

Abstract
α-Aryl-α-diazoamides were synthesized in two steps under mild conditions. This expeditious route employs Pd-catalyzed C–H arylation of N-succinimidyl 2-diazoacetate to obtain N-succinimidyl 2-aryl-2-diazoacetates, followed by aminolysis. The ensuing diazo compounds can esterify carboxyl groups in aqueous solution, and the ester products are substrates for an esterase. The broad scope of the synthetic route enables the continued development of diazo compounds in chemical biology.
Graphical Abstract
Since the discovery of diazomethane by von Pechmann in 1894,1 diazo compounds have become important reagents in synthetic organic chemistry. Often, diazo groups are utilized via thermal, photochemical, or transition metal-mediated carbenoid formation for constructing new C–C, C–O, or C–N bonds.2 Recently, the utility of diazo compounds has been extended into the realm of chemical biology.3,4
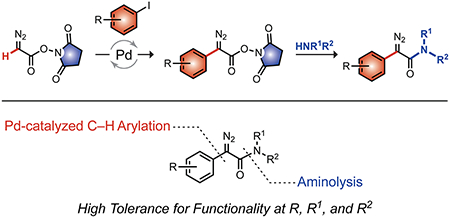
Recent work has shown that α-aryl-α-diazoacetamides can esterify carboxyl groups in proteins (ribonuclease A,5 green fluorescent protein,6 and ribonuclease 17), enabling their delivery across cellular membranes (Figure S1).6,7 This strategy bears analogy to the use of ester prodrugs of small-molecule carboxylic acids.8 The critical attribute of efficacious diazo compounds is their basicity,9 which leads to abstraction of a proton from a carboxylic acid but not water and thereby to the esterification of carboxyl groups in aqueous solution.5,10 Moreover, the ensuing esters are substrates for intracellular esterases.6,7 This bioreversibility11 provides a unique means to “cloak” protein carboxyl groups in a traceless manner (Figure 1A).
Figure 1.
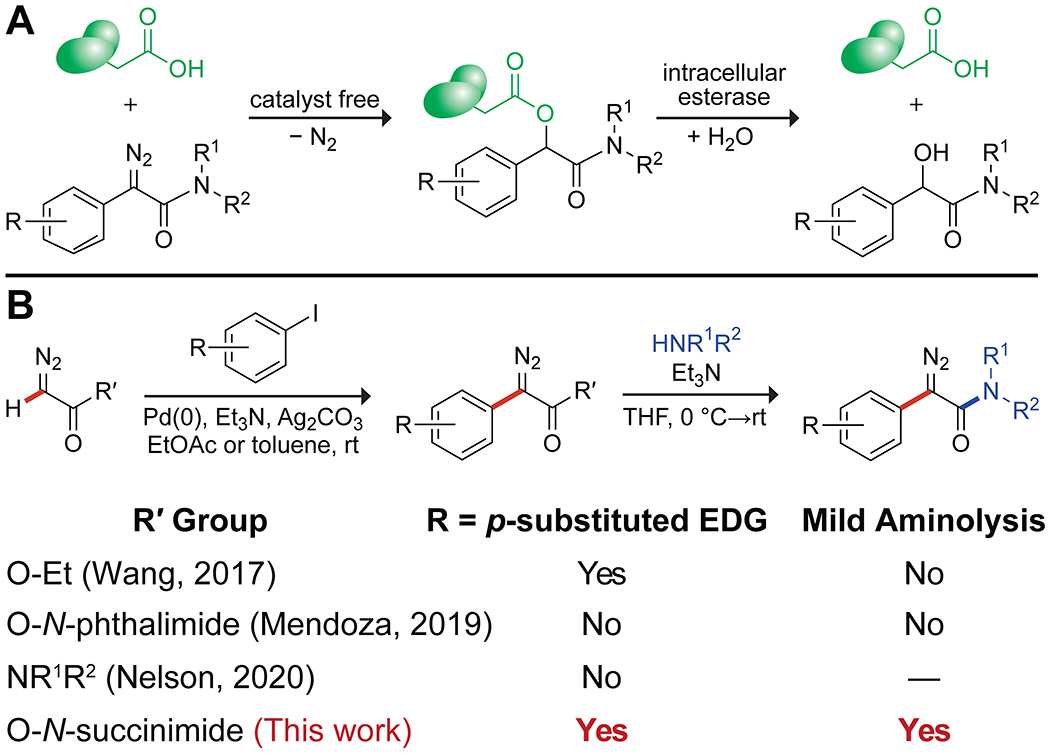
(A) Bioreversibility of protein esterification by an α-aryl-α-diazoacetamide. (B) Two-step synthesis of α-aryl-α-diazoacetamides. EDG, electron-donating group.
