

Abstract
Aim:
Type 1 diabetes (T1D) is a chronic autoimmune disease leading to progressive loss of pancreatic beta cells. Interferon (IFN)-α plays a critical role in the crosstalk between pancreatic beta cells and the immune system in early insulitis. In human beta cells IFNα signals through JAK1 and TYK2, leading to endoplasmic reticulum stress, inflammation and HLA class I overexpression. IFNα, acting synergistically with IL-1β, induces apoptosis. Polymorphisms in TYK2 that decrease its activity are associated with protection against T1D, and we hypothesized that pharmacological inhibitors that specifically target TYK2 could protect human beta cells against the deleterious effects of IFNα.
Materials and Methods:
Two TYK2 inhibitors provided by Nimbus Lakshmi were tested in human insulin-producing EndoC-βH1 cells and human islets to evaluate their effect on IFNα signalling, beta-cell function and susceptibility to viral infection using RT-qPCR, western blot, immunofluorescence, ELISA and nuclear dyes.
Results:
The two TYK2 inhibitors tested prevented IFNα-induced human beta-cell gene expression in a dose-dependent manner. They also protected human islets against IFNα + IL-1β-induced apoptosis. Importantly, these inhibitors did not modify beta-cell function or their survival following infection with the potential diabetogenic coxsackieviruses CVB1 and CVB5.
Conclusions:
The two TYK2 inhibitors tested inhibit the IFNα signalling pathway in human beta cells, decreasing its pro-inflammatory and pro-apoptotic effects without sensitizing the cells to viral infection. The preclinical findings could pave the way for future clinical trials with TYK2 inhibitors for the prevention and treatment of type 1 diabetes.
Keywords: antidiabetic drug, cellular research, drug mechanism, islets, type 1 diabetes
1 |. INTRODUCTION
Interferon-α (IFNα)-mediated signalling is an integral component of type 1 diabetes (T1D) pathophysiology. In line with this concept, type I IFN is expressed in human islets from donors with T1D,1 and children genetically at risk for the disease present a type I IFN transcriptomic signature in the circulation before the development of autoantibodies.2 In addition, laser-captured islets obtained from living donors with recent-onset T1D show a significant increase in the expression of IFN-stimulated genes3 and human islets exposed in vitro to IFNα or to interleukin-1β (IL-1β) + IFNγ have gene signatures that are similar to those observed in islets obtained from patients affected by T1D, but not by type 2 diabetes.4,5 We recently found that exposure of human beta cells to IFNα induces three hallmarks of human T1D, namely, release of chemokines, endoplasmic reticulum (ER) stress and a massive human leukocyte antigen (HLA) class I overexpression. IFNα also triggers human beta-cell apoptosis when combined with IL-1β.6 These effects are mediated via the two kinases, janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2), and the transcription factors, signal transducer and activator of transcription 1 and 2 (STAT1 and STAT2). We identified the TYK2/STAT2/IFN regulatory factor 9 (IRF9) axis as a critical pathway responsible for the action of IFNα in human beta cells.6,7 TYK2 is a candidate gene for T1D and genetic variants that decrease TYK2 activity and which are protective against the disease.8,9 Notably, individuals with the protective variants of TYK2 do not have increased prevalence of infections and are not immunodeficient, as is the case for individuals with a complete TYK2 deficiency.9,10 Consistent with this observation, a partial inhibition of TYK2 expression in human beta cells using siRNAs protects them against the viral by-product, double-stranded RNA (dsRNA) used in the synthetic version poly-IC-induced apoptosis, lowering the production of IFNα and the chemokine, C-X-C motif chemokine ligand 10 (CXCL10), and expression of major histocompatibility complex (MHC) class I proteins.7
Specific small molecule inhibitors of TYK2 have the potential to reproduce the beneficial effects of the genetic variants that protect against T1D. Notably, TYK2 inhibitors are under development and being tested in other immune diseases. A specific TYK2 inhibitor (BMS-986165) has recently shown clinical efficacy in a phase 2 trial for psoriasis11 as well as in preclinical studies for lupus nephritis and inflammatory bowel disease.12 In the current study, we evaluated the ability of two TYK2 inhibitors developed by Nimbus Lakshmi (TYKi2A and TYK2iB) to protect human beta cells against the deleterious effects of IFNα. The results obtained indicate that these inhibitors prevent IFNα-induced HLA class I overexpression, chemokine production and beta-cell apoptosis without modifying human beta-cell function or susceptibility to potentially diabetogenic viruses.
2 |. MATERIALS AND METHODS
2.1 |. Culture of human EndoC-βH1 cells and human islets
The human beta cell line EndoC-βH113 (kindly provided by Dr. R. Scharfmann, University of Paris, France) was cultured in Matrigel fibronectin-coated plates as previously described.14 The MycoAlert Mycoplasma Detection kit (Lonza, Basel, Switzerland) was used to regularly assure that the cells were free from mycoplasma infection.
Human islets from 15 non-diabetic organ donors (see Table S1; beta-cell purity was determined by immunohistochemistry for insulin, as described5) were isolated in Pisa, Italy, based on methods previously developed15 with the approval of the local ethics committee and sent to Brussels for dispersion and experiments.7
All experiments with EndoC-βH1 cells or human islets are shown as independent biological data (i.e. considering EndoC-βH1 cells from different passages or human islets from different donors as n = 1). Cells were treated with TYKiA and TYKiB provided by Nimbus Lakshmi (Cambridge, MA, USA), the JAK1/2 inhibitor Baricitinib (Selleckchem, Munich, Germany), used as a positive control, and human IFNα (PeproTech, Rocky Hill, NJ, USA) 2000 U/mL (concentration selected based on our previous studies6).
2.2 |. TYK2i development and characterization
TYK2iA and TYK2iB were developed by Nimbus Lakshmi as part of a drug development campaign using structure-based drug design. Both inhibitors bind to the JH2 pseudokinase domain of TYK2 and act as allosteric inhibitors of the kinase activity of the enzyme. The JH2-directed allosteric inhibition also provides the high specificity for TYK2 over other JAK family members.16 Summarized information on the two TYK2 inhibitors presently used is provided in Table S2.
Binding affinity for the JH2 pseudokinase domain of human and mouse TYK2 was evaluated by measuring the ability of the TYK2 inhibitors to compete with the binding of an immobilized kinase ligand to a recombinant partial protein consisting of the JH2 pseudokinase domain region of TYK2 (Eurofins DiscoverX, Freemont, CA, USA). The amount of TYK2 JH2 pseudokinase protein blocked from binding to the kinase ligand was measured following incubation with TYK2i at various concentrations. TYK2iA and TYK2iB showed concentration-dependent competitive inhibition of binding of the immobilized kinase ligand to the TYK2 JH2 pseudokinase domains with Kd values shown in Table S2.
