

Abstract
The human CST (CTC1-STN1-TEN1) complex is an RPA-like single-stranded DNA binding protein complex. While its telomeric functions have been well investigated, numerous studies have revealed that hCST also plays important roles in maintaining genome stability beyond telomeres. Here, we review and discuss recent discoveries on CST in various global genome maintenance pathways, including findings on the CST supercomplex structure, its functions in unperturbed DNA replication, stalled replication, double-strand break repair, and the ATR-CHK1 activation pathway. By summarizing these recent discoveries, we hope to offer new insights into genome maintenance mechanisms and the pathogenesis of CST mutation-associated diseases.
Keywords: CTC1-STN1-TEN1, replication stress, DNA replication, fork stalling, DSB repair, genome stability
Introduction
Genome instability is a major driving force for the development of cancer, aging-related diseases and other complex diseases [1–3]. The most common cause of genome instability is the exposure to exogenous and endogenous DNA-damaging agents, including ionizing radiation (IR), UV radiation, reactive oxygen species, DNA-protein crosslinking agents, etc. In addition, DNA replication fork stalling caused by intrinsic and extrinsic insults threatens genome integrity and leads to inaccurate DNA duplication. To protect genome integrity, cells have evolved a complex network of pathways to respond to DNA damage and replication stress, activate cell cycle checkpoints, repair DNA lesions and rescue stalled replication [1, 4–7]. A broad range of genome maintenance proteins in these pathways exert direct and/or indirect functions to mitigate detrimental DNA transactions [8–10].
Mammalian CST complex, composed of CTC1, STN1 and TEN1, is an OB-fold containing heterotrimeric protein complex that binds to ssDNA [11]. It was originally identified as an accessory cofactor of DNA polymerase α (POLα). By interacting with POLα, CST stimulates POLα primase activity and mediates the switch of the primase to polymerase activity [12–14]. For this reason, CTC1 and STN1 were initially known as alpha accessory factor AAF132 and AAF44, respectively. In addition, STN1 has another alias OBFC1 (OB-fold containing 1) [15]. Currently, CST is the widely accepted name for this complex.
CST has been extensively investigated for its role in telomere maintenance especially in promoting telomere replication and regulating telomerase activity [16–19]. However, growing evidence has revealed that CST possesses much more diverse functions in promoting DNA replication genome-wide and repairing double-strand breaks (DSBs) [20–27]. In this review, we summarize recent findings of CST structures and its biochemical properties, briefly discuss the roles of CST in maintaining telomere integrity, and then focus on reviewing its functions in maintaining global genome stability. Finally, we speculate possible applications of CST aberrations in clinical diagnosis and therapy.
Structural and DNA-binding properties of CST
Among the three subunits of CST, STN1 and TEN1 are highly conserved across a wide range of species and can form the STN1-TEN1 subcomplex. The structures of STN1 and TEN1 closely resemble the RPA32 and RPA14 subunits of the RPA complex (Figs. 1A and 1B) [28, 29]. In contrast, orthologs of CTC1 in different species show less similarity. The yeast Cdc13, which was thought to be the mammalian CTC1 homolog, lacks sequence identity with CTC1 [30]. In fact, the cryo-EM structure of human CST indicates that human CTC1 shares more similarity with RPA70 rather than Cdc13. Thus, it is proposed that CST may have evolved from the RPA complex [31].
Figure 1.
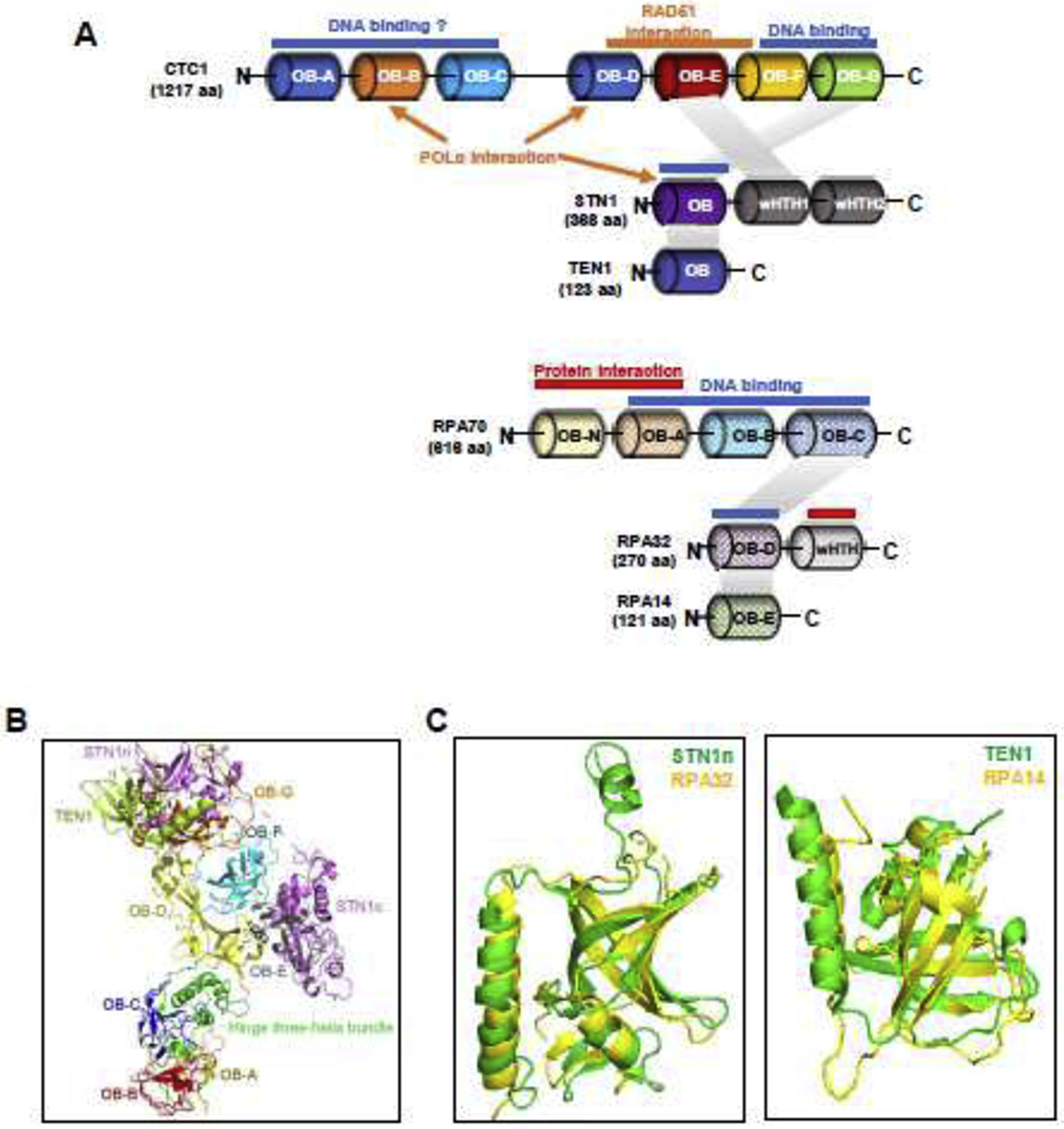
Structures of the human CST complex.
