

Abstract
Liver-related diseases including drug-induced liver injury are becoming increasingly prominent in AIDS patients. Cobicistat (COBI) is the backbone of multiple regimens for antiretroviral therapy. The current work investigated the mechanisms of adverse drug-drug interactions associated with COBI that lead to liver damage. For individuals co-infected with HIV and tuberculosis (TB), the World Health Organization recommends the initiation of TB treatment followed by antiretroviral therapy. Rifampicin (RIF), a first line anti-TB drug, is a human specific activator of pregnane X receptor (PXR). Using PXR-humanized mice, we found that RIF-mediated PXR activation potentiates COBI hepatotoxicity. In contrast, rifabutin, a PXR-neutral analog of RIF, has no impact on COBI hepatotoxicity. Because of the crosstalk between PXR and the constitutive androstane receptor (CAR), the role of CAR in COBI hepatotoxicity was also investigated. Similar to PXR, ligand-dependent activation of CAR also potentiates COBI hepatotoxicity. Our further studies illustrated that PXR and CAR modulate COBI hepatotoxicity through the CYP3A4-dependent pathways. In summary, the current work determined PXR and CAR as key modulators of COBI hepatotoxicity. Given the fact that many prescription drugs and herbal supplements can activate PXR and CAR, these two receptors should be considered as targets to prevent COBI hepatotoxicity in the clinic.
Keywords: drug-induced liver injury, pregnane X receptor, constitutive androstane receptor, humanized mice, translational medicine, cobicistat, HIV
Impact Statement: The current work has illustrated that ligand-dependent activation of human PXR and CAR potentiates COBI hepatotoxicity through the CYP3A4-dependent pathways. The findings from this work can be used to guide the safe use of COBI in the clinic by targeting PXR and CAR.
Polypharmacy is very common in AIDS patients due to the treatment of HIV, comorbid conditions, and/or opportunistic infections (Edelman et al., 2020). Because of polypharmacy, drug-drug interactions frequently occur in AIDS patients (Edelman et al., 2020). Unfortunately, adverse drug-drug interactions during antiretroviral therapy can lead to poor adherence, drug resistance, and even AIDS-related death (Nachega et al., 2012). Therefore, mechanism-based strategies for prevention of adverse drug-drug interactions during antiretroviral therapy are urgently needed for AIDS patients.
Cobicistat (COBI), a pharmacokinetic enhancer, is the backbone in multiple regimens for antiretroviral therapy (Deeks 2014; Xu et al., 2010). COBI has a similar structure and mechanism-of-action as ritonavir (RTV), the first FDA-approved pharmacokinetic enhancer for antiretroviral therapy. Clinical studies have observed adverse drug-drug interactions between RTV-containing regimens and rifampicin (RIF), a first line anti-tuberculosis drug, which caused severe liver damage (Haas et al., 2009; Nijland et al., 2008; Schmitt et al., 2009). Clinical studies also found that lead-in treatment with efavirenz (EFV), an antiretroviral drug, potentiated hepatotoxicity of RTV-containing regimens (Jamois et al., 2009). Both RIF and EFV are activators of human pregnane X receptor (PXR), a xenobiotic nuclear receptor that regulates the expression of drug-metabolizing enzymes including CYP3A4 (Aristoff et al., 2010; Bertilsson et al., 1998; Blumberg and Evans, 1998; Jones et al., 2000; Kliewer et al., 1998; Lehmann et al., 1998). We recently demonstrated that human PXR- and CYP3A4-dependent pathways accounted for the lead-in effect of both RIF and EFV on RTV hepatotoxicity (Shehu et al., 2019). Our previous work also revealed that CYP3A4 plays an important role in COBI metabolism and bioactivation (Wang et al., 2016). These data raise a concern whether PXR activators and CYP3A4 inducers, many of which are frequently prescribed drugs or commonly used herbal supplements (Chang, 2009; Chang and Waxman, 2006; Fisher et al., 2007; Huang et al., 2004; Lynch et al., 2015), will cause adverse drug-drug interactions with COBI and lead to liver damage.
Inter-species differences of PXR in response to its activators are significant (Jones et al., 2000). For example, RIF strongly activates human PXR, but has a very weak effect on mouse Pxr (Bertilsson et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998). However, it is not feasible and ethical to test the potential adverse drug-drug interactions between PXR activators and COBI in humans. The current work used PXR-humanized mouse models to overcome the inter-species differences of PXR in response to its activators. In addition to PXR, we also explored the role of the constitutive androstane receptor (CAR) in COBI hepatotoxicity, because CAR plays a similar role as PXR in CYP3A4 regulation (Moore et al., 2000; Sueyoshi et al., 1999; Wang and LeCluyse, 2003). The findings from this work can be used to guide the safe use of COBI in the clinic.
MATERIALS AND METHODS
Chemicals
RIF, rifabutin (RFB), and NADPH were purchased from Sigma-Aldrich (St. Louis, MO). COBI was provided by Medchem Express (South Brunswick, NJ). TCPOBOP and CDDO-Im were purchased from Tocris Bioscience (Bristol, United Kingdom).
