

Abstract
Advances in genomics are opening new windows into the biology of schizophrenia. Though common variants individually have small effects on disease risk, GWAS provide a powerful opportunity to explore pathways and mechanisms contributing to pathophysiology. Here we highlight an under-appreciated biological theme emerging from GWAS: the role of glycosylation in schizophrenia. The strongest coding variant in schizophrenia GWAS is a missense mutation in the manganese transporter SLC39A8, which is associated with altered glycosylation patterns in humans. Furthermore, variants near several genes encoding glycosylation enzymes are unambiguously associated with schizophrenia: FUT9, MAN2A1, TMTC1, GALNT10, and B3GAT1. Here we summarize the known biological functions, target substrates, and expression patterns of these enzymes as a primer for future studies. We also highlight a subset of schizophrenia-associated proteins critically modified by glycosylation including glutamate receptors, voltage-gated calcium channels, the dopamine D2 receptor, and complement glycoproteins. We hypothesize that common genetic variants alter brain glycosylation and play a fundamental role in the development of schizophrenia. Leveraging these findings will advance our mechanistic understanding of disease and may provide novel avenues for treatment development.
Introduction:
Schizophrenia is a common and debilitating neuropsychiatric condition, with prevalence estimates ranging between 0.4–0.8% and disease onset primarily in adolescence and young adulthood [1]. Clinical presentations are heterogeneous in both severity and symptoms, but most individuals experience significant impairment in functioning due to the lifetime burden of cognitive problems (executive and memory impairment), social withdrawal (avolition, anhedonia), and psychosis (hallucinations and delusions) [2–4]. Aside from agreement on the developmental origins of schizophrenia, advances in our mechanistic understanding of disease and development of novel pharmacologic treatments have been slow over the last half-century [5, 6]. Nearly all of our current treatments are based on decades-old hypotheses and the serendipitous discovery that sedative antihistamines blocking dopamine and serotonin systems provide some symptomatic relief.
Genetic vulnerability has long been recognized as a major factor in the development of schizophrenia, though environmental exposures including maternal infection, perinatal complications and early adversity are also associated with increased risk [7]. Advances in genetics have allowed for analysis of large numbers of patients and increased power to identify genetic variation associated with common polygenic conditions such as schizophrenia. In addition to studies of copy number variants and rare mutations, GWAS have identified a treasure trove of common variants associated with schizophrenia. A landmark 2014 paper by the Psychiatric Genomics Consortium (PGC) identified 108 risk loci [8], and this number has since grown to over 250 (S. Ripke, personal communication). Despite the increasing number of associated loci, translating these findings to mechanistic and therapeutic insights remains a formidable challenge, particularly given that most common variant risk loci exist in the non-coding/regulatory region of the genome.
Nevertheless, GWAS provide novel and unexpected insights into disease pathogenesis. For schizophrenia, the most striking example to date is the discovery that common variants in components of the complement pathway contribute to excessive microglia-mediated synaptic pruning [9]. Here, we discuss a new and promising clue to the biology of schizophrenia emerging from GWAS studies: compelling evidence that glycobiology plays an important role in the etiology of the disorder.
GWAS Implicate Multiple Glycosylation Enzymes in Schizophrenia
Glycosylation is a fundamental biological process implicated in nearly all cellular pathways and most disease states [10, 11]. Glycosylation involves the complex and highly regulated activity of enzymes and chaperones that comprise nearly 2% of all proteins coded in the human genome [12]. These enzymes generate a diverse set of glycans ranging from single monosaccharides to oligosaccharides and polysaccharides covalently attached to proteins and lipids to regulate their structure, function, and localization [11]. Glycosylation-related changes in patients with schizophrenia have been reported in several post-mortem studies (reviewed in linked publication, Williams et al, 2020, submitted for publication). However, it is unclear if these changes are related to genetic vulnerability, environmental factors, or secondary effects of living with a serious mental illness. The recent association of genetic variants in multiple glycosylation enzymes and SLC39A8 support a proximal contribution of this pathway to the development of schizophrenia and necessitates further consideration.
Schizophrenia GWAS have identified genome-wide significant variants within several genes encoding glycosylation enzymes (Table 1): FUT9, MAN2A1, TMTC1, GALNT10, and B3GAT1 [8] (S. Ripke, personal communication). The SNPs with the strongest statistical evidence of association in all of these genes are intronic and do not have a known function. It is possible they are in linkage disequilibrium with true causal SNPs which might be coding variants or regulate gene expression, but future research will be needed to fine-map these loci. In the sections that follow, we describe the basic function, target substrates, and known links to animal phenotypes and human disease of the enzymes encoded by these genes (Figure 1). In addition, we highlight several glycoproteins with compelling evidence including GWAS data supporting their role in schizophrenia, encompassing ionotropic glutamate receptors, voltage-gated calcium channels, the dopamine D2 receptor, and components of the complement system. Understanding how these key glycoproteins are affected by genetic variants in glycosylation enzymes may serve as an approachable next step in connecting the growing list of implicated genes with disease mechanisms and potential therapeutic targets.
Table 1:
Glycosylation enzymes unambiguously associated with schizophrenia through GWAS.
| Gene | Lead SNP | p-value | OR | MAF | Location (chr/bp) |
|---|---|---|---|---|---|
| FUT9 | rs117074560 | 1.66e-08 | 0.86 | 0.05 | Intronic (6:96011775) |
| MAN2A1 | rs4388249 | 1.02e-07 | 1.07 | 0.21 | Intronic (5:109700365) |
| TMTC1 | rs679087 | 7.06e-08 | 0.94 | 0.34 | Intronic (12:29764332) |
| GALNT10 | rs11740474 | 4.46e-09 | 1.05 | 0.35 | Intronic (5:154301187) |
| B3GAT1 | rs892949 | 1.13e-06* | 1.05 | 0.44 | Intronic (11:134426490) |
Abbreviations: SNP, single-nucleotide polymorphisms; OR, odds ratio; MAF, minor allele frequency.
B3GAT1 reaches statistical significance in more recent data freezes with increased sample size (personal communication, S. Ripke).
Figure 1. Glycosylation enzymes unambiguously associated with schizophrenia through GWAS.