(A) Domain organizations of CST and RPA based on the published crystal or cryo-EM structures. STN1 has an extra C-terminal wHTH domain compared to RPA32. DNA binding sites are indicated in blue bars, subunit interactions in shaded parallelograms, and domains for interacting with other proteins in orange bars/arrows (in CST) or red bars (in RPA).
(B) Structure of monomeric CST derived from PDB ID:6W6W.
(C) Human STN1n and TEN1 subunits share structural similarity with RPA32 and RPA14, respectively. STN1n and TEN1 are colored in green, and RPA32 and RPA14 are colored in yellow. Structures are derived from Protein Data Bank with structure codes 4JOI and IQUQ.
CST binding to ssDNA relies on its OB-fold domains and the nature of its three-dimensional structures [31, 32]. The recently resolved overall architecture of the CST heterotrimer indicates that the telomeric ssDNA staples CST monomers to a decamer assembly [31]. Analysis of the CST monomeric structure extracted from the overall structure of the decamer supercomplex suggests that the CST complex has a subunit stoichiometry of 1:1:1 [31]. The largest subunit CTC1 (134 kDa) contains seven tandem OB-fold domains (OB-A to OB-G). These OB-fold domains are divided into two sections, OB-A to OB-C and OB-D to OB-G, by an intramolecular three-helix bundle bridge (Fig. 1A). In the OB-A to OB-C section, OB-C functions as a scaffold for OB-A and OB-B. The OB-B and OB-C domains are structurally similar to Ustilago maydis RPA70 OB-A and OB-B [31]. In the OB-D to OB-G section, OB-D domain works as a scaffold to support OB-E, -F and -G to form a ring-like structure (Fig. 1B). The OB-F and OB-G domains harbor the anchor sites for C ST binding to a short telomeric oligonucleotide, and mutations in this nucleotide-anchor patch abolish CST binding to telomeric DNA [31]. Binding to such an oligonucleotide is found to stabilize the overall CST architecture.
STN1 consists of an N-terminal OB-fold domain and two winged helix-tum-helix (wHTH) motifs at the C-terminus. STN1 works as a bridge connecting CTC1 and TEN1, and no direct interaction between CTC1 and TEN1 has been found from biochemical and structural analysis (Fig. 1A) [11, 31, 33]. STN1 interacts with CTC1 via two interacting interfaces – the N-terminal half of STN1 (STN1n) interacts with OB-G of CTC1 and the first wHTH domain at STN1 C-terminal (STN1c) interacts with OB-E of CTC1. The interaction of STN1n/OB-Gis relative stable, which explains the observation that disease-causing CTC1 mutations L1142H and 1196-Δ7, both residing within OB-G, disrupt CTC1-STN1 interaction [18, 31, 33]. The interaction between STN1c and CTC1 manifests two different binding modes in the presence and absence of ssDNA, hinting that such conformational changes may be necessary for DNA binding and decametric assembly [31]. Although the CST cryo-EM structure provides little information on STN1-ssDNA binding, STN1 alone is known to be able to bind telomeric ssDNA (> 18 bp) and long non-telomeric ssDNA (>30 bp), albeit with a lower affinity compared to that of the whole CST complex [11, 14, 28]. Intriguingly, STN1 OB-fold domain mutations (W89A/R139L/Y141A), which are the residue counterparts in RPA32 that are associated with ssDNA binding, only affect CST binding capacity to short telomeric sequence but not the long telomeric DNA or non-telomeric sequence [34]. This raises the possibility that CST may be able to adopt dynamic DNA binding modes similar to RPA to promote telomere DNA replication and alleviate replication stress.
The third subunit TEN1 is the smallest and contains one OB-fold domain. TEN1 alone is unable to bind ssDNA [28]. TEN1 and STN1 can form a heterodimer via interaction between TEN1 and STN1n (Fig. 1A), and subsequently STN1-TEN1 stabilizes the interaction with CTC1.
The three components form an intact CST complex to avidly bind to telomeric ssDNA [31]. CST binds to 18-nt telomeric ssDNA with observed Kd values in the range of 2.2 to 21.6 nM [11, 22, 34, 35]. The broad range of Kd values may be due to experimental discrepancies during protein purification procedures. Nonetheless, it is considered that CST binds to ssDNA with affinity comparable to its close relative RPA. Unlike RPA which binds ssDNA in a sequence independent manner, the preferred binding substrate for CST is G-rich ssDNA but not necessarily telomeric [11, 35]. Such binding preference quickly decreases with the increase in nucleotide length [11]. CST also binds to ss-dsDNA junctions without sequence specificity [22, 25]. While the CST cryo-EM structure characterizes its decameric status with a short telomeric sequence and helps describe its functions at the telomeric region, it is noticeable that mutations in the nucleotide-anchor patch that abolish telomeric DNA binding appear to have minimal impact on binding to non-telomeric sequences [31]. In addition, it has been reported that removal of the N-terminal half of CTC1, which includes OB-A to OB-C domains, abolishes DNA binding (Fig. 1A) [11], yet the cryo-EM study shows that these domains have no direct contact with the telomeric sequence [31]. Thus, the structural status of CST binding to non-telomeric sequences and ss-dsDNA junctions requires further investigation, which is important for deciphering its roles in maintaining global genome stability.
Aside from providing insights into how CST binds to DNA, the cryo-EM structure also shed lights on other molecular features of CST. One major function of CST is to assist POLα to fill in the telomeric C-strand and replenish telomeres. Chen et al reveal that a subset of CTC1 disease mutations (A227, V259 in OB-B and V665 in OB-D) mediate the association with POLα [33]. Interestingly, these three residues locate at two separate surfaces on the cryo-EM structure [31, 33], indicating that CST may interact with POLα through multiple interfaces, and perhaps involve multiple interaction modes (Fig. 1A). In addition, it has been reported that the STN1n OB-fold physically interacts with the regulatory subunit of POLα, POLA2, and mediates the activation of POLα primase activity [13]. It is possible that STN1 may enable the CST complex to adopt conformational changes that allow CST to access specific sequences of ssDNA and/or interact with downstream factors to execute its cellular functions. Further investigation is needed to determine CST-POLα interaction modes and their respective biological functions.
Roles of CST in telomere replication and telomere protection
Telomeres are specialized DNA segments localized at the ends of chromosomes in eukaryotic cells, which are required for maintaining genome stability and cell proliferation [36]. CST, along with the six-member Shelterin complex (TRF1-TRF2-TPP1-POT1-TIN2-RAP1), aids in overcoming the challenges arising from telomere DNA duplication (Fig. 2A) [37]. The telomeric functions of CST have been well-described (see other reviews for more detailed discussion) [30, 37–40]. In human cells, CST binds to the 3’ ss G-rich overhangs at telomere ends and enacts two functions. First, CST inhibits telomerase activity by interacting with POT1-TPP1 to prevent the over-extension of the G-strand by telomerase [16]. Second, CST promotes C-strand synthesis (also known as C-strand fill-in) via POLα during the late S/G2 phase (Fig. 2A) [17, 19, 24]. Thus, CST coordinates G- and C-strand synthesis to preserve telomere integrity. As for the functions of the individual subunits of CST, Feng et al reported that CTC1-STN1 is sufficient to inhibit telomerase activity and reduce telomeric DNA damage, whereas STN1-TEN1 is mainly responsible for C-strand synthesis [41].