Although this application of α-aryl-α-diazoamides has demonstrated promise, synthetic accessibility (e.g., a lengthy preparation time and a lack of scalability) has been a major deterrent to progress. Previously, such diazo compounds have been accessed via deimidogenation of the corresponding azide (Figure S1).12 This approach has a high tolerance for functional groups, but access to the azide typically required lengthy low-yielding synthetic routes.13 Additionally, the deimidogenation reaction was not compatible with 2-aryl-2-azidoacetamides containing bulky N-substituents.6
We sought a facile and general route to the modular α-aryl-α-diazoamide scaffold. Known synthetic routes can provide access to α-diazo carbonyl compounds. Most, however, focus on stable diazoketones, diazoesters, or aryl diazomethanes14 and employ explosive diazo-transfer reagents, high temperature, or strong base3,15—conditions that can be incompatible with applications in chemical biology. Routes to α-aryl-α-diazoamides are underdeveloped and have limited substrate scope.14,16 Their preparation and isolation is challenging because of insolubility and functional group incompatibility.3,12,15,17
Here, we report on the mild, efficient, and versatile synthesis of α-aryl-α-amides in two steps from a commercially available18 and highly scalable precursor, N-succinimidyl 2-diazoacetate (1).19 Desired α-aryl-α-diazoamides are accessed via palladium-catalyzed C–H arylation followed by aminolysis under mild and safe conditions (Figure 1B). This route encompasses multiple benefits for applications in chemical biology: (1) facility, (2) broad applicability because of available building blocks (i.e., aryl iodides and amines), and (3) compatibility with diverse functionality (e.g., azido and alkynyl groups) that can be useful for late-stage bioconjugation.
Metal-catalyzed C–H arylation in the presence of a diazo group has been reported only sporadically due to the undesired competitive formation of metal–carbene species (Figure 1B).20 Wang and coworkers reported on the C–H functionalization of ethyl diazoacetate using Pd(PPh3)4.20b,21 The product, however, required the use of a strong base or metal catalyst to effect amidation.22 Mendoza and coworkers used another catalytic system, Pd(II) acetate and tris(2-furyl)phosphine (P(2-Fu)3), for the C–H arylation of N-phthalimidoyl diazoacetate to couple (hetero)aryl groups but encountered incompatibility with p-substituted electron-rich aryl groups (e.g., 4-iodoanisole).20c Recently, Nelson and coworkers reported a suite of methods for the synthesis of α-diazoamides, including the C–H arylation of α-diazo N,N-disubstituted acetamides.15 Similarly, this method failed in the coupling of electron-rich substrates (e.g., 4-iodoanisole) and provided no examples of C–H arylation with α-diazo N-monosubstituted acetamides.
To access a large number of target compounds under mild conditions, we investigated diazo compound 1 as a coupling partner for C–H arylation. Diazo compound 1 has been used to install a diazo group via acyl transfer reactions with amines, phenols, thiophenol, and peptides.19 We envisioned that the C–H arylation of diazo compound 1 could enable a rapid entry into more complex succinimidyl diazo compounds (2), and ultimately into diverse α-aryl-α-diazoamides (3).
