

The charge transport–limiting factors in conjugated polymers without single-bond linkages in the backbone have been identified.
Abstract
We investigate the charge transport physics of a previously unidentified class of electron-deficient conjugated polymers that do not contain any single bonds linking monomer units along the backbone but only double-bond linkages. Such polymers would be expected to behave as rigid rods, but little is known about their actual chain conformations and electronic structure. Here, we present a detailed study of the structural and charge transport properties of a family of four such polymers. By adopting a copolymer design, we achieve high electron mobilities up to 0.5 cm2 V−1 s−1. Field-induced electron spin resonance measurements of charge dynamics provide evidence for relatively slow hopping over, however, long distances. Our work provides important insights into the factors that limit charge transport in this unique class of polymers and allows us to identify molecular design strategies for achieving even higher levels of performance.
INTRODUCTION
Conjugated polymers have attracted substantial research attention in recent decades and are being developed for a range of applications in displays, large-area electronics, and bioelectronics (1–7), which make use of their compatibility with large-area, low-temperature solution-based manufacturing deposition imparted by functionalization with solubilizing side chains (8, 9). Unlike inorganic semiconductors for which delocalized electronic states and band structures stem from long-range crystallinity within atomic lattices, the charge transport and optoelectronic properties of conjugated polymers are largely determined at the single molecular level by the conformation of the polymer backbone, which governs the ability of the π-conjugated electronic states in the frontier molecular orbitals (MOs) to delocalize along the polymer backbone. A distribution of different conformations adopted across the network of polymer chains in the solid state is also responsible for energetic disorder and broadening of the electronic density of states. Key factors that determine the chain conformation, backbone planarity, and persistence length include the potentials for torsion and for backbone deflection (10). Characterization of molecular-level chain conformations (11–13) is arguably the starting point for understanding structure-property relationships (14–16) in specific conjugated polymer systems.
In recent years, the persistence length, which is defined as the average projection of the end-to-end vector on the tangent to the chain contour at a chain end in the limit of infinite chain length (17), has been uncovered to intimately correlate with the optoelectronic characteristics of the polymer. A long persistence length has been demonstrated to be desirable for applications such as high-efficiency organic photovoltaics and high-mobility organic field-effect transistors (OFETs) (12). Early examples of the importance of persistence length include studies of the photoluminescence quantum yield of polyphenylene vinylene derivatives, which was found to be positively correlated with the persistence length and stiffness of the backbone (18). Unusually high optical absorption in specific donor-acceptor (D-A) conjugated polymers has been ascribed to long persistence length as well (19). Already from early studies, it has been known that persistence length is related to the degree of conformational and energetic disorder (20). Stiffer chains not only reduce the number of possible backbone configurations and, hence, energetic disorder (21, 22) but also provide straight tie chains bridging neighboring crystallites, forming efficient percolation pathways required for macroscopic charge transport within thin-film devices such as OFETs (23).
One of the factors that limit the persistence length of traditional conjugated polymers is the existence of single-bond linkages that connect rigid conjugated units along the polymer backbone. At these single-bond linkages, cis-trans isomerism or variations of the torsional angle between neighboring conjugated units can occur that limit π-electron delocalization and may change the orientation of the backbone. As a result, most conjugated polymers exhibit limited chain rigidity with random coil or worm-like conformations on length scales above 1 to 10 nm (12) and more flexibility compared with other π-conjugated systems such as carbon nanotubes (24). To date, the conjugated polymers with the longest persistence lengths have been found to be donor-acceptor copolymers with extended, fused-ring conjugated units linked by single bonds designed to exhibit steep torsion potentials (10, 19). In such rigid and low-disorder polymers, the highest carrier mobilities on the order of 1 to 5 cm2 V−1 s−1 have been observed in polymer-based OFETs to date. A wide range of such donor-acceptor polymers have been investigated, but at present, there are no clear molecular design guidelines that would allow going beyond the level of performance that is achievable with such systems, and for the last 5 years, there has been only gradual and slow progress in the performance of conjugated polymers for OFETs (7, 25–27). There is an urgent need for new design motifs for conjugated polymers that might allow further reductions in main-chain conformational flexibility and energetic disorder.
Recently, an aldol condensation reaction between bis-isatin and bis-oxindole monomers has been used to synthesize an unusual series of electron-deficient conjugated polymers with completely fused backbone structures that do not contain any single-bond linkages (28). These polymers comprise fused-ring units that are linked together by double as opposed to single, carbon-carbon bonds. Because the energetic barrier for rotation around a double bond is expected to be an order of magnitude higher than for rotation around a single bond (28), we expect these polymers to have a more rigid backbone with potentially longer persistence lengths. In one such polymer, a homopolymer based on naphthalene-derived monomers, referred to as NN1 within this work, a high electron affinity (EA) of 4.2 eV and air-stable electron transport for up to 300 hours were observed within OFETs with a field-effect mobility (μFET) of 0.03 cm2 V−1 s−1 (28). Given the unique nature of the backbone of these polymers, this level of achievable μFET is maybe somewhat disappointing. However, to date, a detailed study of the structure-property relationships and charge transport physics of this class of polymers has not been reported yet, and it is important to understand the polymer chain conformations that these polymers adopt and the factors that limit their charge transport properties.
In this work, we systematically investigate the chain conformations, persistence lengths, microstructure, energetic disorder, and charge transport properties in four such polymers, herein named NN1, NN2, AN1, and AN2. In addition, we compare the air stability of these four polymers to the widely investigated electron-deficient polymer P(NDI2OD-T2). The chemical structures are shown in Fig. 1A. Compared to the systems first reported in (28), including NN1, we would like to further improve the mobility by modifying the structure using two different strategies: extending the aromatic core and incorporation of branched side chains with branching points further removed from the polymer backbone. By extension of the planar aromatic core, the backbone rigidity and coplanarity could be increased. This strategy has proved useful to improve charge transport properties in other rigid-rod polymers (29). More specifically, we adopted a copolymer design with some units extended from a naphthalene to an anthracene core. By designing derivatives in which the branch point is moved further away from the backbone, we aim to combine the advantages of improved solubility from branched side chains with stronger π-π intermolecular interactions, which are normally associated with linear side chains. This strategy has been shown to promote crystallinity and mobility (30).
Fig. 1. Chemical structures and thin-film UV-Vis-NIR absorption spectra.
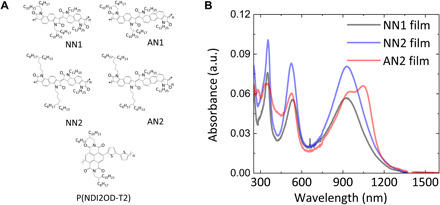
(A) Chemical structure of NN1, NN2, AN1, AN2, and P(NDI2OD-T2) polymers. (B) Thin-film UV-Vis-NIR absorption spectra of the polymers. The films were spun from hot 1,2,4-trichlorobenzene solution.
We present a full characterization, including the molecular weight dependence of charge transport properties as well as film microstructure by grazing-incidence wide-angle x-ray scattering (GIWAXS) and quantification of energetic disorder by photothermal deflection spectroscopy (PDS) and quantum-chemical modeling. We show that optimization of molecular design provides access to electron mobilities of 0.5 cm2/V−1 s−1 that are significantly higher than observed previously in this class of polymers and comparable to those of other state-of-the-art electron-deficient conjugated polymers, such as the widely investigated P(NDI2OD-T2) (31). We use small-angle neutron scattering (SANS) in solution to quantify persistence lengths, use field-induced electron spin resonance (FI-ESR) to quantify charge hopping times and hopping lengths in working OFETs, and show that a long persistence length is key to achieving good charge transport properties. Our main aim in this work is to identify clear strategies for further enhancement of the charge transport properties of this unique class of conjugated polymers.
RESULTS AND DISCUSSION
Figure 1A shows the four fused polymers synthesized and investigated within this work, together with the widely investigated electron-deficient polymer P(NDI2OD-T2). We note that with the AN1 polymer, we encountered some synthetic problems caused by limited solubility; the AN1 polymer exhibited only a small molecular weight (Mn = 32.1 kDa, Mw = 89.5 kDa, as determined by gel permeation chromatography) and is included here for completeness only without the full characterization that was conducted for the other polymers. Figure 1B shows the thin-film UV-Vis-NIR absorption spectra for the NN1, NN2, and AN2 polymers with wavelengths ranging from 250 to 1600 nm. Three absorption peaks appear within this spectral range, one each in the ultraviolet (UV), visible, and near infrared (NIR). The absorption peaks in the UV and visible bands are observed at similar positions for the three polymers, namely, 355 nm for the UV band and 524 nm for the visible band. However, the longest wavelength absorption in the NIR occurs at longer wavelength (1047 nm) in AN2 than in NN1 and NN2 (930 nm). AN2 is the only polymer in which it is possible to observe a well-resolved 0-0 and 0-1 vibronic progressions in the NIR band, suggesting a smaller degree of energetic disorder. Thin-film and solution absorption spectra are quite similar for all three polymers (fig. S1). For AN2, the ratio between the intensities of the 0-0 and 0-1 NIR absorption peaks is slightly higher in solution than in thin films (fig. S1C). The ionization energies and EAs of these polymers are summarized within Table 1, with the optical bandgaps calculated from the onset of the thin-film absorption spectra. While the optical bandgaps of the three fused polymers measured are all around 1 eV, the EAs of the three polymers are between −4.1 and −4.2 eV. The high EA is essential for the good air-stable electron transport in these polymers, as discussed below.
