

Abstract
The two phospholipase C-γ (PLC-γ) isozymes are major signaling hubs and emerging therapeutic targets for various diseases, yet there are no selective inhibitors for these enzymes. We have developed a high-throughput, liposome-based assay that features XY-69, a fluorogenic, membrane-associated reporter for mammalian PLC isozymes. The assay was validated using a pilot screen of the Library of Pharmacologically Active Compounds 1280 (LOPAC1280) in 384-well format; it is highly reproducible and has the potential to capture both orthosteric and allosteric inhibitors. Selected hit compounds were confirmed with secondary assays, and further profiling led to the interesting discovery that adenosine triphosphate potently inhibits the PLC-γ isozymes through noncompetitive inhibition, raising the intriguing possibility of endogenous, nucleotide-dependent regulation of these phospholipases. These results highlight the merit of the assay platform for large scale screening of chemical libraries to identify allosteric modulators of the PLC-γ isozymes as chemical probes and for drug discovery.
Graphical Abstract
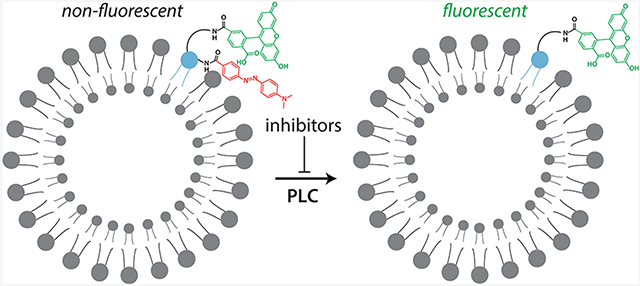
The two PLC-γ isozymes (PLC-γ1 and -γ2) are a major nexus for signals originating from numerous receptor tyrosine kinases and immune receptors, including the B and T cell receptors.1 Like the other PLCs in humans, PLC-γ1 and PLC-γ2 have a conserved catalytic domain and hydrolyze the membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) to generate the second messengers diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3).2,3 Increased concentrations of DAG and IP3 result in activated protein kinase C (PKC) isozymes and the release of Ca2+ from internal stores, respectively. This bifurcated pathway regulates numerous cellular processes such as proliferation and differentiation, chemotaxis, and the fusion of granulocytic vesicles with the plasma membrane. Unlike the other PLCs, the PLC-γ isozymes have several regulatory domains that mediate their phosphorylation-dependent activation.4 Aberrant regulation of the PLC-γ isozymes has been found in various diseases such as cancer, rheumatoid arthritis, and Alzheimer’s disease. A case in point is that ≤37% of patients with adult T cell lymphoma have gain-of-function mutations in PLC-γ1.5 Consequently, the PLC-γ isozymes have emerged as promising new therapeutic targets.
Despite the appreciation that PLC enzymes are widely studied and involved in various pathologies, there are few reported inhibitors (Figure 1) of these phospholipases and none are useful for selectively regulating the PLC-γ isozymes. For example, the aminosteroid U73122 was originally reported to directly inhibit PLCs and is widely used to this effect.6 Unfortunately, it was later demonstrated that U73122 does not inhibit the phospholipase activity of several purified PLCs, including PLC-γ1, PLC-β2, and PLC-δ1.7 This discrepancy likely arises from the fact that most experiments with U73122 monitor events secondary to the activation of PLCs such as intracellular Ca2+ release that likely do not faithfully report direct inhibition of PLCs. This is consistent with the finding that U73122 prevented Ca2+ release by directly inhibiting various Ca2+ pumps.8-11 In addition, the maleimide group within U73122 makes it highly reactive so that most U73122 modifies membrane components and does not effectively enter cells.12 The U73122 that is internalized reacts with and inhibits a variety of unrelated enzymes, including the lipid-modifying enzymes, phosphatidylinositol-4-phosphate kinase and 5-lipoxygenase.13,14 Finally, U73122 was reported to sequester the PLC substrate, PIP2.15 Thus, U73122 is a singularly poor reagent for probing signaling by PLC isozymes. Likewise, D60916 contains a reactive dithiocarbonate that modifies multiple proteins, including sphingomyelin synthase17 and phospholipase A2.18 In addition, D609 chelates Zn2+ and has glutathione mimetic properties.19,20 Although its antiviral and antitumor effects are attributed to inhibition of phosphatidylcholine (PC)-dependent PLC, the direct inhibition of a purified, mammalian phospholipase C by D609 has not been established. Less frequently used to inhibit PLC enzymes are edelfosine,21 CCT129957,22 and small peptides.23 Unfortunately, these compounds also suffer from indirect effects that are mistakenly attributed to PLCs. Small peptides have the additional limitation of being highly cell impermeable.
Figure 1.
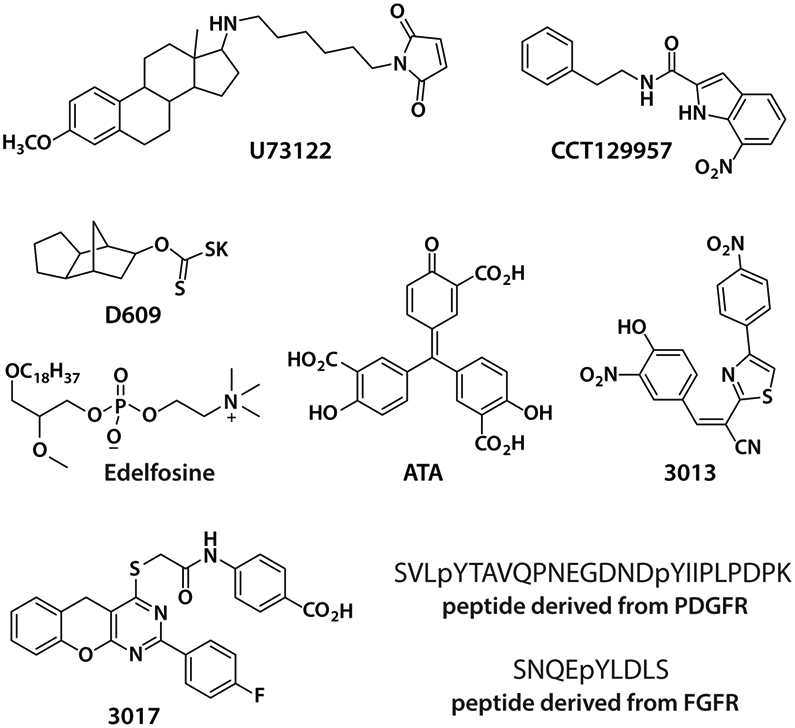
Reported small molecule and peptide inhibitors of PLCs. These inhibitors either do not directly inhibit the PLC-γ isozymes or lack isoform selectivity and sufficient potency to be useful.