We found that diazo compound 1 can undergo arylation with aryl iodides containing a wide variety of functional groups (Scheme 1). To do so, we prepared diazo compound 1 on a gram scale (Figure S2)19,23 and employed a Pd(OAc)2/P(2-Fu)3 catalytic system. Two additives, triethylamine (Et3N) and silver carbonate (Ag2CO3), prevent product decomposition and scavenge iodide, respectively.21,24 The reaction mixture was stirred in EtOAc at room temperature for 6 h. A range of aryl iodides, spanning electron-donating to -withdrawing p-substituted phenyl iodides, bulky m-substituted phenyl iodides, and a heteroaryl iodide, were subjected to the same reaction conditions. Notably, the electron-rich (2a), electron-neutral (2c), and electron-poor (2d) phenyl iodides all afforded high isolated yields (≥77%). Of the sterically hindered phenyl iodides, methoxy (2f) and trifluoromethyl (2h) functional groups at the m-position resulted in >80% isolated yields, whereas the smaller hydroxy group (2g) gave an even higher yield of 90%. We note too that 3-iodophenol (2g) proved to be orthogonal to the N-succinimidyl diazoester moiety, whereas 1-(4-iodophenyl)piperazine did not (Figure S11). The cross-coupling condition was compatible with heteroaryl substrate (2i) and a variety of fluoro groups, including trifluoromethoxy (2b), which is an important functional group for medicinal chemistry because of its high metabolic stability and cell permeability.25 We effected C–H arylation in the presence of a TMS-protected alkynyl (2e) or azido (2j) group in 95% and 66% yields, respectively. Further, compound 2j highlights a convenient means of diversification. This compound was accessed by a condensation reaction with 4-iodophenyl acetic acid. Lastly, we note that previously reported routes failed in arylation with 4-iodoanisole,15 whereas our route provided 2a in 65% yield. Overall, we successfully demonstrated metal-catalyzed C–H arylation in the presence of N-succinimidyl and diazo groups, both of which will serve as important functionality for instilling diversity.
Scheme 1.
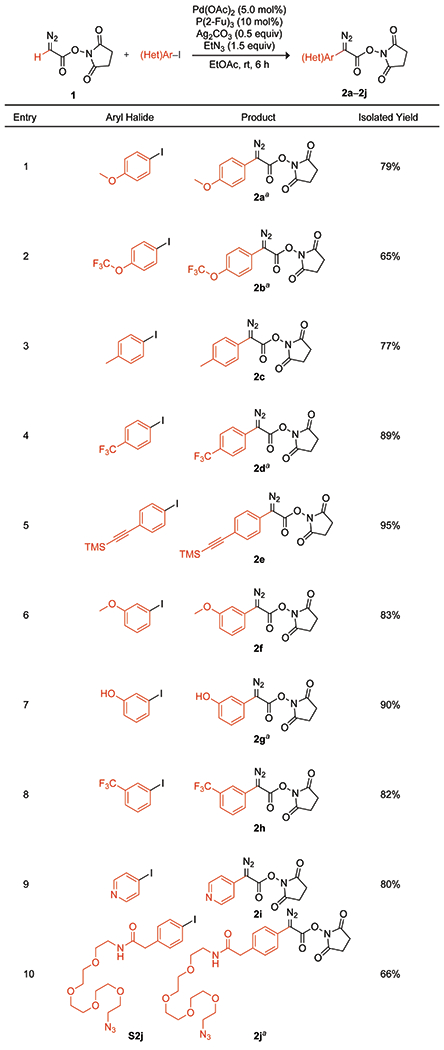
Scope of the C–H Arylation of Diazo Compound 1
aReaction conditions: 10 mol% Pd(OAc)2 20 mol% P(2-Fu)3.
Next, we examined the aminolysis of representative N-succinimidyl α-aryl-α-diazoacetates 2b–2f and 2h. We first tested the aminolysis of analogous N-phthalimidoyl diazoesters S5–S7, which were synthesized by a method reported previously (Figure 2A).20c Those diazoesters yielded only a trace amount of diazoamide product based on liquid chromatography–mass spectrometry (LC–MS) analysis. Even after an extensive screening of reactant concentrations, solvents, and additives, aminolysis at N-phthalimidoyl diazoesters proved to be unattainable, possibly due to rapid decarboxylation (Table S1). An initial evaluation of aminolysis with 2c showed that the use of 1,8-diazabicycloundec-7-ene (DBU) led to degradation, whereas Et3N afforded the desired product (Figure 2A). In these reactions, a solution of the N-succinimidyl diazoester was treated with a secondary amine and Et3N in tetrahydrofuran (THF) at 0 °C. The reaction mixture was stirred for 1–3 h at room temperature to yield the corresponding α-diazoamide (3e–3l) in up to 76% yield. An excess of Et3N was used to prevent product decomposition. Most of the reactions showed quantitative conversion based on analysis with TLC (Figure S12). Due to their apparent degradation on silica, the isolated yields for diazoamide compounds 3a, 3c, 3e, 3i, and 3k were low. Still, a wide range of aryl diazoesters was converted into N,N-disubstituted diazoamides.
