

Abstract
A joint analysis of the NCI60 small molecule screening data, their genetically defective genes, and mechanisms of action (MOA) of FDA approved cancer drugs screened in the NCI60 is proposed for identifying links between chemosensitivity, genomic defects and MOA. Self-Organizing-Maps (SOMs) are used to organize the chemosensitivity data. Student’s t-tests are used to identify SOM clusters with enhanced chemosensitivity for tumor cell lines with versus without genetically defective genes. Fisher’s exact and chi-square tests are used to reveal instances where defective gene to chemosensitivity associations have enriched MOAs. The results of this analysis find a relatively small set of defective genes, inclusive of ABL1, AXL, BRAF, CDC25A, CDKN2A, IGF1R, KRAS, MECOM, MMP1, MYC, NOTCH1, NRAS, PIK3CG, PTK2, RPTOR, SPTBN1, STAT2, TNKS and ZHX2, as possible candidates for roles in chemosensitivity for compound MOAs that target primarily, but not exclusively, kinases, nucleic acid synthesis, protein synthesis, apoptosis and tubulin. These results find exploitable instances of enhanced chemosensitivity of compound MOA’s for selected defective genes. Collectively these findings will advance the interpretation of pre-clinical screening data as well as contribute towards the goals of cancer drug discovery, development decision making, and explanation of drug mechanisms.
Introduction
The emergence of extensive human tumor cell line compound screening data, coupled with advances in cancer genomic technologies, has generated comprehensive and complex databases [1]. Strategies for analyzing this data may identify important links between genetic changes that contribute to the hallmarks of cancer biology [2] and the discovery of leads in the pursuit of small-molecule cancer therapy [3]. The present report examines links between genetically defective genes in the National Cancer Institute’s panel of sixty tumor cell lines (referred to hereafter as NCI60), chemosensitivity, as measured by growth inhibition (GI50NCI60; adopting the convention of an under bar to describe the vector of GI50NCI60 (N = 59) measurements for each screened compound) and preferences for mechanisms of action (MOA) of identified linkages. An elegant study by Ikediobi et al. [4] addressed this goal by examining relationships between mutations in 24 cancer genes in the NCI60 tumor cell lines and the GI50NCI60 activity of ~8k screened compounds. Their finding of a strong association between the BRAF mutation (V600E) and the GI50NCI60 activity of phenothiazines supports important links between altered genes, chemosensitivity and MOAs. The current analysis extends this work, with significant differences.
GI50NCI60 results for ~53k screened compounds are analyzed (DTP database).
A larger set of gene mutations (N = 368) for the NCI60 tumor cell lines are analyzed (CBioPortal database).
A novel analysis of GI50NCI60, based on Self-Organizing-Maps (SOMs), emphasizing FDA approved compounds with assigned MOAs in the NCI60 screened compounds, is used to derive links between tumor cell chemosensitivity, genetically altered genes and MOAs.
Efforts to develop links between pre-clinical tumor cell screening data, genomic defects and drug mechanisms may contribute to advances in small-molecule cancer therapies. An important element of these efforts requires more informed interpretations of small-molecule screening results in the context of genomic profiles and drug action. These associations may yield undiscovered opportunities for drug re-purposing and new applications of gene mutations towards personalized medicine.
Data
Three publicly available data sources are used for this analysis. First, chemosensitivity data consists of the 2019 release of GI50NCI60 measurements from the Developmental Therapeutics Program (DTP) in the National Cancer Institute. Historically the NCI60 screen was designed to identify relationships between chemotypes and cellular responses [5]. Their bulk download(https://dtp.cancer.gov/discovery_development/nci-60) includes GI50 values for 159 tumor cell types. A subset of 70 tumor cell lines, identified previously [6] as representing an information-rich component of this data, consists of ~53k screened compounds, which for this analysis was reduced to 46,798 GI50NCI60 records when filtered for a coefficient of variation above 0.1. Z-score normalized GI50NCI60 measurements of the filtered data (~46K) were used for chemosensitivity analysis. The raw data file is included in the S1 master_appendix sheet GI50.
Second, genetic data is obtained from the cBioPortal database (https://www.cbioportal.org/) [7,8]. A total of 368 altered genes are listed for the NCI60; with either a mutation (MUT), copy number alteration (CNA) or fusion/splice (FUSION). These genomic changes are grouped, so that a gene alteration due to any or all types of variations will be designated as genetically defective. In this context, a defective gene indicates only a modification from the wild-type allele. Genes designated as defective genes can have wide-ranging effects including gain-of and/or loss-of gene function. Defective genes occur within each NCI60 tumor cell individually or as pairs, doublets, triplets, etc. S1 Appendix Fig 1 in S1 File displays a histogram for the frequency of defective genes within the NCI60. The highest frequency exists for tumor cell lines having a single defective gene. This frequency decreases progressively down to less than one percent for tumor cell lines sharing 10 defective genes. The cumulative frequency of tumor cell lines sharing any defective gene is 0.97, an indication that the probability of tumor cell lines sharing any defective gene is approaches one. S2 Appendix Fig 2 in S1 File displays the histogram of defective genes shared between tumor cell lines. These results find that shared defective genes, comprised of doublets and triplets are more common compared to the appearance of only a single defective gene (consistent with Ikediobi et al. [4]). S3 master_appendix sheet appendix_table_I lists the singlets, doublets and triplets of defective genes observed in the NCI60. S4 master_appendix sheet appendix_table_II summarizes their counts. Inspection finds a diverse set of defective genes, some of which are not considered to have important roles in cancer. CDC25A, TP53, CDKN2A, CDKN2B, MYC, BRAF, EP300, KRAS, NOTCH1 and PTK2 are the top ten most frequently occurring defective genes. To summarize, defective genes appearing as doublets or triplets finds these top ten defective genes to appear in combination with themselves and other genes. Collectively these results indicate that shared defective genes, with diverse roles in cellular biology, are common within the NCI60.
Fig 1. Results for selecting optimal cluster size using the elbow method (Panel A), silhouette method (Panel B) and the gap_statistic method (Panel C).

Selection for the elbow method is based on the largest local derivative of the within groups sum of squares (Panel A triangles), the maximum silhouette width (Panel B) and the first non-negative value for Gap(k)-(Gap(k + 1)−sdk+1) (Panel C triangles). These results indicate an optimal number of clusters in the 26–28 range. Panel D displays the GI50codebook dendrogram (Euclidean, Ward’s) with cuts at 28 (red lines) and 7 clusters (green lines), respectively.
Fig 2. Panel A displays a heatmap of GI50codebook, colored spectrally from green(chemoinsensitive) to red(chemosensitive) response.

Dendrogram at the left represents hierarchical clustering (Euclidean, Ward’s) of GI50codebooks (reproduced from Fig 1 Panel D). Panel B displays the SOMDTP colored according to hierarchical cutree [18] specified at the optimal number of 28 meta-clades. The 28 colors appear spectrally from meta-clade 1 (dark blue), at the bottom of the hierarchical dendrogram, to meta-clade 28 (dark red), at the top of the hierarchical dendrogram. Grayscale bar adjacent to the 28 meta-clade spectrally colored bar displays the 7 meta-clades groupings. The NCI60 tumor cell lines clustered in the heatmap are ordered, left to right, as: SK.OV.3.Ovarian, NCI.H322M.Lung, DU.145.Prostate, A549.ATCC.Lung, HOP.62.Lung, OVCAR.5.Ovarian, TK.10.Renal, EKVX.Lung, A498.Renal, NCI.H226.Lung, SK.MEL.28.Melanoma, SK.MEL.2.Melanoma, BT.549.Breast, UACC.257.Melanoma, MALME.3M.Melanoma, SN12C.Renal, OVCAR.8.Ovarian, NCI.H23.Lung, IGROV1.Ovarian, MDA.MB.231.ATCC.Breast, OVCAR.4.Ovarian, CAKI.1.Renal, ACHN.Renal, UO.31.Renal, HS.578T.Breast, RXF.393.Renal, T.47D.Breast, HOP.92.Lung, HCC.2998.Colon, HT29.Colon, COLO.205.Colon, NCI.H460.Lung, KM12.Colon, PC.3.Prostate, OVCAR.3.Ovarian, M14.Melanoma, MDA.MB.435.Breast, UACC.62.Melanoma, SK.MEL.5.Melanoma, SW.620.Colon, HCT.15.Colon, HCT.116.Colon, LOX.IMVI.Melanoma, MCF7.ATCC.Breast, MCF7.Breast, NCI.H522.Lung, K.562.Leukemia, RPMI.8226.Leukemia, HL.60.TB..Leukemia, SR.Leukemia, MOLT.4.Leukemia, CCRF.CEM.Leukemia.
Third, CellMiner [9] (https://discover.nci.nih.gov/cellminer/home.do) provides information about mechanism of action (MOA) for Food and Drug Administration (FDA) approved compounds. CellMiner reports 270 FDA compounds with unique NSC (National Service Center) and Name assignments that have been screened in the NCI60 (ca. 2019). One-hundred and ninety FDA screened compounds appear in the 46,798 GI50NCI60 responses. One-hundred and four MOA assignments exist for this set of compounds. These assignments consist of a primary MOA designation followed by secondary MOAs. For example the assignment BCR-ABL|YK,FYN,LYN indicates BCR-ABL at the primary MOA, with YK (tyrosine kinase), FYN and LYN (both Proto-Oncogene, Src Family Tyrosine Kinases) as secondary MOAs. Thirty primary MOAs are assigned to this data. The complete set of MOAs for FDA screened compounds is listed in S1 S5 master_appendix sheet appendix_table_III. Seven of the most frequent primary MOA classes spanning this data function to target tubulin:Tu, topoisomerase 2:T2, topoisomerase 1:T1, alkylation:A (A2: Alkylating_at_N-2_position_of_guanine, A6: Alkylating_at_O-6_of_guanine, A7: Alkylating_at_N-7_position_of_guanine, AlkAg: Alkylating agent and anti-metabolites:AM), DNA:D (Db:DNA_binder, DDI/R, DNA_damage_repair/inducer, Df:antifols, Ds: DNA_synthesis_inhibitor), kinases:PK and apoptosis:Apo. MOA:PK consists of over 100 kinase targets. FDA compounds screened in the NCI60, and their assigned CellMiner MOA, will be used for the linking MOA to chemosensitivity.
METHODS: Data clustering
The methods for linking chemosensitivity, defective genes and MOA apply a sequential, multi-tiered approach. First, the GI50NCI60 data is organized into clusters. Many statistical tools are now available for clustering GI50NCI60 data [10]. Relying on our prior analysis [6], the results presented here use Self-Organizing-Maps (SOMs) [11,12]. Parameters from prior SOM analyses are selected for clustering (hexagonal nearest neighbors, Epanechnikov Function kernal [13]). SOM dimensions are based on a heuristic using the ratio of the first and second principal components of the data. The y-axis dimension is calculated as round(sqrt(munits/ratio*sqrt(0.75))), where munits = 5*nsamples^0.543. The x-axis dimension is calculated as round(munits/y-dimension). This heuristic is derived from the developers of SomPak, based on their usage. The sqrt(0.75) multiplier is explained as follows, “in the hexagonal lattice, the side lengths are not directly proportional to munits (= 5*nsamples^0.543) since the units on the y-axis are squeezed together by a factor of sqrt(0.75)”. Applying this procedure yields SOM map dimensions of 44 rows and 28 columns.
Each of these 1232 SOM nodes defines a vector representing the average GI50NCI60 for all compounds clustered within each SOMDTP node (referred to hereafter as a node’s GI50codebook). S2 master_appendix sheet SOM_codebook lists the 1232 GI50codebooks. Each compound’s SOMDTP node will be referred to as its projection. The best projection can be extended to include the 2nd, 3rd, 4th, etc. SOMDTP nodes to determine whether a compound’s next best projections appear as SOMDTP neighbors. Prior analyses found GI50codebook patterns to be associated with a compound’s MOA (e.g. alkylating agents, tubulin targeting agents, DNA/RNA damaging agents and agents affecting mitochondrial function [6]). Analysis of GI50codebook patterns has also been proposed for use in the development of clinical strategies based on differentially expressed molecular targets within classes of tumors [14,15]. Other applications include the recent identification of unique GI50codebook patterns within the NCI60 renal subpanel as the basis for further testing of the natural product-derived family of englerins [16].
While each SOMDTP node represents a cluster of GI50NCI60 values, a more global, lower resolution representation, that optimally groups SOMDTP nodes into meta-clades, is proposed. Three state-of-the art procedures are used to determine the optimal number of meta-clades; the elbow method minimizes the within-cluster sum of squares, WSS (a measure of within cluster similarity) and maximizes the between-cluster sum of squares (a measure of how separated each cluster is from the others), the silhouette method computes the average silhouette of observations for different numbers of clusters (selecting the optimal cluster size that maximizes the average silhouette) and the gap_statistic method [17] determines the total within intra-cluster variation for different numbers of clusters (selecting the cluster size that maximizes the gap_statistic). Fig 1 displays the results for these three methods applied to GI50codebooks. The optimal cluster size is indicated by the vertical lines in each plot. The elbow and gap methods rely on an inflection point on each curve for optimal cluster size, while the silhouette method seeks the largest value for silhouette width. Panel A displays WSS as circles for the elbow method and the first derivative of WSS, normalized by the local average WSS, as triangles. The maximum value of the derivative of WSS occurs for 28 clades. The silhouette method yields a maximum value at 26 clusters (cf. Panel B). The criterion for optimal cluster size using the gap statistic seeks the smallest number of clusters such that the gap statistic is within one standard deviation of the next gap statistic: Gap(k)≥Gap(k + 1)−sd(k+1) (displayed as triangles in Panel C), yields 28 clusters as optimal. Panel D displays the GI50codebook cluster dendrogram using the cutree tool [18] to group the dendrogram into 28 meta-clades (red lines) and 7 meta-clades (green lines). Based on these results a value of 28 was selected for the optimal number of meta-clades used in this analysis. The rationale for cutting the dendrogram at 7 clusters will be provided later in the analysis of MOAs.
