

Abstract
Understanding cellular heterogeneity is the holy grail of biology and medicine. Cells harboring identical genomes show a wide variety of behaviors in multicellular organisms. Genetic circuits underlying cell-type identities will facilitate the understanding of the regulatory programs for differentiation and maintenance of distinct cellular states. Such a cell-type-specific gene network can be inferred from coregulatory patterns across individual cells. Conventional methods of transcriptome profiling using tissue samples provide only average signals of diverse cell types. Therefore, reconstructing gene regulatory networks for a particular cell type is not feasible with tissue-based transcriptome data. Recently, single-cell omics technology has emerged and enabled the capture of the transcriptomic landscape of every individual cell. Although single-cell gene expression studies have already opened up new avenues, network biology using single-cell transcriptome data will further accelerate our understanding of cellular heterogeneity. In this review, we provide an overview of single-cell network biology and summarize recent progress in method development for network inference from single-cell RNA sequencing (scRNA-seq) data. Then, we describe how cell-type-specific gene networks can be utilized to study regulatory programs specific to disease-associated cell types and cellular states. Moreover, with scRNA data, modeling personal or patient-specific gene networks is feasible. Therefore, we also introduce potential applications of single-cell network biology for precision medicine. We envision a rapid paradigm shift toward single-cell network analysis for systems biology in the near future.
Subject terms: Gene regulatory networks, Bioinformatics
Systems biology: A single-cell, network-driven approach to disease
Gene regulatory networks reconstructed from single-cell RNA sequencing datasets are allowing researchers to better understand the molecular circuits and cell states that contribute to complex human disease. Junha Cha and Insuk Lee from Yonsei University in Seoul, South Korea, review the concept of ‘single-cell network biology’, which involves using computational algorithms on genetic expression data from thousands of cells to infer functional interactions in various biological contexts. This systems biology approach to analyzing the profiles of messenger RNA in single cells is helping researchers discover new signaling pathways that could serve as disease biomarkers or therapeutic targets. In the future, patient-specific models of personal gene networks could explain why certain genetic variants affect disease risk. This research could also eventually lead to new types of individualized medical treatments.
Introduction
The adult human body is composed of ~37 trillion cells1, which are the functional units of organismal systems. Although each cell contains almost identical genomic information, at least several hundred major cell types with distinct morphology, behavior, and functions are expected to exist in the human body. Deviation from the destined identity of functional cells is a major cause of human diseases. Different cellular compositions of tumor tissue may result in different drug responses and prognoses. Disease-associated genetic variants affect only particular cell types, which makes functional validation of candidate variants derived from genome-wide association studies challenging2. Therefore, understanding human body operation at the cellular resolution is the ultimate goal in biology and medicine.
Investigation of individual cell types in vivo is technically challenging. Flow cytometry analysis has been used for single-cell profiling for the past several decades3, albeit with some limitations. First, it is a targeted analysis method for only a preselected set of molecules. Second, due to the spectral limitation of fluorescent proteins, this method can profile up to 17 proteins simultaneously, which is extended to ~40 proteins by mass cytometry4. Recently, we have witnessed a rapid improvement in single-cell RNA sequencing (scRNA-seq) technology, which is indeed a game changer in the field of single-cell biology. Current scRNA-seq technology can easily generate whole-transcriptome data for hundreds to thousands of cells from a single sequencing reaction and identify key genes associated with each cell type or state by differential expression analysis across distinct cellular groups of similar transcriptome. Therefore, we now characterize individual cell types or states in a tissue that is generally composed of diverse cell types. To date, a wide variety of methods for scRNA-seq data generation and analysis have been developed, and they are extensively described in other excellent reviews4–7. Recent benchmarking studies also showed that scRNA-seq protocols differ substantially in their ability to capture RNA, scalability, and cost effectiveness8,9.
Despite much improvement, single-cell omics may not be sufficient for understanding cellular heterogeneity. Although differential expression analysis of scRNA-seq data may identify genes specific to cell types and states, understanding cellular identity simply from a list of up or downregulated genes would be a daunting task because the functional effects of genes depend on their relationships. Gene functions and the effects of disease-associated variants are largely attributable to the interaction partners of these genes in the given cellular context10,11. From a systems biology perspective, network modeling of genes will be highly useful for understanding functional organizations of key regulators involved in operational pathways of each cell state12. Network biology has shifted our perception of a cell from a system mainly comprised of the linear signaling pathways to one occupied by many highly complex intertwined connections among molecules. In particular, the gene regulatory network (GRN) is an intuitive but versatile graph model for functional analysis that has been extensively utilized over the past decade. GRNs have made significant contributions to identifying disease biomarkers and therapeutic targets and were ultimately realized as a crucial tool for deciphering medical genomics data13. Scrutinizing the regulatory interactions between genes in various biological contexts will provide valuable insights into how the emergent functions of a given living system was designed to be regulated.
In this review article, we introduce the definition of single-cell network biology and present the current methodologies to infer GRNs from scRNA-seq data and determine how they can improve our understanding of regulatory circuits for cellular identity and facilitate the practice of precision medicine.
What is single-cell network biology?
Network biology has served as a useful tool for the study of complex cellular systems by providing a glimpse into the functional organization of genes operating in normal and disease states. The GRN is a particularly useful type of gene network that is composed of regulatory relationships inferred from variations across many sources of expression. Typical approaches to analyze GRNs include the identification of hub genes based on network centrality measures14 and functional modules using algorithms for finding network communities15. Network biology has already proven useful for the study of cellular systems, and here, we present an emerging approach in network biology with single-cell transcriptome data, namely, single-cell network biology.
