

ABSTRACT
Antibody–drug conjugates (ADCs) are targeted therapeutic agents that treat cancers by selective delivery of highly potent cytotoxic drugs to tumor cells via cancer-specific antibodies. However, their clinical benefit is limited by off-target toxicity and narrow therapeutic windows. To overcome these limitations, we have applied reductive alkylation to develop a new type of ADC that has cytotoxic drugs conjugated to the N-terminal of an antibody through amine bonds introduced via reductive alkylation reactions (NTERM). To test whether the NTERM-conjugated ADCs can widen therapeutic windows, we synthesized three different ADCs by conjugating trastuzumab and monomethyl auristatin-F using three different methods, and compared their stability, efficacy, and toxicity. The NTERM-conjugated ADC was more stable in vitro and in vivo than the thiol-conjugated and the lysine-conjugated ADCs. The NTERM-conjugated ADC showed lower toxicity compared to other ADCs, whereas its efficacy was comparable to that of the thiol-conjugated ADC and better than that of the lysine-conjugated ADC. These results suggest that the NTERM conjugation method could widen the therapeutic window of ADCs by enhancing its stability and reducing toxicity.
KEYWORDS: Antibody–drug conjugate, N-terminal conjugation, therapeutic window, toxicity, stability, trastuzumab, monomethyl auristatin-F
Introduction
An antibody-drug conjugate (ADC) is a therapeutic agent composed of a target-specific antibody and a highly potent cytotoxic drug.1 ADCs, along with highly potent payloads, can directly kill targeted tumor cells without causing antibody-dependent cell-mediated cytotoxicity or complement-dependent cytotoxicity on which naked antibodies rely for a therapeutic effect. Consequently, ADCs have shown better objective response rates (ORRs) than naked antibodies during therapy. For example, brentuximab vedotin (Adcetris™) showed 75% ORR, whereas brentuximab alone could not evoke any response in patients with Hodgkin lymphoma (HL). Moreover, the ORR of trastuzumab emtansine (T-DM1; Kadcyla™) was up to 64%, whereas just 26% ORR was achieved by trastuzumab alone in trastuzumab-naïve patients.1 From 2011 to 2020, 10 ADCs were approved owing to their notable ORRs.2 Among them, T-DM1 and brentuximab vedotin highlight the application of ADCs as novel therapeutic agents against cancer by revealing the benefits of ADC at the clinical stage. These products have proved clinically the concept that ADCs can selectively deliver highly potent drugs to target cell and widen the therapeutic window, defined as the range between the maximum tolerable dose (MTD) and the minimum efficacious dose (MED) of the payload. The payloads used in the ADCs have a very narrow or near zero therapeutic window. In other words, their MTD is close to or lower than their MED, so that they cannot be used as therapeutic agents. When conjugated antibodies, the therapeutic window of a payload widens, with higher MTD and lower MED.
The therapeutic window of the ADC itself, however, also needs to be improved. A study of the exposure–response relationship of T-DM1 showed that patients with lower trough concentration (Cmin) experienced less clinical benefit. The study stratified patients according to the quartile of their T-DM1 Cmin values, and patients with the lowest quartile showed 3.2 months shorter progression-free survival (PFS) than the PFS of patient in other quartiles. If more T-DM1 can be administered to patients who showed lower Cmin, then patients might have a better response. However, the clinical dose of T-DM1 has been determined as 3.6 mg per kg (mpk) by its MTD, and the dose cannot be increased. In other words, the clinical benefit of T-DM1 is limited by its narrow therapeutic window.
Brentuximab vedotin also had a narrow therapeutic window owing to the fact that its MTD is 2.3 mpk and MED is 1.8 mpk, despite being an effective treatment option against HL and systemic anaplastic large cell lymphoma.3 Many MMAE-based ADCs against various targets have entered clinical trials and their MTD were usually limited to 1.8 mpk. Of those that advanced to Phase 2 clinical trials, most failed because of lack of efficacy. This result could mean that their MTDs were lower than their potential MED.
The MTD of an ADC is governed by factors such as the nature of target, property of antibody, drug-like property of linker-payload, and conjugation method.4 Here, we focus on how the conjugation method affects the properties of ADCs. Most common dose-limiting toxicities (DLTs) of ADCs observed in human clinical trials are myelotoxicity, including neutropenia and thrombocytopenia, and organ or tissue toxicities such as those exhibited on the peripheral nervous system, liver, kidney, lung, or cornea. The aforementioned toxicities are classified as off-target toxicities, since these organs are not the actual targets of ADCs.5 These off-target toxicities are considered to originate from the nonspecific absorption and catabolism of the ADC, and/or the dissociation of its payload from the intact ADC due to instability. Junutula et al.6 reported that the thiol-conjugated ADC was cleared faster and lost more payload than the corresponding cysteine-engineered (thiomabTM) ADC in rat, whereas the thiomab ADC showed less liver toxicity than the thiol-conjugated ADC. Lewis Phillips et al.7 showed that an ADC with readily cleavable disulfide linker exhibited a shorter half-life and more toxicity than an ADC with the non-cleavable linker. In addition, Hamblett et al.8 showed that an ADC with drug-to-antibody ratio (DAR) of 8 cleared faster than an ADC with DAR of 4. Moreover Lyon et al.9 showed that the conjugation of hydrophobic payload made an ADC hydrophobic, and this hydrophobic ADC could then be taken up by Kupffer cells in the liver.
DS-8201a (trastuzumab deruxtecan), a human epidermal growth factor receptor 2 (HER2)-targeting ADC developed by Daiichi Sankyo Co, Ltd., has shown potent anti-tumor activity and increased stability in plasma and demonstrated favorable safety profiles.10 The half-life of DS-8201a at 6.4 mpk dose was 7.33 days, which is comparable to that of trastuzumab (7.63 days).11,12 Currently, DS-8201a is the only anti-HER2 ADC that has comparable pharmacokinetic (PK) profile with trastuzumab, the naked antibody.
Since we presume that the current limitations of ADCs might be due to its unstable nature, it is necessary to develop an ADC that is more stable under in vitro and in vivo conditions. Additionally, we hypothesize that such an ADC would be safer and more effective. Therefore, to synthesize a more stable and tolerable ADC, we developed the N-terminal conjugation method (NTERM), which conjugates cytotoxic drugs to the N-termini of an antibody through amine bond via reductive alkylation reaction. A schematic diagram of NTERM-conjugated ADC is shown in Figure 1a. In this study, we used trastuzumab as a standard to evaluate the technical feasibility and enhancing properties, including stability and tolerability, of the NTERM-conjugated ADC. Brentuximab, which targets CD30, and lorvotuzumab, which targets CD56, were conjugated and analyzed to test whether the method is applicable to other antibodies.
Figure 1.
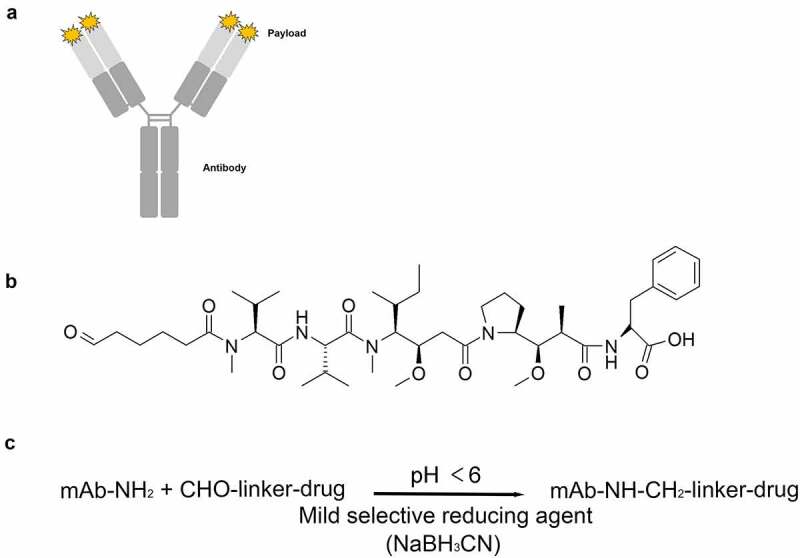
Overview of the NTERM conjugation method. (a) Schematic diagram of NTERM-conjugated ADC. (b) Chemical structure of the aldehyde-tagged MMAF (ald-MMAF). (c) Schematic representation of the NTERM conjugation reaction
NTERM, N-terminal conjugation through amine bond formation by reductive alkylation reaction; ADC, antibody–drug conjugate; MMAF, monomethyl auristatin F.
Results
NTERM conjugation
Each of the four protein chains in an antibody terminates in an alpha amino group that represents a primary amine (Figure 1a). NTERM-conjugated ADCs were synthesized by combining an aldehyde-tagged monomethyl auristatin F (ald-MMAF) (Figure 1b) or monomethyl auristatin E (ald-MMAE) with intact antibodies via reductive alkylation reaction (Figure 1c). Since the aldehyde group can act as an electrophile, it can react with amines that have lone-pair electrons. However, the aldehyde group cannot react with an ammonium ion because the ion has no lone pair electrons. Additionally, since the pKa of N-terminal α-amine is 2–3 units smaller than that of lysine ε-amine, the primary amine of the N-terminus comprises the dominant population of reactive amine over lysine ε-amine that mostly represents an ammonium ion at pH below 7. Grimsley et al. summarized pKa values of the ionizable groups in folded proteins from the literature, and the pKa was 7.7±0.5 for the N-terminus and 10.5±1.1 for lysines.13 At pH 5.49, the ratio of amine and ammonium ion becomes 1:102.21 (162-fold difference) for the N-terminus, and 1:105.01 (102,330-fold difference) for lysine ε-amine. Therefore, the alkylation reaction will happen preferentially on the N-termini of heavy and light chain of the antibody. The reaction then generates reversible, unstable imine bonds. Therefore, the imine bonds need to be reduced selectively by NaBH3CN to form a stable amine bond (Figure 1c).