Table 1. Polymer ionization energies, electron affinities, and optical properties.
| Polymers | Eopt.gap (eV)* | IE (eV)† | EA (eV)‡ |
| NN1 | 1.01 | −5.20 | −4.19 |
| NN2 | 1.04 | −5.16 | −4.12 |
| AN1 | - | −5.39 | - |
| AN2 | 1.03 | −5.13 | −4.10 |
*Estimated optical gap was calculated using onset of the thin-film (spin-coated on glass substrates) absorption spectra (Eopt = 1240/λonset).
†Measured by the Photoelectron Spectroscopy in Air (PESA) system.
‡EA is calculated from EA = IE − Eopt.
To understand the nature of the associated optical transitions, the optical absorption spectrum of the NN and AN polymers has also been simulated for representative model systems at the quantum-chemical TD-DFT (time-dependent density functional theory) level (see Materials and Methods). Our calculations nicely reproduce the main features observed experimentally: (i) a low-energy absorption band peaking at slightly longer absorption wavelength for AN (873 nm) compared to NN (841 nm), (ii) a second band in the visible region (at 497 and 499 nm for AN and NN, respectively), and (iii) a third absorption band in the UV region (387 and 389 nm for AN and NN, respectively). As a proxy for the charge-transfer (CT) character of the relevant electronic excitations, we have calculated the overlap, Φs, between the hole and electron densities (fig. S2). In both polymers, the lowest two optically active electronic transitions involve overlapping hole and electron wave functions (with high Φs indicating small CT) that extend over the entire polymer chains, in line with (28). Close inspection reveals that these two excitations involve the same ending empty MOs but differ by the nature of the starting occupied MOs, being the HOMO (highest occupied molecular orbital) and HOMO-1 orbitals of the anthracene and naphthalene cores, and correspond to the so-called L and B electronic transitions in the perimeter free electron orbital picture of polyaromatic hydrocarbons. The low optical bandgap of the polymers results from a low transition energy for the repeating monomer units combined with strong electronic interactions between the monomers along the polymer chains. This can be appreciated from the large red shift of the lowest excitation energy calculated in oligomers of increasing size (fig. S3).
To investigate the rigidity of single polymer chains and measure persistence lengths, we performed SANS experiments on solutions of NN1, NN2, and AN2 fused polymers and compared them to the widely investigated electron-deficient polymer P(NDI2OD-T2). The polymer samples were dissolved in hot (125°C) dichlorobenzene at dilute concentrations (5 mg/ml) to minimize aggregation effects. Experimental details can be found in Materials and Methods. Figure 2 shows the scattering cross section of these four polymers and the fitting of the scattering profiles with appropriate models (details about fitting procedures are included within Materials and Methods and section S3). The chain parameters extracted from the fitting of the SANS results are summarized in Table 2.
Fig. 2. SANS of NN1, NN2, AN2, and P(NDI2OD-T2) in hot, dichlorobenzene solution.
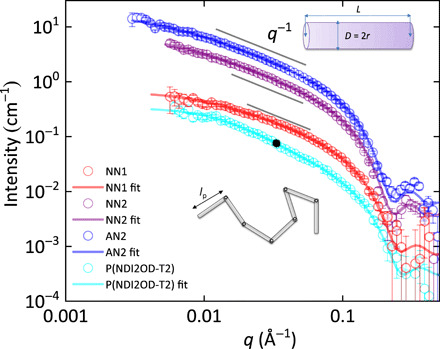
The NN1, NN2, and AN2 fused polymers adopt a rigid-rod shape with a scaling of I ≈ q−1 over a large region, while the semiflexible P(NDI2OD-T2) polymer shows a characteristic worm-like chain behavior. The curves have been shifted vertically for clarity. The incoherent scattering background was subtracted.
Table 2. Chain parameters extracted from the fitting of the SANS curves.
| Polymer | Model | Length (nm) | SD of L (nm) | Radius (nm) | Kuhn length (b) (nm) |
| P(NDI2OD-T2) | Worm-like | 46.8 | 37.4 | 1.40 | 10.2 |
| NN1 | Cylinder | 17.3 | 14.8 | 1.48 | >L |
| NN2 | Cylinder | 30.9 | 26.9 | 1.65 | >L |
| AN2 | Cylinder | 39.2 | 32.1 | 1.65 | >L |
For the NN1, NN2, and AN2 polymers, the general SANS features are similar: A rapid decrease in scattering intensity can be seen for large scattering vectors above 0.1 Å−1. For intermediate scattering vectors in the range between 0.01 and 0.1 Å−1, the scattering intensity decreases approximately as I ∝ q−1. Below 0.01 Å−1, we observe a gradually weaker dependence of intensity on q. Those features of scattering agree well with a rod with a finite length. The sharp decrease of intensity at high q corresponds to a cross-sectional Guinier behavior. If the chain adopts a rod-like conformation, the length L is the contour length (Lmax), which is proportional to the molecular weight. Since the AN2 polymer has the highest molecular weight among the fused polymers, a longer sample-to-detector distance was chosen (8 m), as compared with the NN1, NN2, as well as the P(NDI2OD-T2) reference measurements. Unfortunately, the lowest q of the measurement (~0.003 A−1) is still not low enough to reach the Guinier regime of the whole rod. Therefore, the determination of the length of the rod relies on the medium q data, which is not as accurate. However, according to the dynamic light scattering results (fig. S4) performed on the fully dissolved NN1 polymers, it is highly likely that the chain lengths (~10 nm) have been extracted from the fitting process accurately and well within the limit of errors. The polydispersity has been accounted for by assuming a Schultz-Zimm distribution in molecular weight (details can be found in section S3). The fitting parameters are listed in Table 2. NN2 and AN2 share the same radius (1.65 nm), which is slightly larger than NN1. The difference agrees well with the length of the side chains. The length of the “rod” depends on the molecular weight (table S1). The results confirm that the AN2 polymer has the largest molecular weight. A direct link between the length and molecular weight is not established since the molecular weight is measured on a relative scale. The most important information we obtain is that for the range of molecular weights investigated here, the chain of the three fused polymers is completely rigid and adopts a rod-shaped conformation. In other words, the persistence length is beyond the detection limit and is larger than the contour length (Lmax) of the materials investigated.
For comparison, we measured P(NDI2OD-T2) (31), which has been shown to exhibit low energetic disorder (Eu = 32 meV) (32) in thin films. Although the scattering behavior at large q is almost the same as in the fused polymers, the SANS curve of P(NDI2OD-T2) cannot be well fitted with a rod model (fig. S7). This is because the power law for the scattering intensity I ∝q−α has an exponent α that is clearly larger than 1. We found that the best model to fit the P(NDI2OD-T2) scattering data is a worm-like chain, which describes a chain with rod-like behavior on short length scales but coil-like behavior on longer length scales. The fitting parameters give a Kuhn length (b) (33) of 10.2 nm and a contour length of 46.8 ± 37.4 nm. For sufficiently long chains (Lmax >> b), the Kuhn length is twice the persistence length (lp). The main conclusion from the SANS analysis is, therefore, that the NN1, NN2, and AN2 polymers are significantly more rigid and less flexible than P(NDI2OD-T2). The rigid-rod nature of these polymers is a clear manifestation of their unique double-bond linkages between the fused conjugated units, which distinguishes these materials from other more traditional conjugated polymers.
The SANS experiments in Fig. 2 were performed on polymer chains that were fully dissolved in hot (125°C) and diluted deuterated solvent without observable aggregation and represent the behavior of isolated polymer chains in solution. They leave open the question to which extent the high degree of backbone rigidity can be maintained in the solid state. A first indication that a high chain rigidity is also present in the solid state is that optical absorption spectra between solutions and films show almost no differences or spectral shifts (fig. S1). To gain further insight into the solid-state microstructure of thin films, we performed GIWAXS. Polymer films were spin-coated under the same conditions as used for the fabrication of the active layers of OFET devices (details in Materials and Methods).
For the three fused polymers characterized, GIWAXS patterns demonstrate bimodal textures in general, namely, face-on and edge-on crystallites coexisting within samples, evidenced by the existence of in-plane and out-of-plane lamellar scattering peaks in the two-dimensional (2D) GIWAXS patterns (Fig. 3). However, there are still differences between the three polymers: The NN1 sample shows a predominantly face-on texture, as is evident from the lack of a pronounced in-plane π-π stacking (010) peak, while the obvious in-plane (010) peaks seen in the patterns of the NN2 and AN2 indicated a significant edge-on population in these samples (Fig. 3). Previous work shows that this difference could be related to different degrees of aggregation behavior within the precursor solution (34); in other words, NN2 and AN2 polymers may exhibit a larger ratio of edge-on crystallites due to their stronger aggregation within the precursor solution compared with the NN1 polymer. This could originate from stronger interchain interactions induced by the side-chain branching point being further away from the main chain in NN2 and AN2 than in NN1. The effect of side-chain branching point extension is also evident from an analysis of GIWAXS spacing parameters. The lamellar stacking distance for NN2 and AN2 crystallites is larger than for NN1 crystallites (Table 3), which is consistent with the longer side chain of NN2 and AN2. Higher-order (400) lamellar diffraction peaks are seen in the out-of-plane direction for both NN2 and AN2, indicating a high degree of order in the lamellar stacking. In addition, the π-π stacking distance shrinks from 3.72 Å (NN1) to 3.69 Å (NN2 and AN2), which is consistent with the branching point extension and beneficial for interchain charge transport.
