 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu E, Marin D, Banerjee P et al. Use of CAR‐transduced natural killer cells in CD19‐positive lymphoid tumors. N Engl J Med 2020; 382: 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Freud AG, Mundy‐Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity 2017; 47: 820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Strauss‐Albee DM, Fukuyama J, Liang EC et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci Transl Med 2015; 7: 297ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Y, Wallace DL, de Lara CM et al. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology 2007; 121: 258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dogra P, Rancan C, Ma W et al. Tissue determinants of human NK cell development, function, and residence. Cell 2020; 180: 749–763.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yun HD, Varma A, Hussain MJ, Nathan S, Brunstein C. Clinical relevance of immunobiology in umbilical cord blood transplantation. J Clin Med 2019; 8: 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Horowitz A, Strauss‐Albee DM, Leipold M et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med 2013; 5: 208ra145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li LI, Chen H, Marin D et al. A novel immature natural killer cell subpopulation predicts relapse after cord blood transplantation. Blood Adv 2019; 3: 4117–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Romee R, Rosario M, Berrien‐Elliott MM et al. Cytokine‐induced memory‐like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med 2016; 8: 357ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu H, Blum RH, Bjordahl R et al. Pluripotent stem cell‐derived NK cells with high‐affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020; 135: 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luevano M, Daryouzeh M, Alnabhan R et al. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum Immunol 2012; 73: 248–257. [DOI] [PubMed] [Google Scholar]
- 34. Liu E, Ang SOT, Kerbauy L et al. GMP‐compliant universal antigen presenting cells (uAPC) promote the metabolic fitness and antitumor activity of armored cord blood CAR‐NK cells. Front Immunol 2021. 10.3389/fimmu2021626098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu E, Tong Y, Dotti G et al. Cord blood NK cells engineered to express IL‐15 and a CD19‐targeted CAR show long‐term persistence and potent antitumor activity. Leukemia 2018; 32: 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dolstra H, Roeven MWH, Spanholtz J et al. Successful transfer of umbilical cord blood CD34+ hematopoietic stem and progenitor‐derived NK cells in older acute myeloid leukemia patients. Clin Cancer Res 2017; 23: 4107–4118. [DOI] [PubMed] [Google Scholar]
- 37. Spanholtz J, Tordoir M, Eissens D et al. High log‐scale expansion of functional human natural killer cells from umbilical cord blood CD34‐positive cells for adoptive cancer immunotherapy. PLoS One 2010; 5: e9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spanholtz J, Preijers F, Tordoir M et al. Clinical‐grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed‐system culture process. PLoS One 2011; 6: e20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cany J, van der Waart AB, Tordoir M et al. Natural killer cells generated from cord blood hematopoietic progenitor cells efficiently target bone marrow‐residing human leukemia cells in NOD/SCID/IL2Rg(null) mice. PLoS One 2013; 8: e64384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lehmann D, Spanholtz J, Osl M et al. Ex vivo generated natural killer cells acquire typical natural killer receptors and display a cytotoxic gene expression profile similar to peripheral blood natural killer cells. Stem Cells Dev 2012; 21: 2926–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De Oliveira SN, Ryan C, Giannoni F et al. Modification of hematopoietic stem/progenitor cells with CD19‐specific chimeric antigen receptors as a novel approach for cancer immunotherapy. Hum Gene Ther 2013; 24: 824–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gong JHMG, Klingemann HG. Characterization of a human cell line (NK‐92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994; 8: 652–658. [PubMed] [Google Scholar]
- 43. Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy – advantages of the NK‐92 cell line over blood NK cells. Front Immunol 2016; 7: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tonn T, Schwabe D, Klingemann HG et al. Treatment of patients with advanced cancer with the natural killer cell line NK‐92. Cytotherapy 2013; 15: 1563–1570. [DOI] [PubMed] [Google Scholar]