Figure 2.
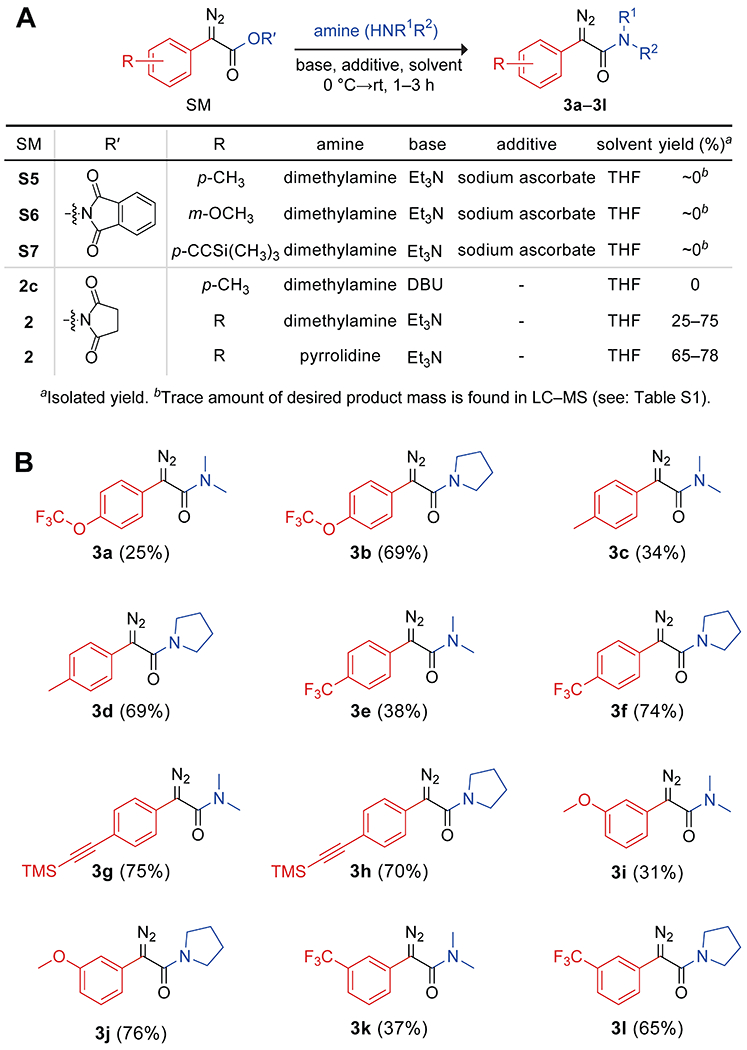
(A) Aminolysis of N-succinimidyl α-aryl-α-diazoacetates with secondary amines. (B) Scope of the ensuing N,N-disubstituted α-aryl-α-diazoamides.
Then, we demonstrated that the aminolysis of N-succinimidyl α-aryl-α-diazoacetates is also effective with various primary amines (Scheme 2). Those containing a pyridinyl (4a), arylhalo (4b), Boc-protected amino (4c), alkynyl (4d), or azido (4e) group displaced the N-hydroxysuccinimide moiety of 2c to yield the desired N-monosubstituted diazoamides. Additional scope for this reaction includes 6 N-succinimidyl α-aryl-α-diazoacetates × 4 primary amines = 24 α-aryl-α-diazoacetamides (see: Scheme S1).
Scheme 2.
