

Abstract
Fluid accumulation in the abdominal cavity is commonly found in advanced-stage ovarian cancer patients, which creates a specialized tumor microenvironment for cancer progression. Using single-cell RNA sequencing (scRNA-seq) of ascites cells from 5 patients with ovarian cancer, we identified 7 cell types, including heterogeneous macrophages and ovarian cancer cells. We resolved a distinct polarization state of macrophages by MacSpectrum analysis and observed subtype-specific enrichment of pathways associated with their functions. The communication between immune and cancer cells was predicted through a putative ligand-receptor pair analysis using NicheNet. We found that CCL5, a chemotactic ligand, is enriched in immune cells (T cells and NK cells) and mediates ovarian cancer cell survival in the ascites, possibly through SDC4. Moreover, SDC4 expression correlated with poor overall survival in ovarian cancer patients. Our study highlights the potential role of T cells and NK cells in long-term survival in patients with ovarian cancer, indicating SDC4 as a potential prognostic marker in ovarian cancer patients.
Keywords: Ascites, Single-cell RNA-seq, Intercellular communication, Ovarian cancer, Tumor microenvironment
1. INTRODUCTION
Ovarian cancer is the most lethal type of gynecological malignancy with a five-year survival rate below 50%.1 For the past three decades, there has been only a slight increase in the ovarian cancer survival outcome. Early detection is difficult because no specific symptoms of ovarian cancer as well as no effective detection markers with high accuracy are available. In addition to the fact that the vast majority of patients are diagnosed at advanced stages, the current gold standard of care for advanced ovarian cancer is a primary debulking surgery followed by taxol/platinum-based chemotherapy, which has not changed for many years. Patients are treated irrespective of histologic subtypes and genetic background, and chemoresistance is highly prevalent in this cancer.2 With the recent advances of sequencing at single-cell resolution, our understanding of molecular mechanisms underlying cancer progression has been significantly enhanced. Accumulating evidence supports the importance of tumor microenvironment (TME) in facilitating the oncogenic behavior of cancer cells, but its precise role in cancer progression remains poorly understood. A deeper understanding of interactions between diverse cells in the cancer tissue may allow the identification of novel diagnostic biomarkers and therapeutic targets of ovarian cancer.
The major route of metastasis in ovarian cancer is transcoelomic dissemination causing the accumulation of fluid, malignant ascites.3 Malignant ascites could act as a vehicle and reservoir for ovarian cancer metastases to various secondary sites. Malignant ascites accumulates in the peritoneal cavity during ovarian cancer progression, providing a niche for disseminated cancer cells.4 The majority of patients in the advanced stage of the disease have malignant ascites, which is associated with poor survival.5 Therefore, it is of prime importance to understand the role of ascites TME in ovarian cancer progression.
In our previous studies, we focused on acelluar components (IL-6 and cholesterol) that are enriched in malignant ascites and their effects on ovarian cancer progression.6,7 In the current study, we postulated that cellular components within the malignant ascites could be highly heterogeneous and asked what types of cellular components and interactions are enriched in the ascites TME. To answer this question, we conducted a scRNA-seq on malignant ascites-derived cells isolated from an ovarian cancer patient with the advanced stage (stage IV). Our scRNA-seq data were analyzed along with previously reported data from Schelker et al. (2017)8 to dissect cellular heterogeneity and key interactions taking place in the malignant ascites. Understanding cellular heterogeneity and ligand-receptor interactions within malignant ascites may yield novel therapeutic targets for cancer metastasis and cell survival in ovarian cancer.
2. MATERIALS AND METHODS
2.1. Patient samples and sample processing
This study was performed after approval from the Institutional Review Board (IRB) of Seoul National University Hospital (IRB no. 1409-154-616). The study was conducted in compliance with the Declaration of Helsinki and informed consent was obtained from the subject. The malignant ascites (a25) was obtained from the 58-year-old high-grade serous ovarian cancer patient (stage IV). Isolation of cellular components from malignant ascites was described previously.9 Briefly, ascitic fluid was centrifuged at 1,400 × g for 10 min at 4°C. To remove erythrocytes, density-gradient centrifugation with Ficoll was performed. Isolated cells were stored in liquid nitrogen until further analysis.
2.2. scRNA-seq library preparation and data pre-processing
A total of 1,086 cells from a25 patient were sequenced to a read depth of ~120,000 per cell and run through the 10x Genomics cell ranger platform. Single-cell suspensions were prepared as outlined by the 10x Genomics Single Cell 3’ v2 Reagent kit user guide. In brief, single cell suspensions were loaded into a 10x Chromium Controller (10x Genomics, Pleasanton, CA, USA) and aimed for 5,000 cells. Following Gem capturing and lysis, cDNA was synthesized and amplified to construct sequencing libraries.
2.3. scRNA-seq Data analysis
The sequenced data were processed into expression matrices with the Cell Ranger Single Cell software 1.3.1 by 10x Genomics (http://10xgenomics.com/). The libraries were sequenced on the Illumina HiSeq 2500 platform, and sequencing reads were aligned with STAR to the human transcriptome using the human reference transcriptome GRCh38. From the gene expression matrix, the downstream analysis was carried out with R version 3.6.3 (2020-02-29). Quality control, data pre-processing, integration of multiple scRNA-seq datasets from two different technologies and visualization, and the differential expression analysis was carried out using Seurat version 3.1.5 package.10,11 For each individual dataset, genes expressed in less than 3 cells as well as cells with < 200 genes and any cells with > 10% UMIs mapped to mitochondrial genes were removed from the gene expression matrix. Based on these criteria, 26,669 genes across 7,343 samples remained for downstream analysis. We performed PCA on the gene expression matrix and used the first 15 principal components for clustering and visualization. Unsupervised shared nearest neighbor (SNN) clustering was performed with a resolution of 0.15 and we employed UMAP (uniform manifold approximation and projection) for dimensionality reduction and visualization. Differentially expressed genes were identified using Seurat’s “FindAllMarkers” to process all clusters combined with the MAST test12 with logFC threshold > 0.25 and expressed in at least 10% of the cells in the cluster. The expression profile of each cluster’s top 10 genes based on average log FC were visualized as a heatmap.
2.4. MacSpectrum analysis
A total of 1,868 differentially expressed genes between macrophage subclusters were selected and expression levels of each genes per cell were extracted to generate MPI and AMDI values using MacSpectrum analyzer Release v1.0.1 (https://macspectrum.uconn.edu/).13 MPI and AMDI values were added to the metadata slot of Seurat objects for further categorization of macrophage subtypes.
2.5. Pathway enrichment analysis
GSEA14 was applied to identify enriched pathways between the groups stated in Figure 3 and Supplementary Figure S2. We used hallmark gene sets (h.all.v7.1.symbols.gmt) from Molecular Signature Database (MSigDB, https://www.gsea-msigdb.org/gsea/downloads.jsp).15 Genes were ranked by the average log fold change and P value was calculated using pre-ranked gene set enrichment analysis method fgsea (https://github.com/ctlab/fgsea)16 package with hundred thousand gene set permutations.
FIGURE 3.
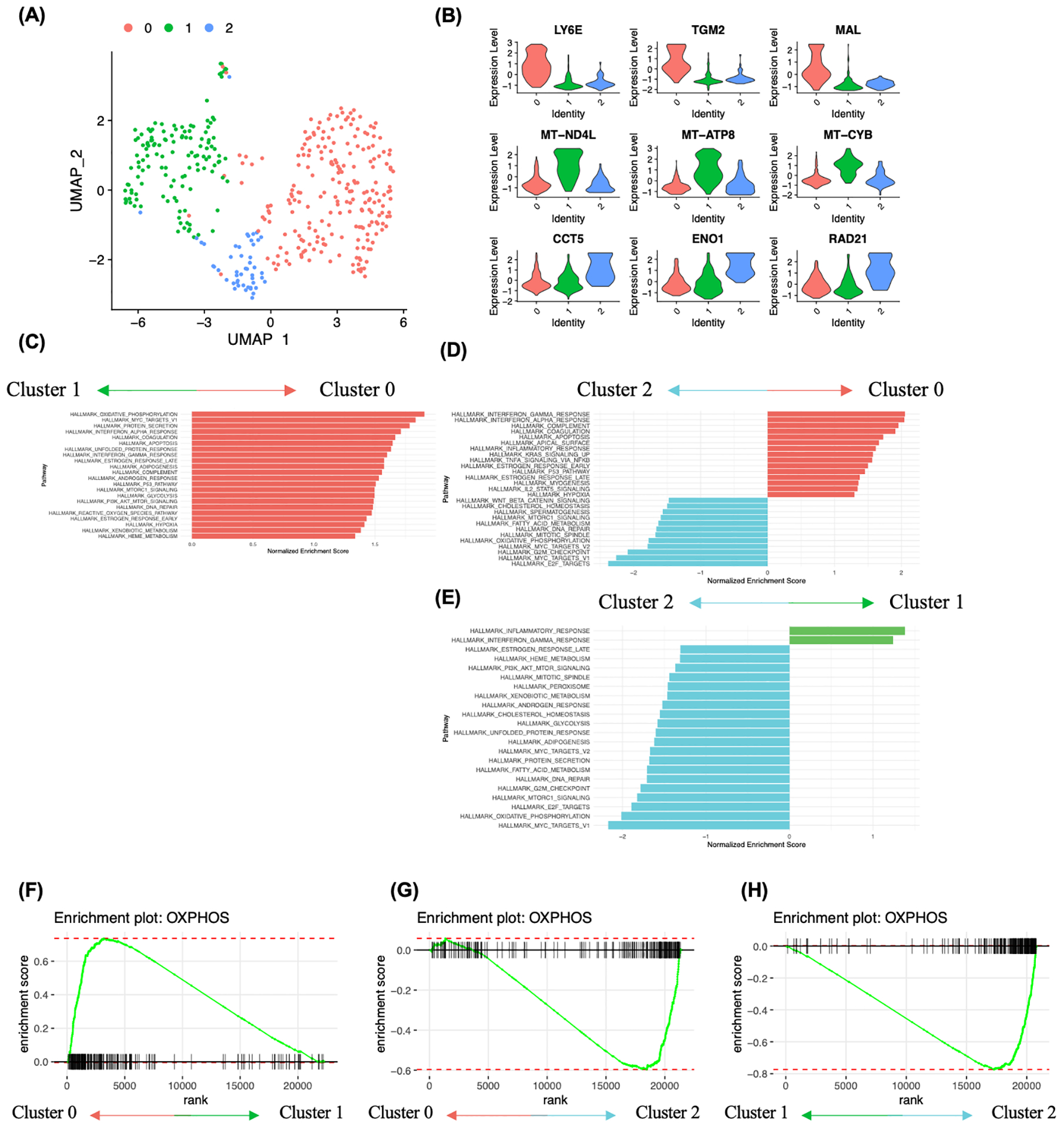
Analysis of cancer cell heterogeneity and enriched molecular pathways. A, Subclustering analysis of ovarian cancer cells identified 3 clusters. B, Ovarian cancer subcluster 0 showed high expression of stem cell related genes LY6E, TGM2 and MAL, subcluster 1 showed high expression of mitochondrial genes MT-ND4L, MT-ATP8 and MT-CYB, and subcluster 2 showed high expression of CCT5, ENO1 and RAD21. C-E, Gene set enrichment analysis between ovarian cancer cell subclusters using hallmark genesets. Pathway enrichment is expressed as Normalized Enrichment Score (NES), and the graphs display statistically significant pathways (FDR < 0.05). F-H, Enrichment plot showing OXPHOS heterogeneity in ovarian cancer cells. F, Enrichment plot of the gene set OXPHOS in cluster 0 vs cluster 1. G, Enrichment plot of the gene set OXPHOS in cluster 0 vs cluster 2. H, Enrichment plot of the gene set OXPHOS in cluster 1 vs cluster 2.