- 45. Snyder KM, Hullsiek R, Mishra HK et al. Expression of a recombinant high affinity IgG Fc receptor by engineered NK cells as a docking platform for therapeutic mAbs to target cancer cells. Front Immunol 2018; 9: 2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Robertson MJ, Cochran KJ, Cameron C, Le JM, Tantravahi R, Ritz J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp Hematol 1996; 24: 406–415. [PubMed] [Google Scholar]
- 47. Yagita M, Huang CL, Umehara H et al. A novel natural killer cell line (KHYG‐1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia 2000; 14: 922–930. [DOI] [PubMed] [Google Scholar]
- 48. Han J, Chu J, Keung Chan W et al. CAR‐engineered NK cells targeting wild‐type EGFR and EGFRvIII enhance killing of glioblastoma and patient‐derived glioblastoma stem cells. Sci Rep 2015; 5: 11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell‐derived NK cells acquire functional receptors and cytolytic activity. J Immunol 2005; 175: 5095–5103. [DOI] [PubMed] [Google Scholar]
- 50. Woll PS, Grzywacz B, Tian X et al. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood 2009; 113: 6094–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ni Z, Knorr DA, Kaufman DS. Hematopoietic and nature killer cell development from human pluripotent stem cells. Methods Mol Biol 2013; 1029: 33–41. [DOI] [PubMed] [Google Scholar]
- 52. Cichocki F, Bjordahl R, Gaidarova S et al. iPSC‐derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti‐PD‐1 therapy. Sci Transl Med 2020; 12: eaaz5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ni Z, Knorr DA, Bendzick L, Allred J, Kaufman DS. Expression of chimeric receptor CD4zeta by natural killer cells derived from human pluripotent stem cells improves in vitro activity but does not enhance suppression of HIV infection in vivo . Stem Cells 2014; 32: 1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC‐derived natural killer cells engineered with chimeric antigen receptors enhance anti‐tumor activity. Cell Stem Cell 2018; 23: 181–192.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bernareggi D, Pouyanfard S, Kaufman DS. Development of innate immune cells from human pluripotent stem cells. Exp Hematol 2019; 71: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Goldenson BH, Zhu H, Wang YM et al. Umbilical cord blood and iPSC‐derived natural killer cells demonstrate key differences in cytotoxic activity and KIR profiles. Front Immunol 2020; 11: 561553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Perna F, Berman SH, Soni RK et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell 2017; 32: 506–519.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Parker KR, Migliorini D, Perkey E et al. Single‐cell analyses identify brain mural cells expressing CD19 as potential off‐tumor targets for CAR‐T immunotherapies. Cell 2020; 183: 126–142.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Müller T, Uherek C, Maki G et al. Expression of a CD20‐specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK‐resistance of lymphoma and leukemia cells. Cancer Immunol Immunother 2008; 57: 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Romanski A, Uherek C, Bug G et al. CD19‐CAR engineered NK‐92 cells are sufficient to overcome NK cell resistance in B‐cell malignancies. J Cell Mol Med 2016; 20: 1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Boissel L, Betancur M, Wels WS, Tuncer H, Klingemann H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res 2009; 33: 1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Oelsner S, Waldmann A, Billmeier A et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3‐positive B‐ALL and inhibit in vivo leukemia growth. Int J Cancer 2019; 145: 1935–1945. [DOI] [PubMed] [Google Scholar]
- 63. Tassev DV, Cheng M, Cheung NK. Retargeting NK92 cells using an HLA‐A2‐restricted, EBNA3C‐specific chimeric antigen receptor. Cancer Gene Ther 2012; 19: 84–100. [DOI] [PubMed] [Google Scholar]
- 64. Quintarelli C, Sivori S, Caruso S et al. Efficacy of third‐party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B‐cell precursor acute lymphoblastic leukemia. Leukemia 2020; 34: 1102–1115. [DOI] [PubMed] [Google Scholar]
- 65. Müller S, Bexte T, Gebel V et al. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19‐specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front Immunol 2019; 10: 3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Herrera L, Santos S, Vesga MA et al. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci Rep 2019; 9: 18729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Goodridge JP, Mahmood S, Zhu H et al. FT596: translation of first‐of‐kind multi‐antigen targeted off‐the‐shelf CAR‐NK cell with engineered persistence for the treatment of B cell malignancies. Blood 2019; 134(Suppl 1): 301. [Google Scholar]