A visual perspective of SOMDTP and the 28 meta-clades appears in Fig 2. The 1232 GI50codebooks appear as a clustered heatmap (Euclidean,Ward’s) in Panel A. The dendrogram at the left edge of the heatmap, displays the dendrogram appearing in Fig 1, Panel D. The vertical ribbon, adjacent to this dendrogram, colored spectrally from blue to red, represents the subdivision of the hierarchal clade tree into 28 meta-clades. The pvclust utility [19], using random resampling, confirms this set of 28 meta-clades with a confidence p-value above 0.995 across resampling (n = 1000 resamples). Panel B in Fig 2 displays the 28 meta-clades on SOMDTP, colored according to the spectral-colored vertical ribbon in the left panel. The data reduction of 1232 SOMDTP nodes to 28 meta-clades yields a lower resolution, more manageable, perspective of the complete SOMDTP. The gray ribbon in Panel B displays the dendrogram cut into 7 major groups. Groupings consist of A: meta-clades 1–6, B: meta-clades 7–9, C: meta-clades 10–15, D: meta-clades 16–18, E: meta-clades 19–20, F: meta-clades 21–24 and G: meta-clades 25–28. The vertical grayscale colored bar in Fig 2 displays these seven groupings from A(bottom:black) to G(top:light gray). SOMDTP meta-clades will be assessed according to the differential chemosensitivity of tumor cell lines with and without defective genes.
Noteworthy is the mapping of the 28 cutree clades to discontinuous SOMDTP regions. Ideally, cutree clades might appear as contiguous regions the 2-dimensional SOMDTP. However, this is not the case. To obtain contiguous SOMDTP regions, an alternative hierarchical clustering algorithm would need to be used that only combines adjacent dendrogram clades that appear beside each other on SOMDTP. Although not pursued here, assigning contiguous SOM regions is an active area of research in dimensionality reduction [20], with specific focus on representing SOMs in one dimension [21]. Many of these efforts use randomized resampling to identify contiguous map regions by consensus. Usually standard hierarchical clustering suffices, and any outlying (noncontiguous) points can be accounted for manually. Towards this end, SOMDTP singletons, appearing as a hierarchical clade that maps to SOMDTP as a node without the same meta-clade neighbors, have been replaced by their neighborhood meta-clade assignments. There are 12 such cases (0.0097 = 12/1232) for this data set.
An additional consideration for non-contiguous SOMDTP meta-clades may result from the assignment of distances used for clustering (Euclidean for hierarchical clustering and Epanechnikov Function [13] for SOMs). Our choice of the Epanechnikov Function for SOM clustering consistently yielded the lowest SOMDTP quantization errors [6]. However, a more likely explanation for non-contiguous SOMDTP meta-clades involves differences in clustering methodology. SOMs organize data by mapping each cluster to its most similar neighbors (six in the case of hexagonal mapping); whereas hierarchical clustering, as used to obtain heatmap dendrograms, builds each branch of the dendrogram by pairwise associations. The failure to map hierarchical clustering methods directly to contiguous SOMDTP regions is not unexpected and points more to the limitations of hierarchical methods to match non-hierarchical methods, regardless of distance metrics.
METHODS: Identification of SOMDTP nodes with enhanced chemosensitivity
SOMDTP nodes are analyzed for enhanced chemosensitivity of tumor cell lines with versus without defective genes. Each GI50codebook is divided into subsets comprising tumor cell lines with (GI50defective) and without (GI50wild-type) a defective gene. A Student’s t-test is used to identify cases of relatively higher chemosensitivity for GI50defective versus GI50wild-type. SOMDTP nodes with Student’s p-values less than or equal to 0.05 were further assessed for statistical significance by bootstrap resampling [22,23]. Each node’s GI50codebook was randomly shuffled and a Student’s t-test performed, while maintaining the tumor cell’s wild-type and defective gene status. One-thousand trials were conducted for each GI50codebook and a p-value was estimated by counting the number of times the shuffled p-value was less than the initial, unshuffled, p-value. Dividing this value by 1000 yields an estimate for the probability of the observed p-value occurring by chance. SOMDTP nodes with measured p-values less than 0.05 and below their estimated chance occurrence were accepted for further analysis. Sixty-five percent (65%, n = 635) of the 1232 SOM nodes pass this criterion and account for 121 defective genes.
Fig 3 summarizes the results for GI50codebook at SOM1,13 (subscripting refers to the SOM node, i.e. SOMrow,column). Five NCI60 tumor cell lines have the defective ABL1 gene; with these tumor cell lines having a mean GI50defective response nine-fold higher than GI50wild-type (p = 6.91e-3). Panel A in Fig 3 displays GI50codebook, ordered from most chemosensitive to least chemosensitive values. NCI60 tumor cell lines with the ABL1 alteration, highlighted in red and representing GI50defective, are ranked at positions 3, 6, 8, 25 and 51. SOMDTP can also be viewed according to each NCI60 tumor cell (referred to as GI50component). Panel B of Fig 3 displays GI50component for each of the 5 tumor cell lines with defective ABL1. Regions of greatest and least chemosensitivity for each tumor cell are displayed spectrally from red to blue, respectively. Noteworthy is the location of Gleevec chemosensitivity to the most sensitive (e.g. red) GI50component SOMDTP regions for K-562, RMPI-8226 and HS578T.
Fig 3. Panel A displays GI50codebook for SOM1,13, ordered from most to least chemosensitivity.
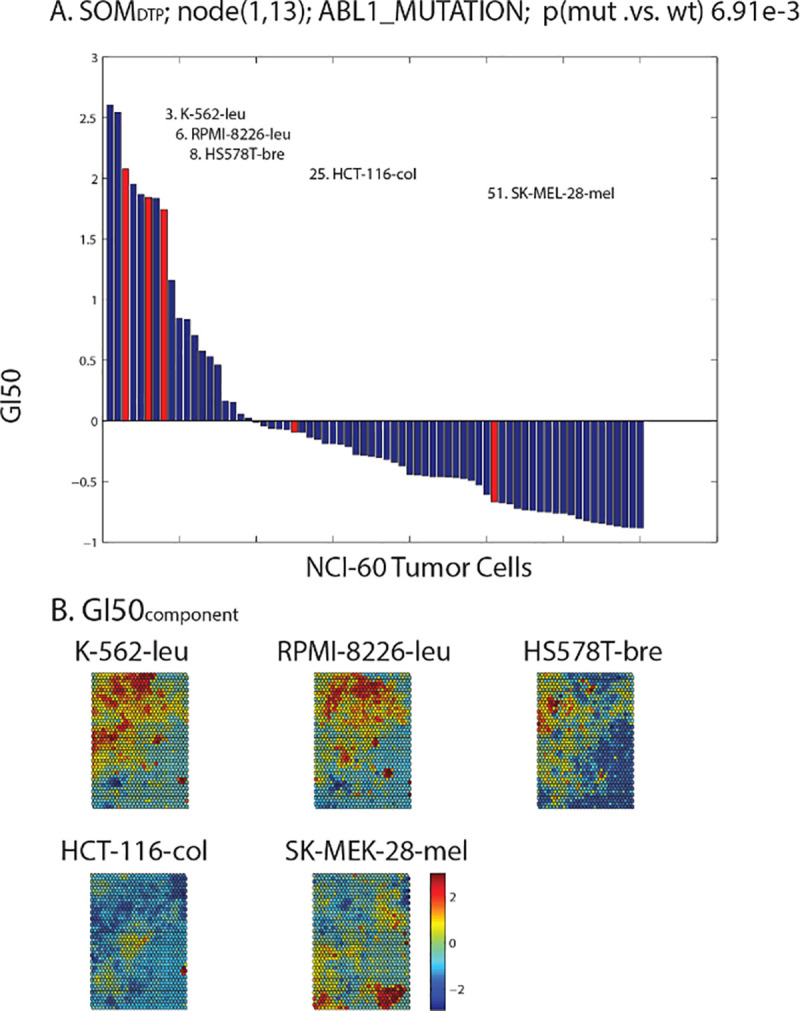
The 5 tumor cell lines with the defective ABL1 gene appear as red bars. Panel B displays GI50component for the 5 tumor cell lines with defective ABL1. SOMDTP nodes are colored spectrally from highest chemosensitivity (red) to lowest chemosensitivity (blue).
Panel A of Fig 4 projects onto the SOMDTP the Students t-statistic for tumor cell lines with defective ABL1; where the t-statistic values are colored spectrally from low(blue) to high(red) significance. SOMDTP nodes without statistical significance (p>0.05) are not colored. The most significant t-statistics for defective ABL1 are located mainly in SOM meta-clades 1, 10, 14 and 26. Gleevec appears as the most significant SOM5,15 node in meta-clade 14. For comparison, the results for KRAS are projected in Panel B of Fig 4. There are 12 tumor cell lines (A549/ATCC-Lung, CCRF-CEM-Leukemia, HCC-2998-Colon, HCT-116-Colon, HCT-15-Colon, HOP-62-Lung, NCI-H23-Lung, NCI-H460-Lung, OVCAR-5-Ovarian, RPMI-8226-Leukemia, SK-OV-3-Ovarian and SW-620-Colon) harboring defective KRAS, with significant chemosensitive SOMDTP nodes appearing in meta-clades 21, 22 and 27. SOM meta-clade 21 is the location of the FDA compound cytarabine (ara-C) and is consistent with the conclusion of Ahmad et al [24] that adult AML patients carrying defective KRAS benefit from higher ara-C doses more than wt KRAS patients.
Fig 4. Panels A and B display significant chemosensitive SOMDTP nodes (projected as their t-statistic from a Student’s t-test; blue:least, red:most significant) for tumor cell lines with defective ABL1 and KRAS, respectively.

Panel C displays the 28 SOMDTP meta-clades.
S3 and S4 Appendix Figures 3 and 4 in S1 File display additional examples for the defective genes PIK3RI and IGF1R, respectively. PIK3R1 (Phosphatidylinositol 3-Kinase Regulatory Subunit Alpha) and a related gene, PIK3CA (PI3-Kinase Subunit Alpha) are lipid kinases capable of phosphorylating the 3’OH of the inositol ring of phosphoinositides. Both are responsible for coordinating a diverse range of cell functions including proliferation and survival. Defective PIK3CA has been documented by Whyte and Holbeck [25] to enhance tamoxifen sensitivity in selected NCI60 tumor cell lines. The results here also find chemosensitivity in NCI60 tumor cell lines having defective PIK3R1. The second example of defective IGF1R supports the importance of evaluating drug sensitivity for compounds targeting leukemia cell lines [26] and the emergence of IGF1R as a potential therapeutic target for the treatment of different types of cancer including plasma cell myeloma, leukemia, and lymphoma [27]. Both examples illustrate potential role of defective genes in chemosensitivity.
The Students t-statistic represents the significance when comparing the chemosensitivity of a SOMDTP node for tumor cell lines with, versus without, defective genes. Parametric tests, such as the Student’s t-test, are applicable over non-parametric tests (Wilcoxon/Whitney/Mann, Kruskal-Wallis) when the underlying sample distribution is known and normal. The data analyzed here represents a strongly normal distribution (p < 0.001, lognormal test) with small deviations at the tails from a linear log normal quantile-quantile plot; supporting the use of a parametric statistic.
The application of a bootstrap procedure to cases with a significant Student’s t-test is applied as a correction against Type I error for the following reasons. First, a bootstrap method can be used to estimate the sampling distribution of GI50codebook for each SOMDTP node. This test utilizes the node’s codebook vector as the initial sample representative and applies a bootstrap procedure to estimate the sampling distribution. Since 1000 samples were used, the p-value estimate corrects the empirical estimate using a division by 1000. This correction parallels multiple test corrections for traditional statistics [28]. Second, in this design there are 1000 statistical tests performed for each of the 1232 SOMDTP nodes. An important caveat of jointly using GI50codebook to create some type of correction for multiple test is their lack of independence [11,12]. This non-independence is disallowed when applying a Bonferroni, Holm or Benjamini-Hochberg [29] corrections. None-the-less, the long-standing debate continues to exist about bootstrap applications, possible overestimation of ‘true’ values, and appropriate corrections for random noise [30].
Methods: Mapping MOA to SOMDTP
SOMDTP projections for the most frequent primary CellMiner MOA assignments (Tu, T1, T2, A, D, Apo and PK) are displayed Panel A of Fig 5. Panel B displays the histogram of SOMDTP node counts for these MOA assignments. Inspection indicates that MOA classes A, D, T1 and T2 appear mainly in the upper right SOMDTP region (SOM meta-clade 21; Group A), while MOA Apo appears mainly in the upper left region (SOMDTP meta-clades 25 and 26; Group G). Tu compounds are found mainly in SOMDTP meta-clades 16, 17 and 18 (Group D). SOMDTP meta-clade 19(Group E) consists of only MOA PK; while MOA PK compounds are in the majority for SOMDTP meta-clades 1 through 6(Group A). The horizontal gray scale bar at the bottom of the right panel identifies the seven meta-clade groups assigned earlier (cf. Figs 1 and 2). Inspection indicates relative similarities of MOA types within each of the seven meta-clade groups A:G. Notable is the majority representation of MOA:PK in Group A and MOA:Tu in Group D. Detailed results for MOAs across meta-clade groups will be presented later.
Fig 5. Panel A: SOMDTP projections for FDA approved compounds for the primary CellMiner assigned MOAs.
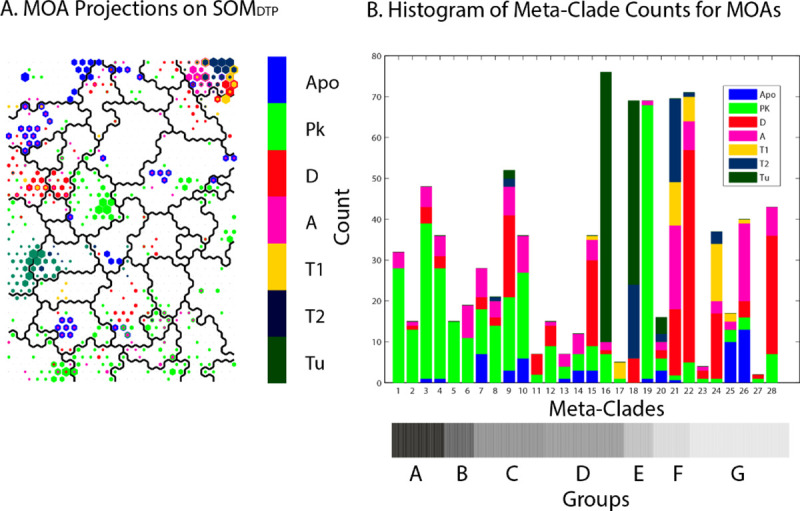
Projections include the top 10th percentile of SOMDTP nodes for each compound. Panel B: histogram of the counts for these primary MOAs across SOM meta-clade groups. Primary MOAs appear color-coded in each vertical bar, with their heights corresponding to MOA counts in each meta-clade. Horizontal grayscale bar below Panel B indicates meta-clade groups A:G (reproduced from Fig 2 Panel A).
