

Abstract
Radical S-adenosyl-l-methionine (SAM) enzymes initiate biological radical reactions with the 5’-deoxyadenosyl radical (5’-dAdo•). A [4Fe-4S]+ cluster reductively cleaves SAM to form the Ω organometallic intermediate in which the 5’-deoxyadenosyl moiety is directly bound to the unique iron of the [4Fe-4S] cluster, with subsequent liberation of 5’-dAdo•. Here we present synthesis of the SAM analog S-adenosyl-l-ethionine (SAE) and show SAE is a mechanistically-equivalent SAM-alternative for HydG, both supporting enzymatic turnover of substrate tyrosine and forming the organometallic intermediate Ω. Photolysis of SAE bound to HydG forms an ethyl radical trapped in the active site. The ethyl radical withstands prolonged storage at 77 K and its EPR signal is only partially lost upon annealing at 100 K, making it significantly less reactive than the methyl radical formed by SAM photolysis. Upon annealing above 77K, the ethyl radical adds to the [4Fe-4S]2+ cluster, generating an ethyl-[4Fe-4S]3+ organometallic species termed ΩE.
Keywords: S-adenosylethionine, ethyl radical, radical SAM, EPR, organometallic
Entry for the Table of Contents
The radical SAM enzyme HydG can use S-adenosyl-l-ethionine (SAE) in place of SAM during catalysis. HydG reacts with SAE and tyrosine to form the organometallic intermediate Ω, similar to reaction with SAM. The SAE-bound [4Fe-4S]+ cluster of HydG undergoes cryogenic blue light photolysis to generate an ethyl radical, which upon annealing forms an organometallic species in which an ethyl is bound to the unique iron of a [4Fe-4S]3+ cluster.
Institute and/or researcher Twitter usernames: BroderickLabFeS
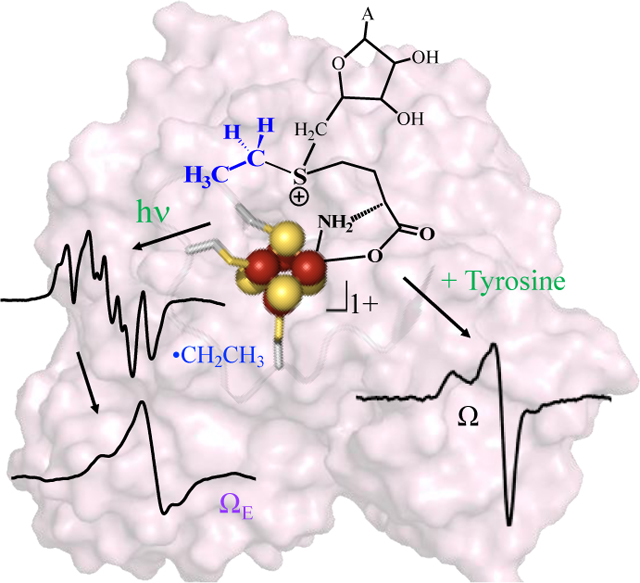
Introduction
Catalysis by radical S-adenosyl-l-methionine (radical SAM or RS) enzymes is accomplished via the reductive cleavage of SAM to initially generate an organometallic intermediate Ω comprising a 5’-deoxyadenosyl ligand covalently bound to the unique iron of the active-site [4Fe-4S]3+ cluster.[1–2] The intermediate Ω then liberates a 5’-deoxyadenosyl radical (5’-dAdo•) via homolytic Fe-C bond cleavage, and this radical abstracts an H-atom from substrate to initiate the wide variety of reactions performed by the RS superfamily.[3–7] Suess and coworkers have recently synthesized a model for alkyl-[4Fe-4S]3+ clusters that shows spectroscopic properties similar to Ω.[8] The discovery of Ω reinforced the remarkable catalytic parallels between RS and adenosylcobalamin (B12) radical enzymes, with both using homolytic metal-carbon bond cleavage of an organometallic species to generate the central 5’-dAdo• intermediate responsible for H-atom abstraction.[1–2] Despite its central role in radical initiation in both enzyme families, the 5’-dAdo• radical remained an elusive species for nearly 60 years, escaping direct observation and characterization. Our recent discovery of novel photochemistry of SAM-bound [4Fe-4S]+ clusters in RS enzyme active sites changed this: we showed that photoinduced electron transfer in the pyruvate formate-lyase activating enzyme (PFL-AE) [4Fe-4S]+:SAM complex cleaves the S-C5’ bond of SAM to generate 5’-dAdo• trapped in the PFL-AE active site, enabling the first direct characterization of this important radical intermediate.[9]
HydG is a RS enzyme essential for the maturation of the [FeFe]-hydrogenase (Fig 1), catalyzing the radical decomposition of tyrosine to produce CO and CN–, both of which end up as ligands in the H-cluster at the active site of [FeFe]-hydrogenase.[10–13] Radical formation occurs at a site-differentiated [4Fe-4S] cluster bound near the N-terminus of HydG, which coordinates SAM and catalyzes reversible reductive cleavage of SAM to generate the 5’-dAdo• species. This ultimately abstracts an H atom from the amino of tyrosine,[13–15] through a mechanism that proceeds via the Ω organometallic intermediate central to RS enzyme reactions.[2] The initial products of tyrosine cleavage are p-cresol and dehydroglycine;[16] the latter migrates towards an auxiliary cluster that is involved in the subsequent chemistry, ultimately forming CN– and CO.[17] These diatomic products, perhaps together with iron in the form of a small-molecule synthon,[18–20] are delivered to the GTPase scaffold protein HydF.[21–24] The RS enzyme HydE is proposed to provide the dithiomethylamine (DTMA) ligand that forms a 2Fe H-cluster precursor on HydF ([2Fe]F), which is then delivered to HydA to produce the active hydrogenase.[25–27] A recent report provides intriguing evidence that the substrate of HydE is a HydG-generated cysteine-coordinated organometallic synthon.[28]
Figure 1.

