

Characterization of the worldwide genetic diversity of 447 P. vivax strains shows evidence for a Southeast Asian origin.
Abstract
Plasmodium vivax is the most common and widespread human malaria parasite. It was recently proposed that P. vivax originates from sub-Saharan Africa based on the circulation of its closest genetic relatives (P. vivax-like) among African great apes. However, the limited number of genetic markers and samples investigated questions the robustness of this hypothesis. Here, we extensively characterized the genomic variations of 447 human P. vivax strains and 19 ape P. vivax-like strains collected worldwide. Phylogenetic relationships between human and ape Plasmodium strains revealed that P. vivax is a sister clade of P. vivax-like, not included within the radiation of P. vivax-like. By investigating various aspects of P. vivax genetic variation, we identified several notable geographical patterns in summary statistics in function of the increasing geographic distance from Southeast Asia, suggesting that P. vivax may have derived from a single area in Asia through serial founder effects.
INTRODUCTION
Plasmodium vivax is the most common human malarial parasite responsible for 12 million to 22 million clinical cases per year (1). It is widespread in the tropical belt where almost 3 billion people are at risk of infection. It is the most frequent cause of human malaria (2), particularly in Central and Southeast Asia. Conversely, populations living in sub-Saharan Africa are protected from P. vivax transmission due to the absence of the Duffy antigen (i.e., Duffy negativity) at the surface of their red blood cells (3, 4). Historically, P. vivax remained understudied compared with Plasmodium falciparum because of the lower mortality rate of malaria caused by P. vivax. However, the recent emergence of new therapeutic resistance and the discovery of fatal cases due to P. vivax have questioned the benign status of P. vivax malaria (5). Moreover, the identification of P. vivax strains that can invade Duffy-negative red blood cells has been raising concerns over the potential spread of malaria in regions that are assumed to be protected (6). Today, P. vivax is considered a major public health threat (7). To put in place effective strategies for malaria control and elimination, we need to accumulate knowledge on the genetic structure of isolates from infected individuals, because this helps to understand the local patterns of malaria transmission and the dynamics of genetic recombination in natural P. vivax populations.
Early work on the patterns of genetic variation in P. vivax populations worldwide was limited to a small set of samples and genetic markers (mainly mitochondrial and autosomal markers) (8–16). Consequently, these genetic analyses yielded an incomplete picture of P. vivax evolutionary history. Recent technological breakthroughs allowed sequencing the whole P. vivax genome after parasite DNA enrichment from clinical blood samples (17, 18). In few years, several projects characterized P. vivax genetic variation at the whole-genome scale (19–22) and highlighted strong signals of recent evolutionary selection, partly due to known drug resistance genes. These studies released hundreds of complete genome sequences, but they restricted their investigation to a very small fraction of the worldwide P. vivax diversity. Consequently, a comprehensive picture of the worldwide genetic diversity and structure of P. vivax populations is still missing, and its evolutionary history and how it spread in the world are still poorly understood. Moreover, despite growing evidence suggesting an underlying widespread presence of P. vivax in all African malaria-endemic regions (23), too few complete genome sequences have been released for this area. Thus, a key challenge is to provide a worldwide understanding of P. vivax genetic variability genome wide with the ultimate aim of better understanding its past demographic history and origin.
The origin of the current P. vivax in humans has stimulated passionate and exciting debates for years. Some studies placed the origin of human P. vivax in Southeast Asia (“out-of-Asia” hypothesis) based on its phylogenetic position in a clade of malaria parasites that infect Asian monkeys (10, 13, 24–28). This scenario is also supported by genotyping data at 11 microsatellite markers collected in four continents, showing the highest microsatellite diversity in Southeast Asia (29). However, the Asian origin has been challenged by an “out-of-Africa” scenario after the recent discovery of a closely related Plasmodium species that circulates in wild-living African great apes (chimpanzees and gorillas) (28, 30–34). It has been suggested that this new lineage, hereafter referred to as P. vivax-like, gave rise to P. vivax in humans following a parasite transfer from African apes (33, 35). This new finding brought to the limelight a 70-year-old hypothesis according to which the high prevalence of Duffy negativity in sub-Saharan African human populations is the consequence of the long interaction between humans and malaria parasites, thus supporting an African origin of P. vivax (36). Yet, despite a century of research, the debate has not been settled yet. Although privileged, the out-of-Africa hypothesis remains debatable because the exact series of events linking the current human P. vivax populations and African great ape P. vivax-like remains unclear (37).
Here, to provide novel insights into the worldwide historical demography of P. vivax populations, we analyzed the genomic variations of 447 human P. vivax strains from 21 different countries. We also included 19 P. vivax-like strains from great apes to explore P. vivax evolutionary history from its origins to contemporary time and to specifically assess whether the parasite origin is consistent with the out-of-Africa or out-of-Asia hypothesis. To our knowledge, this dataset provides the most comprehensive characterization of P. vivax and P. vivax-like population genetic structure and diversity worldwide, with the identification of two distinct non-recombining P. vivax-like clades that circulate in sympatry among great apes. Our results demonstrate that P. vivax is a sister clade of P. vivax-like and is not included within the radiation of P. vivax-like, as previously suggested (33, 35). Last, several lines of evidence from summary statistics on geographic trends in P. vivax worldwide genetic variation support the hypothesis of a serial founder effect from a single origin in Southeast Asia.
RESULTS AND DISCUSSION
P. vivax and P. vivax-like genomic data and diversity
We processed and analyzed genome-wide data from 1154 P. vivax isolates sampled from all around the world. This dataset included 20 newly sequenced African isolates (Mauritania, n = 14; Ethiopia, n = 3; and Sudan, n = 3) because the worldwide sampling lacked P. vivax isolates from Africa [only 3 isolates from Madagascar (20) and 24 from Ethiopia (21)]. P. vivax samples were from 21 countries (769 from Asia, 338 from America, and 47 from Africa) (table S1). To trace P. vivax genetic ancestry, we also included 27 genome sequences of P. vivax-like isolated from African great apes (n = 10 newly sequenced in our laboratory and n = 17 from public databases: Gabon, n = 11; Cameroon, n = 5; and Ivory Coast, n = 1) (35, 37).
Because of the heterogeneity of DNA enrichment methods and sequencing technologies used to obtain the sequences in these datasets, our genome-wide data exhibited a broad range of sequencing depth coverage (fig. S1A). Therefore, to obtain reliable results, we retained only full-genome sequences with a minimum average sequencing depth of at least 5×. This reduced the P. vivax isolates from 1154 to 473 and the P. vivax-like isolates from 27 to 20. Then, we discarded isolates and variants with a fraction of missing call rate of >50% (fig. S1B). The final dataset included 466 full-genome sequences (447 for P. vivax and 19 for P. vivax-like isolates; Fig. 1A) in which we identified 2,005,455 high-quality single-nucleotide polymorphisms (SNPs).
Fig. 1. Geographical origin of Plasmodium isolates and patterns of genomic variation.
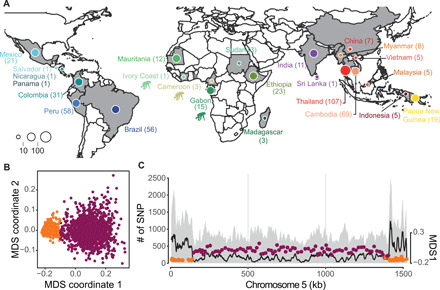
(A) Country of origin of the 447 P. vivax and 19 P. vivax-like isolates used in this study. Within each country, isolates were collected at different locations. The chimpanzee pictogram represents African P. vivax-like isolates. (B) Local variation of genetic relatedness along the genome of P. vivax isolates visualized by multidimensional scaling (MDS) based on the local PCA approach (39). Each point represents a non-overlapping genomic window of 100 SNPs. On the basis of the variation of the MDS-1 coordinate, each window was classified in two groups: subtelomeric hypervariable region (orange) and core region (purple). (C) Genome scan of the SNPs on chromosome 5 (left y axis) identified for both P. vivax and P. vivax-like. The gray area in the background shows the total number of SNPs identified (left y axis), and the black line represents the number of SNPs shared by the two species. The right y axis represents the MDS-1 coordinates against the middle point of each window. Windows were classified in the subtelomeric hypervariable region (orange) or the core region (purple).
The P. vivax genome has a heterogeneous genetic structure
Population structuration and demographic fluctuations have a global impact on genomic variations in natural populations. Conversely, local heterogeneity can be observed in genomic regions due to nonrandom factors, including structural chromosomal features, such as chromosomal inversions and heterochromatin, or due to selective forces that affect the local genetic diversity and recombination (38). These factors can lead to local genome variations in the genetic ancestry and in the relatedness of individual strains, confounding global patterns of genetic diversity and population structure. We identified potential local variations in individual relatedness along the genome using a local principal components analysis (PCA) (39). We detected contrasted patterns of individual relatedness along the P. vivax genome that defined two main genomic partitions: a small partition localized mainly at the subtelomeric ends of each chromosome (in orange in Fig. 1, B and C) and a larger partition at the center of each chromosome that encompassed 80% of the total SNP set (hereafter called the “core region,” in purple in Fig. 1, B and C). The subtelomeric regions that we identified as having distinct ancestry from the rest of the genome also coincided with hypervariable genomic regions (in orange in Fig. 1C and fig. S2). These regions are known to include hypervariable repetitive regions that often cause genotyping errors. Their exact coordinates have only been reported on a former version of the P. vivax genome assembly (19). The differences in genetic ancestry and strain relatedness provided by the two partitions were evident when looking at the local PCA (fig. S3). While the population structure recovered from the core regions displayed interpretable genetic patterns that were consistent with previous studies (19, 20), the genetic picture obtained from the SNPs in the subtelomeric hypervariable regions was much harder to interpret. Therefore, we excluded these regions from the subsequent analyses that focused only on the core regions on the central chromosomal region (table S2). This region represented ~21 Mb of the genome and included 1,610,445 high-quality SNPs (~1 SNP every 13 bp).
Last, we evaluated in each sample the within-sample parasite infection complexity by calculating the within-sample Fws metric (40). Monoclonal infections occurred in approximately 71% of samples, with the highest proportion of mixed infections observed in Thai and Cambodian isolates (fig. S4). The proportion of monoclonal infections was higher in the newly sequenced P. vivax individuals from Mauritania (92%, 11 of 12) but was not significantly different from the percentage found in Ethiopian isolates (61%, 14 of 23; Pearson’s χ2 test, P = 0.13), although it was skewed toward higher Fws scores, suggesting less frequent superinfection and/or cotransmission in this area. To avoid further possible bias due to multiple infections by several strains, we designed an SNP calling procedure to select within each sample the variant with the highest frequency. Therefore, for each site, one single variant was called per sample. In addition, we estimated the genome fraction that exhibited identity by descent (IBD) among pairs of samples from each population and selected only samples with low level of relatedness (IBD of >70%) for the downstream population genetic analyses (fig. S5). Our results provided insights into the parasite recent epidemiology history because populations with high levels of relatedness were considered as coming from regions with lower malaria transmission.