Before the era of single-cell genomics, transcriptomic data were generated from tissue samples using bulk RNA sequencing (bulk RNA-seq). To estimate expression correlation between genes, a large number of expression measurements was generally required, accordingly demanding an equal number of sequencing reactions for tissue-based analysis. Consequently, the correlation of gene expression could be measured through a sample-by-gene matrix (Fig. 1a). Therefore, it is imperative to prepare a large number of samples for network modeling based on bulk RNA-seq data. Conversely, GRNs can be inferred from a single sample preparation followed by a single sequencing reaction with scRNA-seq analysis because it can generate expression measurements for generally hundreds to thousands of individual cells in parallel, generating a cell-by-gene matrix (Fig. 1b). To infer regulatory interactions specific to a particular cell type, we need to divide cells into groups representing cell types using dimension reduction and unsupervised clustering. This procedure provides multiple cell-by-gene matrices for distinct cell types, each of which will be used for building cell-type-specific GRNs. Recently, multiple studies demonstrated that the majority of bulk tissue coregulatory links are explained by “cell-type composition variation” among samples rather than “state variation within a cell type”16,17. Therefore, only a fraction of the network inferred from bulk RNA-seq data might represent true within-cell coregulation between genes (Fig. 1a). In contrast, networks inferred from the cell-by-gene matrix for each cell type mainly represent intra-cell-type coregulatory relations between genes (Fig. 1b).
Fig. 1. Comparison between network inference with bulk RNA-seq and scRNA-seq.

a Network inference with bulk RNA-seq analysis. Multiple tissue samples and sequencing are required to produce a gene-by-sample matrix. Correlation between genes can be detected from both expression variation across cell states and variation of cell-type composition across tissue samples. The resultant coregulatory network is mostly composed of cell-type composition-induced coexpression. b Network inference with scRNA-seq. A single tissue sample is disassociated into cells that are individually analyzed in parallel. Clustering of the cells along with dimension reduction enables the identification of cell populations for each of the major cell types. Using a gene-by-cell count matrix for each cell type, we can infer networks mainly composed of within-cell coregulatory links.
Needless to say, the first benefit of single-cell network biology is its enabling of the reconstruction of cell-type-specific transcriptional regulatory programs. Since the regulatory program specific to each cell type is the core element governing the cellular identity, cell-type-specific GRNs would be key tools for the study of cellular heterogeneity. Furthermore, these cell-type-specific GRNs will reveal key regulatory factors and circuits for specific cell types, facilitating mapping between disease-associated variants and affected cell types. In addition, single-cell network biology provides technical advantages. First, it requires only a small amount of tissue sample for network modeling; even a single biopsy would suffice with adequately high throughput. Second, it can infer regulatory networks from single cells at various levels of cellular identities: major types, subtypes, or states. Third, it can infer regulatory networks from single cells of each person, resulting in personalized GRNs. Thus, in this aspect, single-cell network biology is cost-effective and highly flexible and provides a personalized platform for biomedical research.
Network inference from single-cell gene expression data
Various algorithms for inferring regulatory interactions between genes using bulk transcriptome data have been developed. Popular approaches to network inference from bulk transcriptome data are based on Boolean networks, Bayesian networks, ordinary differential equations (ODEs), information theory, regression, and correlation18–20. Although these methods can be directly applied to single-cell transcriptome data with some adjustment, network inference algorithms specifically developed for single-cell transcriptome data are also available.
Since single-cell transcriptome data can be ordered by pseudotime, many algorithms to infer regulatory networks based on time-ordered transcriptomes have been explicitly developed. The basic assumption of trajectory analysis is that each cell lies in a continuous process of cellular differentiation. The trajectory reconstructed by “pseudotemporal” ordering of cells can then be used for network inference. However, the lack of consensus among resultant trajectories implies that the performance of the network inference with pseudotime information will greatly depend on the trajectory analysis algorithm. Pseudotime information has been used to reconstruct GRNs21–24 from single-cell transcriptome data. A recent benchmarking study, however, showed that the methods that do not require pseudotime information performed better25.
There are a wide variety of metrics that can be used for measuring coregulatory associations between genes, but their application for single-cell transcriptome data was mostly unsatisfactory26. Another benchmarking study concluded that most of the currently available methods for regulatory network inference are not effective for single-cell transcriptome data, even those explicitly developed for single-cell studies27. The high proportion of false-positive network links inferred from single-cell gene expression data may be attributable to the intrinsic sparsity and high technical variation. Although these benchmarking results may suggest a lack of general applicability of network inference methods for single-cell biology, caution is advised in making such conclusions. The true positive regulatory links used for evaluation may not accurately represent the ground truth of the regulatory gene network in the tested cell types or states. In addition, the optimal network inference method for given single-cell data could vary across cell types.
As this review focuses on the application of single-cell network biology, we only provide a brief description of major approaches to GRN inference from single-cell transcriptome data. More extensive reviews about computational algorithms are available from other recent publications28,29.
Boolean models
A Boolean network is the simplest approach to reconstructing regulatory gene networks30. In systems biology, a Boolean network refers to a set of genes with binary states (activated or repressed)31. This approach is often used to describe the interaction between mRNAs and proteins to predict gene patterns32. In this network, each cell is classified into a certain state, and similar cells are then connected. The resulting state-cell graph provides useful information about key regulators that drive certain cellular states. Its simplicity allows the resulting network to be determined with as few assumptions as possible, with one naturally being that all genes must follow a binary law. Single-cell Boolean GRNs have been successfully applied to predict curated models of hematopoiesis33–35. A drawback of this approach is the computational burden. Thus, Boolean-based tools have limited scalability, which will prevent users from building a genome-scale network. Therefore, users must carefully select the genes they wish to model, which is usually no more than 100 genes. The Partially Observed Boolean Dynamical system model is a framework for modeling the behavior of GRNs, and this approach allows indirect and incomplete observation of gene states and has been explored for application to scRNA-seq data36.