The yield and quality of the conjugated antibodies were analyzed using a reverse-phase chromatography with C4 column, and less than 1% unconjugated antibody under optimized reaction conditions were obtained (Fig S1A, B). The presence of aggregates or fragments was analyzed using size-exclusion chromatography and sodium dodecyl sulfate polyacrylamide gel electrophoresis. No aggregations or fragments were detected (Fig S1C). The overall yield obtained was approximately 87%, calculated as the ratio of the amount of input antibody to that of the output ADC. The reaction was scalable, and therefore we could set up a conjugation reaction with up to 600 mg of antibodies or 50 mL of the reaction mixture.
Determination of DAR using liquid chromatography/mass spectroscopy
DAR is defined as the number of drug molecules that are attached to an antibody, and it is the critical quality attribute of an ADC because it determines the potency of the molecule. The degree of conjugation and the DAR were determined using liquid chromatography (LC)/mass spectroscopy (MS). The deconvoluted mass spectrum of trastuzumab-MMAF conjugate synthesized via NTERM conjugation (T-N-F) is shown in Figure 2a. Several species with DAR varying from 0 to 7 were identified. The apparent DAR was determined as 3.2 upon calculating the weighted-average of DAR. The most abundant species identified was DAR 3, whereas DAR 0 and 7 were the least abundant species, with occupancy was less than 3%.
Figure 2.
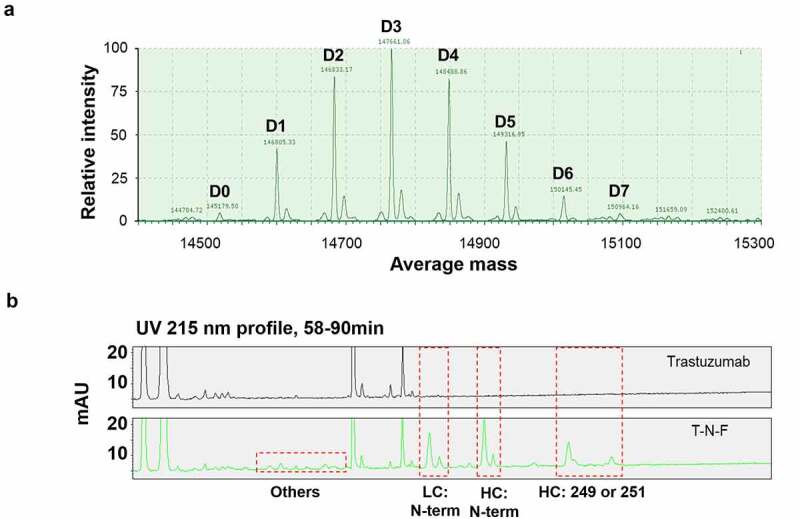
LC/MS analysis of NTERM conjugate. (a) DAR profile of T-N-F. T-N-F was analyzed using Waters Synapt G-2 system after deglycosylation reaction. Species with DAR 0–DAR 7 were observed and marked at D0–D7. The apparent DAR is 3.2 that is calculated as the weighted-average of DAR. (b) Conjugation sites were identified by peptide mapping. Trypsin-treated trastuzumab (top) and T-N-F (down) fragments mixture were resolved using an UPLC system with ACQUITY UPLC PST (BEH) C18 column
LC, liquid chromatography; MS, mass spectroscopy; DAR, drug-to-antibody ratio; T-N-F, trastuzumab-MMAF conjugate synthesized via NTERM conjugation reaction; UPLC, ultra-performance liquid chromatography.
Identification of conjugation sites
To determine whether the NTERM conjugation reaction is more selective to N-terminal α-amine, conjugation sites were identified using peptide mapping. The unconjugated naked antibody and the MMAF-conjugated trastuzumab were fragmented by trypsin digestion, and the fragments were separated using reverse-phase column and identified using multiple reaction monitoring MS. Conjugated fragments were easily identified as newly emerged peaks (Figure 2b) by comparison with the chromatogram of the naked antibody, and the conjugated sites were identified based on mass and diminishing intensity of the originating peaks of unconjugated fragments.
Overall, 15 conjugated fragments were detected and identified (Table 1). The major conjugated fragments detected were the N-terminal fragment of antibody heavy or light chain, which indicates that the major conjugation sites were the primary amines of the N-terminal. The N-terminal fragments of heavy chain contain 45.7% of the conjugated fragments, and 28.5% of that of the light chain. In summary, N-terminal conjugation accounted for 74.2% of the conjugated fragments, suggesting the preferred selectivity toward N-terminal residues. However, unintended conjugation at the Lys residues increased heterogeneity of the overall products. The next most frequent conjugated site was Lys249 (or Lys 251, as per amino acid numbering, not EU numbering,) that is located on the CH2 domain of the heavy chain and accounts for 16.8% of the conjugated fragments. Hence, the top three fragments account for more than 91% of the conjugated fragments, thereby suggesting the site preference of NTERM conjugation reactions. Fragments with conjugation at Lys207 and Lys169 of the light chain comprised only 2.4% and 1.1% of the total conjugated fragments, respectively. The sum of the rest fragments were 5.6% and each fragment occupied less than 1%. The number of conjugated peaks in the NTERM-conjugated ADC were considerably lower than that of T-DM1,14 thereby suggesting that NTERM conjugation generated more homogenous ADCs.
Table 1.
Conjugation sites identified by peptide mapping
| Label | Peptide | Chain | Position |
Relative Population | |
|---|---|---|---|---|---|
| Start | End | ||||
| HC N-term | EVQLVESGGGLVQPGGSLR | HC | 1 | 19 | 45.7% |
| LC N-term | DIQMTQSPSSLSASVGDR | LC | 1 | 18 | 28.5% |
| HC:249 or 251 | THTCPPCPAPELLGGPSVFLFPPKPK | HC | 226 | 251 | 16.8% |
| Others | VYACEVTHQGLSSPVTK | LC | 191 | 207 | 9.1% |
| VDNALQSGNSQESVTEQDSK | LC | 150 | 169 | ||
| VQWK | LC | 146 | 149 | ||
| ASQDVNTAVAWYQQKPGK | LC | 25 | 42 | ||
| GPSVFPLAPSSK | HC | 125 | 136 | ||
| TKPR | HC | 292 | 295 | ||
| VEIK | LC | 104 | 107 | ||
| KVEPK | HC | 217 | 221 | ||
| VSNK | HC | 326 | 329 | ||
| ADYEK | LC | 184 | 188 | ||
| ALPAPIEK | HC | 330 | 337 | ||
| VDK | HC | 214 | 216 | ||
Bold: conjugation site
Since there is no actual observed pKa values for amine in trastuzumab, we predicted their values using PROPKA3.15 The predicted pKa values were 8.07, 7.75, and 9.99 for the heavy chain N-terminus, the light chain N-terminus, and Lys249, respectively. From the predicted pKa values, it is not clear why Lys249 (or Lys251) showed more conjugation than other lysine residues.
Ligand binding affinity
NTERM conjugation could affect affinity, as the N-termini of the antibody heavy and light chains are located near the paratope site, where an antigen will bind. Moreover, an antibody can have lysine residues on its complementarity-determining regions (CDRs). If NTERM conjugation modifies lysine residues on CDRs because of lack of selectivity, the binding affinity will decrease after the conjugation.
To investigate whether N-terminal modification of an antibody alters its binding affinity, the affinity of trastuzumab with or without conjugation was determined using surface plasmon resonance (SPR) with Biacore T200. The association and dissociation rate constants of trastuzumab and the MMAF-conjugated trastuzumab were measured twice, and the average dissociation constants were calculated (Table 2).
Table 2.
Binding affinity and kinetics parameter of trastuzumab and trastuzumab-MMAF conjugates against HER2
| Sample | 1st measurement |
2nd measurement |
Avg. KD (nM) | ||||
|---|---|---|---|---|---|---|---|
| ka (M-1s-1) | kd (s−1) | KD (nM) | ka (M-1s-1) | kd (s−1) | KD (nM) | ||
| Trastuzumab | 3.46 E + 05 | 4.32 E-05 | 0.13 | 1.71 E + 05 | 3.75 E-05 | 0.22 | 0.18 |
| T-N-F(DAR 1.6) | 2.96 E + 05 | 3.51 E-05 | 0.12 | 9.55 E + 04 | 8.17 E-06 | 0.09 | 0.11 |
| T-N-F(DAR 3.2) | 3.81 E + 05 | 3.99 E-05 | 0.11 | 9.44 E + 04 | 6.23 E-06 | 0.07 | 0.09 |
| T-C-F (DAR 3.7) | 4.10 E + 05 | 3.74 E-05 | 0.09 | 1.14 E + 05 | 1.62 E-05 | 0.14 | 0.12 |
| T-K-F (DAR 3.9) | 5.31 E + 05 | 6.32 E-05 | 0.12 | 9.39E+04 | 2.03E-05 | 0.22 | 0.17 |
MMAF: monomethyl auristatin F, HER2: human epidermal growth factor receptor 2, DAR: drug-to-antibody ratio, T-N-F: trastuzumab-MMAF conjugate synthesized via NTERM conjugation, T-C-F: trastuzumab-MMAF conjugate synthesized via thiol conjugation, T-K-F: trastuzumab-MMAF conjugate synthesized via lysine conjugation.