Top: Radical initiation mechanism in RS Enzymes. Lower Left: HydG structure showing the RS [4Fe-4S] cluster and the auxiliary cluster. Lower Right: HydG catalyzed tyrosine cleavage into diatomic CO and CN− ligands.
Reduced HydG with SAM bound to the [4Fe-4S]+ cluster furthermore undergoes cryogenic photoinduced electron transfer from the cluster to SAM, resulting in regiospecific cleavage of the S-CH3 bond, rather than the S-C5’ bond (as in the case of PFL-AE).[29–30] This generates a •CH3 radical in the active site, as characterized by electron paramagnetic resonance (EPR) spectroscopy.[29] We found that this •CH3 undergoes rapid rotational diffusion even at 40 K, and converts to a slower-tumbling state at lower temperatures.[29] The •CH3 radical decays rapidly at 77 K with a half-life of minutes,[29] in contrast to the 5’-dAdo• radical formed by photolysis of [4Fe-4S]+:SAM in PFL-AE, which is indefinitely stable at 77 K.[9] We concluded that the •CH3 is intrinsically more reactive than 5’-dAdo•, primarily because of its small size and ability to undergo rapid translational diffusion in search of a reaction partner.[29]
This report focuses on the reactivity of the ethyl analog of SAM, S-adenosyl-l-ethionine (SAE) with HydG. SAE can form in vivo in yeast and in mammals fed a diet containing ethionine,[31–32] and has been shown to inhibit some methyltransferase reactions,[33–36] but we are not aware of any prior studies of SAE reactivity with RS enzymes. Ji et al. have reported on the use of the SAM analog allyl-SAM in mechanistic investigations of the radical SAM enzyme NosN.[37] We report here the enzymatic synthesis and characterization of SAE, and show that HydG can utilize SAE as an effective alternative cosubstrate to SAM under normal enzymatic turnover conditions, producing the Ω intermediate and allowing efficient catalytic turnover of substrate L-tyrosine. We also report that cryogenic photolysis of the HydG [4Fe-4S]+:SAE complex results in S-CH2CH3 homolytic bond cleavage to form a •CH2CH3 radical trapped in the HydG active site, which we have characterized by EPR spectroscopy. This ethyl radical has significantly greater stability than the methyl radical studied previously, persisting under prolonged storage at 77 K and through annealing at 100 K.[29] Upon annealing to higher temperatures this radical adds to the [4Fe-4S]2+ cluster to generate an ethyl-[4Fe-4S]3+ organometallic species termed ΩE. Our results provide a window into the remarkable versatility of RS enzymes in generating alkyl radicals to initiate diverse biological radical reactions.
Results and Discussion
Synthesis of SAE.
SAE was synthesized in vitro from ATP and ethionine using E. coli SAM synthetase. SAE was purified on a cation exchange column, eluting at the same concentration of HCl eluent as required for elution of SAM.[38] The yield of SAE from the enzymatic synthesis and subsequent purification was approximately 25% that of a typical SAM preparation, most likely reflecting a lower specific activity for SAM synthetase with ethionine vs. methionine. The synthesis of SAE was verified by NMR (Fig S1) and HPLC-MS (Fig S2). The NMR showed characteristic resonances for the ethyl group vs. methyl group of SAM. Close examination of the characteristic triplet for the methyl group of the -CH2CH3 at 1.45 ppm reveals a smaller triplet slightly upfield (1.41 ppm), which is due to the R,S-diastereomer of SAE. Relative integration gives approximately 10% of the R,S product in the synthesized SAE, indicating some racemization occurred during the workup and purification procedures, similar to what is observed for SAM.[39]
EPR Evidence for SAE Coordination to the HydG [4Fe-4S] Cluster.