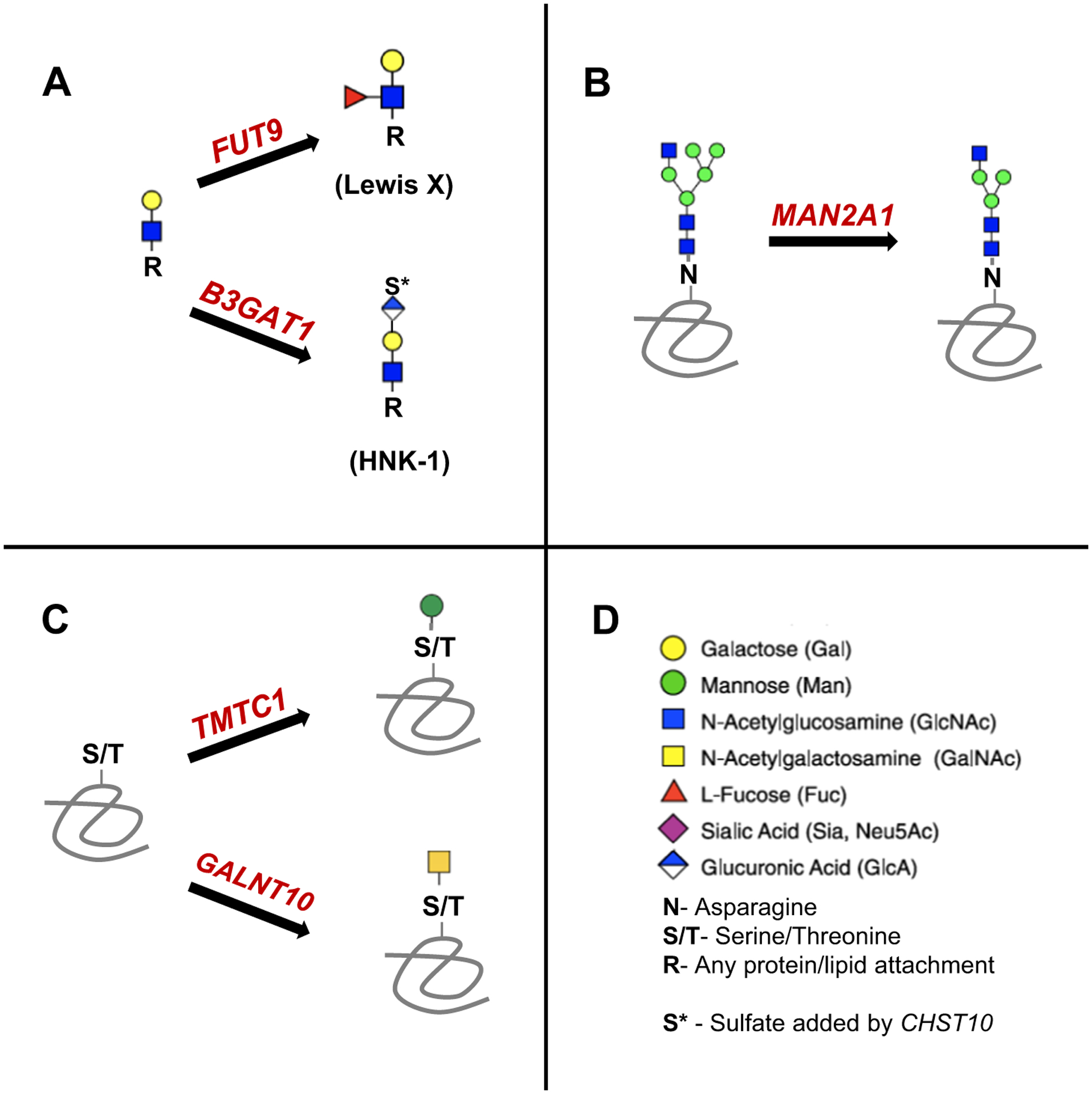
A) FUT9 and B3GAT1*, brain-enriched glycosyltransferases responsible for synthesizing the unique carbohydrate epitopes LewisX and HNK-1, respectively, on N-glycans, O-glycans, and glycolipids. B) MAN2A1, an alpha-mannosidase critical in the formation of complex N-glycans. C) TMTC1 and GALNT10 are involved in the generation of O-glycans. D) Glycan symbols according to Symbol Nomenclature For Glycans (https://www.ncbi.nlm.nih.gov/glycans/snfg.html). *B3GAT1 reaches statistical significance in more recent data with increased sample size (personal communication, S. Ripke).
FUT9
Fucosyltransferase-9 (FUT9) is one of eight α-1,3-fucosyltransferases capable of adding fucose (Fuc) from GDP-Fuc to N-acetylglucosamine (GlcNAc) in an α-1,3-linkage on both glycoproteins and glycolipids [13, 14]. FUT9 is unique among fucosyltransferases as it can only transfer Fuc to GlcNAc in a type-II lactosamine unit (Gal-β1,4-GlcNAc) lacking a terminal sialic acid, generating an important carbohydrate epitope known as the Lewis X (Lex) antigen (also called CD15 and SSEA-1) [15]. Lex is differentially expressed in the developing brain and plays a role in pattern formation in the forebrain [16–18]. Lex is also thought to play a role in neuron-glia interactions and is one of three cell surface markers defining the neural stem cell lineage [19–21]. FUT9 expression is dramatically enriched in the brain [22], where it is the sole fucosyltransferase responsible for synthesizing Lex [15, 23, 24]. In cellular models of neuronal differentiation, knockdown of FUT9 using siRNA led to impaired neurite outgrowth [25]. Fut9 knock-out mice lack Lex expression in the brain and appear grossly normal, but display an increase in anxiety-like behaviors [26].
MAN2A1
Alpha-mannosidase II member 1 (MAN2A1) is one of two α-II mannosidases in the Golgi responsible for the final cleavage of mannose (Man) residues from N-glycan precursors, which is required for the generation of all complex N-glycans [27, 28]. MAN2A1 and the gene encoding its only isoenzyme, MAN2A2, are broadly expressed at relatively high levels [22], likely due to the indispensable requirement of complex N-glycans. Mice lacking Man2a1 show a lack of N-glycans on erythroid cells and anemia, as well as a late onset autoimmune disorder similar to systemic lupus erythematosus, but are otherwise normal [29, 30]. Mice with deletion of Man2a2 display male infertility but are also otherwise normal [31]. While deletion of either Man2a1 or Man2a2 alone results in relatively mild and organ specific phenotypes, deletion of both genes leads to perinatal lethality and a complete lack of complex N-glycans, demonstrating the required but overlapping function of these critical enzymes and their substrates in development [32]. GWAS have linked variants in MAN2A1 to intelligence and general cognitive ability [33, 34], further highlighting the importance of this gene in neurodevelopmental phenotypes. While MAN2A1 is a clear GWAS hit for schizophrenia, MAN2A2 exists in a statistically significant but ambiguous locus containing at least two other genes (FES and FURIN) [8]. A recent study in zebrafish identified MAN2A2 as a good candidate in this multigene locus, as MAN2A2 deletion in zebrafish led to a neurologic phenotype while FURIN and FES deletion did not [35].
TMTC1
TMTC1 is one of four transmembrane and tetratricopeptide repeat domains-containing (TMTC) members originally described as ER proteins involved in calcium homeostasis [36]. Recently, Halim and colleagues identified TMTCs as a novel family of O-mannosyltransferases that utilize dolichol-phospho-mannose as the donor to add α-linked O-mannose to serine and threonine residues in the cadherin superfamily in cell lines [37]. Cadherins and protocadherins represent a substantial portion of the O-mannosylated proteins in the brain [38], and were previously shown to be modified independently from the traditional O-mannosylation enzymes POMT1 and POMT2 [39].
Each TMTC isoenzyme appears to modify different sites on cadherins and protocadherins, suggesting that each likely serves a distinct function [37]. The α-protocadherin gene cluster has been associated with schizophrenia by GWAS [8] and shows dysregulation in an iPSC model of schizophrenia [40]. Given the critical role of cadherins and protocadherins in cell-adhesion, synaptogenesis, and wiring of the developing brain [41–44], understanding the contribution of TMTC1 and its isoenzymes to the cadherin superfamily may provide important insights into the aberrant neural connectivity implicated in schizophrenia.
TMTC1 has diffuse expression but exhibits a two-fold enrichment in the brain [22]. We are unaware of murine knockouts of Tmtc1 or congenital disorders associated with TMTC1 in humans. However, Tmtc3 knockout leads to early neonatal lethality of mice [45], and homozygous mutations of TMTC3 in humans causes a congenital disorder of glycosylation characterized by cobblestone lissencephaly, intellectual disability, and epilepsy, suggesting a critical function of this glycan modification in neurodevelopment [46, 47].
GALNT10
The first step in mucin-type O-glycosylation is mediated by a family of twenty N-acetylgalactosamine (GalNAc) transferases, termed GALNTs, which utilize UDP-GalNAc as a donor. These enzymes catalyze the addition of α-linked GalNAc to serine and threonine residues within proteins, generating the carbohydrate epitope known as Tn-antigen [12]. While first studied in mucins, it is now appreciated that more than 80% of the proteins in the secretory pathway may acquire one or more of such O-GalNAc modifications [48]. Tn-antigen is a precursor and rarely found in normal tissues as it is rapidly modified by subsequent enzymes to generate a diverse array of extended O-glycans (core 1, core 2, etc.), though Tn expression has been associated with tumor cells and Tn syndrome [49, 50].