The binding affinity of trastuzumab was not altered by NTERM conjugation. The equilibrium dissociation constants (KD) of T-N-F (DAR 3.2), T-N-F (DAR 1.6), and trastuzumab were determined as 0.09, 0.11, and 0.19 nM, respectively. Additionally, the association and the dissociation rate constants followed a similar trend, and no considerable differences between those of the antibody and the ADCs were observed. The T-C-F and T-K-F were also analyzed as a comparison and showed similar values of 0.12 nM and 0.17 nM, respectively. To confirm the observation, we performed another SPR analysis where the ligand and the analyte were switched. Trastuzumab or T-N-F (DAR 3.2) were captured on a protein A chip and monomeric HER2 was used as the analyte. In this analysis, trastuzumab and T-N-F gave KD values of 4.6 nM and 5.2 nM, respectively, at the same magnitude of association rate constants and dissociation rate constants (Fig S2). Therefore, NTERM conjugation does not seem to alter or interfere with the antigen-binding activity of the antibody. This result was confirmed by the results of the cell-surface antigen-binding activity of antibodies estimated via flow-cytometry analysis (Fig S3A).
To examine whether NTERM conjugation maintains the same binding affinity in other antibodies as well, the ligand binding properties of brentuximab and lorvotuzumab were determined using enzyme-linked immunosorbent assays (ELISA). The conjugated antibodies showed the same response curves and EC50 values compared to their corresponding naked antibodies (Fig S3B). Therefore, we concluded again that N-terminal modification did not alter the binding properties of the antibodies.
The tested antibodies have different numbers of lysine residues on their CDRs. Trastuzumab has one lysine on CDR-H2, brentuximab has three lysines on CDR-H2, and lorvotuzumab has one lysine on CDR-H3 and another on CDR-L2. However, none of these antibodies showed any meaningful change in binding affinity. Therefore, it seems that NTERM conjugation does not alter lysines on CDRs.
In vitro cytotoxicity
The potency of T-N-F was compared with that of the ADCs synthesized via other conventional techniques, including thiol conjugation and lysine conjugation. T-C-F is a trastuzumab and maleimidocaproyl MMAF (mc-MMAF) conjugate synthesized via thiol conjugation and T-K-F is a trastuzumab and mercaptohexanoic MMAF (SH-MMAF) conjugate synthesized via lysine conjugation, which uses succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) as a bifunctional linker that attaches to the lysine ε-amine and thiol group of the payload. The potency of three ADCs, namely T-N-F (DAR 3.2), T-C-F (DAR 3.7, Fig S4A), and T-K-F (DAR 3.9, Fig S4B), were tested in BT474, a trastuzumab-sensitive HER2-overexpressing (3+) breast cancer cell line, and their estimated IC50 values were 115.5, 49, and 77.2 pM, respectively. Moreover, in HCC1954, a trastuzumab-insensitive HER2-overexpressing (3+) breast cancer cell line, the IC50 values estimated for T-N-F, T-C-F and T-K-F were 39.9, 22.1, and 45 pM, respectively. Hence, for both cell lines, T-C-F showed two-fold higher potency than T-N-F, whereas T-N-F and T-K-F were comparable in terms of potency.
Additionally, to confirm whether NTERM conjugation method can be applied to other ADCs, we synthesized brentuximab–MMAF conjugate by NTERM conjugation and tested its cytotoxicity. Brentuximab is the antibody component of the approved anti-CD30 ADC, AdcetrisTM (brentuximab vedotin). Two brentuximab–MMAF conjugates (B-N-F) with different DARs of 1.8 and 2.6 were synthesized and their cytotoxicity in CD30-positive cell lines (Karpas-299 and L-540) were analyzed. The B-N-F with DAR 1.8 and 2.6 effectively killed Karpas-299 cells with the estimated IC50 values of 30 and 21 pM, respectively. In the case of L-540 cells, the IC50 values were 100 and 97 pM, respectively, for B-N-F DAR 1.8 and DAR 2.6 (Fig S5). Adcetris, which has a DAR of 4, showed IC50 of 24 pM for Karpas-299 and 66 pM for L-540 cells.
In vitro stability
The stability of ADCs in serum was analyzed to determine whether NTERM conjugation resulted in improved serum stability compared to other known ADCs. To compare T-N-F with T-C-F, these ADCs were mixed with human or rat serum and incubated for 7 days. Subsequently, changes in the concentration and the DAR of the ADCs were compared (Table 3). The concentration of the conjugated antibody was measured using ELISA and the change in DAR was analyzed using LC/MS. After 7 days of incubation in human serum, the concentration of T-N-F decreased by 12.7% from the baseline value (day 0), whereas the DAR of T-N-F changed slightly from 1.5 to 1.6. Because the DAR of the T-N-F sample was as low as 1.5, we repeated this experiment with T-N-F of higher DAR, and the DAR changed from 3.5 to 3.4. In contrast, T-C-F, the thiol-conjugated antibody, showed a 21.8% reduction in its concentration and its DAR changed from 3.0 to 2.0. When the concentration of the remaining conjugated T-C-F were measured, the values were 55% on day 3 and 35% on day 7 compared to the baseline. As there is difference between total antibody concentration and the conjugated antibody concentration, it seemed that deconjugation of payload happened in the serum during incubation. A similar trend was observed in the rat serum incubation study as well (Table 3). Therefore, we could conclude that the NTERM-conjugated antibody showed more stability than the thiol-conjugated antibody in both rat and human serum.
Table 3.
In vitro stability of ADCs in serum samples
| |
Remaining mAb (%) |
Relative DAR (%) |
|||
|---|---|---|---|---|---|
| Human serum1) |
Rat serum2) |
Human serum |
Rat serum |
||
| Day 3 | Day 7 | Day 3 | Day 7 | Day 3 | |
| Trastuzumab | 102.8 | 90.7 | 82 | - | - |
| T-N-F | 95.1 (T) | 87.3 (T) | 75 | 101.4 | 98 |
| 95.0 (C) | 86.0 (C) | ||||
| T-C-F | 95.7 (T) | 78.2 (T) | 66 | 65.6 | 58 |
| 55.0 (C) | 35.0 (C) | ||||
1) In vitro stability in human serum: 37°C, 3 days or 7 days incubation, measured by ELISA.
2) In vitro stability in rat serum: 37°C, 3 days incubation, measured by LC/MS.
ADC: antibody–drug conjugate, mAb: monoclonal antibody, DAR: drug-to-antibody ratio, T-N-F: trastuzumab-monomethyl auristatin F (MMAF) conjugate synthesized via NTERM conjugation, T-C-F: trastuzumab-MMAF conjugate synthesized via thiol conjugation.
When the LC/MS data were analyzed, co-purified endogenous human IgGs gave background noise signals on the LC/MS spectra. However, signals from ADCs could be distinguished from the baseline noise signal. The concentration of human IgG would be 10- to 15-fold higher than the concentration of ADC samples, but human serum IgG is a mixture of vastly diverse IgGs and the mole fraction of each individual IgG clone must be very small and give a very small signal. Summation of these small, diverse, and interfering signals would give undistinguishable, averaged, and continuous baseline noise. These baseline signal could be removed by applying baseline correction before mass spectrum deconvolution. Conversely, the ADC samples have high molar fraction and would give higher signal-to-noise ratio against serum IgG. Therefore, it was possible to calculate DAR of ADC samples in human serum.
Single-dose PK study in rats
To compare the stability of ADCs in vivo, we compared the PK profile of ADCs in rats. In this study, ADCs with faster clearance than the parental antibody or that lose their payloads during circulation are defined as unstable. The faster clearance can be described as shorter half-life or clearance (CL) rate. Loss of payload can be observed by comparing the PK profiles of total antibody and conjugated antibody, as described below.
Four groups of female Sprague Dawley (SD) rats were administered trastuzumab, T-N-F (DAR 3.2), T-C-F (DAR 3.7), and T-K-F (DAR 3.9), respectively, at 2.5 mpk dose intravenously and the serum concentration of total antibody and conjugated antibody were measured for 14 days. The total antibody concentration was measured using ELISA with ErbB2 and goat anti-human kappa light chain-horseradish peroxidase (HRP) conjugate antibody. To measure the concentration of the conjugated antibody, rabbit anti-MMAF polyclonal antibody was used to capture the conjugated antibody, and the captured antibody was then detected using biotinylated ErbB2/streptavidin-HRP. The results are shown in Figure 3 and Table 4.
Figure 3.
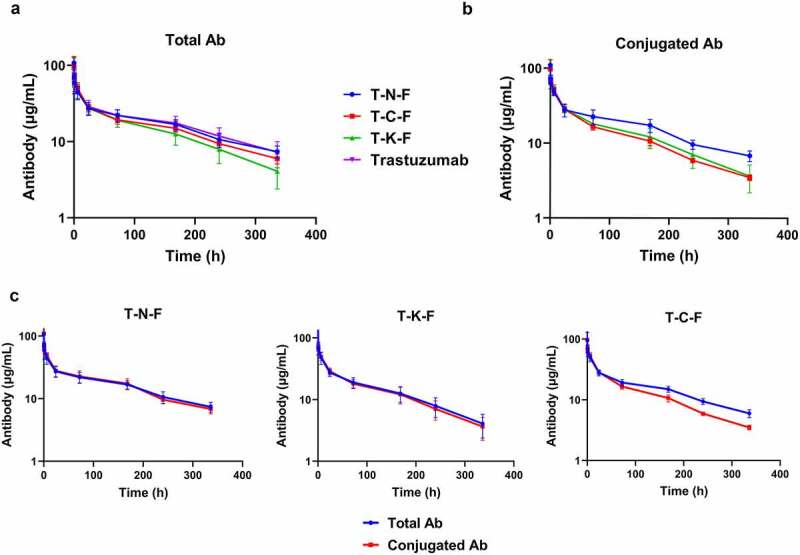
Pharmacokinetic profile of ADCs, synthesized with different conjugation techniques, in rats at a single dose of 2.5 mg/kg. (a) Overview of the total antibody concentration of ADCs. (b) Overview of the conjugated antibody concentration. Blue line represents T-N-F, whereas T-C-F, T-K-F, and trastuzumab are represented by red, green and magenta lines, respectively. (c) Overview of the total and conjugated antibody concentration profiles of T-N-F (left), T-C-F (middle), and T-K-F (right). Blue lines denote total mAb and red lines represent ADC. N = 5 and the error bars represent standard error
ADCs, antibody–drug conjugates; T-N-F, trastuzumab-monomethyl auristatin F (MMAF) conjugate synthesized via NTERM conjugation; T-C-F, trastuzumab-MMAF conjugate synthesized via thiol conjugation; T-K-F, trastuzumab-MMAF conjugate synthesized via lysine conjugation; mAb, monoclonal antibody.