2.6. NicheNet analysis - ligand-receptor communication prediction model
To predict the potential ligand-receptor communication between ovarian cancer cells and niche cells in ascites TME, we used a pre-built prior model, the NicheNet method.17,18 In brief, we defined a ligand or receptor as expressed in a particular cell type when it is expressed in at least 30% of the cells in that cluster. To perform NichNet ligand activity analysis, we’ve defined the geneset of interest by combining two GO terms, negative regulation of anoikis (GO:2000811) and negative regulation of programmed cell death (GO:0043069) for anoikis related ligand-receptor communication prediction model and used all genes expressed in ovarian cancer cells as background of genes.
2.7. TCGA, GEO and GTEx dataset analysis
Patient prognosis depending on the expression of syndecan4 (SYND4, also known as SDC4) was analyzed using KM-plotter. For dichotomization of the sample based on SDC4 expression, ‘split patient by’ tab was set at ‘auto select best cutoff’. TCGA and GEO cohorts (GSE9891, GSE3149, GSE26193 and GSE63885) were utilized for the analysis (http://kmplot.com/analysis/).19 The Pearson correlation between expression of MET and SDC4 and the expression of SDC4 in normal and cancer tissues were analyzed using the GEPIA (Gene Expression Profiling Interactive Analysis) online database. The mRNA expression data from both TCGA (cancer) and GTEx (normal ovary) cohorts were incorporated (http://gepia.cancer-pku.cn).20 The cut-off for P value was set at 0.01. UALCAN is a web-based tool available at http://ualcan.path.uab.edu and was used to compare SDC4 expression in different stages of ovarian cancer.21 The GEO dataset, GSE85296, was used to analyze transcript levels of CCL5 (probe ID: 50545). This dataset contains transcript expression data from primary and metastatic samples of ovarian cancer.
2.8. Cell culture and reagent
Ovarian cancer cell lines, SKOV3, ES-2, PA1 (ATCC, Manassas, VA) and Kuramochi (JCRB Cell Bank, Japan) were grown in RPMI1640 medium (WELGENE, Seoul, Korea) supplemented with 10% FBS (Gibco, Gaithersburg, MD), 100 μg/mL streptomycin and 100 U/mL penicillin (Invitrogen, Carlsbad, CA). Cells were maintained in 5% CO2 humidified incubator at 37°C. All cell lines used in this study were authenticated and tested for mycoplasma. For non-adherent cell culture, cells were seeded in 10-cm ultra-low attachment (ULA; Corning, NY) culture plates at a density of 200,000 to 600,000 cells per plate. Recombinant human CCL5 was purchased from BioLegend (San Diego, CA).
2.9. Western blotting
Immunoblotting was performed as previously described. Briefly, cells were harvested and lysed with lysis buffer containing Tris-HCl, NaCl, EDTA, EGTA, 1% Triton X-100, sodium deoxycholate, EDTA-free protease inhibitor cocktail, phenylmethylsulfonyl fluoride and NA3VO4 for 20 min. Protein concentration was obtained using BCA Protein Assay Kit (Thermo Scientific, Waltham, MA), SDS-PAGE was performed with the 9 and 15% polyacrylamide gel. Separated proteins were then transferred onto nitrocellulose membrane. Membranes were blocked with 5% dry skim milk (w/v) in TBS-T for 1h. Membranes were incubated with specific primary and secondary antibodies. Signals were visualized using chemiluminescence detection kit (AbFrontier, Seoul, Korea). Primary antibodies used in this study are Syndecan 4 (1:500; #12236, Cell Signaling Technology, Beverly, MA), PARP-1 (1:1000; sc-7150, SantaCruz Biotechnology, Santa Cruz, CA) and α-tubulin (1:5000; T5168. Sigma-Aldrich, St. Louis, MO).
2.10. Statistics
Significant differences were defined at P < 0.05 or adj.P < 0.05, where applicable. Survival analysis by KM plotter was performed using a Kaplan-Meier and the hazard ratio with 95% confidence intervals, and logrank P values are computed. Pearson correlation coefficient (Pearson’s r) was used for co-expression test and automatically calculated by the GEPIA tool. Statistical significance was calculated by using GraphPad Prism 5 software for the correlation analysis when applicable.
3. RESULTS
3.1. Clustering of single-cell transcriptomic data identifies cellular heterogeneity of malignant ascites
To investigate the cellular populations of malignant ascites, scRNA-seq was conducted on malignant ascites-derived cells using the 10x Genomics Chromium platform. Malignant ascites (a25) from the high-grade serous patient (stage IV) was collected before the patient received chemotherapy and underwent surgery at Seoul National University Hospital (SNUH). After quality control, 1,082 cells were retained for analysis. Mean reads per cell were 118,862 and median genes per cell was 3,109. scRNA-seq data from a25 and data from a previous study8 (7873M, 7882M, 7892M, 7890M2) were incorporated for the subsequent analysis. From ascites samples, 7,343 cells were analyzed. To eliminate the batch-specific variations between different datasets, batch-correction was executed prior to the subsequent analysis (Figure 1A, left panel). For comprehensive visualization of transcriptomic likeness between cells from the five ascites samples, UMAP analysis was performed with the batch-corrected dataset. Ten major clusters were revealed, indicating the existence of transcriptomically distinct cellular profiles within malignant ascites (Figure 1A, right panel).
FIGURE 1.

Identification of cellular components of ovarian cancer patient-derived ascites. A, Batch corrected unsupervised clustering of ascites cellular components. UMAP dimensional reduction analysis identified 10 major clusters. B-I, Expression of cluster defining genes. B-G, Immune cell signatures (PTPRC, CD3G, CD4, CD14, CD68, KLRB1, KLRF1, CD19, CD79A, CLEC4C, IL3RA). H-I non-immune cell signatures, ovarian cancer cell signatures (WFDC2, EPCAM) and fibroblast signatures (PDPN, THY1), visualized by Violin plots. J, UMAP plots of ascites cellular components from 5 patient samples with cell types labeled. K, Number of cells counted in each cell types.
We then used the expression of known genetic markers to identify cell types within the malignant ascites. Expression levels of T cell, macrophage, NK cell, B cell and dendritic cell signatures were investigated to characterize immune cells. Based on the expression level of the pan-immune marker, PTPRC (also known as CD45), the clusters fell into two large groups, immune-related and non-immune related clusters, with the latter expressing mostly ovarian cancer and fibroblast signatures. We identified 8 distinct immune cell clusters including a T cell cluster (cluster 0 highly expressing CD3G; while low level of CD4+ cells was found), four macrophage clusters (clusters 1, 2, 3, 4 highly expressing CD14 and CD68), an NK cell cluster (cluster 6 highly expressing KLRB1 and KLRF1), a B cell cluster (cluster 9 highly expressing CD19 and CD79A), and a dendritic cell cluster (cluster 7 highly expressing CLEC4C and IL3RA) (Figure 1B–G). However, we could not detect cell clusters expressing polymorphonuclear leukocytes (e.g., mast cells, eosinophils, basophils and neutrophils). The two non-immune cell populations based on low PTPRC levels were characterized by an ovarian cancer cell signature (cluster 5 highly expressing WFDC2 and EPCAM) and a fibroblast signature (cluster 8 highly expressing COL1A1 and THY1) (Figure 1H, I). About 5.5% of cells derived from malignant ascites were expressing ovarian cancer cell signatures (WFDC2 and EPCAM). A small fraction of cells (2.6%) was expressing fibroblast markers (PDPN and THY1) (Figure 1J, K). These results show that different cell types (immune cells, ovarian cancer cells and fibroblasts) exist in the malignant ascites TME.