Glycosyltransferase gene families usually contain one or a few isoenzymes capable of catalyzing the same step of glycan synthesis. The size of the GALNT family is unique among glycosyltransferases and hypothesized to allow for the fine tuning of mucin-type O-glycosylation. GALNT enzymes do not share full redundancy for targets, and the expression profile for each differs widely [51]. GALNT10 is expressed broadly throughout the body including the brain [22]. We are unaware of published results of Galnt10 knockout mice, though the Mouse Genome Informatics database suggests there is no resultant phenotype of gene deletion [52].
B3GAT1
Although B3GAT1 falls short of GWAS significance in the 2014 PGC paper [8], recent data with increased sample size have associated it with schizophrenia (personal communication, S. Ripke), and a 2015 GWAS on schizophrenia in an Ashkenazi Jewish population reported a significant association for B3GAT1 [53]. Beta-1,3-glucuronyltransferase (B3GAT1, or GlcAT-P) is one of two enzymes in the genome capable of transferring glucuronic acid (GlcA) in a β-1,3 linkage to galactose (Gal) on glycoproteins and glycolipids [54]. The GlcA is then modified by a single sulfotransferase CHST10 (also known as HNK1ST) to generate the unique 3-O-sulfated GlcA glycan epitope HNK-1, originally termed CD57 [55–57]. Numerous key neuronal proteins carry the HNK-1 epitope including NCAM and the AMPA receptor GluA2 (GRIA2) [54]. B3GAT1 expression is enriched one hundred-fold in the brain [22]. The importance of this enzyme is illustrated by B3gat1 knockout mice, which lack HNK-1 in the brain and show abnormal hippocampal synaptic plasticity, spatial memory problems, and abnormal spine morphology [58, 59]. In humans, autoantibodies to HNK-1 are found in some forms of neuropathy [60], and HNK-1 levels are reduced in brains of Alzheimer’s patients [61]. B3GAT1 was implicated as a risk factor for schizophrenia-like psychosis through a balanced translocation in one family [62]. The gene encoding its only isoenzyme, B3GAT2, was linked to schizophrenia risk and cortical surface area in a candidate gene analysis [63], but this association has not replicated in more recent and larger samples. Finally, a genetic variant in CHST10 has been associated with intelligence through GWAS [33], further supporting a role for HNK-1 synthesis in neurodevelopment.
SLC39A8
To date, the most significantly associated coding variant in schizophrenia GWAS is in the manganese transporter SLC39A8 [8]. Although SLC39A8 itself is not a glycosylation enzyme, it plays a critical role in the pathway by regulating concentrations of manganese, an irreplaceable co-factor for many glycosylation enzymes [64–66]. Severe homozygous loss of function mutations in SLC39A8 lead to near complete absence of circulating manganese and a severe type-II congenital disorder of glycosylation [67, 68].
Unlike the genetic associations described above, the schizophrenia associated variant in SLC39A8 results in a missense mutation in the coding region of the protein (rs13107325 (C->T), A391T). We recently confirmed that human carriers of the schizophrenia risk (T) allele have decreased serum Mn and reduced complexity of plasma protein N-glycans [69], consistent with prior reports of reduced activity of Mn-dependent glycosylation enzymes in SLC39A8 mutation carriers [67, 70]. The mechanistic link connecting schizophrenia and SLC39A8 variants remains an active area of study and is the topic of several recent reviews [71–75], though the evidence suggesting dysregulated glycosylation is compelling.
Glycosylated Proteins Associated with Schizophrenia through GWAS
The expanding list of loci associated with schizophrenia includes numerous genes encoding proteins that are critically regulated by glycosylation (Figure 2). Notable examples include glutamate receptors, voltage gated calcium channels, the dopamine D2 receptor, and complement-associated proteins, all of which have enriched expression in the brain (Table 2). Glycosylation of these proteins has been shown to impact their function and cellular localization, illustrating the importance of this post-translational modification in several facets of schizophrenia biology. Schizophrenia-associated variants may impact the glycosylation of these key proteins or represent a point of convergence in the pathophysiology of disease.
Figure 2. Select neuronal proteins critically modified through glycosylation and associated with schizophrenia through GWAS.
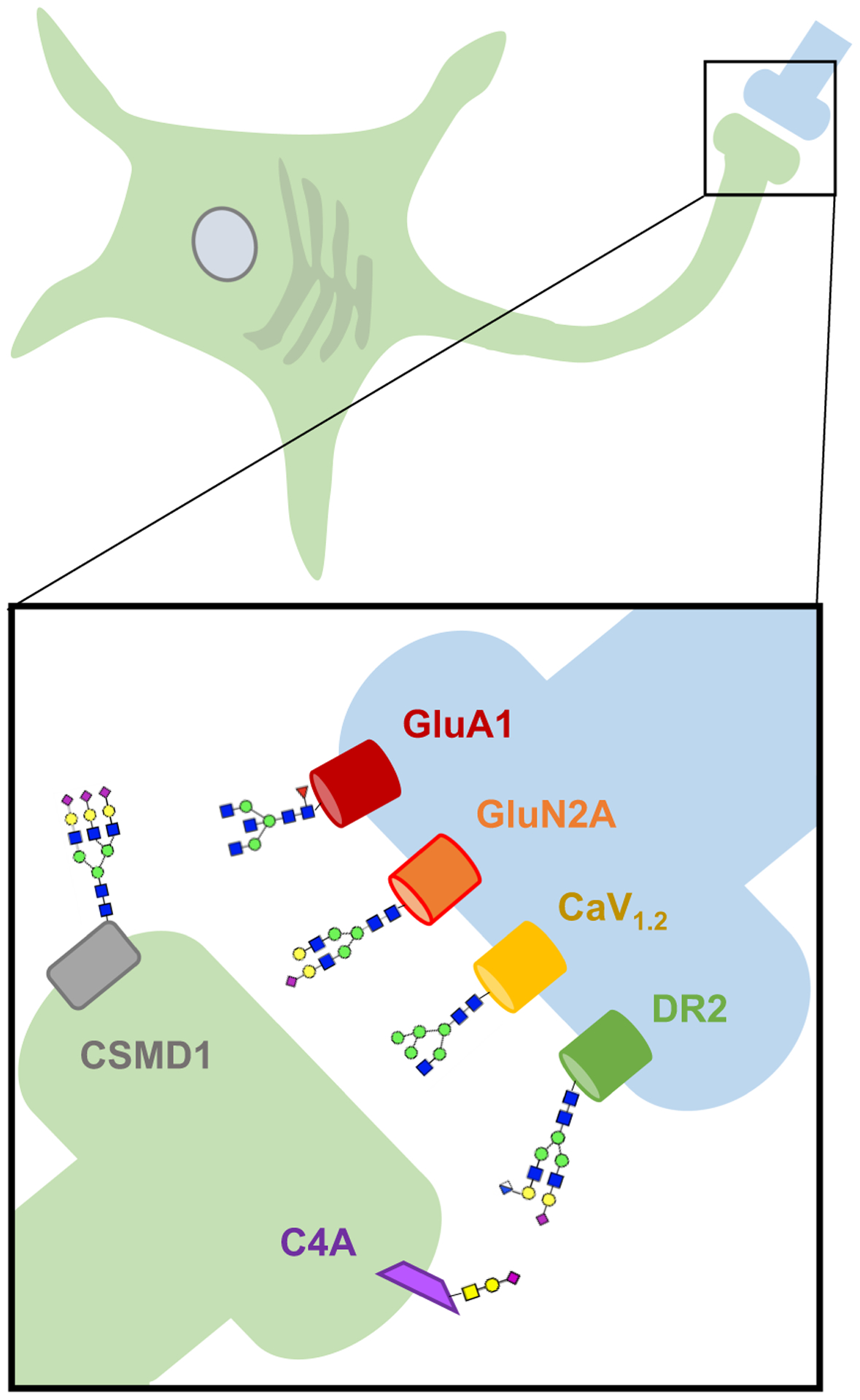
GluA1 (GRIA1), GluN2a (GRIN2A), CaV1.2 (CACNA1C), D2R (DRD2) Complement C4 (C4A), and CSMD1 (CSMD1) are known to be modified by glycosylation and thought to play key roles in the pathogenesis of schizophrenia. Protein names shown and corresponding gene symbols in parenthesis. Presynaptic neuron is shown in green, postsynaptic neuron in blue. Selected glycan structures are shown for illustrative purposes.