Animals
Three genetically engineered PXR and CYP3A4 mouse models were used in the current work, including PXR- and CYP3A4-humanized (hPXR/CYP3A4), PXR-humanized mice deficient in CYP3A (hPXR/Cyp3a-null), and CYP3A4-humanized mice deficient in PXR (Pxr-null/CYP3A4). hPXR/CYP3A4 and hPXR/Cyp3a-null mice were described in a previous report (Shehu et al., 2019), whereas Pxr-null/CYP3A4 mice were developed in the current study. In brief, Pxr-null/CYP3A4 mice were generated by crossing hPXR/CYP3A4 with Pxr-null mice. All mouse models were verified by PCR genotyping of human PXR and CYP3A4, and mouse Pxr and Cyp3a. CYP3A4 expression in the liver was also verified by Western blotting. All mice (male, 2–4 months old) were handled in accordance with study protocols approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Animal Treatment
Mice were maintained under a standard 12-h light/12-h dark cycle with water and chow provided ad libitum. To determine the roles of human PXR and CYP3A4 in COBI hepatotoxicity, hPXR/CYP3A4, Pxr-null/CYP3A4, and hPXR/Cyp3a-null mice were pretreated with either a PXR activator, RIF (50 mg/kg in diet), or its PXR-neutral analog RFB (50 mg/kg in diet) for 7 days followed by COBI (50 mg/kg, two doses, i.p.). To determine the role of CAR-mediated CYP3A4 induction in COBI hepatotoxicity, Pxr-null/CYP3A4 mice were pretreated with a CAR activator, TCPOBOP (3 mg/kg, i.p.), for 3 days followed by COBI. To determine the role of oxidative stress in COBI hepatotoxicity, hPXR/CYP3A4 mice were treated with an antioxidant, CDDO-Im (2 mg/kg, i.p.), on day 6 and 7 during RIF treatment followed by COBI on day 8. All mice were sacrificed 18 h after COBI treatment. Blood and liver samples were collected for evaluation of liver injury.
Assessment of Liver Injury
Biochemical and histological analyses were conducted to evaluate liver damage. In brief, activities of alanine transaminase (ALT) and aspartate transaminase (AST) in serum were analyzed using a standard kit (Point Scientific Inc., Canton, MI). Liver histology was carried out using paraffin embedded liver tissues. Four μM sections of liver tissues were cut and stained with hematoxylin and eosin (H&E). In addition, transmission electron microscopy (TEM) was used to examine the target organelle of COBI hepatotoxicity.
qPCR Analysis
TRIzol reagent (Invitrogen, Carlsbad, CA) was used to extract RNA from liver tissues. One μg of total RNA was used to make complementary DNA (cDNA) using a SuperScript II Reverse Transcriptase kit and random oligonucleotides (Invitrogen, Carlsbad, CA). The qPCR reaction contained 25 ng of cDNA, 150 nM of each primer, and 5 μL of SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) in a total volume of 10 μL. ABI-Prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA) was used for plate analysis, and the values were calculated using the comparative CT method. Cyclophilin was used as the internal control.
Western Blotting
The effects of PXR/CAR activators and CDDO-Im on CYP3A4 expression were determined by Western blotting. In brief, twenty μg of liver proteins were resolved on a 10% SDS-polyacrylamide gel electrophoresis. PVDF membranes were used for protein transfer. Proteins were then probed using primary antibodies against CYP3A4 (Laboratory of Metabolism, NCI, #275-1-2) and Gapdh (EMD Millipore, #MAB374), respectively. A chemiluminescence detection method was used to identify the target protein. Gapdh was used as the internal control.
COBI Metabolism
Liver microsomes were used to determine the effects of RIF-mediated PXR activation and CYP3A4 induction on COBI metabolism. In brief, the incubation was carried out in 1× phosphate buffered saline (pH 7.4) containing 30 μM of COBI and 1 mg of liver microsomes. The mixture was pre-incubated for 5 min and then started by adding NADPH, a co-factor of CYP-mediated reactions. The incubation was continued with gentle shaking for 30 min and terminated by adding acetonitrile: methanol (1:1). The resulting mixture was vortexed for 1 min and centrifuged at 15 000 × g for 10 min. The supernatant containing COBI and its metabolites was transferred to an autosampler vial for analysis by ultra-performance liquid chromatography and quadrupole time-of-flight mass spectrometry (UPLC-qTOFMS, Waters Corporation, Milford, MA).
UPLC-qTOFMS Analysis
UPLC-qTOFMS analysis was conducted to quantify COBI and its metabolites, as well as ophthalmic acid (OA), a biomarker of oxidative stress. In brief, the Acquity UPLC BEH C18 column (2.1 × 100 mm, 1.7 µm, Waters Corporation, Milford, MA) was used for metabolite separation. Acetonitrile/water containing 0.1% formic acid was used as the mobile phase. qTOFMS (50–1000 Da) was operated in positive mode.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7.0 (GraphPad Software, San Diego, CA). One-way or two-way analysis of variance (ANOVA) was used to compare the differences among multiple groups whereas the two-tailed Student’s t test was used for statistical analysis between two groups. All data are shown as mean ± standard error of the mean (SEM). Statistical significance was established when the p value was < .05.