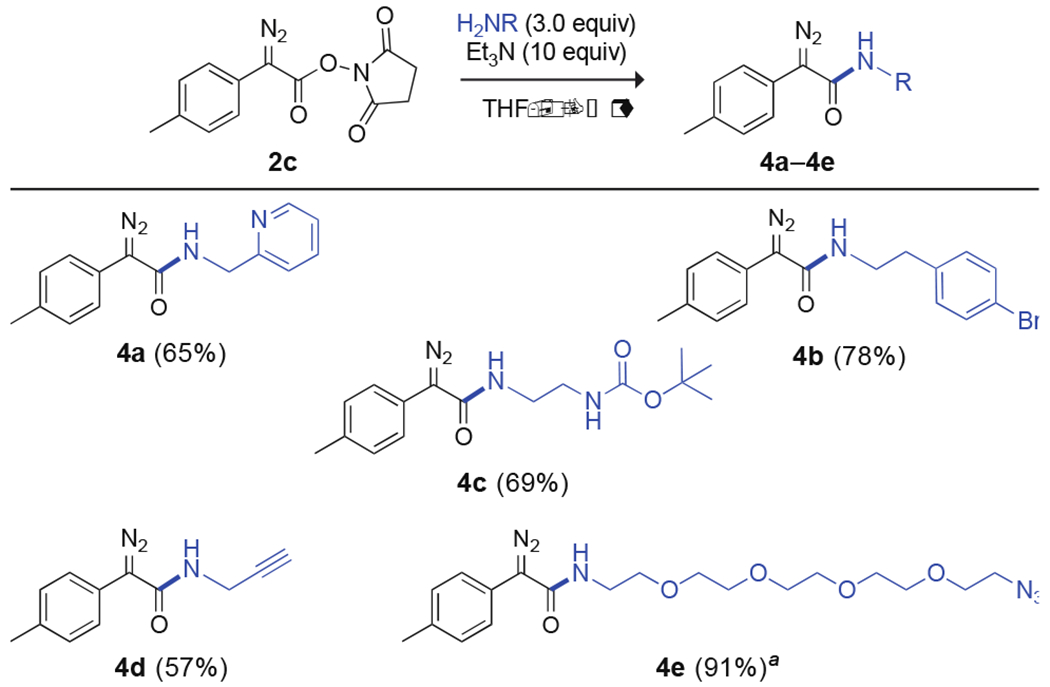
Scope of the Aminolysis of N-Succinimidyl α-Aryl-α-diazoacetates with Primary Amines; Isolated Yields Are Reported
aReaction conditions: 1.0 equiv of H2NR.
Having accomplished the facile synthesis of α-aryl-α-diazoamides, we turned our attention to their esterification of carboxylic acids. Specifically, we screened for the O-alkylation of five structurally diverse small molecules: pivalic acid, rhodamine B, coumarin-3-carboxylic acid, biotin, and HGluOMe by three representative diazo compounds (3h, 3j, and 3l) in 1:1 acetonitrile:MES–NaOH buffer, pH 6.0, at 37 °C for 19 h (Figure S6). Each of the reactions was analyzed by LC–MS to quantify the esterified product as well as the hydrolyzed byproduct, α-aryl-α-hydroxyamide (Table S2). Though hydrolysis is unavoidable due to the excess of water, esterification was successful regardless of the steric and electronic nature of the carboxylic acid or diazo compound.
Finally, we tested the bioreversibility of esterification by our diazo reagents. As a model acid, we used AcGluNH2 (5), which we derived from l-glutamic acid and which represents the most common residue for protein esterification6 and 6.4% of the residues in human proteins.26 In compound 5, the N-terminal amino group is acetylated to prevent aminolysis of a side-chain ester and the C-terminal carboxyl group is amidated to prevent main-chain esterification (Scheme 2). Compound 5 was treated with 3j to yield ester 6, which was then subjected to hydrolysis in the presence or absence of pig liver esterase (PLE) under biomimetic conditions at 37 °C (pH 5.8 for endosomes, pH 7.2 for the cytosol, and pH 8.0 for mitochondria).27 Though stable at pH 5.8, ester 6 hydrolyzed readily at pH 8.0, even in the absence of PLE (Figure S10). The hydrolysis at pH 7.2 was, however, reliant on PLE (Figures 3 and S9). These data suggest that cellular esterases will catalyze the hydrolysis of a nascent ester to reveal the native carboxylic acid of a protein.