Fig. 3. Structural characterization of the representative rigid-rod polymer thin films.
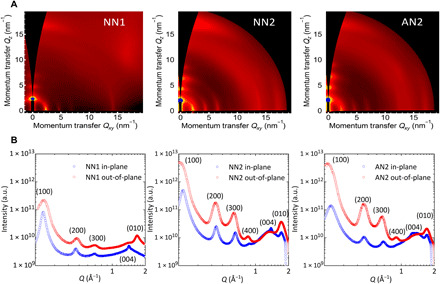
(A) 2D GIWAXS patterns of NN1, NN2, and AN2 thin-film samples. (B) In-plane and out-of-plane 1D linecuts of NN1, NN2, and AN2’s 2D GIWAXS patterns.
Table 3. GIWAXS spacing parameters for the NN1, NN2, and AN2 samples.
N/A denotes not available.
| Crystallographic parameters | NN1 | NN2 | AN2 | |
| Lamella stacking (out-of-plane) | q (Å−1) | 0.240 | 0.217 | 0.232 |
| d spacing (Å) | 26.4 | 29.0 | 27.4 | |
| ΔFWHM (Å−1) | 0.047 | 0.055 | 0.061 | |
| Coherence length (Å) | 135 | 115 | 105 | |
| Lamella stacking (in-plane) | q (Å−1) | 0.240 | 0.220 | 0.230 |
| d spacing (Å) | 26.6 | 28.6 | 27.3 | |
| ΔFWHM (Å−1) | 0.021 | 0.027 | 0.043 | |
| Coherence length (Å) | 300 | 235 | 145 | |
| π-π stacking (out-of-plane) | q (Å−1) | 1.69 | 1.70 | 1.70 |
| d spacing (Å) | 3.72 | 3.69 | 3.69 | |
| ΔFWHM (Å−1) | 0.218 | 0.204 | 0.194 | |
| Coherence length (Å) | 28.8 | 30.8 | 32.4 | |
| π-π stacking (in-plane) | q (Å−1) | N/A | 1.70 | 1.70 |
| d spacing (Å) | N/A | 3.69 | 3.69 | |
| ΔFWHM (Å−1) | N/A | 0.141 | 0.20 | |
| Coherence length (Å) | N/A | 44.5 | 31.4 | |
| Backbone repeat (004) (in-plane) |
q (Å−1) | 1.42 | 1.39 | 1.25 |
| d spacing (Å) | 17.7 | 18.1 | 20.1 | |
| ΔFWHM (Å−1) | 0.064 | 0.054 | N/A | |
| Coherence length (Å) | 100 | 115 | N/A | |
In-plane reflections assigned to backbone stacking peaks are observed for the three polymers. NN1 and NN2 exhibit peaks at roughly the same Q value (1.42/1.39 Å−1 for NN1/NN2), which, if indexed to (004) reflections, correspond to a backbone repeat distance of 1.8 nm, consistent with the length of a single naphthalene–co-naphthalene repeating unit according to the results of force field calculations. The coherence lengths extracted from the full width at half maximum of these (004) peaks provide a measure of the length scale along which crystalline packing is maintained along the backbone direction. The values for NN1 (10 nm) and NN2 (11.5 nm) are comparable and correspond to five to six backbone repeat units. Peaks associated with periodic stacking along the backbone are not universally observed in conjugated polymers but rather are seen for a limited number of rigid polymers including pBTTT (35), P(NDI2OD-T2) (36–38), and C16 IDT-BT (39). The observation of backbone peaks here suggests a relatively high degree of order along the backbone direction. The coherence lengths extracted for NN1 and NN2, however, are slightly smaller than those of other high-performance conjugated polymers such as thermally annealed films of P(NDI2OD-T2) (20.7 nm) (36) and C16 IDT-BT polymer (22 nm) (39). In addition, the (004) peaks observed here show “arcing,” indicating that the backbones are not lying perfectly flat in the plane of the film, which is different to other polymers such as pBTTT (35) and P(NDI2OD-T2) (37) where the backbone peaks shown are vertical in Q space. In the case of the AN2 films, a peak can just be discerned in-plane at 1.25 Å−1, which is consistent with the (004) reflection of this polymer given the larger size of the backbone in this polymer. However, we are not able to extract the coherence length of this peak due to weaker intensity of this peak. We conclude that the GIWAXS results provide evidence for a highly ordered, semicrystalline thin-film microstructure with pronounced interchain interactions and close π-π stacking. The observation of the (004) backbone repeat peaks shows that the rigid-rod chain conformation observed in solution by the SANS is maintained in spin-coated thin films. However, the measured coherence lengths are not exceptionally long compared to other rigid-rod polymers. This may point to the existence of some degree of torsional disorder along the backbone, an issue that is discussed further below.
To investigate energetic disorder in the solid state, we use PDS (40, 41) to measure the broadening of the tail of optical absorption below the band edge (Fig. 4). We extract the Urbach energy, which characterizes the width of the joint excitonic density of states and is a measure of energetic disorder (32, 42), by fitting the exponential tail of the PDS spectra to the equation
Fig. 4. PDS of rigid-rod polymer thin films.
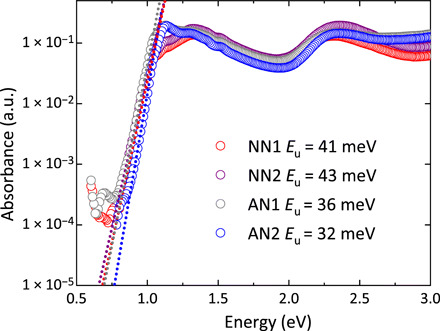
Absorbance of the NN1, NN2, AN1, and AN2 thin films measured by PDS. Dotted lines represent exponential tail fits for extraction of the Urbach energies Eu (inset).
Both anthracene-derived polymers, AN1 and AN2, exhibit lower energetic disorder compared to the naphthalene-derived systems, NN1 and NN2. In particular, AN2 has a low Urbach energy Eu = 32 meV, which is similar to the low value found in P(NDI2OD-T2) (32), but still somewhat larger than the lowest value observed in a conjugated polymer, which was reported for C16 IDT-BT (Eu = 24 meV) and is less than kBT at room temperature (10).
To understand the origin of the low energetic disorder in the NN and AN polymers, we have simulated the conformational potential energy surface of representative systems (see technical details in Materials and Methods). Although the two fragments are connected by a double bond, NN and AN polymers are not perfectly planar because of steric repulsions between the carbonyl oxygens and their adjacent C─H groups (fig. S8) (28). According to a Maxwell-Boltzmann distribution, the average value of the torsion angle in the electronic ground state and its deviation from planarity at room temperature amount to 18.1° ± 6.1° and 18.6° ± 6.1° for NN and AN, respectively. As the chains explore these conformations, the calculated TD-DFT excitation energies vary around mean values of 2.22 eV in NN and 2.04 eV in AN with corresponding SDs significantly smaller in AN (23 meV) compared to NN (32 meV), in line with the measured Urbach energies. We attribute the smaller thermal energetic disorder for AN versus NN to the spreading of the π system in anthracene versus naphthalene, which reduces the variation in excitonic couplings due to conformational motion because of the lower weight of the wave function at the intermonomer connecting atoms.
To investigate the charge transport properties of the different polymers, we fabricated spin-coated, top-gate, bottom-contact OFETs with a 480-nm polymethyl methacrylate (PMMA) dielectric. Details of device fabrication are reported in Materials and Methods; the capacitance-frequency (C-f) characteristics for determination of the dielectric capacitance are shown in fig. S9. The devices exhibit ambipolar transport characteristics with a current modulation of >105 (linear curves) and evidence for hole injection and hole transport when VD = 60 V and VG < 20 V (Fig. 5A). However, better injection and higher currents are obtained in the electron regime, while the hole regime suffers from severe injection issues (fig. S10F), which suggests the formation of an unfavorable interfacial dipole at the organic semiconductor (OSC)–gold interface, making hole injection more difficult than expected given the favorable value of the ionization potential. Hence, we focus on a discussion of the electron transport regime here. We observed the highest mobilities in AN2, for which the best devices exhibit linear and saturation electron mobility of 0.4 and 0.8 cm2 V−1 s−1, respectively (Fig. 5B), when measured at 300 K in air. These are relatively moderate values of charge carrier mobility considering the low effective mass of ~0.13 me calculated by DFT for the electrons (compared to ~0.21 me for the holes). The large dispersion in momentum space of the conduction band (fig. S15) is the primary reason for the high EA of the polymer and its low optical gap. In a real space representation, this is also borne out by the calculated large changes in the LUMO (lowest unoccupied molecular orbital) energy with chain length (fig. S16).
Fig. 5. Temperature-dependent and molecular weight–dependent charge transport investigation of rigid-rod polymer FETs.