Ordinary differential equation (ODE) models
GRN modeling via ODE focuses on a series of discrete states to capture the dynamics of the network in question. While other methods discretize variables, ODE uses continuous variables and is one of the popular methods to map a dynamic system of gene regulation. To date, ODE is the best analyzed approach for nonlinear systems37. In this model, the change in expression over continuous time is characterized by a function that takes the inhibitory or activating influence of other genes as variables18. This approach is most suitable for identifying a process in a system that is assumed to be continuous (e.g., differentiation). The input time scale could be either an inferred pseudotime or metadata from a time-series experiment. SCODE38 is a network construction tool that relies on ODE to map differentiation in single-cell transcriptome data. Some tools based on ODE assume a steady state condition39,40, which makes them suboptimal for differentiation-related analysis.
Regression models
Most regression-based network inference tools follow an underlying assumption that the expression of all genes can be summarized as a simple weighted linear equation. For this assumption to hold true and produce a reliable prediction, the variables of the data must be independent, and the residuals (errors of fitted linear model) must follow a normal distribution, which is not usually the case for current single-cell transcriptome data. Therefore, most network inference tools based on regression models must be adjusted by a statistical trick (e.g., polynomial modeling, data transformation) to bypass these assumptions. Users must be careful so that this preprocessing step does not compromise the overall structure of the data. In this approach, users may need to provide a list of regulators such as transcription factors (TFs) as input data. Then, the network inference algorithm deconstructs the problem of explaining the expression of a certain target gene with a set of regulators. Here, each subproblem is viewed as a feature selection. Regression-based approaches not only estimate the underlying association between regulators and target genes but also infer the association intensities. The success in ensemble of regression trees (random forest) by GENIE341 has led to this approach being widely used for network inference from both bulk and single-cell transcriptome data. However, GENIE3 calculation is not feasible for data from more than several thousand cells. Subsequently, a much faster and more scalable assembly method for regression trees, GRNBoost, was developed42,43. Regulatory networks inferred from single-cell regression analysis tend to have more false-positive links than those inferred by bulk transcriptome regression analysis. To reduce false-positive links, networks inferred from GENIE3 or GRNBoost were filtered for putative direct-binding targets based on TF binding motif enrichment in the SCENIC software package42.
Correlation and other association models
GRNs based on coregulatory interactions are commonly inferred from correlations between genes across sources of expression variation44. Common measures of expression correlation between genes are the Pearson correlation coefficient and rank-based Spearman correlation coefficient. Sources of expression variation are not limited to cell state differences. A large portion of variation can originate from various technical factors, which can easily create confounding effects in correlation inference. Batch effects across samples can also generate nonbiological variation. Because single-cell transcriptomic data are associated with high noise and sparsity, the effect of technical variation could be more critical for single-cell coregulatory network inference. An evaluation of coexpression-based network inference with scRNA-seq data from 31 individual studies comprising 163 cell types showed lower retrieval of known functional links than those inferred from bulk RNA-seq data45. The same study also showed reduced performance of coexpression-based network inference with the normalization of UMI data, probably due to unintended covariation, particularly among low-expressing genes. The improved performance with batch-corrected UMI data45, however, suggests that with single-cell coexpression-based network inference, extra care is needed for handling technical variations.
Mutual information (MI) can also measure associations between genes based on expression profiles, and it is particularly useful for mapping nonlinear associations46. In constructing a coexpression network from scRNA-seq data, users must consider the various technical properties distinct among different sequencing platforms that govern single-cell transcriptome data. An algorithm of MI-based network inference has been explicitly developed for single-cell transcriptome data47.
The coregulatory association between genes with multiple sources of expression variations can be measured by many other metrics. Recently, 17 distinct measures of association for inferring gene networks were evaluated and showed that proportionality measures performed best across multiple scRNA-seq datasets and technologies26. The compositional nature of transcriptomic data, in which only the relative abundance of transcripts is measured per sample48, may contribute to the high performance because scRNA-seq currently only captures a small proportion of the total transcripts per cell. It is, however, noteworthy that all the association measures, including proportionality, assessed in this study barely performed above random expectation, suggesting that the high noise and sparsity of scRNA-seq data must be addressed during data preprocessing before network inference. One such effort recently developed is a method for measuring correlation with scRNA-seq data by pooling cells considered biological replicates and transforming the count matrix to z scores, which dramatically increases correlation between genes and facilitates network inference49.
Network filtration for single-cell gene expression
While the “bottom-up approaches” are mainly used to infer cell-type-specific networks from gene expression data, they can also be constructed by filtration of reference gene networks through single-cell gene expression data (referred to as the “top–down approach”). In this approach, single-cell transcriptome data that contain multiple factors are used to fine-tune the reference network to reflect specific context. Gene network databases, such as STRING50, HumanNet51, and PCNET52 provide high-confidence gene functional links. Filtering the global networks for expressed genes for a distinct cell type will result in a cell-type-specific network. The “top–down approach” for constructing context-specific networks with bulk RNA-seq data has already been applied to cancer research. Prognostic biomarkers of ovarian cancer and leukemia have been identified by filtering the global protein–protein interaction network for disease specificity53. Sample-specific network54 analysis has been shown to be more effective for identifying driver genes in individual tumors55, and aggregating these drivers across cancers may reveal new insights into precision cancer therapy.