Figure 2.
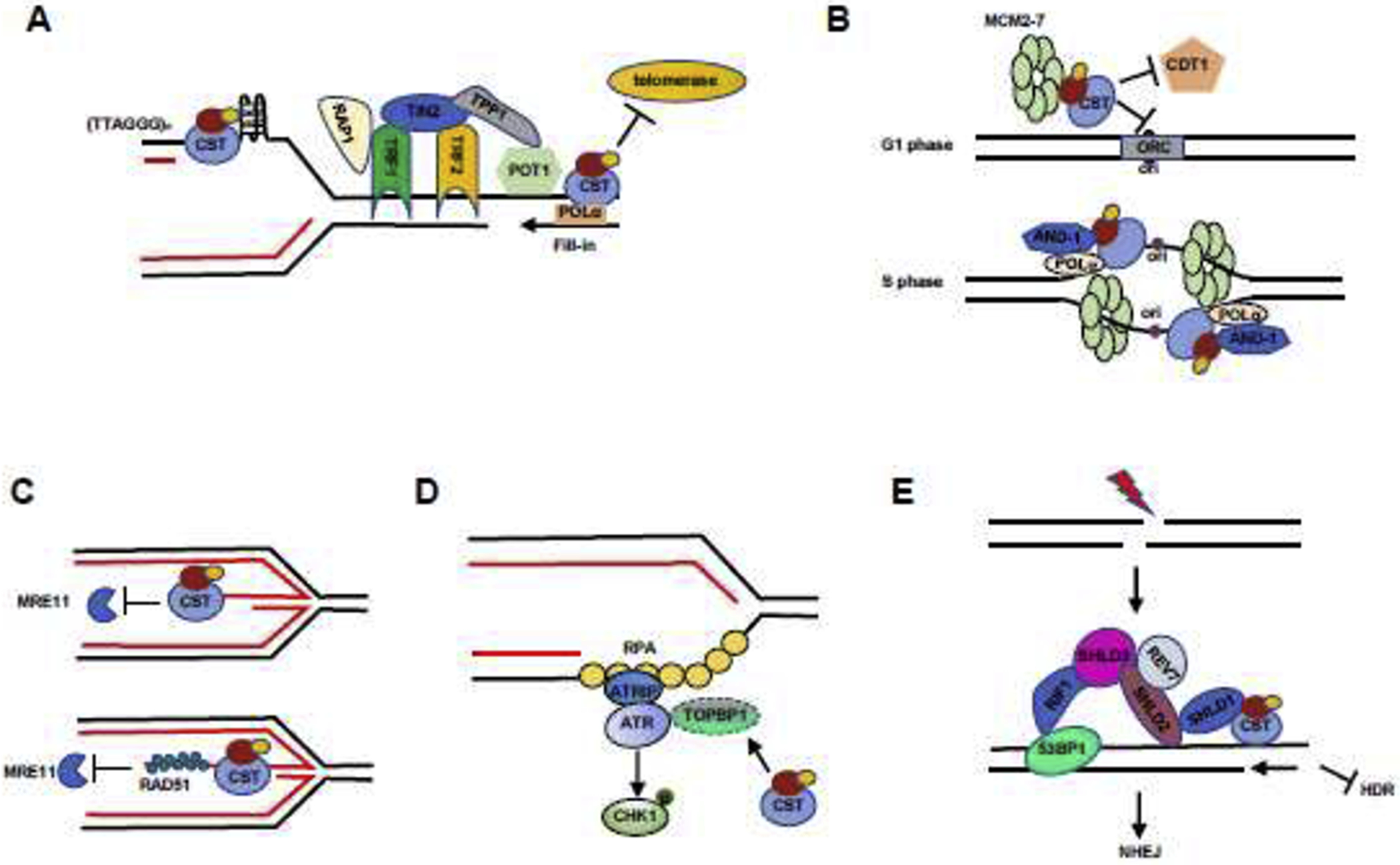
Models illustrating the roles of CST in genome stability maintenance.
(A) CST regulates telomere stability via the telomeric synthesis processes. In the ds telomeric region, CST resolves G4 structures and relieves replication stress in the telomeric region. At telomere ends, CST binds to ss G-overhangs to inhibit the access of telomerase to telomeres. CST also mediates C-strand fill-in to replenish C-strands.
(B) CST regulates DNA replication initiation under normal replication condition. In the G1 phase, CST blocks origin licensing by interacting with MCM to prevent CDT1 interacting with MCM. In the S phase, CST interacts with AND-1 and POLα and then facilitates replisome assembly and subsequent initiation of DNA synthesis.
(C) CST protects reversed fork stability under replication stress. Top: CST directly binds to the regressed arms of reversed forks to inhibit unscheduled MRE11 degradation of nascent strand DNA. In addition, CST indirectly protects reversed forks via recruiting RAD51 (bottom).
(D) CST regulates the ATR-CHK1 pathway under replication stress. CST prevents the degradation of the ATR activator TOPBP1. CST deficiency decreases TOPBP1 protein level, therefore suppressing CHK1 phosphorylation following replication stress.
(E) CST controls end-resection during DSB repair and favors c-NHEJ. At DSB sites, 53BP1-RIF1 recruits the Shieldin complex (SHLD1-SHLD2-SHLD3-REV7), which interacts with and recruits CST to DSB ends to counteract end resection.
Multiple studies have established that CST is indispensable for the complete and efficient replication of telomere duplex DNA. The tandem (TTAGGG)n repeats are prone to forming G4 secondary structures which are well-known obstacles to replication fork progression and tend to form DNA gaps or breaks [42, 43]. Purified CST complex can bind to and melt G4 structures in vitro [22], and STN1 deficiency has been shown to accumulate G4 structures in the telomere duplex region [44]. Thus, by preventing G4 accumulation, CST plays a prominent role in facilitating the replication of the bulk telomeric DNA (Fig. 2A) [19, 45]. CST deficiency increases ssDNA gaps within telomeres and elevates multi-telomere FISH signals (MTSs) – a hallmark of replication perturbation at telomeric repeats, leading to sudden telomere loss [19, 33, 45]. In addition to directly relieving G4-caused replication stress, CST can recruit RAD51 to telomeres and facilitate fork restart and/or protect the stability of stalled forks at telomeres [23].
Roles of CST in unperturbed genomic DNA replication
Faithful duplication and transmission of genetic information to daughter cells is vital for maintaining genome stability and the genetic continuity of species. Starting from DNA replication initiation to termination, various proteins and protein complexes participate in a well-organized manner to ensure the faithful completion of the replication process during the S phase [46]. Beyond telomeres, CST has been uncovered as a multifaceted player in genome-wide DNA replication.