Evolutionary relationships between ape P. vivax-like and human P. vivax
First, we analyzed the phylogenetic relationships between P. vivax and P. vivax-like infecting great apes, the closest relatives of human P. vivax, by building a maximum likelihood (ML) genetic tree based on the SNP data for the 19 P. vivax-like and 447 P. vivax full-genome sequences. This analysis revealed three distinct clades (Fig. 2A). The first bifurcation in the tree splits ape-infecting strains from human-infecting strains and represented the strongest axis of genetic variation on the PCA (Fig. 2B; explaining 28.92% of the total genetic variance). Therefore, the whole-genome ML phylogenetic tree showed that the human P. vivax strains formed a sister clade of ape P. vivax-like strains, unlike previous results based on single-gene phylogenies (mitochondrial and autosomal markers) (33, 35). Liu et al. (33) and Loy et al. (35) suggested that P. vivax diversity was nested within the P. vivax-like radiation (33, 35). Incongruences between whole-genome (37) and single-gene (33, 35) tree topologies are common (41, 42) and may be caused, for instance, by incomplete lineage sorting (ILS) (43), introgression, or selection at a specific locus. ILS denotes the persistence of ancestral polymorphisms across multiple successive speciation events, followed by stochastic allele fixation in each descendant lineage. This process leads to phylogenetic incongruences among individual loci (44, 45). Disagreement between gene tree topologies may also be linked to the usage of mitochondrial markers that poorly represent the population history (46). Loy et al. (35) proposed that the shift in tree topologies—from sister clade to a nested relationship—was due to the incorporation of gorilla-infecting P. vivax-like samples and/or the inclusion of a new outgroup, Plasmodium carteri, a very rare parasite found in chimpanzees (35). However, the presence of two gorilla-infecting P. vivax-like lineages in our phylogeny partly contradicts this assertion. We could not build a whole-genome phylogeny using P. carteri sequences due to the unavailability of whole-genome sequencing data on this rare and elusive parasite. Nevertheless, our finding that P. vivax is a sister clade of P. vivax-like instead of a lineage included within the radiation of P. vivax-like challenges the view that P. vivax is included inside P. vivax-like diversity.
Fig. 2. P. vivax-like strains are structured in two distinct clades that form a sister monophyletic lineage to the human P. vivax.
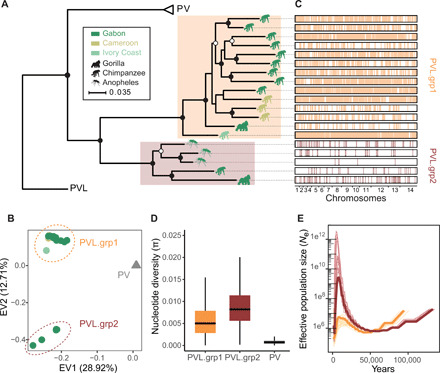
(A) ML phylogenetic tree illustrating the relationships between P. vivax (PV) and P. vivax-like (PVL) strains, rooted using P. cynomolgi (PC, M strain). Two distinct P. vivax-like groups were identified: PVL.grp1 (in orange) and PVL.grp2 (in brown). The animal pictograms on each leaf indicate the primate host, gorilla, chimpanzee, or mosquito (unknown primate host), colored according to the country of origin (Gabon, Cameroon, and Ivory Coast). Open and closed circles indicate nodes with >80 and >90% bootstrap support, respectively. (B) PCA displaying the two first eigen vectors (EVs) and the proportion of genetic variance they explain. (C) Genome-wide visualization of recent recombination events between P. vivax-like individuals. The horizontal black rectangles represent the 19 P. vivax-like genome sequences, and the vertical colored lines represent the recombining genomic segments on the recipient individual. Colors indicate the lineage membership of the donor individual. (D) Differences in nucleotide diversity (π) between P. vivax-like lineages and P. vivax strains. (E) Multiple sequentially Markovian coalescent (MSMC) estimates of the effective population size (Ne) in the two P. vivax-like groups (PVL.grp1 in orange and PVL.grp2 in brown). Lines in lighter color represent 50 bootstrap resampling replicates of randomly sampled segregating sites.
The second split in the phylogenomic tree (Fig. 2A) and on the PCA along the second PC axis (Fig. 2B; explaining 12.71% of the genetic variation) identified two distinct lineages among P. vivax-like strains (PVL.grp1 and PVL.grp2 in Fig. 2), composed of 5 and 14 strains, respectively. As P. vivax-like infects chimpanzees and gorillas, these two lineages might have been associated with host specialization. However, we found both lineages in both host species. Then, we tested the extent of recombination between these two genetically distinct P. vivax-like lineages to assess whether gene flow still occurs between them. We used the fastGEAR software (47) and the 19 P. vivax-like full-genome sequences to identify recently exchanged genomic fragments (i.e., fragments shared between two individuals) and their clade of origin. The fastGEAR clustering method recovered the two major P. vivax-like clades and identified 12,108 recently imported fragments, with a mean imported fragment length of 1775 bp (base pairs) (Fig. 2C). All the donor and recipient individuals of the recently imported fragments belonged to the same lineage, suggesting that no recent interlineage recombination occurred between the P. vivax-like clades. We confirmed this result by building a reticulation network for the 19 P. vivax-like individuals (fig. S6). The lack of reticulation between individuals belonging to different lineages confirmed the absence of recombination between the two P. vivax-like clades. Thus, our results demonstrate that although circulating in sympatry within the same host populations of great apes (i.e., the La Lékédi Park in Gabon), the two P. vivax-like lineages do not recombine, which suggests that they may represent distinct species. A similar subdivision was found in Plasmodium praefalciparum, the closest parasite to P. falciparum, but the status of these two clades was never investigated, as done here for P. vivax-like (48).
The nucleotide diversity (π) values of the two P. vivax-like lineages were approximately nine times higher than the value for P. vivax (Fig. 2D), in agreement with previous reports (35, 37). As it is unlikely that the mutation rates radically differ between P. vivax-like and P. vivax (49), this difference may reflect a higher effective size for P. vivax-like than for P. vivax. The lower nucleotide diversity observed in P. vivax could result from a bottleneck effect that occurred during the host shift, when the pathogen colonized humans (35). Similar differences were observed between African apes and humans (50). Genetic diversity is much lower in humans than in African apes due to the bottleneck effect linked to the modern human populations’ expansion out of Africa from a small number of founders who replaced the archaic forms of humans (e.g., Neanderthals). This could suggest a similar history for P. vivax in humans, and Africa as the point of origin of human P. vivax, as recently proposed (33). However, within the P. vivax-like samples, genetic diversity was higher in the PVL.grp1 than PVL.grp2 lineage. This may reflect sampling differences because PVL.grp1 isolates were collected in three African countries, whereas PVL.grp2 isolates came from a single location in the Park of La Lékédi in Gabon. The identification of two new Plasmodium lineages in African great apes, in such a limited geographic range, highlights the putative underestimated current and ancestral diversity of this genus in the African continent.
Last, we inferred historical fluctuations in the effective population size (Ne) for the two P. vivax-like lineages using a multiple sequentially Markovian coalescent (MSMC) approach. The analysis of genome variations indicated an ancient major expansion in genetic diversity for both clades, followed by a recent important decline (Fig. 2E). Unexpectedly, Ne increase was much higher for the PVL.grp2 lineage than for the PVL.grp1 lineage. The diverging trends in Ne values observed in each lineage (about 50 thousand years ago) suggest a putative ancient split of the two P. vivax-like lineages. This is consistent with the lack of recombination between these lineages and further supports their reproductive isolation.
Genetic structure of human P. vivax isolates collected worldwide
To gain insight into the genetic relationships among P. vivax populations around the world, we investigated the genetic structure of the 447 P. vivax isolates collected from 21 countries. We analyzed biallelic SNPs from the core region of the genome of unrelated samples (IBD of ≤0.7) using complementary population genomic approaches: a PCA that does not rely on any model assumptions (51), a model-based individual ancestry analysis implemented in the ADMIXTURE software (52), an ML phylogenetic tree of individual strains, and an analysis of the amount of genetic differentiation among population pairs using the FST index.
All analyses revealed consistent patterns that split the genetic variation primarily by continent (Fig. 3). The first principal component (PC) of the genetic variation (EV1) split Southeast Asian from African and American P. vivax populations, while the second axis (EV2) split African populations from the rest of the world (Fig. 3A and fig. S7). These three major geographical clusters were also identified on the ML tree (Fig. 3B) and displayed clearly distinct genetic ancestries, as estimated by simulating three ancestral clusters (K = 3) with the ADMIXTURE analysis (fig. S8). Within each cluster/continent, we observed additional subdivisions. The ADMIXTURE analysis (figs. S8 and S9) detected six main distinct genetic pools (three distinct ancestral populations in Asia, one in Africa, and two in America) (Fig. 3, C and D).
Fig. 3. The structure of P. vivax core region of the genome is mostly due to genetic isolation of natural populations.
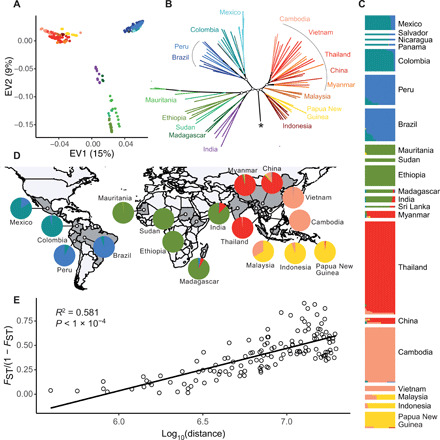
(A) PCA using the SNPs from the core region of the genome of the 447 P. vivax isolates showing the top two EVs and the accounted proportion of genetic variance. (B) ML phylogenetic tree built using SNP data from the core region of the genome. The star indicates the root of the tree, leading to the P. vivax-like outgroup. Specific internal nodes are highlighted with open circles and represent >80% bootstrap support. (C) The genetic ancestry proportion for individual strains, depicted as a vertical bar, was estimated using ADMIXTURE for each of the K = 6 inferred ancestral populations. (D) These same ancestral proportions for each population are displayed as pie charts on the world map. (E) Isolation by distance among populations. Pairwise estimates of FST/1 − FST were plotted against the corresponding geographical distances between countries. The Spearman correlation coefficient (R2) and the P value estimated using a Mantel test with 1000 permutations are shown.