HydG is a RS enzyme with two [4Fe-4S] clusters: the RS [4Fe-4S] cluster that binds near the N-terminal of the protein ([4Fe-4S]RS), and an auxiliary cluster that binds near the C-terminus ([4Fe-4S]aux). Under anoxic and reducing conditions, both clusters are in the EPR-active S=1/2 [4Fe-4S]+ cluster state and have distinct EPR g-values: for the [4Fe-4S]+RS, g = [2.03, 1.935, 1.893], and for the [4Fe-4S]+aux, g = [2.027, 1.919, 1.888] (Fig 2, Fig S3). Upon addition of SAM, the EPR signal of the [4Fe-4S]+RS is perturbed, with g = [1.998 1.88 1.834], while that of the [4Fe-4S]+aux is unchanged, Fig 2. The g-values were obtained by simulations of the observed spectra in Fig 2 with a two cluster model (Figs S3, S4); the obtained g-values agree well with previously reported values.[13]
Figure 2.

EPR spectral evidence for SAE binding to the [4Fe-4S]+ cluster of HydG. CW-EPR spectra of reduced HydG (top) is due to the overlay of signals arising from [4Fe-4S]+RS and [4Fe-4S]+AUX. Addition of SAM to reduced HydG (middle) perturbs the [4Fe-4S]+RS EPR signal while the [4Fe-4S]+AUX signal remains unchanged. Addition of SAE to reduced HydG (lower) also perturbs the [4Fe-4S]+RS EPR signal while the [4Fe-4S]+AUX signal remains unchanged. Conditions: [HydG] = 1 mM, [dithionite] = 3 mM, [SAM] or [SAE] = 5 mM. EPR parameters: microwave frequency, 9.37 GHz; T = 12 K; modulation amplitude, 10 G.
The changes in the EPR spectra of [4Fe-4S]+RS upon addition of SAM arise from coordination of the SAM amino and carboxylate moieties to the unique iron of the [4Fe-4S]+RS,[3, 25] as has been observed in a wide range of RS enzymes, although the details vary from enzyme to enzyme and can depend on buffer conditions as well as the monovalent cation present in the assay buffer.[40] Also impacting the EPR spectra of HydG is the potential for an additional FeII bridged to the [4Fe-4S]aux cluster via a cysteine ligand; the presence of this 5th iron leads to an EPR-silent state for the auxiliary cluster,[41] and therefore the contribution of the auxiliary cluster to the EPR signal can vary depending on iron loading.
Because of the sensitivity of the EPR signature for the [4Fe-4S]+RS cluster to SAM coordination, we used EPR spectroscopy to probe whether the SAM analog SAE would bind to the unique iron of the HydG [4Fe-4S]1+RS as does SAM. When SAE is added to HydG, the EPR spectrum of [4Fe-4S]+RS cluster changes to one with g = [2.012 1.876 1.854], different from the spectrum of unliganded [4Fe-4S]1+RS and similar to the spectrum when SAM binds (Fig 2, S5). This indicates that SAE binds to the [4Fe-4S]1+RS cluster in a manner similar to that of SAM, presumably with chelation of the unique iron through the amino nitrogen and carboxylate oxygen of SAE. The SAE-bound [4Fe-4S]+RS has somewhat different g-values from the SAM-bound [4Fe-4S]+RS, as revealed through simulations (Figs S4, S5); these differences likely reflect perturbations in the unique iron coordination environment due to the greater steric bulk of ethyl vs. methyl. Specifically, replacing the methyl of SAM with an ethyl in SAE would likely change the positioning of the positively-charged sulfonium relative to the [4Fe-4S]+RS, which change would propagate to the Fe chelate and would be expected to alter the EPR signal arising from this cluster.
SAE is a Catalytically Functional Cofactor.
To determine whether SAE can serve as a functional cofactor in RS enzyme turnover, enzymatic activity assays were carried out under anoxic conditions with reduced HydG, SAE, and tyrosine, with deoxymyoglobin serving as a detector of the product CO.[11] The results (Fig 3) show that SAE is able to replace SAM during HydG catalysis, with assays containing SAE showing CO production rates (apparent first-order rate, k = 0.035 ± 0.003 min−1) that were 80% of those observed with SAM (k = 0.044 ± 0.004 min−1). LC-MS analysis confirms that this SAE-supported turnover is accompanied by the production of 5’-deoxyadenosine, a normal product of SAM turnover; thus SAE, like SAM, supports normal catalytic function of HydG, wherein the interaction of SAE (or SAM) with the [4Fe-4S]+RS results in reductive S-C5’ bond cleavage to produce a 5’-dAdo• radical which abstracts a H atom from substrate L-tyrosine to initiate the conversion of tyrosine to p-cresol, CO, and CN−.