Table 1 lists the most frequent primary MOAs for meta-clade groups A:G. These results segregate the primary MOAs into separate regions of the cutree = 7 hierarchical dendrogram (gray bar in Fig 2 Panel A and Fig 5 Panel B); MOA:PK appears at the bottom, MOAs targeting DNA and Apo appear at the top and mixtures of primary MOAs appear in the middle. These meta-clade grouping will be analyzed in greater detail for links of MOAs to defective genes.
Table 1. Most frequent primary MOA assignments within meta-clade groups A:G.
| Meta-clade Groups | MOA |
|---|---|
| A: 1–6 | PK |
| B: 7–9 | PK, A |
| C: 10–15 | PK, A, D |
| D: 16–18 | Tu, T2 |
| E: 19–20 | PK |
| F: 21–24 | A, D, T1 |
| G: 25–28 | Apo, A, D |
Methods: Enrichment of MOA
Fisher’s exact and chi-square tests are used to identify cases where the SOMDTP projections of defective genes are statistically enriched in co-projections of MOA types. These tests are useful for categorical data that result from classifying objects in two different ways; and are used to determine a statistical measure for the random likelihood of the intersection of each classification. For each defective gene the number of SOMDTP nodes with significant Student’s t-statistics are determined (Ngene). FDA approved compounds that co-project to Ngene determine a unique set of MOA’s associated with each defective gene (MOAgene). All FDA compounds that share any MOAgene are collected (NFDA); where the 10th best FDA projections are included in the count. Extending FDA projections beyond only the best node achieves two goals. First, it establishes SOMDTP regions rather than individual nodes for MOA classification. Second, increasing the numbers in the contingency table extends significance testing to include Fisher’s exact and the chi-square testing. The contingency table entries become; p11 = intersection(Ngene,NFDA), p12 = Ngene−p11, p21 = NFDA−p22 and p22 = p11 by default to conserve equal row and column sums.
Fig 6 illustrates a sample result of the steps for calculating the Fisher’s exact statistic using ABL1. Panel A (reproduced from Fig 4) displays the significant SOMDTP nodes for defective ABL1, where Ngene = 48. Collecting the MOA’s for the 11 FDA compounds co-projected to Ngene finds 6 MOAs (Apo, Ho, HSP90, NonCan, PK and BCR-ABL). Panel B in Fig 6 displays the top 10th percentile of all FDA compounds sharing any one of these 6 MOAs, to yield NFDA = 189. Completing the contingency table with their intersection (22, results in a Fisher’s exact score of 1.958262e-09 (logpval(-20.051208)).
Fig 6. Panel A displays the significant SOMDTP nodes for ABL1 (Ngene = 48).

Eleven FDA compounds are co-projected to Ngene; yielding 6 MOAs. The SOMDTP in Panel B displays the top 10th percentile of projections for FDA compounds sharing these MOAs (NFDA = 189). The intersection of Ngene and NFDA = 22, yielding a Fishers exact p-value of 1.958262e-09, log(p-value = -20.05).
Fig 7 displays a bar chart of the log(pvalue) for the 47 genes with significant (p< = 0.05) Fisher’s exact scores, when tested over the complete SOMDTP. Defective genes with the top-most significance scores include MTOR, SPEN, CDC25A, PTK2, TEK, CYP11B1, CYP11B2, NCOR2, PIK3CG, MSH2, APC and MMP1. Note that this subset of defective genes is not exclusively associated with human cancers. Fisher’s exact and chi-square tests for MOAs will be applied to meta-clade groups (A-E). The average log(pvalue) for both statistics will be reported as a contingency score.
Fig 7. Fisher’s exact scores (log(pvalue), pvalue< = 0.05).
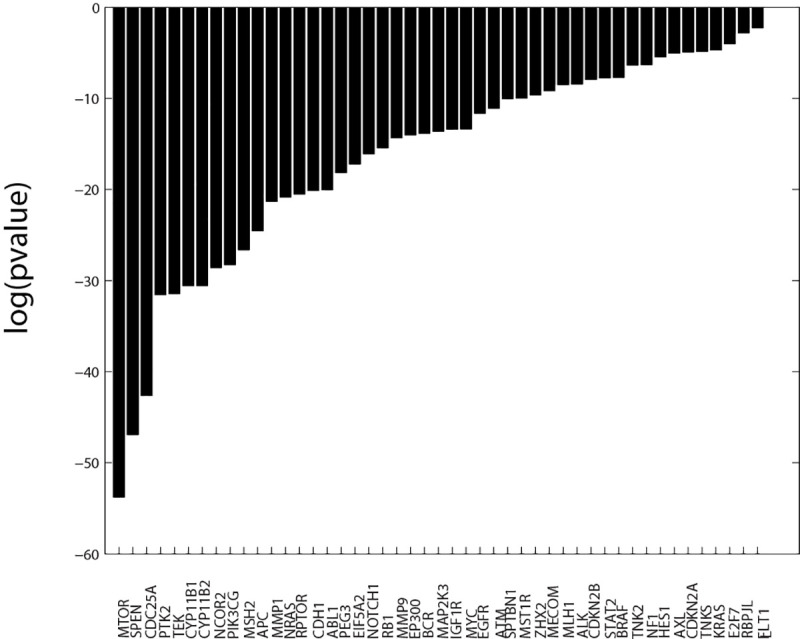
Results are based on classifications using up to the 10th best SOM projection nodes for FDA compounds. Forty-seven defective genes have significant Fisher’s exact scores when tested over the complete SOMDTP.
Results
The multi-tiered approach described in the METHODS builds a framework to achieve the study’s goal of associating chemosensitivity, defective genes and MOA. In summary: chemosensitivity data(n = 46k, GI50NCI60) is clustered as SOMDTP (n = 1232, GI50codebooks), subdivided into meta-clades (n = 28) and 7 meta-clade groups (A-G). Defective genes (n = 368) are analyzed according to significant chemosensitivity on SOMDTP (n = 121, Student’s t/bootstrap) and enrichment for type of MOA of defective genes (n = 47 genes, contingency score; reported as the average log(Fisher’s exact and chi-square scores). Contingency scores will be used to identify significant MOA enrichments for defective genes across meta-clade groups (A-G). The results for SOMDTP clustering will be presented first, followed by the results for MOA enrichments in groups A-G.
Results: SOMDTP
Fig 8 Panel A displays SOMDTP, colored according to similarity of neighboring GI50codebooks; where the most similar GI50codebook neighbors are displayed in deep red and the most dis-similar GI50codebook neighbors appear in bright yellow. The 28 optimal meta-clade boundaries are displayed as a black line, with the boundaries of the 7 meta-clade groups super-imposed as a white line. Two distinctive features characterize SOMDTP. First, the best projections of FDA approved compounds appear as blue hexagons in Fig 8 Panel A, where hexagons are sized according to the number of FDA agents appearing in any SOMDTP node. Inspection finds a general tendency for approved agents to project to SOMDTP nodes with unique patterns (e.g. dissimilar GI50codebooks). Statistical support for this observation is displayed in Fig 8 Panel B, in the form of histograms based on intra-node GI50codebook distances for nodes containing FDA approved agents (top histogram) and lacking FDA approved agents (bottom histogram). A Student’s t-test for the vector distance between these two groups finds a p-value of 5.3e-7, in support of the visual association of FDA compounds and unique (e.g dis-similar) chemosensitivity patterns. Second, compound names for FDA screened agents are listed as a table in Fig 8 Panel C and projected on to SOMDTP in Fig 8 Panel D. A listing of these nodes and their SOMDTP projections also appears in S5 master_appendix sheet appendix_Table_III. In brief, FDA compounds with known MOAs are grouped together, with, for example, nucleic acid targeting agents appearing in the upper right corner of SOMDTP (meta-clade 21), tubulin targeting agents (meta-clades 16 and 18) and defective BRAF targeting agents (meta-clade 19). Collectively, these results support our prior report [6] of associations between NCI60 screened compounds, their MOAs and projections on SOMDTP.
Fig 8. Panel A.

SOMDTP is colored according to similarity of GI50codebooks, where the most similar node neighbors are displayed in deep red and the most dis-similar node neighbors appear in bright yellow (see vertical bar adjacent to SOMDTP). The 28 optimal meta-clade boundaries are displayed as a black line, with the boundaries of the 7 meta-clade groups super-imposed as a white line. FDA approved compounds are projected onto SOMDTP as blue hexagons, where hexagons are sized according to the number of FDA agents appearing in any node. Panel B displays the between node GI50codebook Euclidean distances for nodes with FDA compound projections (top) and without (bottom). Panel C lists FDA compound names grouped by 28 meta-clades. Panel D displays SOMDTP with FDA compounds (blue hexagons), meta-clade boundaries (solid lines) and meta-clade labels as numbers. FDA approved projections to SOMDTP nodes are listed in S5 master_appendix sheet appendix_Table_III.
Results: Group A (meta-clades 1 through 6)
The results for SOM meta-clade group A find twelve defective genes with significant contingency scores (ABL1, ACVR2A, CDC25A, MMP1, MTOR, NCOR2, NF1, PIK3R1, RB1, RPTOR, SOX9 and ZHX2) associated with eleven MOA classes (PK, Ang, Ho, PARP, AM, BCR-ABL, NonCan, Db, HDAC, HYP and Pase). Fig 9 Panel A displays the contingency scores, ordered left to right, from the most to least significance. Fig 9 Panel B displays the SOMDTP projections for these significant defective genes. Projections, colored according the legend, represent instances where significant Student’s t-statistics co-project with compounds having these MOAs. For example, ABL1 projections (blue) appear mainly in the lower right region. Color coding is unique for all defective genes analyzed herein; intended to provide a visual separation for each defective gene. Fig 9 Panel C lists the SOMDTP node counts, ordered from top to bottom and left to right. Fig 9 Panel D displays a histogram for these counts.
Fig 9. Panel A displays the contingency scores, ordered left to right, from the most to least significance.

The horizontal dashed lines represent significance thresholds of p< = 0.05 (lower line) and p< = 0.1 (upper line). Panel B displays the SOMDTP co-projections of significant defective genes and MOAs for FDA compound. Only co-occurrences for SOMDTP projections of FDA compounds are displayed. The SOMDTP region displayed in Panel B represents the boundary for meta-clades 1 through 6 (see the white border in Fig 8 Panel A). Panel C lists the counts for co-occurrence (see S6 master_appendix sheet gp_A). Panel D displays the tabular results in Panel C as a histogram. Node colors for defective genes correspond to the legend inserted into the upper left panel. The counts displayed in Panel C represent the top 10th percentile of SOMDTP co-projections for FDA compounds. A consistent coloring scheme is used for this and all subsequent figures, such that all defective genes presented in the RESULTS are assigned a unique color. S13 master_appendix_sheet gp_A_FDA list the counts for each FDA and MOA entry for these significant genes.
The most frequently appearing defective genes are MMP1; associated mainly with MOAs PK, AM and Ho, and NF1; associated with MOAs PK, BCR-ABL, PARP and Ho. MOA:PK occurs most frequently with MMP1, NF1 and PIK3R1 as the most frequent defective genes. The second highest count is for MOA:Ang, which is associated with defective genes RPTOR, SOX9 and MTOR. The next most frequent counts are associated with MOA:HO(MMP1, NF1, PIK3R1, NCOR2 and ABL1), MOA:PARP(NF1, NCOR2, PIK3R1 and ACVR2A), MOA:AM(MMP1 and ZHX2) and MOA:BCR-ABL(NF1 and PIK3R1). Inspection of Fig 9 Panel D summarizes these results. For example, MOA:PK has MMP1 (teal) and NF1 (light red) as representing the majority of co-projections, while MOA:BCR-ABL is dominated by NF1 (light red).
The common feature of the defective genes associated with SOM meta-clade Group A is their potential to influence the Ras/Raf/MEK/ERK and the PI3K/AKT pathways. The Ras/Raf/MEK/ERK mitogen activated protein kinase (MAPK) cascade is constitutively active and is the dominant pathway driving the production MMP1 [31], the defective gene with the highest SOMDTP node count. MMP1 also modulates cytoskeleton organization, cell motility and additional metastasis signature genes [32] which in turn are mediated by the ERK pathway [33]. In general, the expression of the family of matrix metalloproteinases (MMP) is broadly affected by intracellular signaling via the MAPK family. Targeting the RAF-MEK-ERK mitogen-activated protein kinase cascade is being actively pursued for the treatment of cancer [34].
A direct role of MMP1 on chemosensitivity has not been reported. However, Zhou et al. [35] identify MMP1 as a potential gene conferring resistance of EGFR drugs targeting in non-small cell lung cancer. Rapamycin significantly enhanced the expression of interstitial collagenase (MMP1) at the protein and mRNA levels [36]. An assessment of upregulated expression levels in serous ovarian cancer cell lines by Zhang et al. [37] find matrix metalloproteinase 1 (MMP1) to be among the most upregulated mRNAs in the chemoresistant cell lines. Given that MMP1 is the most frequent defective gene associated with MOA:PK (cf. Fig 9), combined with its role in chemosensitivity, suggests that defective MMP1 may play a role in the weak GI50NCI60 responses to PIK3 and EGFR targeting agents screened in the NCI60.
NF1 has the 2nd highest node count in group A and has links to the MAPK cascade. For example, loss of NF1 gene expression leads to increased RAS activation and hyperactivation of the downstream RAS effectors, including the RAF/MEK/ERK and the PI3K/AKT pathways [38]. Abnormal activation of RAS by defective NF1 is a central driver event in some soft-tissue sarcomas (MPNST). Receptor tyrosine kinases (RTKs), including PDGFRA and EGFR, can activate RAS signaling and downstream factors such as MEK and mTOR. Ki et al. [39] find the addition of mTOR inhibitors to cell lines harboring defective NF1 enhance the activity of DNA targeting agents. Defective genes that impact PI3K-Akt-mTOR signaling could weaken the tumor cell and enhance susceptibility to chemotherapeutic drugs.