A series of in vitro studies investigated the functional consequences of the binding of the TYK2 inhibitors to the TYK2 JH2 pseudokinase domain in isolated human peripheral blood mononuclear cells (PBMCs; Confluence Discovery Technologies, St. Louis, MO, USA). In this system, TYK2iA and TYK2iB inhibited TYK2-dependent IL-12-induced STAT4 phosphorylation with an IC50 of 36.3 and 13.3 nM, respectively. The potential ‘off-target’ effects of the TYK2 inhibitors were also examined in the same human PBMC system using JAK1/3-dependent IL-2-induced STAT5 phosphorylation and JAK2-dependent GM-CSF-induced STAT5 phosphorylation. In these studies, both TYK2 inhibitors showed no inhibition at concentrations of up to 50 μM.
The effects of the TYK2 inhibitors on TYK2-dependent production and release of CXCL10 in response to IFNα receptor activation was tested in freshly isolated heparinized human and mouse whole blood samples (HD Biosciences, San Diego, CA, USA). TYK2iB inhibited TYK2-dependent IFNα-induced CXCL10 protein production in human whole blood with an IC50 of 325 nM. TYK2iA was not tested in the human whole blood assay. TYK2iA and TYK2iB inhibited TYK2-dependent IFNα-induced CXCL10 protein production in mouse whole blood with IC50s of 1.3 uM and 460 nM, respectively.
2.3 |. mRNA extraction and real-time PCR
Polyadenylated mRNA was isolated from cultured cells using the Dynabeads mRNA DIRECT purification kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, and reverse transcription was performed using the Reverse Transcriptase Core kit (Eurogentec, Liège, Belgium). The real-time PCR amplification reactions were performed using SYBR Green and the data were compared with a standard curve. Gene expression values were corrected by the housekeeping gene β-actin, as its expression is not affected by the conditions used in this study.7 The primers used in this study are listed in Table S3.
2.4 |. Protein extraction and western blot analysis
Cells were washed with PBS and lysed using 1X Laemmli Sample Buffer (60 mM tris[hydroxymethyl]aminomethane pH 6.8, 10% glycerol, 1.5% dithiothreitol, 1.5% 2-mercaptoethanol, 2% sodium dodecyl sulphate and 0.005% bromophenol blue). Immunoblot analyses were performed using antibodies against pSTAT1, pSTAT2, VP1, α-tubulin and β-actin as housekeeping. We used a secondary antibody coupled with the horseradish peroxidase. Immunoreactive bands were visualized using the SuperSignal West Femto chemiluminescent substrate (Thermo Scientific, Waltham, MA, USA), detected using ChemiDoc XRS+ (Bio-Rad, Hercules, CA, USA), and quantified with Image Studio Lite version 5.2 software (LI-COR Biosciences). A list of all the antibodies used in this study is provided in Table S4.
2.5 |. Immunofluorescence
Immunofluorescence was performed as previously described,6 using anti-MHC class I antibody, anti-insulin antibody, Hoechst 33342 (14 533, Sigma-Aldrich, Bornem, Belgium) and Alexa Fluor-conjugated secondary antibodies (Table S4).
2.6 |. ELISA
An enzyme-linked immunosorbent assay (Quantikine ELISA kit, R&D Systems, Minneapolis, MN, USA) was used to determine the release of CXCL10 to the culture medium (by 30 000 human islet cells/200 μL; culture time as indicated in the legends for the figures).
2.7 |. Assessment of cell viability
Cell viability was evaluated by nuclear dye staining (Hoechst 33342 and propidium iodide [P4170, Sigma-Aldrich, Bornem, Belgium]), as previously described.7 These measurements were executed by two different observers, one of them blinded to sample identity.
2.8 |. Viral infection and titration
The prototype strains of enterovirus (CVB5/Faulkner; CVB1/Conn-5) were obtained from the American Type Culture Collection (Old Town Manassas, VA, USA). These viruses were passaged in green monkey kidney (GMK) cells and their identity was confirmed by a plaque neutralization assay with type-specific anti-sera. EndoC-βH1 cells and dispersed human islets were infected with CVB1 (multiplicity of infection [MOI] 0.1) or CVB5 (MOI 5) in culture medium; these MOIs were selected based on our previous studies.17 After adsorption for 1 hour at 37°C, the inoculum virus was removed and the culture medium containing fœtal bovine serum was added to the plates. The virus was then allowed to replicate for 24 hours.17
Infectivity was evaluated in the medium of infected EndoC-βH1 cells and human islets using endpoint dilutions in microwell cultures of GMK cells. After 6 days, cytopathic effects were determined by microscopy, and 50% tissue infection dose titers were calculated using the Kärber formula.18 We used relative viral titers obtained from the ratio between the viral titers measured in the treated condition and those measured under the control condition.
2.9 |. Insulin secretion
EndoC-βH1 cells were preincubated with culture medium containing 2.8 mM glucose for 24 hours. Cells were then incubated in Krebs-Ringer buffer (β-KREBS serum-free medium, Univercell-Biosolutions, Toulouse, France) for 1 hour and sequentially stimulated with 0 mM glucose, 20 mM glucose, or 20 mM glucose + 20 μM forskolin for 40 minutes (modified from19,20). Insulin release and insulin content were measured using the human insulin ELISA kit (Mercodia, Uppsala, Sweden) in cell-free supernatants and acid-ethanol extracted cell lysates, respectively. Results were normalized by total protein content.20
2.10 |. Statistical analysis
Data are expressed as means ± SEM. A normality test was performed to assess the Gaussian distribution. In case of normality, parametric ANOVA tests were used, followed by paired or unpaired t test with Bonferroni correction. When the distribution was considered not normal, a non-parametric ANOVA test was used. Results with P < .05 were considered statistically significant.
3 |. RESULTS
3.1 |. TYK2 inhibitors block IFNα-induced upregulation of inflammatory markers and chemokines in EndoC-βH1 cells in a dose-dependent manner
Because IFNαsignals via both JAK1 and TYK2, baricitinib, a JAK1/2 inhibitor21 was used as a positive control. Pretreatment of EndoC-βH1 cells with different doses of the two TYK2 inhibitors prevented IFNα-induced STAT1 and STAT2 phosphorylation (Figure 1A–C and Figure S1A–C) and induction of HLA-ABC (Figure 1D and Figure S1D), the chemokine CXCL10 (Figure 1E and Figure S1E) and the antiviral MX dynamin-like GTPase 1 (MX1) (Figure 1F and Figure S1F) expression in a dose-dependent manner. However, TYK2 inhibitors did not prevent IFNα-induced expression of the ER stress marker CCAAT/enhancer-binding protein homologous protein (CHOP,also known as DDIT3) (Figure 1G and Figure S1G). Based on these results, we selected 1 μM as the dose of TYK2 inhibitors to be tested in subsequent experiments.