Strains from India and Sri Lanka were genetically closer to African P. vivax populations than to other neighboring Asiatic groups (Fig. 3, A, B, and D). Such genetic exchange may result from the different trades that took place between the Indian subcontinent and Africa over the last two millennia. Moreover, movements of populations, and of diseases, occurred from India to East Africa and vice versa (8). The most recent exchange has been the migration of the human Karana population from the northwest of India to Madagascar at the end of the 17th century (53).
Across the Americas, P. vivax strains were structured in two distinct ancestral populations (Fig. 3, B to D). This is consistent with two putative evolutionary scenarios of the parasite in America. The first scenario hypothesizes that both ancestral populations originated from the isolation between Amazonian and non-Amazonian populations, after a single introduction during the European colonization in the 15th century (54). In the second scenario, two distinct waves of introduction occurred from the same or from different sources (15). Disentangling these two hypotheses will require more samples and statistical population genetic modeling (55).
P. vivax individuals collected in the Southeast Asia/Pacific region were structured in three distinct ancestral populations. In each ancestral population, the genetic differentiation between countries, estimated with the FST index, was weak (fig. S10), consistent with a relatively unrestricted gene flow between countries. Among these three ancestral populations, one was shared by P. vivax populations located in Indonesia and Papua New Guinea and was explained by their insular isolation. The other two ancestral populations may reflect the effect of the malaria-free corridor previously established through central Thailand, consistent with observations in P. falciparum (56). The Malaysian population was an admixture between two ancestral populations, at a contact zone in which admixture proportions progressively changed from one cluster to the other.
Last, at the global scale, we observed a very significant relationship between the genetic differentiation among populations [estimated using FST/(1 − FST)] and the logarithm of the geographic distance (Mantel test r2 = 0.56, P < 10−4; Fig. 3E). This is consistent with an isolation by distance in a two-dimensional habitat and suggests limited P. vivax dispersal across space (57).
Genetic evidences in favor of an Asian origin of human P. vivax
If P. vivax originated in Africa (33, 35), the phylogeny of Plasmodium individuals should have displayed a topology where the P. vivax-like root was located within or near the monophyletic clade of African P. vivax individuals. However, our ML tree based on whole-genome SNP data did not display such topology (Fig. 3B). On the contrary, the branching of P. vivax-like lineages splits P. vivax populations from Asia on one side and from India, Africa, and America on the other side. The most common ancestor to all P. vivax populations was closely related to populations from Indonesia and Papua New Guinea (Fig. 3B and fig. S11). Although P. vivax-like is the closest outgroup to all P. vivax populations, the exact branching position of P. vivax-like into P. vivax populations could be affected by various caveats: (i) the relatively large genetic divergence between P. vivax and P. vivax-like (net average genetic divergence da = 0.43 ± 0.051) compared with the divergence among P. vivax populations (average da = 0.015 ± 0.0025) could make P. vivax-like branching position uncertain; (ii) at the recent evolutionary scale, ILS and gene flow among populations could also influence the tree topology of P. vivax populations, making it more reticulated than a simple model of bifurcating populations.
Therefore, we estimated the historical relationships among P. vivax populations by modeling shared and population-specific genetic drifts among populations along a reticulated evolutionary tree using TreeMix (58). This allowed us to model how the sampled populations were related to their common ancestor through a graph of ancestral populations that included split and migration edges. The ML population tree topology rooted with P. vivax-like without migrations (fig. S12) displayed a similar topology as the individual-based ML phylogenetic tree (Fig. 3B). The addition of sequential migration events increased significantly the model likelihood with an optimum found with four migration edges (fig. S13). In this model, the TreeMix graph suggested that gene flow or admixture events might have occurred between neighboring territories (i.e., between Myanmar and India) (Fig. 4, A and B). Unlike the model without migration events, the four migration edges changed slightly the branching of the tree root and placed the most recent common ancestor to all P. vivax populations within populations from Southeast Asia. Despite the low node support (≤50% of the bootstrap replicates) on the branching of the ancestral group leading to China, Thailand, Vietnam, and Cambodia, these results are still consistent with the ML trees showing that the outgroup branch was closer to Southeast Asian populations than to African or American populations. This suggests that P. vivax Asian populations have kept a higher proportion of ancestral alleles compared with the African and American populations, which appeared more derived. However, similar to the ML phylogenetic analysis, the large genetic distances of the P. vivax-like outgroup call for caution in the placement of the root in P. vivax population tree built by TreeMix.
Fig. 4. Relationships and gene flow between P. vivax populations.
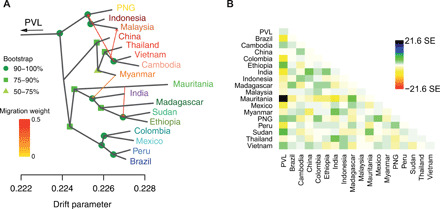
(A) TreeMix ML tree of P. vivax populations with four migration edges (arrows) rooted with P. vivax-like (PVL), including bootstrap node support. (B) TreeMix residual matrix from the tree in (A). PNG, Papua New Guinea.
To further explore P. vivax origin, we analyzed the distribution of genetic variability among populations in a spatial context. In a model of population range expansion and progressive colonization of new areas from a unique origin with serial founder events, several genetic trends are expected as a function of the increasing geographical distance from the origin (59–62): (i) The population genetic diversity should decline; (ii) the linkage disequilibrium (LD) should increase; (iii) the ancestral allele frequency (AAF) spectrum should flatten, indicating that derived alleles tend to be more frequent in populations at a greater distance; and (iv) the time to the most recent common ancestor (TMRCA) estimates within each population should decrease with the distance from the origin.
First, analysis of the geographical distribution of nucleotide diversity (π) showed that most P. vivax populations from Southeast Asia exhibited the highest genetic diversities, while African populations and American populations displayed intermediate and low values, respectively (figs. S14 and S15). Moreover, we observed the highest decrease in genetic diversity with increasing geographic distance when we considered Southeast Asia as the putative origin of P. vivax (Fig. 5, A and B). However, this geographic trend in genetic diversity could also be related to variations in the local population size or to very recent population dynamics (e.g., malaria eradication treatments, such as artemisinin). As the reported cases of P. vivax infections are higher in Asia than in the rest of the world [World Health Organization 2010 report (63)], we used the estimated number of cases as a covariate in our model. Nevertheless, the correlation between genetic diversity and geographic distance from Asia remained highly significant after controlling for the number of infections [generalized linear models (GLM), P < 2.37 × 10−5].
Fig. 5. The Southeast Asian origin of P. vivax is supported by patterns of nucleotide diversity, LD, and AAF.
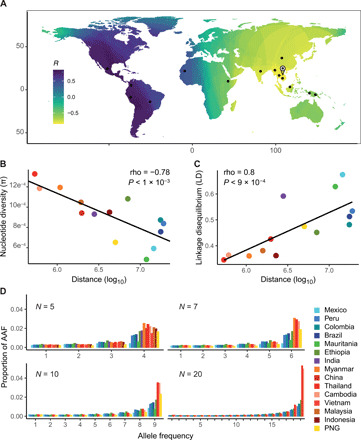
(A) Genetic diversity of P. vivax regressed on the great circle distance across the world. The value at each pixel of the map corresponds to the Spearman correlation coefficient (R) between the expected genetic diversity in each population and the geographic distance between this population and the pixel. Black dots represent the sampling sites used in the regression analysis (where n ≥ 5 individuals). The landmark on the map represents the most negative correlation coefficient indicative of the most probable source of the range expansion. (B) Correlation between nucleotide diversity (π) and geographic distance, measured as the distance between the milestone indicated in (A) and each population. (C) Correlation between LD at 100 bp (measured as the normalized R2) and the geographic distance. (D) Histograms of AAF proportions in P. vivax populations with an increasing sample size (N = 5, 7, 10, and 20).
Next, analysis of LD variations among P. vivax populations showed a very strong positive correlation between LD at 100 bp within each population and the geographic distance to the previously identified putative Asian origin (Fig. 5C and fig. S15). This pattern remained significant when controlling for variations in the number of cases per country (GLM, P = 0.0396).
The patterns of AAF distributions can also inform on P. vivax origin and its worldwide colonization routes (60). Using P. vivax-like and Plasmodium cynomolgi as outgroups, we could polarize the allele frequency spectrum and identify the ancestral and derived allelic states for ~94,000 SNPs. Figure 5D shows the corresponding AAF spectrum when considering an increasing sample size (5, 7, 10, and 20 genomes) in each population to compare our heterogeneously sized populations. Regardless of the number of full-genome sequences considered, populations located in Southeast Asia (red-orange shades) always displayed more SNPs with high AAFs and fewer SNPs with low AAFs. Conversely, in non-Asian populations, AAF distributions progressively flattened. This is consistent with the phylogenetic and population tree analyses done with TreeMix (Figs. 3B and 4A).
Last, we used the MSMC approach and five individuals from 14 P. vivax populations to obtain estimates of the historical population size changes and of the TMRCA. The MSMC curves revealed that P. vivax populations displayed two effective population size dynamics (Fig. 6A). The effective population sizes (Ne) of Southeast Asian populations were relatively stable, and some experienced a steady increase in a more distant past, followed by a quick decline. Conversely, in African and American populations, Ne have mainly steadily decreased since the TMRCA. The MSMC analysis showed that most populations from Southeast Asia exhibited much older TMRCA values than the African and American populations (Fig. 6B and fig. S16).
Fig. 6. Coalescent-based inference of the demographic history in each P. vivax population.
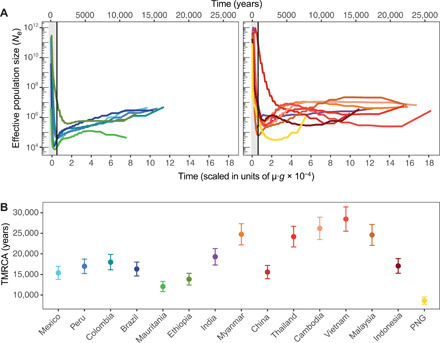
(A) Effective population size (Ne) variations back to the TMRCA inferred using MSMC and (B) inferred TMRCA in each population. The analysis was performed using five individuals from each population, assuming a mutation rate per generation (μ·g) of 1.158 × 10−9 and a generation time (g) of 0.18. The gray area represents the first 1000 generations, at which low resolution of the recent past effective population size is observed, likely due to the presence of sites with high probability of being called inaccurately.