A noteworthy entry in S6 master_appendix sheet gp_A_FDA is for Olaparib, MOA:PARP and defective gene NF1. Combination treatment with olaparib and various inhibitors of PD-L1, VEGFR, PI3K, and AKT may effectively inhibit the growth of rapidly proliferating triple negative breast cancer cell lines [40]. A review of candidate synthetic lethality partners to PARP inhibitors in the treatment of ovarian clear cell cancer by Kawahara et al. [41] finds PARP and NF1 to be synthetic lethality pairs [42]. Synthetic lethality (SL) describes the genetic interaction by which the combination of two separately non-lethal mutations results in lethality [43]. Generally, the ablation of two genes located in parallel pathways (leading to cell survival or a common essential product) is one of the important patterns causing synthetic lethality. Synthetic lethality appears to be achieved with combined EGFR and PARP inhibition [44]. SL has recently emerged as a promising new approach to cancer therapy [45].
MOA:Ang ranks 2nd among the MOA’s listed for group A and is associated with defective RPTOR, SOX9 and MTOR. Oncogenic activation of the phosphatidylinositol-3-kinase (PI3K), and mammalian target of rapamycin (MTOR) facilitates tumor formation, disease progression, therapeutic resistance, and the sensitivity of prostate cancer cell lines to PI3K-AKT-mTOR-targeted therapies [46]. SOX9 is reported to promote of tumor growth, proliferation, migration and invasion and the metastasis and regulation of Wnt/β-catenin signalling [47]. Inhibition of SOX9 expression in led to a significant reduction in primary tumor growth, angiogenesis, and metastasis [48]. The full extent of the PI3K-AKT-mTOR signaling network during tumor/angiogenesis, invasive progression and disease recurrence remains to be determined. The existing results link chemosensitivity of MOA:Ang agents to a selective set of defective genes.
Results: Group B(meta-clades 7 through 9)
Eleven MOA classes (PK, Ho, Db, NonCan, Ds, Apo, AM, T2, A7, HDAC, and PARP) are associated with eight defective genes (ZHX2, MECOM, MMP1, EP300, MTOR, BMP7, CYP11B1 and CYP11B2) for SOM meta-clades 7 through 9. ZHX2 has the most and EP300 the least significant contingency scores (Fig 10 Panel A). ZHX2 projects to the central region of group A, while CYP11B2, MECOM and MMP1 project to the perimeter regions (Fig 10 Panel B). Fig 10 Panels C and D indicate that defective genes ZHX2, MECOM, MMP1 and PTK2 and MOAs PK, Ho and Db occupy the most SOMDTP nodes. These defective genes are associated with the GO ontology pathway Regulation_of_Response_to_Stress, with a potential to influence cellular functions such as differentiation and translation. ZHX2 is a member of the zinc fingers and homeoboxes gene family that acts as a transcriptional repressor. Ontology (GO) annotations related to ZHX2 also include DNA-binding_transcription_factor_activity. MECOM (MDS and EVI1 complex locus protein), with the 2nd highest SOMDTP counts, is found to be commonly enriched in cancer cell lines. Makondi et al. [49] suggest that targeting the MAPK signal transduction pathway through the targeting of the MECOM might increase tumor responsiveness to irinotecan treatment. Saito et al. [50] notes that EVI1 alters metabolic programming associated with leukemogenesis and increases sensitivity to L-asparaginase. The 3rd most frequent gene, MMP1, is in the family of matrix metalloproteinases that are involved in the breakdown of extracellular matrix and contribute to metastasis, as noted above. S7 master_appendix sheet gp_B (meta-clades 7 through 9) lists the defective genes with a significant contingency score (p<0.1) for each meta-clade in Group B. Row entries in S14 master_appendix sheet gp_B_FDA list the counts for each FDA and MOA entry for significant defective genes.
Fig 10. Results for group B(meta-clades 7 through 9).

The SOMDTP region displayed in Panel B represents the boundary for meta-clades 7 through 9 (see the white border in Fig 8 Panel A). S7 master_appendix sheet gp_B lists the table in Panel C. See the legend of Fig 9 for details. S14 master_appendix sheet gp_B_FDA lists the FDA compounds associated with these defective genes.
Results: Group C (meta-clades 10 through 15)
Contingency scores order the defective genes as: NOTCH1, RBPJ, IGF1R, PIK3CG, CDKN2A, ATM, NRAS, MSH2, CDKN2B, CDC25A, NCOR2, RPTOR, STAT2, EIF5A2, MYC, SPEN and MTOR (Fig 11 Panel A). IGF1R projects mainly at the perimeter of SOMDTP for group C, while the remaining defective genes project to more central regions (Fig 11 Panel B). Seventeen MOA classes, ordered from most to least node counts, are Ds, PK, HDAC, Apo, Ho, AM, BCR-ABL, A7, NFkB, BRD, Mito, NonCan, PARP, KLF4, PSM, T1 and SMO (Fig 11 Panel C and D).
Fig 11. Results for group C(meta-clades 10 through 15).

The SOMDTP region displayed in Panel B represents the boundary for meta-clades 10 through 15 (see the white border in Fig 8 Panel A). S8 master_appendix sheet gp_C lists the table in Panel C. See the legend of Fig 9 for details. S15 master_appendix sheet gp_C_FDA lists the FDA compounds associated with these defective genes.
The ten most frequent defective genes, IGF1R, CDC25A, NOTCH1, NCOR2, RPTOR, CDKN2A, MSH2, NRAS are associated with MOAs Ds, PK, HDAC, Apo, Ho, AM and BCR-ABL. The salient feature of these defective genes is their role in arresting the cell cycle. Cellular processes involving phosphorylation function to interrupt the cell-cycle, particularly from members of the family of tyrosine kinases. For example insulin-like growth factor 1 receptor (IGF1R) belongs to the large family of tyrosine kinase receptors and is activated by a hormone called insulin-like growth factor 1 (IGF-1) and by a related hormone called IGF-2 [51]. SOMDTP nodes in meta-clades 10 through 15 that are associated with defective IGF1R exist for chemosensitivity mainly to leukemia cell lines. IGF1R is often overexpressed by tumors and mediates proliferation and apoptosis protection [52,53]. As noted earlier [27], evaluation of drug sensitivity for compounds targeting leukemia cell lines has prompted the emergence of IGF1R as a potential therapeutic target for the treatment of leukemia. Weisberg, et al. [54] report that IGF1R protein expression/activity was substantially increased in mutant RAS-expressing cell lines, and suppression of RAS led to decreases in IGF1R. Synergy between MEK and IGF1R inhibitors correlated with induction of apoptosis, inhibition of cell cycle progression, and decreased phospho-S6 and phospho-4E-BP1. They suggested that given the complexity of RAS signaling, it is likely that combinations of targeted agents will be more effective than single agents, inclusive of IGF1R inhibitors.
CDC25A, with the 2nd highest node counts, affects cell proliferation and its expression is thought to be controlled through the PI3K-AKT-MTOR signaling pathway [55]. Sadeghi et al. [56] suggest that CDC25A controls the cell proliferation and tumorigenesis by a change in expression of proteins involved in cyclin D1 regulation and G1/S transition. The finding that defective CDC25A is associated with MOA:PK is consistent with the appearance of pazopanib and axitinib in the FDA compounds listed in S15 master_appendix sheet gp_C_FDA.
The evolutionarily conserved NOTCH family of receptors regulates a myriad of fundamental cellular processes including development, tissue patterning, cell-fate determination, proliferation, differentiation and cell death [57]. The crosstalk among Notch1 (3rd highest node counts) and other prominent molecules/signaling pathways includes DNA damage repair(DDR) [58]. DDR is a complex protein kinase based signaling pathway which is conducted by the members of the phosphoinositide 3-kinase-like kinase (PIKK) family, such as ataxia telangiectasia mutated (ATM). NOTCH1 is a major oncogenic driver in T cell acute lymphoblastic leukemia [59]. NOTCH1 siRNA can effectively inhibit the expression of NOTCH1 gene, inhibit the proliferation of lung cancer A549 cell lines and increase the sensitivity to chemotherapeutic drugs [60]. Of specific interest is the intersection of defective NOTCH1 and the projection for imatinib (S15 master_appendix sheet gp_C_FDA). Aljedai et al. [61] explored the role of NOTCH1 signaling in chronic myeloid leukemia cell lines to find cross-talk between NOTCH1 and BCR-ABL. Their results revealed that imatinib induced BCR-ABL inhibition results in upregulation of NOTCH1 activity. In contrast, inhibition of NOTCH1 leads to hyperactivation of BCR-ABL. They proposed that the antagonistic relationship between NOTCH1 and BCR-ABL in CML suggests a combined inhibition of NOTCH1 and BCR-ABL may provide superior clinical response over tyrosine-kinase inhibitor monotherapy.
CDKN2A, MSH2 and ATM (with the next most frequent node counts) have roles in cell cycle arrest. CDKN2A is capable of inducing cell cycle arrest in the G1 and G2 phases. Gene Ontology (GO) annotations related to CDKN2A include transcription factor binding. MSH2 and ATM are components of the post-replicative DNA mismatch repair system (MMR), whereby activation of checkpoint arrest and homologous DNA repair are necessary for maintenance of genomic integrity during DNA replication [62]. Germ-line mutations of the ataxia telangiectasia mutated (ATM) gene result in the well-characterized ataxia telangiectasia syndrome, which manifests with an increased cancer predisposition. Somatic ATM mutations or deletions are commonly found in lymphoid malignancies. Such mutations may be exploited by existing or emerging targeted therapies that produce synthetic lethal states. Cancers with mutations in genes encoding proteins involved in DNA repair may be more sensitive to treatments that induce synthetic lethality by inducing DNA damage or inhibiting complementary DNA repair mechanisms.
Results: Group D(meta-clades 16 through 18)
Contingency scores order the defective genes as: MAP2K3, PTK2, BRAF, CYP11B1, CYP11B2, MMP9, MYC, FLT1 and RBPJL (Fig 12 Panel A). MYC, BRAF and FLT1 project to mainly non-overlapping SOMDTP regions (Fig 12 Panel B). Eight MOA classes, ordered from most to least node counts, are Tu, HSP90, NonCan, PSM, DB, T1, PK, T2 and Pase (Fig 12 Panels C and D). MOA:Tu dominates these results, while MOA:HSP90, MOA:NonCan and MOA:PSM appear with the next highest node counts. The most frequent defective genes include MYC, RBLJL, FLT1 and MMP9. S16 master_appendix sheet gp_D_FDA lists the FDA compounds associated with these defective genes.
Fig 12. Results for group D(meta-clades 16 through 18).

The SOMDTP region displayed in Panel B represents the boundary for meta-clades 16 through 18 (see the white border in Fig 8 Panel A). S9 master_appendix sheet gp_D lists the table in Panel C. See legend of Fig 9 for additional details. S16 master_appendix sheet gp_D_FDA lists the FDA compounds associated with these defective genes.
Most of the defective genes in group D are involved with the mitotic component of tumor cell proliferation. For example, MYC encodes a nuclear phosphoprotein that has been implicated in the regulation of cell proliferation and the development of human tumors [63] and is regarded as a major determinant of mitotic cell fate [64]. Inhibition of microtubule polymerization has been reported to block mitosis and induce cell death [65]. Conacci-Sorell et al. [66] report the expression of MYC results in the induction of the actin-bundling protein fascin, formation of filopodia, and plays a role in cell survival, autophagy, and motility. MYC also recruits acetyltransferases that modify cytoplasmic proteins, including α-tubulin. Marzo-Mas et al. [67] find the antiproliferative activity of colchicine to inhibit tubulin polymerization to be modulated by the downregulation of c-MYC expression. Alexandrova et al. [68] report that the N-terminal domain of c-MYC associates with alpha-tubulin and microtubules. Marzo-Mas et al. [67] also found that tubulin binding compounds were able to downregulate the expression of the VEGF, hTERT and c-MYC genes. Others [69] have proposed targeting oncogenic MYC as a strategy for cancer treatment, proposing the destruction of a microtubule-bound MYC reservoir during mitosis contributes to vincristine´s anti-cancer activity [70]. Collectively these results support a role of defective MYC in chemosensitivity to tubulin targeting agents.
The 2nd most frequent defective gene is RBPJL. RBPJL binds to DNA sequences almost identical to that bound by the Notch receptor signaling pathway transcription factor recombining binding protein J (RBP-J). A related family member RITA (RBPJ Interacting And Tubulin Associated 1) also acts as a negative regulator of the Notch signaling pathway that induces apoptosis and cell cycle arrest in human hepatocellular carcinoma [71]. Structural and biophysical studies demonstrate that RITA binds RBP-J and biochemical and cellular assays suggest that RITA interacts with additional regions on RBP-J [72]. Emerging evidence reveals Notch as a microtubule dynamics regulator and that activation of Notch signaling results in increased microtubule stability [73]. The RBPJL/RITA association raises the possibility that RITA-mediated regulation of Notch signaling may be influenced by RBPJL and potentially play a role in the chemosensitivity of Tu agents.
The 3rd most frequent defective gene is FLT1 (Fms-related tyrosine kinase (FLT) or VEGF receptor 1). The role of FLT1 in the chemosensitivity of tubulin agents would appear to be unexpected. However, the blockade of VEGFR-1 and VEGFR-2 enhances paclitaxel sensitivity in gastric cancer cell lines [74]. Microtubule-targeted drugs inhibit VEGF Receptor-2 expression by both transcriptional and post-transcriptional mechanisms [75]. Novel anti-mitotics, which target the mitotic spindle through interactions with non-microtubule mitotic mediators like mitotic kinases and kinesins, have been identified and are now in clinical testing [76]. Included in clinical testing are compounds that have low nanomolar potency against ABL, FLT1 and PDGFR [77]. Tumor endothelial cell lines demonstrate a strong activation of VEGF and Notch signaling [78]. VEGF-B is a growth factor that binds FLT1 and is considered the odd member of the VEGF family, with mainly angiogenic and lymphangiogenic activities. VEGF-B has protective effects on neuropathy [79]. FLT1 has been proposed as a prognostic indicator in endometrial carcinoma [80].