FIGURE 1.
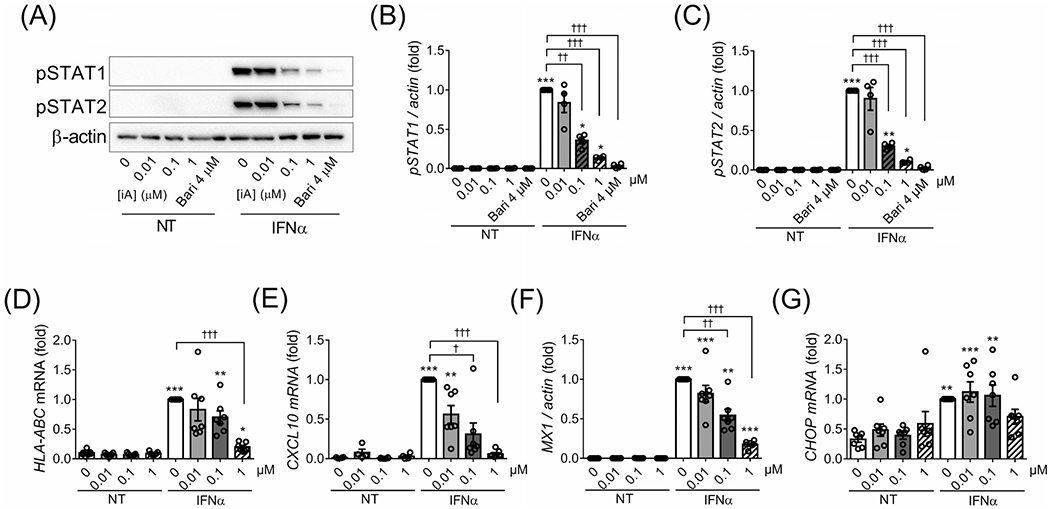
The TYK2iA prevents IFNα-induced STAT1/2 phosphorylation and gene expression in EndoC-βH1 cells in a dose-dependent manner. EndoC-βH1 cells were pretreated for 2 hours with the TYK2iA (0.01,0.1 and 1 μM) or with the JAK inhibitor baricitinib (4 μM; used as a positive control). IFNα (2000 U/mL) was then added for 8 (A-C) or 24 hours (D-G) in the continuous presence of the inhibitors. (A) Protein expression was measured by western blot and representative images of four independent experiments are shown. Densitometry results are shown for pSTAT1 (B) and pSTAT2 (C). The mRNA expression of HLA-ABC (D), CXCL10 (E), MX1 (F) and CHOP (G) was analysed by RT-qPCR. In all experiments values were normalized by β-actin and then by cells treated with IFNα without TYK2iA or baricitinib, considered as 1. Results are mean ± SEM of four (A-C) or seven (D-G) independent experiments. *P < .05, **P < .01 and ***P < .001 vs. control (no inhibitor and no IFNα); †P < .05, ††P < .01 and †††P < .001 vs. IFNα; one-way ANOVA
Next, we evaluated the potential of these inhibitors to reverse IFNα effects. EndoC-βH1 cells were pretreated with IFNα for 24 hours, and then TYK2 inhibitors were added (still in the presence of IFNα) for an additional 24 hours. Interestingly, TYK2iA and TYK2iB completely reversed the expression of the chemokine CXCL10 and the antiviral protein MX1 (Figure 2A,B). By contrast, HLA-ABC expression remained high in the presence of the inhibitors (Figure 2C), which is in agreement with our previous data showing the long-lasting expression of HLA-ABC after IFNα washout from the cell culture.22 In agreement with the finding that TYK2 inhibitors did not prevent CHOP induction by IFNα (Figure 1G and Figure S1G), they also failed to reverse IFNα-induced CHOP expression (Figure 2D).
FIGURE 2.
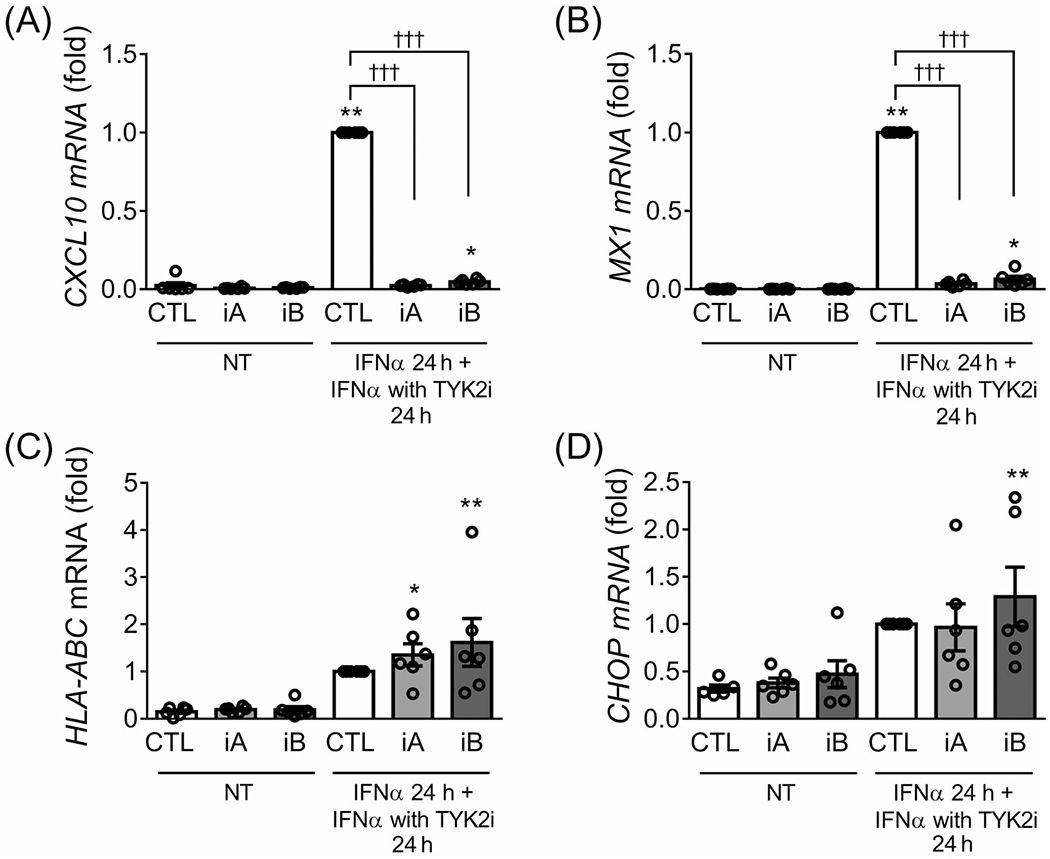
TYK2 inhibitors revert IFNα-induced CXCL10 and MX1 but not HLA-ABC and CHOP expression in EndoC-βH1 cells. EndoC-βH1 cells were pretreated for 24 hours with IFNα (2000 U/mL). The TYK2 inhibitors (iA and iB for TYK2iA and B, respectively, 1 μM) were then added for an additional 24 hours in the continuous presence of IFNα. The mRNA expression of CXCL10 (A), MX1 (B), HLA-ABC (C) and CHOP (D) was analysed by RT-qPCR and the values were normalized by β-actin and then by cells treated with IFNα without TYK2 inhibitors, considered as 1. Results are mean ± SEM of six independent experiments. *P < .05, **P < .01 and ***P < .001 vs. control (CTL NT); †††P < .001 vs. CTL IFNα; one-way ANOVA
3.2 |. TYK2 inhibition reduces IFNα-mediated upregulation of inflammatory and ER stress markers in dispersed human islets