An early study shows that ~80% of STN1 foci localize at non-telomeric regions [11], providing the initial evidence implicating the participation of CST in global DNA replication. Recently, it has been reported that CST directly interacts with two subunits of the MCM complex, MCM4 and MCM7, and is likely an active component at replication origins [47]. In agreement with this, we have observed the presence of both CTC1 and STN1 at unperturbed replication forks using the assay known as in situ analysis of protein interactions at DNA replication forks (SIRF) [25]. By interacting with the MCM complex, CST disrupts the interaction between MCM and CDT1, thereby negatively regulating origin licensing to avoid DNA re-replication in S-phase (Fig. 2B, top) [47]. Additionally, CST physically interacts with AND-1, a ssDNA-binding protein previously reported to collaborate with POLα for primer synthesis (Fig. 2B, bottom) [47]. CST deficiency in HeLa cells decreases the amount of chromatin-bound AND-1 [47]. It has been proposed that CST facilitates AND-1/POLα loading and replisome assembly on chromatin. Under unperturbed conditions, depletion of CST proteins leads to genome instability, including excessive ssDNA accumulation, metaphase chromosome aberration, and increase of γH2AX foci [25, 27]. It is likely that CST deficiency may disrupt the regulation of DNA re-replication and replisome assembly, thus contributing to the observed genome instability.
Roles of CST during perturbed DNA replication
DNA synthesis is constantly challenged by various endogenous and exogenous agents that slow down or transiently stall replication fork progression, leading to replication stress and genome instability. To preserve genome stability, a number of pathways including fork reversal, re-priming, translesion synthesis, and perhaps others, are engaged in stabilizing and protecting stalled forks, and subsequently restarting stalled replication in a timely manner (reviewed in [48–50]).
CST has been demonstrated to be an important player in antagonizing genome instabilities arising from replication stress. CST localization at replication forks is enhanced upon fork stalling [25]. CST depleted cells show reduced cell viability after treatment with replication stress inducers such as hydroxyurea (HU), aphidicolin (APH) and methyl methanesulphonate (MMS), whereas CST overexpression increases cell fitness in response to these stresses [26]. Recently, ChIP-seq analysis of STN1 in S-phase cells treated with 2 mM HU reveals that nearly 90% of STN1-enriched sequences contain repetitive features including long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs), as well as regions of low complexity and simple repeats, suggesting a role of CST in overcoming replication obstacles formed in repetitive sequences [23]. In agreement with its DNA binding properties in vitro, STN1 is found to be enriched at GC-rich repetitive sequences genome-wide upon HU treatment [23]. STN1 depletion induces instabilities of G-rich sequences, in particular after fork stalling [23]. Given that G-rich sequences are prone to G4 formation, and CST help unwind G4 [22, 44]. CST may help in resolving G4 structures genome-wide (including telomeres), thereby directly reducing fork barriers caused by G4 formation. This notion is further supported by the observation that the amount of G4 in cells is inversely proportional to the level of CST complex [44] and STN1-depleted cells confer sensitivity to G4-stabilizing agent [51].
In addition to directly resolving G4, CST plays a unique role in protecting stalled forks from unscheduled nuclease degradation [25]. Upon fork stalling, the canonical three-way forks can regress and be remodeled by translocases like SMARCAL1, ZRANB3, HLTF to form a four-way chicken foot-like structure [52–54]. While fork reversal is an important mechanism for restarting stalled DNA synthesis, reversed forks can be attacked by nucleases including MRE11, DNA2, EXO1 and CtIP. If unprotected, nascent DNA strands at reversed forks are degraded by these nucleases, leading to fork degradation and genome instability [55–62]. CST localizes at stalled forks, and CST deficiency increases MRE11 association at forks, leading to unscheduled MRE11 degradation of nascent-strand DNA in a fork reversal-dependent manner [25]. In vitro biochemical analysis using purified CST proteins reveals that CST binds to the ss-dsDNA junction substrate mimicking the regressed arm of the reversed fork and specifically protects this substrate from MRE11 degradation (Fig. 2C, top) [25]. While previous studies suggest that CST prefers binding to G-rich or long random ssDNA sequences (described above), CST binds and protects the [ISP CHK]5’ overhang of the ss-dsDNA substrate with no sequence specificity [25]. The DNA binding ability of CST is essential for fork protection, reinforcing the importance of DNA binding in modulating CST functions [25]. It will be interesting to determine whether CST can adopt flexible conformations when binding to ssDNA or ss-dsDNA junctions in a way reminiscent to RPA that depends on ssDNA length [63, 64], and if so, whether the flexible DNA binding modes play a role in remodeling or reshaping the fork structure.
Upon exposure to HU, CST proteins form distinct foci [23]. A subset of these replication stress-induced CST foci co-colocalize with RAD51 [23] – an essential player for promoting fork reversal and protecting reversed forks from degradation [65–67]. CST physically interacts with RAD51 in response to HU or APH treatment in an ATR-dependent manner [23]. Truncational analysis of CTC1 shows that the RAD51-interacting regions are located at the C-terminal half of CTC1 (Fig. 1A) [32]. Depletion of CST subunits partially reduces the RAD51 recruitment to stalled forks and RAD51 foci formation [23, 25], indicating that nucleation of the RAD51 filament may be affected by CST deficiency. Thus, it is possible that RAD51 recruitment by CST offers additional protection against unscheduled nuclease attack at stalled forks (Fig. 2C, bottom).
It has been reported that CST promotes genome-wide replication restart after HU treatment [24, 27]. DNA fiber analysis reveals that CST facilitates new origin firing [26, 27, 34], albeit how CST achieves this function remains unclear. Since RAD51 promotes replication fork restart by both strand exchange-dependent and strand exchange-independent mechanisms [68], it remains to be determined whether CST recruiting RAD51 plays a role in the fork restart process.
Interplay between CST and ATR-CHK1 activation
ATR kinase activation is a vital process for maintaining genome stability during S phase [69–71]. In response to replication stress, the rapidly emerging ssDNA is coated by RPA, which recruits ATRIP-ATR to the stalled site. Subsequently, two ATR activation proteins TOPBP1 and ETAA activate ATR. The activated ATR then phosphorylates its downstream effector CHK1 kinase, which exerts S/G2 checkpoint arrest [71–73]. It has been shown that CST deficiency activates ATR-CHK1 in both human and mouse cells. STN1 knockdown induces CHK1 phosphorylation at later passages in telomerase-negative normal human fibroblasts [19]. Similarly, CTC1-deleted MEF cells also display elevated CHK1 phosphorylation [45]. The DDR marker γ-H2AX is found at chromosome ends missing telomere signals in CTC1 null MEF cells, suggesting that ATR-CHK1 activation is at least in part attributed to telomeric defects [17]. Two additional situations caused by CST loss can contribute to ATR activation. One is that in the absence of CST, overextended telomeric G-overhangs may exhaust the pool of POT1 and subsequently recruit RPA to activate ATR, and the other is that simultaneously G4 structures-associated stalled replication forks can accumulate within telomere duplex [17, 44].