As an improvement, ATA, 3013, and 3017 were demonstrated as direct inhibitors of PLCs, both with purified enzymes and in intact cells.24 However, these compounds are paninhibitors and unlikely to be useful for specifically regulating the PLC-γ isozymes given the diverse and sometimes opposing roles of different PLC isoforms in biology and disease.25-27 In addition, ATA is not cell permeable and inhibits multiple enzymes other than PLCs.28-30 The moderate potency and solubility of 3013 and 3017 are also reasons for caution when using them to modulate the phospholipase activity of PLCs. Consequently, there is a substantial need for the development of small molecules that directly and selectively modulate the PLC-γ isozymes.
The catalytic core of PLCs is highly conserved. Therefore, inhibitors that target the active site are unlikely to be selective among different PLC isozymes. However, in contrast to the other PLCs, the PLC-γ isozymes possess an array of regulatory domains that are autoinhibitory until phosphorylated at specific tyrosines during regulated activation. Recently, Sondek and co-workers determined the first structure of an essentially full-length, autoinhibited PLC-γ1 that offers an excellent framework for explaining activation due to phosphorylation or mutation.31 In this framework, there are three states of PLC-γ1 in equilibrium (Figure 2A). In the autoinhibited state, the regulatory array sits atop the core domains to prevent membrane engagement and PIP2 hydrolysis. This state is greatly favored until phosphorylation of Tyr783 or mutations shift the equilibrium toward an open state that is subsequently stabilized at membranes to produce the fully active form.
Figure 2.

Design of a high-throughput assay for selective, allosteric inhibitors of PLC-γ1. (A) Model for the activation of PLC-γ1. There are three major states that are in equilibrium. The autoinhibited state cannot bind PIP2 in membranes and is greatly favored. Phosphorylation of Tyr783 or oncogenic mutations shift the equilibrium toward the open state to varying degrees, and only the membrane-associated active state can access PIP2 or XY-69 in vesicles. (B) Four major classes of inhibitors are expected from high-throughput screens of PLC-γ isozymes. (C) XY-69 is a membrane-associated, fluorogenic reporter for PLC isozymes. Inhibitors are expected to block hydrolysis of XY-69 catalyzed by PLC-γ1. (D) Chemical structure of a soluble PIP2 analogue, WH-15, that functions as a PLC substrate. (E) WH-15 and XY-69 can discern among the classes of inhibitors indicated in panel B.
This framework underpins the rationale and promise for identifying selective, allosteric inhibitors of the PLC-γ isozymes. For example, there are many concavities between the regulatory and core domains that could readily interact with compounds to prevent the necessary rearrangements needed for phospholipase activation (Figure 2B). Alternatively, surfaces of PLC-γ1 needed to interact with membranes could also bind compounds to inhibit phospholipase activity. In both scenarios, inhibitors would not target the conserved catalytic core and so would be much more likely to be isozyme specific. We also expect these types of inhibitors to be much more frequent than orthosteric inhibitors because the potential surface area for interaction with allosteric inhibitors is immense relative to the surface area of the active site.
We previously developed XY-69 (Figure 2C) to robustly monitor PLC activity at membranes in real time.32 XY-69 is a PIP2 analogue with fluorescein introduced at the sn-1 position and a 4-(dimethylaminoazo)benzene-4-carboxylic acid (DABCYL) moiety incorporated into the 6-hydroxyl position of the headgroup. This arrangement effectively quenches the intrinsic fluorescence of fluorescein by the DABCYL moiety. However, once XY-69 is cleaved by PLCs, the fluorophore/quencher pair separate, resulting in a dramatic increase in the quantum yield of the fluorescein group. XY-69 was also designed to retain a long C15H31 acyl chain at the sn-2 position to favor the partitioning of XY-69 into lipid membranes. Therefore, much like PIP2 and DAG, XY-69 and the fluorescent product of XY-69 hydrolysis remain in lipid membranes. Importantly, XY-69 reconstituted into liposomes readily reports the activation of PLC-γ1 upon tyrosine phosphorylation or mutations31 with either purified proteins or cell lysates (Figure S1).
We therefore propose to develop a high-throughput assay based on XY-69 embedded in liposomes to identify allosteric inhibitors of PLC-γ1 that are also likely to be selective. In contrast, a previously developed high-throughput assay using WH-15 (Figure 2D),24 a water-soluble analogue of PIP2, is expected to identify mainly orthosteric inhibitors of PLC-γ1. Consequently, the complementary use of both membrane-embedded XY-69 and soluble WH-15 will facilitate the rapid identification of selective, allosteric inhibitors of PLC-γ1 (Figure 2E) using a workflow that can also be applied to PLC-γ2.
MATERIALS AND METHODS
General.
XY-69 and purified PLC-γ1 (D1165H) protein were produced as previously reported. All other reagents, including liver phosphatidylethanolamine (PE) and phosphatidylinositol 4,5-bisphosphate (PIP2), were purchased from commercial sources and used directly. All buffer stock solutions were prefiltered with a 0.45 μm PES syringe tip filer and stored at 4 °C unless otherwise indicated. All microplate assays were performed using ProxiPlate-384 Plus F black plates.
High-Throughput Screen of the LOPAC1280 Library.
The assay plate was prepared by transferring individual compounds (100 nL, 1 mM in DMSO) to a small-volume 384-well microplate using a Mosquito HTS nanoliter dispensing instrument. Appropriate controls, including DMSO, EGTA (500 mM, pH 10.3), and ATA (5 mM in DMSO), were added to empty wells prior to the high-throughput screen. Liposomes containing liver PE (440 μM), brain PIP2 (40 μM), and XY-69 (1 μM) were generated by mixing the lipids, drying the mixture under a stream of nitrogen, and resuspending the dried lipid mixture in HEPES (20 mM, pH 7.4) through sonication. Such vesicles were then mixed with 6× assay buffer to form a liposome solution. In parallel, purified PLC-γ1 (D1165H) protein was diluted to 2.5 nM with a buffer containing HEPES (20 mM, pH 7.4), NaCl (50 mM), dithiothreitol (DTT, 2 mM), and fatty acid-free BSA (1 mg/mL). The diluted PLC solution (4 μL) was then added to each well of the assay plate at room temperature. After 15 min, the vesicle solution (6 μL) was added to initiate the enzymatic reaction. After 1 h at room temperature, the quenching buffer (5 μL) containing EGTA (50 mM, pH 10.3) was added. Thirty minutes later, the assay plate was analyzed by fluorescence intensity (λex = 485 nm, and λem = 520 nm) with a plate reader.