In dispersed human islets, the two TYK2 inhibitors used at the selected dose of 1 μM prevented IFNα-mediated STAT1 and STAT2 activation (Figure 3A–C) and upregulation of HLA-ABC (Figure 3D), CXCL10 (Figure 3E) and MX1 (Figure 3F) mRNA expression, with the same efficacy as observed in EndoC-βH1 cells. In addition, and differently from the observations in EndoC-βH1 cells, TYK2iA and iB prevented IFNα-induced CHOP expression in human islets (Figure 3G). Inhibition of TYK2 also prevented IFNα-mediated CXCL10 secretion to the cell culture medium (Figure 3H) and MHC class I expression at the cell surface (Figure 3I). A higher dose of the TYK2 inhibitors (4 μM) induced similar effects as observed with 1 μM (Figure 3A; data not shown). We also evaluated the effect of lower concentrations of the TYK2 inhibitors—namely, 0.1 and 0.5 μM—in dispersed human islets and observed a clear protection with 0.1 and particularly 0.5 μM against IFNα-induced STAT1 and STAT2 phosphorylation (Figure S2A–C) and HLA-ABC (Figure S2D), CXCL10 (Figure S2E), MX1 (Figure S2G), and CHOP (Figure S2H) mRNA expression. These low concentrations of TYK2 inhibitors also prevented IFNα-mediated CXCL10 secretion into the cell culture medium (Figure S2F).
FIGURE 3.
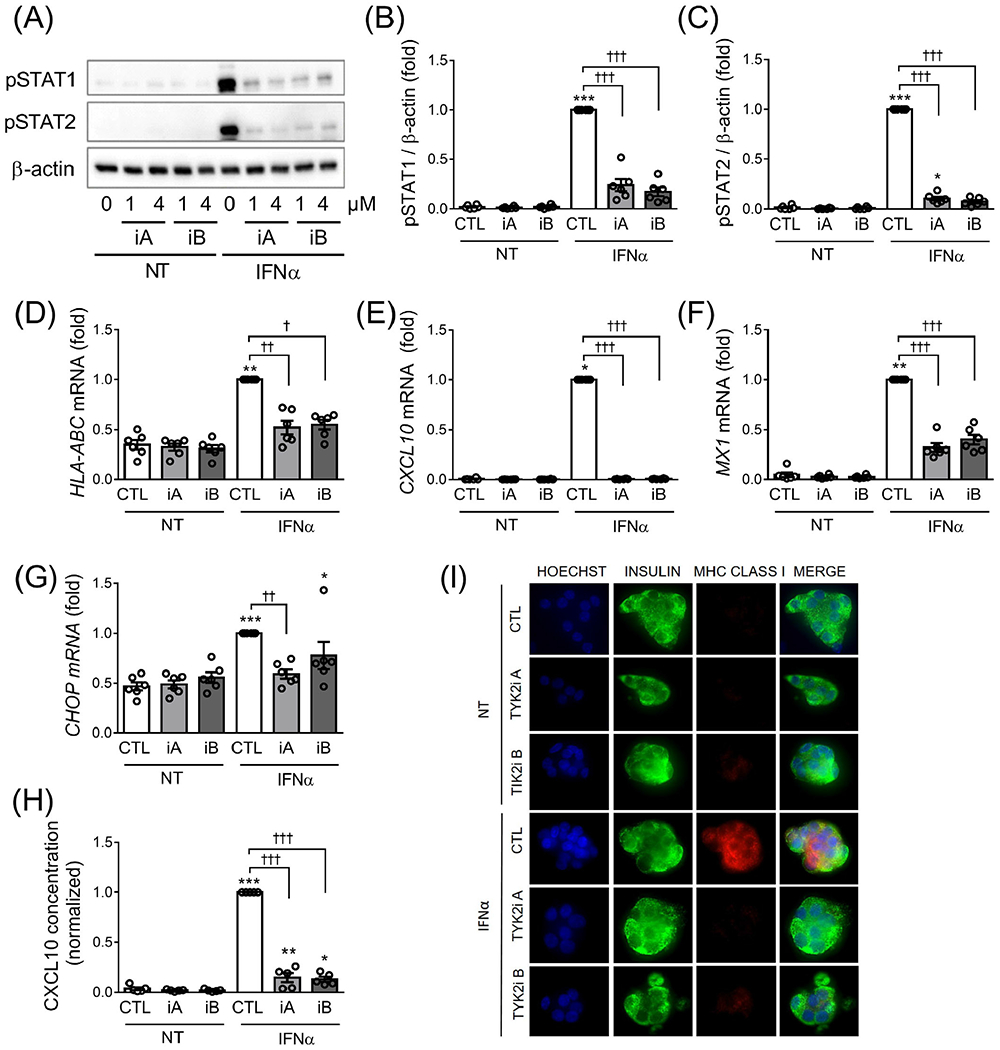
TYK2 inhibition prevents IFNα-induced STAT1/2 phosphorylation and HLA-ABC, CXCL10, MX1 and CHOP upregulation in human islet cells. Dispersed human islets were left untreated (CTL) or pretreated with 1 μM of TYK2 inhibitor (iA and iB for TYK2iA and B, respectively) for 2 hours. Then IFNα (2000 U/mL) was added for 24 hours in the continuous presence of the inhibitors. (A) Protein expression was measured by western blot and representative images of six independent experiments are shown. Densitometry results are shown for pSTAT1 (B) and pSTAT2 (C) and the values were normalized by β-actin. The mRNA expression of HLA-ABC (D), CXCL10 (E), MX1 (F) and CHOP (G) was analysed by RT-qPCR and the values were normalized by β-actin. (H) CXCL10 protein secretion in the supernatant fraction was determined by ELISA. In all experiments, the values were normalized by cells treated with IFNα without TYK2 inhibitors, considered as 1. Results are mean ± SEM of six (A-G) or five (H) independent experiments. *P < .05, **P < .01 and ***P < .001 vs. control (CTL NT); †P < .05, ††P < .01 and †††P < .001 vs. CTL IFNα; one-way ANOVA. (I) ICC of MHC class I (red), insulin (green) and Hoechst (blue) in human islets treated with IFNα with or without TYK2 inhibitors as described above (magnification ×40)
3.3 |. TYK2 inhibitors prevent apoptosis and the expression of inflammatory and ER stress markers induced by IFNα + IL-1β in dispersed human islets