The 4th most frequent defective gene, MMP9 (matrix metalloproteinases 9) and its associated vascular endothelial growth factor (VEGF) are critical for tumor vascularization and invasion. A recent study of the expression of MMP-9 and VEGF(FLT1) in breast cancer patients found their correlation significant enough to propose these genes as prognostic indicators [81]. Inspection of these SOM meta-clades finds MOA:PK agents to be located mainly in the upper portion of SOM meta-clades 16 through 18, where defective genes MMP9 and RBPJL also appear. Crizitonib is co-projected to these SOM nodes. Cizitonib is a small molecule TKI that inhibits the activity of the ALK fusion proteins, MET, ROS1, and MST1R (RON) [82,83]. Noteworthy is the impressive clinical responses to crizotinib and other small-molecule drugs inhibiting the ALK tyrosine kinase [84]. Defective MMP9 or RBPLJ may contribute to enhanced crizitonib chemosensitivity.
MOA:HSP90 is the 2nd most frequent MOA class in SOM meta-clades 16 through 18. Several studies have suggested a possible connection between HSP90 and the microtubule cytoskeleton. Weis et al. [85] find that HSP90 protects tubulin against thermal denaturation. Anti-tumor selectivity of a novel Tubulin and HSP90 dual-targeting inhibitor has been identified in non-small cell lung cancer model [86]. The presence of geldanamycin within the list of agents in this SOM region is consistent with this observation. Liu et al. ([87]) find evidence that mis-regulated HSP90 can affect drug sensitivity, an effect proposed to be due the altered regulation of HSP90 client proteins, inclusive of tubulin.
Group E(meta-clades 19 through 20)
Contingency scores order the defective genes as: BRAF, EGFR, JAK3, RPTOR, PIK3CG and SPTBN1 (Fig 13 Panel A). BRAF dominates the central region of SOMDTP for group E (Fig 13 Panel B). Nine MOA classes, ordered from most to least node counts, are Pk, BCR-ABL, HDAC, NonCan, PSM, A7, Ds, HSP90 and Ho (Fig 13 Panels C and D). The most frequent defective gene is BRAF and is associated with the most frequent MOA:PK, followed by MOA:BCR-ABL, MOA_HDAC. PIK3CG, RPTOR and JAK3 are the 2nd, 3rd and 4th ranking defective genes. This SOMDTP region corresponds to the projection of known FDA approved BRAF targeting agents; dabrafenib, hypomethicin, selmutinub and vemurafenib (S17 master_appendix sheet gp E_FDA). These results are consistent with the findings of Ikediobi et al. [4].
Fig 13. Results for group E(meta-clades 19 through 20).

The SOMDTP region displayed in Panel B represents the boundary for meta-clades 19 through 20 (see the white border in Fig 8 Panel A). S10 master_appendix sheet gp_E lists the table in Panel C. See legend to Fig 9 for additional details. S17 master_appendix sheet gp_E_FDA lists the FDA compounds associated with these defective genes.
The association of defective BRAF with compounds that target this condition are well documented [4,88]. Mutant BRAF (v-Raf murine sarcoma viral oncogene homolog B1) inhibitors such as vemurafenib and dabrafenib have achieved unprecedented clinical responses in the treatment of melanomas [89,90]. The association of defective BRAF to MOA:HDAC is consistent with literature reports. Recent studies have shown that histone deacetylase (HDAC) and mutant BRAF (v-Raf murine sarcoma viral oncogene homolog B1) inhibitors synergistically kill melanoma cell lines with activating mutations in BRAF by induction of necrosis [91].
A role for defective PIK3CG is indicated in SOM meta clades 19 through 20 for MOA:PK and MOA:BCR-ABL. The publications from Shi et al. [92], Van Allen et al. [93] and Rizos et al. [94] addressed the roles of PI3K pathway gene’s mutations. Resistance to BRAF inhibitors can be associated with upregulation of the PI3K/AKT pathway, resulting from AKT1/3 mutations and mutations in positive (PIK3CA, PIK3CG) and negative (PIK3R2, PTEN and PHLPP1) regulatory genes [95]. The results in Fig 13 Panels C and D indicate a role for HDAC in BRAF chemosensitivity. Gallagher et al. [96] find that HDAC inhibitors affect BRAF-inhibitor sensitivity by altering PI3K activity.
A role for defective RPTOR is indicated for MOA:BCR-ABL. Drugs simultaneously targeting two or more pathways essential for cancer growth could slow or prevent the development of resistant clones. Puausova et al. [97] identify dual inhibitors of proliferative pathways in human melanoma cell lines bearing the V600E activating mutation of BRAF kinase. They found these inhibitors to simultaneously disrupt the BRAF V600E-driven extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase (MAPK) activity and the mechanistic target of rapamycin complex 1 (mTORC1) signaling in melanoma cell lines, yielding dynamic changes in mTOR(RPTOR) signaling.
The non-receptor tyrosine Janus kinases (JAK) are involved in various processes such as cell growth, development, or differentiation. The result presented here finds an enhanced chemosensitivity to HDAC inhibitors for tumor cell lines with defective JAK3. DUAL kinase and HDAC inhibitors have been developed based on the reasoning that specifically blocking more than one oncogenic pathway simultaneously with a combination of different drugs may be a more effective cancer treatment [98]. Dual inhibitors of Janus kinases and HDAC have been developed [99,100], As an example, Dymock’s group has designed and synthesized a novel series of dual JAK and HDAC inhibitors based on the core features of ruxolitinib [101]. Upregulation of JAK3 has been observed in response to increases of oxygen-containing species following HDAC inhibition [102]. Although the design of dual JAK/HDAC inhibitors was based on simultaneously targeting different oncogenic pathways, a role for defective JAK in chemosensitivity may be important.
Results: Group F(meta-clades 21 through 24)
Contingency scores order the defective genes as: AXL, STAT2, CDKN2A, RPTOR, KRAS and MYC(Fig 14 Panel A). CDKN2A is the dominant defective gene in meta-clade 21 while RPTOR and STAT2 are located primarily in meta-clade 22 (Fig 14 Panel B). Eighteen MOA classes exist, with MOAs appearing with the highest counts all targeting DNA (Ds, T2, A7, Db, Df and A2)(Fig 14 Panels C and D). CDKN2A as the most frequent defective gene, followed by AXL, MYC, STAT2 and KRAS. Meta-clades 21 through 24 represent, by far, the largest number of FDA approved agents. These defective genes affect proliferation largely resulting from their role in targeting DNA, DNA damage repair and phosphorylation. These genes do not overlap with a prior analysis of DNA repair genes in the NCI60 and their predictive value for anticancer drug activity [103].
Fig 14. Results for group F(meta-clades 21 through 24).

The SOMDTP region displayed in Panel B represents the boundary for meta-clades 21 through 24 (see the white border in Fig 8 Panel A). S11 master_appendix sheet gp_F lists the table in Panel C. See legend to Fig 9 for additional details. S18 master_appendix sheet gp_F_FDA lists the FDA compounds associated with these defective genes.
Su et al. [104] report that CDKN2A loss is significantly associated with the sensitivity of CDK4/6 inhibitors (also projected to SOM meta-clade 14). Evidence supports the role of CDKN2A in cell cycle independent functions such as DNA damage repair [105]. CDKN2A also provides instructions for making several proteins, including p16(INK4A) and p14(ARF), which function as tumor suppressors that keep cell lines from growing and dividing too rapidly or in an uncontrolled way. Overexpression of CDKN2A inhibits cell proliferation and invasion, to cause cell cycle arrest in the G1 phase. CDKN2A mediates the AKT–mTOR (RPTOR) signaling pathway by suppressing lactate dehydrogenase (LDHA) [106]. Taken together, these results suggest therapeutic agents that target CDKN2A and RPTOR in cancers that share these defective genes. Consistent with chemosensitivity for FDA compounds in these meta-clades, recent observations report that long term survivorship after high dose DNA damaging chemotherapy with melphalan is compatible with an increased chemosensitivity due to impairment of the DNA repair pathway [107]. Loss of CDK2 presents a different challenge to cell lines, aside from the more conventional role to regulate cyclins, which in turn might lead to altered DNA damage response and checkpoint activation, mutations in DNA repair genes drive cancer development [108,109]. Ras proteins play a crucial role as a central component of the cellular networks controlling a variety of signaling pathways that regulate growth, proliferation, survival, differentiation, adhesion, cytoskeletal rearrangements and motility of a cell [110]. KRAS (Kirsten-rat sarcoma viral oncogene homolog) is a prominent oncogene that has been proven to drive tumorigenesis, modulate numerous genetic regulatory mechanisms including the induction of DNA damage repair pathways [111,112]. Mutant RAS-driven tumorigenesis arises independently of wild-type RAS isoforms, but recent evidence indicates wild-type isoforms are involved. Grabocka and colleagues [113] report how the loss of wild-type RAS alters oncogenic signaling and dampens the DNA-damage response, thereby affecting tumor progression and chemosensitivity. Since the MOA agents listed for SOM meta-clades 21 through 24 have roles in DNA damage, defective CDKN2A, RPTOR and KRAS may contribute to chemosensitivity of tumor cell lines to these agents. While targeting defective KRAS remains elusive [114], small molecule inhibitors are in the pipeline [115]. Exploration of NCI60 screened compounds that project to meta-clades 21 through 24 may provide a starting point for lead discovery.
Pyrazoloacridine, palbociclib, methotrexate, fluorouracil, 8-Chloro-adenosine, pralatrexate, pemetrexed, pelitrexol, by-product_of_CUDC-305, 6-Mercaptopurine and oxaliplatin appear most frequently in the SOM region for group F (S18 master_appendix sheet gp_F_FDA). A study of gastric cancer patients detected a high frequency of mutations in MLL4, ERBB3, FBXW7, MLL3, mtor(RPTOR), NOTCH1, PIK3CA, KRAS, ERBB4 and EGFR [116]. KRAS mutations have been reported as predictors of the response of lung adenocarcinoma patients receiving platinum-based chemotherapy [117,118]. NOTCH1 mutations target KRAS mutant CD8+ cells to contribute to their leukemogenic transformation [119,120]. Notable in the list of FDA approved agents associated with SOM meta-clades 21 through 24 is oxaliplatin. Oxaliplatin-based chemotherapy is more beneficial in KRAS mutant than in KRAS wild-type metastatic colorectal cancer [121]. SOM meta-clade 21 is the location of cytarabine (ara-C) and is consistent with the conclusion of Ahmad et al [24] that adult AML patients carrying defective KRAS benefit from higher ara-C doses more than wt KRAS patients. Enhanced chemosensitivity of tumor cell lines with defective KRAS may represent a link to these observations.
Results: Group G(meta-clades 25 through 28)
Contingency scores order the defective genes as PEG3, ABL1 and PIK3CG as the most significant and MTOR, KRAS and NRAS as the genes with the least significance (Fig 15 Panel A). PIK3CG represents the largest count of SOMDTP projections, located mainly in the central region of meta-clades 25 through 28. MTOR, KRAS and NRAS are located mostly in the bottom of this region (Fig 15 Panel B). Twelve MOA classes exist, with MOAs appearing with the highest counts as Ds, Apo, and PK (Fig 15 Panels C and D). The most frequently occurring defective genes are PIK3CG, NRAS, PTK2 and ABL1 (Fig 15 Panels C and D). The defective genes in meta-clade 25 through 28 represent an amalgamation of many of the previous meta-clade groups, where sets of defective genes were involved in cellular processes of phosphorylation and progression through the cell cycle for proliferation. Consequently, many of these defective genes have been previously discussed, with the exceptions of NRAS and PTK2.
Fig 15. Results for group G(meta-clades 25 through 28).

The SOMDTP region displayed in Panel B represents the boundary for meta-clades 25 through 28 (see the white border in Fig 8 Panel A). S12 master_appendix sheet gp_G lists the table in Panel C. See legend to Fig 9 for additional details. S19 master_appendix sheet gp_G_FDA lists the FDA compounds associated with these defective genes.
NRAS (ranked 2nd by node counts) is one of the most common targets of oncogenic signaling mutations in hematologic malignancies. Even with the challenge of directly targeting mutant RAS oncoproteins, mitogen-activated protein kinase (MAPK) inhibition has been shown to reduce leukocytosis by targeting the downstream pathway of NRAS [122]. As noted earlier, combinations of targeted agents may not supersede conventional cytotoxic regimens, however combinations may enhance treatment efficacy. Identifying compounds that target defective NRAS and other compounds that target defective PTK2, ABL1 or PIK3CG, in the case of meta-clades 25 through 28, may offer effective combination therapies. Without doubt, large numbers of molecular pathways are likely to be synergistically involved in cancer biology, the contribution of each pathway may be different and identifying which combinations to select will be experimentally exhausting. Bioinformatic approached as discussed herein may offer useful clues.
PTK2 (ranked 3rd by node counts) is a non-receptor protein-tyrosine kinase with functions that include cell migration, reorganization of the actin cytoskeleton, cell cycle progression, cell proliferation and apoptosis through kinase-dependent and -independent mechanisms [123]. It is a member of the FAK (focal adhesion kinase) subfamily of protein tyrosine kinases and is listed as a transcriptional regulator. FAKs are reported to modulate chemosensitivity by altering chemokine production [31]. Enhanced chemosensitivity to gemcitabine has been reported with interference of FAKs [124]. Because of the involvement of PTK2(FAK) in many cancers, drugs that inhibit FAK are being sought and evaluated [125]. A screen to identify mechanisms of bleomycin resistance identified Sky1, PTK2 and Agp2 as determinants of chemosensitivity [126].
Discussion
The development of rational strategies for targeted cancer therapy will require integrative analysis of data derived from diverse sources including, but not exclusive to, large-scale, publicly available, pre-clinical and clinical small-molecule screening and genomic data. A widely accepted challenge of linking screening and genomic data is how to gain molecular insight into the MOA(s) of active compounds. Not unexpectedly, the range of potentially important links is enormous; yielding massive challenges to the development of statistical/computational bioinformatic tools that assist integrative analyses. Advances have been made by focusing studies on fewer compounds (24 compounds in the CCLE [127] or approved FDA compounds [128]) or by studying small numbers of driver or mutated genes [129].