RESULTS
Inter-Species Differences of PXR in COBI Hepatotoxicity
To investigate the role of human PXR in COBI hepatotoxicity, we utilized RIF, a human specific PXR activator (Bertilsson et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998), together with the hPXR/CYP3A4 mouse model, in which mouse Pxr and Cyp3a were genetically replaced by human PXR and CYP3A4, respectively (Shehu et al., 2019). Pretreatment with RIF followed by COBI resulted in hepatotoxicity in hPXR/CYP3A4 mice as shown by a marked elevation of liver injury biomarkers including ALT and AST (Figs. 1A and 1B). In addition, cell death was observed in the liver of hPXR/CYP3A4 mice treated with RIF+COBI, but not in the control, COBI, or RIF groups (Figs. 1C–F). Histological analysis also revealed massive hepatocyte degeneration with cell enlargement and the formation of large vacuoles in hPXR/CYP3A4 mice treated with RIF+COBI (Fig. 1F). A similar study was conducted in wild-type (WT) mice, but no notable liver injury was observed (Figs. 1A and 1B). These results suggest that RIF potentiates COBI hepatotoxicity through human PXR.
Figure 1.

Roles of human PXR and CYP3A4 in COBI hepatotoxicity. A–F, Inter-species differences in the hepatotoxicity caused by adverse drug-drug interactions between RIF and COBI. WT and hPXR/CYP3A4 mice were pretreated with RIF for 7 days followed by COBI for 1 day. A, B, Serum activities of ALT and AST in hPXR/CYP3A4 and WT mice. C–F, Histological analysis of liver samples from control, COBI, RIF, and RIF+COBI groups of hPXR/CYP3A4 mice. Hepatocyte degeneration (*) and cell death (^) were observed in RIF+COBI group. H&E staining. CV, central vein. Scale bars: 50 µm. G–J, RIF potentiates COBI hepatotoxicity in a PXR- and CYP3A4-dependent manner. G, PCR genotyping results of hPXR/CYP3A4, Pxr-null/CYP3A4, and hPXR/Cyp3a-null mice. Pxr-null/CYP3A4 mice are positive for CYP3A4 but negative for both Pxr and PXR. hPXR/Cyp3a-null mice are positive for human PXR (hPXR) but negative for both Cyp3a and CYP3A4. H, Expression of CYP3A4 in the liver of hPXR/CYP3A4, Pxr-null/CYP3A4, and hPXR/Cyp3a-null mice pretreated with or without RIF. Total proteins in the liver were used for analysis of CYP3A4 by Western blotting. Gapdh was used as a loading control. I, J, Serum activities of ALT and AST in hPXR/CYP3A4, Pxr-null/CYP3A4 mice, and hPXR/Cyp3a-null mice pretreated with RIF followed by COBI. All data are shown as mean ± SEM (n = 3–7). Statistical significance was determined by one-way or two-way ANOVA with Tukey’s post hoc test. **p < .01, ***p < .001, ****p < .0001.
Roles of PXR and CYP3A4 in COBI Hepatotoxicity
To further determine the role of PXR in COBI hepatotoxicity, we developed and used a mouse model that expresses CYP3A4 but is deficient in both Pxr and PXR (Pxr-null/CYP3A4) (Fig. 1G). As expected, treatment with RIF did not induce CYP3A4 expression in the liver of Pxr-null/CYP3A4 mice, indicating that PXR is not functional in these mice (Fig. 1H). Compared with hPXR/CYP3A4 mice, lead-in treatment with RIF followed by COBI did not cause liver injury in Pxr-null/CYP3A4 mice (Figs. 1I and 1J and Supplementary Figure 1 A, B). These data confirm that human PXR is a key modulator of COBI hepatotoxicity. As a PXR target gene-encoded enzyme, CYP3A4 is essential in COBI metabolism and bioactivation (Wang et al., 2016). Therefore, we hypothesized that RIF-mediated PXR activation potentiates COBI hepatotoxicity through the CYP3A4-dependent pathway. To test this hypothesis, we utilized a PXR-humanized mouse model deficient in both Cyp3a and CYP3A4 (hPXR/Cyp3a-null) (Figs. 1G and 1H). Compared with hPXR/CYP3A4 mice, the liver injury was abolished in hPXR/Cyp3a-null mice pretreated with RIF followed by COBI (Figs. 1I and 1J, and Supplementary Figure 1 C, D). These results indicate that PXR-mediated CYP3A4 induction is critical for the development of COBI hepatotoxicity.
Rifabutin (RFB), a PXR-Neutral Analog of RIF, Does Not Potentiate COBI Hepatotoxicity
Co-infection of tuberculosis (TB) in AIDS patients is common, and in these subjects, the World Health Organization (WHO) recommends the initiation of TB treatment followed by antiretroviral therapy (World Health Organization, 2016). RFB belongs to the same class of drugs as RIF and is frequently used in individuals co-infected with HIV and TB (Blumberg et al., 2003; World Health Organization, 2016). We therefore assessed whether lead-in treatment with RFB potentiates COBI hepatotoxicity (Fig. 2A). The effect of RFB on PXR activation and CYP3A4 induction in the liver was compared with that of RIF in hPXR/CYP3A4 mice. As expected, RIF-mediated PXR activation strongly induced CYP3A4 expression in the liver of hPXR/CYP3A4 mice, but RFB had a very weak effect on CYP3A4 induction (Fig. 2B), indicating that RFB is a weak PXR activator, which agreed with previous reports (Reinach et al., 1999; Zhu et al., 2007). In hPXR/CYP3A4 mice pretreated with RFB or RIF followed by COBI, liver injury was observed in the RIF+COBI group, but not in the RFB+COBI group (Figs. 2C and 2D), suggesting that RFB is safer than RIF for lead-in treatment in HIV and TB co-infected individuals receiving COBI-containing regimens. These data provide additional evidence for the roles of human PXR and CYP3A4 in COBI hepatotoxicity.