Given that CST is involved in unperturbed DNA replication as described above, it remains to be determined whether the genome-wide function of CST may affect ATR-CHK1 activation. Surprisingly, a recent study indicates that CST may regulate the ATR-CHK1 pathway in response to global replication stress by targeting TOPBP1. While CTC1 deletion in a human cell line HCT116 does not affect ATR recruitment and RPA phosphorylation, cytoplasmic and chromatin-associated TOPBP1 is profoundly decreased by CTC1 deletion, thereby inhibiting CHK1 phosphorylation following HU treatment [20]. The insult of ATR-CHK1 pathway results in decreased cell proliferation and a G2 arrest (Fig. 2D) [20]. It is currently unclear whether this observation is specific to cell types. More investigation is needed to explore the detailed mechanism underlying how CST affects TOPBP1 stability, which will have important clinical applications in cancer treatment that targets the ATR-CHK1 pathway.
Reciprocally, ATR has been shown to regulate the cellular functions of CST. Upon HU or APH treatment, CST physically interacts with and recruits RAD51 to G-rich fragile sites in an ATR-dependent manner [23]. However, ATR phosphorylation sites on human CST have not been reported. The budding yeast Cdc13 can be phosphorylated on serines 225, 249, 255, 306 by Mec1 (related to ATR) to promote the telomerase recruitment, and thus, these Mec1-mediated Cdc13 phosphorylation sites are critical for telomere protection [74]. No STN1 or TEN1 phosphorylation modifications by ATR have been identified yet. More investigation is needed to study the CST phosphorylation by ATR kinase and their corresponding cellular functions.
CST in DSB repair
DSB is considered the most deleterious type of DNA damage. Nonhomologous end joining (NHEJ) and homology-directed repair (HDR) are the two primary pathways for repairing DSBs and have been extensively studied [75, 76]. NHEJ occurs throughout the cell cycle. In contrast, HDR functions in the S and G2 phases on DSB sites with 3’ overhang ends, which result from the 5’ to 3’ resection of DSB ends conducted by several nucleases including MRE11, CtIP, EXO1 or DNA2 with BLM1 helicase [77–80]. Such end resection directs the choice of NHEJ and HR [81, 82]. BRCA1 directs the repair pathway to HDR by stimulating end resection and blocking 53BP1 function, while 53BP1 promotes NHEJ-mediated DSB repairs [82–84]. The Shieldin complex (SHLD1-SHLD2-SHLD3-REV7) is a newly identified 53BP1 effector complex that is recruited to DSBs in a 53BP1-RIF1-dependent manner [85–87]. Interestingly, CST physically interacts with the Shieldin complex and is recruited to DSB sites via the 53BP1-RIF1-Shieldin axis (Fig. 2E) [88]. Genetic screening for the PARP inhibitor (PARPi) resistant factors in BRCA1-deficient cells reveals that CST deficiency suppresses such synthetic lethality [21, 88]. It is thought that CST may recruit POLα to DSB ends to fill in the resected ends in a manner reminiscent to C-strand fill-in at telomere ends, therefore antagonizing DSB end resection and facilitating the canonical NHEJ repair of DSBs [88]. Removal of CST promotes end resection, restoring HDR in BRCA1-deficient cells and thus leading to PARPi resistance [21]. It remains to be determined whether POLα-independent mechanisms participate in antagonizing end resection.
Post-translational modifications (PTMs) of CST
Proteins PTMs modulate protein functions and influence almost all aspects of biology. Identifying and understanding PTMs is critical in the study of disease treatment and prevention. To date, PTMs of CST proteins have mainly been characterized in yeasts. In budding yeast, phosphorylation of Cdc13 and Stn1 plays a very important role in its telomere protection function. The phosphorylation of Cdc13 at T308 by Cdk1 promotes its interaction with the Est1 subunit of the telomerase holoenzyme and facilitates the recruitment of telomerase to telomeres in late S to G2 phases to ensure proper telomere replication [89]. The Mec1 (related to ATR) and Tel1 (related to ATM) phosphorylation of Cdc13 also promotes Cdc13-Est1 interaction [74]. The protein phosphatase 2A (PP2A) dephosphorylation of Cdc13 and Ipl1 phosphorylation helps in telomerase dissociation from telomeres [90]. Following Cdc13 phosphorylation, Cdk1 then phosphorylates Stn1 at T223 and S250, which promotes its interaction with Cdc13 and Ten1 to form a stable CST complex, thus inhibiting telomerase [91, 92]. PTMs on Ten1 have not been reported so far.
In fission yeast S. pombe, the Ssu72 phosphatase is reported to regulate the recruitment of Stn1 to telomeres by dephosphorylating Stn1 at Ser74, a residue located within the OB-fold domain of Stn1. Defective Stn1 dephosphorylation caused by ssu72Δ or Stn1 S74D mutation abolishes Stn1 recruitment to telomeres and telomerase inhibition, leading to telomerase-dependent telomere elongation [93]. Ssu72 is required for Polα activation. Interestingly, Cdk1 activity counteracts Ssu72 phosphatase in telomere length regulation and Stn1 recruitment [93]. Such regulation appears to be conserved in human cells. Human SSU72 is required for hSTN1 recruitment to human telomeres, and down-regulation of hSSU72 results in telomere elongation and fragility [93]. It is thus proposed that Cdk1-dependent phosphorylation prevents Stn1 association with telomeres and the Ssu72 phosphatase reverses phosphorylation, allowing Stn1 recruitment to telomeres and consequently telomerase inhibition and (C)ST-Polα activation to fill in C-strands [93].
Unlike yeast CST, PTMs of human CST remain largely unexplored. It will be critically important to investigate PTMs of human CST in regulating its genome-wide functions.
CST deficiency in mice and human diseases
Coat Plus syndrome (CP) and dyskeratosis congenita (DC) are two diseases associated with mutations in CTC1 and STN1 genes [94–98]. CP is clinically characterized by retinal telangiectasia and exudates, intracranial calcifications, leukodystrophy and brain cysts, vascular ectasias and other neurologic signs [99, 100]. DC is a telomere-associated disease characterized by bone marrow failure and reticulate hyperpigmentation, nail dystrophy, oral leukoplakia and other premature aging manifestations [101, 102]. In addition, STN1 variants are associated with risks of age-related diseases including cardiovascular disease mortality [103] and idiopathic interstitial pneumonia [104]. Given the role of CST in telomere maintenance, it is not surprising that some CP patients carrying CTC1 variants exhibit shorter telomeres and telomere defective symptoms [18, 33, 96]. Interestingly, telomere lengths in a subset of CTC1 mutant-carrying patients have no significant changes [96, 97]. CTC1 and STN1 mutations identified in CP patients cause increased DSB foci in the non-telomeric regions and induce genome instability [32, 98, 105], in agreement with the global genome maintenance functions of CST [23, 98, 105].