SCINET56 is a recent computational framework that allows optimal filtering of the reference network to obtain a cell-type-specific network according to the input single-cell data. Using these cell-type-specific networks, the authors showed that disease-associated genes tend to interact with each other with cell-type specificity, with marker genes showing higher cell-type-specific centralities than those in the global network by integration of cell-type-specific networks. This analytical framework, which can be generally applied to any reference network and any single-cell expression dataset, enables researchers to infer cell types and cell-type-specific modules governing certain disorders.
Hypothesis generation in single-cell network biology
Global gene networks inferred from diverse biological contexts have proven useful in generating hypotheses of the functions and phenotypic effects of genes via network centrality and information propagation through the network. Moreover, analysis of network communities can elucidate pathways or functional modules for complex phenotypes such as diseases57. Cell-type-specific networks along with single-cell gene expression data can extend the power of network biology to explain the cellular heterogeneity underlying phenotypes of multicellular organisms such as human diseases. Major strategies for hypothesis generation in single-cell network biology (summarized in Table 1) are based on identifying context-associated subnetworks and utilizing topological dynamics. In addition, analysis of personalized gene networks along with genotype information can elucidate network-mediated effects of disease-associated genetic variants.
Table 1.
Summary of hypothesis generation through single-cell network biology.
| Approaches | Advantages | Limitations | References |
|---|---|---|---|
| Subnetwork analysis of module and regulon activities (Fig. 2) | Enabling the identification of key regulators that are associated with a disease-associated phenotype at cell-type resolutions |
1. Various parameters to adjust for module identification. Difficult to choose optimal parameters without some form of prior knowledge of functional gene sets. 2. No definitive method exists for detecting regulatory links. Inferred links will vary depending on applied network-inference algorithm. |
42,58 |
| Topology analysis of disease-associated cell-type-specific network (Fig. 3) | Graph-based, intuitive, and comprehensive methods for prioritization of genes associated with a disease-associated phenotype at cell-type resolutions |
1. Various measures of network centrality (hubness). Different centrality measures may predict different candidate genes. 2. Various experimental and technical factors must be taken into consideration that might affect network topological changes. |
49,56 |
| Genotype-network association and coexpression QTL analysis (Fig. 4) |
1. Considerable amount of false-positive SNPs may be removed from cell-type specificity. 2. Significantly fewer number of samples needed compared to eQTL studies through the bulk counterpart. |
1. Compared to other types of single-cell studies, relatively large number of patient samples may be necessary. 2. Doublets (two genotypes barcoded in a single cell) must be taken into consideration and processed during demultiplexing of data. |
77 |
Hypothesis from subnetwork analysis
Pathways rather than individual genes are the functional units of cells. Thus, pathway-based functional interpretation of cellular states is more intuitive than gene-based interpretation. Weighted correlation network analysis (WGCNA)58 has been a popular tool for identifying functional modules based on coexpression networks inferred from a large number of gene expression profiles. WGCNA with single-cell transcriptome data for a cell type may identify functional modules that are associated with a particular state (e.g., disease-related state) of the cell type. Often, by using topological properties (e.g., centrality) or external functional information, we may be able to identify key regulators of functional modules and, in turn, the associated cellular states (Fig. 2a). For example, WGCNA along with scRNA-seq data from early embryo cells revealed that each stage of the early development of mouse and human embryos can be delineated by a few functional modules59. WGCNA on single-cell transcriptome data also enabled the discovery of signals that activate dormant neural stem cells in nonneurogenic brain regions60, regulators of chemotherapy resistance in esophageal squamous cell carcinoma61 and prognostic markers for prostate cancer62. The WGCNA package requires users to adjust various parameters so that appropriate modules are defined, and this may often become a potential difficulty in the absence of prior knowledge of disease-associated gene sets.
Fig. 2. Hypothesis generation from subnetwork analysis in single-cell network biology.

a Weighted correlation network analysis (WGCNA) of scRNA-seq data generally reveals multiple modules (M1-5) of coexpressed genes of various sizes. The activity of modules can be measured by the average gene expression level. Module activity may significantly differ between cells from different states (e.g., cells of disease samples versus those of healthy controls), which suggests that this coexpressed module is associated with the disease state and may contain key regulators for the disease, often those with high network centrality. b Transcription factor (TF)-target interaction inference generates a set of regulons (R1-5) that are genes regulated by each TF. Comparison of regulon activity between healthy and disease states, similar to module activity, can suggest its association with the disease state. Then, the TF for the associated regulon is predicted to be a key regulator. These candidate regulators are often subjected to experimental validation and gene set enrichment analysis (GSEA) for functional interpretation.
Subnetworks composed of a TF and its target genes are also useful for functional analysis in single-cell network biology. Here, a set of target genes regulated by each TF is called a regulon. SCENIC42 is a popular software tool for the generation of TF-regulon subnetworks for given scRNA-seq data and their downstream analysis. In this analytical platform, individual cells or subpopulations that represent a particular cell state can be depicted by the activity of each regulon. Because each regulon is considered a regulatory unit, regulon activity profiles across cellular states can suggest GRNs governing cellular identity or transitions. Moreover, regulon analysis facilitated the identification of key regulators for cellular states and interpretation of their target pathways by gene set enrichment analysis for the regulon genes (Fig. 2b). In a recent study, regulon-based analysis of scRNA-seq data of patient-derived melanoma cultures revealed key regulators and GRNs specific for intermediate states during the epithelial–mesenchymal transition of melanoma cells63, which may provide new therapeutic targets to prevent the acquisition of metastatic potential and drug resistance due to cell state switching.