Table 4.
Pharmacokinetic parameters of trastuzumab and MMAF-conjugated ADCs
| |
AUC |
Beta half-life |
Clearance rate |
||
|---|---|---|---|---|---|
| h·µg/mL | % vs. trastuzumab | h | % vs. trastuzumab | mL/h/kg | |
| Trastuzumab | 6964.9 | 100% | 115.7 | 100% | 0.37 |
| T-N-F (T) | 6795.7 | 98% | 122.1 | 106% | 0.37 |
| T-N-F (C) | 6813.2 | 98% | 118.3 | 102% | 0.38 |
| T-C-F (T) | 5933.5 | 85% | 111.6 | 96% | 0.43 |
| T-C-F (C) | 4315.4 | 62% | 84.6 | 73% | 0.60 |
| T-K-F (T) | 4324.3 | 62% | 65.1 | 56% | 0.64 |
| T-K-F (C) | 3718.7 | 54% | 53.2 | 46% | 0.69 |
ADCs: antibody–drug conjugates, AUC: area under the curve, T-N-F: trastuzumab-monomethyl auristatin F (MMAF) conjugate synthesized via NTERM conjugation, (T): total antibody concentration, (C): conjugate antibody concentration, T-C-F: trastuzumab-MMAF conjugate synthesized via thiol conjugation, T-K-F: trastuzumab-MMAF conjugate synthesized via lysine conjugation.
The total antibody concentration profiles for all the test subjects are shown in Figure 3a. T-N-F(T) [“(T)” denotes the total antibody concentration] showed a profile similar to that of the naked antibody (trastuzumab), whereas the other two ADCs (T-C-F and T-K-F) showed lower concentration profiles than that of the naked antibody over different time points, thereby suggesting faster CL. T-N-F(T) showed the same CL rate of 0.37 mL/h/kg as trastuzumab; however, T-K-F(T) showed the fastest CL rate (0.64 mL/h/kg), followed by T-C-F(T) with a CL rate of 0.43 mL/h/kg. The half-life of trastuzumab and T-N-F(T) were 115.7 h and 122.1 h, respectively, whereas T-C-F(T) and T-K-F(T) had comparatively shorter half-lives of 111.6 and 65.1 h, respectively (Table 4).
The concentration profiles of the conjugated ADCs are shown in Figure 3b. These data clearly demonstrate prominent differences in CL rates and half-lives of the ADCs. The CL rate of T-N-F(C) [“(C)” denotes the conjugated antibody concentration] is 0.38 mL/h/kg, whereas T-C-F(C) and T-K-F(C) showed comparatively faster CL rates of 0.60 and 0.69 mL/h/kg, respectively. In addition, the half-lives of T-C-F(C), T-K-F(C), and T-N-F(C) are 84.6, 53.2 and 118.3 h, respectively. These data indicated that T-N-F(C) has a half-life that is better than other ADCs and similar to that of the naked antibody.
The graphs shown in Figure 3a and Figure 3b were re-plotted for each ADC and are shown in Figure 3c to demonstrate the differences between the total and the conjugated antibody profiles. The total and conjugated antibody profiles of T-N-F were almost identical, thereby suggesting that T-N-F did not lose its payload during blood circulation. In contrast, considerable differences between the total and conjugated antibody concentration for T-C-F were observed, thereby indicating that T-C-F had lost its payload during blood circulation. This observation was consistent with the fact that the deconjugation of the maleimide-MMAF payload in serum can happen via the retro-Michael reaction, and a thiol-conjugated ADC could, therefore, gradually lose its payload during blood circulation.16 For T-K-F, the total and conjugated antibody profiles were almost identical, thereby suggesting minimal loss of payload. However, the total antibody of T-K-F itself is eliminated from serum faster than the naked antibody, thus suggesting nonspecific uptake of T-K-F, which was not observed in T-N-F.
Collectively, these differences resulted in different exposure of ADCs as determined by area under the curve (AUC). The AUCs of T-N-F(T) and T-N-F(C) were very similar to the AUC of the naked antibody. However, the AUC of T-C-F(T) showed 85% of the AUC of the naked antibody, whereas the AUC of T-C-F(C) was just 62%. The lysine-conjugated antibody, T-K-F, showed considerable decrease of AUC, to 62% and 54% for T-K-F(T) and T-K-F(C), respectively (Table 4). The smaller value of AUCs and shorter half-lives of ADCs suggest that the conjugated antibody is degraded and absorbed faster in tissues than the naked antibody.
Toxicity study in rats
To confirm whether the tolerability of ADC is improved by its stabilization, a single-dose toxicity study was performed in rats. Female SD rats were injected with 50, 100 or 200 mpk of each ADC (T-N-F and T-C-F) intravenously, while T-K-F was given at 40, 80, or 160 mpk. Because T-K-F had higher DAR of 3.9 than T-N-F ADC (DAR 3.2), 80% of doses were administered to match the dose of the payload. T-C-F (DAR 3.7) also had higher DAR than T-N-F, but T-C-F was given at the same doses as T-N-F (50, 100, 200 mpk) because the incorrect DAR of T-C-F (DAR 3.1) was reported when the animal experiment was planned and performed. Therefore, cautious interpretation of results is required because T-C-F-treated animal were dosed 15% more MMAF than T-N-F treated group, while the same amounts of ADCs were administrated. MMAF (18 mg/m2 or 3 mpk) or trastuzumab (200 mpk) were administered in a single dose that is equivalent to 200 mpk dose of T-N-F. Because no considerable toxicity in the MMAF- or trastuzumab-treated group was observed, we assumed that the observed effect hereafter was due to the adverse effect caused by ADC.
Weight loss was observed only in the T-C-F- (200 mpk) and the T-K-F-treated (160 mpk) groups (Figure 4a). All but one animal in the T-C-F-treated (200 mpk) group died after 8 days. In the T-K-F-treated groups, one animal administered a 80 mpk dose died after day 5. The T-N-F-treated (200 mpk) group showed considerable delay in weight gain, but after 7 days, they gained similar weights compared to that of the other groups. Moreover, no animal from the T-N-F-treated group died.
Figure 4.
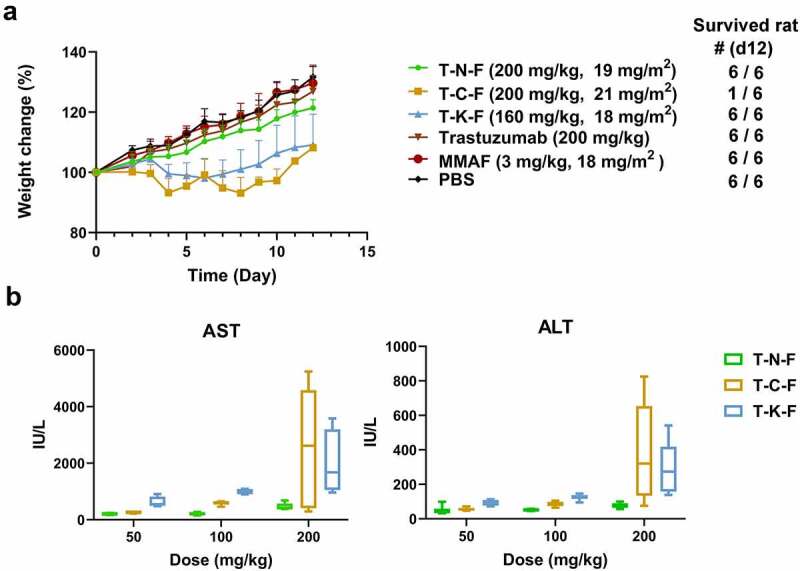
(a) Weight change of rats after administering a single dose of test samples. The numbers of survived animals are indicated on the right. (b) Dose-related hepatotoxicity induced by ADCs. AST and ALT activity in rat serum were assessed at day 5 after ADC administration. Each point denotes the average value obtained from five animals. Box plots denote mean value and 25–75% quartile
ADCs, antibody–drug conjugate; AST, aspartate aminotransferase; ALT, alanine aminotransferase.
Liver toxicity was assessed by determining aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels in the serum. The thiol- and lysine-conjugated ADCs showed an increase in the AST and ALT levels, thereby suggesting severe liver toxicity. In contrast, T-N-F was well-tolerated, as there was no surging increase in the AST and ALT levels (Figure 4b). The relative changes in AST levels of T-N-F groups were as follows: −26% (50 mpk), −25% (100 mpk), and 69% (200 mpk) at day 5 post-administration, compared to the control groups whose AST levels were 276.7 IU/L (PBS) or 182.22 IU/L (trastuzumab 200 mpk). However, the T-C-F groups showed a dose-dependent increase of up to 839% in the AST level. In addition, the T-K-F-treated group showed an increase in AST of up to 130% even at the lowest dose (40 mpk). The highest dose of T-C-F and T-K-F (200 mpk or 160 mpk) increased AST levels up to 916% and 690%, respectively, whereas T-N-F showed 109% increase in AST as compared to that of the control. The ALT levels also followed a similar trend. At the highest dose of T-N-F (200 mpk), the ALT level was 78.01 IU/L on day 5 post-administration compared to the control groups whose ALT levels were 37.31 IU/L (phosphate-buffered saline (PBS)) or 36.15 IU/L (trastuzumab 200 mpk). However, the ALT level of the T-C-F-treated group (200 mpk) surged to 379.15 IU/L and that of the T-K-F-treated group (180 mpk) was 294.62 IU/L. While T-N-F increased the ALT level by approximately 2-fold, T-C-F and T-K-F increased the ALT levels by 10-fold and 8-fold, respectively. Therefore, these results suggest that T-N-F has less severe liver toxic effects than those of the T-C-F and T-K-F.