Figure 3.

HydG activity assays with SAE and tyrosine. Top, UV-visible spectral changes as CO produced by HydG catalysis binds to H64L deoxy-Mb. Bottom, kinetic traces for the HydG-catalyzed formation of CO with SAM (squares) and SAE (circles). The red lines represent exponential fits to the data used to determine kcat values provided in the text.
Rapid freeze-quench (RFQ) experiments were carried out to define the catalytic pathway for the reaction of HydG with SAE and tyrosine. The RFQ experiments were performed by rapidly mixing reduced HydG with a solution containing (tyrosine+SAM) or (tyrosine+SAE) and freeze-quenching at 500 ms. The EPR spectrum of HydG+(tyrosine+SAM) quenched 500 ms after mixing shows the disappearance of the cluster signal of [4Fe-4S]1+RS+SAM at g⊥=1.864, and the appearance of the organometallic intermediate Ω with g|| = 2.035, g⊥ = 2.004, Fig 4, as previously reported.[2] The EPR spectrum of freeze quenched HydG+(tyrosine+SAE) is virtually identical to the Ω captured during RFQ with SAM and HydG (Fig 4), as well as the organometallic Ω intermediates previously shown to be central to catalysis in a wide range of radical SAM enzymes.[1–2]
Figure 4.

X-band CW-EPR of the catalytically central Ω trapped in HydG (250 μM) on reaction with tyrosine and SAM (red) or tyrosine and SAE (black) quenched at 500 ms. Conditions: T = 12 K; microwave power, 2 mW; modulation amplitude, 5 G; microwave frequency, 9.37 GHz; both have been annealed to 150K for 1 min to remove a very weak impurity signal.
Together, the enzymatic and spectroscopic results indicate that in the presence of substrate and at optimal temperatures for catalysis the ethyl-for-methyl substitution does not perturb the local active site environment sufficiently to disrupt the normal catalytic function of HydG. SAE in the HydG active site is bound in a catalytically competent state and oriented in a manner that favors reductive cleavage of the SAE S-5C’ bond, followed by formation of Ω, liberation of 5’-dAdo•, and efficient catalytic reaction with substrate to initiate the conversion of substrate tyrosine to p-cresol, CO, and CN−.
Photolysis of SAE bound to HydG.
We recently demonstrated that blue-light photolysis of SAM bound to the active site [4Fe-4S]+ cluster of PFL-AE in the absence of PFL results in photoinduced electron transfer from the reduced iron-sulfur cluster to SAM, reductively cleaving the SAM to produce a 5’-deoxyadenosyl radical (5’-dAdo•) trapped in the active site.[9] Subsequent studies showed that photolysis of HydG [4Fe-4S]+-SAM complex in the absence of substrate tyrosine instead led to a methyl radical (•CH3) trapped in the active site; that is, in HydG the photoinduced electron transfer led to homolytic S-CH3 bond cleavage, rather than S-C5’ bond cleavage as in PFL-AE.[29]
To test the sensitivity of photoinduced S-C cleavage to variations in SAM, we carried out cryogenic (12 K) 450 nm photolysis of the [4Fe-4S]+-SAE complex of HydG in the absence of tyrosine. The EPR spectrum subsequent to 5 mins of photolysis reveals the appearance of a new radical species concomitant with the loss of a majority of the [4Fe-4S]1+ cluster signal (Fig 5). This contrasts with photolysis of the [4Fe-4S]+-SAM complex of HydG, which requires about 1 hour to go to completion.[29]
Figure 5.

X-band CW-EPR spectra of •CH2CH3 (top) and •CH2CH3* (bottom) after 450 nm photolysis of [4Fe-4S]1+RS:SAE complex of HydG for 5 mins and 1 hr. The simulation (red) parameters for •CH2CH3 and •CH2CH3* are supported in Table 1. EPR parameters: temperature, 40 K; microwave frequency, 9.37 GHz; modulation amplitude, 3 G.