Table 2:
Select neuronal proteins critically modified through glycosylation and associated with schizophrenia through GWAS.
| Pathway | Gene | Protein | Putative Glycosylation Sites | Function of Glycans |
|---|---|---|---|---|
| Glutamate Signaling | GRIA1 | GluA1 | 6 (N) | Cell surface expression Tetramer formation Protein trafficking |
| GRIN2A | GluN2A | 6 (N) | Receptor Localization | |
| Dopamine Signaling | DRD2 | D2R | 3 (N) | Cell surface expression Receptor internalization |
| Calcium Signaling | CACNA1C | CaV1.2 | 4 (N) | Cell surface expression Current density |
| CACNA1D | CaV1.3 | 3 (N) | Unknown | |
| CACNA1I | CaV3.3 | 5 (N) | Cell surface expression Current density Rebound bursting | |
| Complement Cascade | C4A | C4A | 4 (N), 1 (O) | Unknown |
| C4B | C4B | 4 (N) | Unknown | |
| CSMD1 | CSMD1 | 40 (N) | Unknown |
Glutamate Receptors
Glutamate signaling has been associated with schizophrenia through a variety of studies including post-mortem protein analysis, pharmacological observations, in vivo imaging, and clinical trials of glutamate receptor modulators [76]. Two ionotropic glutamate receptors, encoded by GRIA1 and GRIN2A, are associated with schizophrenia by GWAS [8]. GRIA1 and GRIN2A each possesses six putative N-glycosylation sites based on sequence analysis [77]. The AMPA receptor subunit GluA1, encoded by GRIA1, relies on glycosylation at N63 and N363 for proper tetramer formation, intracellular trafficking and cell surface expression [78]. Glycosylation of the NMDA receptor 2A (GluN2A, encoded by GRIN2A) has not been studied extensively, but insights from related NMDAR subunits highlight the functional importance of N-glycans to this receptor complex. An N-glycosylation site present in GluN2B was demonstrated to be necessary and sufficient for the localization of receptors into synapses, although GluN2A lacks this glycosylation site [79]. Computational analysis of GluN1 and GluN2B predict that the N-glycan stabilizes the closed conformation of the ligand binding domain, and the same glycan (N440) enhanced glycine co-agonist binding on GluN1 [80]. Glycosylation of two other asparagine residues on GluN1 (N-203 and N-368) are required for the glycoprotein to exit the ER [81], and are necessary along with three residues in the GluN3A subunit for cell-surface expression of GluN3A-containing NMDA receptors [82].
Dopamine Receptors
The dopamine system has long been implicated in the pathophysiology of schizophrenia, and half a century of studies focused on this pathway were validated by the PGC GWAS in 2014 which presented convincing association of the D2 dopamine receptor (DRD2) with schizophrenia [8]. N-glycosylation of DRD2 was predicted by sequence analysis and supported by de-glycosylation assays [83]. Further analysis of the protein revealed that the N-glycans on the N-terminus repress internalization of the receptor into the cytosol, as they are required for interactions with caveolin-1, a negative regulator of endocytosis. The same study demonstrated that N-glycans are involved in the desensitization and cell surface expression of the dopamine D3 receptor, as well as its clathrin-dependent internalization from the plasma membrane.
Voltage-Gated Calcium Channels
N-glycosylation is critical to the function and localization of voltage-gated calcium channels, which have been implicated in a wide-range of neuropsychiatric phenotypes including schizophrenia [84]. Genes encoding three distinct voltage-gated calcium channels have emerged as GWAS hits for schizophrenia: CACNA1C, CACNA1D, and CACNA1I. The pore-forming subunit of the L-type calcium channel CaV1.2 is encoded by CACNA1C and has four putative N-glycosylation sites. Mutating one individual site does not change the biophysical properties of the channel, but a quadruple mutant shows reduced surface expression as well as current density [85]. A different subunit of the same channel, CaVα2δ1, has 16 putative glycosylation sites, and combined mutation of six of these decreased the protein stability, cell surface localization, and functionality of the subunit [86]. CACNA1D encodes the pore-forming subunit of L-type channel CaV1.3, which has three putative glycosylation sites. The α2δ1 subunit of this channel interacts with the pore-forming subunit CaV1.3α1 and enhances channel current density, but this enhancement is diminished when N-glycan sites are mutated [87]. CACNA1I encodes the pore-forming subunit of the CaV3.3 T-type voltage-gated calcium channel, which has five putative N-glycosylation sites. A rare missense variant in this gene, R1346H, is one and four amino acids away from two putative N-glycosylation sites on the protein [88]. This mutant subunit has impaired glycosylation and reduced cell surface expression, leading to decreased current density. This functional change is predicted to interfere with rebound bursting and sleep spindle generation in thalamic reticular nucleus neurons, a phenotype previously observed in patients with schizophrenia. N-glycosylation impacts the functionality and localization of an additional T-type calcium channel, CaV3.2, further emphasizing the critical role of N-glycosylation on voltage-gated calcium channels associated with schizophrenia [89].
Complement-Associated Proteins
The strongest association from the landmark 2014 schizophrenia GWAS by the PGC [8] - the major histocompatibility complex (MHC) locus - contains over 200 genes, but genetic variation within the C4 gene has since been identified as a key contributor to schizophrenia risk [9]. Increased levels of complement component 4A (C4A) correlate with increased risk for the disease as well as excess synaptic pruning in mice through the microglia-mediated pathway, providing the first link between an unbiased genetic association and potential disease mechanism. All of the five putative N-glycosylation sites on C4A have been confirmed by mass spectrometry, as have three out of four putative sites on the alternative isotype complement component 4B (C4B) [90–95]. The complement cascade has been further implicated in schizophrenia through the genetic association of CUB and sushi domain-containing protein-1 (CSMD1) [8]. CSMD1 encodes a complement inhibitor that acts on C4 and is predicted to have 40 putative N-glycosylation sites [96]. The functional implications of N-glycan attachments on C4A, C4B, and CSMD1 in the brain are not fully characterized, but N-glycan attachments on many other complement-associated proteins are known to regulate their function by mediating folding, trafficking, conformational stability, and protein-protein interactions [97]. Sialic acid, a charged monosaccharide present in both glycoproteins and glycolipids, is a critical nutrient for brain development [98, 99] and may represent an important connection between complement and schizophrenia. Sialic acid regulates complement binding and opsonization in several contexts including in utero [100, 101], and also prevents microglia from binding and removing neurites in culture [102–104].
Discussion
Despite the prevalence and socio-economic burden of schizophrenia, treatment options have changed little over the last half-century. A major obstacle is the complexity and heterogeneity of the disorder, which has prevented the development of reliable biomarkers and therapeutics. The classification of many different disorders into a single diagnostic entity, or the existence of subclasses of schizophrenia with unique endophenotypes, may account for some of the heterogeneity [105, 106]. Similar to other polygenic disorders, thousands of genetic variants in each individual create a unique set of vulnerabilities, and only under certain environmental conditions will this manifest in expression of disease.