The CTC1 null mouse model has provided a better understanding of pathogenesis caused by CTC1 loss. CTC1 deleted mice are born smaller, grow slower, have significant smaller thymi and spleens than their wild-type littermates, and die prematurely with a median lifespan of only 24 days [45]. Hematopoietic stem cells (HSCs) from the CTC1 null mice shows a profound G2/M arrest and then significantly depleted, leading to bone marrow failure [45]. CTC1 null mice display an overall decrease in cellular proliferation in highly proliferative tissues. CTC1 deletion cells exhibits significant defects in telomere end protective functions, such as end-to-end chromosome fusions, catastrophic telomere loss, abnormal telomeric G-overhang extension, and an increase in extrachromosomal telomeric repeat DNA. In addition, CTC1 deletion significantly impairs efficient telomere replication [45]. The defective telomeres caused by CTC1 deletion are sensed as damaged DNA that activates a DNA damage response (DDR), leading to increased p53 and p21 expression [45]. While the proliferation deficiency observed in CTC1 null mice can be explained by telomere defects, genome-wide replication defects should also be considered and studied in vivo.
Elevated genome instabilities are observed in CST deficient cells, including micronuclei, aberrant chromosome segregation, chromosome fragmentation and γH2AX accumulation, suggesting that DNA damage response and/or repair activities are defective upon CST deficiency [25, 27]. Thus, CST dysregulation might be linked to carcinogenesis and/or clinical response to radiotherapy and chemotherapy. Indeed, genetic variants of STN1 and STN1 downregulation are associated with a number of cancers including epithelial ovarian cancer, colorectal cancer, leukemia, thyroid cancer, melanoma, and uterine cancer [106–111]. In addition, higher expression of CTC1 and STN1 appears to correlate with better disease-free survival in breast cancer patients [25, 112]. It will be interesting to investigate whether CST can serve as a clinical target for cancer diagnosis or therapy.
Concluding remarks and future directions
The CST complex is evolutionarily conserved across a wide range of eukaryotic species. The cryo-EM structure of CST has provided new insights into its functional mechanisms at telomeres. Recent studies highlight the engagement of CST in protecting global genome stability, including replication origin firing, stalled forks protection and restart, DSB repair, and ATR signaling. Despite these observations, the molecular mechanisms underlying CST-mediated genome maintenance remain largely unclear, and more studies in the following areas will be helpful to better understand the roles of this complex in genome protection. First, while the cryo-EM structure of CST has revealed the mechanism underlying its preferential binding to telomeric sequence, its non-telomeric sequence binding modes, including binding to ds-ssDNA junction and long nonspecific sequence substrates, still requires further investigation. Second, a number of key questions on the regulatory network surrounding CST need to be answered. Does CST participate in RAD51-mediated HDR pathway and/or stalled fork restart process? Since CST structure is closely related to RPA, what is the functional and molecular relationship between CST and RPA in these pathways? Can CST resolve other abnormal DNA structures in addition to G4? Does CST binding to DNA serve as a platform for recruiting other proteins to stalled forks or DSB sites? Third, CST has been found to interact with POT1-TPP1, POLα, Shieldin, RAD51 and MCMs. Yet much of these interactions have not been fully characterized. Pinpointing the specific amino acid residues mediating these protein interactions will be essential to understand the complex functions of CST. Moreover, given that CST participates in multiple genome maintenance pathways, it is highly likely that the actual number of CST interacting partners surpasses the above list. Discovering these unidentified binding partners will be important to fully understand the molecular mechanisms for maintaining genome integrity. In addition, PTMs of human CST proteins are largely unknown. Identifying and characterizing PTMs of hCST proteins will be crucial for understanding the regulation of CST in various genome protection pathways and disease development caused by CST mutations. Lastly, while pathogenic CST mutations have been investigated in the context of telomere maintenance, the molecular basis of CST pathological mutations in genome maintenance pathways beyond telomeres remains to be further investigated in order to obtain a better understanding of pathological processes and provide potential therapeutic strategies.
Acknowledgments
Funding
Research in Chai Lab is supported by R01CA234266 and R01GM 112864 to W.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Abbas T, Keaton MA, and Dutta A, Genomic instability in cancer. Cold Spring Harb Perspect Biol, 2013. 5(3): p. a012914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hou Y, et al. , Genome instability in Alzheimer disease. Mechanisms of Ageing and Development, 2017. 161: p. 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vijg J and Suh Y, Genome Instability and Aging. Annual Review of Physiology, 2013. 75(1): p. 645–668. [DOI] [PubMed] [Google Scholar]
- 4.Ceccaldi R, Rondinelli B, and D’Andrea AD, Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol, 2016. 26(1): p. 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson SP and Bartek J, The DNA-damage response in human biology and disease. Nature, 2009. 461(7267): p. 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quinet A, Lemaçon D, and Vindigni A, Replication Fork Reversal: Players and Guardians. Mol Cell, 2017. 68(5): p. 830–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith J, et al. , The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res, 2010. 108: p. 73–112. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen DD, et al. , Roles of OB-Fold Proteins in Replication Stress. Front Cell Dev Biol, 2020. 8: p. 574466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rickman K and Smogorzewska A, Advances in understanding DNA processing and protection at stalled replication forks. J Cell Biol, 2019. 218(4): p. 1096–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sirbu BM, et al. , Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem, 2013. 288(44: p. 31458–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyake Y, et al. , RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell, 2009. 36(2): p. 193–206. [DOI] [PubMed] [Google Scholar]
- 12.Nakaoka H, et al. , Xenopus laevis Ctc1-Stn1-Ten1 (xCST) protein complex is involved in priming DNA synthesis on single-stranded DNA template in Xenopus egg extract. J Biol Chem, 2012. 287(1): p. 619–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganduri S and Lue NF, STN1-POLA2 interaction provides a basis for primase-pol α stimulation by human STN1. Nucleic Acids Res, 2017. 45(16): p. 9455–9466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casteel DE, et al. , A DNA polymerase-{alpha}{middle dot}primase cofactor with homology to replication protein A-32 regulates DNA replication in mammalian cells. J Biol Chem, 2009. 284(9): p. 5807–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wan M, et al. , OB fold-containing protein 1 (OBFC1), a human homolog of yeast Stn1, associates with TPP1 and is implicated in telomere length regulation. The Journal of biological chemistry, 2009. 284(39): p. 26725–26731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen LY, Redon S, and Lingner J, The human CST complex is a terminator of telomerase activity. Nature, 2012. 488(7412): p. 540–4. [DOI] [PubMed] [Google Scholar]