The new radical species exhibits a multiline EPR signal at 40 K, clearly different than the distinct 1:3:3:1 methyl radical signal observed upon photolysis of HydG complexed to SAM,[29] and also different from the signal observed for the 5’-dAdo• generated on photolysis of PFL-AE/SAM.[9] This multiline signal is associated with the •CH2CH3 radical liberated by photolytic cleavage of SAE in HydG. This is established by simulation of the EPR spectrum on the basis of a carbon spin center coupled to the two •CH2- α-protons, each with anisotropic hyperfine tensors, A(1H) = [80, 40, 60] MHz, and to three β-protons from a non-rotating methyl group, the three having isotropic couplings, aiso(1Ha, 1Hb, 1Hc) = (110 MHz, 20 MHz and 20 MHz), Fig 5, upper. The photoinduced S-CH2CH3 cleavage of SAE occurs much more rapidly under steady-state photolysis than does the photoinduced S-CH3 cleavage of SAM, which suggests more efficient photoinduced electron transfer within [4Fe-4S]1+RS:SAE than [4Fe-4S]1+RS:SAM.
On extending the time of radiation up to 1hr, the EPR signal of •CH2CH3 gradually evolves into a radical signal with sharper and more resolved 1H-hyperfine-split peaks, while maintaining the same EPR breadth and total spin integration, Fig 5, lower. The spectrum of the 1hr-radiation generated radical is well simulated with only slight adjustment of the spin-Hamiltonian parameters for •CH2CH3 (Fig 5, lower, Table 1), showing that the properties of the radical itself are not altered. Instead, the enhanced resolution results from a decreased linewidth (decreased homogeneous linewidth, but with the introduction of a heterogeneous linewidth component in simulations; Table 1). This change suggests that the prolonged illumination has relaxed the environment of the •CH2CH3 radical, and we denote the resulting radical center, •CH2CH3*. We attribute this phenomenon to local photothermal annealing in the vicinity of the cluster caused by the release as heat of the energy of multiple photons absorbed during the extended illumination period by the [4Fe-4S]2+ cluster proximate to the ethyl radical. It is well known that absorption of a single photon of this energy can cause extremely large transient increases in temperature of the absorbing cofactor, with smaller but substantial temperature increases in the protein vicinity as the energy flows through the protein.[42–43] Such a temperature increase would be greatly enhanced at the cryogenic temperatures employed here, as the protein heat capacity is strongly decreased at such temperatures.
Table 1.
| Spin Hamiltonian | •CH2CH3 | •CH2CH3* |
|---|---|---|
| g | 2.003,2.002,2.001 | 2.005,2.004,2.003 |
| A(α−1Ha) MHz[b] | 80,40,60 | 80,40,60 |
| A(α−1Hb) MHz | 80,40,60 | 80,40,60 |
| A(β−1Ha) MHz | 110,110,110 | 100,100,115 |
| A(β−1Hb) MHz | 20,20,20 | 25,25,30 |
| A(β−1Hc) MHz | 20,20,20 | 25,25,30 |
The homogenous EPR linewidth for the simulation of •CH2CH3 is 30 MHz, and for the simulation of •CH2CH3
is 6 MHz; this simulation further includes inhomogeneous line-broadening with HStrain = (40, 40, 10) MHz.
Euler angle (α,β,γ) = (60,0,0) for 1Ha of •CH2-, and (α,β,γ) = (−60,0,0) for 1Hb of •CH2-, corresponding to ∠1Ha-C-1Hb = 120°.
Thermal-annealing of •CH2CH3 and •CH2CH3*:
The •CH2CH3 radical formed directly by photolysis of the HydG [4Fe-4S]+-SAE complex at 12 K (Fig 5 upper) is indefinitely stable at 77 K, as is the photothermally annealed •CH2CH3* radical. These results are in sharp contrast to our observation that the methyl radical generated by photolysis of the HydG/SAM complex decays at 77 K with a half-life of minutes.[29] Thus, the modest difference in steric bulk between the 2-carbon ethyl and the 1-carbon methyl radicals affords a major increase in stability at cryogenic temperatures, likely a result of decreased translational freedom of the radical in the active site leading to a diminished susceptibility to quenching reactions. Indeed, the ethyl radical behaves similarly to the much larger 5’-dAdo• radical generated by photolysis of SAM bound to reduced PFL-AE, which is stable indefinitely at 77 K.[9]
When a sample that exhibits the •CH2CH3 signal formed by photolysis at 12 K (Fig 5 upper) was annealed for 1 minute at 150K and then at 200 K for 1 min, its intensity decreases and the signal becomes distorted, Fig 6, in a manner suggesting the conversion of the •CH2CH3 radical into a second paramagnetic species that shows no 1H splittings. Upon further annealing at 220K for 1 min, •CH2CH3 is completely gone, and the spectrum exhibits the signal from the new paramagnetic species. The new signal, with a substantial g-anisotropy (g = [2.02 2.002 1.984]); Fig 6) and an absence of hyperfine splittings is analogous to, but more rhombic than, the signal of the Ω organometallic intermediate (g = [2.035, 2.004, 2.004]) that is observed widely across the RS superfamily, in which the SAM-derived 5’-dAdo moiety is covalently bound to the unique iron of the [4Fe-4S] cluster. This new signal is also similar to the methyl-[4Fe-4S]3+ ΩM observed upon annealing of •CH3 in HydG (g = [2.02 2.005 1.987]),[29] as well as the methyl-[4Fe-4S]3+ model complex recently synthesized by Suess and coworkers (g = [2.101, 2.050, 2.042]).[8] We therefore assign the new signal (Fig 6) to an organometallic species formed by reaction of •CH2CH3 with the [4Fe-4S]2+ cluster to create a CH3CH2-[4Fe-4S]3+ adduct, denoted ΩE. Equivalent annealing of the •CH2CH3* sample (Fig 7) likewise causes the loss of the •CH2CH3* radical signal and the appearance a signal whose substantial g anisotropy again indicates the formation of an CH3CH2-[4Fe-4S]3+ organometallic, but with g-values (g = [2.035, 2.001, 1.984]) and linewidths slightly different from those of ΩE; it is thus denoted ΩE* (Fig 7). The difference presumably reflects a conformational difference arising from the photothermal relaxation of the protein environment induced by the prolonged irradiation.