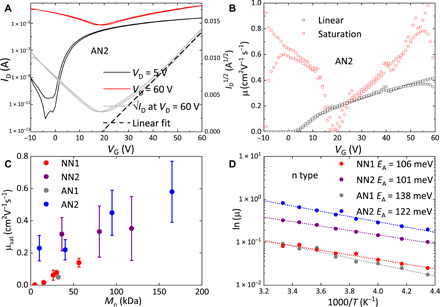
(A) Transfer curves measured on top-gate, bottom-contact FETs (L = 20 μm, W = 1 mm) fabricated from the spin-coated AN2 film. (B) Linear and saturation mobility extracted from transfer curves measured on the same device. (C) Molecular weight–dependent saturation mobility for spin-coated NN1, NN2, AN1, and AN2 top-gate, bottom-contact FETs (L = 20 μm, W = 1 mm). (D) Temperature-dependent saturation electron mobility (at VG = 60 V, VD = 60 V) for spin-coated NN1, NN2, AN1, and AN2 top-gate, bottom-contact FETs (L = 20 μm, W = 1 mm).
We note that neither the n-type linear nor saturation mobility exhibits a plateau at high gate voltage but continues to increase with increasing VG. This is not likely to be a contact resistance effect but could be ascribed to residual energetic disorder, trap filling, or potentially electron-electron interactions that are commonly observed in semicrystalline polymer OFETs (43, 44). The output curves of the mentioned AN2 OFET in the electron regime in fig. S10C show well-defined linearity at low drain voltage, largely free from contact resistance and injection issues. The increase of the drain current in the VG = 0 V curve above VD = 35 V is a clear signature of ambipolar hole injection and transport. Figure 5C compares the molecular weight dependence of the four polymers. At a fixed molecular weight, AN2 and NN2 exhibit higher carrier mobilities than AN1 and NN1. This is consistent with the more pronounced interchain interactions in AN2 and NN2 that are facilitated by moving the branching point away from the polymer backbone. We also find that the molecular weight dependence is weaker in AN2/NN2 than in AN1/NN1: When the number average molecular weight in AN2 increases more than 15 times, from 10 kDa to more than 150 kDa, the μsat increases by less than a factor of 3. In contrast, in NN1, high mobilities approaching 0.1 cm2 V−1 s−1 can only be achieved for molecular weights above 50 kDa; for smaller molecular weights, mobilities were found to drop markedly. One possible explanation for such reduced molecular weight dependency of NN2 and AN2 devices compared with NN1 devices could be the increased crystallinity and closer interchain interactions, making NN2 and AN2 polymers less reliant on high–molecular weight tie chains interconnecting between few close-contact points/aggregates to form effective charge transport networks than in NN1 and AN1.
Temperature-dependent OFET charge transport measurements were also performed. For these, we used samples with the highest molecular weight/mobility for each of the four polymers. The performances of representative devices at 300 K are presented in fig. S11. The saturation transfer curves as a function of temperature are presented in fig. S12. The transistor current for both electron and hole transport decreases monotonically with decreasing temperature, but the shape of the ambipolar transfer characteristics does not change, which suggests that electron and hole transport exhibit similar activation energies. Because of the issues with hole contact injection, we focus on a discussion of electron transport here. Activation energies extracted according to are shown for the four polymers in Fig. 5D, for which we used the mobility values at VG = 60 V. For all four polymers, activation energies are around 100 meV; these are comparable with those of other high-performance semicrystalline polymeric OFETs (15) and only slightly higher than values reported for the electron-deficient polymer P(NDI2OD-T2) OFETs with the same top-gate, bottom-contact architecture and PMMA dielectric (EA = 86 meV) (45). The EA is significantly higher than found in some of the highest-performing OFETs (15, 46), based on conjugated polymers with a very low degree of energetic disorder combined with gate dielectrics of low relative permittivity (39). Given the relatively low value of the Urbach energy extracted from the PDS measurements for both AN2 (32 meV; Fig. 4) and P(NDI2OD-T2) [32 meV (10, 32)], we might attribute, on one hand, the comparatively high activation energy found in our polymers at least partly due to the usage of PMMA as a dielectric layer. Its relative permittivity of 3.6 tends to induce larger dipolar disorder at the OSC-dielectric interface compared with other lower relative permittivity dielectric such as CYTOP and polystyrene (45, 47–49). Cytop is not used because it is hard to spin-coat uniform films on annealed fused polymer films reproducibly.
Another factor that is likely to contribute to the high activation energy is the reorganization energy. DFT calculations reveal that the reorganization energies associated with electron hopping are extremely large: 0.81 and 0.87 eV for the sum of the relaxation energies in the neutral and charged states in NN and AN, respectively. A major fraction of that reorganization stems from high-frequency (namely, carbon-carbon stretching) vibrations that do not contribute to the observed temperature dependence (as their population is constant across the T range explored) but, to first-order approximation, renormalize (to lower values) the effective electronic bandwidth, thereby reducing the mobility values. Our calculations suggest the formation of small polarons extending over a few repeating units and where the central carbon-carbon double bonds in the neutral ground state (~1.36 Å) considerably elongate (to ~1.42 Å at the center of the polaron) in the anionic state (fig. S17). We believe that this is the main reason for the somewhat disappointing electron mobility values measured in NN and AN in view of their low-electron effective masses. If we subtract the contribution of the high-frequency modes to the total reorganization energy λ (by relaxing versus freezing out the conformational degrees of freedom), we are left with that due to soft (low-frequency) vibrations that amount to 109 and 116 meV for NN and AN, respectively. This would translate into an activation energy, λ/4, of ~25 to 30 meV. According to Bässler et al. (50), the activation energy for transport in disordered materials is the sum of polaron and disorder contributions, . For a disorder with an SD σ of 40 meV, this translates into an activation energy of ~90 meV, in good agreement with the experiment.
These high activation energies find their origin in large-amplitude fluctuations in twist angles between the subunits upon charging. While conventional polymers tend to planarize in the ionized state, the opposite behavior is observed here because the connecting bonds between the fused rings acquire an increased single-bond character when adding an extra electron, and, as a result, the torsion potential energy surface is significantly softened and displaced toward larger equilibrium torsion angles (fig. S18).
In our previous work, we compared the air stability of NN1 OFETs with P(NDI2OD-T2) OFETs and found NN1 to exhibit more air-stable electron transport than P(NDI2OD-T2). Here, we expand the air stability evaluation to all four polymers synthesized. For these environmental stability tests, the bias was merely applied during measurements. Between measurements, unbiased devices are stored in a dark environment fully exposed to air.
Figure 6A shows that the incorporation of the anthracene-derived molecular units into the backbone, which tends to slightly reduce the EA, does not compromise the air stability of electron transport. AN2 OFETs remain stable after continuous air exposure for 350 hours without further degradation and retain a saturation electron mobility of around 0.2 cm2 V−1 s−1 after prolonged air exposure. This is in clear contrast to P(NDI2OD-T2), the performance of which plummets after more than 250-hour air exposure. In Fig. 6B, the operational stability of a representative AN2 device after 450-hour continuous air exposure is characterized, showing stable ambipolar operation under both p- and n-channel accumulation. As far as we know, this is the first observation of such a high level of stability in the ambipolar charge transport characteristics for a conjugated polymer OFET. Threshold voltage VT shift of air-exposed AN2 devices is shown in fig. S13.
Fig. 6. Environmental and stress stability investigation of rigid-rod polymer FETs.
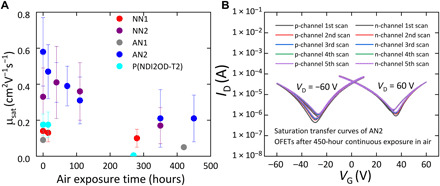
(A) Saturation electron mobility of NN1, NN2, AN1, AN2, and P(NDI2OD-T2) top-gate, bottom-contact OFETs (L = 20 μm, W = 1 mm) measured under various air exposure time, highlighting the air stability of electron transport of fused polymer OFETs in comparison to the P(NDI2OD-T2) OFET. (B) Saturation transfer curves (with p- and n-channel accumulation) for a representative AN2 OFET measured in the air after 450-hour continuous air exposure to demonstrate the operational stability of the sufficiently air-exposed device.
Last, we used continuous-wave FI-ESR to study polaron spin dynamics and to obtain microscopic insight into the corresponding charge dynamics. By measuring the power saturation characteristics and linewidths of the FI-ESR spectra, we extracted the spin-lattice relaxation time, T1, and the coherence time, T2, as a function of temperature, as can be seen in Fig. 7B for NN1 and Fig. 7E for AN2 (51, 52). In these, we compare FI-ESR data for NN1 and AN2 devices from 5 to 290 K; we focus on the electron transport regime and show data extracted at a fixed gate voltage, VG = 60 V. The transfer and output characteristics of NN1 and AN2 ESR FET devices at 170 K are shown in fig. S14. The spin lifetimes show three distinguishable temperature regimes. For the NN1 device below 120 K and for the AN2 device below 100 K, spins hop very slowly and experience inhomogeneous magnetic environments with stochastic fluctuations in the magnetic field amplitude δBrms, which are due to, for example, local variations of the hyperfine magnetic fields. On the basis of a procedure described in our previous work (36), we extracted from the 5 K spectra values of δBrms = 0.54 G for NN1 and 0.84 G for AN2. For the NN1 device from 120 to 170 K, and for the AN2 device from 100 to 185 K, we observed a “motional narrowing” regime (36): In this temperature range, the motion of charges/spins is sufficiently fast such that the ESR spectrum reflects an average of the magnetic field environments. This averaging effect due to charge motion leads to a single Lorentzian line shape (Fig. 7, A and D), whose linewidth narrows inversely with T2 as temperature increases. At even higher temperatures (>200 K in NN1 and AN2), a different spin relaxation mechanism manifests and leads to a peak in T2 and a subsequent decrease with increasing temperature. In this regime, coupling between the vibrational dynamics and the microscopic charge/spin motion determines the spin relaxation behavior. The latter regime provides information on the coupling between electron spins and vibrational modes (36) and will not be discussed in detail here.