GWAS have identified over 250 associated loci to date (S. Ripke, personal communication), providing new opportunities to study what goes awry in neurodevelopment to contribute to risk for schizophrenia. The results highlight an enrichment of genes expressed in the brain, but no single pathway or neurotransmitter system provides a unifying mechanism for disease. Most variants are in non-coding regions and likely contribute to risk of disease through alterations in gene expression levels. Less common are coding variants, with the A391T missense mutation in SLC39A8 representing the most strongly associated to date [8]. We and others have shown that the A391T mutation affects Mn concentration and glycosylation patterns, further suggesting a link between schizophrenia and glycosylation [69, 70, 107].
Because of the stepwise assembly of glycans, even a subtle change in activity at any part of the pathway could affect the final product and functionality. The glycosylation enzymes associated with schizophrenia by GWAS have a wide variety of functions across the glycosylation pathway. FUT9 and B3GAT1 synthesize the unique carbohydrate epitopes LeX and HNK-1, respectively, in the brain, both of which can be found on N-glycans, O-glycans, and glycolipids. MAN2A1 plays a critical role in the generation of all complex N-glycans. TMTC1 is a recently identified O-mannosyltransferase specifically for the cadherin superfamily. GALNT10 is one of twenty N-acetylgalactosamine transferases that catalyze the first step of mucin-type O-glycosylation necessary for the extension of all complex branched O-glycans.
Our observation of at least five genes directly involved in glycan synthesis is based on the original 108 reported loci [8]. As the sample size of GWAS increases, more genes will be identified, and we predict this will include many glycosylation genes whose statistical association fell short of significance in the 2014 study. Furthermore, we are aware of several additional glycogenes that are located in statistically significant but complex regions containing many genes, such as MAN2A2, making it difficult to ascribe the association to a single gene without detailed fine mapping [8]. We are unaware of copy number variants or exome-wide associations in glycosylation enzymes being associated with schizophrenia. Most glycosylation enzymes are relatively small genes with one or a few exons, presumably making them less likely to accrue and tolerate variation found in larger genes. Of note, many examples exist of glycosylation enzymes associated with schizophrenia through candidate gene studies, such as ST8SIA2 [71, 108]. ST8SIA2 is one of two enzymes responsible for generating polysialylated-NCAM, an important cell adhesion molecule studied extensively in brain development and schizophrenia (reviewed in linked publication, Williams et al, 2020, submitted for publication) [109]. As GWAS and exome data sets grow larger, such genes may show an association with schizophrenia, but the current data does not support a link through genetics [8].
With the increasing number of loci associated with schizophrenia, distinct pathways have begun to emerge. However, translating these variants into biological mechanisms remains a complex task, with the first major success showing that schizophrenia-associated genetic variation in C4 haplotypes alter synaptic pruning in a mouse model of neurodevelopment [9]. Here we highlight results from the most recent genomic studies of schizophrenia suggesting that altered glycosylation may play a role in schizophrenia. A “variant to function” approach similar to the study by Sekar and colleagues [9] should be applied to schizophrenia-associated variants including glycogenes. Important considerations include determining which genes are responsible for the change in risk, how such variants affect protein expression or function, and what pathways are dysregulated as a result. Such studies will further our mechanistic understanding of schizophrenia and ideally inform the design of rational therapeutics, as genetically-validated targets are more likely to succeed in therapeutic development [110]. If dysfunctional glycosylation contributes to the development of schizophrenia, small molecule pharmaceuticals or precursors for specific enzymes (Gal, Fuc, GalNAc, manganese, etc.) may lead towards novel treatments in schizophrenia which are already employed successfully in some congenital disorders of glycosylation [111, 112]. We propose that glycobiology is a promising frontier in schizophrenia research with the potential to develop novel and targeted therapeutics, a central goal of precision medicine.
Acknowledgements:
We would like to thank Stephan Ripke of MGH and Broad Institute for allowing inclusion of the current estimate of schizophrenia GWAS associations and B3GAT1 as a personal communication. This work was supported by a foundation grant from the Stanley Center for Psychiatric Research at the Broad Institute of Harvard/MIT (awarded to RGM).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest: The authors have declared that no conflicts of interest exist.
References:
- 1.Moreno-Küstner B, Martín C, Pastor L. Prevalence of psychotic disorders and its association with methodological issues. A systematic review and meta-analyses. PLOS ONE. 2018;13:e0195687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Owen MJ, Sawa A, Mortensen PB. Schizophrenia. Lancet Lond Engl. 2016;388:86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.American Psychiatric Association, American Psychiatric Association, DSM-5 Task Force. Diagnostic and statistical manual of mental disorders: DSM-5 2013.
- 4.Kahn RS, Sommer IE, Murray RM, Meyer-Lindenberg A, Weinberger DR, Cannon TD, et al. Schizophrenia. Nat Rev Dis Primer. 2015;1:15067. [DOI] [PubMed] [Google Scholar]
- 5.Birnbaum R, Weinberger DR. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat Rev Neurosci. 2017;18:727–740. [DOI] [PubMed] [Google Scholar]
- 6.Millan MJ, Andrieux A, Bartzokis G, Cadenhead K, Dazzan P, Fusar-Poli P, et al. Altering the course of schizophrenia: progress and perspectives. Nat Rev Drug Discov. 2016;15:485–515. [DOI] [PubMed] [Google Scholar]
- 7.Avramopoulos D Recent Advances in the Genetics of Schizophrenia. Mol Neuropsychiatry. 2018;4:35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lauc G, Pezer M, Rudan I, Campbell H. Mechanisms of disease: The human N-glycome. Biochim Biophys Acta. 2016;1860:1574–1582. [DOI] [PubMed] [Google Scholar]
- 11.Varki A Biological roles of glycans. Glycobiology. 2017;27:3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al. , editors. Essentials of Glycobiology. 3rd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015. [PubMed] [Google Scholar]
- 13.de Vries T, Knegtel RM, Holmes EH, Macher BA. Fucosyltransferases: structure/function studies. Glycobiology. 2001;11:119R–128R. [DOI] [PubMed] [Google Scholar]
- 14.Schneider M, Al-Shareffi E, Haltiwanger RS. Biological functions of fucose in mammals. Glycobiology. 2017;27:601–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kudo T, Ikehara Y, Togayachi A, Kaneko M, Hiraga T, Sasaki K, et al. Expression cloning and characterization of a novel murine alpha1, 3-fucosyltransferase, mFuc-TIX, that synthesizes the Lewis x (CD15) epitope in brain and kidney. J Biol Chem. 1998;273:26729–26738. [DOI] [PubMed] [Google Scholar]