- 17.Feng X, et al. , CTC1-mediated C-strand fill-in is an essential step in telomere length maintenance. Nucleic Acids Res, 2017. 45(8): p. 4281–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu P and Chang S, Functional characterization of human CTC1 mutations reveals novel mechanisms responsible for the pathogenesis of the telomere disease Coats plus. Aging Cell, 2013. 12(6): p. 1100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang C, Dai X, and Chai W, Human Stn1 protects telomere integrity by promoting efficient lagging-strand synthesis at telomeres and mediating C-strand fill-in. Cell Res, 2012. 22(12): p. 1681–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ackerson SM, Gable CI, and Stewart JA, Human CTC1 promotes TopBP1 stability and CHK1 phosphorylation in response to telomere dysfunction and global replication stress. Cell Cycle, 2020: p. 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barazas M, et al. , The CST Complex Mediates End Protection at Double-Strand Breaks and Promotes PARP Inhibitor Sensitivity in BRCA1-Deficient Cells. Cell Rep, 2018. 23(7): p. 2107–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhattacharjee A, et al. , Dynamic DNA binding, junction recognition and G4 melting activity underlie the telomeric and genome-wide roles of human CST. Nucleic Acids Res, 2017. 45(21): p. 12311–12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chastain M, et al. , Human CST Facilitates Genome-wide RAD51 Recruitment to GC-Rich Repetitive Sequences in Response to Replication Stress. Cell Rep, 2016. 16(7): p. 2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kasbek C, Wang F, and Price CM, Human TEN1 maintains telomere integrity and functions in genome-wide replication restart. J Biol Chem, 2013. 288(42): p. 30139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lyu X, et al. , Human CST complex protects stalled replication forks by directly blocking MRE11 degradation of nascent-strand DNA. Embo j, 2020: p. e103654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang F, Stewart J, and Price CM, Human CST abundance determines recovery from diverse forms of DNA damage and replication stress. Cell Cycle, 2014. 13(22): p. 3488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stewart JA, et al. , Human CST promotes telomere duplex replication and general replication restart after fork stalling. Embo j, 2012. 31(17): p. 3537–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bryan C, et al. , Structure of the human telomeric Stn1-Ten1 capping complex. PLoS One, 2012. 8(6): p. e66756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun J, et al. , Stn1-Ten1 is an Rpa2-Rpa3-like complex at telomeres. Genes Dev, 2009. 23(24): p. 2900–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rice C and Skordalakes E, Structure and function of the telomeric CST complex. Comput Struct Biotechnol J, 2016. 14: p. 161–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim CJ, et al. , The structure of human CST reveals a decameric assembly bound to telomeric DNA. Science, 2020. 368(6495): p. 1081–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y and Chai W, Pathogenic CTC1 mutations cause global genome instabilities under replication stress. Nucleic Acids Res, 2018. 46(8): p. 3981–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen LY, Majerská J, and Lingner J, Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev, 2013. 27(19): p. 2099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhattacharjee A, et al. , STN1 OB Fold Mutation Alters DNA Binding and Affects Selective Aspects of CST Function. PLoS Genet, 2016. 12(9): p. e1006342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hom RA and Wuttke DS, Human CST Prefers G-Rich but Not Necessarily Telomeric Sequences. Biochemistry, 2017. 56(32): p. 4210–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Sullivan RJ and Karlseder J, Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol, 2010. 11(3): p. 171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giraud-Panis M-J, et al. , CST Meets Shelterin to Keep Telomeres in Check:. Molecular Cell, 2010. 39(5): p. 665–676. [DOI] [PubMed] [Google Scholar]
- 38.Ishikawa F, CST Complex and Telomere Maintenance. In: Hanaoka F., Sugasawa K. (eds) DNA Replication, Recombination, and Repair. Springer, Tokyo, 2016. [Google Scholar]
- 39.Lue NF, Evolving Linear Chromosomes and Telomeres: A C-Strand-Centric View. Trends Biochem Sci, 2018. 43(5): p. 314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim CJ and Cech TR, Shaping human telomeres: from shelterin and CST complexes to telomeric chromatin organization. Nature Reviews Molecular Cell Biology, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng X, et al. , CTC1-STN1 terminates telomerase while STN1-TEN1 enables C-strand synthesis during telomere replication in colon cancer cells. Nat Commun, 2018. 9(1): p. 2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bryan TM, G-Quadruplexes at Telomeres: Friend or Foe? Molecules, 2020. 25(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petraccone L, et al. , Structure and stability of higher-order human telomeric quadruplexes. J Am Chem Soc, 2011. 133(51): p. 20951–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang M, et al. , Mammalian CST averts replication failure by preventing G-quadruplex accumulation. Nucleic Acids Res, 2019. 47(10): p. 5243–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gu P, et al. , CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. Embo j, 2012. 31(10): p. 2309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burgers PMJ and Kunkel TA, Eukaryotic DNA Replication Fork. Annu Rev Biochem, 2017. 86: p. 417–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, et al. , Human CST suppresses origin licensing and promotes AND-1/Ctf4 chromatin association. Life Sci Alliance, 2019. 2(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mazouzi A, Velimezi G, and Loizou JI, DNA replication stress: Causes, resolution and disease. Experimental Cell Research, 2014. 329(1): p. 85–93. [DOI] [PubMed] [Google Scholar]
- 49.Zeman MK and Cimprich KA, Causes and consequences of replication stress. Nature Cell Biology, 2014. 16(1): p. 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cortez D, Replication-Coupled DNA Repair. Mol Cell, 2019. 74(5): p. 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Q and Chai W, Suppression of STN1 enhances the cytotoxicity of chemotherapeutic agents in cancer cells by elevating DNA damage. Oncol Lett, 2016. 12(2): p. 800–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bansbach CE, et al. , The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev, 2009. 23(20): p. 2405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciccia A, et al. , Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell, 2012. 47(3): p. 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blastyák A, et al. , Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol Cell Biol, 2010. 30(3): p. 684–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schlacher K, et al. , Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell, 2011. 145(4): p. 529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Przetocka S, et al. , CtIP-Mediated Fork Protection Synergizes with BRCA1 to Suppress Genomic Instability upon DNA Replication Stress. Mol Cell, 2018. 72(3): p. 568–582.e6. [DOI] [PubMed] [Google Scholar]
- 57.Lemaçon D, et al. , MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun, 2017. 8(1): p. 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cotta-Ramusino C, et al. , Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol Cell, 2005. 17(1): p. 153–9. [DOI] [PubMed] [Google Scholar]
- 59.Ying S, Hamdy FC, and Helleday T, Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res, 2012. 72(11): p. 2814–21. [DOI] [PubMed] [Google Scholar]
- 60.Thangavel S, et al. , DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol, 2015. 208(5): p. 545–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karanja KK, et al. , DNA2 and EXO1 in replication-coupled, homology-directed repair and in the interplay between HDR and the FA/BRCA network. Cell Cycle, 2012. 11(21): p. 3983–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yeo JE, et al. , CtIP mediates replication fork recovery in a FANCD2-regulated manner. Hum Mol Genet, 2014. 23(14): p. 3695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dueva R and Iliakis G, Replication protein A: a multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer, 2020. 2(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fanning E, Klimovich V, and Nager AR, A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Research, 2006. 34(15): p. 4126–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bhat KP and Cortez D, RPA and RAD51: fork reversal, fork protection, and genome stability. Nat Struct Mol Biol, 2018. 25(6): p. 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hashimoto Y, et al. , Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol, 2010. 17(11): p. 1305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zellweger R, et al. , Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol, 2015. 208(5): p. 563–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mason JM, et al. , Non-enzymatic roles of human RAD51 at stalled replication forks. Nat Commun, 2019. 10(1): p. 4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saxena S, et al. , ATR Signaling Uncouples the Role of RAD51 Paralogs in Homologous Recombination and Replication Stress Response. Cell Rep, 2019. 29(3): p. 551–559.e4. [DOI] [PubMed] [Google Scholar]