The results of the present study demonstrate the power of combining genomic data and small-molecule screens of FDA compounds in the NCI60 to provide mechanistic clues about compound activity. These results reveal coarse-grained associations between chemosensitivity of target-directed FDA agents towards tumor cell lines harboring specific genetic defects. SOM clustering finds seven regions of GI50NCI60 responses, broadly assigned to FDA MOA classes that target, not exclusively, tubulin, BRAF mutations, RAF/MEK/ERK/mTOR and the PI3K/AKT pathways, DNA or protein synthesis pathways, the cell cycle and are associated with a relatively unique set of defective genes for each MOA class. Salient associations include the role of defective MYC for tubulin targeting agents, defective CDKN2A, NRAS and KRAS for DNA damaging/targeting agents and the role of defective NOTCH1 for mutant BRAF targeting agents. Remarkably, nearly half of the defective genes reported herein also appear in Ikediobi et al. [4], albeit using very different methods.
The results described here may be applied to future pre-clinical studies. Notably there are exploitable instances of enhanced chemosensitivity of compound MOA’s for a few defective genes. Specifically, there is support for synthetically lethal defective genes as contributing to chemosensitivity. Defective genes exist withing the NCI60 as doublets, triplets, quartets, etc., and a subset of these genes are associated with tumor cell lines that exhibit chemosensitivity. Exploiting chemosensitive SOMDTP nodes associated with tumor cell lines having more than one defective gene, that are also associated with numerous screened compounds, may identify additional synthetic lethal strategies. The notion of targeting parallel pathways can be extend beyond synthetically lethal genes. Combining agents with enhanced chemosensitivity against one defective gene, and its related cellular pathways, with other agents showing enhanced chemosensitivity towards other defective genes in alternative pathways, may enhance the efficacy of each agent. For example, each SOMDTP node with significant chemosensitivity for one defective gene includes many NSCs with similar GI50NCI60 responses, inclusive of FDA compounds. Combinations of NSCs from SOMDTP nodes also exhibiting differential chemosensitivity for one or more defective gene in parallel pathways may be considered for experimental testing. The goal would be to identify combinations of NSCs that separately target parallel cellular pathways to determine whether their combination would enhance individual efficacies. The bioinformatic analysis described herein may provide clues for experimental pre-clinical testing of possible drug combinations.
Important caveats underly the interpretation of the results presented here. First, links of defective genes to chemosensitivity are not revealed in a clear-cut manner. Rarely is chemosensitivity associated only to tumor cell lines harboring defective genes. Chemosensitivity also exists within tumor cell lines lacking defective genes (cf. Fig 3). Consequently, while gene-drug associations may provide a genetic basis for drug selection [130], there is clear evidence herein that additional, not well understood, factors are in play. Second, combinations of defective genes appear to play a role in chemosensitivity. For example, 44 defective genes are listed in the tables provided in each of the RESULTS subsections. Eighty-eight percent of these genes are listed only once (N = 24) or twice (N = 14) across the seven meta-clade groups. This result is an indication that relatively few defective genes contribute to enhanced chemosensitivity across meta-clade groups. In contrast, only two (RPTOR and MTOR) and three genes (PIK3CG, MYC and CDC25A) appear jointly in four or three of the seven meta-clade groups, respectively. Consequently, identifying a single defective gene as responsible for chemosensitivity may be rare; while combinations with genes commonly labeled as cancer genes may be more likely. Third, the 44 defective genes listed in the RESULTS subsections can be compared to current compendia of cancer gene mutations derived from human studies. The Cancer Genome Interpreter [131] has been developed to classify protein-coding somatic mutations and copy number variants into predicted passenger or known/predicted oncogenic mutations. Half (N = 22) of the defective genes listed here are identified by the Cancer Genome Interpreter’s encyclopedia of patient-derived tumor xenografts (PDX) as driver mutations. A recent report using driver mutation patterns for prioritization of personalized cancer therapy [132] finds nearly 20% of their 39 tumor biomarkers to be included in this set of defective genes. Although the defective genes listed here were derived from novel applications of bioinformatic tools, these results find support within other databases. The absence of overlapping genes suggests potentially important roles for non-driver genes in chemosensitivity. Fourth, global analysis of modest to large scale genomic and screening data offers only one perspective. The genetic make-up of the NCI60 represents only a snapshot of data for a small number of tumor cell lines. The universal application of results derived from the NCI60 may be relevant only in the rare instance that another tumor cell matches the genetic makeup of any NCI60 cell. This does not, however, rule out analyses, parallel to that presented here, that jointly examine existing and new data. Fourth, the absence of defective TP53 in these results has not gone undetected. Most NCI60 tumor cell lines harbor defective TP53. As a result, establishing a statistically significant Student’s t-test for selective chemosensitivity fails mainly due to too few responses of tumor cell lines lacking defective TP53. Extending the data analysis to more tumor cell lines, lacking mutant TP53, may prove helpful. While addressing each of these caveats is massively challenging, resolution of each issue contributes to the understanding of preclinical screening results derived from a small set of human tumor cell lines.
In summary, the challenge of finding meaningful results within complex and noisy data has been proposed using contemporary data and state-ot-the-art statistical tools. This global analysis of multiple datasets, overlapping in their origins within the NCI60, has provided a unique perspective for associations of chemosensitivity, defective genes and MOAs.
Supporting information
(TIF)
(TIF)
(TIF)
(TIF)
(DOCX)
(DOCX)
Acknowledgments
I would like to thank Drs. Ruili Huang and John Beutler for their extremely helpful comments provided during the preparation of this manuscript. I would also extend a special note of appreciation to the reviewers for their thoughtful suggestions.
Abbreviations
- NCI
National Cancer Institute
- FDA
Food and Drug Administration
- GI50
tumor cell Growth Inhibition
- SOM
Self Organizing Map
- DTP
Developmental Therapeutics Program
- MOA
Mechanism of Action
- NSC
Cancer Chemotherapy National Service Center
- MUT
gene mutation
- CN
copy number alteration
- FUSION
gene fusion or splice
- Tu
tubulin
- T1
topoisomerase I
- T2
topoisomerase II
- A
alkylating agent
- D
DNA interacting agent
- PK
protein kinase
- Apo
apoptosis
- Ho
hormone
- HDAC
histone deacetylase
- HSP90
heat shock protein 90
- PSM
proteasome
- GI50codebook
vector of tumor cell responses for each SOM node
- GI50component
SOM response for each tumor cell
Data Availability
All data are publicly available. Specific data related to this paper appear in the Supporting Information files.
Funding Statement
This project was funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contracts No. HHSN261200800001E and HHSN261201700007I. There was no additional external funding received for this study.
References
- 1.Poliakov E, Cooper DN, Stepchenkova EI, Rogozin IB. Genetics in genomic era. Genet Res Int. 2015;2015:364960. Epub 2015/04/18. 10.1155/2015/364960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. Epub 2011/03/08. 10.1016/j.cell.2011.02.013 . [DOI] [PubMed] [Google Scholar]
- 3.Hoelder S, Clarke PA, Workman P. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol Oncol. 2012;6(2):155–76. Epub 2012/03/24. 10.1016/j.molonc.2012.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O’Meara S, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5(11):2606–12. Epub 2006/11/08. 10.1158/1535-7163.MCT-06-0433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chabner BA. NCI-60 Cell Line Screening: A Radical Departure in its Time. J Natl Cancer Inst. 2016;108(5). Epub 2016/01/13. 10.1093/jnci/djv388 . [DOI] [PubMed] [Google Scholar]
- 6.Rabow AA, Shoemaker RH, Sausville EA, Covell DG. Mining the National Cancer Institute’s tumor-screening database: identification of compounds with similar cellular activities. J Med Chem. 2002;45(4):818–40. Epub 2002/02/08. 10.1021/jm010385b . [DOI] [PubMed] [Google Scholar]
- 7.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. Epub 2012/05/17. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. Epub 2013/04/04. 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reinhold WC, Sunshine M, Varma S, Doroshow JH, Pommier Y. Using CellMiner 1.6 for Systems Pharmacology and Genomic Analysis of the NCI-60. Clin Cancer Res. 2015;21(17):3841–52. Epub 2015/06/07. 10.1158/1078-0432.CCR-15-0335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manjunath M, Zhang Y, Yeo SH, Sobh O, Russell N, Followell C, et al. ClusterEnG: an interactive educational web resource for clustering and visualizing high-dimensional data. PeerJ Comput Sci. 2018;4. Epub 2018/01/01. 10.7717/peerj-cs.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohonen T. The self-organizing map. Proceedings of the IEEE. 1990;78(9):1464–80. 10.1109/5.58325 [DOI] [Google Scholar]
- 12.Kohonen T. Essentials of the self-organizing map. Neural Netw. 2013;37:52–65. Epub 2012/10/17. 10.1016/j.neunet.2012.09.018 . [DOI] [PubMed] [Google Scholar]
- 13.Epanechnikov VA. Non-Parametric Estimation of a Multivariate Probability Density. 1969;14(1):153–8. 10.1137/1114019 [DOI] [Google Scholar]
- 14.Monks A, Zhao Y, Hose C, Hamed H, Krushkal J, Fang J, et al. The NCI Transcriptional Pharmacodynamics Workbench: A Tool to Examine Dynamic Expression Profiling of Therapeutic Response in the NCI-60 Cell Line Panel. Cancer Res. 2018;78(24):6807–17. Epub 2018/10/26. 10.1158/0008-5472.CAN-18-0989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinstein JN. Spotlight on molecular profiling: "Integromic" analysis of the NCI-60 cancer cell lines. Mol Cancer Ther. 2006;5(11):2601–5. Epub 2006/11/08. 10.1158/1535-7163.MCT-06-0640 . [DOI] [PubMed] [Google Scholar]
- 16.Ratnayake R, Covell D, Ransom TT, Gustafson KR, Beutler JA. Englerin A, a selective inhibitor of renal cancer cell growth, from Phyllanthus engleri. Org Lett. 2009;11(1):57–60. Epub 2008/12/09. 10.1021/ol802339w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tibshirani R, Walther G, Hastie T. Estimating the number of clusters in a data set via the gap statistic. J Roy Stat Soc B. 2001;63:411–23. 10.1111/1467-9868.00293 WOS:000168837200013. [DOI] [Google Scholar]
- 18.Bodenhofer U, Kothmeier A, Hochreiter S. APCluster: an R package for affinity propagation clustering. Bioinformatics. 2011;27(17):2463–4. Epub 2011/07/09. 10.1093/bioinformatics/btr406 . [DOI] [PubMed] [Google Scholar]
- 19.Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22(12):1540–2. Epub 2006/04/06. 10.1093/bioinformatics/btl117 . [DOI] [PubMed] [Google Scholar]
- 20.Lawrence ND. A Unifying Probabilistic Perspective for Spectral Dimensionality Reduction: Insights and New Models2010 October 01, 2010:[arXiv:1010.4830 p.]. Available from: https://ui.adsabs.harvard.edu/abs/2010arXiv1010.4830L.
- 21.Gansner ER, Hu Y, Kobourov S, editors. GMap: Visualizing graphs and clusters as maps. 2010 IEEE Pacific Visualization Symposium (PacificVis); 2010 2–5 March 2010.