- 68. Daher M, Basar R, Gokdemir E et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR‐NK cells. Blood 2020; 137: 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mikkilineni L, Kochenderfer JN. CAR T cell therapies for patients with multiple myeloma. Nat Rev Clin Oncol 2021; 18: 71–84. [DOI] [PubMed] [Google Scholar]
- 70. Akhmetzyanova I, McCarron MJ, Parekh S, Chesi M, Bergsagel PL, Fooksman DR. Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia 2020; 34: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jiang H, Zhang W, Shang P et al. Transfection of chimeric anti‐CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol 2014; 8: 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hsi ED, Steinle R, Balasa B et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res 2008; 14: 2775–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chu J, Deng Y, Benson DM et al. CS1‐specific chimeric antigen receptor (CAR)‐engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014; 28: 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McMahon CM, Luger SM. Relapsed T cell ALL: current approaches and new directions. Curr Hematol Malig Rep 2019; 14: 83–93. [DOI] [PubMed] [Google Scholar]
- 75. Chen KH, Wada M, Pinz KG et al. Preclinical targeting of aggressive T‐cell malignancies using anti‐CD5 chimeric antigen receptor. Leukemia 2017; 31: 2151–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen KH, Wada M, Firor AE et al. Novel anti‐CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 2016; 7: 56219–56232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. You F, Wang Y, Jiang L et al. A novel CD7 chimeric antigen receptor‐modified NK‐92MI cell line targeting T‐cell acute lymphoblastic leukemia. Am J Cancer Res 2019; 9: 64–78. [PMC free article] [PubMed] [Google Scholar]
- 78. Oprita A, Baloi S‐C, Staicu G‐A et al. Updated insights on EGFR signaling pathways in glioma. Int J Mol Sci 2021; 22: 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Brennan C, Verhaak R, McKenna A et al. The somatic genomic landscape of glioblastoma. Cell 2013; 155: 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Parsons DW, Jones S, Zhang X et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321: 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Genßler S, Burger MC, Zhang C et al. Dual targeting of glioblastoma with chimeric antigen receptor‐engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016; 5: e1119354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Murakami T, Nakazawa T, Natsume A et al. Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells. Anticancer Res 2018; 38: 5049–5056. [DOI] [PubMed] [Google Scholar]
- 83. Muller N, Michen S, Tietze S et al. Engineering NK cells modified with an EGFRvIII‐specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF‐1alpha‐secreting glioblastoma. J Immunother 2015; 38: 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Koka V, Potti A, Forseen SE et al. Role of Her‐2/neu overexpression and clinical determinants of early mortality in glioblastoma multiforme. Am J Clin Oncol 2003; 26: 332–335. [DOI] [PubMed] [Google Scholar]
- 85. Zhang C, Burger MC, Jennewein L et al. ErbB2/HER2‐specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst 2016; 108: djv375. [DOI] [PubMed] [Google Scholar]
- 86. Patriarca C, Macchi RM, Marschner AK, Mellstedt H. Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treat Rev 2012; 38: 68–75. [DOI] [PubMed] [Google Scholar]
- 87. Sahm C, Schonfeld K, Wels WS. Expression of IL‐15 in NK cells results in rapid enrichment and selective cytotoxicity of gene‐modified effectors that carry a tumor‐specific antigen receptor. Cancer Immunol Immunother 2012; 61: 1451–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Liu H, Yang B, Sun T et al. Specific growth inhibition of ErbB2‐expressing human breast cancer cells by genetically modified NK92 cells. Oncol Rep 2015; 33: 95–102. [DOI] [PubMed] [Google Scholar]
- 89. Chen X, Han J, Chu J et al. A combinational therapy of EGFR‐CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016; 7: 27764–27777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu Y, Zhou Y, Huang K‐H et al. Targeting epidermal growth factor‐overexpressing triple‐negative breast cancer by natural killer cells expressing a specific chimeric antigen receptor. Cell Prolif 2020; 53: e12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hu Z. Tissue factor as a new target for CAR‐NK cell immunotherapy of triple‐negative breast cancer. Sci Rep 2020; 10: 2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Uherek C, Tonn T, Uherek B et al. Retargeting of natural killer‐cell cytolytic activity to ErbB2‐expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002; 100: 1265–1273. [PubMed] [Google Scholar]