We next evaluated the effect of TYK2 inhibitors on a combination of IFNα + IL-1β, two cytokines that are probably present in the islet milieu during insulitis,23 and observed a clear protection by the TYK2 inhibitors against IFNα + IL-β–induced human islet apoptosis (Figure 4A). The TYK2 inhibitors by themselves did not affect islet cell viability. Moreover, similar to the effects on IFNα signalling (see above), TYK2 inhibitors prevented IFNα + IL-1β-induced STAT1 and STAT2 phosphorylation (Figure 4B–D) and HLA-ABC (Figure 4E), CXCL10 (Figure 4F), MX1 (Figure 4H), CHOP (Figure 4I) and the activating transcription factor 3 ATF3 (Figure 4J) mRNA expression. IFNα + IL-1β-induced CXCL10 secretion to the medium (Figure 4G) and ATF3 protein expression (Figure 4K,L) were also reduced by the TYK2 inhibitors. These observations indicate that TYK2 inhibitors also have beneficial effects in the context of human islet exposure to both IFNα and IL-1β.
FIGURE 4.
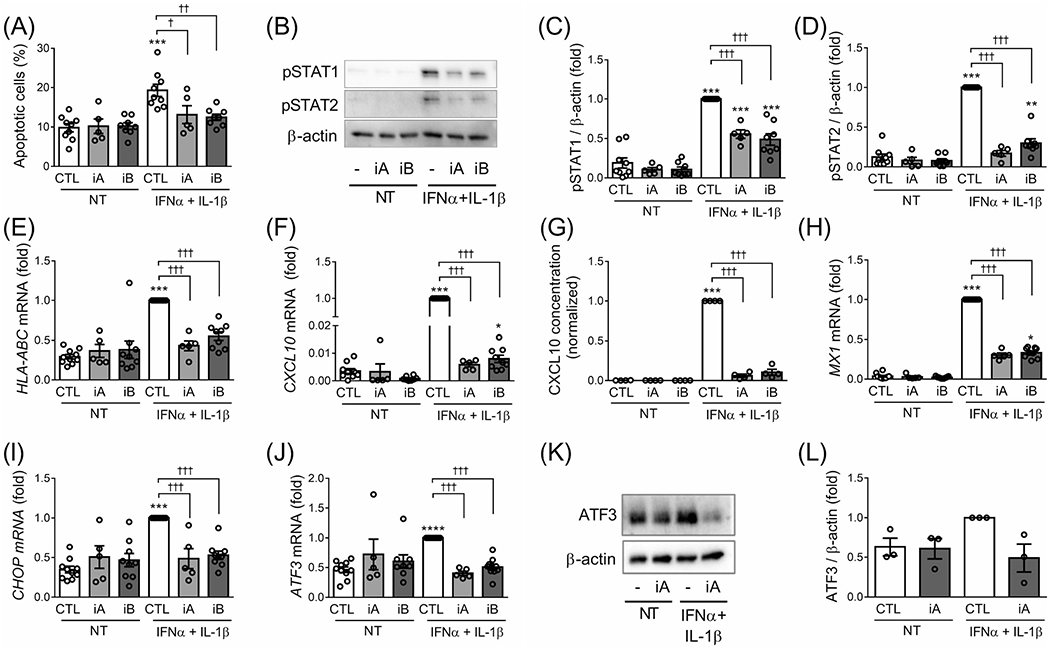
TYK2 inhibition prevents apoptosis and the expression of inflammatory and ER stress markers induced by IFNα + IL-1β in dispersed human islets. Dispersed human islets were left untreated (CTL) or pretreated with 1 μM of TYK2 inhibitor (iA and iB for TYK2iA and B, respectively) for 2 hours. Afterwards, IFNα (2000 U/mL) + IL-1β (50 U/mL) were added for 24 hours in the continuous presence of the inhibitors. (A) Apoptosis was evaluated using HO/PI staining. (B, K) Protein expression was measured by western blot and representative images of three (K) or five to eight (B) independent experiments are shown. Densitometry results normalized by β-actin are shown for pSTAT1 (C), pSTAT2 (D) and ATF3 (L). The mRNA expression of HLA-ABC (E), CXCL10 (F), MX1 (H), CHOP (I) and ATF3 (J) was analysed by RT-qPCR and the values were normalized by β-actin. (G) CXCL10 protein secretion in the supernatant fraction was determined by ELISA. In all experiments, values were normalized by cells treated with IFNα + IL-1β without TYK2 inhibitors, considered as 1. Results are mean ± SEM of three to nine independent experiments. *P < .05, **P < .01 and ***P < .001 vs. control (CTL NT); †P < .05, ††P < .01 and †††P < .001 vs. CTL IFNα + IL-1β; one-way ANOVA
3.4 |. TYK2 inhibitors do not sensitize human beta cells to viral infection
TYK2 plays an essential role in the cellular antiviral responses, as supported by the present observation that TYK2 blockade prevents IFNα-induced expression of the antiviral protein MX1 (see above), and individuals with a complete TYK2 deficiency are immunodeficient.10 By contrast, individuals with a protective single nucleotide polymorphism (SNP) in the TYK2 gene that decreases its activity by ~ 50% are healthy.9 We thus evaluated if TYK2 inhibitors sensitize human islets to CVB1 and CVB5, two viruses that may contribute to trigger autoimmunity and T1D.24 Inhibition of TYK2 did not augment CVB1- and CVB5-induced apoptosis in both EndoC-βH1 cells (Figure 5A) or dispersed human islets (Figure 5B). In agreement with these data, TYK2 inhibition did not increase viral titers (Figure 5C,D) or the expression of the viral capsid protein VP1 (Figure 5E–H) in CVB1- and CVB5-infected EndoC-βH1 cells and dispersed human islets, showing that TYK2 inhibitors do not worsen the outcome of a viral infection in human beta cells.