Together, these results indicate that P. vivax populations display a decrease in genetic diversity and an increase in LD with the increasing geographic distance from Southeast Asia, and are consistent with a historical model of colonization with serial founder events from an origin located in Southeast Asia. The higher proportion of ancestral alleles and older TMRCA found in the Southeast Asian populations are additional evidences supporting this scenario. These four different analyses constitute only two independent lines of evidence because our genetic trends based on π, LD, and TMRCA are interrelated and rely on genetic diversity. On the basis of P. vivax and P. vivax-like phylogenetic position within a radiation of Plasmodium parasites infecting Asian monkeys, we hypothesize that these two parasite lineages resulted from two independent waves of colonization from Asia. The first wave gave rise to P. vivax-like in African apes (with some unknown host species bridging Africa to Asia), and the second wave gave rise to the current P. vivax populations.
However, our conclusions need to be considered with caution for the following reasons. Although population genetic theories offer testable hypotheses to investigate P. vivax origin, the recent epidemiological history of each population might have influenced the local patterns of genetic variation. For instance, malaria eradication campaigns, such as the programs carried out in the 1950s and 1960s to stop malaria transmission (64), could have generated strong bottlenecks that affected local populations, thus preventing us from accurately inferring the evolutionary history of the affected populations based on their summary statistics. Populations from Mexico, Mauritania, and Papua New Guinea may be examples of populations in which the high level of recent common ancestry is associated with low genetic diversity and high LD. To take this issue into account, we corrected all our genetic trends for the current demographical variations between populations. As all correlations remained significant, it is unlikely that the local epidemiology might have affected the full global patterns at the world scale (i.e., local epidemiology is different for each P. vivax population). Furthermore, we used the powerful MCMC2 method to trace the evolutionary history of the entire population back to the most recent common ancestor, without being negatively influenced by very recent demographic changes.
An additional limitation of our study was the heterogeneity in sample sizes and sample collection schemes that prevented us to compute summary statistics with the same resolution for all populations. Concerning sample sizes, we calculated nucleotide diversity and LD for populations with at least five individuals, as a trade-off between including the highest possible number of populations and minimizing the bias in nucleotide diversity and LD estimation. Consequently, as presented in our rarefaction analysis, small-size populations exhibited high variation around their expectation. We think that this issue is not a major limitation because the large number of used genetic markers should offset the small sample sizes (65, 66). Nevertheless, increasing the size of the small populations and collecting new populations, especially those lying between the supposed source populations in Asia and in Africa (e.g., populations from the Middle East), would be particularly useful to better characterize the patterns of P. vivax genetic variation worldwide and to refine the question of the origin of P. vivax range expansion. In addition, collecting samples using the same method, at the same time and during the same period, and in similar geographic areas would be the best sampling approach to study the evolutionary origin and history of a pathogen. Unfortunately, such ideal sampling is currently unavailable and extremely complex to put in place. Therefore, this study presents results obtained using up-to-date population genomic tools that can efficiently identify the origin of organisms (e.g., P. falciparum) (61). Last, to determine the impact of closely related isolates on nucleotide diversity, LD, and TMRCA estimations, we analyzed our dataset using a stringent IBD cutoff of 0.25 (instead of 0.7). This new analysis gave similar patterns as before, with a stronger coefficient of regression (figs. S17 and S18).
Loy et al. (35) proposed an alternative hypothesis according to which P. vivax spread from Africa to Asia, was then eliminated from Africa due to the evolution of Duffy negativity, and was reintroduced in Africa by more recent migration waves from Asia. Although this scenario provides a potential explanation of Duffy negativity appearance in Africa, unfortunately it does not fit with the genetic variability patterns of P. vivax populations. According to the scenario by Loy et al. (35), populations from the Middle East and India (located on the migratory route between Africa and Southeast Asia) should display traces of admixture from both migration waves and higher genetic diversity. Our current results on the genetic variation of populations from India (Figs. 3C and 4B) and from the Middle East (29) do not support this scenario. Nevertheless, this hypothesis remains possible if all ancient populations disappeared from the world, except the Southeast Asian populations. However, testing this scenario can be very difficult in the absence of ancient P. vivax strains to study.
This study provides one of the most detailed views of the worldwide distribution of P. vivax population genetic diversity and demographic history. Our work investigated not only the phylogenetic relationship between P. vivax infecting human and apes but also different features of P. vivax genetic variation worldwide to test different hypotheses of an African or Asian origin. We showed that P. vivax is a sister group and not a sublineage of P. vivax-like. Genetic diversity in P. vivax-like is richer than in P. vivax, consistent with a strong bottleneck in this lineage that gave rise to P. vivax. Our results based on whole-genome sequencing data support an out-of-Asia origin, rather than an African origin, for the world populations of P. vivax, with a signal of stepping-stone colonization events accompanied by serial founder effects. Efforts should now be focused on precisely describing P. vivax demographic and selective history in specific regions (e.g., South America, Africa, and Europe) and on estimating when and how these different regions were colonized. The question of the host of origin still remains open.
MATERIALS AND METHODS
African P. vivax sample collection and ethical statements
Twenty P. vivax isolates from Mauritania (N = 14), Ethiopia (N = 3), and Sudan (N = 3) were sequenced. P. vivax infection was detected by microscopy analysis, polymerase chain reaction amplification of the cytochrome b gene, and/or rapid diagnostic test. Samples were collected from P. vivax–infected patients after informed consent and ethical approval by the local institutional review board of each country. The informed consent procedure for the study consisted in the presentation of the study aims to the community followed by inviting adults for enrollment. At the time of sample collection, the study purpose and design were explained to each individual and a study information sheet was provided before collection of the oral informed consent. Only oral consent was required because the study did not present any harm for the subjects and did not involve procedures that needed a written consent. The oral consent process was consistent with the ethical expectations for each country at the time of enrollment and was approved by each country, and these procedures were approved by local ethics committees. The privacy and confidentiality of the collected data were ensured by the sample anonymization before the study start. For samples from Mauritania, the study was approved by the pediatric services of the National Hospital, the Cheikh Zayed Hospital, and the Direction régionale à l’Action Sanitaire de Nouakchott (DRAS)/Ministry of Health in Mauritania. No ethics approval number was given. For samples from Sudan, no specific consent was required; the human clinical, epidemiological, and biological data were collected in the Centre National de Réferénce du Paludisme (CNRP) database and analyzed in accordance with the common public health mission of all French National Reference Centers (www.legifrance.gouv.fr/affichTexte.do?cidTexte=JORFTEXT000000810056&dateTexte=&categorieLien=id). The study of biological samples obtained in the framework of medical care was considered as noninterventional research (article L1221-1.1 of the French public health code) and only required the patient’s non-opposition during sampling (article L1211-2 of the French public health code). All data collected were anonymized before analyses. For samples from Ethiopia, this study was approved by the Ethical Clearance Committee of the Haramaya University—College of Health and Medical Sciences, and from the Harari and Oromia Regional State Health Bureau in Ethiopia.
P. vivax-like sample collection and ethical statements
P. vivax-like samples were obtained during a continuous survey of great ape Plasmodium infections carried out in the Park of La Lékédi, in Gabon, in collaboration with the Centre International de Recherches Médicales de Franceville (CIRMF). The La Lékédi Park is a sanctuary for ape orphans in Gabon. During sanitary controls, blood samples were collected and leukocyte depletion was carried by CF11 cellulose column filtration (67) in the field, before storage at −20°C at the CIRMF. The animal well-being was guaranteed by the veterinarians of the “Park of La Lékédi” and the CIRMF who were responsible for all sanitary procedures (including blood collection). All animal work was performed according to the relevant national and international guidelines. These investigations were approved by the government of the Republic of Gabon and by the Animal Life Administration of Libreville, Gabon (no. CITES 00956). Our study did not involve randomization or blinding.
P. vivax-like samples were also obtained from sylvatic anopheles mosquitoes collected with Centers for Disease Control (CDC) light traps in the forest of the Park of La Lékédi during a longitudinal study (34). Anopheles mosquitoes were morphologically identified using standard keys (68), stored in liquid nitrogen, and then kept at −80°C at the CIRMF until analysis.
Genomic DNA was extracted from each sample using the DNeasy Blood and Tissue Kit (Qiagen, France) according to the manufacturer’s recommendations. P. vivax-like samples were identified by amplifying and sequencing the Plasmodium cytochrome b (Cytb) or cytochrome oxidase 1 (Cox1) gene, as described elsewhere (24, 69). In total, 10 P. vivax-like samples were selected: 3 from gorillas, 4 from chimpanzees, and 3 from anopheles mosquitoes.
African P. vivax and P. vivax-like genome sequencing
To avoid host DNA contamination, selective whole-genome amplification (sWGA) was used to enrich submicroscopic DNA levels, as already described elsewhere (70). This technique preferentially amplifies P. vivax and P. vivax-like genomes from a set of target DNAs. For each sample, DNA amplification was carried out using the strand-displacing phi29 DNA polymerase and two sets of P. vivax–specific primers that target short (6 to 12 nucleotides) motifs commonly found in the P. vivax genome (set1920 and PvSet1) (69, 70). For each set of primers, 30 ng of input DNA was added to a 50-μl reaction mixture containing 3.5 μM of each sWGA primer, 30 U of phi29 DNA polymerase enzyme (New England Biolabs), 1× phi29 buffer (New England Biolabs), 4 mM deoxynucleoside triphosphates (Invitrogen), 1% bovine serum albumin, and sterile water. DNA amplifications were carried out in a thermal cycler with the following program: a ramp down from 35° to 30°C (10 min per degree), 16 hours at 30°C, 10 min at 65°C, and hold at 4°C. For each sample, the products of the two amplifications (one per primer set) were purified with AMPure XP beads (Beckman Coulter) at a 1:1 ratio according to the manufacturer’s recommendations and pooled at equimolar concentrations. Last, each sWGA library was prepared using the two pooled amplification products and a Nextera XT DNA kit (Illumina) according to the manufacturer’s protocol. Samples were then pooled and clustered on a HiSeq 2500 sequencer in Rapid Run mode with 2 × 250-bp paired end reads.