IC50 Measurement with XY-69.
Assay buffer was prepared with HEPES (20 mM, pH 7.4), KCl (70 mM), CaCl2 (2.35 mM), EGTA (3 mM), and DTT (2 mM). Individual hit compounds were prepared as 10 mM stock solutions in DMSO or H2O according to the manufacturer’s instructions. A 5 μL aliquot of either Hit-1 or Hit-3 was diluted with assay buffer to make a working stock of 500 μM inhibitor. This working stock was used to make 1:3 serial dilutions containing 5% (v/v) DMSO that were each added to wells of a microplate in quadruplicate aliquots of 2 μL. Solutions of purified PLC protein were diluted to the appropriate concentrations with assay buffer containing fatty acid-free BSA (1 mg/mL). Two microliters of the PLC solution was added to each well of inhibitor solution, and the mixtures were incubated at room temperature for 15 min. Liposomes containing liver PE (367 μM), brain PIP2 (33.3 μM), and XY-69 (0.83 μM) were generated by mixing the lipids, drying the mixture under a stream of nitrogen, and resuspending the dried lipid film using sonication in HEPES (20 mM, pH 7.4), followed by mixing with a 6× concentrated assay buffer to give the vesicle solution containing HEPES (38 mM, pH 7.4), KCl (70 mM), CaCl2 (2.35 mM), EGTA (3 mM), and DTT (2 mM). To initiate the enzymatic reaction, 6 μL of the liposome solution was added to each PLC/inhibitor mixture. The microplate was immediately placed in the plate reader, and the fluorescence intensity was recorded every minute. The linear range of each reaction curve was used to calculate the initial reaction rate for each inhibitor concentration, and the resulting plot was analyzed using GraphPad Prism to calculate the IC50 value.
The final assay mixtures contained various concentrations of inhibitors (100, 33.3, 11.1, 3.70, 1.23, 0.411, 0.137, 0.046, 0.015, or 0.005 μM) or inhibitor-free assay buffer for controls of 100% enzyme activity. Baseline correction was performed using replicates that excluded both the inhibitor and PLC (BSA only). The final concentrations for wild-type PLC-γ1, PLC-γ1 (D1165H), PLC-γ1 (P867R), and wild-type PLC-γ2, -β2, -β3, and -δ1 were 2.0, 0.5, 2.0, 2.0, 0.5, 10, and 0.003 nM, respectively. The volumes of all final assay mixtures were 10 μL, and all of the mixtures contained PE (220 μM), PIP2 (20 μM), XY-69 (0.5 μM), 1% (v/v) DMSO, HEPES (30 mM, pH 7.4), KCl (70 mM), CaCl2 (2.35 mM), EGTA (3 mM), DTT (2 mM), and fatty acid-free BSA (0.2 mg/mL). For experiments containing lower concentrations of PIP2 (4 or 2 μM), phosphatidylinositol (PI) was included (16 or 18 μM, respectively) to balance the total final lipid content back to 240 μM.
Kinetic Studies of Inhibition of XY-69 Hydrolysis by ATP.
ATP dilutions were prepared using a cholate micelle buffer containing HEPES (50 mM, pH 7.4), KCl (70 mM), CaCl2 (3 mM), EGTA (3 mM), and 0.5% (w/v) sodium cholate and then added to a microplate in quadruplicate aliquots of 2 μL. Purified, wild-type PLC-γ1 was diluted with micelle buffer that included fatty acid-free BSA (1 mg/mL). The diluted enzyme solution (4 μL) was added to each well, and the microplate was incubated at room temperature for 15 min. Subsequently, XY-69 at various concentrations in micelle buffer (4 μL) containing DTT (5 mM) was added to initiate the reaction. The microplate was immediately placed in the plate reader, and the fluorescence intensity was recorded every 30 s. All final assay mixtures contained wild-type PLC-γ1 (0.8 nM), 1% (v/v) DMSO, HEPES (50 mM, pH 7.4), KCl (70 mM), CaCl2 (3 mM), EGTA (3 mM), DTT (2 mM), 0.5% (w/v) sodium cholate, and fatty acid-free BSA (0.4 mg/mL) in a volume of 10 μL. Kinetic assays were performed with final ATP concentrations of 0, 1.5, and 5 μM in combination with 0.5, 0.75, 1.0, 1.5, 5, and 10 μM XY-69. The initial velocities were calculated as the fluorescence intensity increase per minute, and a double-reciprocal plot was used to analyze the data.
Measurement of the Binding Affinity of ATP with Wild-Type PLC-γ1.
ITC studies were carried out on an Auto-iTC200 instrument at 25 °C. Briefly, purified PLC-γ1 (D1165H) was dialyzed against ITC buffer {50 mM HEPES (pH 7.4), 150 mM NaCl, 1 μM CaCl2, and 2 mM TCEP [tris(2-carboxyethyl)phosphine]}, and ATP was dissolved in the same ITC buffer. To initiate titrations, ATP (500 μM) was injected into a sample cell containing PLC-γ1 (D1165H) (50 μM, 200 μL) while being stirred at 850 rpm. An initial injection (0.2 μL) was followed by 19 serial injections (2 μL each) with a 3 min interval between injections to allow for signals to return to baseline. Data were analyzed by integrating the peaks using Origin software (MicroCal). Heats of dilution were subtracted from heats of binding, and the data were fit to a one-site binding model.
IC50 Measurement with WH-15.
Micelle buffer was prepared with HEPES (50 mM, pH 7.4), KCl (70 mM), CaCl2 (3 mM), EGTA (3 mM), 0.5% (w/v) sodium cholate, and DTT (2 mM). Inhibitor dilutions containing 5% (v/v) DMSO were prepared as described above using micelle buffer and added to a microplate in quadruplicate aliquots of 2 μL. Solutions of purified PLC-γ1 (D1165H) were diluted to the appropriate concentration with micelle buffer containing fatty acid-free BSA (1 mg/mL). Four microliters of a PLC solution was added, and the microplate was incubated at room temperature for 15 min. Subsequently, 4 μL of micelle buffer containing 25 μM WH-15 was added to initiate the reaction. The microplate was immediately placed in the plate reader, and the fluorescence intensity (λex = 355 nm, and λem = 530 nm) was recorded every minute. The volumes of all final assay mixtures were 10 μL, and the mixtures contained PLC-γ1 (D1165H) (0.5 nM), WH-15 (10 μM), 1% (v/v) DMSO, HEPES (50 mM, pH 7.4), KCl (70 mM), CaCl2 (3 mM), EGTA (3 mM), DTT (2 mM), 0.5% (w/v) sodium cholate, and fatty acid-free BSA (0.4 mg/mL).