- 22.Diaconis P, Efron B. Computer-intensive methods in statistics. Sci Am; (United States). 1983:Medium: X; Size: Pages: 116–30. [Google Scholar]
- 23.Langham J. Ranking small molecules by how much they preferentially inhibit the growth of cancer cell lines with either BRAF or KRAS oncogene mutations. PeerJ Preprints; 2014. [Google Scholar]
- 24.Ahmad EI, Gawish HH, Al Azizi NM, Elhefni AM. The prognostic impact of K-RAS mutations in adult acute myeloid leukemia patients treated with high-dose cytarabine. Onco Targets Ther. 2011;4:115–21. Epub 2011/07/28. 10.2147/OTT.S12602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whyte DB, Holbeck SL. Correlation of PIK3Ca mutations with gene expression and drug sensitivity in NCI-60 cell lines. Biochem Biophys Res Commun. 2006;340(2):469–75. Epub 2005/12/27. 10.1016/j.bbrc.2005.12.025 . [DOI] [PubMed] [Google Scholar]
- 26.Vishwamitra D, George SK, Shi P, Kaseb AO, Amin HM. Type I insulin-like growth factor receptor signaling in hematological malignancies. Oncotarget. 2017;8(1):1814–44. Epub 2016/09/24. 10.18632/oncotarget.12123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang F, Chang H, Greer A, Hillerman S, Reeves KA, Hurlburt W, et al. IRS2 copy number gain, KRAS and BRAF mutation status as predictive biomarkers for response to the IGF-1R/IR inhibitor BMS-754807 in colorectal cancer cell lines. Mol Cancer Ther. 2015;14(2):620–30. Epub 2014/12/21. 10.1158/1535-7163.MCT-14-0794-T . [DOI] [PubMed] [Google Scholar]
- 28.Jafari M, Ansari-Pour N. Why, When and How to Adjust Your P Values? Cell J. 2019;20(4):604–7. Epub 2018/08/21. 10.22074/cellj.2019.5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological). 1995;57(1):289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 30.Efron B. A 250-year argument: Belief, behavior, and the bootstrap. Bulletin of the American Mathematical Society. 2012;50(1):129–46. 10.1090/s0273-0979-2012-01374-5 [DOI] [Google Scholar]
- 31.FAK Modulates Chemosensitivity via NF-κB–Dependent Cytokine Production. Cancer Discovery. 2014;4(10):OF7–OF. 10.1158/2159-8290.Cd-rw2014-167 [DOI] [Google Scholar]
- 32.Choi C, Helfman DM. The Ras-ERK pathway modulates cytoskeleton organization, cell motility and lung metastasis signature genes in MDA-MB-231 LM2. Oncogene. 2014;33(28):3668–76. 10.1038/onc.2013.341 [DOI] [PubMed] [Google Scholar]
- 33.Huntington JT, Shields JM, Der CJ, Wyatt CA, Benbow U, Slingluff CL, et al. Overexpression of Collagenase 1 (MMP-1) Is Mediated by the ERK Pathway in Invasive Melanoma Cells: ROLE OF BRAF MUTATION AND FIBROBLAST GROWTH FACTOR SIGNALING*. Journal of Biological Chemistry. 2004;279(32):33168–76. 10.1074/jbc.M405102200 [DOI] [PubMed] [Google Scholar]
- 34.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–310. 10.1038/sj.onc.1210422 [DOI] [PubMed] [Google Scholar]
- 35.Zhou H, Xiang Q, Hu C, Zhang J, Zhang Q, Zhang R. Identification of MMP1 as a potential gene conferring erlotinib resistance in non-small cell lung cancer based on bioinformatics analyses. Hereditas. 2020;157(1):32. Epub 2020/07/25. 10.1186/s41065-020-00145-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poulalhon N, Farge D, Roos N, Tacheau C, Neuzillet C, Michel L, et al. Modulation of collagen and MMP-1 gene expression in fibroblasts by the immunosuppressive drug rapamycin. A direct role as an antifibrotic agent? J Biol Chem. 2006;281(44):33045–52. Epub 2006/08/18. 10.1074/jbc.M606366200 . [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Jiang H, Xie T, Zheng J, Tian Y, Li R, et al. Differential Expression and Alternative Splicing of Transcripts Associated With Cisplatin-Induced Chemoresistance in Nasopharyngeal Carcinoma. Front Genet. 2020;11:52. Epub 2020/03/13. 10.3389/fgene.2020.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ricker CA, Pan Y, Gutmann DH, Keller C. Challenges in Drug Discovery for Neurofibromatosis Type 1-Associated Low-Grade Glioma. Front Oncol. 2016;6:259. Epub 2017/01/10. 10.3389/fonc.2016.00259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ki DH, Oppel F, Durbin AD, Look AT. Mechanisms underlying synergy between DNA topoisomerase I-targeted drugs and mTOR kinase inhibitors in NF1-associated malignant peripheral nerve sheath tumors. Oncogene. 2019;38(39):6585–98. Epub 2019/08/25. 10.1038/s41388-019-0965-5 . [DOI] [PubMed] [Google Scholar]
- 40.Hwang SY, Park S, Kwon Y. Recent therapeutic trends and promising targets in triple negative breast cancer. Pharmacol Ther. 2019;199:30–57. Epub 2019/03/03. 10.1016/j.pharmthera.2019.02.006 . [DOI] [PubMed] [Google Scholar]
- 41.Kawahara N, Ogawa K, Nagayasu M, Kimura M, Sasaki Y, Kobayashi H. Candidate synthetic lethality partners to PARP inhibitors in the treatment of ovarian clear cell cancer. Biomed Rep. 2017;7(5):391–9. Epub 2017/11/08. 10.3892/br.2017.990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye H, Zhang X, Chen Y, Liu Q, Wei J. Ranking novel cancer driving synthetic lethal gene pairs using TCGA data. Oncotarget. 2016;7(34):55352–67. Epub 2016/07/21. 10.18632/oncotarget.10536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nijman SM. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011;585(1):1–6. Epub 2010/11/26. 10.1016/j.febslet.2010.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nowsheen S, Cooper T, Stanley JA, Yang ES. Synthetic lethal interactions between EGFR and PARP inhibition in human triple negative breast cancer cells. PLoS One. 2012;7(10):e46614. Epub 2012/10/17. 10.1371/journal.pone.0046614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nijman SM, Friend SH. Cancer. Potential of the synthetic lethality principle. Science. 2013;342(6160):809–11. Epub 2013/11/16. 10.1126/science.1244669 . [DOI] [PubMed] [Google Scholar]
- 46.Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int J Mol Sci. 2020;21(12). ARTN 4507 10.3390/ijms21124507. WOS:000549441500001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grimm D, Bauer J, Wise P, Krüger M, Simonsen U, Wehland M, et al. The role of SOX family members in solid tumours and metastasis. Seminars in Cancer Biology. 2020;67:122–53. 10.1016/j.semcancer.2019.03.004 [DOI] [PubMed] [Google Scholar]
- 48.Camaj P, Jäckel C, Krebs S, De Toni EN, Blum H, Jauch KW, et al. Hypoxia-independent gene expression mediated by SOX9 promotes aggressive pancreatic tumor biology. Mol Cancer Res. 2014;12(3):421–32. Epub 2013/12/05. 10.1158/1541-7786.MCR-13-0351 . [DOI] [PubMed] [Google Scholar]
- 49.Makondi PT, Chu CM, Wei PL, Chang YJ. Prediction of novel target genes and pathways involved in irinotecan-resistant colorectal cancer. PLoS One. 2017;12(7):e0180616. Epub 2017/07/28. 10.1371/journal.pone.0180616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saito Y, Sawa D, Kinoshita M, Yamada A, Kamimura S, Suekane A, et al. EVI1 triggers metabolic reprogramming associated with leukemogenesis and increases sensitivity to L-asparaginase. Haematologica. 2020;105(8):2118–29. Epub 2019/10/28. 10.3324/haematol.2019.225953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Badzio A, Wynes MW, Dziadziuszko R, Merrick DT, Pardo M, Rzyman W, et al. Increased insulin-like growth factor 1 receptor protein expression and gene copy number in small cell lung cancer. J Thorac Oncol. 2010;5(12):1905–11. Epub 2010/12/03. 10.1097/JTO.0b013e3181f38f57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riedemann J, Macaulay VM. IGF1R signalling and its inhibition. Endocr Relat Cancer. 2006;13 Suppl 1:S33–43. Epub 2007/01/30. 10.1677/erc.1.01280 . [DOI] [PubMed] [Google Scholar]
- 53.Yuan J, Yin Z, Tao K, Wang G, Gao J. Function of insulin-like growth factor 1 receptor in cancer resistance to chemotherapy. Oncol Lett. 2018;15(1):41–7. Epub 2017/12/30. 10.3892/ol.2017.7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weisberg E, Nonami A, Chen Z, Nelson E, Chen Y, Liu F, et al. Upregulation of IGF1R by mutant RAS in leukemia and potentiation of RAS signaling inhibitors by small-molecule inhibition of IGF1R. Clin Cancer Res. 2014;20(21):5483–95. Epub 2014/09/05. 10.1158/1078-0432.CCR-14-0902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brenner AK, Reikvam H, Lavecchia A, Bruserud O. Therapeutic targeting the cell division cycle 25 (CDC25) phosphatases in human acute myeloid leukemia—the possibility to target several kinases through inhibition of the various CDC25 isoforms. Molecules. 2014;19(11):18414–47. Epub 2014/11/15. 10.3390/molecules191118414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sadeghi H, Golalipour M, Yamchi A, Farazmandfar T, Shahbazi M. CDC25A pathway toward tumorigenesis: Molecular targets of CDC25A in cell-cycle regulation. J Cell Biochem. 2019;120(3):2919–28. Epub 2018/11/18. 10.1002/jcb.26838 . [DOI] [PubMed] [Google Scholar]
- 57.Previs RA, Coleman RL, Harris AL, Sood AK. Molecular pathways: translational and therapeutic implications of the Notch signaling pathway in cancer. Clin Cancer Res. 2015;21(5):955–61. Epub 2014/11/13. 10.1158/1078-0432.CCR-14-0809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Majidinia M, Darband SG, Kaviani M, Nabavi SM, Jahanban-Esfahlan R, Yousefi B. Cross-regulation between Notch signaling pathway and miRNA machinery in cancer. DNA Repair (Amst). 2018;66–67:30–41. Epub 2018/05/04. 10.1016/j.dnarep.2018.04.002 . [DOI] [PubMed] [Google Scholar]
- 59.Choi SH, Severson E, Pear WS, Liu XS, Aster JC, Blacklow SC. The common oncogenomic program of NOTCH1 and NOTCH3 signaling in T-cell acute lymphoblastic leukemia. PLoS One. 2017;12(10):e0185762. Epub 2017/10/13. 10.1371/journal.pone.0185762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng F, Yang ZF, Sun CQ. The role of Notch1 genes in lung cancer A594 cells and the impact on chemosensitivity. Eur Rev Med Pharmacol Sci. 2017;21(11):2659–64. Epub 2017/07/06. . [PubMed] [Google Scholar]
- 61.Aljedai A, Buckle AM, Hiwarkar P, Syed F. Potential role of Notch signalling in CD34+ chronic myeloid leukaemia cells: cross-talk between Notch and BCR-ABL. PLoS One. 2015;10(4):e0123016. Epub 2015/04/08. 10.1371/journal.pone.0123016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi M, Kipps T, Kurzrock R. ATM Mutations in Cancer: Therapeutic Implications. Mol Cancer Ther. 2016;15(8):1781–91. Epub 2016/07/15. 10.1158/1535-7163.MCT-15-0945 . [DOI] [PubMed] [Google Scholar]
- 63.Niklinski J, Claassen G, Meyers C, Gregory MA, Allegra CJ, Kaye FJ, et al. Disruption of Myc-tubulin interaction by hyperphosphorylation of c-Myc during mitosis or by constitutive hyperphosphorylation of mutant c-Myc in Burkitt’s lymphoma. Mol Cell Biol. 2000;20(14):5276–84. Epub 2000/06/24. 10.1128/mcb.20.14.5276-5284.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Topham C, Tighe A, Ly P, Bennett A, Sloss O, Nelson L, et al. MYC Is a Major Determinant of Mitotic Cell Fate. Cancer Cell. 2015;28(1):129–40. Epub 2015/07/16. 10.1016/j.ccell.2015.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996;56(4):816–25. Epub 1996/02/15. . [PubMed] [Google Scholar]
- 66.Conacci-Sorrell M, Ngouenet C, Anderson S, Brabletz T, Eisenman RN. Stress-induced cleavage of Myc promotes cancer cell survival. Genes Dev. 2014;28(7):689–707. Epub 2014/04/04. 10.1101/gad.231894.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marzo-Mas A, Falomir E, Murga J, Carda M, Marco JA. Effects on tubulin polymerization and down-regulation of c-Myc, hTERT and VEGF genes by colchicine haloacetyl and haloaroyl derivatives. Eur J Med Chem. 2018;150:591–600. Epub 2018/03/20. 10.1016/j.ejmech.2018.03.019 . [DOI] [PubMed] [Google Scholar]
- 68.Alexandrova N, Niklinski J, Bliskovsky V, Otterson GA, Blake M, Kaye FJ, et al. The N-terminal domain of c-Myc associates with alpha-tubulin and microtubules in vivo and in vitro. Mol Cell Biol. 1995;15(9):5188–95. Epub 1995/09/01. 10.1128/mcb.15.9.5188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5. Epub 2018/03/13. 10.1038/s41392-018-0008-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Becker S, Kiecke C, Schafer E, Sinzig U, Deuper L, Trigo-Mourino P, et al. Destruction of a Microtubule-Bound MYC Reservoir during Mitosis Contributes to Vincristine’s Anticancer Activity. Mol Cancer Res. 2020;18(6):859–72. Epub 2020/03/13. 10.1158/1541-7786.MCR-19-1203 . [DOI] [PubMed] [Google Scholar]
- 71.Wang H, Yang Z, Liu C, Huang S, Wang H, Chen Y, et al. RBP-J-interacting and tubulin-associated protein induces apoptosis and cell cycle arrest in human hepatocellular carcinoma by activating the p53-Fbxw7 pathway. Biochem Biophys Res Commun. 2014;454(1):71–7. Epub 2014/12/03. 10.1016/j.bbrc.2014.10.023 . [DOI] [PubMed] [Google Scholar]
- 72.Tabaja N, Yuan Z, Oswald F, Kovall RA. Structure-function analysis of RBP-J-interacting and tubulin-associated (RITA) reveals regions critical for repression of Notch target genes. J Biol Chem. 2017;292(25):10549–63. Epub 2017/05/11. 10.1074/jbc.M117.791707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonini SA, Ferrari-Toninelli G, Montinaro M, Memo M. Notch signalling in adult neurons: a potential target for microtubule stabilization. Ther Adv Neurol Disord. 2013;6(6):375–85. Epub 2013/11/15. 10.1177/1756285613490051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hwang JE, Lee JH, Park MR, Kim DE, Bae WK, Shim HJ, et al. Blockade of VEGFR-1 and VEGFR-2 enhances paclitaxel sensitivity in gastric cancer cells. Yonsei Med J. 2013;54(2):374–80. Epub 2013/02/01. 10.3349/ymj.2013.54.2.374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meissner M, Pinter A, Michailidou D, Hrgovic I, Kaprolat N, Stein M, et al. Microtubule-targeted drugs inhibit VEGF receptor-2 expression by both transcriptional and post-transcriptional mechanisms. J Invest Dermatol. 2008;128(8):2084–91. Epub 2008/03/08. 10.1038/jid.2008.37 . [DOI] [PubMed] [Google Scholar]