Genomic data from public and/or published data archives
For the worldwide comparative study of P. vivax genetic diversity, population structure, and evolution, a large literature search was carried out to identify previously published genomic datasets. This allowed the identification and extraction of the fastq files of 1134 P. vivax isolates from the following 12 projects: PRJEB10888 (21), PRJEB2140 (19), PRJNA175266 (71), PRJNA240356 (20), PRJNA240452, PRJNA240531, PRJNA271480 (20), PRJNA284437 (72), PRJNA350554 (73), PRJNA420510 (74), PRJNA432819 (75), and PRJNA65119 (76). In addition, 17 P. vivax-like sequenced genomes from Cameroon, Ivory Coast, and Gabon were from two other projects (PRJNA474492 and PRJEB2579) (35, 37). Published genomes were downloaded from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) (77) and converted into fastq files using the NCBI SRA Toolkit (fastq-dump –split-3). Details about the genomic samples are provided in table S1.
P. vivax and P. vivax-like read mapping and variant calling
Newly generated and previously published sequencing reads were trimmed to remove adapters and preprocessed to eliminate low-quality reads (--quality-cutoff=30) using the program cutadapt (78). Reads shorter than 50 bp and containing “N” were discarded (--minimum-length=50 --max-n=0). Sequenced reads were aligned to the PVP01 (79) P. vivax reference genome using bwa-mem (80). Here, a first filter was applied to exclude isolates with an average genome coverage depth lower than 5×. The Genome Analysis Toolkit (GATK, version 3.8.0) (81) was used to identify SNPs in each isolate following the GATK best practices. Duplicate reads were marked using the MarkDuplicates tool from the Picard tools (82) with default options. Local realignment around indels was performed using the IndelRealigner tool from GATK. Variant calling was carried out using the HaplotypeCaller module in GATK on reads mapped with a “reads minimum mapping quality” of 30 (-mmq 30) and a minimum base quality of >20 (--min_base_quality_score 20). Low-quality SNPs were eliminated according to GATK best-practice recommendations, and SNPs at sites covered by at least five reads were kept. Last, the VCF (variant call format) files for the different isolates were merged using the GATK module GenotypeGVCFs. Because of our tolerant genome coverage depth cutoff (5×), a second round of data filtering was carried out to exclude variants and isolates with a missing call rate higher than 50%. The within-host infection complexity was assessed by calculating the FWS (40) with the R package moimix (http://bahlolab.github.io/moimix/). An FWS threshold of >0.95 was used as a proxy of monoclonal infection.
Heterogenous patterns of relatedness among Plasmodium genomes
Highly related samples and clones can generate spurious population structure signals, bias estimators of population genetic variation, and violate the assumptions of the model-based population genetic approaches used in this study (e.g., ADMIXTURE, TreeMix, and MSMC2) (83). Therefore, the relatedness between haploid genotype pairs was measured by estimating the pairwise IBD between strains within populations using the hmmIBD program, with the default parameters for recombination rate, and genotyping error rate, and using the allele frequencies estimated by the program (84). Isolate pairs that shared >70% of IBD were considered highly related. Only one strain of each related pair was kept for the downstream analyses.
To identify heterogenous patterns of relatedness among Plasmodium genomes, a local PCA was performed, as described by Li and Ralph (39). Briefly, the P. vivax genome was divided in 1439 contiguous and nonoverlapping windows of 100 SNPs. On each window, the PCA was applied and the individual isolate scores for the first two PC were stored. To measure the similarity among windows, a Euclidian distance matrix was computed among windows based on the PC scores. Last, multidimensional scaling (MDS) was used to visualize the relationships among windows. A set of three coordinates was used to visualize the patterns of relatedness shared by windows. Because the local PCA was sensitive to missing data, the top 304 P. vivax genomes with an SNP missing discovery rate lower than 25% were selected for this analysis.
Analysis of genetic recombination among P. vivax-like genomes
Recombination among P. vivax-like strains was inferred using the fastGEAR software (47). From the SNPs present in the core region of the genomes, a multi-fasta file was created and used as input for fastGEAR that was launched with the iteration number set to 15 (default). Recent recombination events were detected with the Bayesian factor (BF) > 1 (default) and referred to as interlineage recombination for which the donor-recipient relation can be inferred. A phylogenetic network was generated using the SplitsTree 4 software (85) and the polymorphic sites present in the core region of the genome.
Phylogenetic analysis
ML phylogenetic trees were inferred with IQ-TREE (86) using the general time reversible (GTR) model with ascertainment bias correction. The amount of missing data present in the genome alignment was minimized by selecting 85 P. vivax individuals with the lowest missing call rate (up to 5 individuals per population). For P. vivax-like, all 19 isolates were used in the phylogenetic analysis presented in Fig. 2A, but only the 12 isolates with a missing call rate lower than 5% were used for the phylogeny presented in Fig. 3B. Consequently, the results presented in Fig. 2A were inferred on the basis of 28,000 SNPs, and the results in Fig. 3B were inferred on the basis of 87,000 SNPs. Phylogenies were plotted with FigTree v.1.4.3 (87), and the node reliability was assessed by performing 100 bootstrap replicates. P. cynomolgi [strain M (88)] was used as outgroup, and P. cynomolgi genomic coordinates were converted to P. vivax coordinates using the liftOver genome tool (89).
Population structure analysis and population evolutionary history
PCA and ADMIXTURE analyses were performed after selecting only biallelic SNPs present in the P. vivax core region of the genome and excluding singletons from the dataset. The variants were LD-pruned to obtain a set of unlinked variants using the option --indep-pairwise 50 5 0.7 in PLINK (90). PCA was performed using PLINK and plotted in R. The population structure and the estimation of individual genetic ancestry to various numbers (K) of ancestral populations were performed using ADMIXTURE (52). Each ADMIXTURE analysis was repeated 100 times using a subset of 100,000 SNPs randomly selected throughout the core region of the genome. Each run was launched with a random seed, with a K value ranging from 2 to 20. The most likely number of ancestral populations (K) was determined using the cross-validation error rate (52). Then, CLUMPAK (91) was used to analyze ADMIXTURE outputs and compute ancestry proportions.
Genetic differentiation among populations was estimated with the Weir and Cockerham’s estimator of the FST index using VCFtools (92). This metric accounts for sample size differences among populations.
Demographic history analysis and range expansion
TreeMix was used to investigate the historical population relationships by estimating an ML population tree, the amount of genetic drift in each population, and the number of migration events that best fitted the data (58). The number of migration events (m) that best fitted the data was calculated by running TreeMix 15 times for each m value, with m ranging from 1 to 12. LD among SNPs was taken into account by grouping SNPs by blocks of 500 (-k 500). The optimal m value (m = 4) was estimated using the OptM R package (https://cran.r-project.org/web/packages/OptM/index.html). Then, a consensus ML tree including bootstrap node support was obtained by running TreeMix 100 times (m = 4 -k 500) and post-processing using the BITE R package (93).
To determine which model (out-of-Asia or out-of-Africa) of population range expansion best described the demographic history of P. vivax, summary statistics were computed to capture different features of genetic variation within populations. Each analysis was computed for each of the populations defined by their country of origin using the SNPs present in the reference core region of the genome. To minimize the impact of missing calls in our analyses, only individuals with <5% of missing data were included in the following analysis. The genome-wide nucleotide diversity (π) was calculated per site using VCFtools and divided by the number of nucleotides in the core region of the genome (20,844,131 bp) to derive an approximation of the genome-wide average nucleotide diversity (π) for each population, as described in (21, 22). To take into account the sample size heterogeneity effect on π estimation, the rarefaction approach was used with five genomes resampled in each population to estimate and compare values among populations. This number provided a trade-off between minimizing the π value estimation variance due to sample size heterogeneity and keeping as many populations as possible in the analysis (fig. S14).
The nucleotide diversity regression in function of the geographic distance was calculated by dividing the world map into a 400 pixel × 500 pixel two-dimensional lattice and by considering each pixel as a potential source of P. vivax geographic range expansion. The geographic distances between populations and the focal pixel were determined using the R package geosphere to calculate the Spearman correlation coefficient between nucleotide diversity and geographic distance. The pixel with the lowest negative correlation coefficient was considered as indicative of the origin of the expansion range (94, 95).
LD decay was estimated by randomly sampling five individuals from each population using the PopLDdecay tool (96) that calculates the genotype correlation coefficient R2 for pairs of SNPs at a maximum distance range of 10 kb. Regression between LD at 100 bp and the geographic distance to the potential source was calculated using the R package geosphere. Last, the biallelic SNPs of each P. vivax population were polarized using the shared alleles identified between P. vivax-like individuals and P. cynomolgi (custom scripts). The final unfolded site frequency spectrum (SFS) was computed for a total of 93,652 SNPs.
Estimates of historical variation in effective population size
The MSMC2 program (97) and the core region of the genome of all chromosomes were used to infer historical changes in Ne and to estimate the TMRCA in the P. vivax and P. vivax-like populations. Using the mutation rate (μ) per generation (g) previously estimated for P. falciparum (49), the estimates were scaled to years and Ne, assuming μ·g = 1.158 × 10−9 and a generation time of g = 0.18. MSMC2 was run for 20 iterations with a fixed recombination rate. The MSMC2 method was applied by randomly selecting five individuals with a missing call rate lower than 5% from each population. The error around the parameter estimates was estimated using 50 bootstrapped replicates by randomly resampling the segregating sites used in the analysis.