Fig. 7. FI-ESR characterization of rigid-rod polymers.
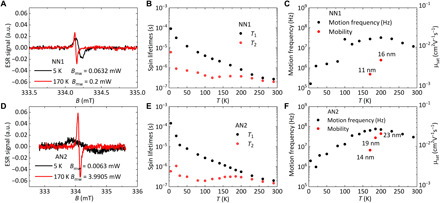
FI-ESR spectra of the top-gate, bottom-contact sample (L = 100 μm, W = 243 mm) fabricated from spin-coated (A) NN1 and (D) AN2 film at 5 and 170 K. Spin lifetimes T1 and T2 for electron polarons in (B) NN1 and (E) AN2 FI-ESR sample at VG = 60 V. Motion frequency of charges determined from T2 (left axis) and saturation mobilities from FET measurements at VG = 60 V (right axis) of (C) NN1 and (F) AN2 FI-ESR samples. Labels show the calculated hopping distances in the motional narrowing regime by relating the motion frequency and saturation electron mobility with the Einstein relation for charge transport.
Instead, we focus on the motional narrowing regime, in which quantitative information on charge carrier motion frequencies and hopping distances can be extracted. Motion frequencies, ν, can be computed directly from the measured ESR linewidths (36). From the Einstein relationship using the measured OFET mobility μ, it is then possible to compute the average hopping distance (Fig. 7, C and F). Since the calculation of is reliant on both μ and v, we can only extract reliable values in a narrow temperature range between 170 K (where charge motion becomes sufficiently fast to reliably determine a FET mobility) and below 200 K (the highest temperature for which motional narrowing holds). In the motional narrowing regime, T2 relaxation is determined by local changes in the magnetic field parallel to the external field B0. The reported coherence times were measured with a field perpendicular to the sample and are therefore dominated by hyperfine fields within crystallites. Between-crystallite motion can additionally cause variations in the g-tensor component that is parallel to B0 if the backbone tilt angle relative to B0 changes. This would occur, for example, for measurements taken with B0 parallel to the substrate and an isotropic distribution of in-plane crystallite orientations. Because the hopping distances reported here correspond to local motion while the measured mobilities may well be limited by slower intercrystallite hopping (23), the values of reported here should be interpreted as lower limit estimates of the true hopping distance. While this does not affect the conclusion of this work, it is important to keep this in mind when comparing absolute values of to those obtained in other, morphologically different systems, which might be affected to a different extent by differences between FET and local mobilities. Nevertheless, it is worth noting that the extracted is within the same order of magnitude as the chain contour lengths obtained from the fitting of the SANS curves. Although of course a direct comparison between measurements performed in solution and in the solid state is not possible, this similarity of values suggests at least that chain conformations do not change dramatically from solution to the solid state. This is also consistent with the close similarity between the optical absorption spectra in solution and in the solid state (fig. S1).
The extracted values of v are compared in Table 4 with those of other, high-mobility polymers that we have investigated recently by the same technique. The motion frequencies for both NN1 and AN2 are significantly slower than those of PBTTT, P(NDI2OD-T2), and C16 IDT-BT. This could be a manifestation of significant residual energetic disorder that slows down motion of carriers through the network of polymer chains. This is consistent with the comparatively high Urbach energy of NN1, the comparatively short coherence lengths for the backbone (004) diffractions observed in GIWAXS, and the theoretical predictions of significant polaronic relaxation effects in the anionic state prompting the connecting double bonds with an increased single-bond character and translating into large conformational motions around nonplanar equilibrium backbone geometries. It is actually remarkable to notice that, in contrast to “conventional” conjugated polymers (i.e., with single bonds connecting the repeating units), electron trapping in our polymers is expected to occur in the most twisted segments of the polymer chains. Because the relevant conformational motion occurs around the double bonds that feature an antibonding pattern in the LUMO (rather than a bonding pattern for single bonds in conventional polymers), increasing the torsion angle decreases the antibonding character, thus stabilizing the electron.
Table 4. Motion frequency comparison between different polymers.
| Materials | Motion frequency (MHz) |
| PBTTT-C14 (150 K) | 100 (p-channel) |
| P(NDI2OD-T2) (150 K) | 100 (n-channel) |
| IDTBT-C16 (150 K) | 1000 (p-channel) |
| NN1 (170 K) | 28 (n-channel) |
| AN2 (170 K) | 65 (n-channel) |
On the other hand, the extracted lower limit estimates of the hopping distances for both NN1 and AN2 (16 and 23 nm, respectively, at 200 K) are longer than those of PBTTT and P(NDI2OD-T2) and are even comparable to the record values observed in C16 IDT-BT (36). This is consistent with the rigid-rod nature of the polymers that is evident from the solution SANS measurements and that is preserved in thin films, as evident from GIWAXS. From the FI-ESR measurements, we arrive at the important conclusion that charge transport in the NN/AN polymers is characterized by a relatively slow hopping motion between sites that are separated by long distances. Those large hopping distances are ultimately responsible for the respectable electron mobility values of up to 0.5 cm2 V−1 s−1 that can be achieved in AN2.
Our study has provided direct experimental evidence that it is possible to realize rigid-rod behavior in conjugated polymers synthesized by aldol condensation with only double-bond and no single-bond linkages between fused-ring conjugated units. We have shown evidence that the persistence length of these polymers matches/exceeds their contour length. We have also demonstrated that by side-chain optimization and by adopting a copolymer design, it is possible to achieve respectably high electron mobilities of 0.5 cm2 V−1 s−1 that are within an order of magnitude to those of the best conjugated polymers. The level of performance achieved to date is already of interest for large-area electronics because of the excellent stability for n-channel OFET operation in air. We have also uncovered some of the transport-limiting factors that prevent achieving even higher carrier mobilities: In the systems investigated to date, the polymer backbone is not expected to be planar due to steric hindrance between a carbonyl carbon and an adjacent C─H group, and the torsion potentials are sufficiently flat around the equilibrium configurations that they allow significant variations in torsion angles. Besides, the torsional fluctuations are facilitated in the ionized state where the double-bond linkages largely stretch out compared to the neutral ground state. These polaronic effects combined with the increased torsional energetic disorder act against the large electronic dispersion and slow down electron transport. In the future, molecular designs should be sought within this class of polymers that allow planar backbone conformations without steric hindrance and exhibit a narrower distribution of torsion angles. Such systems could potentially retain the unique rigid-rod nature of these polymers but achieve lower energetic disorder and reorganization energies and allow reaching even higher charge carrier mobilities, potentially exceeding those of state-of-the-art systems.
MATERIALS AND METHODS
Device fabrication
Top-gate, bottom-contact architectures are used to fabricate all the field-effect devices, including OFETs and FI-ESR samples. Photolithographically patterned electrodes (15-nm gold layer on top on 3-nm chromium adhesion layer) were evaporated on low-alkali 1737F Corning glass substrates under high vacuum (<5 × 10−6 mbar). For standard OFET devices, interdigitated electrode patterns (20 μm channel length and 1000 μm channel width) are used, while for FI-ESR samples, larger-area elongated substrates are used [sample geometry details within (36)] in order to accumulate more charges/spins. After the lift-off process, substrates are cleaned with acetone and isopropanol, blow-dried with a nitrogen gun, and then ready to use. Around 30-nm conjugated polymer films are spin-coated on top of clean substrates from hot precursor solutions (140°C 1,2,4-trichlorobenzene) at 1200 rpm for 240 s and then thermally annealed in nitrogen atmosphere at 200°C for half an hour before quenching down to room temperature. Around 500 nm of PMMA layer was then spin-coated on top of the annealed semiconductor layer from orthogonal solvent n-butyl-acetate and then annealed at 90°C for 20 min. After this step, samples are transferred from a glove box to an evaporator for the gate electrode evaporation, and a 30-nm aluminum layer is thermally evaporated on top of the channel area within shadow mask–defined regions.
Device characterization
Transfer and output curves of OFET devices are measured with an Agilent 4155 semiconductor parameter analyzer. For temperature-dependent characterization, a Lake Shore TTPX cryogenic probe station was used to characterize the electronic performance of related OFETs from 230 to 300 K, with 10 K chosen as the temperature interval between measurements.
Both linear and saturation mobility are extracted from the related transfer curves based on the equation derived from the MOSFET Gradual Channel Approximation (53)
The activation energy of the field-effect mobility is extracted from the fitting of an Arrhenius plot of the temperature-dependent saturation mobility.