Overall, seven distinct cell types were identified by the expression levels of cell type-specific signatures in the malignant ascites TME (Fig. 1J, K). Preprocessing of the samples using Ficoll-density gradient centrifugation, enriched mononuclear cells in our ascites samples. Data from analysis of immune cell signatures suggest that the vast majority of the cells within ascites are immune cells. Notably, macrophages (55.5%) were the most abundant cell type, followed by T cells (23.9%), NK cells (5.4%), dendritic cells (4.2%) and B cells (2.6%). Among these immune cells, macrophages are the dominant cells in the ascites TME and are noticeably scattered in the UMAP plot, implying that the cells expressing macrophage markers are highly heterogeneous.
3.2. Profiling of macrophage polarization states in ascites TME of ovarian cancer patients with MacSpectrum analysis
As mentioned above, macrophages comprised the most abundant population found in multiple UMAP clusters in our dataset. This may imply that macrophages are the most heterogeneous immune cell population within the malignant ascites. The heterogeneity of macrophages in ascites TME is not simply characterized by the conventional M1 and M2 subtypes (Figure S1). To characterize macrophage heterogeneity in ascites TME, macrophages were re-clustered, and 13 Seurat defined clusters were identified (Figure 2A, left panel). There was no notable difference in transcriptomic expression pattern in macrophages across the patient samples (Figure 2A, right panel).
FIGURE 2.
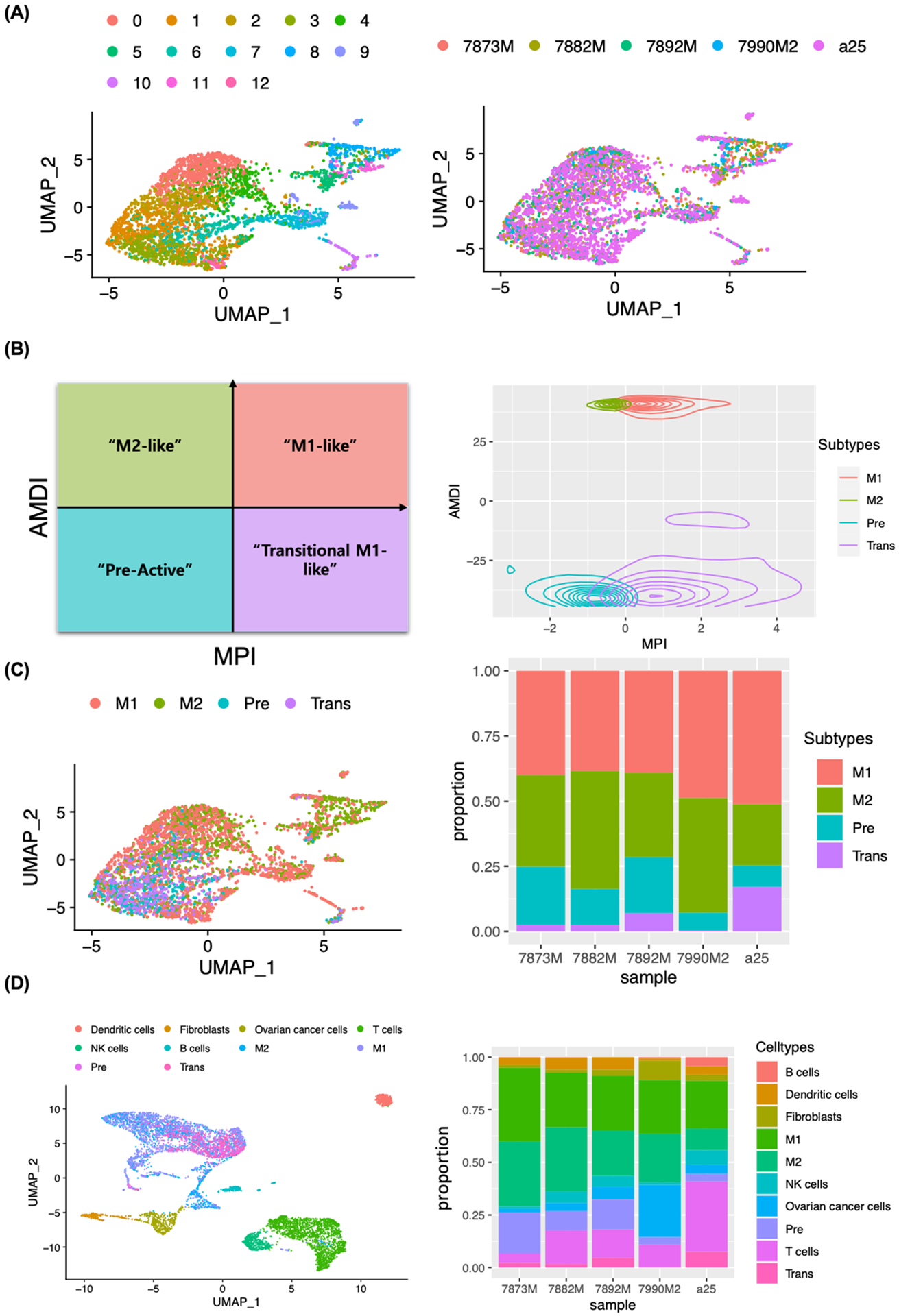
Macrophage subtype annotation using MacSpectrum. A, Unsupervised clustering of macrophages (4,073 cells) of ascites from five patients (colored by patient; right panel). UMAP dimensional reduction analysis identified 13 clusters (colored by subclusters; left panel). B, Macrophage Polarization Index (MPI)/Activation induced Macrophage Differentiation Index (AMDI) plot illustrating separation of M1 (M1-like), M2 (M2-like), Pre (Pre-active) and Trans (Transitional M1-like) macrophages by MacSpectrum. C, Macrophages colored by subtypes (M1, M2, Pre and Trans) and displayed with UMAP (left) and proportional barplot of macrophage subtypes from 5 patient samples (right). D, UMAP embedding macrophage subtypes with all cells, colored by cell types (left) and proportional barplot of cell types present in ascites for each patient sample (right).
Macrophage polarization status in ascites TME was further characterized using the MacSpectrum analysis tool, which allows a comprehensive analysis of multifaceted activation states of macrophages under various conditions. We have used differentially expressed gene (DEG) lists from 13 Seurat clusters (2552 genes in total) to generate a Macrophage Polarization Index (MPI) and an Activation-induced Macrophage Differentiation Index (AMDI) (Figure 2B). MacSpectrum analysis defined macrophages into four subtypes, Pre- (low MPI/low AMDI), Trans- (high MPI/low AMDI), M2- (low MPI/high AMDI) and M1- (high MPI/high AMDI) (Figure 2B, left panel). MacSpectrum-defined macrophage subtypes were superimposed on a Seurat-defined UMAP plot (Figure 2C, left panel), which revealed that M1 polarized macrophages comprised the largest subtype in ascites TME followed by M2, Pre and Trans macrophages (Figure 2C, right panel).
To further characterize the macrophage subpopulation in ascites TME, we performed DEG analysis and gene set enrichment (GSEA) analysis on each macrophage subtype. The M1 macrophage cluster expressed relatively higher levels of antigen presentation genes (HLA class II genes), HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1 and classic inflammatory pathways, such as interferon-gamma, IL6 and NFkB were strongly upregulated (Figure S2A, E). The M2 macrophage cluster was enriched with anti-apoptotic genes, MTRNR2L1, MTRNR2L6, MTRNR2L8, MTRNR2L11, MTRNR2L12 and the Wnt/beta-catenin signaling pathway was strongly enriched (Figure S2B, F), which has been previously reported to promote M2 macrophage polarization.22,23 The Pre-macrophage cluster was enriched with PI3K-Akt signaling pathway, which regulates macrophage survival, migration and proliferation24, whereas the Trans-macrophage cluster was in an intermediate state enriched with innate immune response-related pathways, including interferon alpha, interferon gamma and complement pathways (Figure S2C, D, G, H). GSEA analysis of subtype-specific genes supports MacSpectrum analysis based macrophage subtype annotation and reveals the existence of unique, macrophage subtype-specific functions in ascites TME.
Adaptive immune cells (T cells and B cells), innate immune cells (NK cells, dendritic cells and macrophage subtypes), fibroblasts and ovarian cancer cells were re-plotted with UMAP analysis. The proportion of each cell type was compared among the samples. Markedly higher proportion of T cells was observed in the a25 sample, while the percentage of M2 in a25 was the lowest among the samples analyzed (Figure 2D). These results suggest that there is significant heterogeneity in the distribution of cell types in malignant ascites.
3.3. Comparison of molecular pathways enriched in ovarian cancer cell clusters in malignant ascites reveals intra- and inter-patient heterogeneity
We identified the intratumor-heterogeneity of ovarian cancer cells in ascites TME, showing 3 Seurat-defined subclusters (Figure 3A). We analyzed a list of putative ovarian cancer stem cell markers documented in a recent review.25 We found that all ovarian cancer cell sub-clusters showed high expression levels of CD24 and EPCAM (Figure S3), which were recently highlighted as unique markers to cancer cells with the highest sensitivity.26 Noteworthy was the low expression of other putative stem cell markers, including the most frequently used ovarian cancer biomarker, CA12527 and the highly documented side-population of ovarian cancer stem-like cell markers, ALDH1A1 and ABCG2.28 These observations suggest that ovarian cancer stem-like cells are rare and highly heterogeneous at the transcript level in the malignant ascites.