Figure 6.

X-band EPR spectra of •CH2CH3 radical formed after photolysis of HydG/SAE for 5 mins (top, same as the top spectrum in Figure 5), and its behavior under step-annealing at 150 K for 1 min, 200 K for 1 min, and 220 K for 1min. Simulation of ΩE: g = [2.02, 2.002, 1.983], LW = 36 MHz, HStrain = [115, 20, 45] MHz. Conditions: microwave frequency, 9.37 GHz; modulation amplitude, 5 G; T = 40 K.
Figure 7.

X-band EPR spectra of •CH2CH3* radical formed under prolonged photolysis of HydG/SAE and its behavior under step-annealing at 100 K for 25 min, 210 K for 5 min, and 220 K for 1min. The EPR intensity is normalized to the gain level. Simulation of ΩE*: g = [2.035, 2.001, 1.984], LW = 30 MHz, HStrain = [65, 25, 70] MHz. Conditions: microwave frequency, 9.37 GHz; modulation amplitude 2 G; T = 40 K.
The signals from both these organometallic ΩE and ΩE* species are more intense than that of ΩM formed in HydG upon photolysis/annealing of the HydG/SAM complex. This suggests that the larger ethyl radical is more constrained within the active site than •CH3, and so more of the ethyl radical is converted into an organometallic intermediate instead of diffusing away to undergo nonspecific reactions within the protein matrix.
The involvement of Ω as an intermediate in the RS reductive cleavage mechanism was surprising when first reported,[1] but it was subsequently shown to be central in catalysis across the RS superfamily.[2, 4, 7] However, the present observation of an ΩE species (CH3CH2-[4Fe-4S]3+) in HydG provides now the third organometallic, alkyl-[4Fe-4S]3+ complex, along with Ω and ΩM,[29] observed in RS enzymes. The photolysis/annealing method utilized to generate ΩM[29] and ΩE appears to be versatile, and may allow for the synthesis of other alkyl-[4Fe-4S]3+ species. While ΩM and ΩE are not involved in catalysis, they highlight the ability of site-differentiated [4Fe-4S] clusters to form stable alkyl-complexes. As expanded by the recent synthesis and characterization of a CH3-[4Fe-4S]3+ complex by Suess and coworkers,[8] these results reveal an unexpectedly rich organometallic chemistry of [4Fe-4S] clusters.
Conclusion
SAM is widely distributed in all organisms, and plays essential roles not only in the chemistry of the RS superfamily, but also in methyltransferases and other reactions.[44] SAM is synthesized in vivo from ATP and methionine by the enzyme SAM synthetase. While SAE has been detected in living systems under conditions where ethionine is available, few studies have focused on the impact of SAE on enzymatic systems that use SAM as a cofactor, such as the RS enzymes. Here we provide a study of SAE as an analog of SAM in a RS enzyme, demonstrating that SAE is a catalytically functional cofactor that supports the same catalytic chemistry, via the same mechanism, as the natural cofactor SAM.
We also show that photoinduced electron transfer of the SAE-HydG complex results in homolytic cleavage of a different sulfonium S-C bond (S-CH2CH3) than is cleaved catalytically (S-C5’), leading to formation of an ethyl radical. Interestingly, prolonged irradiation of [4Fe-4S]2+RS after SAE cleavage causes a photothermal local annealing of the protein environment near •CH2CH3, which results in better-resolved and sharper 1H-hyperfine split peaks for the •CH2CH3* radical (Fig 5). Contrary to the rotationally and translationally mobile methyl radical, which decays in hours at 77K,[29] the larger ethyl radical is less mobile and is indefinitely stable at liquid nitrogen temperatures.