FIGURE 5.
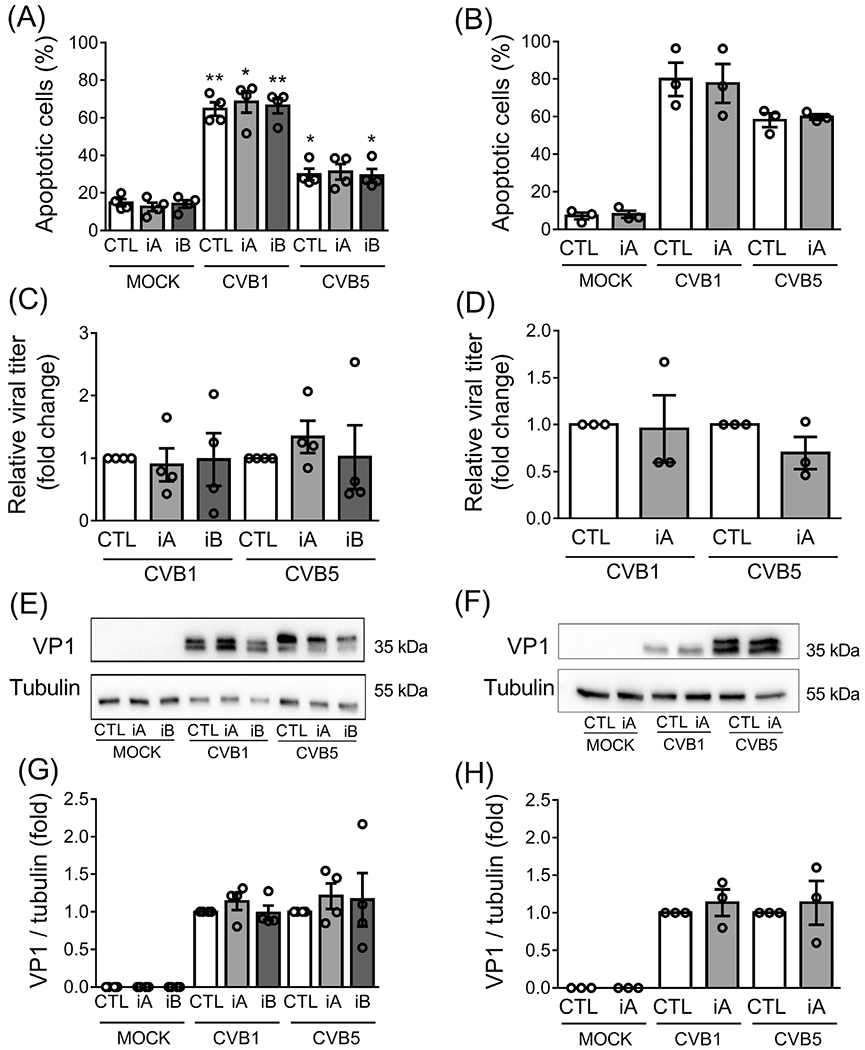
TYK2 inhibitors do not sensitize human beta cells to viral infection. EndoC-βH1 cells (A, C, E, G) and dispersed human islets (B, D, F, H) were left untreated (CTL) or pretreated with 1 μM of TYK2 inhibitor (iA and iB for TYK2iA and B, respectively) for 2 hours. Afterwards, cells were infected or not (MOCK) with CVB1 (MOI 0.1) or CVB5 (MOI 5) for 24 hours in the continuous presence of the inhibitors. (A, B) Apoptosis was evaluated using HO/PI staining. (C, D) Viral replication was quantified by viral titration and presented as relative viral titer compared with the control condition (CTL) for each virus. (E, F) The expression of the viral capsid protein (VP1) was evaluated by western blot and representative images of four (E) and three (F) independent experiments are shown. Densitometry results are shown (G, H) and the values were normalized by α-tubulin and then by each control condition (without TYK2 inhibitor), considered as 1. Results are mean ± SEM of four (EndoC-βH1 cells) and three (dispersed human islets) independent experiments. *P < .05 and **P < .01 vs. CTL MOCK; one-way ANOVA
3.5 |. TYK2 inhibitors do not affect human beta-cell function and insulin secretory capacity
In dispersed human islets, IFNα and the combination of IFNα + IL-1β had no effect on the expression of insulin (INS) and the pancreatic duodenal homeobox 1 (PDX1), a key regulator of INS expression; and TYK2 inhibition had no effect as well (Figure S3A–D), indicating that these inhibitory compounds have no impact on human beta-cell function. In addition, IFNα or IFNα + IL-1β reduced the expression of the pro-insulin processing enzyme PC1/3 (PCSK1) (Figure S4A,D) and PC2 (PCSK2) (Figure S4B,E), as previously described,25 but not the carboxypeptidase E (CPE) (Figure S4C,F); again, TYK2 inhibition did not alter the expression of these genes.
In line with the observations with INS and PDX1 mRNA expression (Figure S3A–D), neither IFNα nor the TYK2 inhibitors affected glucose or glucose + forskolin-induced insulin release in EndoC-βH1 cells (Figure S5). These observations indicate that IFNα has a mild/limited effect on human beta-cell function, and that, importantly, TYK2 inhibitors have no deleterious impact on the functional phenotype of these cells.
4 |. DISCUSSION
We presently show that TYK2 inhibitors can prevent the key deleterious effects of IFNα in human beta cells, namely, HLA class I overexpression, inflammation and ER stress, and, together with IL-1β induction of apoptosis. Importantly, these compounds neither alter the beta-cell function nor the susceptibility to infections by CVB1 and CVB5. These data provide proof of principle that TYK2 inhibitors block the IFNα pathway in human pancreatic beta cells and may therefore prevent inflammatory signalling in the beta cells and amplification of the innate immune response during the initiation and evolution of T1D.23
T1D is a complex disease that remains clinically silent for many years before appearance of symptoms.26 IFNα is present in the islets of individuals with T1D and is believed to play a crucial role at the very early stages of disease pathogenesis by mediating MHC class I overexpression, ER stress and inflammation.1,6 In children at risk of developing T1D, a type I IFN transcriptional signature is detected ahead of seroconversion for autoantibodies, suggesting that activation of the innate type I IFN pathway occurs very early in the development of the disease.2 Furthermore, islets obtained from patients affected by T1D show a gene signature similar to human islets treated in vitro with pro-inflammatory cytokines.4,5 It remains to be proven by clinical trials, however, whether and/or to what extent INFα is a key cytokine for the induction of insulitis and the autoimmune killing of beta cells in T1D.