The most differentially expressed genes between ovarian cancer cell subclusters were closely related to the self-renewal, respiration and proliferation. Subcluster 0 exhibited enrichment of many stem cell-related genes, such as LY6E, TGM2, MAL and CD55 (Figure 3B). Mitochondrial genes, for example MT-ATP8, MT-CYB, MT-ND4L, MT-ND3 and MT-ND6, are expressed throughout the majority of ovarian cancer cell subclusters, but are particularly enriched in subcluster 1 (Figure 3B and Figure S4A). Subcluster 2 expressed genes such as CCT5, HSP90AB1, PPIA and ENO1 and was transcriptionally the most unique subcluster. Hallmark gene sets were used for the analysis of pathways enriched between subclusters of ovarian cancer cells (Figure 3C–E). Ovarian cancer cell subcluster 0 showed enrichment of several metabolic pathways such as ‘oxidative phosphorylation’, ‘unfolded protein response’, ‘adipogenesis’, ‘xenobiotic metabolism’ and ‘heme metabolism’, when compared to the ovarian cancer cell subcluster 1 (Figure 3C, F). Additionally, we observed an enrichment of inflammation-related pathways in subcluster 0, including ‘interferon gamma response’, ‘interferon alpha response’, ‘complement’, ‘inflammatory response’, ‘TNFA signaling via NFKB’ and ‘IL2-STAT5’ signaling when compared to subcluster 2 (Figure 3D, G). Subcluster 1 exhibited enrichment of two gene sets compared to the subcluster 2, including inflammation-related signaling pathways (‘inflammatory response’ and ‘interferon gamma response) (Figure 3E). Subcluster 2 showed relatively higher expression levels of genes related to several pathways including ‘E2F targets’, ‘G2M checkpoint’, ‘MYC targets’, ‘oxidative phosphorylation’, ‘mitotic spindle’, ‘DNA repair’, ‘fatty acid metabolism’ and ‘MTORC1 signaling’ than subcluster 0 or 1 (Figure 3D, E, G, H). Overall, GSEA analysis of ovarian cancer cell subclusters further highlighted the presence of metabolic heterogeneity in relation to oxidative phosphorylation (Figure 3C–H). These data support the presence of ovarian cancer cell subgroups, with varying degrees of disparities in metabolic programming of cancer cells.
Though the subclustering analysis showed a low degree of inter-patient variability among the samples we have analyzed and cluster 0 was over-represented by the sample 7990M2, there were several genes significantly upregulated in every sample (Figure S4C). To identify genes from cancer cells that vary among the ascites samples, a heatmap was generated for comprehensive visualization of differences at the transcript level (Figure S4B).
Cancer cells from the 7873M sample showed enrichment of several genes related to inflammatory responses (SPP1, C1QA, FCER1G, CD14, LAPTM5, CD68, SRGN and S100A8), translation and cell cycle (CDC123) and a RNA polymerase (POLR2D). We found that the cancer cells in the 7873M sample showed higher expression of CD68 than those from other samples. As observed in the UMAP plots presenting different cell types within the ascites sample, cancer cells in the ascites samples were transcriptomically similar to macrophage populations. This may suggest the presence of trans-differentiated or macrophage-ovarian cancer hybrid-like cells in the ascites TME, reported in previous studies.29,30 Cancer cells in 7882M sample showed enrichment of a subunit of mitochondrial ATP synthase (MT-ATP8), a nuclear pore complex gene (NPIPB5), an anti-viral gene (RSAD2), a renin inhibiting gene (RENBP), a transcription factor (ZNF224) and a gene coding for an acetylcholine receptor subunit (CHRNB1). The cancer cells from the 7892M sample showed higher expression of tumor suppressor genes CDKN2A, which codes for two proteins, p16 and p14. Along with CDKN2A, other genes enriched in the 7892M sample were a cancer-fibroblast interaction mediator (TSPAN12), a metal cation transporter gene (SLC39A8), an ubiquitin-producing gene (UCHL1), a creatine kinase (CKB), a cell adhesion gene (ADGRL2), a tissue repairing gene (CTHRC1), a chaperone (HSP90AA1), a colon carcinoma-associated gene (C12orf75) and a cytokinesis regulator (NUDC). Interestingly, the cancer cells in the 7890M2 sample, which contained the highest proportion of fibroblasts, showed increased expression of extracellular proteases, kallikreins (KLK6 and KLK7). Aside from kallikreins, genes coding for a membrane-integrated protein (MAL), an oncogenic enzyme (TGM2), a homeobox protein (HOXB2), a mediator of protein-protein interaction (LMO7), bromine metabolism (PXDN), a cysteine protease inhibitor (CST6), a regulator of the complement system (CD55) and an inhibitor of CaMKII (CAMK2N1) were upregulated in the 7890M2 (Figure S4B). The increased kallikreins might be due to the adaptive responses of cancer cells to retain their invasiveness in the fibroblast-rich ascites TME of the sample 7890M2.
We observed that the a25 sample overlapped and spanned all three subclusters found in ovarian cancer cells (Figure S4C). The differences may reflect sampling bias due to the differences in scRNA-seq techniques used across the patient samples. The SNUH a25 sample was processed with 10X genomics, which outperforms InDrop technique in terms of sensitivity and coverage. This could also be due to the true differences between patient samples reflecting the inter-patient heterogeneity. Altogether, we observed that each patient sample harbored cancer cells expressing distinct gene that may play a role in sample-specific molecular mechanisms of cancer cell dissemination, survival and invasiveness in the ascites TME and the cancer cells from the a25 sample showed the most diverse gene expression pattern.
3.4. Elevated expression of ligands related to the anoikis-resistant phenotype may provide survival advantages to cancer cells in the malignant ascites
To examine cellular crosstalk between ovarian cancer cells and the niche cells in ascites TME, NicheNet was used to predict the ligand-receptor pairs that could potentially induce the transcriptomic changes in ovarian cancer cells. We were interested in the anoikis resistance process, as it has been previously reported that the acquisition of anoikis resistance is a critical cellular process driving cancer progression.31 We combined two gene sets, negative regulation of programmed cell death (GO:0043069) and negative regulation of anoikis (GO:2000811) for NicheNet analysis. Potential immune cell-derived ligands included CCL5 (a chemokine involved in an inflammatory response, cancer cell migration, survival and proliferation), TNF family members TNFSF13B and LTB, and immune response genes ITGB2 and ICAM1 (Figure 4A). CCL5 was the most highly expressed ligand by T cells and NK cells (Figure 4B) and across patients (Figure S5). The top 5 potential receptors for CCL5 in ovarian cancer cells included ACKR3, LPAR2, SDC1, CDH1 and SDC4 (Figure 4C). Finally, among the potential receptors in ovarian cancer cells, SDC4 was the most highly-expressed across the patient samples (Figure 4D). A Circos plot shows the overall ligand-receptor links between predicted ligands from immune cells and receptors found on ovarian cancer cells, where the transparency of the inner lines represents the degree of ligand activity (Figure 4E). Expression levels of CCL5 and SDC4 were visualized using Violin plots and Feature plots (Figure 4F, G), demonstrating that the ligand CCL5 is expressed exclusively in T cells and NK cells and that the receptor SDC4 is expressed in ovarian cancer cells and fibroblasts. To evaluate CCL5-SDC4 axis, we tested our model with recently published scRNA-seq data from 6 additional ovarian cancer patient-derived ascites (GSE146026). Of those, one patient had no ovarian cancer cells and was excluded from the analysis. Intriguingly, the evaluation of CCL5 and SDC4 expression across 5 patients, further supports our identification (Figure S6A–C). Overall, NicheNet highlighted the CCL5-SDC4 axis as a potential mechanism for ovarian cancer cells to acquire anoikis resistance in ascites TME.
FIGURE 4.