Thermal annealing of the active-site trapped ethyl radical yields an organometallic species ΩE in which the ethyl group is covalently bound to the unique iron of the active site [4Fe-4S] cluster. This center is slightly different when produced by photo-annealing the •CH2CH3 and •CH2CH3* environment, hence the two are termed ΩE and ΩE*. Thus, SAE is capable of forming organometallic alkyl-[4Fe-4S]3+ species in HydG with two different alkyl groups, Ω and ΩE, depending on the method of S-C bond cleavage (photolytic or enzymatic turnover). Given that a third alkyl-[4Fe-4S]3+ species, with a methyl-group bound to Fe, was formed in the RS enzyme HydG when SAM was utilized,[29] these results demonstrate a remarkable versatility of RS enzymes in generating alkyl radicals and organometallic alkyl-[4Fe-4S]3+ species.
Supplementary Material
Acknowledgements
Preparation and characterization of HydG was funded by the U.S. Department of Energy, Office of Basic Energy Sciences grant DE-SC0005404 (to JBB and EMS). All other work was funded by the NIH (GM 111097 to BMH and GM 131889 to JBB). The authors thank Dr. Maike Blakeley for assistance with NMR spectroscopy and analysis.
References
- [1].Horitani M, Shisler KA, Broderick WE, Hutcheson RU, Duschene KS, Marts AR, Hoffman BM, Broderick JB, Science 2016, 352, 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Byer AS, Yang H, McDaniel EC, Kathiresan V, impano S, Pagnier A, Watts H, Denler C, Vagstad AL, Piel J, Duschene KS, Shepard EM, Shields TP, Scott LG, Lilla EA, Yokoyama K, Broderick WE, Hoffman BM, Broderick JB, J. Am. Chem. Soc 2018, 140, 8634–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Broderick JB, Duffus BR, Duschene KS, Shepard EM, Chem. Rev 2014, 114, 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Broderick WE, Hoffman BM, Broderick JB, Acc. Chem. Res 2018, 51, 2611–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Frey PA, Annu. Rev. Biochem 2001, 70, 121–148. [DOI] [PubMed] [Google Scholar]
- [6].Frey PA, Magnusson OT, Chem. Rev 2003, 103, 2129–2148. [DOI] [PubMed] [Google Scholar]
- [7].Broderick WE, Broderick JB, J. Biol. Inorg. Chem 2019, 24, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McSkimming A, Sridharan A, Thompson NB, Müller P, Suess DLM, J. Am. Chem. Soc 2020, 142, 14314–14323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yang H, McDaniel EC, Impano S, Byer AS, Jodts RJ, Yokoyama K, Broderick WE, Broderick JB, Hoffman BM, J. Am. Chem. Soc 2019, 141, 12139–12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Driesener RC, Challand MR, McGlynn SE, Shepard EM, Boyd ES, Broderick JB, Peters JW, Roach PL, Angew. Chem. Int. Ed. Engl 2010, 49, 1687–1690. [DOI] [PubMed] [Google Scholar]
- [11].Shepard EM, Duffus BR, McGlynn SE, Challand MR, Swanson KD, Roach PL, Peters JW, Broderick JB, J. Am. Chem. Soc 2010, 132, 9247–9249. [DOI] [PubMed] [Google Scholar]
- [12].Kuchenreuther JM, George SJ, Grady-Smith CS, Cramer SP, Swartz JR, PLoS ONE 2011, 6, e20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Driesener RC, Duffus BR, Shepard EM, Bruzas IR, Duschene KS, Coleman NJ-R, Marrison APG, Salvadori E, Kay CWM, Peters JW, Broderick JB, Roach PL, Biochemistry 2013, 52, 8696–8707. [DOI] [PubMed] [Google Scholar]
- [14].Duffus BR, Ghose S, Peters JW, Broderick JB, J. Am. Chem. Soc 2014, 136, 13086–13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nicolet Y, Zeppieri L, Amara P, Fontecilla-Camps JC, Angew. Chem 2014, 126, 12034–12038. [DOI] [PubMed] [Google Scholar]
- [16].Pilet E, Nicolet Y, Mathevon C, Douki T, Fontecilla-Camps JC, Fontecave M, FEBS Lett. 2009, 583, 506–511. [DOI] [PubMed] [Google Scholar]