Figure 2.
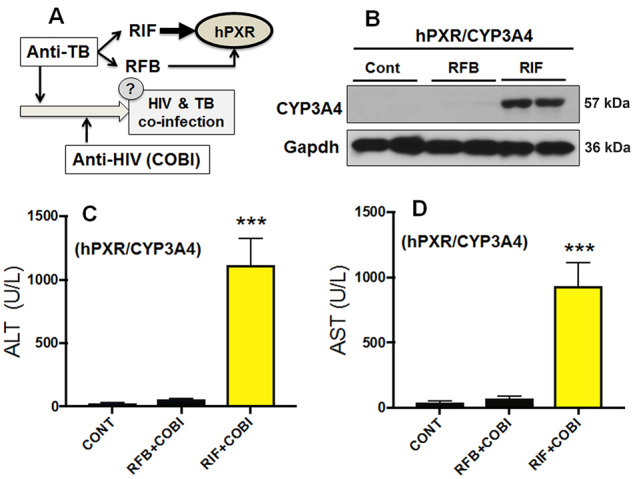
RFB, a PXR-neutral analog of RIF, does not potentiate COBI hepatotoxicity in hPXR/CYP3A4 mice. A, A scheme showing the WHO guideline for the treatment of HIV and TB co-infection, in which lead-in treatment with anti-TB drugs is recommended. Both RIF and RFB are commonly used anti-TB drugs that can interact with human PXR (hPXR). We therefore tested whether lead-in treatment with RFB will potentiate COBI hepatotoxicity through hPXR. B, CYP3A4 induction in the liver of hPXR/CYP3A4 mice pretreated with RIF or RFB. Expression of CYP3A4 was analyzed by Western blotting. Gapdh was used as a loading control. C, D, Serum activities of ALT and AST in hPXR/CYP3A4 mice pretreated with RIF or RFB followed by COBI. All data are shown as mean ± SEM (n = 3–7). Statistical significance was determined by one-way ANOVA with Tukey’s post hoc test. ***p < .001.
PXR-Mediated CYP3A4 Induction Increases COBI Metabolism and Bioactivation
To further explore the mechanisms of COBI hepatotoxicity, we profiled the impact of PXR-mediated CYP3A4 induction on COBI metabolism and bioactivation. M2 and M9 are the major metabolites of COBI (Fig. 3A). The byproduct through the M2 pathway of COBI is an N-acetyl cysteine conjugated metabolite, suggesting the formation of a reactive intermediate that interacts with glutathione, an important antioxidant in cells (Wang et al., 2016). In addition, the M9 pathway of COBI produces an unstable metabolite, 2-isopropylthiazole-4-carbaldehyde (Fig. 3A). Furthermore, M18 and M19 are thiazole ring-opening metabolites of COBI (Fig. 3A), which can undergo oxidation and cause oxidative stress (Mizutani and Suzuki, 1996; Wang et al., 2016). Using liver microsomes, the vesicles derived from the endoplasmic reticulum (ER) where CYPs are situated, we observed dramatic decreases of COBI and increases of COBI metabolites M2, M9, M18, and M19 in hPXR/CYP3A4 mice pretreated with RIF, but not in Pxr-null/CYP3A4 or hPXR/Cyp3a-null mice with the same pretreatment (Supplementary Figure 2, and Figs. 3B–E). These data indicate that lead-in treatment with RIF increases COBI metabolism and bioactivation through the PXR- and CYP3A4-dependent pathways.
Figure 3.

PXR-mediated CYP3A4 induction increases COBI metabolism and bioactivation. A, A scheme showing the roles of CYP3A4 in COBI metabolism and bioactivation. B–E, The abundances of M2, M9, M18, and M19 produced in the incubations of COBI with liver microsomes of hPXR/CYP3A4, Pxr-null/CYP3A4, and hPXR/Cyp3a-null mice pretreated with or without RIF. COBI metabolites were analyzed by UPLC-qTOFMS. All data are expressed as mean ± SEM (n = 3–4). Data in control groups of hPXR/CYP3A4 mice are set as 1, respectively. Statistical significance was determined by two-way ANOVA with Tukey’s post hoc test. ****p < .0001. ND, not detected.
Adverse Drug-Drug Interactions between RIF and COBI Cause Oxidative Stress in the Liver
We next investigated oxidative stress in the liver because metabolic activation of drugs by CYPs can lead to oxidative stress and cellular injury (Guengerich, 2008). Genetic analyses revealed a substantial up-regulation of antioxidant responsive genes in the liver of hPXR/CYP3A4 mice pretreated with RIF followed by COBI (Figs. 4A–C, and Supplementary Figure 3), indicating the occurrence of oxidative stress. Using OA as a biomarker (Soga et al., 2006), oxidative stress was confirmed because hepatic OA levels increased >100-fold in RIF+COBI group when compared with the control group (Supplementary Figure 4A). To further determine the role of oxidative stress in COBI hepatotoxicity, hPXR/CYP3A4 mice were pretreated with CDDO-Im, an antioxidant (Mehta et al., 2018). CDDO-Im had no impact on PXR-mediated CYP3A4 induction (Supplementary Figure 4B), but it significantly decreased oxidative stress in the liver and protected against the hepatotoxicity in hPXR/CYP3A4 mice pretreated with RIF followed by COBI (Supplementary Figure 4C-E). These results indicate that oxidative stress plays an important role in PXR-mediated COBI hepatotoxicity.