Inhibition of the Phospholipase Activity in Live Cells.
HEK293A cells were plated at a density of ~75000 cells/well in 12-well tissue culture dishes in growth medium [DMEM containing 10% (v/v) FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B]. Twenty-four hours later, cells were transiently transfected with 100 ng of a plasmid encoding HA-tagged wild-type PLC-γ1 or PLC-γ1 (D1165H) using Continuum transfection reagent according to the manufacturer’s protocol (GeminiBio). Twenty-four hours post-transfection, growth medium was removed and cells were metabolically labeled overnight with 1 μCi of [3H]-myo-inositol in serum-free, inositol-free DMEM. Radiolabeling medium was then removed and replaced with vehicle [serum-free, inositol-free DMEM containing 1 μCi of [3H]-myo-inositol and 1% (v/v) DMSO] or a vehicle containing 30 μM Hit-1, and cells were incubated for 1 h. Cells were then treated with 10 mM LiCl (final concentration) for an additional 1 h, and accumulation of [3H]inositol phosphates was quantified by Dowex column chromatography as described previously. Expression of each version of PLC-γ1 was confirmed by immunoblotting of cell lysates using a monoclonal antibody against the HA epitope (BioLegend, clone 16B12; RRID: AB_2565335). A monoclonal antibody against β-actin (SigmaAldrich, clone AC-15; RRID: AB_476692) was used as a loading control.
RESULTS AND DISCUSSION
Development of a High-Throughput Assay Based on XY-69 in Liposomes.
We have chosen to use oncogenic and constitutively active PLC-γ1 (D1165H) as the screening target. This choice is driven by both practical and theoretical considerations. First, the basal activity of wild-type PLC-γ1 with membrane-embedded XY-69 is very low, and it would be difficult to produce a reasonable signal-to-noise ratio with this form of the enzyme. Alternatively, phosphorylated PLC-γ1 could be used, but it is difficult to produce a homogeneous amount of this form sufficient for large scale screens. Instead, the enzymatic activity of PLC-γ1 (D1165H) with membrane-embedded XY-69 is high enough to produce a robust signal-to-noise ratio.31 This is likely due to D1165H-induced shifts in the equilibrium population of PLC-γ1 toward a more open distribution predisposed to engage membranes. However, the equilibrium likely continues to favor the autoinhibited state because PLC-γ1 (D1165H) retains phosphorylation-induced activation31 and its intrinsic activity remains slow enough that initial rates of hydrolysis of XY-69 in liposomes remain linear (Figure S1). Consequently, screened compounds are expected to have opportunities to interact with populations of PLC-γ1 (D1165H) in the autoinhibited and open states before addition of liposomes containing XY-69.
To optimize a high-throughput assay using membrane-embedded XY-69 to screen compounds for inhibitors of PLC-γ1, we systematically assessed (i) the concentration of XY-69, (ii) the production and composition of liposomes, (iii) the concentration of PLC-γ1 (D1165H), (iv) the amount and order of reagent addition, and (v) the quench conditions. For the sake of brevity, only studies assessing the concentrations of XY-69 and PLC-γ1 (D1165H) are discussed in detail.
In previous experiments,31 0.5 nM PLC-γ1 (D1165H) was used in liposomes incorporated with 5 μM XY-69. This concentration of XY-69 was deemed prohibitive for large screens and was decreased over a 10-fold range while the concentration of PLC-γ1 (D1165H) was maintained at 0.5 nM (Figure 3A). The hydrolysis of XY-69 was continuously monitored using fluorescence (λex = 485 nm, and λem = 520 nm), and the extent of activation was calculated. XY-69 embedded in liposomes produced robust changes in fluorescence over a wide range of concentrations. To minimize XY-69 consumption while maintaining a robust signal-to-noise ratio, 0.5 μM XY-69 was used in all subsequent experiments.
Figure 3.

Assay optimization using XY-69 embedded in liposomes. (A) Assay window assessed as a function of XY-69 concentration. The concentration of PLC-γ1 (D1165H) was 0.5 nM. (B) The level of hydrolysis of XY-69 increases with an increase in the concentration of PLC-γ1 (D1165H). The concentration of XY-69 was 0.5 μM. (C) Membrane-embedded XY-69 highlights the preferential interfacial activation of constitutively active PLC-γ1 (D1165H). PLCs at 1 nM; XY-69 at 0.5 μM. (D) Hydrolysis of XY-69 by PLC-γ1 (D1165H) tolerates up to 5% DMSO with no degradation of the signal.
With the concentration of XY-69 fixed, amounts of PLC-γ1 (D1165H) were varied (Figure 3B). The assay was linear over the course of 1 h for several concentrations of PLC-γ1 (D1165H). Consequently, 1 nM PLC-γ1 was chosen for subsequent experiments. This choice was designed to minimize the consumption of PLC-γ1 (D1165H) while maintaining a robust, linear signal that allowed sufficient time for the numerous plate manipulations required for high-throughput screens.
The final protocol consisted of incubation of 1 nM PLC-γ1 (D1165H) with 0.5 μM XY-69 embedded in liposomes composed of 220 μM liver phosphoethanolamine (PE) and 20 μM PIP2. The final volumes were 10 μL per well, and reactions were monitored at room temperature (~23 °C) for 60 min prior to the addition of 50 mM EGTA to chelate the Ca2+ cofactor needed for phospholipase activity. Importantly, this final assay format readily reads out the preferential interfacial activation of PLC-γ1 (D1165H) relative to that of the wild type (Figure 3C). In addition, the assay is immune to concentrations of DMSO of ≤5% (v/v) (Figure 3D). The fluorescence values (λex = 485 nm, and λem = 520 nm) for the final assay conditions were measured for 16 parallel reactions in a 384-well plate for three consecutive days. In parallel, experiments under identical conditions were performed but with PLC-γ1 (D1165H) (high control) substituted for BSA (low control). From these data, the average Z′-factor33 was calculated to be 0.77 (Figure S2), indicating a highly robust high-throughput assay.
Proof-of-Concept High-Throughput Screen with the LOPAC1280 Library.
Using essentially the same conditions as described above, we completed a pilot screen in duplicate with the 1280 compounds comprising the Library of Pharmacologically Active Compounds 1280 (LOPAC1280) (Figure 4). A correlation coefficient of approximately 0.86 for the two runs attests to the reproducibility of the format, and Z′ factors were maintained above 0.7 for both runs. The screen produced 12 hits (Figure 5) flagged as having >50% inhibition in both runs for an estimated hit rate of 0.94%.