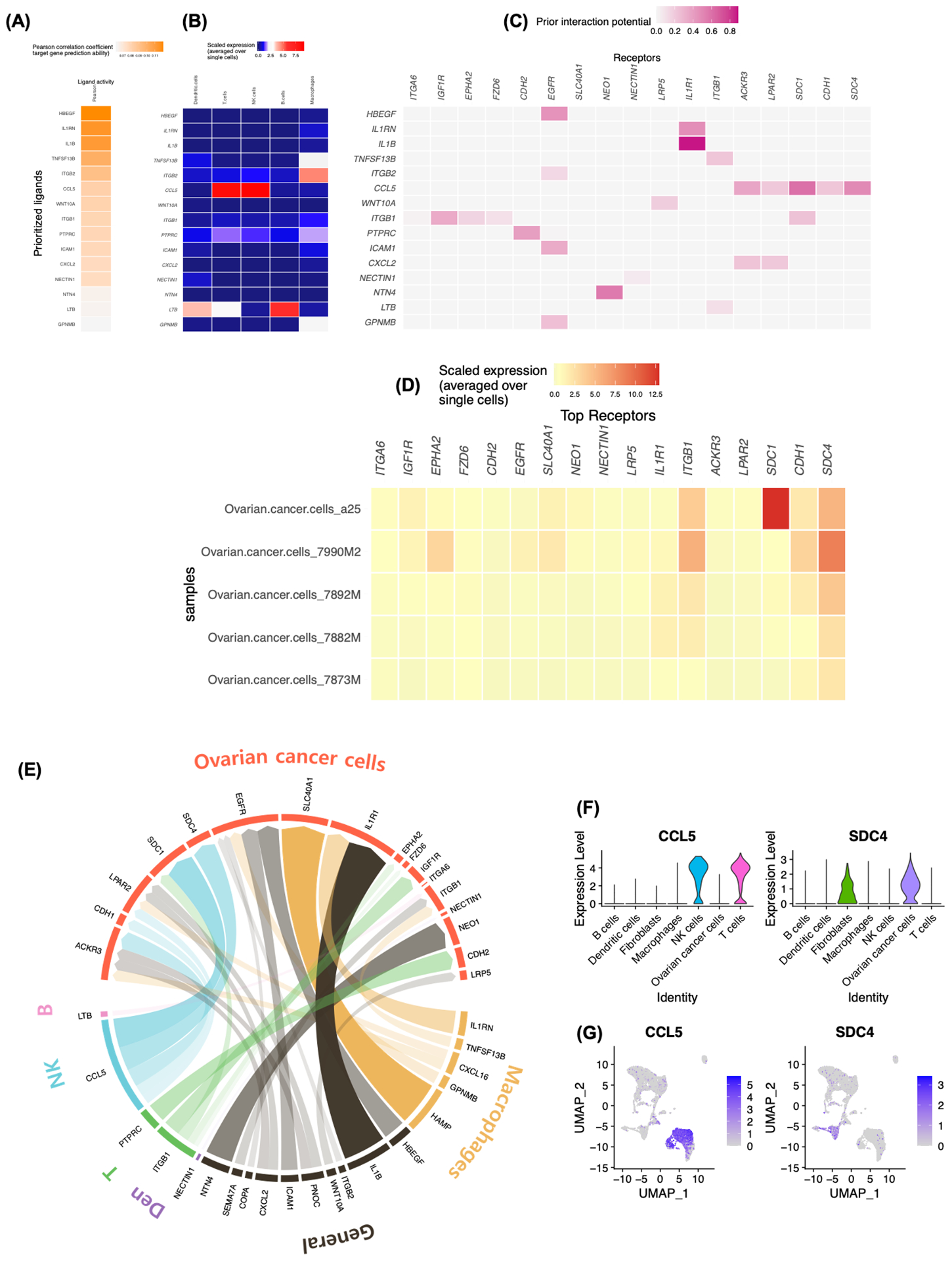
CCL5-SDC4 axis serve as a potential upstream ligand-receptor signal inducing anoikis resistance in ovarian cancer cells. A, B, NicheNet analysis of ligand-receptor pairs inducing the anoikis resistance in ovarian cancer. Ligand activity analysis (A) Average expression of the ligands (B) by the different immune cell types. C, Receptors of prioritized ligands expressed by ovarian cancer cells. D, Average expression of potential receptors in ovarian cancer cells across patient samples. E, Circos plot showing links between predicted ligands from immune cell types with their associated receptors found on ovarian cancer cells. The degree of transparency indicates the prior interaction weight of the ligand-receptor interaction. F-G, Expression of CCL5 and SDC4 in cellular components of ascites TME visualized by Violin plots (F) and Feature plots (G).
3.5. Effector and exhausted CD8+ T cells and NK cells highly express CCL5 in the malignant ascites
To examine the complexity of T cell and NK cell subsets found in ascites TME, each cell types were re-clustered, and 5 and 3 Seurat defined clusters were identified in T cell and NK cell populations, respectively (Figure 5A and Figure S7A). Expression profiles were similar across subclusters in both T cell and NK cell (Figure 5B and Figure S7B). Interestingly, co-expression analysis of CD4 and CD8A with CCL5 in T cell subsets revealed that cluster 0 and 1 are CD8+ T cell with high CCL5 co-expression, whereas clusters 2 and 3 are CD4+ T cell with low CCL5 co-expression (Figure 5C, D). T cell subsets were further characterized by functional markers reported in a recent study.32 Expression profiles of T cell subclusters revealed that subcluster 0 is CD8+ effector T cell cluster, with high expression of cytokines and effector molecules, GZMA, GNLY, PRFA, GZMB, GZMK, NKG7. Whereas, subcluster 1 is CD8+ exhausted T cell cluster, with exhaustion marker expression, LAG3, TIGIT, PDCD1 and HAVCR213 (Figure 5E). NK cell subsets analysis show that all subclusters highly express KLRB1 with high CCL5 co-expression (Figure S7C). These results suggest that CCL5 is exclusively expressed in effector and exhausted CD8+ T cell subsets and NK cells in malignant ascites TME.
FIGURE 5.
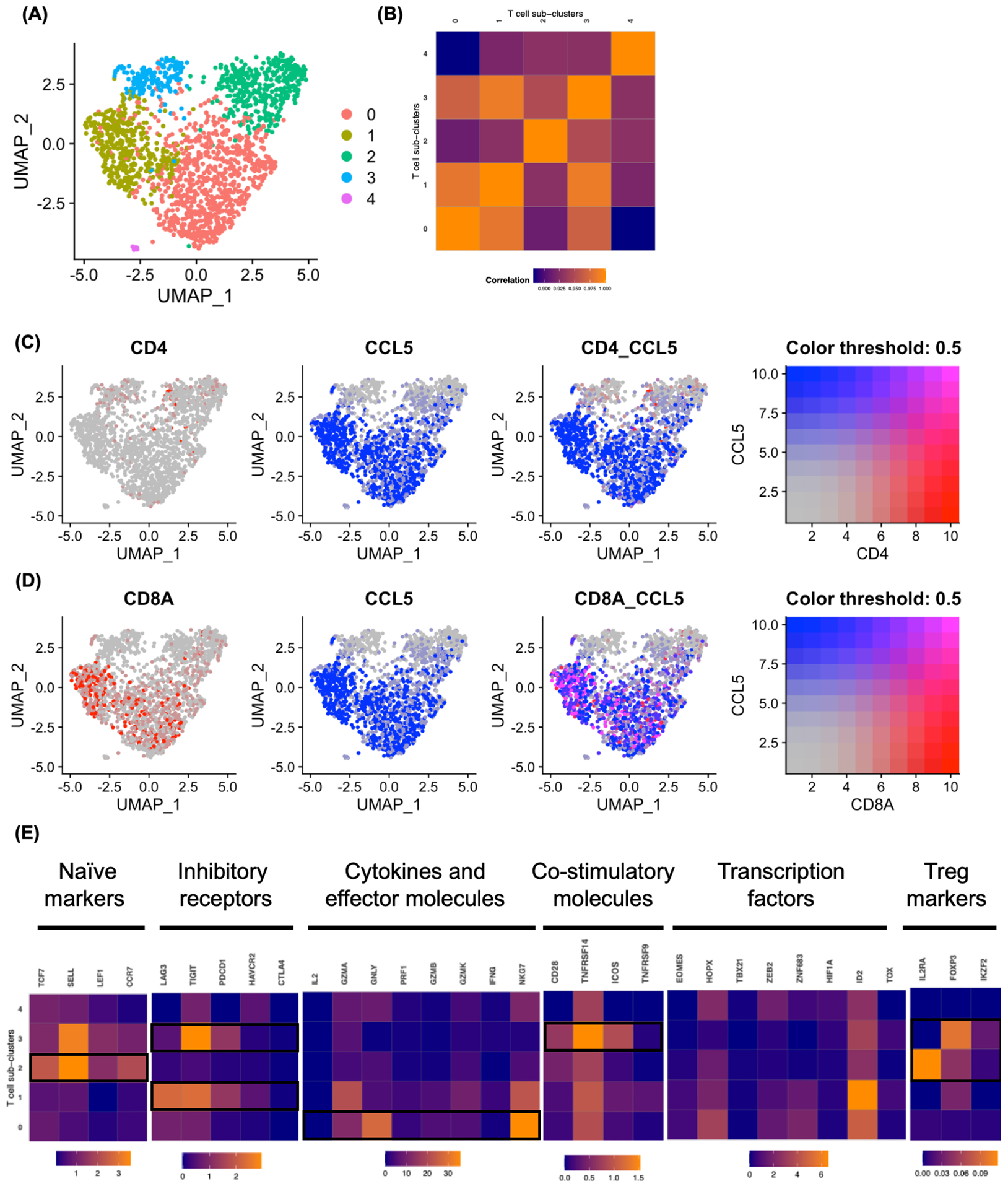
Clustering T cell subsets in ascites TME. A, The UMAP projection of T cell subclusters identified 5 clusters (colored by subclusters). B, Expression profiles were examined by Pearson correlation coefficient between the mean profiles of T cell subclusters. C-D, Co-expression of CCL5 with CD4 (C) and CD8A (D) in T cell subclusters visualized by Feature plots. E, Heatmap showing mean expression of T cell function associated genes in each T cell subclusters. Black boxes highlight the prominent patterns used to define T cell subsets.
3.6. SDC4 expression correlates with poor prognosis in ovarian cancer patients
We next assessed the prognostic value of SDC4 in ovarian cancer patients. Using datasets from TCGA, the patients were divided into two groups according to SDC4 (also known as SYDN4) expression. High expression levels of SDC4 was associated with significantly poor overall survival (OS) in ovarian cancer patients (Figure 6A). For further validation, we additionally analyzed GEO datasets (GSE9891, GSE3149, GSE26193 and GSE63885). Kaplan-Meier and log rank test analysis with GEO datasets also suggested that the SDC4 expression level was negatively correlated with OS in ovarian cancer patients (Figure 6B–E). In addition, we compared expressions of SDC4 at the transcript level in cancer and normal tissues, and we found significantly elevated transcript levels of SDC4 in ovarian cancer tissues (Figure 6F). Moreover, the SDC4 expression increased across the tumor stage (Figure 6G).
FIGURE 6.