Figure 3.
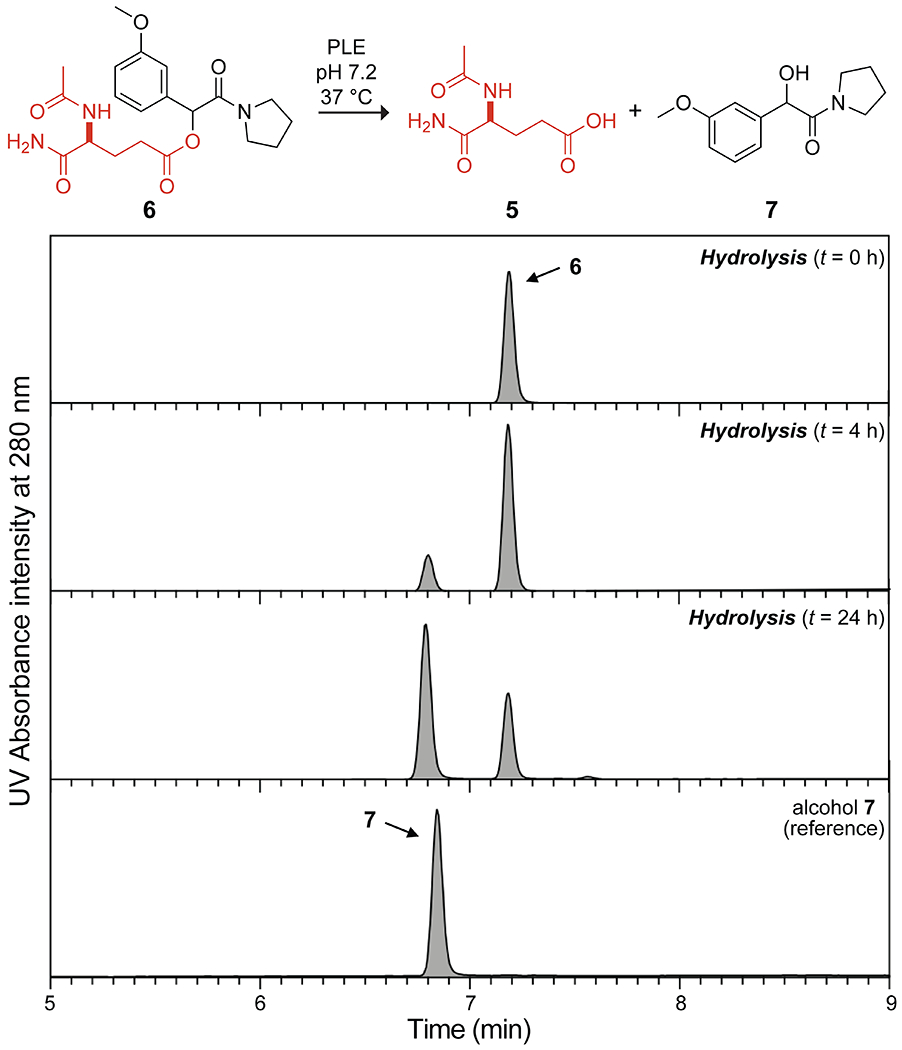
LC–MS analysis of the enzyme-catalyzed hydrolysis of γ-glutamyl ester 6 at pH 7.2 and 37 °C.
In conclusion, we demonstrated a facile two-step synthesis of α-aryl-α-diazoamides, which are modular reagents. This route will expedite the ongoing exploration of diazo compounds as reagents in chemical biology. We anticipate that the bioreversibility of our modification will enable applications in chemical biology, including the cellular delivery of proteins.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Dr. W. W. Massefski (Department of Chemistry, Massachusetts Institute of Technology) for analyzing NMR spectra of diazo compounds. We thank Dr. K. J. Hetrick (Department of Chemistry, Massachusetts Institute of Technology) for helpful discussions about cellular esterases.