Hypothesis from network topology analysis
Emergent cellular phenotypes depend not only on genotypes but also on edgotypes, context-specific networks of molecular interactions64, implying that the dynamics of regulatory interactions underlie cellular heterogeneity. Comparisons between cell-type-specific networks for different states, such as disease and healthy states, will show topological changes for each gene in centrality (hubness) and neighbors (targets). Genes that show significant changes in one of these topological properties would be candidate regulators involved in the cellular state of interest (Fig. 3). For example, a recent study generated healthy and type 2 diabetes (T2D) regulatory networks using scRNA-seq data from pancreatic islet cells49. The study demonstrated that many genes with significant changes in centrality are involved in T2D. Another study generated GRNs for self-renewing cells, erythroid-committed progenitors, and myeloid-committed progenitors and demonstrated that the lineage regulator DDIT3 changes its targets in three different GRNs65. Gene sets involved in particular biological processes or diseases may also change their modularity (intraconnectivity) between different cellular states, which suggests their association with a particular cellular state (e.g., disease-related state). For example, gene networks were generated for six brain cell types56, in which neuropsychiatric disorder genes were found to preferentially interact in neuronal cells, whereas genes for neurodegenerative diseases do so in glial cells. Another recent study demonstrated that modularity measures based on the enrichment of coexpression among genes associated with specific neurodevelopmental disorders increased in specific cell types66. These results suggest that disease-related genes tend to preferentially interact with cell types for different disease classes. Although network topology analysis offers an intuitive method for observing a cell-type-specific system, a large number of links and the associated complexity potentially cause difficulty in interpretation. Researchers must also take into account that many experimental and technical factors must be controlled to accurately compare different networks.
Fig. 3. Hypothesis generation from network topology analysis in single-cell network biology.
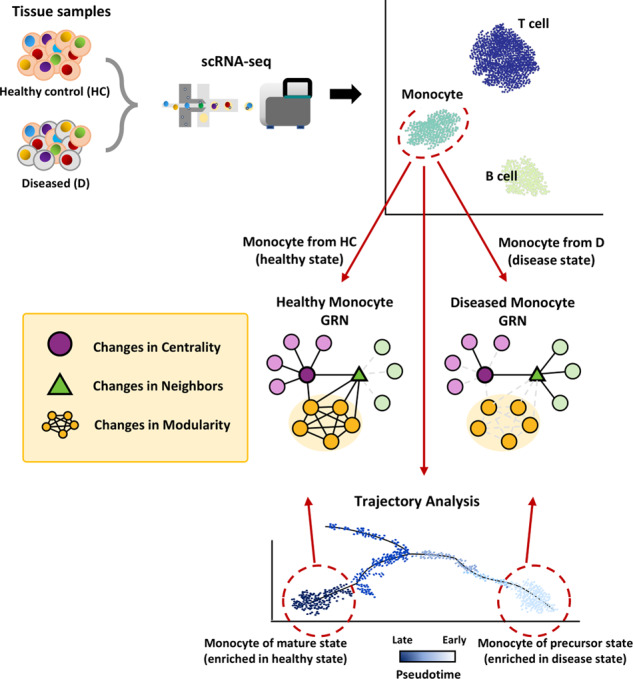
Inferences in coregulatory transcriptome profiles of cells from two distinct states (healthy control versus disease state) lead to the construction of different GRNs. Genes that show changes in three types of network topology are likely to be associated with the state: centrality, neighbors, and modularity. For example, correlation analyses for monocytes from healthy and diseased samples may generate different networks, and changes in three types of topology between them will be examined for every gene. Similarly, networks for different developmental times along with topological analysis would suggest disease-associated genes because many disease states are associated with defects in development. For example, defects in the maturation of monocytes into functional dendritic cells would result in immune disorders.
Hypothesis from genotype-network association
A major problem in health care today is imprecision medicine, wherein only a small portion of patients respond to routinely prescribed drugs67. This may be because patients have different genetic variations that influence the functional effects of genes involved in pathogenesis or pharmacodynamics. The majority of such variations exert phenotypic effects via the action of expression quantitative trait loci (eQTLs)68 because most of them are located within noncoding regions69. The eQTLs have long been suggested to exert their influence in a cell-specific manner, and the large portion of unresolved eQTLs may be attributable to the cell-type dependent effects of these eQTLs70,71. Cell-type-specific eQTL analysis can be conducted by sorting each cell type, which generally has a high cost. As scRNA-seq can provide transcriptome data for multiple cell types of a given tissue simultaneously, it can greatly facilitate cell-type-specific eQTL analysis72–75 (Fig. 4a). Cell-type-specific eQTL studies may possibly reduce the detection of false-positive SNPs associated with disease that have often emerged as potential limitations of bulk RNA-seq-based eQTL research (e.g., Simpson’s paradox). Moreover, utilizing a large number of cells in single-cell datasets may significantly reduce the number of samples required for eQTL detection. It is noteworthy, however, that for more accurate analysis, this approach will require a larger number of donors than typical single-cell-based studies.
Fig. 4. Hypothesis generation from genotype-network association in single-cell network biology.