IFNα acts via the activation of JAK1 and TYK2 and the subsequent phosphorylation and activation of STAT1 and STAT2. Thus, targeting the key downstream effectors of IFNα, i.e. JAK1 and TYK2, for the early treatment of T1D is a logical strategy. Four different JAK inhibitors have been approved for the treatment of different autoimmune diseases, including rheumatoid arthritis, systemic lupus erythematosus and dermatitis.27 Repurposing drugs that are already in use for other diseases is of great interest as the safety profiles of these molecules have already been evaluated, and this reduces the delay from the laboratory to the clinic.28 This possibility was evaluated in non-obese diabetic (NOD) mice treated with the JAK1/JAK2 inhibitor AZD1480 that targets the same active site as ruxolitinib and baricitinib, two clinically approved JAK inhibitors.21,29 AZD1480 blocked IFNγ signalling in mouse beta cells in vitro and in vivo, reducing beta-cell MHC class I expression and immune infiltration. Mice were protected from autoimmune diabetes and the inhibitor also reverted disease in newly diagnosed mice.29 AZD1480 is not in clinical use, and we evaluated the efficiency of ruxolitinib, baricitinib and cerdulatinib, another JAK inhibitor, and showed that they block IFNα signalling in a dose-dependent manner in EndoC-βH1 cells22 (present data and unpublished data). These results support the use of JAK inhibitors for the treatment of diabetes. It is of concern, however, that these first-generation JAK inhibitors are non-selective for JAK1 as they target the highly homologous catalytic domain of all JAKs. Therefore, there is a risk of important side effects such as infections, anaemia, neutropenia and thrombocytopenia. Despite these side effects, the clinically approved JAK inhibitors seem to be comparatively well tolerated,30,31 but the discovery of more selective JAK inhibitors should narrow the spectrum of action and improve safety.21
In the context of T1D, it may be more interesting to target TYK2 instead of JAK1. Indeed, SNPs in the TYK2 gene have been associated with protection against autoimmune diseases, including T1D.8,9 These SNPs reduce TYK2 activity, but, importantly, individuals homozygous for the minor allele (protective allele) are healthy and not prone to develop more infectious diseases,9,10 suggesting that a drug-induced partial inhibition of TYK2 could have beneficial effects in autoimmune diseases without increasing the risk of secondary infections. Recently, pharmaceutical compounds were found to bind to the autoinhibitory pseudokinase (JH2) domain of TYK2, stabilizing it and maintaining the autoinhibition on the catalytic domain of TYK2, even under the stimulation condition.32 This effect might be unique for TYK2 and thus provides the opportunity to develop new and very specific TYK2 inhibitors, such as the presently tested ones (see Table S2).
Recently, the TYK2 inhibitor BMS-986165—which also acts via stabilization of JH2—showed positive results in the treatment of psoriasis in a phase 2 clinical trial. Importantly, during the 12 weeks of the study there were no important side effects on blood counts, immunoglobulins, liver enzymes, electrocardiograms and vital signs, as is the case for JAK inhibitors.11 The same compound also showed positive effects in preclinical models of lupus nephritis and inflammatory bowel disease.12 In addition, preclinical evaluation of the TYK2 inhibitor NDI-031301 showed significant activity against T-cell acute lymphoblastic leukaemia both in vitro and in an in vivo mouse model, reducing tumour burden and improving survival.33 These promising first results of TYK2 inhibitors in different diseases, together with the beneficial effects that we presently observed in human pancreatic beta cells, suggest that TYK2 inhibition might also be efficient for the treatment of T1D. Target populations would be high-risk individuals in stage 1 or 2 diabetes (i.e. autoantibody positivity) or in individuals with recent-onset stage 3 T1D, as it is crucial to intervene when there are still enough beta cells remaining to enable clinical improvement.
In conclusion, we have shown that TYK2 inhibitors protect human pancreatic beta cells against IFNα- and IFNα + IL-1β-induced MHC class I overexpression, inflammation, ER stress and apoptosis. The safety and efficacy of this class of inhibitors for other diseases has already been shown, and the present data thus provide a proof of principle for considering TYK2 inhibitors in future clinical trials for T1D.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to M. Pangerl, A. M. Musuaya, M. Depessemier and I. Millard of the ULB Center for Diabetes Research, Université Libre de Bruxelles, Belgium, for excellent technical support. This work was supported by grants from the Juvenile Diabetes Research Foundation International (2-SRA-2019-834-S-B) to D.L.E., R.G.M. and C.E.-M. D.L. E. was also funded by Welbio and Fonds National de la Recherche Scientifique (FNRS), Belgium, grants CR-2015A-06 and CR-2019C-04; Innovate2CureType1-Dutch Diabetes Research Fundation (DDRF) and start-up funding provided by the Indiana Biosciences Research Institute. D.L.E and P.M. have received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement numbers 115 797 and 945 268 (INNODIA and INNODIA HARVEST). This Joint Undertaking receives support from the Union’s Horizon 2020 research and innovation programme and EFPIA, Juvenile Diabetes Research Foundation International and The Leona M. and Harry B. Helmsley Charitable Trust. R.G.M. receives funding from National Institutes of Health grants R01 DK060581 and R01 DK105588, C.E.-M. receives funding from National Institutes of Health grants R01 DK093954 and P30 DK097512 and VA Merit Award 2I01BX001733.
Funding information
European Federation of Pharmaceutical Industries and Associations; Fonds De La Recherche Scientifique - FNRS, Grant/Award Numbers: CR-2015A-06, CR-2019C-04; Indiana Biosciences Research Institute; INNODIA, Grant/Award Number: 115797; INNODIA HARVEST, Grant/Award Number: 945268; Innovate2CureType1-Dutch Diabetes Research Fundation; Juvenile Diabetes Research Foundation International, Grant/Award Number: 2-SRA-2019-834-S-B; National Institutes of Health, Grant/Award Numbers: P30 DK097512, R01 DK060581, R01 DK093954, R01 DK105588; The Leona M. and Harry B. Helmsley Charitable Trust; Union’s Horizon 2020 research and innovation programme; VA Merit Award, Grant/Award Number: 2I01BX001733; Université Libre de Bruxelles; ULB Center for Diabetes Research
Footnotes
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1111/dom.14104.