Figure 4.

PXR- and CYP3A4-dependent oxidative stress and ER stress in COBI hepatotoxicity. A, A heatmap of genes related to oxidative stress, ER stress, and cell death in the liver of hPXR/CYP3A4, Pxr-null/CYP3A4, and hPXR/Cyp3a-null mice pretreated with RIF followed by COBI. B, C, Expression of genes (Gpx2 and Cbr3) associated with oxidative stress. D–F, Expression of genes (Bip, Chop, and Atf3) associated with ER stress. G, A TEM image showing ER dilation in the liver of hPXR/CYP3A4 mice pretreated with RIF followed by COBI. N, nucleus. H, I, Expression of genes (Bax and Dr5) associated with cell death. mRNAs were analyzed by qPCR. All data are expressed as mean ± SEM (n = 3–7). Data in control groups are set as 1, respectively. Statistical significance was determined by two-way ANOVA with Tukey’s post hoc test. *p < .05, **p < .01, ***p < .001, ****p < .0001.
ER is a Target Organelle in PXR-Mediated COBI Hepatotoxicity
Following CYP3A4-mediated COBI bioactivation and oxidative stress, we explored the target organelle in PXR-mediated COBI hepatotoxicity. We focused on the ER because (i) CYP3A4 predominantly localizes in the ER of hepatocytes (Guengerich, 2008), and (ii) CYP-mediated oxidative stress can lead to ER stress and cell death (Malhotra and Kaufman, 2007; Sano and Reed, 2013). As expected, a notable increase in ER stress biomarkers was observed in the liver of hPXR/CYP3A4 mice pretreated with RIF followed by COBI, but not in Pxr-null/CYP3A4 or hPXR/Cyp3a-null mice with the same treatment (Figs. 4A and 4D–F). TEM provided additional evidence of ER damage, as remarkable ER dilation was observed in the liver of hPXR/CYP3A4 mice treated with RIF+COBI (Fig. 4G), which is consistent with histological analysis showing hepatocyte enlargement and large vacuoles (Fig. 1F). Prolonged ER stress causes cell death (Malhi and Kaufman, 2011; Sano and Reed, 2013). Indeed, the expression of genes associated with cell death including death receptor 5 (Dr5) and BCL2-associated X (Bax) was significantly increased in the liver of hPXR/CYP3A4 mice pretreated with RIF followed by COBI (Figs. 4H and 4I). These data suggest that the ER is a target organelle in PXR-mediated COBI hepatotoxicity.
Ligand-Dependent Activation of CAR Potentiates COBI Hepatotoxicity
No liver injury was observed in Pxr-null/CYP3A4 mice pretreated with RIF followed by COBI because PXR-mediated CYP3A4 induction was abrogated in these mice (Figs. 1H–J, and Supplementary Figure 1 A, B). In addition to PXR, activation of CAR up-regulates CYP3A4 expression in the liver (Moore et al., 2000; Sueyoshi et al., 1999; Wang and LeCluyse, 2003). We therefore hypothesized that CAR activators will re-sensitize Pxr-null/CYP3A4 mice to COBI hepatotoxicity. We administered Pxr-null/CYP3A4 mice with TCPOBOP, a potent CAR activator (Lodato et al., 2018). As expected, TCPOBOP significantly induced CYP3A4 expression in the liver of Pxr-null/CYP3A4 mice (Fig. 5A). In addition, lead-in treatment with TCPOBOP followed by COBI caused a significant elevation of liver injury biomarkers in the serum of Pxr-null/CYP3A4 mice (Figs. 5B and 5C). Similar to the phenotypes observed in the livers of hPXR/CYP3A4 mice pretreated with RIF followed by COBI, severe hepatocyte degeneration, ER stress, and cell death were observed in the livers of Pxr-null/CYP3A4 mice pretreated with TCPOBOP followed by COBI (Fig. 5D–G). These results suggest that CAR is also a modulator of COBI hepatotoxicity and it is mediated by the CYP3A4-dependent pathways.
Figure 5.

Roles of xenobiotic nuclear receptors PXR and CAR in COBI hepatotoxicity. A, CYP3A4 expression in the liver of Pxr-null/CYP3A4 mice pretreated with RIF (PXR activator) or TCPOBOP (CAR activator). CYP3A4 was analyzed by Western blotting. Gapdh was used as a loading control. B, C, Serum activities of ALT and AST in Pxr-null/CYP3A4 mice pretreated with TCPOBOP followed by COBI. D, E, Histological analysis of liver samples from Pxr-null/CYP3A4 mice treated with TCPOBOP and TCPOBOP+COBI. Hepatocyte degeneration (*) and cell death (^) were observed in TCPOBOP+COBI group (E). CV, central vein. Scale bars: 50 µm. F, G, The expression of genes related to ER stress in the liver of Pxr-null/CYP3A4 mice pretreated with TCPOBOP followed by COBI. mRNAs of Bip (F) and Chop (G) were analyzed by qPCR. All data are expressed as mean ± SEM (n = 3–7). Statistical significance was determined by the two-tailed Student’s t test. *p < .05, **p < .01, ***p < .001. H, A scheme showing the roles of PXR and CAR in COBI hepatotoxicity. Activation of PXR and CAR leads to CYP3A4 induction in hepatocytes. Overexpressed CYP3A4 in the ER increases the metabolism and bioactivation of COBI to form reactive metabolites, which can directly target the ER leading to oxidative stress, ER stress, and hepatocellular injury.