a Many disease-associated single nucleotide polymorphisms (SNPs), which are called expression QTLs (eQTLs), exert phenotypic effects through the regulation of gene expression in a cell type-specific manner. Therefore, eQTL analysis needs to be conducted for specific cell types, particularly for minor cell types. The recently developed multiplexed scRNA-seq technology along with demultiplexing based on genotype information will facilitate cell-type-specific eQTL mapping. b Some eQTL effects are dependent on the expression of other genes. This dependency is detected by genotype-specific coexpression, called coexpression QTL (cxQTL). Here, a disease gene X is coexpressed with gene Y only if its eQTL has a homozygous major allele (AA). c If the gene Y is a target of drug A that eventually inhibits the activity of disease gene X via interaction with gene Y, the genotype-dependent coregulatory interaction between genes X and Y is critical for drug action. Then, for prescription of drug A, the cxQTL genotype information can be utilized for precision medicine (e.g., prescribing it only for patients with SNP AA).
Interestingly, some eQTL effects of a gene can be modified by the expression of another gene76 (Fig. 4b). For example, the effect of a FADS2 eQTL is modulated by the expression of the sterol binding factor gene SREBF2. Therefore, these genetic variants are called coexpression QTLs, because they affect the coregulatory relationship between two genes76,77. Single-cell transcriptome data from each person can be sufficient to infer gene–gene correlation, building personalized GRNs77,78. Given that personal- and cell-type-specific coregulatory relationships between genes can be modeled using scRNA-seq data, we may test whether personal genetic variants affect disease risk or drug response by altering coregulatory interactions. If a coregulatory interaction between a disease gene and a drug target that affects the disease gene activity is modulated by a coexpression QTL, this genotype information could be utilized in tailored prescription for individual patients in the future (Fig. 4c).
Challenges and future perspectives
The major challenges in single-cell network biology are associated with the single-cell omics technology, as the quality of inferred networks relies largely on the quality of single-cell transcriptome data. Single-cell profiling technologies are rapidly evolving. However, various technical hurdles, such as low capture efficiency, high dropout rates, and high noise in signals, must be considered and overcome to observe true biological variations in gene expression6. New computational methods need to be developed to overcome those intrinsic limitations of scRNA-seq data. For example, imputation of dropouts in single-cell transcriptome data will vary in the probability of false gene–gene correlations79, and the methods need to be further improved in the future. In addition, the integration of multimodal single-cell omics data80 and multiomics data81 would contribute in improving network inference and interpretations.
Many statistical approaches have been developed to address these issues, and depending on the basic assumptions that the researchers are willing to adhere to an appropriate method must be chosen for different datasets. Each algorithm with its own preprocessing steps will result in different networks. Therefore, preprocessing of the single-cell dataset will be the critical step of the network inference algorithm. Moreover, network inference tools with different algorithmic concepts will perform optimally for different sets of data (e.g., time series, developmental, perturbation). Therefore, researchers must choose their methods depending on the data that they have collected and the system that they wish to evaluate. Different types of networks (regulatory or functional) will provide different insights, and it is important to extract reasonable conclusions allowed from numerous types of networks and make suitable predictions.
In this paper, we highlighted the effectiveness of using network-based studies in resolving cellular heterogeneity. Personalized gene networks obtained from single-cell transcriptome data will facilitate the development of novel applications based on personal genetic variation for precision medicine. For translation of single-cell network analysis to clinical settings, user-friendly analytical pipelines must be established for different types of diseases. These efforts together will improve our ability to accurately diagnose and predict disease risks and ultimately lead to the development of precision medicine.
Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2018M3C9A5064709, 2018R1A5A2025079, and 2019M3A9B6065192).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bianconi E, et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013;40:463–471. doi: 10.3109/03014460.2013.807878. [DOI] [PubMed] [Google Scholar]
- 2.Cano-Gamez E, Trynka G. From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front Genet. 2020;11:424. doi: 10.3389/fgene.2020.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKinnon KM. Flow cytometry: an overview. Curr. Protoc. Immunol. 2018;120:5 1 1–5 1 11. doi: 10.1002/cpim.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papalexi E, Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018;18:35–45. doi: 10.1038/nri.2017.76. [DOI] [PubMed] [Google Scholar]
- 5.Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol. Cell. 2015;58:610–620. doi: 10.1016/j.molcel.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 2019;15:e8746. doi: 10.15252/msb.20188746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018;50:96. doi: 10.1038/s12276-018-0071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding J, et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020;38:737–746. doi: 10.1038/s41587-020-0465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mereu E, et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat. Biotechnol. 2020;38:747–755. doi: 10.1038/s41587-020-0469-4. [DOI] [PubMed] [Google Scholar]
- 10.Kachroo AH, et al. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science. 2015;348:921–925. doi: 10.1126/science.aaa0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sahni N, et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell. 2015;161:647–660. doi: 10.1016/j.cell.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat. Rev. Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 13.Emmert-Streib F, Dehmer M, Haibe-Kains B. Gene regulatory networks and their applications: understanding biological and medical problems in terms of networks. Front Cell Dev. Biol. 2014;2:38. doi: 10.3389/fcell.2014.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang W, et al. Identification of novel hub genes associated with liver metastasis of gastric cancer. Int J. Cancer. 2009;125:2844–2853. doi: 10.1002/ijc.24699. [DOI] [PubMed] [Google Scholar]