Acknowledgments
We acknowledge the IRD itrop HPC (South Green Platform) at IRD Montpellier for providing HPC resources that have contributed to the research results reported within this paper. URL: https://bioinfo.ird.fr and www.southgreen.fr. We are grateful to E. Andermarcher for proofreading this article. Funding: This study was supported by ANR T-ERC EVAD, ANR JCJC GENAD, PEPS ECOMOB MOV 2019, CNRS, and MGX Montpellier sequencing facility. J.D. was supported by the Fondation pour la recherche Médicale (FRM, ARF20170938823) as well as by the Marie-Curie EU Horizon 2020 Marie-Skłodowska-Curie research and innovation program grant METHYVIREVOL (contract number 800489). Author contributions: Conception: V.R. and F.P.; funding acquisition: V.R. and J.D.; biological data acquisition and management: V.R., A.B., B.N., L.B., S.H., C.A., C.S., J.-F.T., and P.D.; sequence data acquisition: A.B. and V.R.; method development and data analysis: J.D. and M.C.F.; interpretation of the results: J.D., M.C.F., F.P., and V.R.; drafting of the manuscript: J.D. and V.R.; reviewing and editing of the manuscript: J.D., F.R., M.C.F., F.P., and V.R. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The Illumina sequence reads generated on the new P. vivax and P. vivax-like samples have been deposited in the National Center for Biotechnology Information (NCBI) under the bioproject name PRJNA720520. The Illumina sequence reads generated on the new P. vivax samples from Mauritania, Sudan, and Ethiopia have been deposited in the European Nucleotide Archive (ENA) under the accession codes listed in table S1. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/18/eabc3713/DC1
REFERENCES AND NOTES
- 1.Battle K. E., Lucas T. C. D., Nguyen M., Howes R. E., Nandi A. K., Twohig K. A., Pfeffer D. A., Cameron E., Rao P. C., Casey D., Gibson H. S., Rozier J. A., Dalrymple U., Keddie S. H., Collins E. L., Harris J. R., Guerra C. A., Thorn M. P., Bisanzio D., Fullman N., Huynh C. K., Kulikoff X., Kutz M. J., Lopez A. D., Mokdad A. H., Naghavi M., Nguyen G., Shackelford K. A., Vos T., Wang H., Lim S. S., Murray C. J. L., Price R. N., Baird J. K., Smith D. L., Bhatt S., Weiss D. J., Hay S. I., Gething P. W., Mapping the global endemicity and clinical burden of Plasmodium vivax, 2000–17: A spatial and temporal modelling study. Lancet 394, 332–343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howes R. E., Battle K. E., Mendis K. N., Smith D. L., Cibulskis R. E., Baird J. K., Hay S. I., Global epidemiology of Plasmodium vivax. Am. J. Trop. Med. Hyg. 95, 15–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horuk R., Chitnis C. E., Darbonne W. C., Colby T. J., Rybicki A., Hadley T. J., Miller L. H., A receptor for the malarial parasite Plasmodium vivax: The erythrocyte chemokine receptor. Science 261, 1182–1184 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Chaudhuri A., Zbrzezna V., Polyakova J., Pogo A. O., Hesselgesser J., Horuk R., Expression of the Duffy antigen in K562 cells. Evidence that it is the human erythrocyte chemokine receptor. J. Biol. Chem. 269, 7835–7838 (1994). [PubMed] [Google Scholar]
- 5.R. N. Price, E. Tjitra, C. A. Guerra, S. Yeung, N. J. White, N. M. Anstey, Vivax Malaria: Neglected and Not Benign (American Society of Tropical Medicine and Hygiene, 2007). [PMC free article] [PubMed] [Google Scholar]
- 6.Ménard D., Barnadas C., Bouchier C., Henry-Halldin C., Gray L. R., Ratsimbasoa A., Thonier V., Carod J.-F., Domarle O., Colin Y., Bertrand O., Picot J., King C. L., Grimberg B. T., Mercereau-Puijalon O., Zimmerman P. A., Plasmodium vivax clinical malaria is commonly observed in Duffy-negative Malagasy people. Proc. Natl. Acad. Sci. U.S.A. 107, 5967–5971 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baird J. K., Evidence and implications of mortality associated with acute Plasmodium vivax malaria. Clin. Microbiol. Rev. 26, 36–57 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Culleton R., Coban C., Zeyrek F. Y., Cravo P., Kaneko A., Randrianarivelojosia M., Andrianaranjaka V., Kano S., Farnert A., Arez A. P., Sharp P. M., Carter R., Tanabe K., The origins of African Plasmodium vivax; insights from mitochondrial genome sequencing. PLOS ONE 6, e29137 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunawardena S., Karunaweera N. D., Ferreira M. U., Phone-Kyaw M., Pollack R. J., Alifrangis M., Rajakaruna R. S., Konradsen F., Amerasinghe P. H., Schousboe M. L., Galappaththy G. N. L., Abeyasinghe R. R., Hartl D. L., Wirth D. F., Geographic structure of Plasmodium vivax: Microsatellite analysis of parasite populations from Sri Lanka, Myanmar, and Ethiopia. Am. J. Trop. Med. Hyg. 82, 235–242 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jongwutiwes S., Putaporntip C., Iwasaki T., Ferreira M. U., Kanbara H., Hughes A. L., Mitochondrial genome sequences support ancient population expansion in Plasmodium vivax. Mol. Biol. Evol. 22, 1733–1739 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leclerc M. C., Durand P., Gauthier C., Patot S., Billotte N., Menegon M., Severini C., Ayala F. J., Renaud F., From The Cover: Meager genetic variability of the human malaria agent Plasmodium vivax. Proc. Natl. Acad. Sci. U.S.A. 101, 14455–14460 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miao M., Yang Z., Patch H., Huang Y., Escalante A. A., Cui L., Plasmodium vivax populations revisited: Mitochondrial genomes of temperate strains in Asia suggest ancient population expansion. BMC Evol. Biol. 12, 22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mu J., Joy D. A., Duan J., Huang Y., Carlton J., Walker J., Barnwell J., Beerli P., Charleston M. A., Pybus O. G., Su X., Host switch leads to emergence of Plasmodium vivax malaria in humans. Mol. Biol. Evol. 22, 1686–1693 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Orjuela-Sánchez P., Sá J. M., Brandi M. C. C., Rodrigues P. T., Bastos M. S., Amaratunga C., Duong S., Fairhurst R. M., Ferreira M. U., Higher microsatellite diversity in Plasmodium vivax than in sympatric Plasmodium falciparum populations in Pursat, Western Cambodia. Exp. Parasitol. 134, 318–326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodrigues P. T., Valdivia H. O., de Oliveira T. C., Alves J. M. P., Duarte A. M. R. C., Cerutti-Junior C., Buery J. C., Brito C. F. A., de Souza J. C. Jr., Hirano Z. M. B., Bueno M. G., Catão-Dias J. L., Malafronte R. S., Ladeia-Andrade S., Mita T., Santamaria A. M., Calzada J. E., Tantular I. S., Kawamoto F., Raijmakers L. R. J., Mueller I., Pacheco M. A., Escalante A. A., Felger I., Ferreira M. U., Human migration and the spread of malaria parasites to the New World. Sci. Rep. 8, 1993 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor J. E., Pacheco M. A., Bacon D. J., Beg M. A., Machado R. L., Fairhurst R. M., Herrera S., Kim J.-Y., Menard D., Póvoa M. M., Villegas L., Mulyanto M., Snounou G., Cui L., Zeyrek F. Y., Escalante A. A., The evolutionary history of Plasmodium vivax as inferred from mitochondrial genomes: Parasite genetic diversity in the Americas. Mol. Biol. Evol. 30, 2050–2064 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Auburn S., Marfurt J., Maslen G., Campino S., Ruano Rubio V., Manske M., MacHunter B., Kenangalem E., Noviyanti R., Trianty L., Sebayang B., Wirjanata G., Sriprawat K., Alcock D., MacInnis B., Miotto O., Clark T. G., Russell B., Anstey N. M., Nosten F., Kwiatkowski D. P., Price R. N., Effective preparation of Plasmodium vivax field isolates for high-throughput whole genome sequencing. PLOS ONE 8, e53160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melnikov A., Galinsky K., Rogov P., Fennell T., Van Tyne D., Russ C., Daniels R., Barnes K. G., Bochicchio J., Ndiaye D., Sene P. D., Wirth D. F., Nusbaum C., Volkman S. K., Birren B. W., Gnirke A., Neafsey D. E., Hybrid selection for sequencing pathogen genomes from clinical samples. Genome Biol. 12, R73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pearson R. D., Amato R., Auburn S., Miotto O., Almagro-Garcia J., Amaratunga C., Suon S., Mao S., Noviyanti R., Trimarsanto H., Marfurt J., Anstey N. M., William T., Boni M. F., Dolecek C., Hien T. T., White N. J., Michon P., Siba P., Tavul L., Harrison G., Barry A., Mueller I., Ferreira M. U., Karunaweera N., Randrianarivelojosia M., Gao Q., Hubbart C., Hart L., Jeffery B., Drury E., Mead D., Kekre M., Campino S., Manske M., Cornelius V. J., MacInnis B., Rockett K. A., Miles A., Rayner J. C., Fairhurst R. M., Nosten F., Price R. N., Kwiatkowski D. P., Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat. Genet. 48, 959–964 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hupalo D. N., Luo Z., Melnikov A., Sutton P. L., Rogov P., Escalante A., Vallejo A. F., Herrera S., Arévalo-Herrera M., Fan Q., Wang Y., Cui L., Lucas C. M., Durand S., Sanchez J. F., Baldeviano G. C., Lescano A. G., Laman M., Barnadas C., Barry A., Mueller I., Kazura J. W., Eapen A., Kanagaraj D., Valecha N., Ferreira M. U., Roobsoong W., Nguitragool W., Sattabonkot J., Gamboa D., Kosek M., Vinetz J. M., González-Cerón L., Birren B. W., Neafsey D. E., Carlton J. M., Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat. Genet. 48, 953–958 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auburn S., Getachew S., Pearson R. D., Amato R., Miotto O., Trimarsanto H., Zhu S. J., Rumaseb A., Marfurt J., Noviyanti R., Grigg M. J., Barber B., William T., Goncalves S. M., Drury E., Sriprawat K., Anstey N. M., Nosten F., Petros B., Aseffa A., McVean G., Kwiatkowski D. P., Price R. N., Genomic analysis of Plasmodium vivax in Southern Ethiopia reveals selective pressures in multiple parasite mechanisms. J. Infect. Dis. 220, 1738–1749 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auburn S., Benavente E. D., Miotto O., Pearson R. D., Amato R., Grigg M. J., Barber B. E., William T., Handayuni I., Marfurt J., Trimarsanto H., Noviyanti R., Sriprawat K., Nosten F., Campino S., Clark T. G., Anstey N. M., Kwiatkowski D. P., Price R. N., Genomic analysis of a pre-elimination Malaysian Plasmodium vivax population reveals selective pressures and changing transmission dynamics. Nat. Commun. 9, 2585 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Twohig K. A., Pfeffer D. A., Baird J. K., Price R. N., Zimmerman P. A., Hay S. I., Gething P. W., Battle K. E., Howes R. E., Growing evidence of Plasmodium vivax across malaria-endemic Africa. PLOS Negl. Trop. Dis. 13, e0007140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carlton J. M., Das A., Escalante A. A., Genomics, population genetics and evolutionary history of Plasmodium vivax. Adv. Parasitol. 81, 203–222 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Cornejo O. E., Escalante A. A., The origin and age of Plasmodium vivax. Trends Parasitol. 22, 558–563 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Escalante A. A., Freeland D. E., Collins W. E., Lal A. A., The evolution of primate malaria parasites based on the gene encoding cytochrome B from the linear mitochondrial genome. Proc. Natl. Acad. Sci. U.S.A. 95, 8124–8129 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Escalante A. A., Cornejo O. E., Freeland D. E., Poe A. C., Durrego E., Collins W. E., Lal A. A., A monkey’s tale: The origin of Plasmodium vivax as a human malaria parasite. Proc. Natl. Acad. Sci. U.S.A. 102, 1980–1985 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prugnolle F., Rougeron V., Becquart P., Berry A., Makanga B., Rahola N., Arnathau C., Ngoubangoye B., Menard S., Willaume E., Ayala F. J., Fontenille D., Ollomo B., Durand P., Paupy C., Renaud F., Diversity, host switching and evolution of Plasmodium vivax infecting African great apes. Proc. Natl. Acad. Sci. U.S.A. 110, 8123–8128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rougeron V., Elguero E., Arnathau C., Hidalgo B. A., Durand P., Houze S., Berry A., Zakeri S., Haque R., Alam M. S., Nosten F., Severini C., Woldearegai T. G., Mordmüller B., Kremsner P. G., González-Cerón L., Fontecha G., Gamboa D., Musset L., Legrand E., Noya O., Pumpaibool T., Harnyuttanakorn P., Lekweiry K. M., Albsheer M. M., Hamid M. M. A., Boukary A. O. M. S., Trape J.