Figure 4.

Pilot screen of the LOPAC1280 library. The high-throughput assay based on XY-69 was used to screen 1280 compounds in the Library of Pharmacologically Active Compounds 1280 (LOPAC1280, 10 μM) for inhibitors of PLC-γ1 (D1165H). The screen was run in duplicate, and the correlation between runs was plotted. Full inhibition was obtained using either the promiscuous inhibitor aurintricarboxylic acid (ATA, 50 μM) or EGTA (10 mM) to chelate the Ca2+ cofactor. Twelve compounds produced >50% inhibition in both runs for an estimated hit rate of approximately 1%; the Z′-factor > 0.75 for both runs.
Figure 5.

Chemical structures of the hit compounds from the pilot screen. The 12 hits are categorized into five groups (A–E) based on structural features and possible mechanisms of action. Although hits in groups D and E were also identified from a previous screen based on the soluble PIP2 analogue, WH-15, the other hits are unique to the screen based on XY-69. The number in parentheses represents the percentage inhibition of PLC-γ1 (D1165H) by the corresponding hit compound at 10 μM.
U73122 is widely accepted as an inhibitor of PLCs and also included in LOPAC1280. However, it was not flagged as an inhibitor of PLC-γ1 in this screen. This result is consistent with our published data showing that U73122 does not inhibit a panel of purified PLCs, including PLC-γ1.7
The 12 hit compounds (Figure 5) were categorized into five groups (A–E) based on structural similarities and possible mechanisms of action. We previously used WH-15 to screen LOPAC1280 for PLC inhibitors,24 and under these conditions, only hit compounds in groups D and E, including ATA (Hit-7), were identified. Clearly, the new assay format based on XY-69 can identify new classes of inhibitors, possibly allosteric, that are invisible to assays using WH-15.
Inhibitory Profiles of Two Hit Compounds from the Pilot Screen.
To further validate the hits and establish secondary assays, we measured the concentration-dependent inhibition of PLC-γ1 (D1165H) by Hit-1 in group A and Hit-3 in group B. The IC50 value for Hit-1 was 13.9 μM when XY-69 was used in the assay but increased to 36.0 μM when WH-15 was used (Figure 6A). Similarly, Hit-3 inhibited PLC-γ1 with differing IC50 values (0.26 μM vs 7.3 μM) when XY-69 and WH-15 were used (Figure 6A). When the substrate PIP2 concentration was varied in the liposome (Figure 6B), Hit-3 showed a constant IC50 (~0.25 μM) indicative of a noncompetitive and likely allosteric mode of action.34-36 Another cancer-associated mutant, PLC-γ1 (P867R), can also be inhibited by Hit-3 with an IC50 of 0.85 μM (Figure S3). Furthermore, Hit-3 demonstrated important isoform selectivity, showing strong inhibition of the wild-type PLC-γ isozymes but no significant inhibition of either PLC-β (β2 or β3) or PLC-δ1 isozymes (Figure 6C).
Figure 6.

Validation of two hit compounds in secondary assays. (A) The concentration-dependent inhibition of phospholipase activity of PLC-γ1 (D1165H) by Hit-1 or Hit-3 was quantified using either XY-69 or WH-15. (B) The inhibition of PLC-γ1 (D1165H) by Hit-3 is independent of the concentration of the substrate PIP2 in liposomes, indicative of noncompetitive inhibition. (C) Isoform selectivity. The inhibition of the indicated wild-type PLC isozymes by Hit-3 was measured. (D) Inhibition of PLC-γ1 (D1165H) by ATP, ADP, and GTP.
Hit-3 is an ATP analogue and is negatively charged. There are eight other ribonucleotides or derivatives (Figure S4) in LOPAC1280. Despite the entire set being negatively charged, only the two ATP analogues significantly inhibited the phospholipase C activity of PLC-γ1 (D1165H), suggesting that the inhibition by Hit-3 is likely specific. This intriguing inhibitory profile prompted us to investigate whether ATP also inhibits the phospholipase activity of PLC-γ1 (D1165H). As shown in Figure 6D, ATP indeed inhibited with a potency (IC50 = 0.19 μM) similar to that of Hit-3. In contrast, ADP and GTP showed much weaker potency, with IC50 values of 4.3 and 12.7 μM, respectively.
To further understand the mechanism of inhibition by ATP, we carried out kinetic analysis of XY-69 hydrolysis by wild-type PLC-γ1 (Figure 7). The initial velocities at various substrate concentrations with or without ATP (1.5 and 5 μM) were measured, and the data were analyzed using a double-reciprocal plot (Lineweaver–Burk plot). The Vmax of the enzymatic reaction was reduced by 1.9- and 4.1-fold when 1.5 and 5 μM ATP was present, respectively, while the KM remained essentially unchanged. This result is consistent with noncompetitive inhibition. This outcome is also consistent with the unchanged IC50 when the PIP2 concentration is varied in the presence of ATP analogue Hit-3 (Figure 6C). Given these results, isothermal titration calorimetry (ITC) was used to measure the affinity of ATP and wild-type PLC-γ1 (Figure 8). The titration showed unequivocal interaction (KD of 2.6 μM) of ATP and wild-type PLC-γ1, suggesting the intriguing possibility of endogenous regulation of PLC-γ1 by ATP. In summary, the pilot screen and follow-up studies highlight the potential value of XY-69 for the identification of novel, allosteric inhibitors of PLCs.
Figure 7.

Kinetic analysis of inhibition of wild-type PLC-γ1 by ATP. The concentration of wild-type PLC-γ1 was 0.8 nM. (A) Hydrolysis of XY-69 at the indicated concentrations was monitored by fluorescence. (B) Hydrolysis of XY-69 at the indicated concentrations in the presence of 1.5 μM ATP. (C) Hydrolysis of XY-69 at the indicated concentrations in the presence of 5 μM ATP. (D) Double-reciprocal plot. The initial velocities were calculated as fluorescence per minute, and its reciprocal was plotted against the reciprocal of XY-69 concentration. The data were fitted to linear regression in Prism 8.
Figure 8.

ATP binds wild-type PLC-γ1 as measured by isothermal titration calorimetry. Thermogram (top) and binding isotherm (bottom) for ATP titrated into wild-type PLC-γ1 in a buffer containing 50 mM HEPES (pH 7.4), 150 mM NaCl, 1 μM CaCl2, and 2 mM TCEP.