DISCUSSION
Adverse drug-drug interactions during antiretroviral therapy are of great concern to HIV clinicians, pharmacists, and AIDS patients (Edelman et al., 2020). Therefore, understanding the mechanisms of drug-drug interactions associated with antiretroviral therapy will help reduce the risk of adverse effects and improve the outcomes of therapy. Our current work illustrated that lead-in treatment with PXR and CAR activators potentiates COBI hepatotoxicity by up-regulating CYP3A4 expression in the ER, which increases the production of reactive metabolites of COBI and in turn causes oxidative stress, ER stress, and hepatocellular injury (Fig. 5H).
Liver-related diseases are becoming increasingly prominent and are one of the leading causes of non-AIDS-related death in HIV-infected patients (Kaspar and Sterling, 2017). Inter-species differences of PXR between mice and humans limit the use of WT mice for preclinical studies on human PXR-mediated drug-drug interactions and toxicities (Bertilsson et al., 1998; Jones et al., 2000; Kliewer et al., 1998; Lehmann et al., 1998). Using PXR-humanized mouse models, we demonstrated that RIF-mediated PXR activation potentiates COBI hepatotoxicity. PXR is a ligand-dependent transcription factor that regulates drug metabolism and disposition (Bertilsson et al., 1998; Blumberg and Evans, 1998; Kliewer et al., 1998; Lehmann et al., 1998). In addition to RIF, many commonly prescribed drugs are PXR ligands, such as EFV (anti-retroviral), nifedipine (anti-hypertensive), atorvastatin (lipid lowering agent), and artemisinin (anti-malaria) (Chang and Waxman, 2006; Lehmann et al., 1998; Shukla et al., 2011). Furthermore, the use of herbal supplements alongside antiretroviral therapy is common in AIDS patients (Duggan et al., 2001; Furler et al., 2003), and many herbs such as St John’s Wort and Sutherlandia contain constituents as PXR activators (Furler et al., 2003; Mills et al., 2005). Because of the potential adverse drug-drug interactions between PXR activators and COBI, we suggest that clinicians and pharmacists screen and educate their patients on the danger of and need for avoiding such drug combinations.
The current study also demonstrated that CAR activation potentiates COBI hepatotoxicity. Similar to PXR, the xenobiotic nuclear receptor CAR is a ligand-dependent transcription factor that regulates CYP3A4 expression in the liver (Moore et al., 2000; Sueyoshi et al., 1999; Wang and LeCluyse, 2003). Many commonly used prescription drugs (eg, phenobarbital and phenytoin) and herbal supplements (eg, Artemisia capillaris and Allium sativum) can activate CAR (Chang, 2009; Chang and Waxman, 2006; Fisher et al., 2007; Huang et al., 2004; Lynch et al., 2015). Because CAR activation increases the risk of COBI hepatotoxicity, we suggest that CAR activating drugs and herbs should not be administered before starting COBI-containing regimens. PXR and CAR antagonists are under development to minimize potential drug-drug interactions mediated by these two xenobiotic nuclear receptors (Banerjee et al., 2015; Chai et al., 2019), which shed light on the management of COBI hepatotoxicity in circumstances where the use of PXR and CAR activators is inevitable.
In summary, our work has illustrated the crucial roles of the xenobiotic nuclear receptors PXR and CAR in the adverse drug-drug interactions associated with COBI that lead to hepatotoxicity, suggesting that lead-in treatment with PXR and CAR activators should be monitored with caution in AIDS patients receiving COBI-containing regimens. Our work has also highlighted the power of using humanized mouse models for preclinical studies on drug-drug interactions and toxicities.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
AUTHOR CONTRIBUTIONS
X.M. and A.I.S. conceived the project and wrote the manuscript. A.I.S., J.Z., J.Li, and J.Lu performed the experiments and data analysis. X.M., W.X., and F.J.G. contributed to the new reagents, analytic tools, and animal models. X.M., A.I.S., D.M., W.X., and F.J.G. contributed to the scientific discussion and experimental design.
FUNDING
This work was supported in part by the National Institute of Allergy and Infectious Diseases (R01AI131983) and the National Center for Complementary and Integrative Health (R21AT011088).
DECLARATION OF CONFLICTING INTERESTS
The authors have declared that no conflict of interest exists.