- 16.Cailleau-Thomas A, Coullin P, Candelier JJ, Balanzino L, Mennesson B, Oriol R, et al. FUT4 and FUT9 genes are expressed early in human embryogenesis. Glycobiology. 2000;10:789–802. [DOI] [PubMed] [Google Scholar]
- 17.Mai JK, Andressen C, Ashwell KW. Demarcation of prosencephalic regions by CD15-positive radial glia. Eur J Neurosci. 1998;10:746–751. [DOI] [PubMed] [Google Scholar]
- 18.Mai JK, Krajewski S, Reifenberger G, Genderski B, Lensing-Höhn S, Ashwell KW. Spatiotemporal expression gradients of the carbohydrate antigen (CD15) (Lewis X) during development of the human basal ganglia. Neuroscience. 1999;88:847–858. [DOI] [PubMed] [Google Scholar]
- 19.Gotz M, Wizenmann A, Reinhardt S, Lumsden A, Price J. Selective adhesion of cells from different telencephalic regions. Neuron. 1996;16:551–564. [DOI] [PubMed] [Google Scholar]
- 20.Sajdel-Sulkowska EM. Immunofluorescent detection of CD15-fucosylated glycoconjugates in primary cerebellar cultures and their function in glial-neuronal adhesion in the central nervous system. Acta Biochim Pol. 1998;45:781–790. [PubMed] [Google Scholar]
- 21.Pruszak J, Ludwig W, Blak A, Alavian K, Isacson O. CD15, CD24, and CD29 define a surface biomarker code for neural lineage differentiation of stem cells. Stem Cells Dayt Ohio. 2009;27:2928–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics MCP. 2014;13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaneko M, Kudo T, Iwasaki H, Ikehara Y, Nishihara S, Nakagawa S, et al. Alpha1,3-fucosyltransferase IX (Fuc-TIX) is very highly conserved between human and mouse; molecular cloning, characterization and tissue distribution of human Fuc-TIX. FEBS Lett. 1999;452:237–242. [DOI] [PubMed] [Google Scholar]
- 24.Nishihara S, Iwasaki H, Nakajima K, Togayachi A, Ikehara Y, Kudo T, et al. Alpha1,3-fucosyltransferase IX (Fut9) determines Lewis X expression in brain. Glycobiology. 2003;13:445–455. [DOI] [PubMed] [Google Scholar]
- 25.Gouveia R, Schaffer L, Papp S, Grammel N, Kandzia S, Head SR, et al. Expression of glycogenes in differentiating human NT2N neurons. Downregulation of fucosyltransferase 9 leads to decreased Lewis(x) levels and impaired neurite outgrowth. Biochim Biophys Acta. 2012;1820:2007–2019. [DOI] [PubMed] [Google Scholar]
- 26.Kudo T, Fujii T, Ikegami S, Inokuchi K, Takayama Y, Ikehara Y, et al. Mice lacking alpha1,3-fucosyltransferase IX demonstrate disappearance of Lewis x structure in brain and increased anxiety-like behaviors. Glycobiology. 2007;17:1–9. [DOI] [PubMed] [Google Scholar]
- 27.Misago M, Liao YF, Kudo S, Eto S, Mattei MG, Moremen KW, et al. Molecular cloning and expression of cDNAs encoding human alpha-mannosidase II and a previously unrecognized alpha-mannosidase IIx isozyme. Proc Natl Acad Sci U S A. 1995;92:11766–11770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moremen KW, Robbins PW. Isolation, characterization, and expression of cDNAs encoding murine alpha-mannosidase II, a Golgi enzyme that controls conversion of high mannose to complex N-glycans. J Cell Biol. 1991;115:1521–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chui D, Oh-Eda M, Liao YF, Panneerselvam K, Lal A, Marek KW, et al. Alpha-mannosidase-II deficiency results in dyserythropoiesis and unveils an alternate pathway in oligosaccharide biosynthesis. Cell. 1997;90:157–167. [DOI] [PubMed] [Google Scholar]
- 30.Chui D, Sellakumar G, Green R, Sutton-Smith M, McQuistan T, Marek K, et al. Genetic remodeling of protein glycosylation in vivo induces autoimmune disease. Proc Natl Acad Sci U S A. 2001;98:1142–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akama TO, Nakagawa H, Sugihara K, Narisawa S, Ohyama C, Nishimura S-I, et al. Germ cell survival through carbohydrate-mediated interaction with Sertoli cells. Science. 2002;295:124–127. [DOI] [PubMed] [Google Scholar]
- 32.Akama TO, Nakagawa H, Wong NK, Sutton-Smith M, Dell A, Morris HR, et al. Essential and mutually compensatory roles of {alpha}-mannosidase II and {alpha}-mannosidase IIx in N-glycan processing in vivo in mice. Proc Natl Acad Sci U S A. 2006;103:8983–8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill WD, Marioni RE, Maghzian O, Ritchie SJ, Hagenaars SP, McIntosh AM, et al. A combined analysis of genetically correlated traits identifies 187 loci and a role for neurogenesis and myelination in intelligence. Mol Psychiatry. 2018. 11 January 2018. 10.1038/s41380-017-0001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Savage JE, Jansen PR, Stringer S, Watanabe K, Bryois J, de Leeuw CA, et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet. 2018;50:912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thyme SB, Pieper LM, Li EH, Pandey S, Wang Y, Morris NS, et al. Phenotypic Landscape of Schizophrenia-Associated Genes Defines Candidates and Their Shared Functions. Cell. 2019. 26 March 2019. 10.1016/j.cell.2019.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sunryd JC, Cheon B, Graham JB, Giorda KM, Fissore RA, Hebert DN. TMTC1 and TMTC2 are novel endoplasmic reticulum tetratricopeptide repeat-containing adapter proteins involved in calcium homeostasis. J Biol Chem. 2014;289:16085–16099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larsen ISB, Narimatsu Y, Joshi HJ, Siukstaite L, Harrison OJ, Brasch J, et al. Discovery of an O-mannosylation pathway selectively serving cadherins and protocadherins. Proc Natl Acad Sci U S A. 2017;114:11163–11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vester-Christensen MB, Halim A, Joshi HJ, Steentoft C, Bennett EP, Levery SB, et al. Mining the O-mannose glycoproteome reveals cadherins as major O-mannosylated glycoproteins. Proc Natl Acad Sci U S A. 2013;110:21018–21023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larsen ISB, Narimatsu Y, Joshi HJ, Yang Z, Harrison OJ, Brasch J, et al. Mammalian O-mannosylation of cadherins and plexins is independent of protein O-mannosyltransferases 1 and 2. J Biol Chem. 2017;292:11586–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shao Z, Noh H, Bin Kim W, Ni P, Nguyen C, Cote SE, et al. Dysregulated protocadherin-pathway activity as an intrinsic defect in induced pluripotent stem cell-derived cortical interneurons from subjects with schizophrenia. Nat Neurosci. 2019;22:229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Wit J, Ghosh A. Specification of synaptic connectivity by cell surface interactions. Nat Rev Neurosci. 2016;17:22–35. [DOI] [PubMed] [Google Scholar]
- 42.Basu R, Duan X, Taylor MR, Martin EA, Muralidhar S, Wang Y, et al. Heterophilic Type II Cadherins Are Required for High-Magnitude Synaptic Potentiation in the Hippocampus. Neuron. 2017;96:160–176.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen WV, Nwakeze CL, Denny CA, O’Keeffe S, Rieger MA, Mountoufaris G, et al. Pcdhαc2 is required for axonal tiling and assembly of serotonergic circuitries in mice. Science. 2017;356:406–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mountoufaris G, Chen WV, Hirabayashi Y, O’Keeffe S, Chevee M, Nwakeze CL, et al. Multicluster Pcdh diversity is required for mouse olfactory neural circuit assembly. Science. 2017;356:411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yun EJ, Vu TH. mSmile is necessary for bronchial smooth muscle and alveolar myofibroblast development. Anat Rec Hoboken NJ 2007. 2012;295:167–176. [DOI] [PubMed] [Google Scholar]