- 15.Vlaic S, et al. ModuleDiscoverer: identification of regulatory modules in protein-protein interaction networks. Sci. Rep. 2018;8:433. doi: 10.1038/s41598-017-18370-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farahbod, M. & Pavlidis, P. Untangling the effects of cellular composition on coexpression analysis. Genome Res. 30, 849–859 (2020). [DOI] [PMC free article] [PubMed]
- 17.Zhang, Y., Cuerdo, J., Halushka, M. K. & McCall, M. N. The effect of tissue composition on gene co-expression. Brief Bioinform. bbz135 (2019). [DOI] [PMC free article] [PubMed]
- 18.Lee WP, Tzou WS. Computational methods for discovering gene networks from expression data. Brief. Bioinform. 2009;10:408–423. doi: 10.1093/bib/bbp028. [DOI] [PubMed] [Google Scholar]
- 19.Delgado FM, Gomez-Vela F. Computational methods for gene regulatory networks reconstruction and analysis: a review. Artif. Intell. Med. 2019;95:133–145. doi: 10.1016/j.artmed.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Marbach D, et al. Wisdom of crowds for robust gene network inference. Nat. Methods. 2012;9:796–804. doi: 10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ocone A, Haghverdi L, Mueller NS, Theis FJ. Reconstructing gene regulatory dynamics from high-dimensional single-cell snapshot data. Bioinformatics. 2015;31:i89–i96. doi: 10.1093/bioinformatics/btv257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papili Gao N, Ud-Dean SMM, Gandrillon O, Gunawan R. SINCERITIES: inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics. 2018;34:258–266. doi: 10.1093/bioinformatics/btx575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Specht AT, Li J. LEAP: constructing gene co-expression networks for single-cell RNA-sequencing data using pseudotime ordering. Bioinformatics. 2017;33:764–766. doi: 10.1093/bioinformatics/btw729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamey FK, et al. Reconstructing blood stem cell regulatory network models from single-cell molecular profiles. Proc. Natl Acad. Sci. USA. 2017;114:5822–5829. doi: 10.1073/pnas.1610609114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pratapa A, Jalihal AP, Law JN, Bharadwaj A, Murali TM. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods. 2020;17:147–154. doi: 10.1038/s41592-019-0690-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skinnider MA, Squair JW, Foster LJ. Evaluating measures of association for single-cell transcriptomics. Nat. Methods. 2019;16:381–386. doi: 10.1038/s41592-019-0372-4. [DOI] [PubMed] [Google Scholar]
- 27.Chen S, Mar JC. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinforma. 2018;19:232. doi: 10.1186/s12859-018-2217-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fiers M, et al. Mapping gene regulatory networks from single-cell omics data. Brief. Funct. Genomics. 2018;17:246–254. doi: 10.1093/bfgp/elx046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blencowe M, et al. Network modeling of single-cell omics data: challenges, opportunities, and progresses. Emerg. Top. Life Sci. 2019;3:379–398. doi: 10.1042/ETLS20180176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akutsu, T., Miyano, S. & Kuhara, S. Identification of genetic networks from a small number of gene expression patterns under the Boolean network model. Pac Symp Biocomput. 17–28. (1999). [DOI] [PubMed]
- 31.Lahdesmaki H, Shmulevich I, Yli-Harja O. On learning gene regulatory networks under the Boolean network model. Mach. Learn. 2003;52:147–167. [Google Scholar]
- 32.Saadatpour A, Albert R. Boolean modeling of biological regulatory networks: a methodology tutorial. Methods. 2013;62:3–12. doi: 10.1016/j.ymeth.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 33.Lim CY, et al. BTR: training asynchronous Boolean models using single-cell expression data. Bmc Bioinforma. 2016;17:355. doi: 10.1186/s12859-016-1235-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moignard V, et al. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat. Biotechnol. 2015;33:269–276. doi: 10.1038/nbt.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen H, et al. Single-cell transcriptional analysis to uncover regulatory circuits driving cell fate decisions in early mouse development. Bioinformatics. 2015;31:1060–1066. doi: 10.1093/bioinformatics/btu777. [DOI] [PubMed] [Google Scholar]
- 36.Bahadorinejad, A., Imani, M. & Braga-Neto, U. Adaptive particle filtering for fault detection in partially-observed boolean dynamical systems. IEEE/ACM Trans Comput Biol Bioinform. 17, 1105–1114 (2018). [DOI] [PubMed]
- 37.Chai LE, et al. A review on the computational approaches for gene regulatory network construction. Comput Biol. Med. 2014;48:55–65. doi: 10.1016/j.compbiomed.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 38.Matsumoto H, et al. SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics. 2017;33:2314–2321. doi: 10.1093/bioinformatics/btx194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gardner TS, di Bernardo D, Lorenz D, Collins JJ. Inferring genetic networks and identifying compound mode of action via expression profiling. Science. 2003;301:102–105. doi: 10.1126/science.1081900. [DOI] [PubMed] [Google Scholar]
- 40.Polynikis A, Hogan SJ, di Bernardo M. Comparing different ODE modelling approaches for gene regulatory networks. J. Theor. Biol. 2009;261:511–530. doi: 10.1016/j.jtbi.2009.07.040. [DOI] [PubMed] [Google Scholar]
- 41.Huynh-Thu, V. A., Irrthum, A., Wehenkel, L. & Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS One5, e12776 (2010). [DOI] [PMC free article] [PubMed]
- 42.Aibar S, et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moerman T, et al. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics. 2019;35:2159–2161. doi: 10.1093/bioinformatics/bty916. [DOI] [PubMed] [Google Scholar]