Supplementary Material
REFERENCES
- Aristoff P. A., Garcia G. A., Kirchhoff P. D., Showalter H. D. (2010). Rifamycins–obstacles and opportunities. Tuberculosis (Edinb) 90, 94–118. [DOI] [PubMed] [Google Scholar]
- Banerjee M., Robbins D., Chen T. (2015). Targeting xenobiotic receptors pxr and car in human diseases. Drug Discov. Today 20, 618–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertilsson G., Heidrich J., Svensson K., Åsman M., Jendeberg L., Sydow-Bäckman M., Ohlsson R., Postlind H., Blomquist P., Berkenstam A. (1998). Identification of a human nuclear receptor defines a new signaling pathway for cyp3a induction. Proc. Natl. Acad. Sci. U S A 95, 12208–12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg B., Evans R. M. (1998). Orphan nuclear receptors—new ligands and new possibilities. Genes Dev. 12, 3149–3155. [DOI] [PubMed] [Google Scholar]
- Blumberg H. M., Burman W. J., Chaisson R. E., Daley C. L., Etkind S. C., Friedman L. N., Fujiwara P., Grzemska M., Hopewell P. C., Iseman M. D., et al. (2003). American thoracic society/centers for disease control and prevention/infectious diseases society of America: treatment of tuberculosis. Am. J. Respir. Crit. Care Med. 167, 603–662. [DOI] [PubMed] [Google Scholar]
- Chai S. C., Lin W., Li Y., Chen T. (2019). Drug discovery technologies to identify and characterize modulators of the pregnane x receptor and the constitutive androstane receptor. Drug Discov. Today 24, 906–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T. K. (2009). Activation of pregnane x receptor (pxr) and constitutive androstane receptor (car) by herbal medicines. AAPS J 11, 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T. K. H., Waxman D. J. (2006). Synthetic drugs and natural products as modulators of constitutive androstane receptor (car) and pregnane x receptor (pxr). Drug Metab. Rev. 38, 51–73. [DOI] [PubMed] [Google Scholar]
- Deeks E. D. (2014). Cobicistat: a review of its use as a pharmacokinetic enhancer of atazanavir and darunavir in patients with hiv-1 infection. Drugs 74, 195–206. [DOI] [PubMed] [Google Scholar]
- Duggan J., Peterson W. S., Schutz M., Khuder S., Charkraborty J. (2001). Use of complementary and alternative therapies in hiv-infected patients. AIDS Patient Care STDs 15, 159–167. [DOI] [PubMed] [Google Scholar]
- Edelman E. J., Rentsch C. T., Justice A. C. (2020). Polypharmacy in hiv: recent insights and future directions. Curr. Opin. HIV AIDS 15, 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C. D., Augustine L. M., Maher J. M., Nelson D. M., Slitt A. L., Klaassen C. D., Lehman-McKeeman L. D., Cherrington N. J. (2007). Induction of drug-metabolizing enzymes by garlic and allyl sulfide compounds via activation of constitutive androstane receptor and nuclear factor e2-related factor 2. Drug Metab. Dispos. 35, 995–1000. [DOI] [PubMed] [Google Scholar]
- Furler M. D., Einarson T. R., Walmsley S., Millson M., Bendayan R. (2003). Use of complementary and alternative medicine by hiv-infected outpatients in ontario, canada. AIDS Patient Care STDs 17, 155–168. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. (2008). Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 21, 70–83. [DOI] [PubMed] [Google Scholar]
- Haas D. W., Koletar S. L., Laughlin L., Kendall M. A., Suckow C., Gerber J. G., Zolopa A. R., Bertz R., Child M. J., Hosey L., et al. (2009). Hepatotoxicity and gastrointestinal intolerance when healthy volunteers taking rifampin add twice-daily atazanavir and ritonavir. J. Acquir. Immune Defic. Syndr. 50, 290–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Zhang J., Moore D. D. (2004). A traditional herbal medicine enhances bilirubin clearance by activating the nuclear receptor car. J. Clin. Invest. 113, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamois C., Riek M., Schmitt C. (2009). Potential hepatotoxicity of efavirenz and saquinavir/ritonavir coadministration in healthy volunteers. Arch. Drug Inf. 2, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S. A., Moore L. B., Shenk J. L., Wisely G. B., Hamilton G. A., McKee D. D., Tomkinson N. C., LeCluyse E. L., Lambert M. H., Willson T. M., et al. (2000). The pregnane x receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol. Endocrinol. 14, 27–39. [DOI] [PubMed] [Google Scholar]
- Kaspar M. B., Sterling R. K. (2017). Mechanisms of liver disease in patients infected with hiv. BMJ Open Gastroenterol. 4, e000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer S. A., Moore J. T., Wade L., Staudinger J. L., Watson M. A., Jones S. A., McKee D. D., Oliver B. B., Willson T. M., Zetterström R. H., et al. (1998). An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92, 73–82. [DOI] [PubMed] [Google Scholar]