- [17].Pagnier A, Martin L, Zeppieri L, Nicolet Y, Fontecilla-Camps JC, Proc. Natl. Acad. Sci. U. S. A 2016, 113, 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kuchenreuther JM, Myers WK, Suess DLM, Stich TA, Pelmenschikov V, Shiigi SA, Cramer SP, Swartz JR, Britt RD, George SJ, Science 2014, 343, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Suess DLM, Pham CC, Bürstel I, Swartz JR, Cramer SP, Britt RD, J. Am. Chem. Soc 2016, 138, 1146–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rao GD, Tao LZ, Suess DLM, Britt RD, Nat. Chem 2018, 10, 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shepard EM, McGlynn SE, Bueling AL, Grady-Smith C, George SJ, Winslow MA, Cramer SP, Peters JW, Broderick JB, Proc. Natl. Acad. Sci. U.S.A 2010, 107, 10448–10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Czech I, Silakov A, Lubitz W, Happe T, FEBS Lett. 2010, 584, 638–642. [DOI] [PubMed] [Google Scholar]
- [23].Scott AG, Szilagyi RK, Mulder DW, Ratzloff MW, Byer AS, King PW, Broderick WE, Shepard EM, Broderick JB, Dalton Trans. 2018, 47, 9521–9535. [DOI] [PubMed] [Google Scholar]
- [24].Byer AS, Shepard EM, Ratzloff MW, Betz JN, King PW, Broderick WE, Broderick JB, J. Biol. Inorg. Chem 2019, 24, 783–792. [DOI] [PubMed] [Google Scholar]
- [25].Betz JN, Boswell NW, Fugate CJ, Holliday GL, Akiva E, Scott AG, Babbitt PC, Peters JW, Shepard EM, Broderick JB, Biochemistry 2015, 54, 1807–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shepard EM, Mus F, Betz J, Byer A, Duffus BR, Peters JW, Broderick JB, Biochemistry 2014, 53, 4090–4104. [DOI] [PubMed] [Google Scholar]
- [27].Broderick JB, Byer AS, Duschene KS, Duffus BR, Betz JN, Shepard EM, Peters JW, J. Biol. Inorg. Chem 2014, 19, 747–757. [DOI] [PubMed] [Google Scholar]
- [28].Tao L, Pattenaude SA, Joshi S, Begley TP, Rauchfuss TB, Britt RD, J. Am. Chem. Soc 2020, 142, 10841–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yang H, Impano S, Shepard EM, James CD, Broderick WE, Broderick JB, Hoffman BM, J. Am. Chem. Soc 2019, 141, 16117–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Impano S, Yang H, Jodts RJ, Pagnier A, Swimley R, McDaniel EC, Shepard EM, Broderick WE, Broderick JB, Hoffman BM, in review 2020. [Google Scholar]
- [31].Schlenk F, Dainko JL, Stanford SM, Arch. Biochem. Biophys 1959, 83, 28–34. [DOI] [PubMed] [Google Scholar]
- [32].Smith RC, Salmon WD, Arch. Biochem. Biophys 1965, 111, 191–196. [DOI] [PubMed] [Google Scholar]
- [33].Finkelstein JD, Martin JJ, Biochem. Biophys. Res. Comm 1984, 118, 14–19. [DOI] [PubMed] [Google Scholar]
- [34].Moore BG, Smith RC, Can. J. Biochem 1969, 47, 561–565. [DOI] [PubMed] [Google Scholar]
- [35].Cox R, Irving CC, Cancer Res. 1977, 37, 222–225. [PubMed] [Google Scholar]
- [36].Moore BG, Can. J. Biochem 1970, 48, 702–705. [DOI] [PubMed] [Google Scholar]
- [37].Ji X, Mandalapu D, Cheng J, Ding W, Zhang Q, Angew. Chem. Int. Ed 2018, 57, 6601–6604. [DOI] [PubMed] [Google Scholar]
- [38].Byer AS, McDaniel EC, impano S, Broderick WE, Broderick JB, Methods Enzymol. 2018, 606, 269–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang J, Klinman JP, Anal. Biochem 2015, 476, 81–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shisler KA, Hutcheson RU, Horitani M, Duschene KS, Crain AV, Byer AS, Shepard EM, Rasmussen A, Yang J, Broderick WE, Vey JL, Drennan CL, Hoffman BM, Broderick JB, J. Am. Chem. Soc 2017, 139, 11803–11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dinis P, Suess DLM, Fox SJ, Harmer JE, Driesener RC, De La Paz L, Swartz JR, Essex JW, Britt RD, Roach PL, Proc. Natl. Acad. Sci. U. S. A 2015, 112, 1362–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li P, Champion PM, Biophys. J 1994, 66, 430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mara MW, Hadt RG, Reinhard ME, Kroll T, Lim H, Hartsock RW, Alonso-Mori R, Chollet M, Hodgson KO, Hedman B, Bergmann U, Gaffney KJ, Solomon EI, Science 2017, 356, 1276–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Markham GD, in Nature Encyclopedia of Life Sciences, Nature Publishing Group, London, 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.