However, neither Hit-3 nor ATP is cell permeable, and neither can be profiled in cell-based experiments. To further demonstrate that the primary hits identified in the pilot screen can be confirmed in secondary assays, we investigated the effect of the less potent but cell permeable Hit-1 in intact cells. Briefly, HEK293A cells were transfected with PLC-γ1 (D1165H) and then treated with Hit-1. Indeed, Hit-1 (30 μM) inhibited the phospholipase activity of PLC-γ1 (D1165H) by approximately 80% in intact cells (Figure 9).
Figure 9.

Hit-1 inhibits PLC-γ1 (D1165H) in intact cells. HEK293 cells were transfected with PLC-γ1 (D1165H), wild-type PLC-γ1, or an empty vector control prior to metabolic labeling with [3H]inositol and incubation with either DMSO or 30 μM Hit-1 for 1 h. Cells were subsequently lysed, and the radioactivity of the lysate was quantified. The expression of PLC-γ1 was confirmed by a Western blot (inset).
CONCLUSIONS
We have developed a liposome-based, high-throughput assay to identify selective inhibitors of the PLC-γ isozymes. The assay is based on the fluorogenic, membrane-associated PLC reporter, XY-69, and is highly reproducible. The new format is further validated by a pilot screen of LOPAC1280 in 384-well format with a Z′-factor of >0.7. The screen results demonstrate that the liposome-based assay can identify novel types of inhibitors, presumably allosteric inhibitors, that assays based on a soluble PIP2 analogue will miss. These results also highlight the importance of liposomes in measuring the activity of the PLC-γ isozymes, consistent with the mechanism of PLC-γ activation as illustrated by structural studies. Liposome-based high-throughput screens have been underexplored, and examples in the literature are very limited,37,38 possibly because of the concern about nonspecific disruption of liposomes by different compounds. The moderate hit rate (0.94%) of the pilot screen suggests that such a concern does not prevent practical use of our assay format.
Ribonucleotides including ATP have been shown to inhibit PLC-β4 at a high concentration (1 mM),39 but their effects on the PLC-γ isozymes are not known. We have demonstrated that ATP, but not ADP or GTP, potently and selectively inhibits the phospholipase activity of the PLC-γ isozymes. The cellular concentration of ATP is typically in the range of 1–10 mM, raising the question of how the PLC-γ isozymes can be activated in cells. We have suggested several possibilities. For example, receptor tyrosine kinases such as Src have been shown to activate ATPases.40 Because ADP and AMP have less or no inhibition of PLC-γ1, it is possible that ATP inhibition could be released through ATP hydrolysis in response to activated receptor tyrosine kinases. Alternatively, cellular cations such as Mg2+ and Ca2+ bind to ATP to form different complexes.41-43 It is possible those complexes, as well as free ATP, have a heterogeneous distribution inside cells and differential capacities to inhibit PLC-γ1, especially at the membranes where PLC-γ1 is activated. The negative charges on membranes could also facilitate the dissociation of bound ATP from the enzyme. Although a detailed mechanism of action needs more extensive studies and is beyond the scope of this work, the discovery of ATP’s inhibition of wild-type PLC-γ1 suggests that the assay platform might be suitable for discovering additional endogenous regulators of PLCs.
Hit-1 and Hit-3 were identified from a LOPAC that contains only 1280 compounds, including many newly appreciated pan assay interference compounds (PAINS).44-46 Aggregator Advisor47 is a widely used database for assessing whether screen hits are similar to known aggregators. Indeed, among the 12 hit compounds in Figure 5, five (including Hit-1) are considered as aggregators or similar to a known aggregator in the database. Some other considerations also preclude Hit-1 and Hit-3 from being good candidate inhibitors worthy of further development. For example, Hit-1 [6-bromoindirubin-3′-oxime (BIO)] has multiple other pharmacological activities, including inhibition of GSK-3β,48 and also has poor solubility in aqueous buffer, while both Hit-3 and ATP are not cell permeable. Nonetheless, the pilot screen with this library is intended solely for the validation of the assay format for subsequent high-throughput screens. Our results suggest that the novel liposome-based assay platform developed in this work provides a unique opportunity to potentially identify allosteric modulators of the PLC-γ isozymes as chemical probes and for drug discovery.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the University of North Carolina Lineberger Comprehensive Cancer Center for general support of facilities and resources (CA016086).
Funding
The authors thank the National Institutes of Health (GM057391 and CA177993) and the North Carolina Biotechnology Center for supporting this work.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00511.
Figures S1–S4 (PDF)
The authors declare no competing financial interest.
Contributor Information
Weigang Huang, Division of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Adam J. Carr, Division of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Nicole Hajicek, Department of Pharmacology, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Miri Sokolovski, Department of Pharmacology, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Edhriz Siraliev-Perez, Department of Biochemistry and Biophysics, School of Medicine, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
P. Brian Hardy, Division of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Kenneth H. Pearce, Division of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy and Lineberger Comprehensive Cancer Center, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
John Sondek, Department of Pharmacology, Department of Biochemistry and Biophysics, School of Medicine, and Lineberger Comprehensive Cancer Center, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Qisheng Zhang, Division of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, Department of Pharmacology, and Lineberger Comprehensive Cancer Center, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