- Lehmann J. M., McKee D. D., Watson M. A., Willson T. M., Moore J. T., Kliewer S. A. (1998). The human orphan nuclear receptor pxr is activated by compounds that regulate cyp3a4 gene expression and cause drug interactions. J. Clin. Invest. 102, 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato N. J., Rampersaud A., Waxman D. J. (2018). Impact of car agonist ligand tcpobop on mouse liver chromatin accessibility. Toxicol. Sci. 164, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch C., Zhao J., Huang R., Xiao J., Li L., Heyward S., Xia M., Wang H. (2015). Quantitative high-throughput identification of drugs as modulators of human constitutive androstane receptor. Sci. Rep. 5, 10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhi H., Kaufman R. J. (2011). Endoplasmic reticulum stress in liver disease. J. Hepatol. 54, 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra J. D., Kaufman R. J. (2007). Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid. Redox Signal. 9, 2277–2294. [DOI] [PubMed] [Google Scholar]
- Mehta J., Rayalam S., Wang X. (2018). Cytoprotective effects of natural compounds against oxidative stress. Antioxidants (Basel) 7, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills E., Foster B. C., van Heeswijk R., Phillips E., Wilson K., Leonard B., Kosuge K., Kanfer I. (2005). Impact of african herbal medicines on antiretroviral metabolism. AIDS 19, 95–97. [DOI] [PubMed] [Google Scholar]
- Mizutani T., Suzuki K. (1996). Relative hepatotoxicity of 2-(substituted phenyl)thiazoles and substituted thiobenzamides in mice: evidence for the involvement of thiobenzamides as ring cleavage metabolites in the hepatotoxicity of 2-phenylthiazoles. Toxicol. Lett. 85, 101–105. [DOI] [PubMed] [Google Scholar]
- Moore L. B., Parks D. J., Jones S. A., Bledsoe R. K., Consler T. G., Stimmel J. B., Goodwin B., Liddle C., Blanchard S. G., Willson T. M., et al. (2000). Orphan nuclear receptors constitutive androstane receptor and pregnane x receptor share xenobiotic and steroid ligands. J. Biol. Chem. 275, 15122–15127. [DOI] [PubMed] [Google Scholar]
- Nachega J. B., Hsu A. J., Uthman O. A., Spinewine A., Pham P. A. (2012). Antiretroviral therapy adherence and drug–drug interactions in the aging hiv population. AIDS 26, S39–S53. [DOI] [PubMed] [Google Scholar]
- Nijland H. M., Rafaëlla F., Rongen G. A., van Uden P., van Crevel R., Boeree M. J., Aarnoutse R. E., Koopmans P. P., Burger D. M. (2008). High incidence of adverse events in healthy volunteers receiving rifampicin and adjusted doses of lopinavir/ritonavir tablets. AIDS 22, 931–935. [DOI] [PubMed] [Google Scholar]
- Reinach B., de Sousa G., Dostert P., Ings R., Gugenheim J., Rahmani R. (1999). Comparative effects of rifabutin and rifampicin on cytochromes p450 and udp-glucuronosyl-transferases expression in fresh and cryopreserved human hepatocytes. Chem. Biol. Interact. 121, 37–48. [DOI] [PubMed] [Google Scholar]
- Sano R., Reed J. C. (2013). Er stress-induced cell death mechanisms. Biochim. Biophys. Acta 1833, 3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt C., Riek M., Winters K., Schutz M., Grange S. (2009). Unexpected hepatotoxicity of rifampin and saquinavir/ritonavir in healthy male volunteers. Arch. Drug Inform. 2, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehu A. I., Lu J., Wang P., Zhu J., Wang Y., Yang D., McMahon D., Xie W., Gonzalez F. J., Ma X. (2019). Pregnane x receptor activation potentiates ritonavir hepatotoxicity. J. Clin. Invest. 129, 2898–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S. J., Sakamuru S., Huang R. L., Moeller T. A., Shinn P., VanLeer D., Auld D. S., Austin C. P., Xia M. H. (2011). Identification of clinically used drugs that activate pregnane x receptors. Drug Metab. Dispos. 39, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga T., Baran R., Suematsu M., Ueno Y., Ikeda S., Sakurakawa T., Kakazu Y., Ishikawa T., Robert M., Nishioka T., et al. (2006). Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 281, 16768–16776. [DOI] [PubMed] [Google Scholar]
- Sueyoshi T., Kawamoto T., Zelko I., Honkakoski P., Negishi M. (1999). The repressed nuclear receptor car responds to phenobarbital in activating the human cyp2b6 gene. J. Biol. Chem. 274, 6043–6046. [DOI] [PubMed] [Google Scholar]
- Wang H., LeCluyse E. L. (2003). Role of orphan nuclear receptors in the regulation of drug-metabolising enzymes. Clin. Pharmacokinet. 42, 1331–1357. [DOI] [PubMed] [Google Scholar]
- Wang P., Shehu A. I., Liu K., Lu J., Ma X. (2016). Biotransformation of cobicistat: metabolic pathways and enzymes. Drug Metab. Lett. 10, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. (2016). Consolidated guidelines on the use of antiretroviral drugs for treating and preventing hiv infection: Recommendations for a public health approach. World Health Organization, Geneva, Switzerland. [PubMed] [Google Scholar]
- Xu L., Liu H., Murray B. P., Callebaut C., Lee M. S., Hong A., Strickley R. G., Tsai L. K., Stray K. M., Wang Y., et al. (2010). Cobicistat (gs-9350): A potent and selective inhibitor of human cyp3a as a novel pharmacoenhancer. ACS Med. Chem. Lett. 1, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z. R., Puglisi J., Connors D., Stewart J., Herbst J., Marino A., Sinz M., O'Connell J., Banks M., Dickinson K., et al. (2007). Use of cryopreserved transiently transfected cells in high-throughput pregnane x receptor transactivation assay. J. Biomol. Screen. 12, 248–254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.