SANS experiments
SANS experiments were carried out using the Sans2D instrument at the ISIS Facility, Rutherford Appleton Laboratory, UK. As a time-of-flight instrument, Sans2D uses the neutrons with an incident wavelength of 2.0 to 14.0 Å at 10 Hz. The polymers were dissolved into a deuterated solvent (dichlorobenzene-d4) with a concentration of ~5 mg/ml. The sample-to-detector distance is 4 and 8 m (8-m setup measurement only for the AN2 polymer), which results in a q range of 0.006 to 0.45 Å−1 [where q is the scattering vector q = 4π sin (θ)/λ, 2θ is the scattering angle]. The data were corrected for background scattering, detector response, and the spectral distribution of the incident beam and converted to an absolute scattering cross section (in cm−1) using standard procedures (54).
For a monodisperse collection of particles, the scattering cross section can be written as
where ϕ is the volume fraction of the particle, V is the volume of the particle, Δρ is the scattering length density difference between the particle and the solvent, P(q) is the particle form factor, and S(q) is the structure factor. P(q) describes the structure of the particles and is equal to unity at q = 0. The structure factor describes the interferences of scattering from different particles and contains information on the interaction between the particles. For dilute solution systems, S(q) = 1 and can be neglected. The influence of different form factors corresponding to different models of chain shape on the scattering curves is summarized in the Supplementary Materials.
Grazing-incidence wide-angle x-ray scattering
GIWAXS measurements were performed at the WAXS beamline at the Australian Synchrotron (55). Details of the GIWAXS measurement and analysis can be found elsewhere (56). For the GIWAXS measurements, films were deposited on Si wafers with a native oxide layer on the surface.
UV-Vis-NIR absorption spectroscopy
A SHIMADZU UV-3600i Plus UV-VIS-NIR spectrophotometer is used to measure the UV-Vis-NIR absorption spectroscopy of thin-film and solution samples. Thin-film samples are spin-coated in the same way as for OFETs fabrication, while solution samples are held and measured within quartz cuvettes (1-cm-long light path) at room temperature.
Field-induced electron spin resonance
FI-ESR measurements were carried out on a Bruker E500 spectrometer with an Oxford Instruments ESR900 helium cryostat providing temperature control and a Keithley 2602b performing electrical measurements. All measurements were automatically controlled by a self-developed code available at https://github.com/OE-FET/CustomXepr.
FI-ESR devices were fabricated in a top-gate, bottom-contact architecture with interdigitated electrodes. Electrical contact was made through contact pads outside of the EPR spectrometer cavity, and the electrode thickness of 18 nm was chosen to be well below the skin depth of microwaves at 9.4 GHz. To increase the ESR signal from accumulated charges and to accommodate the geometry of the X-band cavity, samples were fabricated on 3 mm × 40 mm, high-purity quartz substrates with a large active area of 2.45 mm × 20 mm and a channel length and width of L = 100 μm and W = 235 mm, respectively. The initial patterning of electrodes and spin-coating was carried out on 40 mm × 40 mm substrates with eight devices each before separating the substrates into individual devices, following the procedure described in (36). The performance of the FI-ESR FETs on quartz substrates was very similar to that of regular FETs fabricated on glass substrates.
PDS measurements
PDS measurements are performed to investigate the sub-bandgap absorption of the fused polymers film investigated in this work. PDS probes weak sub-bandgap absorption transitions by measuring the deflection of a probe beam due to refractive index change at the surface of the sample caused by heating of the surface upon light absorption. Details about the setup and models for the deflection medium could be found in (57).
Impedance measurement
C-f measurement is performed by an HP 4912A Impedance Analyzer (50 to 100 Hz). C-f curves are measured for the (ca. 500 nm) spin-coated PMMA dielectric layer sandwiched between two overlapping aluminum electrodes. The bottom aluminum electrode was evaporated on top of a prepatterned indium tin oxide layer (in the middle of the glass substrates). The overlapping area between the two electrodes is 0.045 cm2.
Theoretical calculations
To generate the torsion potentials, the ground-state geometries of the monomer unit bearing two naphthalene (NN polymer) or one naphthalene and one anthracene (AN polymer) have first been optimized at the DFT level (LC-ωHPBE/cc-pvdz) while freezing the torsion angle between the two subunits from 0° to 180° by a step of 5°. Then, those geometries have been used as input for single-point calculations with a larger basis set (LC-ωHPBE/cc-pvtz) from which energies have been extracted to build the torsion potentials. The torsion angle distribution at room temperature has been calculated from Maxwell-Boltzmann statistics and used to assess thermally averaged deviations from planarity (58). To probe the electronic (energies of the HOMO and LUMO levels) and optical (energy of the first excited state) properties of the oligomers, we first tuned, for each of them, the ω parameter of the LC-ωHPBE functional in gas phase on the basis of the ground-state optimized LC-ωHPBE/cc-pvtz geometries. The “gap tuning” approach adopted here consists in adjusting the ω value in order to match the HOMO energy of the neutral fragment with the ionization potential and the HOMO energy of the anion with the EA (59, 60). This procedure leads to values of 0.1375 and 0.1290 bohr−1 for the NN and AN fragment, respectively. The energies of the first excited state have been simulated as a function of torsion angle at the LC-ωHPBE/cc-pvtz level on the basis of LC-ωHPBE/cc-pvdz ground-state optimized geometries. Note that the ω values have been kept constant for all the torsion angles.
To simulate the absorption spectra reported in Fig, 1, we have first tuned the ω parameter of the LC-ωHPBE functional in gas phase for oligomers of increasing size and adopting torsion angles between successive units equal to the thermally averaged deviation from planarity values, i.e., 18.1° and 18.6° for NN and AN, respectively. Then, we have calculated the vertical transition energies and oscillator strengths of the electronic excitations at the TD-DFT level (OT-LC-ωHBPE/cc-pvdz). The calculations are performed while accounting implicitly for a solvent continuum environment (mimicking toluene) by adjusting the β parameter (figs. S2 and S3). The nature of the electronic excitations has been characterized from the overlap Φs between the hole and electron densities calculated in the attachment/detachment formalism (61–63). Pure CT excitations correspond to nonoverlapping hole and electron densities resulting in Φs = 0 while Frenkel excitations lead to Φs = 1.
The band structure of the polymers reported in fig. S15 has been estimated from calculations for model systems at the DFT level using the B3LYP functional and a 6-31G(d,p) basis set where the unit cell, containing one monomer, was replicated in one direction to yield infinite isolated polymer chains. These monomer units are extracted from the central part of an optimized dimer (LC-ωHPBE/cc-pvdz) for which the torsion angles have been frozen to thermally averaged deviation from planarity values. The effective masses corresponding to hole (electron) transport have been extracted from the curvature of the highest (lowest) electronic state of the valence (conduction) band.
The reorganization energies have been estimated for oligomers containing two monomers as the sum of two relaxation energy terms: (i) the difference between the energies of the neutral molecule in its equilibrium geometry and in the relaxed geometry characteristic of the ion and (ii) the difference between the energies of the radical ion in its equilibrium geometry and in the neutral geometry. A major fraction of the reorganization stems from high-frequency (namely, carbon-carbon stretching) vibrations that do not contribute to the temperature dependence in contrast to soft (low-frequency) vibrations. The contribution of the high-frequency modes has been estimated similarly to the reorganization energy except that the torsion angles have been frozen to neutral values during the optimization of the anion. The low-frequency contribution, i.e., inter-ring torsions, has then been estimated as the energy difference between the reorganization energy and its high-frequency contribution. All the calculations have been performed using the Gaussian 16/A03 package (64).