REFERENCES
- (1).Gresset A, Sondek J, and Harden TK (2012) The phospholipase C isozymes and their regulation. Subcell. Biochem 58, 61–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nakamura Y, and Fukami K (2017) Regulation and physiological functions of mammalian phospholipase C. J. Biochem 161, 315–321. [DOI] [PubMed] [Google Scholar]
- (3).Kadamur G, and Ross EM (2013) Mammalian phospholipase C. Annu. Rev. Physiol 75, 127–154. [DOI] [PubMed] [Google Scholar]
- (4).Gresset A, Hicks SN, Harden TK, and Sondek J (2010) Mechanism of phosphorylation-induced activation of phospholipase C-γ isozymes. J. Biol. Chem 285, 35836–35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, Totoki Y, Chiba K, Sato-Otsubo A, Nagae G, Ishii R, Muto S, Kotani S, Watatani Y, Takeda J, Sanada M, Tanaka H, Suzuki H, Sato Y, Shiozawa Y, Yoshizato T, Yoshida K, Makishima H, Iwanaga M, Ma G, Nosaka K, Hishizawa M, Itonaga H, Imaizumi Y, Munakata W, Ogasawara H, Sato T, Sasai K, Muramoto K, Penova M, Kawaguchi T, Nakamura H, Hama N, Shide K, Kubuki Y, Hidaka T, Kameda T, Nakamaki T, Ishiyama K, Miyawaki S, Yoon SS, Tobinai K, Miyazaki Y, Takaori-Kondo A, Matsuda F, Takeuchi K, Nureki O, Aburatani H, Watanabe T, Shibata T, Matsuoka M, Miyano S, Shimoda K, and Ogawa S (2015) Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet 47, 1304–1315. [DOI] [PubMed] [Google Scholar]
- (6).Bleasdale JE, Bundy GL, Bunting S, Fitzpatrick FA, Huff RM, Sun FF, and Pike JE (1989) Inhibition of phospholipase C dependent processes by U-73122. Adv. Prostaglandin, Thromboxane, Leukotriene Res. 19, 590–593. [PubMed] [Google Scholar]
- (7).Klein RR, Bourdon DM, Costales CL, Wagner CD, White WL, Williams JD, Hicks SN, Sondek J, and Thakker DR (2011) Direct activation of human phospholipase C by its well known inhibitor U73122. J. Biol. Chem 286, 12407–12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hollywood MA, Sergeant GP, Thornbury KD, and McHale NG (2010) The PI-PLC inhibitor U-73122 is a potent inhibitor of the SERCA pump in smooth muscle. Br. J. Pharmacol 160, 1293–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pulcinelli FM, Gresele P, Bonuglia M, and Gazzaniga PP (1998) Evidence for separate effects of U73122 on phospholipase C and calcium channels in human platelets. Biochem. Pharmacol 56, 1481–1484. [DOI] [PubMed] [Google Scholar]
- (10).Berven LA, and Barritt GJ (1995) Evidence obtained using single hepatocytes for inhibition by the phospholipase C inhibitor U73122 of store-operated Ca2+ inflow. Biochem. Pharmacol 49, 1373–1379. [DOI] [PubMed] [Google Scholar]
- (11).Wang JP (1996) U-73122, an aminosteroid phospholipase C inhibitor, may also block Ca2+ influx through phospholipase C-independent mechanism in neutrophil activation. Naunyn-Schmiedeberg′s Arch. Pharmacol 353, 599–605. [DOI] [PubMed] [Google Scholar]
- (12).Wilsher NE, Court WJ, Ruddle R, Newbatt YM, Aherne W, Sheldrake PW, Jones NP, Katan M, Eccles SA, and Raynaud FI (2007) The phosphoinositide-specific phospholipase C inhibitor U73122 (1-(6-((17beta-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrol e-2,5-dione) spontaneously forms conjugates with common components of cell culture medium. Drug Metab. Dispos 35, 1017–1022. [DOI] [PubMed] [Google Scholar]
- (13).Burgdorf C, Schafer U, Richardt G, and Kurz T (2010) U73122, an aminosteroid phospholipase C inhibitor, is a potent inhibitor of cardiac phospholipase D by a PIP2-dependent mechanism. J. Cardiovasc. Pharmacol 55, 555–559. [DOI] [PubMed] [Google Scholar]
- (14).Feisst C, Albert D, Steinhilber D, and Werz O (2005) The aminosteroid phospholipase C antagonist U-73122 (1-[6-[[17-beta-3-methoxyestra-1,3,5(10)-trien-17-yl] amino]hexyl]-1H-pyrrole-2,5-dione) potently inhibits human 5-lipoxygenase in vivo and in vitro. Mol. Pharmacol 67, 1751–1757. [DOI] [PubMed] [Google Scholar]
- (15).Vickers JD (1993) U73122 affects the equilibria between the phosphoinositides as well as phospholipase C activity in unstimulated and thrombin-stimulated human and rabbit platelets. J. Pharmacol. Exp. Ther 266, 1156–1163. [PubMed] [Google Scholar]
- (16).Kato M, Hammam MA, Taniguchi T, Suga Y, and Monde K (2016) What is the true structure of D609, a widely used lipid related enzyme inhibitor? Org. Lett 18, 768–771. [DOI] [PubMed] [Google Scholar]
- (17).Perry RJ, and Ridgway ND (2004) The role of de novo ceramide synthesis in the mechanism of action of the tricyclic xanthate D609. J. Lipid Res 45, 164–173. [DOI] [PubMed] [Google Scholar]
- (18).Kang MS, Jung SY, Jung KM, Kim SK, Ahn KH, and Kim DK (2008) D609, an inhibitor of phosphatidylcholine-specific phospholipase C, inhibits group IV cytosolic phospholipase A2. Mol. Cells 26, 481–485. [PubMed] [Google Scholar]
- (19).Gonzalez-Roura A, Casas J, and Llebaria A (2002) Synthesis and phospholipase C inhibitory activity of D609 diastereomers. Lipids 37, 401–406. [DOI] [PubMed] [Google Scholar]
- (20).Sultana R, Newman SF, Abdul HM, Cai J, Pierce WM, Klein JB, Merchant M, and Butterfield DA (2006) Protective effect of D609 against amyloid-beta1–42-induced oxidative modification of neuronal proteins: redox proteomics study. J. Neurosci. Res 84, 409–417. [DOI] [PubMed] [Google Scholar]
- (21).Powis G, Seewald MJ, Gratas C, Melder D, Riebow J, and Modest EJ (1992) Selective inhibition of phosphatidylinositol phospholipase C by cytotoxic ether lipid analogues. Cancer Res. 52, 2835–2840. [PubMed] [Google Scholar]
- (22).Reynisson J, Court W, O’Neill C, Day J, Patterson L, McDonald E, Workman P, Katan M, and Eccles SA (2009) The identification of novel PLC-γ inhibitors using virtual high throughput screening. Bioorg. Med. Chem 17, 3169–3176. [DOI] [PubMed] [Google Scholar]
- (23).Blum S, and Dash PK (2004) A cell-permeable phospholipase C-γ1-binding peptide transduces neurons and impairs long-term spatial memory. Learn. Mem 11, 239–243. [DOI] [PubMed] [Google Scholar]