- 70.Paulsen RD and Cimprich KA, The ATR pathway: Fine-tuning the fork. DNA Repair, 2007. 6(7): p. 953–966. [DOI] [PubMed] [Google Scholar]
- 71.Shiotani B and Zou L, ATR signaling at a glance. 2009. 122(3): p. 301–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burrows AE and Elledge SJ, How ATR turns on: TopBP1 goes on ATRIP with ATR. Genes & development, 2008. 22(11): p. 1416–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee Y-C, et al. , RPA-Binding Protein ETAA1 Is an ATR Activator Involved in DNA Replication Stress Response. Current Biology, 2016. 26(24): p. 3257–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tseng SF, Lin JJ, and Teng SC, The telomerase-recruitment domain of the telomere binding protein Cdc13 is regulated by Mec1p/Tel1p-dependent phosphorylation. Nucleic Acids Res, 2006. 34(21): p. 6327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang HHY, et al. , Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol, 2017. 18(8): p. 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wright WD, Shah SS, and Heyer WD, Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem, 2018. 293(27): p. 10524–10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liao S, Tammaro M, and Yan H, The structure of ends determines the pathway choice and Mre11 nuclease dependency of DNA double-strand break repair. Nucleic Acids Research, 2016. 44(12): p. 5689–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tomimatsu N, et al. , Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signaling decisions. DNA Repair, 2012. 11(4): p. 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ceppi I, et al. , CtIP promotes the motor activity of DNA2 to accelerate long-range DNA end resection. 2020. 117(16): p. 8859–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Daley JM, et al. , Enhancement of BLM-DNA2-Mediated Long-Range DNA End Resection by CtIP. Cell Reports, 2017. 21(2): p. 324–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Callen E, et al. , 53BP1 Enforces Distinct Pre- and Post-resection Blocks on Homologous Recombination. Molecular cell, 2020. 77(1): p. 26–38.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Isono M, et al. , BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell Rep, 2017. 18(2): p. 520–532. [DOI] [PubMed] [Google Scholar]
- 83.Adams MM and Carpenter PB, Tying the loose ends together in DNA double strand break repair with 53BP1. Cell Div, 2006. 1: p. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen C-C, et al. , Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annual review of cancer biology, 2018. 2: p. 313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Noordermeer SM, et al. , The shieldin complex mediates 53BP1-dependent DNA repair. Nature, 2018. 560(7716): p. 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ghezraoui H, et al. , 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature, 2018. 560(7716): p. 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dev H, et al. , Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol, 2018. 20(8): p. 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mirman Z, et al. , 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature, 2018. 560(7716): p. 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li S, et al. , Cdk1-dependent phosphorylation of Cdc13 coordinates telomere elongation during cell-cycle progression. Cell, 2009. 136(1): p. 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shen ZJ, et al. , PP2A and Aurora differentially modify Cdc13 to promote telomerase release from telomeres at G2/M phase. Nat Commun, 2014. 5: p. 5312. [DOI] [PubMed] [Google Scholar]
- 91.Gopalakrishnan V, Tan CR, and Li S, Sequential phosphorylation of CST subunits by different cyclin-Cdk1 complexes orchestrate telomere replication. Cell Cycle, 2017.16(13): p. 1271–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu C-C, et al. , Cdk1 Regulates the Temporal Recruitment of Telomerase and Cdc13-Stn1-Ten1 Complex for Telomere Replication. 2014. 34(1): p. 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Escandell JM, et al. , Ssu72 phosphatase is a conserved telomere replication terminator. Embo j, 2019. 38(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.AlSabbagh MM, Dyskeratosis congenita: a literature review. J Dtsch Dermatol Ges, 2020. 18(9): p. 943–967. [DOI] [PubMed] [Google Scholar]
- 95.Savage SA and Alter BP, Dyskeratosis congenita. Hematol Oncol Clin North Am, 2009. 23(2): p. 215–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Anderson BH, et al. , Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nature Genetics, 2012. 44(3): p. 338–342. [DOI] [PubMed] [Google Scholar]
- 97.Polvi A, et al. , Mutations in CTC1, Encoding the CTS Telomere Maintenance Complex Component 1, Cause Cerebroretinal Microangiopathy with Calcifications and Cysts. The American Journal of Human Genetics, 2012. 90(3): p. 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Simon AJ, et al. , Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. The Journal of experimental medicine, 2016. 213(8): p. 1429–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Crow YJ, et al. , Coats’ plus: a progressive familial syndrome of bilateral Coats’ disease, characteristic cerebral calcification, leukoencephalopathy, slow pre- and post-natal linear growth and defects of bone marrow and integument. Neuropediatrics, 2004. 35(1): p. 10–9. [DOI] [PubMed] [Google Scholar]
- 100.Morgado F, et al. , Coats plus syndrome (cerebroretinal microangiopathy with calcifications and cysts-1): A case report. Pediatr Dermatol, 2020. [DOI] [PubMed] [Google Scholar]
- 101.Ward SC, et al. , Progressive reticulate skin pigmentation and anonychia in a patient with bone marrow failure. Journal of the American Academy of Dermatology, 2017. 77(6): p. 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Noto Z, et al. , Dyskeratosis congenita associated with leukoplakia of the tongue. Int J Oral Maxillofac Surg, 2016. 45(6): p. 760–3. [DOI] [PubMed] [Google Scholar]
- 103.Burnett-Hartman AN, et al. , Telomere-associated polymorphisms correlate with cardiovascular disease mortality in Caucasian women: The Cardiovascular Health Study. Mechanisms of Ageing and Development, 2012. 133(5): p. 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fingerlin TE, et al. , Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nature genetics, 2013. 45(6): p. 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sargolzaeiaval F, et al. , CTC1 mutations in a Brazilian family with progeroid features and recurrent bone fractures. Mol Genet Genomic Med, 2018. 6(6): p. 1148–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Phelan CM, et al. , Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat Genet, 2017. 49(5): p. 680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li C, et al. , Relationship between the TERT, TNIP1 and OBFC1 genetic polymorphisms and susceptibility to colorectal cancer in Chinese Han population. Oncotarget, 2017. 8(34): p. 56932–56941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ojha J, et al. , Genetic Variation Associated with Longer Telomere Length Increases Risk of Chronic Lymphocytic Leukemia. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology, 2016. 25(7): p. 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gudmundsson J, et al. , A genome-wide association study yields five novel thyroid cancer risk loci. Nat Commun, 2017. 8: p. 14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Duffy DL, et al. , Novel pleiotropic risk loci for melanoma and nevus density implicate multiple biological pathways. Nature Communications, 2018. 9(1): p. 4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Välimäki N, et al. , Genetic predisposition to uterine leiomyoma is determined by loci for genitourinary development and genome stability. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Györffy B, et al. , An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat, 2010. 123(3): p. 725–31. [DOI] [PubMed] [Google Scholar]