Acknowledgments
M.X. acknowledges King’s College, University of Cambridge, and Cambridge Philosophical Society for studentship. Funding: S.P.S. and I.J. acknowledge funding through the Royal Society Newton Alumni Fellowship. M.NI and S.H. acknowledge funding through the award of a Marie Curie Global Fellowship. We gratefully acknowledge financial support by the Engineering and Physical Sciences Research Council (EPSRC) through a program grant (EP/M005143/1) and by the European Research Council (ERC) through a Synergy grant (SC2 610115). This work was undertaken, in part, at the SAXS/WAXS beamline at the Australian Synchrotron, part of ANSTO. The work in Mons was supported by the European Commission/Région Wallonne (FEDER-SOLIDYE project), the Consortium des Équipements de Calcul Intensif (CÉCI), funded by the Fonds National de la Recherche Scientifique (F.R.S.-FNRS) under grant no. 2.5020.11 as well as the Tier-1 supercomputer of the Fédération Wallonie-Bruxelles, infrastructure funded by the Walloon Region under Grant Agreement n1117545, and FRS-FNRS. D.B. is FNRS Research Director. M.A.-J. thanks Cambridge Materials Limited, Wolfson College, University of Cambridge, and the Royal Society for funding and technical support. A.S. acknowledges funding and support from the SUNRISE project (EP/P032591/1), EPSRC, and UKIERI project. Author contributions: M.X. and R.L.C. fabricated and measured all the FET samples to investigate the charge transport property and spin dynamics of related polymers. M.NI, S.P.S., Y.Z., L.L., E.S., Q.W., and I.J. assisted M.X. in the initial characterization of polymer samples. H.C., C.J., A.O., W.Y., and I.M. synthesized all the related polymers. X.J. and C.R.M. measured all the GIWAXS samples. V.L. and D.B. performed theoretical simulations. S.S. contributes to the ESR analysis. A.S. and M.A.-J. performed PDS measurements. M.X., G.L., S.R., and N.M. performed SANS measurements and analysis. M.X., S.H., and Z.Z. performed UV-Vis spectroscopy measurements. M.X. and M.NG. performed stability test. T.H.T. and X.R. helped with data interpretation and manuscript revision. M.X. and H.S. wrote most of the manuscript (except for the “Theoretical calculations” section and “Synthetic information” section in the Supplementary Materials), with input from all the coauthors. V.L. and D.B. wrote the “Theoretical calculations” section. H.C. and I.M. wrote the “Synthetic information” section in the Supplementary Materials. H.S. supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/18/eabe5280/DC1
REFERENCES AND NOTES
- 1.Sirringhaus H., 25th anniversary article: Organic field-effect transistors: The path beyond amorphous silicon. Adv. Mater. 26, 1319–1335 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bao Z., Dodabalapur A., Lovinger A. J., Soluble and processable regioregular poly(3-hexylthiophene) for thin film field-effect transistor applications with high mobility. Appl. Phys. Lett. 69, 4108–4110 (1996). [Google Scholar]
- 3.Liu Y., Zhao J., Li Z., Mu C., Ma W., Hu H., Jiang K., Lin H., Ade H., Yan H., Aggregation and morphology control enables multiple cases of high-efficiency polymer solar cells. Nat. Commun. 5, 5293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burroughes J. H., Bradley D. D. C., Brown A. R., Marks R. N., Mackay K., Friend R. H., Burns P. L., Holmes A. B., Light-emitting diodes based on conjugated polymers. Nature 347, 539–541 (1990). [Google Scholar]
- 5.Arias A. C., MacKenzie J. D., McCulloch I., Rivnay J., Salleo A., Materials and applications for large area electronics: Solution-based approaches. Chem. Rev. 110, 3–24 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Inal S., Rivnay J., Suiu A.-O., Malliaras G. G., McCulloch I., Conjugated polymers in bioelectronics. Acc. Chem. Res. 51, 1368–1376 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Kim M., Ryu S. U., Park S. A., Choi K., Kim T., Chung D., Park T., Donor–acceptor-conjugated polymer for high-performance organic field-effect transistors: A progress report. Adv. Funct. Mater. 30, 1904545 (2020). [Google Scholar]
- 8.Yang Y., Liu Z., Zhang G., Zhang X., Zhang D., The effects of side chains on the charge mobilities and functionalities of semiconducting conjugated polymers beyond solubilities. Adv. Mater. 31, 1903104 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Mei J., Bao Z., Side chain engineering in solution-processable conjugated polymers. Chem. Mater. 26, 604–615 (2014). [Google Scholar]
- 10.Venkateshvaran D., Nikolka M., Sadhanala A., Lemaur V., Zelazny M., Kepa M., Hurhangee M., Kronemeijer A. J., Pecunia V., Nasrallah I., Romanov I., Broch K., McCulloch I., Emin D., Olivier Y., Cornil J., Beljonne D., Sirringhaus H., Approaching disorder-free transport in high-mobility conjugated polymers. Nature 515, 384–388 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Zhang W., Gomez E. D., Milner S. T., Predicting chain dimensions of semiflexible polymers from dihedral potentials. Macromolecules 47, 6453–6461 (2014). [Google Scholar]
- 12.Kuei B., Gomez E. D., Chain conformations and phase behavior of conjugated polymers. Soft Matter 13, 49–67 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Noriega R., Salleo A., Spakowitz A. J., Chain conformations dictate multiscale charge transport phenomena in disordered semiconducting polymers. Proc. Natl. Acad. Sci. U.S.A. 110, 16315–16320 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salleo A., Kline R. J., DeLongchamp D. M., Chabinyc M. L., Microstructural characterization and charge transport in thin films of conjugated polymers. Adv. Mater. 22, 3812–3838 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Noriega R., Rivnay J., Vandewal K., Koch F. P. V., Stingelin N., Smith P., Toney M. F., Salleo A., A general relationship between disorder, aggregation and charge transport in conjugated polymers. Nat. Mater. 12, 1038–1044 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Rivnay J., Mannsfeld S. C. B., Miller C. E., Salleo A., Toney M. F., Quantitative determination of organic semiconductor microstructure from the molecular to device scale. Chem. Rev. 112, 5488–5519 (2012). [DOI] [PubMed] [Google Scholar]
- 17.S. J. Chalk, IUPAC Compendium of Chemical Terminology (IUPAC, Research Triagle Park, NC, 2014); http://goldbook.iupac.org/P04515.html.
- 18.Gettinger C. L., Heeger A. J., Drake J. M., Pine D. J., A photoluminescence study of poly(phenylene vinylene) derivatives: The effect of intrinsic persistence length. J. Chem. Phys. 101, 1673–1678 (1994). [Google Scholar]
- 19.Vezie M. S., Few S., Meager I., Pieridou G., Dörling B., Ashraf R. S., Goñi A. R., Bronstein H., McCulloch I., Hayes S. C., Campoy-Quiles M., Nelson J., Exploring the origin of high optical absorption in conjugated polymers. Nat. Mater. 15, 746–753 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Rossi G., Chance R. R., Silbey R., Conformational disorder in conjugated polymers. J. Chem. Phys. 90, 7594–7601 (1989). [Google Scholar]
- 21.Coropceanu V., Cornil J., da Silva Filho D. A., Olivier Y., Silbey R., Brédas J.-L., Charge transport in organic semiconductors. Chem. Rev. 107, 926–952 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Snyder C. R., DeLongchamp D. M., Glassy phases in organic semiconductors. Curr. Opin. Solid State Mater. Sci. 22, 41–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka H., Wakamatsu A., Kondo M., Kawamura S., Kuroda S., Shimoi Y., Park W.-T., Noh Y.-Y., Takenobu T., Microscopic observation of efficient charge transport processes across domain boundaries in donor-acceptor-type conjugated polymers. Commun. Phys. 2, 96 (2019). [Google Scholar]
- 24.Zaumseil J., Semiconducting single-walled carbon nanotubes or very rigid conjugated polymers: A comparison. Adv. Electron. Mater. 5, 1800514 (2019). [Google Scholar]
- 25.Paterson A. F., Singh S., Fallon K. J., Hodsden T., Han Y., Schroeder B. C., Bronstein H., Heeney M., McCulloch I., Anthopoulos T. D., Recent progress in high-mobility organic transistors: A reality check. Adv. Mater. 30, 1801079 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Bronstein H., Nielsen C. B., Schroeder B. C., McCulloch I., The role of chemical design in the performance of organic semiconductors. Nat. Rev. Chem. 4, 66–77 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Fratini S., Nikolka M., Salleo A., Schweicher G., Sirringhaus H., Charge transport in high-mobility conjugated polymers and molecular semiconductors. Nat. Mater. 19, 491–502 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Onwubiko A., Yue W., Jellett C., Xiao M., Chen H.-Y., Ravva M. K., Hanifi D. A., Knall A.-C., Purushothaman B., Nikolka M., Flores J.-C., Salleo A., Bredas J.-L., Sirringhaus H., Hayoz P., McCulloch I., Fused electron deficient semiconducting polymers for air stable electron transport. Nat. Commun. 9, 416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen H., Hurhangee M., Nikolka M., Zhang W., Kirkus M., Neophytou M., Cryer S. J., Harkin D., Hayoz P., Abdi-Jalebi M., McNeill C. R., Sirringhaus H., McCulloch I., Dithiopheneindenofluorene (TIF) semiconducting polymers with very high mobility in field-effect transistors. Adv. Mater. 29, 1702523 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Fu B., Baltazar J., Sankar A. R., Chu P.-H., Zhang S., Collard D. M., Reichmanis E., Enhancing field-effect mobility of conjugated polymers through rational design of branched side chains. Adv. Funct. Mater. 24, 3734–3744 (2014). [Google Scholar]