Funding
J.V.J. was supported by a postdoctoral fellowship from the Ludwig Center at the Koch Institute for Integrative Cancer Research at MIT. This work was supported by Grants R01 CA073808 and GM044783 (NIH).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).(a) von Pechmann H Ueber Diazomethan. Ber. Dtsch. Chem. Ges 1894, 27, 1888–1891. [Google Scholar]; (b) von Pechmann H Ueber Diazomethan. Ber. Dtsch. Chem. Ges 1895, 28, 855–861. [Google Scholar]
- (2).a () Regitz M; Maas G, Diazo Compounds. Academic Press: London, UK, 1986. [Google Scholar]; b () Padwa A; Weingarten MD Cascade Processes of Metallo Carbenoids. Chem. Rev 1996, 96, 223–270. [DOI] [PubMed] [Google Scholar]; c () Davies HML; Beckwith REJ Catalytic Enantioselective C–H Activation by Means of Metal–Carbenoid-Induced C–H Insertion. Chem. Rev 2003, 103, 2861–2904. [DOI] [PubMed] [Google Scholar]
- (3).Mix KA; Aronoff MR; Raines RT Diazo Compounds: Versatile Tools for Chemical Biology. ACS Chem. Biol 2016, 11, 3233–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Caution! Exposure to heat, light, pressure, or shock can effect the exothermic decomposition of some diazo compounds. The diazo compounds used in this work are, however, predicted to be safe for use in the contexts of chemical biology. See:; Green SP; Wheelhouse KM; Payne AD; Hallett JP; Miller PW; Bull JA Thermal Stability and Explosive Hazard Assessment of Diazo Compounds and Diazo Transfer Reagents. Org. Process Res. Dev 2020, 24, 67–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mix KA; Raines RT Optimized Diazo Scaffold for Protein Esterification. Org. Lett 2015, 17, 2358–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mix KA; Lomax JE; Raines RT Cytosolic Delivery of Proteins by Bioreversible Esterification. J. Am. Chem. Soc 2017, 139, 14396–14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ressler VT; Mix KA; Raines RT Esterification Delivers a Functional Enzyme into a Human Cell. ACS Chem. Biol 2019, 14, 599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rautio J; Meanwell NA; Di L; Hageman MJ The Expanding Role of Prodrugs in Contemporary Drug Design and Development. Nat. Rev. Drug Discov 2018, 17, 559–587. [DOI] [PubMed] [Google Scholar]
- (9).For example, 2-phenyl-N,N-dimethylacetamide has pKa 26.6 in DMSO; (Bordwell FG; Fried HE Acidities of the H–C Protons in Carboxylic Esters, Amides, and Nitriles. J. Org. Chem 1981, 46, 4327–4331). [Google Scholar]
- (10).McGrath NA; Raines RT Diazo Compounds as Highly Tunable Reactants in 1,3-Dipolar Cycloaddition Reactions with Cycloalkynes. Chem. Sci 2012, 3, 3237–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kosa NM; Haushalter RW; Smith AR; Burkart MD Reversible Labeling of Native and Fusion-Protein Motifs. Nat. Methods 2012, 9, 981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Myers EL; Raines RT A Phosphine-Mediated Conversion of Azides into Diazo Compounds. Angew. Chem., Int. Ed 2009, 48, 2359–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chou H-H; Raines RT Conversion of Azides into Diazo Compounds in Water. J. Am. Chem. Soc 2013, 135, 14936–14939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ford A; Miel H; Ring A; Slattery CN; Maguire AR; McKervey MA Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev 2015, 115, 9981–10080. [DOI] [PubMed] [Google Scholar]
- (15).Chow S; Green AI; Artera C; Liver S; Leggott A; Trask L; Karageorgis G; Warriner S; Nelson A Efficient Approaches for the Synthesis of Diverse α-Diazo Amides. Synthesis 2020, 52, 1695–1706. [Google Scholar]
- (16).a () Chen Z; Popp BV; Bovet CL; Ball ZT Site-Specific Protein Modification with a Dirhodium Metallopeptide Catalyst. ACS Chem. Biol 2011, 6, 920–925. [DOI] [PubMed] [Google Scholar]; b () Zhang B; Wee AGH Conformational, Steric and Electronic Effects on the Site- and Chemoselectivity of the Metal-Catalyzed Reaction of N-Bis(trimethylsilyl)methyl, N-(2-Indolyl)methyl α-Diazoamides. Org. Biomol. Chem 2012, 10, 4597–4608. [DOI] [PubMed] [Google Scholar]; c () Shin SH; Baek EH; Hwang G-S; Ryu DH Enantioselective Synthesis of syn-α-Aryl-β-Hydroxy Weinreb Amides: Catalytic Asymmetric Roskamp Reaction of α-Aryl Diazo Weinreb Amides. Org. Lett 2015, 17, 4746–4749. [DOI] [PubMed] [Google Scholar]
- (17).a () Villalgordo JM; Linden A; Heimgartner H A Novel Amination Reaction with Diphenyl Phosphorazidate: Synthesis of α-Amino-Acid derivatives. Helv. Chim. Acta 1996, 79, 213–219. [Google Scholar]; b () Kägi M; Linden A; Mlostoń G; Heimgartner H 1,3-Oxathiole and Thiirane Derivatives from the Reactions of Azibenzil and α-Diazo Amides with Thiocarbonyl Compounds. Helv. Chim. Acta 1998, 81, 285–302. [Google Scholar]
- (18).Current vendors include Atomax Chemicals, Chemieliva Pharmaceutical, and Hong Kong Chemhere.