Since neutropenia and thrombocytopenia are the major DLTs observed upon the administration of approved ADCs, hematological analyses were performed, and neutrophil and platelet counts were measured at day 5 and 12 post-administration. As shown in Table 5, T-N-F dosed at a 50 mpk resulted in a slight increase in neutrophil counts and relatively moderate changes were observed at higher doses. The T-C-F-treated group showed a dose-dependent decrease in neutrophil counts, and 200 mpk dose of T-C-F showed a 77% decrease in neutrophils on day 5 as compared to that of the control groups. In contrast, all T-K-F-treated groups showed an increase in neutrophils on day 5. However, all T-C-F-treated groups showed an increase in neutrophils on day 12. The average neutrophil count of control groups (PBS, MMAF, trastuzumab) was 3.9 × 1000 cells/μL (or hereafter k/μL), whereas T-K-F-treated groups showed 7.53, 6.64, and 7.59 k/μL at 40, 80, and 160 mpk doses, respectively. The neutrophil count of the T-K-F-treated groups increased by 93%, 70%, and 103% as compared to that of the control group. On day 12, the T-K-F treated group showed different trends in the neutrophil count. The 40 mpk treated group showed a decrease in the neutrophil count by 32%, but the 80 mpk and 160 mpk treated groups showed an increase in the neutrophil count as compared to those observed on day 5.
Table 5.
Hematological analysis of rat blood on day 5 and day 12 after administration of test samples
| |
Neutrophil (k/μL) |
Platelet (k/μL) |
||||
|---|---|---|---|---|---|---|
| 50 mpk | 100 mpk | 200 mpk | 50 mpk | 100 mpk | 200 mpk | |
| Day 5 | ||||||
| PBS | - | - | 3.83 (0.74) | - | - | 857.17 (42.85) |
| trastuzumab | - | - | 3.90(0.85) | - | - | 847.60 (114.75) |
| MMAF | - | - | 3.73 (0.99) | - | - | 904.83 (60.79) |
| T-N-F | 5.84 (1.54) | 4.42 (0.59) | 4.82 (1.74) | 623.67 (292.17) | 1027.83 (124.40) | 680.20 (113.27) |
| T-C-F | 4.01 (1.17) | 2.75 (0.48) | 1.28 (0.77) | 585.67 (284.16) | 495.80 (83.21) | 113.80(15.45) |
| T-K-F | 7.53 (3.91) | 6.64 (2.40) | 7.95 (1.79) | 586.00 (53.81) | 344.17 (83.08) | 208.17(44.75) |
| Day 12 | ||||||
| PBS | - | - | 1.91 (0.36) | - | - | 495 (141.22) |
| trastuzumab | - | - | 2.86 (1.13) | - | - | 477.50 (85.32) |
| MMAF | - | - | 1.94 (0.36) | - | - | 588.83 (91.03) |
| T-N-F | 3.31 (1.42) | 5.86 (1.63) | 5.92 (2.21) | 532.67 (92.86) | 566.83 (221.19) | 871.17 (152.08) |
| T-C-F | 4.72 (0.94) | 3.58 (0.87) | 27.13* | 484.50 (112.61) | 535.50 (167.18) | 335* |
| T-K-F | 5.09 (1.70) | 12.32** (4.14) | 18.96 (3.4) | 581.17 (206.21) | 537.80** (148.33) | 364.00 (113.70) |
* Data from only one survived animal
** One animal died after day 5
Numbers in parentheses denote standard deviation
mpk: mg per kg, MMAF: monomethyl auristatin F, T-N-F: trastuzumab-MMAF conjugate synthesized via NTERM conjugation, T-C-F: trastuzumab-MMAF conjugate synthesized via thiol conjugation, T-K-F: trastuzumab-MMAF conjugate synthesized via lysine conjugation, PBS: phosphate-buffered saline.
Further, the effects of different ADCs on platelet count were determined. The T-N-F-treated group showed −26%, 21%, and −20% changes in the platelet count at 50, 100, and 200 mpk doses, respectively, as compared to that of the control group. The platelet counts were perturbed, but were within the normal range (685–1436 k/μL), and the platelet count of the PBS-treated group was 857 k/μL. The T-K-F-treated groups showed significant dose-dependent decreases in platelet counts (from 586 k/μL to 208 k/μL) that are lower than the normal range. Similarly, the T-C-F-treated groups also showed a dose-dependent decrease in platelets (from 585 k/μL to 114 k/μL). Both the T-K-F- and T-C-F-treated groups had decreased numbers of platelets at the 200 mpk dose, thereby resulting in thrombocytopenia. However, the 200 mpk dose of T-N-F did not show considerable changes and resulted in moderate changes (~ 20%) without dose–response effect.
In vivo efficacy in rats
To test the hypothesis that the improved stability of ADCs synthesized via NTERM conjugation can increase the exposure of ADCs and lead to better efficacy, we compared the anti-tumor efficacy of three ADCs in nude rats that had been injected with trastuzumab-resistant HCC1954, a HER2-overexpressing breast cancer cell line. Immunocompromised mouse is the most suitable xenograft model, but we decided to use the immunocompromised rat model so that both the efficacy and PK studies were conducted in the same species, since our purpose was to test whether the difference in PK can lead to a different response in efficacy. It has been reported that the PK of an ADC differs between mouse and rat models.17
A single dose of ADCs (1 mpk or 2.5 mpk) was administered in the HCC1954 rat-xenograft model, when the average tumor size was 300 mm3, and the growth in tumors was monitored (Figure 5).
Figure 5.
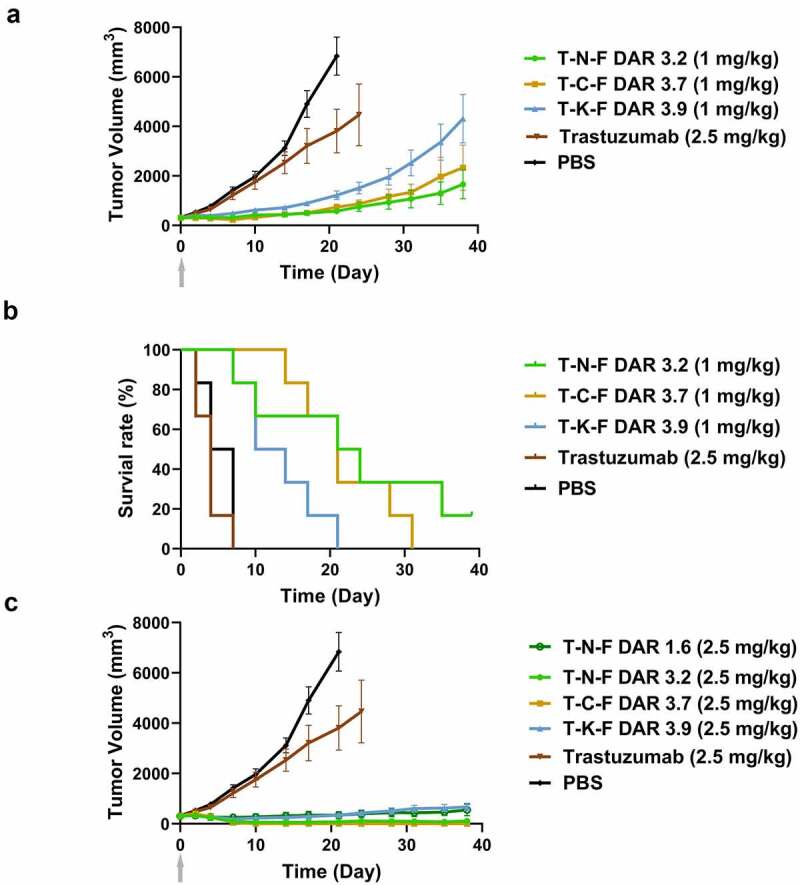
In vivo efficacy of ADCs in nude rat xenograft model. (a) Tumor growth curves of 1 mg/kg ADC-treated groups and PBS- or trastuzumab-treated groups. Tumor volume was calculated as 0.5 × (longest axis) × (shortest axis)2. N = 6, and the error bar represents the standard error. (b) Survival plots of 1 mg/kg ADC-treated groups and the control groups. Animals were removed from the study when their tumor size became 200%. (c) Tumor growth curve of 2.5 mg/kg ADC-treated groups. A single dose was intravenously injected on day 0
ADC, antibody–drug conjugate; PBS, phosphate-buffered saline.
T-N-F (DAR 3.1) showed better efficacy than T-C-F (DAR 3.7) at 1 mpk. The T-N-F-treated group showed 92% of tumor growth inhibition from day 4 to day 21. The growth of tumor was slower than that of the T-C-F-treated groups for a prolonged time and such inhibitory effect on tumor growth was sustained until day 21, when the rats from the control groups were euthanized. The T-C-F-treated group showed better tumor regression (17% reduction from the baseline) until day 7, but the tumor relapsed and grew after day 7 at a rate faster than that of the T-N-F-treated group. On day 14, the size of the tumors in the T-C-F-treated group were similar to those of the T-N-F-treated group, but increased by 26% on day 21. At the end of the experiment, the T-C-F-treated group showed 40% larger tumor size than that of the T-N-F-treated group. The T-K-F-treated group (DAR 3.9, 1 mpk) showed 80% of tumor growth inhibition on day 21, but the tumor size increased by 398% and the tumor was two times bigger than that of the T-N-F-treated group. The T-N-F-treated groups showed better survival rates than that of the other ADC- or trastuzumab-treated groups (Figure 5b).