- 31.Yan H., Chen Z., Zheng Y., Newman C., Quinn J. R., Dötz F., Kastler M., Facchetti A., A high-mobility electron-transporting polymer for printed transistors. Nature 457, 679–686 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Kronemeijer A. J., Pecunia V., Venkateshvaran D., Nikolka M., Sadhanala A., Moriarty J., Szumilo M., Sirringhaus H., Two-dimensional carrier distribution in top-gate polymer field-effect transistors: Correlation between width of density of localized states and urbach energy. Adv. Mater. 26, 728–733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.P. J. Flory, Principles of Polymer Chemistry (Cornell Univ. Press, 1953). [Google Scholar]
- 34.Kim H. G., Kang B., Ko H., Lee J., Shin J., Cho K., Synthetic tailoring of solid-state order in diketopyrrolopyrrole-based copolymers via intramolecular noncovalent interactions. Chem. Mater. 27, 829–838 (2015). [Google Scholar]
- 35.Chabinyc M. L., Toney M. F., Kline R. J., McCulloch I., Heeney M., X-ray scattering study of thin films of poly(2,5-bis(3-alkylthiophen-2-yl)thieno[3,2-b]thiophene). J. Am. Chem. Soc. 129, 3226–3237 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Schott S., Chopra U., Lemaur V., Melnyk A., Olivier Y., Di Pietro R., Romanov I., Carey R. L., Jiao X., Jellett C., Little M., Marks A., McNeill C. R., McCulloch I., McNellis E. R., Andrienko D., Beljonne D., Sinova J., Sirringhaus H., Polaron spin dynamics in high-mobility polymeric semiconductors. Nat. Phys. 15, 814–822 (2019). [Google Scholar]
- 37.Rivnay J., Toney M. F., Zheng Y., Kauvar I. V., Chen Z., Wagner V., Facchetti A., Salleo A., Unconventional face-on texture and exceptional in-plane order of a high mobility n-Type polymer. Adv. Mater. 22, 4359–4363 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Rivnay J., Steyrleuthner R., Jimison L. H., Casadei A., Chen Z., Toney M. F., Facchetti A., Neher D., Salleo A., Drastic control of texture in a high performance n-type polymeric semiconductor and implications for charge transport. Macromolecules 44, 5246–5255 (2011). [Google Scholar]
- 39.Zhang X., Bronstein H., Kronemeijer A. J., Smith J., Kim Y., Kline R. J., Richter L. J., Anthopoulos T. D., Sirringhaus H., Song K., Heeney M., Zhang W., McCulloch I., DeLongchamp D. M., Molecular origin of high field-effect mobility in an indacenodithiophene–benzothiadiazole copolymer. Nat. Commun. 4, 2238 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Jackson W. B., Amer N. M., Boccara A. C., Fournier D., Photothermal deflection spectroscopy and detection. Appl. Optics 20, 1333 (1981). [DOI] [PubMed] [Google Scholar]
- 41.Jackson W. B., Amer N. M., Direct measurement of gap-state absorption in hydrogenated amorphous silicon by photothermal deflection spectroscopy. Phys. Rev. B 25, 5559–5562 (1982). [Google Scholar]
- 42.Urbach F., The long-wavelength edge of photographic sensitivity and of the electronic absorption of solids. Phys. Rev. 92, 1324–1324 (1953). [Google Scholar]
- 43.Di Pietro R., Nasrallah I., Carpenter J., Gann E., Kölln L. S., Thomsen L., Venkateshvaran D., O’Hara K., Sadhanala A., Chabinyc M., McNeill C. R., Facchetti A., Ade H., Sirringhaus H., Neher D., Coulomb enhanced charge transport in semicrystalline polymer semiconductors. Adv. Funct. Mater. 26, 8011–8022 (2016). [Google Scholar]
- 44.Blom P. W. M., de Jong M. J. M., van Munster M. G., Electric-field and temperature dependence of the hole mobility in poly(p-phenylene vinylene). Phys. Rev. B 55, R656–R659 (1997). [Google Scholar]
- 45.Caironi M., Bird M., Fazzi D., Chen Z., Di Pietro R., Newman C., Facchetti A., Sirringhaus H., Very low degree of energetic disorder as the origin of high mobility in an n-channel polymer semiconductor. Adv. Funct. Mater. 21, 3371–3381 (2011). [Google Scholar]
- 46.Luzio A., Nübling F., Martin J., Fazzi D., Selter P., Gann E., McNeill C. R., Brinkmann M., Hansen M. R., Stingelin N., Sommer M., Caironi M., Microstructural control suppresses thermal activation of electron transport at room temperature in polymer transistors. Nat. Commun. 10, 3365 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ogier S. D., Veres J., Cupertino D. C., Mohialdin Khaffaf S., Leeming S. W., Low-k insulators as the choice of dielectrics in organic field-effect transistors. Adv. Funct. Mater. 13, 199–204 (2003). [Google Scholar]
- 48.Veres J., Ogier S., Lloyd G., de Leeuw D., Gate insulators in organic field-effect transistors. Chem. Mater. 16, 4543–4555 (2004). [Google Scholar]
- 49.Richards T., Bird M., Sirringhaus H., A quantitative analytical model for static dipolar disorder broadening of the density of states at organic heterointerfaces. J. Chem. Phys. 128, 234905 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Bässler H., Borsenberger P. M., Perry R. J., Charge transport in poly(methylphenylsilane): The case for superimposed disorder and polaron effects. J. Polym. Sci. B Polym. Phys. 32, 1677–1685 (1994). [Google Scholar]
- 51.Matsui H., Mishchenko A. S., Hasegawa T., Distribution of localized states from fine analysis of electron spin resonance spectra in organic transistors. Phys. Rev. Lett. 104, 056602 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Tsurumi J., Matsui H., Kubo T., Häusermann R., Mitsui C., Okamoto T., Watanabe S., Takeya J., Coexistence of ultra-long spin relaxation time and coherent charge transport in organic single-crystal semiconductors. Nat. Phys. 13, 994–998 (2017). [Google Scholar]
- 53.Pao H. C., Sah C. T., Effects of diffusion current on characteristics of metal-oxide (insulator)-semiconductor transistors. Solid State Electron. 9, 927–937 (1966). [Google Scholar]
- 54.Heenan R. K., Rogers S. E., Turner D., Terry A. E., Treadgold J., King S. M., Small angle neutron scattering using Sans2d. Neutron News 22, 19–21 (2011). [Google Scholar]
- 55.Kirby N. M., Mudie S. T., Hawley A. M., Cookson D. J., Mertens H. D. T., Cowieson N., Samardzic-Boban V., A low-background-intensity focusing small-angle X-ray scattering undulator beamline. J. Appl. Cryst. 46, 1670–1680 (2013). [Google Scholar]
- 56.Gann E., Caironi M., Noh Y.-Y., Kim Y.-H., McNeill C. R., Diffractive X-ray waveguiding reveals orthogonal crystalline stratification in conjugated polymer thin films. Macromolecules 51, 2979–2987 (2018). [Google Scholar]
- 57.Senanayak S. P., Yang B., Thomas T. H., Giesbrecht N., Huang W., Gann E., Nair B., Goedel K., Guha S., Moya X., McNeill C. R., Docampo P., Sadhanala A., Friend R. H., Sirringhaus H., Understanding charge transport in lead iodide perovskite thin-film field-effect transistors. Sci. Adv. 3, e1601935 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lemaur V., Cornil J., Lazzaroni R., Sirringhaus H., Beljonne D., Olivier Y., Resilience to conformational fluctuations controls energetic disorder in conjugated polymer materials: Insights from atomistic simulations. Chem. Mater. 31, 6889–6899 (2019). [Google Scholar]
- 59.Garza A. J., Osman O. I., Asiri A. M., Scuseria G. E., Can gap tuning schemes of long-range corrected hybrid functionals improve the description of hyperpolarizabilities? J. Phys. Chem. B 119, 1202–1212 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Do K., Ravva M. K., Wang T., Brédas J.-L., Computational methodologies for developing structure–morphology–performance relationships in organic solar cells: A protocol review. Chem. Mater. 29, 346–354 (2017). [Google Scholar]
- 61.Etienne T., Assfeld X., Monari A., Toward a quantitative assessment of electronic transitions’ charge-transfer character. J. Chem. Theory Comput. 10, 3896–3905 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Etienne T., Assfeld X., Monari A., New insight into the topology of excited states through detachment/attachment density matrices-based centroids of charge. J. Chem. Theory Comput. 10, 3906–3914 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Londi G., Dilmurat R., D’Avino G., Lemaur V., Olivier Y., Beljonne D., Comprehensive modelling study of singlet exciton diffusion in donor–acceptor dyads: When small changes in chemical structure matter. Phys. Chem. Chem. Phys. 21, 25023–25034 (2019). [DOI] [PubMed] [Google Scholar]
- 64.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. a. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. a. Petersson, H. Nakatsuji, X. Li, M. Caricato, a. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, a. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. a. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. a. Keith, R. Kobayashi, J. Normand, K. Raghavachari, a. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian 16, Revision C.01 (Gaussian Inc., G16_C01, 2016).
- 65.Pedersen J. S., Analysis of small-angle scattering data from colloids and polymer solutions: Modeling and least-squares fitting. Adv. Colloid Interface Sci. 70, 171–210 (1997). [Google Scholar]
- 66.des Cloizeaux J., Form factor of an infinite kratky-porod chain. Macromolecules 6, 403–407 (1973). [Google Scholar]
- 67.Pedersen J. S., Schurtenberger P., Scattering functions of semiflexible polymers with and without excluded volume effects. Macromolecules 29, 7602–7612 (1996). [Google Scholar]
- 68.Kotlarchyk M., Chen S.-H., Analysis of small angle neutron scattering spectra from polydisperse interacting colloids. J. Chem. Phys. 79, 2461–2469 (1983). [Google Scholar]
- 69.Chen H., Moser M., Wang S., Jellett C., Thorley K., Harrison G. T., Jiao X., Xiao M., Purushothaman B., Alsufyani M., Bristow H., De Wolf S., Gasparini N., Wadsworth A., McNeill C. R., Sirringhaus H., Fabiano S., McCulloch I., Acene ring size optimization in fused lactam polymers enabling high n-type organic thermoelectric performance. J. Am. Chem. Soc. 143, 260–268 (2021). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/18/eabe5280/DC1