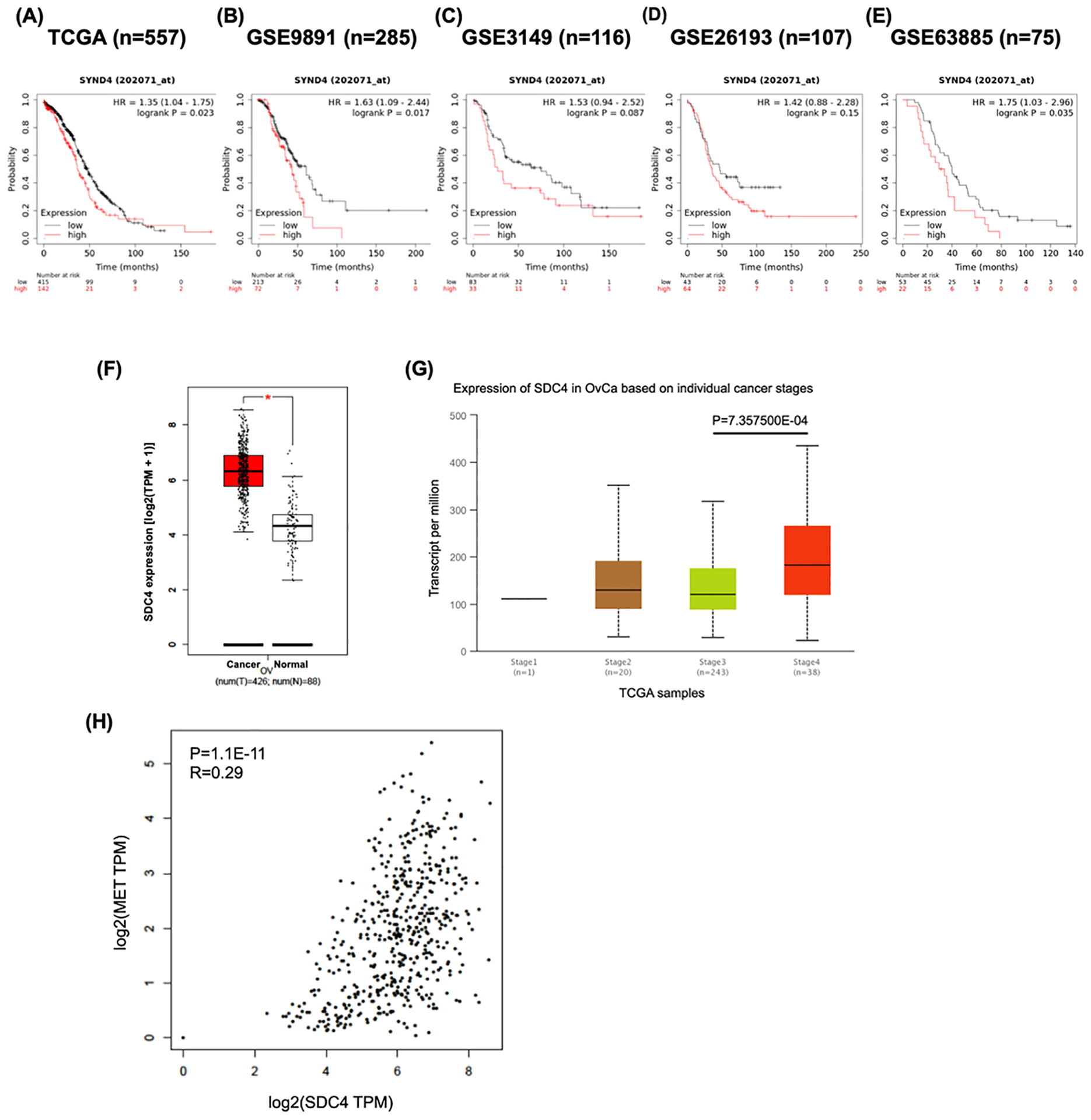
SDC4 expression is associated with the survival of ovarian cancer patients. A-E, Higher expression of SDC4 is associated with poor overall survival of ovarian cancer patients. Ovarian cancer public datasets from TCGA (A) and GEO database (B-E) were used to examine the association between SDC4 expression level and overall survival of the patients. Kaplan-Meier survival curves were plotted by splitting patients into two groups (SDC4 high and SDC4 low). Groups were divided by ‘split patients by’ option set at ‘auto select best cutoff’. The Kaplan-Meier plotter (KM plotter) tool was used. F, The expression of SDC4 was elevated in ovarian cancer tissues compared to the normal tissues. TCGA and GTEx expression data were analyzed using GEPIA. G, The SDC4 expression is significantly upregulated in the cancer tissues from stage IV patients. UALCAN was used to analyze the association of stage with SDC4 expression. H, A significant positive correlation was found between MET and SDC4. The correlation between mRNA expression levels of MET and SDC4 was analyzed with GEPIA using data from TCGA and GTEx database.
To examine the involvement of SDC4 in the anoikis resistance of ovarian cancer cells in the malignant ascites, we investigated a correlation between the average SDC4 expression and the proportion of cancer cells in each sample. Intriguingly, the proportion of cancer cells in the ascites TME positively correlates with average SDC4 expression (Figure S8A). This may imply that SDC4 expression could be vital for cancer cell survival in the ascites. cMet has often been cited in the literature as a key regulator of anoikis resistance in ovarian cancer.33 We observed that SDC4 is positively correlated with MET expression both in the tissue samples from TCGA and GTEx and in the malignant ascites-derived cancer cells (Figure 6H and Figure S8B), further supporting our hypothesis that the expression of SDC4 in ovarian cancer cells may promote cancer progression.
3.7. CCL5 treatment enhances survival of ovarian cancer cells under non-adherent culture conditions
Data from clinical samples (GSE85296) suggest that mRNA expression levels of CCL5 is elevated in the ascites compared to primary sites (left and right ovary; Figure 7A). Both cell lines of ascites origin (SKOV3, Kuramochi and PA1) and tissue origin (ES-2) expressed SDC4 at the basal level. Protein expression of SDC4 was slightly higher in the ascites-derived cell lines than that in the tissue-derived cell line (Figure 7B). We then examined the effect of CCL5 on the anoikis resistance of ovarian cancer cells using non-adherent culture method. ULA plates were used to mimic non-adherent ascites TME in vitro. Spheroid sizes were larger in non-adherent cancer cells cultured in the presence of 50 ng/mL CCL5 (Figure 7C, D). Also, increased PARP-1 cleavage induced by non-adherent culture was suppressed by CCL5 treatment (50 ng/mL, 72h; Figure 7E). These results suggest that CCL5 can increase survival of SDC4-expressing ovarian cancer cells under non-adherent conditions.
FIGURE 7.
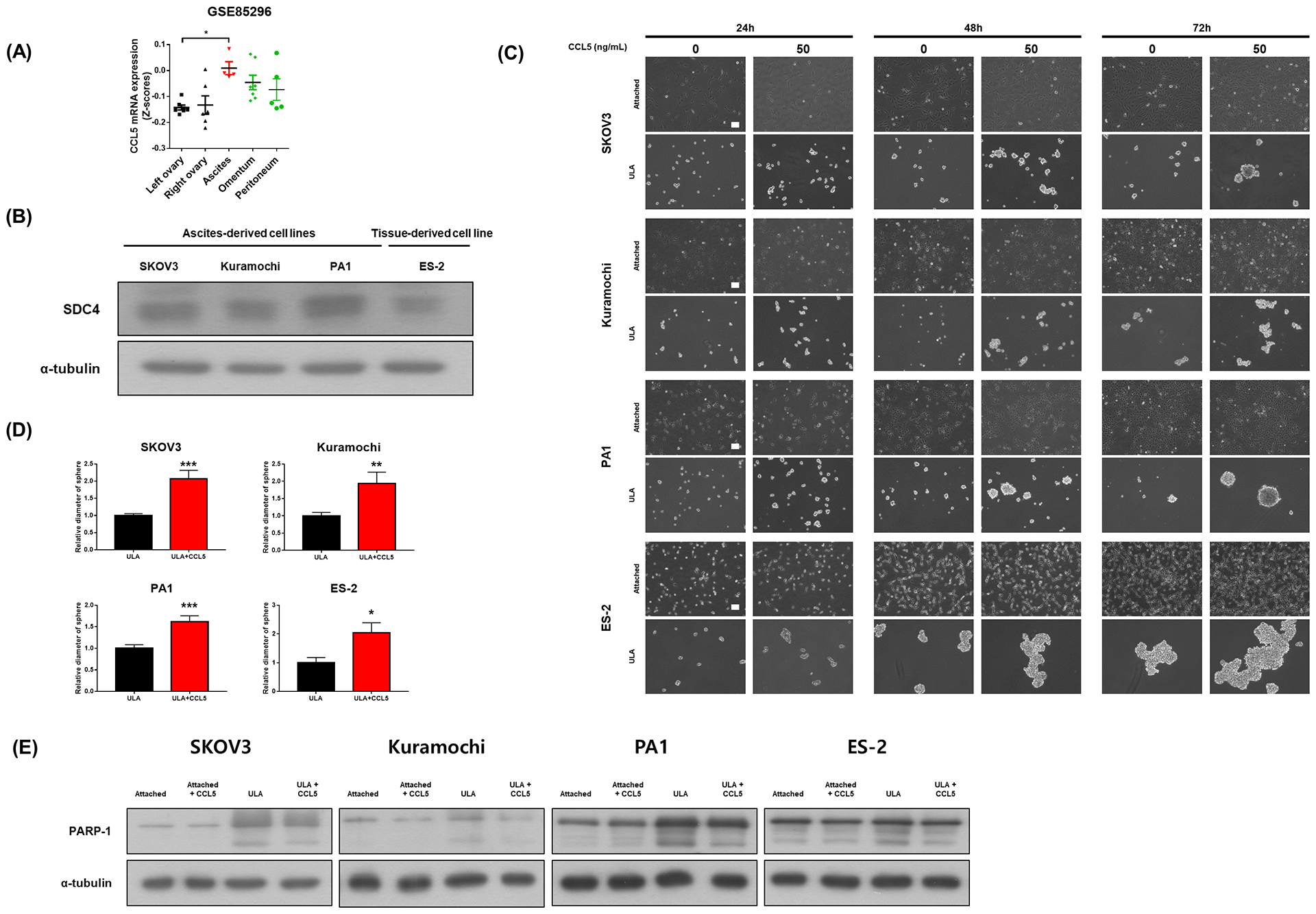
CCL5 treatment increases anoikis resistance. A, Transcript levels of CCL5 in left ovary (n=7), right ovary (n=6), ascites (n=5), omentum (n=7) and peritoneum (n=5) were compared. B, Protein expression of SDC4 in different ovarian cancer cell lines (SKOV3, Kuramochi, PA1 and ES-2). α-tubulin was used as a loading control. C, Microscopic images of attached cells or aggregates/spheroids of ovarian cancer cell lines with or without 50 ng/mL CCL5 treatment for 24, 48 and 72h. Length of the scale bar is 100 μm. D, Diameters of randomly chosen tumor spheroids were measured using ImageJ software (30–80 spheroids per group). E, PARP-1 cleavage was determined by western blotting analysis in different culture conditions of ovarian cancer cells with or without CCL5 (50 ng/mL, 72h) treatment. Values were presented as mean ± SEM. *P<0.05, **P<0.005, ***P<0.001
4. DISCUSSION
The current study uncovered inter- and intra-patient heterogeneity of ovarian cancer and captured the landscape of both immune and non-immune cells enriched in ascites TME, using scRNA-seq combined with a computational approach. We determined a ligand-receptor intercellular communication model by analysis of a putative ligand-receptor interactions between receptors enriched in ovarian cancer cells and ligands expressed by immune cells in ascites TME. Although our computational model does not consider the spatial proximity between the cells of interest, ascites from ovarian cancer patients creates a unique TME as a reservoir of pro-tumorigenic soluble factors including CCL5 and other cytokines reported in our previous work.6 In this study, we found that CCL5-SDC4 axis may play a crucial role in ovarian cancer cell survival and poor patient prognosis. To validate our model, we analyzed the recently published scRNA-seq study of malignant ascites from patients with advanced-stage ovarian cancer.34 Also this analysis highlighted the putative role of intercellular communication through CCL5-SDC4 axis in immune cells and ovarian cancer cells (Figure S6). Moreover, TCGA and GEO analysis further supports the role of SDC4 in ovarian cancer progression (Figure 6).