At a higher dose (2.5 mpk), T-C-F showed the best efficacy, and the efficacy of T-N-F was comparable to that of T-C-F (Figure 5c). In the T-C-F-treated group, complete remission was achieved in all animals, and tumors did not grow back until the end of the study (Day 38). Moreover, in T-N-F-treated group, 5 of 6 rats showed complete remission. Although T-K-F showed modest efficacy and was able to suppress tumor growth to less than 200% of the baseline until the end of the study, the regression of tumor, however, was not observed. Interestingly, T-N-F with low DAR of 1.6 showed a comparable efficacy with T-K-F, despite the fact that its DAR is less than half of T-K-F (DAR 3.9).
Discussion
In this study, we showed that the NTERM-conjugated ADC was more stable and less toxic than other ADCs. The rat PK study showed that the NTERM-conjugated ADC was more stable than other ADCs, and its improved tolerability was clearly demonstrated in the single-dose toxicity study. Moreover, the potency of the NTERM-conjugated ADC in vivo was not compromised by its reduced toxicity. A lower toxicity could mean a lower potency of the molecule; however, the NTERM-conjugated ADC showed the best efficacy in the rat xenograft model, suggesting that the NTERM conjugation did not weaken the potency of the ADC. Conversely, T-C-F showed the highest potency in vitro and in vivo, but it also showed the highest toxicity in the rat model. A higher potency in vitro could mean higher toxicity in vivo if the potency is not selective enough against the target.
We used the same species for PK, efficacy, and toxicology studies because it allowed us to collect consistent information to understand the association of efficacy with PK properties and toxicities. Additionally, we believed that rats could be a good toxicological model to assess the stability and off-target toxicity of an ADC because rats appeared more sensitive than mice in both toxicity and PKs studies used to assess the chemical properties of ADCs. Although rat is devoid of HER2, which is the target of trastuzumab, and is not suitable for studying on-target toxicity, we believe that rat is a suitable toxicological model for studying ADC-related toxicity because the major toxicities of approved or investigated ADCs are off-target toxicities, including thrombocytopenia, neutropenia, and liver toxicity.
Although the antibody component of ADCs target tumor-specific antigens that are expressed on tumor cells abundantly and/or exclusively, most of the toxicity related to ADCs in clinical use is related to normal tissues. ADCs are intended to have target-specific activity, but their toxicity is nonspecific. Thrombocytopenia, neutropenia, peripheral neuropathy, liver toxicities, and ocular toxicity are more frequently occurring dose-limiting toxicities of approved or clinical stage ADCs, and none of them are related to on-target toxicity. Therefore, comparing off-toxicity of ADCs in rat has some meaning for understanding which property of ADCs are related with the clinical toxicity. Since the rat model is devoid of antigens that can bind to trastuzumab, receptor-mediated activity or specific absorption by trastuzumab was likely not involved in the rat model. Therefore, we can conclude that the toxicity and metabolism of the trastuzumab-based ADCs in rats is dependent only on their properties as a potent cytotoxic therapeutic molecule rather than as a target-specific antibody, and the observation in the rat model can give insights about mechanism of nonspecific toxicity.
The absorption or degradation of an ADC could indicate nonspecific absorption of the drug at off-target sites where the payload can cause toxicity, and the absorption or degradation of ADCs can be described by the CL rates in PK profiles. The absorption of an ADC at off-target sites can be nonspecific uptake, which depends on the ADC’s molecular properties, including hydrophobicity, charge distribution, or altered receptor binding affinity.9,18,19 The degradation of an ADC is related to how the linker-payload was conjugated to the antibody. It is necessary to consider these points when we interpret our toxicity data. Because we used the same antibody, same payload, and similar DAR, the different toxic effect among the ADCs could only be due to the different conjugation methods. Since the CL of an ADC may imply the systemic absorption of the molecule, including toxin, it could be hypothesized that lower in vivo stability (faster clearance or deconjugation) results in higher off-target or systemic toxicity.
The key aim of our NTERM method is to synthesize less toxic ADCs by improving their stability, rather than synthesizing homogenous ADCs. Our results showed that the in vivo stability of an ADC could be improved by changing the conjugation method, and the improved stability could then be linked with the enhancement of toxicity and efficacy. In our data, the MTD of T-N-F would be 200 mpk because there was no death of animals until the 200 mpk dose was reached. The MTD of T-C-F would be 100 mpk because there were five dead animals out of six in the 200 mpk group. The MTD of T-K-F would be 40 mpk because one animal died in the 80 mpk group and the 160 mpk group showed 17% less weight than the PBS group at the end. The MED of T-N-F and T-C-F would be 2.5 mpk, in which the ADCs showed tumor reduction. The MED of T-K-F could not be determined because tumor stasis was not achieved at 2.5 mpk dose. Therefore, the therapeutic window of T-N-F would be 80 and that of T-C-F would be 40. The therapeutic window of T-K-F would be less than 16. Because the actual MTD of T-N-F was not reached unlike T-C-F, the therapeutic window of T-N-F would be higher than 80.
Several studies using N-terminal modification of a protein have been reported. Thompson et al. demonstrated selective labeling of alkoxyamine-derivatized MMAE to a serine residue engineered at the N-terminal of light chain.20 The method oxidizes the serine to an aldehyde group by sodium periodate and the linker-payload conjugated by oxime ligation reaction. They only demonstrated that they could make the ADC, and the ADC was stable in serum and showed in vivo potency. Because the N-terminal aldehyde is the only active site, the resulting ADC is homogeneous. This method seems to be limited by two points: the need for protein engineering and use of toxic p-anisidine. In comparison with the N-terminal modification method, an advantage of our approach is that there is no need for antibody engineering. Addition of serine at the N-terminus of an antibody is quite easy, but sometimes it is not possible. For example, ADCs can be made from an antibody that is already manufactured using an established process and cell bank, but that antibody cannot be further engineered unless the manufacturing process is also changed, which can be costly and time-consuming.
Although we have demonstrated improved tolerability of the NTERM-conjugated ADC, the reason for its reduced toxicity remains to be elucidated. Several studies have been conducted to explain which attribute of the ADCs is responsible for toxicity, and hydrophobicity and Fc-receptor mediated absorption of ADCs have been suggested as a cause of its toxicity.9,19 If we accept this statement as it is, the off-target toxicity becomes an unavoidable effect for any ADCs as long as they are made of the same class of payload and the same antibody regardless of conjugation method. However, the NTERM-conjugated ADC showed less toxicity compared to other ADCs, even though the NTERM-conjugated ADC used the same payload and the same antibody. Therefore, our results showed that the key factor to differentiate the toxicity could vary with different conjugation methods. Our results also imply that the conjugation site, the conjugation method, and the toxicity are all associated with each other. The reasons why different conjugation methods demonstrated different degrees of toxicity needs to be further elucidated. One possible explanation is that hydrophobicity at a molecular level is important. For thiol-conjugate ADCs, payloads are crowded around the hinge area of Fc, while payloads are spatially distributed in NTERM-conjugated ADCs. At a molecular level, their hydrophobic patch distribution is different, and this might change their nonspecific uptake. T-C-F also has a smaller number of inter-chain disulfide bonds so that its heavy and light might separate from each other and lead to degradation. T-K-F has less positively charged residues than others, which might alter its nonspecific uptake. Differences in metabolites could also be a reason. Both T-C-F and T-K-F will release payload as MMAF-conjugated amino acid: cysteine-MMAF or lysine-MMAF. These end-products are zwitterions and have both amino and carboxyl group. The metabolic product of NTERM ADC would be a carboxylic acid because the amino group was converted to amine group during conjugate. The biological activity assessment of these metabolic product may be needed.
Although ADCs have opened a new era of cancer therapy by delivering highly potent chemotherapeutic agents via cancer-specific antibody to kill the target cells more directly and efficiently, their toxicity presents a challenge to clinical success. Many ADCs in clinical trials have proceeded to Phase 2 clinical trials, but few have shown efficacy at this stage.18 The lack of efficacy does not mean that the therapeutics failed just to show proof of concept, but it would mean there was insufficient exposure at the administered Phase 2 dose. Generally, there is a dose–response relationship and higher doses will induce more efficacy. However, a recommended Phase 2 dose of a cytotoxic drug has been determined based on MTD, and so the dose cannot be increased when there is little or no response in the Phase 2 trial. Therefore, reducing toxicity and increasing exposure are important for the success of an ADC. For example, Phase 3 trial results have shown that patients could be classified using T-DM1 trough concentration (Cmin) in Kaplan-Meier survival analysis.21 This suggests that some patients did not get enough clinical benefit from T-DM1 treatment because of the limited exposure. However, the dose could not be increased so as to allow enough exposure because the dose was determined on the basis of MTD and the toxicities were probably determined based on the maximum serum concentration or Cmax.21,22
Recently, trastuzumab deruxtecan (DS-8201a), an ADC consisting of an HER2-targeting antibody (trastuzumab), a cleavable tetrapeptide-based linker, and a potent topoisomerase I inhibitor, which acts as the cytotoxic drug (payload), revealed promising clinical benefits by reducing toxicity and increasing exposure.23 Unlike other anti-HER2 ADCs in clinical trials, trastuzumab deruxtecan has higher MTD, favorable PK and safety profiles, and potent anti-tumor activity owing to the pharmacological property of the payload, which is a derivative of the camptothecin analog exatecan. Its clinical dose (5.4 mpk) is close to the clinical dose of the naked antibody, trastuzumab, which can be given at 6 mpk on a every 3-week schedule. The target HER2/ErbB2 would be saturated under the dose and the clinical benefit would be maximized. However, the clinical dose of T-DM1 is only 3.6 mpk, which is lower than that of trastuzumab. In other words, the dose of T-DM1 cannot be increased until the target can be saturated, while DS-8201a can be increased owing to its tolerability and in vivo stability. The clinical success of DS-8201a showed how the drug has overcome the problem of T-DM1 and why the therapeutic window is important. Our study may provide a hint about how to increase the therapeutic window by means of making stable ADCs that have conjugated payload on the N-termini of an antibody.