- 46.Jerber J, Zaki MS, Al-Aama JY, Rosti RO, Ben-Omran T, Dikoglu E, et al. Biallelic Mutations in TMTC3, Encoding a Transmembrane and TPR-Containing Protein, Lead to Cobblestone Lissencephaly. Am J Hum Genet. 2016;99:1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farhan SMK, Nixon KCJ, Everest M, Edwards TN, Long S, Segal D, et al. Identification of a novel synaptic protein, TMTC3, involved in periventricular nodular heterotopia with intellectual disability and epilepsy. Hum Mol Genet. 2017;26:4278–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KT-BG, et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013;32:1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ju T, Cummings RD. Protein glycosylation: chaperone mutation in Tn syndrome. Nature. 2005;437:1252. [DOI] [PubMed] [Google Scholar]
- 50.Ju T, Otto VI, Cummings RD. The Tn antigen-structural simplicity and biological complexity. Angew Chem Int Ed Engl. 2011;50:1770–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, Tabak LA. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology. 2012;22:736–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bult CJ, Blake JA, Smith CL, Kadin JA, Richardson JE, the Mouse Genome Database Group, et al. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019;47:D801–D806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goes FS, McGrath J, Avramopoulos D, Wolyniec P, Pirooznia M, Ruczinski I, et al. Genome-wide association study of schizophrenia in Ashkenazi Jews. Am J Med Genet Part B Neuropsychiatr Genet Off Publ Int Soc Psychiatr Genet. 2015;168:649–659. [DOI] [PubMed] [Google Scholar]
- 54.Morise J, Takematsu H, Oka S. The role of human natural killer-1 (HNK-1) carbohydrate in neuronal plasticity and disease. Biochim Biophys Acta Gen Subj. 2017;1861:2455–2461. [DOI] [PubMed] [Google Scholar]
- 55.Bakker H, Friedmann I, Oka S, Kawasaki T, Nifant’ev N, Schachner M, et al. Expression cloning of a cDNA encoding a sulfotransferase involved in the biosynthesis of the HNK-1 carbohydrate epitope. J Biol Chem. 1997;272:29942–29946. [DOI] [PubMed] [Google Scholar]
- 56.Abo T, Balch CM. A differentiation antigen of human NK and K cells identified by a monoclonal antibody (HNK-1). J Immunol Baltim Md 1950. 1981;127:1024–1029. [PubMed] [Google Scholar]
- 57.Chou DK, Ilyas AA, Evans JE, Costello C, Quarles RH, Jungalwala FB. Structure of sulfated glucuronyl glycolipids in the nervous system reacting with HNK-1 antibody and some IgM paraproteins in neuropathy. J Biol Chem. 1986;261:11717–11725. [PubMed] [Google Scholar]
- 58.Yamamoto S, Oka S, Inoue M, Shimuta M, Manabe T, Takahashi H, et al. Mice deficient in nervous system-specific carbohydrate epitope HNK-1 exhibit impaired synaptic plasticity and spatial learning. J Biol Chem. 2002;277:27227–27231. [DOI] [PubMed] [Google Scholar]
- 59.Morita I, Kakuda S, Takeuchi Y, Kawasaki T, Oka S. HNK-1 (human natural killer-1) glycol-epitope is essential for normal spine morphogenesis in developing hippocampal neurons. Neuroscience. 2009;164:1685–1694. [DOI] [PubMed] [Google Scholar]
- 60.Nobile-Orazio E, Manfredini E, Carpo M, Meucci N, Monaco S, Ferrari S, et al. Frequency and clinical correlates of anti-neural IgM antibodies in neuropathy associated with IgM monoclonal gammopathy. Ann Neurol. 1994;36:416–424. [DOI] [PubMed] [Google Scholar]
- 61.García-Ayllón M-S, Botella-López A, Cuchillo-Ibañez I, Rábano A, Andreasen N, Blennow K, et al. HNK-1 Carrier Glycoproteins Are Decreased in the Alzheimer’s Disease Brain. Mol Neurobiol. 2017;54:188–199. [DOI] [PubMed] [Google Scholar]
- 62.Jeffries AR, Mungall AJ, Dawson E, Halls K, Langford CF, Murray RM, et al. beta-1,3-Glucuronyltransferase-1 gene implicated as a candidate for a schizophrenia-like psychosis through molecular analysis of a balanced translocation. Mol Psychiatry. 2003;8:654–663. [DOI] [PubMed] [Google Scholar]
- 63.Kähler AK, Djurovic S, Rimol LM, Brown AA, Athanasiu L, Jönsson EG, et al. Candidate gene analysis of the human natural killer-1 carbohydrate pathway and perineuronal nets in schizophrenia: B3GAT2 is associated with disease risk and cortical surface area. Biol Psychiatry. 2011;69:90–96. [DOI] [PubMed] [Google Scholar]
- 64.Ramakrishnan B, Ramasamy V, Qasba PK. Structural Snapshots of β-1,4-Galactosyltransferase-I Along the Kinetic Pathway. J Mol Biol. 2006;357:1619–1633. [DOI] [PubMed] [Google Scholar]
- 65.Breton C, Šnajdrová L, Jeanneau C, Koča J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology. 2006;16:29R–37R. [DOI] [PubMed] [Google Scholar]
- 66.Chang A, Singh S, Phillips GN, Thorson JS. Glycosyltransferase structural biology and its role in the design of catalysts for glycosylation. Curr Opin Biotechnol. 2011;22:800–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park JH, Hogrebe M, Grüneberg M, DuChesne I, von der Heiden AL, Reunert J, et al. SLC39A8 Deficiency: A Disorder of Manganese Transport and Glycosylation. Am J Hum Genet. 2015;97:894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boycott KM, Beaulieu CL, Kernohan KD, Gebril OH, Mhanni A, Chudley AE, et al. Autosomal-Recessive Intellectual Disability with Cerebellar Atrophy Syndrome Caused by Mutation of the Manganese and Zinc Transporter Gene SLC39A8. Am J Hum Genet. 2015;97:886–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mealer RG, Jenkins BG, Chen C-Y, Daly MJ, Ge T, Lehoux S, et al. A schizophrenia risk locus alters brain metal transport and plasma glycosylation. 2019. [DOI] [PMC free article] [PubMed]
- 70.Lin W, Vann DR, Doulias P-T, Wang T, Landesberg G, Li X, et al. Hepatic metal ion transporter ZIP8 regulates manganese homeostasis and manganese-dependent enzyme activity. J Clin Invest. 2017;127:2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li M, Wu D-D, Yao Y-G, Huo Y-X, Liu J-W, Su B, et al. Recent Positive Selection Drives the Expansion of a Schizophrenia Risk Nonsynonymous Variant at SLC39A8 in Europeans. Schizophr Bull. 2016;42:178–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Costas J The highly pleiotropic gene SLC39A8 as an opportunity to gain insight into the molecular pathogenesis of schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2018;177:274–283. [DOI] [PubMed] [Google Scholar]
- 73.Fujishiro H, Himeno S. New Insights into the Roles of ZIP8, a Cadmium and Manganese Transporter, and Its Relation to Human Diseases. Biol Pharm Bull. 2019;42:1076–1082. [DOI] [PubMed] [Google Scholar]
- 74.Zang Z-S, Xu Y-M, Lau ATY. Molecular and pathophysiological aspects of metal ion uptake by the zinc transporter ZIP8 (SLC39A8). Toxicol Res. 2016;5:987–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nebert DW, Liu Z. SLC39A8 gene encoding a metal ion transporter: discovery and bench to bedside. Hum Genomics. 2019;13:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Howes O, McCutcheon R, Stone J. Glutamate and dopamine in schizophrenia: an update for the 21st century. J Psychopharmacol Oxf Engl. 2015;29:97–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.The UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kandel MB, Yamamoto S, Midorikawa R, Morise J, Wakazono Y, Oka S, et al. N-glycosylation of the AMPA-type glutamate receptor regulates cell surface expression and tetramer formation affecting channel function. J Neurochem. 2018;147:730–747. [DOI] [PubMed] [Google Scholar]