Among immune cell-related clusters, we observed a highly heterogeneous macrophage population at the transcriptomic level, but we found that only subtle differences in macrophage subtype proportions exist across the five malignant ascites samples analyzed. Previous studies report the differential role of M1 and M2 macrophage subtypes in ovarian cancer progression. M2 subtypes has received the most attention in cancer biology due to their pro-tumorigenic role including stemness maintenance, promoting the chemoresistance and invasiveness in a number of cancer models, including ovarian cancer.23 The prognostic role of M1 subtypes in ovarian cancer show conflicting results. Our group reported a pro-tumorigenic role of in vitro polarized M1 macrophages in ovarian cancer.35 Similar results were also reported in in vitro studies of hepatocellular and bladder cancer.36,37 In contrast, a plentiful number of reports suggest an anti-tumorigenic role of M1 macrophages in ovarian cancer.38,39
A recent study observed the prevalence of both M1 and M2 macrophages in ovarian cancer patient-derived ascites TME. The authors noted that the ratio of these subtypes are important, where a higher M1/M2 macrophage ratio was correlated with a higher overall survival and progression-free survival.40 An earlier study reported that platinum-based chemotherapy increases ovarian cancer cell potency to induce M2 macrophage polarization, resulting in an imbalance in macrophage subtypes in the TME.41 In a murine model, the re-balancing of macrophage subtypes toward M1 in the TME decreased tumor burden and prolonged survival.39 Although vast majority of studies suggest oncogenic roles of M2 macrophage in cancer progression and tumor growth, our data from MacSpectrum analysis suggest that there were comparable proportions of M1 and M2 in the malignant ascites across ovarian cancer patients.42–44 Moreover, previous study from Reinartz et al. suggests that mixed polarization phenotype distinct from the M1/M2 classification was found in the ovarian cancer ascites.42 Hence, we incorporated MacSpectrum tool which uses two indices (macrophage polarization index and activation-induced macrophage differentiation index) because traditional M1 (CD86, IL6 and TNF) and M2 (CD163, ARG1 and IL10) markers poorly separated the two subtypes at single cell level. These results suggest that macrophage subtypes may have a stage-specific role and the balance of macrophage subtypes may play a crucial role in cancer progression. Future longitudinal studies of single-cell profiling of macrophage subtypes will give us a more definitive understanding of their role in ovarian cancer progression.
We showed molecular heterogeneity of T cell and NK cell population (Figure 5 and Figure S7). As presented in Figure 2D, the proportion of T cells was higher in the a25 sample than that in the other four patient samples. However, there is a possibility that difference may be due to the sample preparation method or clinical parameters of patients (e.g., treatment status, histology, etc.). Further investigation on the association between clinical parameters and the cellular constituents is warranted to understand the complex cellular interactions taking place in the malignant ascites niche.
Anoikis resistance is a primary feature of ovarian cancer cells to survive in ascites TME. We hypothesize that niche signals from ascites TME regulate or induce anoikis resistance in ovarian cancer cells. Though macrophages have received the most attention in cancer progression due to their abundance, our prediction model highlights the potential role of T cells and NK cells in anoikis resistance in ascites TME. Since we have only focused on this single process, the various roles of macrophages may have been underrepresented in our analysis. We observed the global overexpression of SDC4, a known receptor of CCL5, in ovarian cancer cells from all samples analyzed at the single cell level (Figure 4D). SDC4 is a heparan sulfate proteoglycan that modulates focal adhesion and has been implicated in cancer progression and metastasis.45 Elevated SDC4 expression has been reported in hepatocellular cancer and mesotheliomas.46,47 Previous ovarian cancer study observed that SDC4 is expressed in normal ovary and in both benign and malignant ovarian tumors.48 CCL5 is elevated in various types of cancers, including ovarian cancer.49 It has been shown that binding of CCL5 to SDC4 induces activation of PKCα50,51, which is involved in multiple pathways including cell cycle progression, tumorigenesis and metastatic dissemination52, but the mechanistic role of SDC4-CCL5 in ovarian cancer progression remains poorly understood.
Given that CCL5 is enriched in T cells and NK cells in ascites TME and that SDC4 expression is restricted to ovarian cancer cells in our scRNA-seq data (Figure 4F–G), the aberrant levels of CCL5 in ascites TME is the critical determinant of ovarian cancer progression and poor prognosis. The expression of SDC4 in ovarian cancer cells was strongly correlated with the proportion of cancer cells found in ascites TME as well as with MET expression at single-cell level (Figure S8). Furthermore, TCGA and GEO data suggest that SDC4 is expressed in ovarian cancer cells and correlates with poor overall survival in ovarian cancer patients (Figure 6) and CCL5 is elevated in the ascites compared to the primary sites (Figure 7A). Our in vitro anoikis resistance analysis further supports the role of CCL5-SDC4 axis in ovarian cancer cell survival in ascites TME (Figure 7).
scRNA-seq has revolutionized our approach to understanding cancer progression. We have utilized this powerful technique, modeling the pathological mechanisms underlying ovarian cancer progression. Despite these findings, a number of challenges and limitations still remain in our model. We had limited access to patient information (e.g., survival status, stage, prognosis, treatment, histology, etc.), small sample size and sample processing bias (filtered tumor spheroids/clusters, our procedure enriched cancer cells and lymphocytes, and scRNA-seq targeted low total cell reads). Profiling a larger number of cancer cells with multiple samples in parallel would be required to validate the presence of inter-tumoral heterogeneity and to identify rare cancer stem cell populations. In addition, longitudinal sampling of the cancer cells over the course of treatment could facilitate our understanding of tumor evolution and chemoresistance.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by BK21 Plus Program of the Department of Agricultural Biotechnology, Seoul National University (Seoul, Korea), a grant of Health Technology R&D Project through the Korea Health Industry Development R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea [grant number: HI16C2037] and a grant from the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea [grant number: HA17C0037]. Also, we would like to thank Dr. Hae-Ock Lee for the technical assistance and Dr. Andreas Raue for the data provision.
Abbreviations
- scRNA-seq
Single-cell RNA sequencing
- TME
Tumor microenvironment
- SNN
Shared nearest neighbor
- UMAP
Uniform manifold approximation and projection
- SDC4
Syndecan-4
- GEPIA
Gene Expression Profiling Interactive Analysis
Footnotes
CONFLICT OF INTERESTS
The authors have declared that no competing interest exists.
DATA AVAILABILITY STATEMENT
scRNA-seq data from four patients which were previously published were kindly provided by Prof. Andreas Raue8. SNUH scRNA-seq data can be provided from the corresponding author upon reasonable request.