Materials and methods
Preparation of ADCs
Antibodies
Trastuzumab (anti-HER2) was expressed in a stable Chinese hamster ovary (CHO)-K1 cell line. Brentuximab (anti-CD30) was expressed in a stable CHO cell line pool. Lorvortuzumab (anti-CD56) was transiently expressed in CHO-S cells by transfecting the cells with protein-encoding plasmid using polyethylenimine. All the antibody sequences were derived from corresponding patent applications and the expressed antibodies were purified from the culture medium using affinity chromatography with MabSelect SuRe™ resin (17543801, Cytiva). Their identities were confirmed using LC/MS and the purities were examined using size-exclusion chromatography with Tosoh TSKgel SuperSW3000 column on the Agilent 1200 series high-performance LC (HPLC) system.
Linker-drug
MMAF with aldehyde group (ald-MMAF, Figure 1b) or maleimide group (mc-MMAF, PubChem CID 56949327) were synthesized by Xcess Bioscience Inc. (IL, USA) MMAF with thiol group (SH-MMAF, Fig S6) was prepared by Concortis, Inc. (CA, USA) Their identities were confirmed by MS and purities were examined using reverse-phase HPLC.
NTERM conjugation
Antibodies were exchanged with the reaction buffer (pH 5.49, 100 mM potassium phosphate, 20 mM NaBH3CN) at the concentration of 5 mg/mL. The conjugation reaction was initiated by adding ald-MMAF (10 mg/mL, 100% acetonitrile) to the antibody solution at approximately 1:9–1:12 molar ratio. The mixture was incubated at 4°C for 15 h. The reaction was terminated by changing the buffer to PBS.
Thiol conjugation
Thiol-conjugated antibodies were prepared as previously described.24 Antibodies were dissolved in a reduction buffer (pH 8.0, 30 mM sodium borate, 100 mM NaCl) at 5 mg/mL. Tris(2-carboxyethyl) phosphine was added in the molar ratio 1:2.75. The mixture was incubated at 37°C for 2 h to reduce disulfide bonds. After the reduction reaction, pH was adjusted by adding an equal volume of the alkylation buffer (pH 6.0, 100 mM sodium phosphate, 100 mM NaCl). The alkylation was initiated by adding mc-MMAF over antibody in 1:10 molar ratio. The tube was incubated on ice for 3 h for the alkylation reaction. The reaction was quenched by adding 50 mM cysteine at the final concentration of 5 mM and incubating on ice for 30 min. To remove small molecules, the reactant was transferred to a HiPrep desalting column (17508701, Cytiva) and the buffer was changed to PBS.
Lysine conjugation
Lysine-conjugated trastuzumab were prepared as previously described.25 Trastuzumab was dissolved in a lysine-conjugation buffer (pH 6.5, 50 mM potassium phosphate, 50 mM NaCl,2 mM ethylene-diamine-tetra-acetic acid). SMCC (22360, Thermo Fisher) was added in 1:7.5 molar ratio and incubated at 25°C for 2 h. To remove small molecules, the reactant was transferred to a HiPrep desalting column (17508701, Cytiva) and the buffer was changed to the lysine-conjugation buffer. The SH-MMAF was added in 1:10 molar ratio and the mixture was incubated at 25°C for about 16.5 h for the conjugation reaction to take place. The buffer was then changed to PBS to stop the reaction.
DAR analysis
DAR was calculated as the weighted-average of each DAR species identified on the MS spectrum. ADC samples were treated with PNGaseF (P0704S, New England Biolab) and incubated at 37°C overnight. The samples (1.5 mL of 1 mg/mL) were injected into Synapt G2-S system (Waters) through an ACQUITY UPLC system (Waters) with BEH 200 SEC column (4.6 × 150 mm, 1.7 mm) (176003906, Waters) at 0.2 mL/min flow rate. The mobile phase consisted of 70% water, 30% acetonitrile, 0.1% formic acid, and 0.05% trifluoroacetic acid (TFA). For the analysis of thiol-conjugated ADC sample, 200 mM ammonium acetate buffer at pH 6.7 was used as mobile phase. DAR of each peak was identified by its mass, and the DAR of the sample was calculated by the sum of (DAR) × (relative population) values.
Identification of conjugated sites
ADC samples (100 µg) were treated with 1% of RapiGest SF (186008090, Waters), followed by dithiothreitol and iodoacetamide treatment. The treated samples were incubated with trypsin (1:20 w/w) (11418475001, Roche) at 37°C for 15 h for digestion. The resulting fragments were separated with ACQUITY UPLC PST (BEH) C18 column (300A, 1.7 µm, 2.1 × 150 mm, P/N 186993687, Waters). Mobile phases used were water or acetonitrile with 0.05% TFA (v/v). The peaks were detected by UV absorbance at 214 nm. Peaks for conjugated fragments were identified by comparing their UV profile with that of the unconjugated trastuzumab. Each fragment was identified using multiple-reaction monitoring MS with Synapt G2-S system (Waters).
Ligand binding assay
ELISA was performed to assess the binding activity of antibodies or to measure their concentration. Corresponding antigens (human ErbB2, CD30, or CD56) were coated on Nunc MaxiSorp™ 96-well immunoplates (Thermo Fisher Scientific) with 100 µL solution in PBS at ~ 1–10 µg/mL concentration for overnight. The plates were blocked with 1% bovine serum albumin (BSA) in PBS. ADCs or unconjugated antibodies were added in a series of concentration from 1000 ng/mL to 5.6 ng/mL. The plate was washed three times with PBS-tween (pH 7.4, PBS with 0.05% polysorbate 20) between each step. To detect the concentration of bound ADC or unconjugated antibodies, goat anti-human kappa light chain-HRP conjugate antibody (A7164, Sigma-Aldrich) was added. Finally, 100 µL of 3,3ʹ,5,5ʹ-tetramethylbenzidine solution (T0440, Sigma-Aldrich) was added to amplify the signal, which was stopped by adding 50 mL of 1 N H2SO4. The optical densities were estimated at 450 nm using SpectraMax 190 microplate reader (Molecular Device), whereas the background signal was eliminated by subtracting the absorbance values at 650 nm. The data were analyzed using SoftMax Pro (version 5.4.1, Molecular Device) to obtain dose–response curves.
SPR spectroscopy
Both the binding affinity and dissociation rate constants of trastuzumab and ADCs were measured using SPR spectroscopy with Biacore™ T200 (Cytiva). ErbB2 (1129-ER, R&D systems) was immobilized on a CM5 sensor chip (BR100530, Cytiva) using amine coupling method. The analyte (trastuzumab or ADCs) was injected with 50, 16.67, 5.56, 1.85, and 0.62 nM concentrations at 30 µL/min flow rate. The running buffer was HBS-EP buffer (BR100669, Cytiva). The injection time was 120 s and the dissociation time was 180 s. The sensorgrams were fitted using a bivalent model to evaluate the kinetic parameters. Trastuzumab and T-N-F (DAR 3.2) were immobilized on a protein A chip (29127556, Cytiva) at capture level of 40 RU. The analyte, monomeric HER2 (10126-ER-050, R&D systems) was injected with 100, 50, 25, 12.5, 6.25 and 0 nM concentrations at 30 µL/min flow rate. The association time and the dissociation time were 180 s and 540 s, respectively. The sensorgrams were fitted using 1:1 binding model.
In vitro cytotoxicity assay
The BT474 (Cat#: HTB-20) and HCC1954 (Cat#: CRL-2338) cell lines were purchased from American Type Culture Collection (USA). The karpas-299 (Cat#: ACC 31) and L540 (Cat#: ACC 72) cell lines were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Germany). The cells were cultured in Dulbecco’s modified Eagle medium with 10% fetal bovine serum (FBS) (BT474), RPMI with 10% FBS (HCC1954), or RPMI with 20% FBS (Karpas-299, L540). For the assay, 2000–10,000 cells/well were cultured in 96-well culture plates with their growth media. ADCs or payloads at various concentrations were prepared in their corresponding culture media and loaded onto the 96-well culture plates. The plates were incubated at 37°C in an atmosphere of 5% CO2. After 96 h, WST-8 (Dojindo) was added to each plate and the plates were incubated for 1–4 h at 37°C to intensify the signal. The absorbance was read at 450 nm using SpectraMax 190 microplate reader and the data was analyzed using SoftMax Pro (version 5.4.1) to obtain IC50 values.
In vitro stability test
Rat serum (Equitech-Bio) or human serum (Sigma-Aldrich) were purchased. ADCs were sterile-filtered and added into the serum at a concentration of 1 mg/mL and each sample was incubated at 37°C for 3 days or 7 days. For day 0 sample, an ADC sample was processed immediately after its addition to the serum. The incubated serum was added to MabSelect SuRe resin column to purify human IgG. The concentration of remaining sample was assessed using the ELISA method as described previously. DAR of ADCs was analyzed using the LC-MS method as described above.
PK study in rats
All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of the Hanwha Chemical Bio R&D Center according to the government guidelines and carried out in the animal facility at the Hanwha Chemical Bio R&D Center.