-F., Renaud F., Prugnolle F., Human Plasmodium vivax diversity, population structure and evolutionary origin. PLOS Negl. Trop. Dis. 14, e0008072 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krief S., Escalante A. A., Pacheco M. A., Mugisha L., André C., Halbwax M., Fischer A., Krief J.-M., Kasenene J. M., Crandfield M., Cornejo O. E., Chavatte J.-M., Lin C., Letourneur F., Grüner A. C., McCutchan T. F., Rénia L., Snounou G., On the diversity of malaria parasites in African apes and the origin of Plasmodium falciparum from Bonobos. PLOS Pathog. 6, e1000765 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu W., Li Y., Learn G. H., Rudicell R. S., Robertson J. D., Keele B. F., Ndjango J.-B. N., Sanz C. M., Morgan D. B., Locatelli S., Gonder M. K., Kranzusch P. J., Walsh P. D., Delaporte E., Mpoudi-Ngole E., Georgiev A. V., Muller M. N., Shaw G. M., Peeters M., Sharp P. M., Rayner J. C., Hahn B. H., Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature 467, 420–425 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaiser M., Löwa A., Ulrich M., Ellerbrok H., Goffe A. S., Blasse A., Zommers Z., Couacy-Hymann E., Babweteera F., Zuberbühler K., Metzger S., Geidel S., Boesch C., Gillespie T. R., Leendertz F. H., Wild chimpanzees infected with 5 Plasmodium species. Emerg. Infect. Dis. 16, 1956–1959 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu W., Li Y., Shaw K. S., Learn G. H., Plenderleith L. J., Malenke J. A., Sundararaman S. A., Ramirez M. A., Crystal P. A., Smith A. G., Bibollet-Ruche F., Ayouba A., Locatelli S., Esteban A., Mouacha F., Guichet E., Butel C., Ahuka-Mundeke S., Inogwabini B.-I., Ndjango J.-B. N., Speede S., Sanz C. M., Morgan D. B., Gonder M. K., Kranzusch P. J., Walsh P. D., Georgiev A. V., Muller M. N., Piel A. K., Stewart F. A., Wilson M. L., Pusey A. E., Cui L., Wang Z., Färnert A., Sutherland C. J., Nolder D., Hart J. A., Hart T. B., Bertolani P., Gillis A., LeBreton M., Tafon B., Kiyang J., Djoko C. F., Schneider B. S., Wolfe N. D., Mpoudi-Ngole E., Delaporte E., Carter R., Culleton R. L., Shaw G. M., Rayner J. C., Peeters M., Hahn B. H., Sharp P. M., African origin of the malaria parasite Plasmodium vivax. Nat. Commun. 5, 3346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Makanga B., Yangari P., Rahola N., Rougeron V., Elguero E., Boundenga L., Moukodoum N. D., Okouga A. P., Arnathau C., Durand P., Willaume E., Ayala D., Fontenille D., Ayala F. J., Renaud F., Ollomo B., Prugnolle F., Paupy C., Ape malaria transmission and potential for ape-to-human transfers in Africa. Proc. Natl. Acad. Sci. U.S.A. 113, 5329–5334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loy D. E., Plenderleith L. J., Sundararaman S. A., Liu W., Gruszczyk J., Chen Y.-J., Trimboli S., Learn G. H., MacLean O. A., Morgan A. L. K., Li Y., Avitto A. N., Giles J., Calvignac-Spencer S., Sachse A., Leendertz F. H., Speede S., Ayouba A., Peeters M., Rayner J. C., Tham W.-H., Sharp P. M., Hahn B. H., Evolutionary history of human Plasmodium vivax revealed by genome-wide analyses of related ape parasites. Proc. Natl. Acad. Sci. U.S.A. 115, E8450–E8459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haldane J., Disease and evolution. Ric. Sci. 19, 68–76 (1949). [Google Scholar]
- 37.Gilabert A., Otto T. D., Rutledge G. G., Franzon B., Ollomo B., Arnathau C., Durand P., Moukodoum N. D., Okouga A.-P., Ngoubangoye B., Makanga B., Boundenga L., Paupy C., Renaud F., Prugnolle F., Rougeron V., Plasmodium vivax-like genome sequences shed new insights into S biology and evolution. PLOS Biol. 16, e2006035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.The Anopheles gambiae 1000 Genomes Consortium , Genetic diversity of the African malaria vector Anopheles gambiae. Nature 552, 96–100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H., Ralph P., Local PCA shows how the effect of population structure differs along the genome. Genetics 211, 289–304 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manske M., Miotto O., Campino S., Auburn S., Almagro-Garcia J., Maslen G., O’Brien J., Djimde A., Doumbo O., Zongo I., Ouedraogo J.-B., Michon P., Mueller I., Siba P., Nzila A., Borrmann S., Kiara S. M., Marsh K., Jiang H., Su X.-Z., Amaratunga C., Fairhurst R., Socheat D., Nosten F., Imwong M., White N. J., Sanders M., Anastasi E., Alcock D., Drury E., Oyola S., Quail M. A., Turner D. J., Rubio V. R., Jyothi D., Amenga-Etego L., Hubbart C., Jeffreys A., Rowlands K., Sutherland C., Roper C., Mangano V., Modiano D., Tan J. C., Ferdig M. T., Amambua-Ngwa A., Conway D. J., Takala-Harrison S., Plowe C. V., Rayner J. C., Rockett K. A., Clark T. G., Newbold C. I., Berriman M., MacInnis B., Kwiatkowski D. P., Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature 487, 375–379 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Degnan J. H., Rosenberg N. A., Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24, 332–340 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Degnan J. H., Rosenberg N. A., Discordance of species trees with their most likely gene trees. PLOS Genet. 2, e68 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avise J. C., Robinson T. J., Hemiplasy: A new term in the lexicon of phylogenetics. Syst. Biol. 57, 503–507 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Rosenberg N. A., Discordance of species trees with their most likely gene trees: A unifying principle. Mol. Biol. Evol. 30, 2709–2713 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suh A., Smeds L., Ellegren H., The dynamics of incomplete lineage sorting across the ancient adaptive radiation of neoavian birds. PLOS Biol. 13, e1002224 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galtier N., Nabholz B., Glémin S., Hurst G. D. D., Mitochondrial DNA as a marker of molecular diversity: A reappraisal. Mol. Ecol. 18, 4541–4550 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Mostowy R., Croucher N. J., Andam C. P., Corander J., Hanage W. P., Marttinen P., Efficient inference of recent and ancestral recombination within bacterial populations. Mol. Biol. Evol. 34, 1167–1182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boundenga L., Ollomo B., Rougeron V., Mouele L. Y., Mve-Ondo B., Delicat-Loembet L. M., Moukodoum N. D., Okouga A. P., Arnathau C., Elguero E., Durand P., Liégeois F., Boué V., Motsch P., Le Flohic G., Ndoungouet A., Paupy C., Ba C. T., Renaud F., Prugnolle F., Diversity of malaria parasites in great apes in Gabon. Malar. J. 14, 111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otto T. D., Gilabert A., Crellen T., Böhme U., Arnathau C., Sanders M., Oyola S. O., Okouga A. P., Boundenga L., Willaume E., Ngoubangoye B., Moukodoum N. D., Paupy C., Durand P., Rougeron V., Ollomo B., Renaud F., Newbold C., Berriman M., Prugnolle F., Genomes of all known members of a Plasmodium subgenus reveal paths to virulent human malaria. Nat. Microbiol. 3, 687–697 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prado-Martinez J., Sudmant P. H., Kidd J. M., Li H., Kelley J. L., Lorente-Galdos B., Veeramah K. R., Woerner A. E., O’Connor T. D., Santpere G., Cagan A., Theunert C., Casals F., Laayouni H., Munch K., Hobolth A., Halager A. E., Malig M., Hernandez-Rodriguez J., Hernando-Herraez I., Prüfer K., Pybus M., Johnstone L., Lachmann M., Alkan C., Twigg D., Petit N., Baker C., Hormozdiari F., Fernandez-Callejo M., Dabad M., Wilson M. L., Stevison L., Camprubí C., Carvalho T., Ruiz-Herrera A., Vives L., Mele M., Abello T., Kondova I., Bontrop R. E., Pusey A., Lankester F., Kiyang J. A., Bergl R. A., Lonsdorf E., Myers S., Ventura M., Gagneux P., Comas D., Siegismund H., Blanc J., Agueda-Calpena L., Gut M., Fulton L., Tishkoff S. A., Mullikin J. C., Wilson R. K., Gut I. G., Gonder M. K., Ryder O. A., Hahn B. H., Navarro A., Akey J. M., Bertranpetit J., Reich D., Mailund T., Schierup M. H., Hvilsom C., Andrés A. M., Wall J. D., Bustamante C. D., Hammer M. F., Eichler E. E., Marques-Bonet T., Great ape genetic diversity and population history. Nature 499, 471–475 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McVean G., A genealogical interpretation of principal components analysis. PLOS Genet. 5, e1000686 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alexander D. H., Novembre J., Lange K., Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kootker L. M., Mbeki L., Morris A. G., Kars H., Davies G. R., Dynamics of Indian Ocean slavery revealed through isotopic data from the colonial era Cobern Street Burial Site, Cape Town, South Africa (1750-1827). PLOS ONE 11, e0157750 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Dorp L., Gelabert P., Rieux A., de Manuel M., de Dios T., Gopalakrishnan S., Carøe C., Sandoval-Velasco M., Fregel R., Olalde I., Escosa R., Aranda C., Huijben S., Mueller I., Marquès-Bonet T., Balloux F., Gilbert M. T. P., Lalueza-Fox C., Plasmodium vivax Malaria viewed through the lens of an eradicated European strain. Mol. Biol. Evol. 37, 773–785 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Estoup A., Guillemaud T., Reconstructing routes of invasion using genetic data: Why, how and so what? Mol. Ecol. 19, 4113–4130 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Miotto O., Amato R., Ashley E. A., MacInnis B., Almagro-Garcia J., Amaratunga C., Lim P., Mead D., Oyola S. O., Dhorda M., Imwong M., Woodrow C., Manske M., Stalker J., Drury E., Campino S., Amenga-Etego L., Thanh T.-N. N., Tran H. T., Ringwald P., Bethell D., Nosten F., Phyo A. P., Pukrittayakamee S., Chotivanich K., Chuor C. M., Nguon C., Suon S., Sreng S., Newton P. N., Mayxay M., Khanthavong M., Hongvanthong B., Htut Y., Han K. T., Kyaw M. P., Faiz M. A., Fanello C. I., Onyamboko M., Mokuolu O. A., Jacob C. G., Takala-Harrison S., Plowe C. V., Day N. P., Dondorp A. M., Spencer C. C. A., McVean G., Fairhurst R. M., White N. J., Kwiatkowski D. P., Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat. Genet. 47, 226–234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rousset F., Genetic differentiation and estimation of gene flow from F-Statistics under isolation by distance. Genetics 145, 1219–1228 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pickrell J., Pritchard J., Inference of population splits and mixtures from genome-wide allele frequency data. PLOS Genet. 8, e1002967 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DeGiorgio M., Jakobsson M., Rosenberg N. A., Explaining worldwide patterns of human genetic variation using a coalescent-based serial founder model of migration outward from Africa. Proc. Natl. Acad. Sci. U.S.A. 106, 16057–16062 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li J. Z., Absher D. M., Tang H., Southwick A. M., Casto A. M., Ramachandran S., Cann H. M., Barsh G. S., Feldman M., Cavalli-Sforza L. L., Myers R. M., Worldwide human relationships inferred from genome-wide patterns of variation. Science 319, 1100–1104 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Tanabe K., Mita T., Jombart T., Eriksson A., Horibe S., Palacpac N., Ranford-Cartwright L., Sawai H., Sakihama N., Ohmae H., Nakamura M., Ferreira M. U., Escalante A. A., Prugnolle F., Björkman A., Färnert A., Kaneko A., Horii T., Manica A., Kishino H., Balloux F., Plasmodium falciparum accompanied the human expansion out of Africa. Curr. Biol. 20, 1283–1289 (2010). [DOI] [PubMed] [Google Scholar]
- 62.Excoffier L., Foll M., Petit R. J., Genetic consequences of range expansions. Annu. Rev. Ecol. Evol. Syst. 40, 481–501 (2009). [Google Scholar]
- 63.World Health Oranization, World Malaria Report 2010 (World Health Organization, 2010); www.who.int/malaria/publications/atoz/9789241564106/en/.