CONFLICT OF INTEREST
C.M., W.M. and S.L. are employees of Nimbus Therapeutics, Cambridge, MA, USA. The other authors report no dualities of interest for the present project.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Foulis AK, Farquharson MA, Meager A. Immunoreactive alpha-interferon in insulin-secreting beta cells in type 1 diabetes mellitus. Lancet. 1987;2:1423–1427. [DOI] [PubMed] [Google Scholar]
- 2.Ferreira RC, Guo H, Coulson RM, et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes. 2014;63:2538–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lundberg M, Krogvold L, Kuric E, Dahl-Jorgensen K, Skog O. Expression of interferon-stimulated genes in Insulitic pancreatic islets of patients recently diagnosed with type 1 diabetes. Diabetes. 2016;65: 3104–3110. [DOI] [PubMed] [Google Scholar]
- 4.Eizirik DL, Pasquali L, Cnop M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol. 2020;16:349–362. [DOI] [PubMed] [Google Scholar]
- 5.Colli ML, Ramos-Rodriguez M, Nakayasu ES, et al. An integrated multi-omics approach identifies the landscape of interferon-α-mediated responses of human pancreatic beta cells. Nat Commun. 2020; 11:2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marroqui L, Dos Santos RS, Op de Beeck A, et al. Interferon-alpha mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia. 2017;60:656–667. [DOI] [PubMed] [Google Scholar]
- 7.Marroqui L, Lopes M, dos Santos RS, et al. Differential cell autonomous responses determine the outcome of coxsackievirus infections in murine pancreatic alpha and beta cells. Elife. 2015;4:e06990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tao JH, Zou YF, Feng XL, et al. Meta-analysis of TYK2 gene polymorphisms association with susceptibility to autoimmune and inflammatory diseases. Mol Biol Rep. 2011;38:4663–4672. [DOI] [PubMed] [Google Scholar]
- 9.Dendrou CA, Cortes A, Shipman L, et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci Transl Med. 2016; 8:363ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minegishi Y, Saito M, Morio T, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25:745–755. [DOI] [PubMed] [Google Scholar]
- 11.Papp K, Gordon K, Thaci D, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018;379:1313–1321. [DOI] [PubMed] [Google Scholar]
- 12.Burke JR, Cheng L, Gillooly KM, et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci Transl Med. 2019;11:eaaw1736. [DOI] [PubMed] [Google Scholar]
- 13.Ravassard P, Hazhouz Y, Pechberty S, et al. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J Clin Invest. 2011;121:3589–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brozzi F, Nardelli TR, Lopes M, et al. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia. 2015;58:2307–2316. [DOI] [PubMed] [Google Scholar]
- 15.Marchetti P, Suleiman M, Marselli L. Organ donor pancreases for the study of human islet cell histology and pathophysiology: a precious and valuable resource. Diabetologia. 2018;61:770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moslin R, Zhang Y, Wrobleski ST, et al. Identification of N-methyl nicotinamide and N-methyl Pyridazine-3-Carboxamide Pseudokinase domain ligands as highly selective allosteric inhibitors of tyrosine kinase 2 (TYK2). J Med Chem. 2019;62:8953–8972. [DOI] [PubMed] [Google Scholar]
- 17.Colli ML, Paula FM, Marselli L, et al. Coxsackievirus B tailors the unfolded protein response to favour viral amplification in pancreatic beta cells. J Innate Immun. 2019;11:375–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lennette EH. General principles underlying laboratory diagnosis of viral and rickettsial infections. In: Lennette EH, Schmidt NJ, eds. Diagnostic Procedures for Viral and Rickettsial Diseases. 3rd ed. New York: American Public Health Association; 1979:1–63. [Google Scholar]
- 19.Andersson LE, Valtat B, Bagge A, et al. Characterization of stimulus-secretion coupling in the human pancreatic EndoC-betaH1 beta cell line. PLoS One. 2015;10:e0120879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Juan-Mateu J, Alvelos MI, Turatsinze JV, et al. SRp55 regulates a splicing network that controls human pancreatic beta-cell function and survival. Diabetes. 2018;67:423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;16:843–862. [DOI] [PubMed] [Google Scholar]
- 22.Coomans de Brachene A, Dos Santos RS, Marroqui L, et al. IFN-alpha induces a preferential long-lasting expression of MHC class I in human pancreatic beta cells. Diabetologia. 2018;61:636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–226. [DOI] [PubMed] [Google Scholar]
- 24.Op de Beeck A, Eizirik DL. Viral infections in type 1 diabetes mellitus-why the beta cells? Nat Rev Endocrinol. 2016;12:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hostens K, Pavlovic D, Zambre Y, et al. Exposure of human islets to cytokines can result in disproportionately elevated proinsulin release. J Clin Invest. 1999;104:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans-Molina C, Sims EK, DiMeglio LA, et al. Beta cell dysfunction exists more than 5 years before type 1 diabetes diagnosis. JCI Insight. 2018;3(15):e120877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He X, Chen X, Zhang H, Xie T, Ye XY. Selective Tyk2 inhibitors as potential therapeutic agents: a patent review (2015-2018). Expert Opin Ther Pat. 2019;29:137–149. [DOI] [PubMed] [Google Scholar]
- 28.Kingsmore KM, Grammer AC, Lipsky PE. Drug repurposing to improve treatment of rheumatic autoimmune inflammatory diseases. Nat Rev Rheumatol. 2020;16:32–52. [DOI] [PubMed] [Google Scholar]
- 29.Trivedi PM, Graham KL, Scott NA, et al. Repurposed JAK1/JAK2 inhibitor reverses established autoimmune insulitis in NOD mice. Diabetes. 2017;66:1650–1660. [DOI] [PubMed] [Google Scholar]
- 30.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012; 366:787–798. [DOI] [PubMed] [Google Scholar]
- 31.Keystone EC, Taylor PC, Drescher E, et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann Rheum Dis. 2015; 74:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tokarski JS, Zupa-Fernandez A, Tredup JA, et al. Tyrosine kinase 2-mediated signal transduction in T lymphocytes is blocked by pharmacological stabilization of its Pseudokinase domain. J Biol Chem. 2015; 290:11061–11074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akahane K, Li Z, Etchin J, et al. Anti-leukaemic activity of the TYK2 selective inhibitor NDI-031301 in T-cell acute lymphoblastic leukaemia. Br J Haematol. 2017;177:271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.