- 76.Harrison MR, Holen KD, Liu G. Beyond taxanes: a review of novel agents that target mitotic tubulin and microtubules, kinases, and kinesins. Clin Adv Hematol Oncol. 2009;7(1):54–64. Epub 2009/03/11. [PMC free article] [PubMed] [Google Scholar]
- 77.Jackson JR, Patrick DR, Dar MM, Huang PS. Targeted anti-mitotic therapies: can we improve on tubulin agents? Nat Rev Cancer. 2007;7(2):107–17. Epub 2007/01/26. 10.1038/nrc2049 . [DOI] [PubMed] [Google Scholar]
- 78.Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun. 2020;11(1):2285. Epub 2020/05/10. 10.1038/s41467-020-16164-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guaiquil VH, Pan Z, Karagianni N, Fukuoka S, Alegre G, Rosenblatt MI. VEGF-B selectively regenerates injured peripheral neurons and restores sensory and trophic functions. Proc Natl Acad Sci U S A. 2014;111(48):17272–7. Epub 2014/11/19. 10.1073/pnas.1407227111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fine BA, Valente PT, Feinstein GI, Dey T. VEGF, flt-1, and KDR/flk-1 as prognostic indicators in endometrial carcinoma. Gynecol Oncol. 2000;76(1):33–9. Epub 2000/01/06. 10.1006/gyno.1999.5658 . [DOI] [PubMed] [Google Scholar]
- 81.Jobim FC, Schwartsmann G, Xavier NL, Uchoa Dde M, Saciloto M, Chemello N. [Expression of MMP-9 and VEGF in breast cancer: correlation with other prognostic indicators]. Rev Bras Ginecol Obstet. 2008;30(6):287–93. Epub 2009/01/15. 10.1590/s0100-72032008000600004 . [DOI] [PubMed] [Google Scholar]
- 82.Clark JW, Camidge DR, Kwak EL, Maki RG, Shapiro GI, Chen I, et al. Dose-escalation trial of the ALK, MET & ROS1 inhibitor, crizotinib, in patients with advanced cancer. Future Oncol. 2020;16(1):4289–301. Epub 2019/11/30. 10.2217/fon-2019-0653 . [DOI] [PubMed] [Google Scholar]
- 83.Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371(21):1963–71. Epub 2014/09/30. 10.1056/NEJMoa1406766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Workman P, van Montfort R. EML4-ALK fusions: propelling cancer but creating exploitable chaperone dependence. Cancer Discov. 2014;4(6):642–5. Epub 2014/06/04. 10.1158/2159-8290.CD-14-0409 . [DOI] [PubMed] [Google Scholar]
- 85.Weis F, Moullintraffort L, Heichette C, Chretien D, Garnier C. The 90-kDa heat shock protein Hsp90 protects tubulin against thermal denaturation. J Biol Chem. 2010;285(13):9525–34. Epub 2010/01/30. 10.1074/jbc.M109.096586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang Q, Zhai S, Li L, Li X, Zhou H, Liu A, et al. Anti-tumor selectivity of a novel tubulin and HSP90 dual-targeting inhibitor in non-small cell lung cancer models. Biochem Pharmacol. 2013;86(3):351–60. Epub 2013/06/08. 10.1016/j.bcp.2013.05.019 . [DOI] [PubMed] [Google Scholar]
- 87.Liu XL, Xiao B, Yu ZC, Guo JC, Zhao QC, Xu L, et al. Down-regulation of Hsp90 could change cell cycle distribution and increase drug sensitivity of tumor cells. World J Gastroenterol. 1999;5(3):199–208. Epub 2002/01/31. 10.3748/wjg.v5.i3.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pan JH, Zhou H, Zhu SB, Huang JL, Zhao XX, Ding H, et al. Development of small-molecule therapeutics and strategies for targeting RAF kinase in BRAF-mutant colorectal cancer. Cancer Manag Res. 2018;10:2289–301. Epub 2018/08/21. 10.2147/CMAR.S170105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov. 2012;11(11):873–86. Epub 2012/10/13. 10.1038/nrd3847 . [DOI] [PubMed] [Google Scholar]
- 90.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. Epub 2011/06/07. 10.1056/NEJMoa1103782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lai F, Guo ST, Jin L, Jiang CC, Wang CY, Croft A, et al. Cotargeting histone deacetylases and oncogenic BRAF synergistically kills human melanoma cells by necrosis independently of RIPK1 and RIPK3. Cell Death Dis. 2013;4:e655. Epub 2013/06/08. 10.1038/cddis.2013.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93. Epub 2013/11/23. 10.1158/2159-8290.CD-13-0642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4(1):94–109. Epub 2013/11/23. 10.1158/2159-8290.CD-13-0617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20(7):1965–77. Epub 2014/01/28. 10.1158/1078-0432.CCR-13-3122 . [DOI] [PubMed] [Google Scholar]
- 95.Amaral T, Sinnberg T, Meier F, Krepler C, Levesque M, Niessner H, et al. MAPK pathway in melanoma part II-secondary and adaptive resistance mechanisms to BRAF inhibition. Eur J Cancer. 2017;73:93–101. Epub 2017/02/07. 10.1016/j.ejca.2016.12.012 . [DOI] [PubMed] [Google Scholar]
- 96.Gallagher SJ, Gunatilake D, Beaumont KA, Sharp DM, Tiffen JC, Heinemann A, et al. HDAC inhibitors restore BRAF-inhibitor sensitivity by altering PI3K and survival signalling in a subset of melanoma. Int J Cancer. 2018;142(9):1926–37. Epub 2017/12/07. 10.1002/ijc.31199 . [DOI] [PubMed] [Google Scholar]
- 97.Palusova V, Renzova T, Verlande A, Vaclova T, Medkova M, Cetlova L, et al. Dual Targeting of BRAF and mTOR Signaling in Melanoma Cells with Pyridinyl Imidazole Compounds. Cancers (Basel). 2020;12(6). Epub 2020/06/14. 10.3390/cancers12061516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Luan Y, Li J, Bernatchez JA, Li R. Kinase and Histone Deacetylase Hybrid Inhibitors for Cancer Therapy. Journal of Medicinal Chemistry. 2019;62(7):3171–83. 10.1021/acs.jmedchem.8b00189 [DOI] [PubMed] [Google Scholar]
- 99.Huang Y, Dong G, Li H, Liu N, Zhang W, Sheng C. Discovery of Janus Kinase 2 (JAK2) and Histone Deacetylase (HDAC) Dual Inhibitors as a Novel Strategy for the Combinational Treatment of Leukemia and Invasive Fungal Infections. J Med Chem. 2018;61(14):6056–74. Epub 2018/06/26. 10.1021/acs.jmedchem.8b00393 . [DOI] [PubMed] [Google Scholar]
- 100.Chu-Farseeva YY, Mustafa N, Poulsen A, Tan EC, Yen JJY, Chng WJ, et al. Design and synthesis of potent dual inhibitors of JAK2 and HDAC based on fusing the pharmacophores of XL019 and vorinostat. Eur J Med Chem. 2018;158:593–619. Epub 2018/09/23. 10.1016/j.ejmech.2018.09.024 . [DOI] [PubMed] [Google Scholar]
- 101.Yang EG, Mustafa N, Tan EC, Poulsen A, Ramanujulu PM, Chng WJ, et al. Design and Synthesis of Janus Kinase 2 (JAK2) and Histone Deacetlyase (HDAC) Bispecific Inhibitors Based on Pacritinib and Evidence of Dual Pathway Inhibition in Hematological Cell Lines. J Med Chem. 2016;59(18):8233–62. Epub 2016/08/20. 10.1021/acs.jmedchem.6b00157 . [DOI] [PubMed] [Google Scholar]
- 102.Laporte AN, Poulin NM, Barrott JJ, Wang XQ, Lorzadeh A, Vander Werff R, et al. Death by HDAC Inhibition in Synovial Sarcoma Cells. Mol Cancer Ther. 2017;16(12):2656–67. Epub 2017/09/08. 10.1158/1535-7163.MCT-17-0397 . [DOI] [PubMed] [Google Scholar]
- 103.Sousa FG, Matuo R, Tang SW, Rajapakse VN, Luna A, Sander C, et al. Alterations of DNA repair genes in the NCI-60 cell lines and their predictive value for anticancer drug activity. DNA Repair (Amst). 2015;28:107–15. Epub 2015/03/12. 10.1016/j.dnarep.2015.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Su D, Zhang D, Jin J, Ying L, Han M, Chen K, et al. Identification of predictors of drug sensitivity using patient-derived models of esophageal squamous cell carcinoma. Nat Commun. 2019;10(1):5076. Epub 2019/11/09. 10.1038/s41467-019-12846-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Satyanarayana A, Kaldis P. A dual role of Cdk2 in DNA damage response. Cell Div. 2009;4:9. Epub 2009/05/19. 10.1186/1747-1028-4-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Luan Y, Zhang W, Xie J, Mao J. CDKN2A inhibits cell proliferation and invasion in cervical cancer through LDHA-mediated AKT/mTOR pathway. Clin Transl Oncol. 2020. Epub 2020/07/01. 10.1007/s12094-020-02409-4 . [DOI] [PubMed] [Google Scholar]
- 107.Shah V, Boyd KD, Houlston RS, Kaiser MF. Constitutional mutation in CDKN2A is associated with long term survivorship in multiple myeloma: a case report. BMC Cancer. 2017;17(1):718. Epub 2017/11/08. 10.1186/s12885-017-3715-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220–8. Epub 2010/02/24. 10.1038/nrm2858 . [DOI] [PubMed] [Google Scholar]
- 109.Basu AK. DNA Damage, Mutagenesis and Cancer. Int J Mol Sci. 2018;19(4). Epub 2018/03/24. 10.3390/ijms19040970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Murugan AK, Grieco M, Tsuchida N. RAS mutations in human cancers: Roles in precision medicine. Semin Cancer Biol. 2019;59:23–35. Epub 2019/07/01. 10.1016/j.semcancer.2019.06.007 . [DOI] [PubMed] [Google Scholar]
- 111.Jinesh GG, Sambandam V, Vijayaraghavan S, Balaji K, Mukherjee S. Molecular genetics and cellular events of K-Ras-driven tumorigenesis. Oncogene. 2018;37(7):839–46. 10.1038/onc.2017.377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Grabocka E, Commisso C, Bar-Sagi D. Molecular pathways: targeting the dependence of mutant RAS cancers on the DNA damage response. Clin Cancer Res. 2015;21(6):1243–7. Epub 2014/11/27. 10.1158/1078-0432.CCR-14-0650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grabocka E, Pylayeva-Gupta Y, Jones MJ, Lubkov V, Yemanaberhan E, Taylor L, et al. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell. 2014;25(2):243–56. Epub 2014/02/15. 10.1016/j.ccr.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Matikas A, Mistriotis D, Georgoulias V, Kotsakis A. Targeting KRAS mutated non-small cell lung cancer: A history of failures and a future of hope for a diverse entity. Crit Rev Oncol Hematol. 2017;110:1–12. Epub 2017/01/23. 10.1016/j.critrevonc.2016.12.005 . [DOI] [PubMed] [Google Scholar]
- 115.Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A. 2019;116(32):15823–9. Epub 2019/07/25. 10.1073/pnas.1904529116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pan X, Ji X, Zhang R, Zhou Z, Zhong Y, Peng W, et al. Landscape of somatic mutations in gastric cancer assessed using next-generation sequencing analysis. Oncol Lett. 2018;16(4):4863–70. Epub 2018/09/27. 10.3892/ol.2018.9314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ghimessy AK, Gellert A, Schlegl E, Hegedus B, Raso E, Barbai T, et al. KRAS Mutations Predict Response and Outcome in Advanced Lung Adenocarcinoma Patients Receiving First-Line Bevacizumab and Platinum-Based Chemotherapy. Cancers (Basel). 2019;11(10). Epub 2019/10/12. 10.3390/cancers11101514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ghimessy A, Radeczky P, Laszlo V, Hegedus B, Renyi-Vamos F, Fillinger J, et al. Current therapy of KRAS-mutant lung cancer. Cancer Metastasis Rev. 2020. Epub 2020/06/18. 10.1007/s10555-020-09903-9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kong G, Du J, Liu Y, Meline B, Chang YI, Ranheim EA, et al. Notch1 gene mutations target KRAS G12D-expressing CD8+ cells and contribute to their leukemogenic transformation. J Biol Chem. 2013;288(25):18219–27. Epub 2013/05/16. 10.1074/jbc.M113.475376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dail M, Li Q, McDaniel A, Wong J, Akagi K, Huang B, et al. Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents. Proc Natl Acad Sci U S A. 2010;107(11):5106–11. Epub 2010/03/03. 10.1073/pnas.1001064107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lin YL, Liang YH, Tsai JH, Liau JY, Liang JT, Lin BR, et al. Oxaliplatin-based chemotherapy is more beneficial in KRAS mutant than in KRAS wild-type metastatic colorectal cancer patients. PLoS One. 2014;9(2):e86789. Epub 2014/02/08. 10.1371/journal.pone.0086789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gao W, Estey E. Moving toward targeted therapies in acute myeloid leukemia. Clin Adv Hematol Oncol. 2015;13(11):748–54. Epub 2016/04/09. . [PubMed] [Google Scholar]
- 123.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14(9):598–610. Epub 2014/08/08. 10.1038/nrc3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Duxbury MS, Ito H, Benoit E, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting focal adhesion kinase enhances pancreatic adenocarcinoma gemcitabine chemosensitivity. Biochem Biophys Res Commun. 2003;311(3):786–92. Epub 2003/11/19. 10.1016/j.bbrc.2003.10.060 . [DOI] [PubMed] [Google Scholar]
- 125.Dunn KB, Heffler M, Golubovskaya VM. Evolving therapies and FAK inhibitors for the treatment of cancer. Anticancer Agents Med Chem. 2010;10(10):722–34. Epub 2011/02/05. 10.2174/187152010794728657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Aouida M, Page N, Leduc A, Peter M, Ramotar D. A genome-wide screen in Saccharomyces cerevisiae reveals altered transport as a mechanism of resistance to the anticancer drug bleomycin. Cancer Res. 2004;64(3):1102–9. Epub 2004/02/12. 10.1158/0008-5472.can-03-2729 . [DOI] [PubMed] [Google Scholar]
- 127.Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569(7757):503–8. Epub 2019/05/10. 10.1038/s41586-019-1186-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer. 2020;1(2):235–48. Epub 2020/07/03. 10.1038/s43018-019-0018-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zsakai L, Sipos A, Dobos J, Eros D, Szantai-Kis C, Banhegyi P, et al. Targeted drug combination therapy design based on driver genes. Oncotarget. 2019;10(51):5255–66. Epub 2019/09/17. 10.18632/oncotarget.26985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dienstmann R, Jang IS, Bot B, Friend S, Guinney J. Database of genomic biomarkers for cancer drugs and clinical targetability in solid tumors. Cancer Discov. 2015;5(2):118–23. Epub 2015/02/07. 10.1158/2159-8290.CD-14-1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tamborero D, Rubio-Perez C, Deu-Pons J, Schroeder MP, Vivancos A, Rovira A, et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018;10(1):25. Epub 2018/03/30. 10.1186/s13073-018-0531-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mateo L, Duran-Frigola M, Gris-Oliver A, Palafox M, Scaltriti M, Razavi P, et al. Personalized cancer therapy prioritization based on driver alteration co-occurrence patterns. Genome Med. 2020;12(1):78. Epub 2020/09/11. 10.1186/s13073-020-00774-x [DOI] [PMC free article] [PubMed] [Google Scholar]