- 64.Nájera J. A., González-Silva M., Alonso P. L., Some lessons for the future from the global malaria eradication programme (1955–1969). PLOS Med. 8, e1000412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Willing E.-M., Dreyer C., van Oosterhout C., Estimates of genetic differentiation measured by FST do not necessarily require large sample sizes when using many SNP markers. PLOS ONE 7, e42649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nazareno A. G., Bemmels J. B., Dick C. W., Lohmann L. G., Minimum sample sizes for population genomics: An empirical study from an Amazonian plant species. Mol. Ecol. Resour. 17, 1136–1147 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Venkatesan M., Amaratunga C., Campino S., Auburn S., Koch O., Lim P., Uk S., Socheat D., Kwiatkowski D. P., Fairhurst R. M., Plowe C. V., Using CF11 cellulose columns to inexpensively and effectively remove human DNA from Plasmodium falciparum-infected whole blood samples. Malar. J. 11, 41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gillies M., Coetzee M., A supplement to the Anophelinae of Africa South of the Sahara (Afrotropical Region). Publ. Afr. Inst. Med. Res. 55, 1–143 (1987). [Google Scholar]
- 69.Sundararaman S. A., Liu W., Keele B. F., Learn G. H., Bittinger K., Mouacha F., Ahuka-Mundeke S., Manske M., Sherrill-Mix S., Li Y., Malenke J. A., Delaporte E., Laurent C., Mpoudi Ngole E., Kwiatkowski D. P., Shaw G. M., Rayner J. C., Peeters M., Sharp P. M., Bushman F. D., Hahn B. H., Plasmodium falciparum-like parasites infecting wild apes in southern Cameroon do not represent a recurrent source of human malaria. Proc. Natl. Acad. Sci. U.S.A. 110, 7020–7025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cowell A. N., Loy D. E., Sundararaman S. A., Valdivia H., Fisch K., Lescano A. G., Baldeviano G. C., Durand S., Gerbasi V., Sutherland C. J., Nolder D., Vinetz J. M., Hahn B. H., Winzeler E. A., Selective whole-genome amplification is a robust method that enables scalable whole-genome sequencing of Plasmodium vivax from unprocessed clinical samples. MBio 8, e02257-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chan E. R., Menard D., David P. H., Ratsimbasoa A., Kim S., Chim P., Do C., Witkowski B., Mercereau-Puijalon O., Zimmerman P. A., Serre D., Whole genome sequencing of field isolates provides robust characterization of genetic diversity in Plasmodium vivax. PLOS Negl. Trop. Dis. 6, e1811 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen S.-B., Wang Y., Kassegne K., Xu B., Shen H.-M., Chen J.-H., Whole-genome sequencing of a Plasmodium vivax clinical isolate exhibits geographical characteristics and high genetic variation in China-Myanmar border area. BMC Genomics 18, 131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Oliveira T. C., Rodrigues P. T., Menezes M. J., Gonçalves-Lopes R. M., Bastos M. S., Lima N. F., Barbosa S., Gerber A. L., Loss de Morais G., Berná L., Phelan J., Robello C., de Vasconcelos A. T. R., Alves J. M. P., Ferreira M. U., Genome-wide diversity and differentiation in New World populations of the human malaria parasite Plasmodium vivax. PLOS Negl. Trop. Dis. 11, e0005824 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Popovici J., Friedrich L. R., Kim S., Bin S., Run V., Lek D., Cannon M. V., Menard D., Serre D., Genomic analyses reveal the common occurrence and complexity of Plasmodium vivax relapses in Cambodia. MBio 9, e01888-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Delgado-Ratto C., Gamboa D., Soto-Calle V. E., Van den Eede P., Torres E., Sánchez-Martínez L., Contreras-Mancilla J., Rosanas-Urgell A., Rodriguez Ferrucci H., Llanos-Cuentas A., Erhart A., Van Geertruyden J.-P., D’Alessandro U., Population genetics of Plasmodium vivax in the Peruvian Amazon. PLOS Negl. Trop. Dis. 10, e0004376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Neafsey D. E., Galinsky K., Jiang R. H. Y., Young L., Sykes S. M., Saif S., Gujja S., Goldberg J. M., Young S., Zeng Q., Chapman S. B., Dash A. P., Anvikar A. R., Sutton P. L., Birren B. W., Escalante A. A., Barnwell J. W., Carlton J. M., The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat. Genet. 44, 1046–1050 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leinonen R., Sugawara H., Shumway M.; International Nucleotide Sequence Database Collaboration , The sequence read archive. Nucleic Acids Res. 39, D19–D21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 17, 10–12 (2011). [Google Scholar]
- 79.Auburn S., Böhme U., Steinbiss S., Trimarsanto H., Hostetler J., Sanders M., Gao Q., Nosten F., Newbold C. I., Berriman M., Price R. N., Otto T. D., A new Plasmodium vivax reference sequence with improved assembly of the subtelomeres reveals an abundance of pir genes. Wellcome Open Res. 1, 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Picard Toolkit (Broad Institute, 2019); http://broadinstitute.github.io/picard/.
- 83.Wang J., Effects of sampling close relatives on some elementary population genetics analyses. Mol. Ecol. Resour. 18, 41–54 (2018). [DOI] [PubMed] [Google Scholar]
- 84.Schaffner S. F., Taylor A. R., Wong W., Wirth D. F., Neafsey D. E., hmmIBD: Software to infer pairwise identity by descent between haploid genotypes. Malar. J. 17, 196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huson D. H., Bryant D., Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Nguyen L.-T., Schmidt H. A., von Haeseler A., Minh B. Q., IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.A. Rambaut, FigTree, a graphical viewer of phylogenetic trees (2007).
- 88.Tachibana S.-I., Sullivan S. A., Kawai S., Nakamura S., Kim H. R., Goto N., Arisue N., Palacpac N. M. Q., Honma H., Yagi M., Tougan T., Katakai Y., Kaneko O., Mita T., Kita K., Yasutomi Y., Sutton P. L., Shakhbatyan R., Horii T., Yasunaga T., Barnwell J. W., Escalante A. A., Carlton J. M., Tanabe K., Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade. Nat. Genet. 44, 1051–1055 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhao H., Sun Z., Wang J., Huang H., Kocher J.-P., Wang L., CrossMap: A versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A. R., Bender D., Maller J., Sklar P., de Bakker P. I. W., Daly M. J., Sham P. C., PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kopelman N. M., Mayzel J., Jakobsson M., Rosenberg N. A., Mayrose I., Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 15, 1179–1191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R.; 1000 Genomes Project Analysis Group , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.M. Milanesi, S. Capomaccio, E. Vajana, L. Bomba, J. F. Garcia, P. Ajmone-Marsan, L. Colli, BITE: An R package for biodiversity analyses. bioRxiv 181610 [Preprint]. 29 August 2017. 10.1101/181610. [DOI]
- 94.François O., Blum M. G. B., Jakobsson M., Rosenberg N. A., Demographic history of european populations of Arabidopsis thaliana. PLOS Genet. 4, e1000075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fontaine M. C., Austerlitz F., Giraud T., Labbé F., Papura D., Richard-Cervera S., Delmotte F., Genetic signature of a range expansion and leap-frog event after the recent invasion of Europe by the grapevine downy mildew pathogen Plasmopara viticola. Mol. Ecol. 22, 2771–2786 (2013). [DOI] [PubMed] [Google Scholar]
- 96.Zhang C., Dong S.-S., Xu J.-Y., He W.-M., Yang T.-L., PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Schiffels S., Wang K., MSMC and MSMC2: The multiple sequentially markovian coalescent. Methods Mol. Biol. 2090, 147–166 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/18/eabc3713/DC1