Reference
- 1.Torre LA, Trabert B, DeSantis CE, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68(4):284–296. doi: 10.3322/caac.21456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim S, Han Y, Kim SI, Kim H-S, Kim SJ, Song YS. Tumor evolution and chemoresistance in ovarian cancer. npj Precis Oncol. 2018;2(1):1–9. doi: 10.1038/s41698-018-0063-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.T DS, A R, K SB. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006;7(11):925–934. http://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=reference&D=emed7&NEWS=N&AN=2006519275 [DOI] [PubMed] [Google Scholar]
- 4.Kim S, Kim B, Song YS. Ascites modulates cancer cell behavior, contributing to tumor heterogeneity in ovarian cancer. Cancer Sci. 2016;107(9). doi: 10.1111/cas.12987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ford CE, Werner B, Hacker NF, Warton K. The untapped potential of ascites in ovarian cancer research and treatment. Br J Cancer. 2020;(September 2019). doi: 10.1038/s41416-020-0875-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim S, Gwak HR, Kim HS, Kim B,Dhanasekaran DN, Song YS. Malignant ascites enhances migratory and invasive properties of ovarian cancer cells with membrane bound IL-6R in vitro. Oncotarget. 2016;7(50):83148–83159. doi: 10.18632/oncotarget.13074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim S, Lee M, Dhanasekaran DN, Song YS. Activation of LXRα/β by cholesterol in malignant ascites promotes chemoresistance in ovarian cancer. BMC Cancer. 2018;18(1). doi: 10.1186/s12885-018-5152-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schelker M, Feau S, Du J, et al. Estimation of immune cell content in tumour tissue using single-cell RNA-seq data. Nat Commun. 2017;8(1):1–12. doi: 10.1038/s41467-017-02289-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han Y, Kim B, Cho U, et al. Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells. Oncogene. 2019;38(45):7089–7105. doi: 10.1038/s41388-019-0949-5 [DOI] [PubMed] [Google Scholar]
- 10.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–420. doi: 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888–1902.e21. doi: 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finak G, McDavid A, Yajima M, et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16(1):1–13. doi: 10.1186/s13059-015-0844-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C, Menoret A, Farragher C, et al. Single-cell transcriptomics-based MacSpectrum reveals macrophage activation signatures in diseases. JCI Insight. 2019;4(10):1–21. doi: 10.1172/jci.insight.126453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–1740. doi: 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korotkevich G, Sukhov V, Sergushichev A. Fast gene set enrichment analysis. bioRxiv. Published online January 2019:60012. doi: 10.1101/060012 [DOI] [Google Scholar]
- 17.Browaeys R, Saelens W, Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods. 2020;17(2):159–162. doi: 10.1038/s41592-019-0667-5 [DOI] [PubMed] [Google Scholar]
- 18.Bonnardel J, T’Jonck W, Gaublomme D, et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity. 2019;51(4):638–654.e9. doi: 10.1016/j.immuni.2019.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gyorffy B, Surowiak P, Budczies J, Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013;8(12). doi: 10.1371/journal.pone.0082241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia (United States). 2017;19(8):649–658. doi: 10.1016/j.neo.2017.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. doi: 10.1093/nar/gkx247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Ye YC, Chen Y, et al. Crosstalk between hepatic tumor cells and macrophages via Wnt/β-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018;9(8). doi: 10.1038/s41419-018-0818-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raghavan S, Mehta P, Xie Y, Lei YL, Mehta G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J Immunother Cancer. 2019;7(1):1–15. doi: 10.1186/s40425-019-0666-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J Immunol. 2017;198(3):1006–1014. doi: 10.4049/jimmunol.1601515 [DOI] [PubMed] [Google Scholar]
- 25.Roy L, Dahl KDC. Can stemness and chemoresistance be therapeutically targeted via signaling pathways in ovarian cancer? Cancers (Basel). 2018;10(8):1–23. doi: 10.3390/cancers10080241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peterson VM, Castro CM, Chung J, et al. Ascites analysis by a microfluidic chip allows tumor-cell profiling. Proc Natl Acad Sci U S A. 2013;110(51). doi: 10.1073/pnas.1315370110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scholler N, Urban N, Gene C a. CA125 in Ovarian Cancer Nathalie. Biomark Med. 2010;1(4):513–523. doi: 10.2217/17520363.1.4.513.CA125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parte SC, Batra SK, Kakar SS. Characterization of stem cell and cancer stem cell populations in ovary and ovarian tumors. J Ovarian Res. 2018;11(1):1–16. doi: 10.1186/s13048-018-0439-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Powell AE, Anderson EC, Davies PS, et al. Fusion between intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011;71(4):1497–1505. doi: 10.1158/0008-5472.CAN-10-3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramakrishnan M, Mathur SR, Mukhopadhyay A. Fusion-derived epithelial cancer cells express hematopoietic markers and contribute to stem cell and migratory phenotype in ovarian carcinoma. Cancer Res. 2013;73(17):5360–5370. doi: 10.1158/0008-5472.CAN-13-0896 [DOI] [PubMed] [Google Scholar]
- 31.Cai Q, Yan L, Xu Y. Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. 2015;(July 2014):3315–3324. doi: 10.1038/onc.2014.264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo X, Zhang Y, Zheng L, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018;24(7):978–985. doi: 10.1038/s41591-018-0045-3 [DOI] [PubMed] [Google Scholar]
- 33.Tang MKS, Zhou HY, Yam JWP, Wong AST. c-Met overexpression contributes to the acquired apoptotic resistance of nonadherent ovarian cancer cells through a cross talk mediated by phosphatidylinositol 3-kinase and extracellular signal-regulated kinase 1/2. Neoplasia. 2010;12(2):128–138. doi: 10.1593/neo.91438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Izar B, Tirosh I, Stover EH, et al. A single-cell landscape of high-grade serous ovarian cancer. Nat Med. Published online 2020. doi: 10.1038/s41591-020-0926-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho U, Kim B, Kim S, Han Y, Song YS. Pro-inflammatory M1 macrophage enhances metastatic potential of ovarian cancer cells through NF-κB activation. Mol Carcinog. 2018;57(2). doi: 10.1002/mc.22750 [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Wang X, Li X, et al. CD68+HLA-DR+ M1-like macrophages promote motility of HCC cells via NF-κB/FAK pathway. Cancer Lett. 2014;345(1):91–99. doi: 10.1016/j.canlet.2013.11.013 [DOI] [PubMed] [Google Scholar]
- 37.Dufresne M, Dumas G, Asselin É, Carrier C, Pouliot M, Reyes-Moreno C. Pro-inflammatory type-1 and anti-inflammatory type-2 macrophages differentially modulate cell survival and invasion of human bladder carcinoma T24 cells. Mol Immunol. 2011;48(12–13):1556–1567. doi: 10.1016/j.molimm.2011.04.022 [DOI] [PubMed] [Google Scholar]
- 38.Wanderley CW, Colón DF, Luiz JPM, et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res. 2018;78(20):5891–5900. doi: 10.1158/0008-5472.CAN-17-3480 [DOI] [PubMed] [Google Scholar]
- 39.Travers M, Brown SM, Dunworth M, et al. DFMO and 5-azacytidine increase M1 macrophages in the tumor microenvironment of murine ovarian cancer. Cancer Res. 2019;79(13):3445–3454. doi: 10.1158/0008-5472.CAN-18-4018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macciò A, Gramignano G, Cherchi MC, Tanca L, Melis L, Madeddu C. Role of M1-polarized tumor-associated macrophages in the prognosis of advanced ovarian cancer patients. Sci Rep. 2020;10(1):1–8. doi: 10.1038/s41598-020-63276-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dijkgraaf EM, Heusinkveld M, Tummers B, et al. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting m2 macrophages in the tumor microenvironment. Cancer Res. 2013;73(8):2480–2492. doi: 10.1158/0008-5472.CAN-12-3542 [DOI] [PubMed] [Google Scholar]
- 42.Reinartz S, Schumann T, Finkernagel F, et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: Correlation of CD163 expression, cytokine levels and early relapse. Int J Cancer. 2014;134(1):32–42. doi: 10.1002/ijc.28335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta V, Yull F, Khabele D. Bipolar tumor-associated macrophages in ovarian cancer as targets for therapy. Cancers (Basel). 2018;10(10):1–13. doi: 10.3390/cancers10100366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takaishi K, Komohara Y, Tashiro H, et al. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Sci. 2010;101(10):2128–2136. doi: 10.1111/j.1349-7006.2010.01652.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woods A, Couchman JR. Syndecan-4 and focal adhesion function. Curr Opin Cell Biol. 2001;13(5):578–583. doi: 10.1016/S0955-0674(00)00254-4 [DOI] [PubMed] [Google Scholar]
- 46.Roskams T, De Vos R, David G, Van Damme B, Desmet V. Heparan sulphate proteoglycan expression in human primary liver tumours. J Pathol. 1998;185(3):290–297. doi: 10.1002/(SICI)1096-9896(199807)185:3<290::AID-PATH91>3.0.CO;2-I [DOI] [PubMed] [Google Scholar]
- 47.Gulyás M, Hjerpe A. Proteoglycans and WTI as markers for distinguishing adenocarcinoma, epithelioid mesothelioma, and benign mesothelium. J Pathol. 2003;199(4):479–487. doi: 10.1002/path.1312 [DOI] [PubMed] [Google Scholar]
- 48.Davies EJ, Blackhall FH, Shanks JH, et al. Distribution and Clinical Significance of Heparan Sulfate Proteoglycans in Ovarian Cancer; 2004. [DOI] [PubMed]
- 49.Aldinucci D, Colombatti A. The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm. 2014;2014. doi: 10.1155/2014/292376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Charnaux N, Brule S, Chaigneau T, et al. RANTES (CCL5) induces a CCR5-dependent accelerated shedding of syndecan-1 (CD138) and syndecan-4 from HeLa cells and forms complexes with the shed ectodomains of these proteoglycans as well as with those of CD44. Glycobiology. 2005;15(2):119–130. doi: 10.1093/glycob/cwh148 [DOI] [PubMed] [Google Scholar]
- 51.Maillard L, Saito N, Hlawaty H, et al. RANTES/CCL5 mediated-biological effects depend on the syndecan-4/PKC signaling pathway. Biol Open. 2014;3(10):995–1004. doi: 10.1242/bio.20148227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garg R, Benedetti LG, Abera MB, Wang H, Abba M, Kazanietz MG. Protein kinase C and cancer: What we know and what we do not. Oncogene. 2014;33(45):5225–5237. doi: 10.1038/onc.2013.524 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.