- 44.Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- 45.Crow M, Paul A, Ballouz S, Huang ZJ, Gillis J. Exploiting single-cell expression to characterize co-expression replicability. Genome Biol. 2016;17:101. doi: 10.1186/s13059-016-0964-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song L, Langfelder P, Horvath S. Comparison of co-expression measures: mutual information, correlation, and model based indices. BMC Bioinforma. 2012;13:328. doi: 10.1186/1471-2105-13-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan TE, Stumpf MPH, Babtie AC. Gene regulatory network inference from single-cell data using multivariate information Measures. Cell Syst. 2017;5:251–267 e253. doi: 10.1016/j.cels.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quinn TP, Richardson MF, Lovell D, Crowley T. M. propr: an R-package for identifying proportionally abundant features using compositional data analysis. Sci. Rep. 2017;7:16252. doi: 10.1038/s41598-017-16520-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iacono G, Massoni-Badosa R, Heyn H. Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol. 2019;20:110. doi: 10.1186/s13059-019-1713-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szklarczyk D, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hwang S, et al. HumanNet v2: human gene networks for disease research. Nucleic Acids Res. 2019;47:D573–D580. doi: 10.1093/nar/gky1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang JK, et al. Systematic evaluation of molecular networks for discovery of disease genes. Cell Syst. 2018;6:484–495 e485. doi: 10.1016/j.cels.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan X, et al. Network biomarkers constructed from gene expression and protein-protein interaction data for accurate prediction of leukemia. J. Cancer. 2017;8:278–286. doi: 10.7150/jca.17302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu X, Wang Y, Ji H, Aihara K, Chen L. Personalized characterization of diseases using sample-specific networks. Nucleic Acids Res. 2016;44:e164. doi: 10.1093/nar/gkw772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo WF, et al. Discovering personalized driver mutation profiles of single samples in cancer by network control strategy. Bioinformatics. 2018;34:1893–1903. doi: 10.1093/bioinformatics/bty006. [DOI] [PubMed] [Google Scholar]
- 56.Mohammadi S, Davila-Velderrain J, Kellis M. Reconstruction of cell-type-specific interactomes at single-cell resolution. Cell Syst. 2019;9:559–568 e554. doi: 10.1016/j.cels.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shim JE, Lee T, Lee I. From sequencing data to gene functions: co-functional network approaches. Anim. Cells Syst. 2017;21:77–83. doi: 10.1080/19768354.2017.1284156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xue Z, et al. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature. 2013;500:593–597. doi: 10.1038/nature12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luo Y, et al. Single-cell transcriptome analyses reveal signals to activate dormant neural stem cells. Cell. 2015;161:1175–1186. doi: 10.1016/j.cell.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu H, et al. Single-cell transcriptome analyses reveal molecular signals to intrinsic and acquired paclitaxel resistance in esophageal squamous cancer cells. Cancer Lett. 2018;420:156–167. doi: 10.1016/j.canlet.2018.01.059. [DOI] [PubMed] [Google Scholar]
- 62.Chen, X., Hu, L., Wang, Y., Sun, W. & Yang, C. Single cell gene co-expression network reveals FECH/CROT signature as a prognostic marker. Cells8, 698 (2019). [DOI] [PMC free article] [PubMed]
- 63.Wouters, J. et al. Single-cell gene regulatory network analysis reveals new melanoma cell states and transition trajectories during phenotype switching. bioRxiv 715995 (2019)
- 64.Sahni N, et al. Edgotype: a fundamental link between genotype and phenotype. Curr. Opin. Genet Dev. 2013;23:649–657. doi: 10.1016/j.gde.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pina C, et al. Single-cell network analysis identifies DDIT3 as a nodal lineage regulator in hematopoiesis. Cell Rep. 2015;11:1503–1510. doi: 10.1016/j.celrep.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pang, K. et al. Coexpression enrichment analysis at the single-cell level reveals convergent defects in neural progenitor cells and their cell-type transitions in neurodevelopmental disorders. Genome Res.30, 835–548 (2020). [DOI] [PMC free article] [PubMed]
- 67.Schork NJ. Personalized medicine: time for one-person trials. Nature. 2015;520:609–611. doi: 10.1038/520609a. [DOI] [PubMed] [Google Scholar]
- 68.Nica AC, Dermitzakis ET. Expression quantitative trait loci: present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013;368:20120362. doi: 10.1098/rstb.2012.0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brown CD, Mangravite LM, Engelhardt BE. Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eQTLs. PLoS Genet. 2013;9:e1003649. doi: 10.1371/journal.pgen.1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fairfax BP, et al. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat. Genet. 2012;44:502–510. doi: 10.1038/ng.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kang HM, et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018;36:89–94. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van der Wijst M, et al. The single-cell eQTLGen consortium. Elife. 2020;9:e52155. doi: 10.7554/eLife.52155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sarkar AK, et al. Discovery and characterization of variance QTLs in human induced pluripotent stem cells. PLoS Genet. 2019;15:e1008045. doi: 10.1371/journal.pgen.1008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cuomo ASE, et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat. Commun. 2020;11:810. doi: 10.1038/s41467-020-14457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhernakova DV, et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nat. Genet. 2017;49:139–145. doi: 10.1038/ng.3737. [DOI] [PubMed] [Google Scholar]
- 77.van der Wijst MGP, et al. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet. 2018;50:493–497. doi: 10.1038/s41588-018-0089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van der Wijst MGP, de Vries DH, Brugge H, Westra HJ, Franke L. An integrative approach for building personalized gene regulatory networks for precision medicine. Genome Med. 2018;10:96. doi: 10.1186/s13073-018-0608-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Andrews TS, Hemberg M. False signals induced by single-cell imputation. F1000Res. 2018;7:1740. doi: 10.12688/f1000research.16613.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stuart T, Satija R. Integrative single-cell analysis. Nat. Rev. Genet. 2019;20:257–272. doi: 10.1038/s41576-019-0093-7. [DOI] [PubMed] [Google Scholar]
- 81.Jung GT, Kim KP, Kim K. How to interpret and integrate multi-omics data at systems level. Anim. Cells Syst. 2020;24:1–7. doi: 10.1080/19768354.2020.1721321. [DOI] [PMC free article] [PubMed] [Google Scholar]