The test samples (trastuzumab, T-N-F, T-C-F, or T-K-F) were intravenously administered at 2.5 mpk into a female SD rat (6-week-old, Orient Bio). Blood samples were collected at 0.5, 1, 2, 4, 6, 24, 72, 144, 336, 400, and 504 h after the administration. The concentrations of each sample were measured using two methods. Total antibody concentration was measured using an ELISA method that captured the sample with an ErbB2 ectodomain (1129-ER, R&D biosystems) and the captured samples were then detected using a goat anti-human kappa light chain-HRP conjugate antibody. The concentrations of drug-conjugated antibodies were measured using a sandwich ELISA method that captures the sample with a polyclonal rabbit anti-MMAF antibody and the captured samples were then detected using biotinylated ErbB2 (HE2-H8225, ACROBiosystems) and streptavidin-HRP. Both the samples and the standards were diluted with PBS containing 1% BSA and 10% (v/v) rat serum. The other ELISA method is described in the “ligand binding assay” subsection. PK parameters were analyzed using WinNonlin software (ver. 6.1, Certara) and statistical analysis was performed using Minitab software (version 16, Minitab LLC).
Single-dose toxicity study
SD rats (6-week-old, N = 6/group) were used for the toxicity study. Each ADC sample was administered intravenously at 50, 100, or 200 mpk doses, respectively. Trastuzumab (200 mpk) and MMAF (18 mg/m2) were used as the controls, and their doses were equivalent to 200 mpk of ADCs. The weight and activities of the rats were monitored every 3 or 4 days. Blood samples were collected on day 5 and 12. Serum biochemistry was analyzed using AU480 chemistry analyzer (Beckman Coulter) and hematological analysis was performed using Hemavet 950 FS (Drew Scientific). The animals that survived were euthanized on day 12, and liver tissues were preserved for histopathological analysis (Korea Experimental Pathology Inc.).
Comparison of in vivo efficacy using rat xenograft models
HCC1954 cells were trypsinized and washed with PBS. Following this, 5 × 106 cells were mixed with Matrigel® (Corning) and transplanted into 7-week-old Hsd:RH-Foxn1rnu nude rats (N = 6/group, Harlan). The rats were irradiated with 4 Gy of gamma rays 7 days before the transplantation to enhance tumor formation. The test samples were injected intravenously into the rats 7 days after the transplantation when the average tumor size was 300 mm3. Tumor size was defined as V = 0.5 × W × L2, where W is the length of the longest axis and L represents the shortest axis of the tumor. The length was measured using a caliper twice a week. The control group animals (PBS, trastuzumab) were euthanized on 3 weeks after treatment, and ADC-treated animals were monitored for another 3 weeks before being euthanized.
Supplementary Material
Funding Statement
Nothing to declare
List of abbreviations
| ADC | Antibody–drug conjugate |
| ald-MMAF | MMAF with aldehyde group |
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| AUC | area under the curve |
| B-N-F | Brentuximab-MMAF conjugate synthesized via NTERM conjugation |
| BSA | Bovine serum albumin |
| (C) | Conjugated antibody concentration |
| CL | Clearance |
| DAR | Drug-to-antibody ratio |
| DLT | Dose-limiting toxicity |
| ELISA | Enzyme-linked immunosorbent assay |
| FBS | Fetal bovine serum |
| HER2 | Human epidermal growth factor receptor 2 |
| HL | Hodgkin lymphoma |
| HPLC | High-performance liquid chromatography |
| HRP | Horseradish peroxidase |
| LC | Liquid chromatography |
| mc-MMAF | MMAF with maleimide group |
| MED | Minimum efficacious dose |
| MMAF | Monomethyl auristatin F |
| mpk | mg per kg |
| MS | Mass spectroscopy |
| MTD | Maximum tolerable dose |
| NTERM | N-terminal conjugation through amine bond formation by reductive alkylation reaction |
| ORR | Objective response rate |
| PBS | Phosphate-buffered saline |
| PK | Pharmacokinetic |
| PFS | Progression-free survival |
| SD | Sprague Dawley |
| SMCC | Succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate |
| SPR | Surface plasmon resonance |
| SH-MMAF | MMAF with thiol group |
| (T) | Total antibody concentration |
| T-C-F | Trastuzumab-MMAF conjugate synthesized thiol-conjugation |
| TFA | Trifluoroacetic acid |
| T-K-F | Trastuzumab-MMAF conjugate synthesized via lysine conjugation |
| T-N-F | Trastuzumab-MMAF conjugate synthesized via NTERM conjugation |
Disclosure of interest
MJK, DS, JK, JYK, JE, BS, YMK, and JJ are the inventor of a patent (PCT/KR2014/005589) related to this manuscript. ABL Bio Inc. is the assignee of the patent. MJK, DS, JK, JE, BS, YGS, WKY, and JJ are employee and stockholders of ABL Bio Inc. SHL is the founder and the CEO of ABL Bio Inc.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Sievers EL, Senter PD.. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–15. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 2.Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat Rev Clin Oncol. 2021. doi: 10.1038/s41571-021-00470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363:1812–21. doi: 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- 4.Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat Rev Drug Discov. 2017;16:315–37. Available from. doi: 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 5.Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs. 2016;8:659–71. doi: 10.1080/19420862.2016.1156829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–32. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 7.Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blättler WA, Lambert JM, Chari RVJ, Lutz RJ, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68:9280–90. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 8.Hamblett KJ, Senter PD, Chace DF, Sun MMC, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10:7063–70. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 9.Lyon RP, Bovee TD, Doronina SO, Burke PJ, Hunter JH, Neff-laford HD, Jonas M, Anderson ME, Setter JR, Senter PD. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015;33:733–35. doi: 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- 10.Doi T, Shitara K, Naito Y, Shimomura A, Fujiwara Y, Yonemori K, Shimizu C, Shimoi T, Kuboki Y, Matsubara N, et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncol. 2017;18:1512–22. doi: 10.1016/S1470-2045(17)30604-6. [DOI] [PubMed] [Google Scholar]
- 11.Baselga J, Tripathy D, Mendelsohn J, Baughman S, Benz CC, Dantis L, Sklarin NT, Seidman AD, Hudis CA, Moore J, et al. Phase II study of weekly intravenous recombinant humanized anti-p185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J Clin Oncol. 1996;14:737–44. doi: 10.1200/JCO.1996.14.3.737. [DOI] [PubMed] [Google Scholar]
- 12.Esteva FJ, Stebbing J, Wood-Horrall RN, Winkle PJ, Lee SY, Lee SJ. A randomised trial comparing the pharmacokinetics and safety of the biosimilar CT-P6 with reference trastuzumab. Cancer Chemother Pharmacol. 2018;81:505–14. doi: 10.1007/s00280-017-3510-7. [DOI] [PubMed] [Google Scholar]
- 13.Grimsley GR, Scholtz JM, Pace CN. A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci. 2009;18:247–51. doi: 10.1002/pro.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu G, Gao Y, Liu D, Tan X, Hu L, Qiu Z, Liu J, He H, Liu Y. Study on the heterogeneity of T-DM1 and the analysis of the unconjugated linker structure under a stable conjugation process. ACS Omega. 2019;4:8834–45. doi: 10.1021/acsomega.9b00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7:525–37. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 16.Doronina SO, Mendelsohn BA, Bovee TD, Cerveny CG, Alley SC, Meyer DL, Oflazoglu E, Toki BE, Sanderson RJ, Zabinski RF, et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjug Chem. 2006;17:114–24. doi: 10.1021/bc0502917. [DOI] [PubMed] [Google Scholar]
- 17.Strop P, Liu S-H, Dorywalska M, Delaria K, Dushin RG, Tran -T-T, Ho W-H, Farias S, Casas MG, Abdiche Y, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol. 2013;20:161–67. doi: 10.1016/j.chembiol.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Zhao H, Atkinson J, Gulesserian S, Zeng Z, Nater J, Ou J, Yang P, Morrison K, Coleman J, Malik F, et al. Modulation of macropinocytosis-mediated internalization decreases ocular toxicity of antibody-drug conjugates. Cancer Res. 2018;78:2115–26. doi: 10.1158/0008-5472.CAN-17-3202. [DOI] [PubMed] [Google Scholar]
- 19.Uppal H, Doudement E, Mahapatra K, Darbonne WC, Bumbaca D, Shen B-Q, Du X, Saad O, Bowles K, Olsen S, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res. 2015;21:123–33. doi: 10.1158/1078-0432.CCR-14-2093. [DOI] [PubMed] [Google Scholar]
- 20.Thompson P, Bezabeh B, Fleming R, Pruitt M, Mao S, Strout P, Chen C, Cho S, Zhong H, Wu H, et al. Hydrolytically stable site-specific conjugation at the N-terminus of an engineered antibody. Bioconjug Chem. 2015;26:2085–96. doi: 10.1021/acs.bioconjchem.5b00355. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Song P, Schrieber S, Liu Q, Xu Q, Blumenthal G, Amiri Kordestani L, Cortazar P, Ibrahim A, Justice R, et al. Exposure-response relationship of T-DM1: insight into dose optimization for patients with HER2-positive metastatic breast cancer. Clin Pharmacol Ther. 2014;95:558–64. doi: 10.1038/clpt.2014.24. [DOI] [PubMed] [Google Scholar]
- 22.Diéras V, Harbeck N, Budd GT, Greenson JK, Guardino AE, Samant M, Chernyukhin N, Smitt MC, Krop IE. Trastuzumab emtansine in human epidermal growth factor receptor 2-positive metastatic breast cancer: an integrated safety analysis. J Clin Oncol. 2014;32:2750–57. doi: 10.1200/JCO.2013.54.4999. [DOI] [PubMed] [Google Scholar]
- 23.Modi S, Saura C, Yamashita T, Park YH, Kim S-B, Tamura K, Andre F, Iwata H, Ito Y, Tsurutani J, et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N Engl J Med. 2020;382:610–21. doi: 10.1056/NEJMoa1914510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21:778–84. doi: 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- 25.Chari RV, Martell BA, Gross JL, Cook SB, Shah SA, Blättler WA, McKenzie SJ, Goldmacher VS. Immunoconjugates containing novel maytansinoids: promising anticancer drugs. Cancer Res. 1992;52:127–31. Available from: https://www.ncbi.nlm.nih.gov/pubmed/1727373 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.