- 79.Storey GP, Opitz-Araya X, Barria A. Molecular determinants controlling NMDA receptor synaptic incorporation. J Neurosci Off J Soc Neurosci. 2011;31:6311–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sinitskiy AV, Stanley NH, Hackos DH, Hanson JE, Sellers BD, Pande VS. Computationally Discovered Potentiating Role of Glycans on NMDA Receptors. Sci Rep. 2017;7:44578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lichnerova K, Kaniakova M, Park SP, Skrenkova K, Wang Y-X, Petralia RS, et al. Two N-glycosylation Sites in the GluN1 Subunit Are Essential for Releasing N-methyl-d-aspartate (NMDA) Receptors from the Endoplasmic Reticulum. J Biol Chem. 2015;290:18379–18390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Skrenkova K, Lee S, Lichnerova K, Kaniakova M, Hansikova H, Zapotocky M, et al. N-Glycosylation Regulates the Trafficking and Surface Mobility of GluN3A-Containing NMDA Receptors. Front Mol Neurosci. 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Min C, Zheng M, Zhang X, Guo S, Kwon K-J, Shin CY, et al. N-linked Glycosylation on the N-terminus of the dopamine D2 and D3 receptors determines receptor association with specific microdomains in the plasma membrane. Biochim Biophys Acta. 2015;1853:41–51. [DOI] [PubMed] [Google Scholar]
- 84.Lazniewska J, Weiss N. Glycosylation of voltage-gated calcium channels in health and disease. Biochim Biophys Acta Biomembr. 2017;1859:662–668. [DOI] [PubMed] [Google Scholar]
- 85.Park H-J, Min S-H, Won Y-J, Lee J-H. Asn-Linked Glycosylation Contributes to Surface Expression and Voltage-Dependent Gating of Cav1.2 Ca2+ Channel. J Microbiol Biotechnol. 2015;25:1371–1379. [DOI] [PubMed] [Google Scholar]
- 86.Tétreault M-P, Bourdin B, Briot J, Segura E, Lesage S, Fiset C, et al. Identification of Glycosylation Sites Essential for Surface Expression of the CaVα2δ1 Subunit and Modulation of the Cardiac CaV1.2 Channel Activity. J Biol Chem. 2016;291:4826–4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Andrade A, Sandoval A, González-Ramírez R, Lipscombe D, Campbell KP, Felix R. The alpha(2)delta subunit augments functional expression and modifies the pharmacology of Ca(V)1.3 L-type channels. Cell Calcium. 2009;46:282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Andrade A, Hope J, Allen A, Yorgan V, Lipscombe D, Pan JQ. A rare schizophrenia risk variant of CACNA1I disrupts CaV3.3 channel activity. Sci Rep. 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weiss N, Black SAG, Bladen C, Chen L, Zamponi GW. Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch. 2013;465:1159–1170. [DOI] [PubMed] [Google Scholar]
- 90.Bunkenborg J, Pilch BJ, Podtelejnikov AV, Wiśniewski JR. Screening for N-glycosylated proteins by liquid chromatography mass spectrometry. Proteomics. 2004;4:454–465. [DOI] [PubMed] [Google Scholar]
- 91.Ramachandran P, Boontheung P, Xie Y, Sondej M, Wong DT, Loo JA. Identification of N-linked glycoproteins in human saliva by glycoprotein capture and mass spectrometry. J Proteome Res. 2006;5:1493–1503. [DOI] [PubMed] [Google Scholar]
- 92.Zhang H, Li X-J, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol. 2003;21:660–666. [DOI] [PubMed] [Google Scholar]
- 93.Liu T, Qian W-J, Gritsenko MA, Camp DG, Monroe ME, Moore RJ, et al. Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J Proteome Res. 2005;4:2070–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen R, Jiang X, Sun D, Han G, Wang F, Ye M, et al. Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J Proteome Res. 2009;8:651–661. [DOI] [PubMed] [Google Scholar]
- 95.Halim A, Rüetschi U, Larson G, Nilsson J. LC-MS/MS characterization of O-glycosylation sites and glycan structures of human cerebrospinal fluid glycoproteins. J Proteome Res. 2013;12:573–584. [DOI] [PubMed] [Google Scholar]
- 96.Escudero-Esparza A, Kalchishkova N, Kurbasic E, Jiang WG, Blom AM. The novel complement inhibitor human CUB and Sushi multiple domains 1 (CSMD1) protein promotes factor I-mediated degradation of C4b and C3b and inhibits the membrane attack complex assembly. FASEB J Off Publ Fed Am Soc Exp Biol. 2013;27:5083–5093. [DOI] [PubMed] [Google Scholar]
- 97.Ritchie GE, Moffatt BE, Sim RB, Morgan BP, Dwek RA, Rudd PM. Glycosylation and the complement system. Chem Rev. 2002;102:305–320–19. [DOI] [PubMed] [Google Scholar]
- 98.Wang B Sialic acid is an essential nutrient for brain development and cognition. Annu Rev Nutr. 2009;29:177–222. [DOI] [PubMed] [Google Scholar]
- 99.Schnaar RL, Gerardy-Schahn R, Hildebrandt H. Sialic acids in the brain: gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol Rev. 2014;94:461–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Abeln M, Albers I, Peters-Bernard U, Flächsig-Schulz K, Kats E, Kispert A, et al. Sialic acid is a critical fetal defense against maternal complement attack. J Clin Invest. 2019;129:422–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ferreira VP, Pangburn MK, Cortés C. Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol. 2010;47:2187–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nomura K, Vilalta A, Allendorf DH, Hornik TC, Brown GC. Activated Microglia Desialylate and Phagocytose Cells via Neuraminidase, Galectin-3, and Mer Tyrosine Kinase. J Immunol Baltim Md 1950. 2017;198:4792–4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Linnartz B, Kopatz J, Tenner AJ, Neumann H. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J Neurosci Off J Soc Neurosci. 2012;32:946–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Linnartz-Gerlach B, Schuy C, Shahraz A, Tenner AJ, Neumann H. Sialylation of neurites inhibits complement-mediated macrophage removal in a human macrophage-neuron Co-Culture System. Glia. 2016;64:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lawrie SM, O’Donovan MC, Saks E, Burns T, Lieberman JA. Improving classification of psychoses. Lancet Psychiatry. 2016;3:367–374. [DOI] [PubMed] [Google Scholar]
- 106.Lawrie SM, O’Donovan MC, Saks E, Burns T, Lieberman JA. Towards diagnostic markers for the psychoses. Lancet Psychiatry. 2016;3:375–385. [DOI] [PubMed] [Google Scholar]
- 107.Ng E, Lind PM, Lindgren C, Ingelsson E, Mahajan A, Morris A, et al. Genome-wide association study of toxic metals and trace elements reveals novel associations. Hum Mol Genet. 2015;24:4739–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Arai M, Yamada K, Toyota T, Obata N, Haga S, Yoshida Y, et al. Association between polymorphisms in the promoter region of the sialyltransferase 8B (SIAT8B) gene and schizophrenia. Biol Psychiatry. 2006;59:652–659. [DOI] [PubMed] [Google Scholar]
- 109.Sato C, Hane M. Mental disorders and an acidic glycan-from the perspective of polysialic acid (PSA/polySia) and the synthesizing enzyme, ST8SIA2. Glycoconj J. 2018;35:353–373. [DOI] [PubMed] [Google Scholar]
- 110.Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47:856–860. [DOI] [PubMed] [Google Scholar]
- 111.Ng BG, Freeze HH. Perspectives on Glycosylation and Its Congenital Disorders. Trends Genet TIG. 2018;34:466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Verheijen J, Tahata S, Kozicz T, Witters P, Morava E. Therapeutic approaches in Congenital Disorders of Glycosylation (CDG) involving N-linked glycosylation: an update. Genet Med Off J Am Coll Med Genet. 2019. 19 September 2019. 10.1038/s41436-019-0647-2. [DOI] [PMC free article] [PubMed] [Google Scholar]