- (24).Huang W, Barrett M, Hajicek N, Hicks S, Harden TK, Sondek J, and Zhang Q (2013) Small molecule inhibitors of phospholipase C from a novel high-throughput screen. J. Biol. Chem 288, 5840–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Behjati S, Tarpey PS, Sheldon H, Martincorena I, Van Loo P, Gundem G, Wedge DC, Ramakrishna M, Cooke SL, Pillay N, Vollan HK, Papaemmanuil E, Koss H, Bunney TD, Hardy C, Joseph OR, Martin S, Mudie L, Butler A, Teague JW, Patil M, Steers G, Cao Y, Gumbs C, Ingram D, Lazar AJ, Little L, Mahadeshwar H, Protopopov A, Al Sannaa GA, Seth S, Song X, Tang J, Zhang J, Ravi V, Torres KE, Khatri B, Halai D, Roxanis I, Baumhoer D, Tirabosco R, Amary MF, Boshoff C, McDermott U, Katan M, Stratton MR, Futreal PA, Flanagan AM, Harris A, and Campbell PJ (2014) Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat. Genet 46, 376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Martins M, McCarthy A, Baxendale R, Guichard S, Magno L, Kessaris N, El-Bahrawy M, Yu P, and Katan M (2014) Tumor suppressor role of phospholipase C-ε in Ras-triggered cancers. Proc. Natl. Acad. Sci. U. S. A 111, 4239–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hu XT, Zhang FB, Fan YC, Shu XS, Wong AH, Zhou W, Shi QL, Tang HM, Fu L, Guan XY, Rha SY, Tao Q, and He C (2009) Phospholipase C-δ1 is a novel 3p22.3 tumor suppressor involved in cytoskeleton organization, with its epigenetic silencing correlated with high-stage gastric cancer. Oncogene 28, 2466–2475. [DOI] [PubMed] [Google Scholar]
- (28).Milanovic M, Radtke S, Peel N, Howell M, Carriere V, Joffre C, Kermorgant S, and Parker PJ (2012) Anomalous inhibition of c-Met by the kinesin inhibitor aurintricarboxylic acid. Int. J. Cancer 130, 1060–1070. [DOI] [PubMed] [Google Scholar]
- (29).Benchokroun Y, Couprie J, and Larsen AK (1995) Aurintricarboxylic acid, a putative inhibitor of apoptosis, is a potent inhibitor of DNA topoisomerase II in vitro and in Chinese hamster fibrosarcoma cells. Biochem. Pharmacol 49, 305–313. [DOI] [PubMed] [Google Scholar]
- (30).Li M, Shandilya SM, Carpenter MA, Rathore A, Brown WL, Perkins AL, Harki DA, Solberg J, Hook DJ, Pandey KK, Parniak MA, Johnson JR, Krogan NJ, Somasundaran M, Ali A, Schiffer CA, and Harris RS (2012) First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol 7, 506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hajicek N, Keith NC, Siraliev-Perez E, Temple BRS, Huang W, Zhang Q, Harden TK, and Sondek J (2019) Structural basis for the activation of PLC-γ isozymes by phosphorylation and cancer-associated mutations. eLife 8, e51700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Huang WG, Wang XY, Endo-Streeter S, Barrett M, Waybright J, Wohlfeld C, Hajicek N, Harden TK, Sondek J, and Zhang QS (2018) A membrane-associated, fluorogenic reporter for mammalian phospholipase C isozymes. J. Biol. Chem 293, 1728–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhang JH, Chung TD, and Oldenburg KR (1999) A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screening 4, 67–73. [DOI] [PubMed] [Google Scholar]
- (34).Nussinov R, and Tsai CJ (2013) Allostery in disease and in drug discovery. Cell 153, 293–305. [DOI] [PubMed] [Google Scholar]
- (35).van Westen GJ, Gaulton A, and Overington JP (2014) Chemical, target, and bioactive properties of allosteric modulation. PLoS Comput. Biol 10, e1003559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Motlagh HN, Wrabl JO, Li J, and Hilser VJ (2014) The ensemble nature of allostery. Nature 508, 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Su Z, Brown EC, Wang W, and MacKinnon R (2016) Novel cell-free high-throughput screening method for pharmacological tools targeting K+ channels. Proc. Natl. Acad. Sci. U. S. A. 113, 5748–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Holme MN, Rana S, Barriga HMG, Kauscher U, Brooks NJ, and Stevens MM (2018) A robust liposomal platform for direct colorimetric detection of sphingomyelinase enzyme and inhibitors. ACS Nano 12, 8197–8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lee CW, Lee KH, Lee SB, Park D, and Rhee SG (1994) Regulation of phospholipase C-β4 by ribonucleotides and the alpha subunit of Gq. J. Biol. Chem 269, 25335–25338. [PubMed] [Google Scholar]
- (40).Wang XQ, and Yu SP (2005) Novel regulation of Na, K-ATPase by Src tyrosine kinases in cortical neurons. J. Neurochem 93, 1515–1523. [DOI] [PubMed] [Google Scholar]
- (41).Kargacin ME, and Kargacin GJ (1997) Predicted changes in concentrations of free and bound ATP and ADP during intracellular Ca2+ signaling. Am. J. Physiol 273, C1416–1426. [DOI] [PubMed] [Google Scholar]
- (42).Pecoraro VL, Hermes JD, and Cleland WW (1984) Stability constants of Mg2+ and Cd2+ complexes of adenine nucleotides and thionucleotides and rate constants for formation and dissociation of MgATP and MgADP. Biochemistry 23, 5262–5271. [DOI] [PubMed] [Google Scholar]
- (43).Lacapere JJ, Bennett N, Dupont Y, and Guillain F (1990) pH and magnesium dependence of ATP binding to sarcoplasmic reticulum ATPase. Evidence that the catalytic ATP-binding site consists of two domains. J. Biol. Chem 265, 348–353. [PubMed] [Google Scholar]
- (44).David L, Walsh J, Sturm N, Feierberg I, Nissink JWM, Chen H, Bajorath J, and Engkvist O (2019) Identification of compounds that interfere with high-throughput screening assay technologies. ChemMedChem 14, 1795–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Chakravorty SJ, Chan J, Greenwood MN, Popa-Burke I, Remlinger KS, Pickett SD, Green DVS, Fillmore MC, Dean TW, Luengo JI, and Macarron R (2018) Nuisance compounds, PAINS filters, and dark chemical matter in the GSK HTS Collection. SLAS Discov 23, 532–545. [DOI] [PubMed] [Google Scholar]
- (46).Dahlin JL, Nissink JW, Strasser JM, Francis S, Higgins L, Zhou H, Zhang Z, and Walters MA (2015) PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem 58, 2091–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Irwin JJ, Duan D, Torosyan H, Doak AK, Ziebart KT, Sterling T, Tumanian G, and Shoichet BK (2015) An aggregation advisor for ligand discovery. J. Med. Chem 58, 7076–7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Polychronopoulos P, Magiatis P, Skaltsounis AL, Myrianthopoulos V, Mikros E, Tarricone A, Musacchio A, Roe SM, Pearl L, Leost M, Greengard P, and Meijer L (2004) Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J. Med. Chem 47, 935–946. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.