- (19).Ouihia A; Rene L; Guilhem J; Pascard C; Badet B A New Diazoacylating Reagent: Preparation, Structure, and Use of Succinimidyl Diazoacetate. J. Org. Chem 1993, 58, 1641–1642. [Google Scholar]
- (20).a () Barluenga J; Moriel P; Valdés C; Aznar F N-Tosylhydrazones as Reagents for Cross-Coupling Reactions: A Route to Polysubstituted Olefins. Angew. Chem., Int. Ed 2007, 46, 5587–5590. [DOI] [PubMed] [Google Scholar]; b () Peng C; Cheng J; Wang J Palladium-Catalyzed Cross-Coupling of Aryl or Vinyl Iodides with Ethyl Diazoacetate. J. Am. Chem. Soc 2007, 129, 8708–8709. [DOI] [PubMed] [Google Scholar]; c () Yu Z; Mendoza A Enantioselective Assembly of Congested Cyclopropanes using Redox-Active Aryldiazoacetates. ACS Catal 2019, 9, 7870–7875. [Google Scholar]
- (21).Ye F; Qu S; Zhou L; Peng C; Wang C; Cheng J; Hossain ML; Liu Y; Zhang Y; Wang Z-X; Wang J Palladium-Catalyzed C–H Functionalization of Acyldiazomethane and Tandem Cross-Coupling Reactions. J. Am. Chem. Soc 2015, 137, 4435–4444. [DOI] [PubMed] [Google Scholar]
- (22).a () Gnanaprakasam B; Milstein D Synthesis of Amides from Esters and Amines with Liberation of H2 under Neutral Conditions. J. Am. Chem. Soc 2011, 133, 1682–1685. [DOI] [PubMed] [Google Scholar]; b () Li G; Szostak M Highly Selective Transition-Metal-Free Transamidation of Amides and Amidation of Esters at Room Temperature. Nat. Commun 2018, 9, 4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gupta AK; Yin X; Mukherjee M; Desai AA; Mohammadlou A; Jurewicz K; Wulff WD Catalytic Asymmetric Epoxidation of Aldehydes with Two VANOL-Derived Chiral Borate Catalysts. Angew. Chem., Int. Ed 2019, 58, 3361–3367. [DOI] [PubMed] [Google Scholar]
- (24).Fu L; Mighion JD; Voight EA; Davies HML Synthesis of 2,2,2,-Trichloroethyl Aryl- and Vinyldiazoacetates by Palladium-Catalyzed Cross-Coupling. Chem.—Eur. J 2017, 23, 3272–3275. [DOI] [PubMed] [Google Scholar]
- (25).a () Shah P; Westwell AD The Role of Fluorine in Medicinal Chemistry. J. Enzyme Inhib. Med. Chem 2007, 22, 527–540. [DOI] [PubMed] [Google Scholar]; b () Miller MA; Sletten EM Perfluorocarbons in Chemical Biology. ChemBioChem 2020, 21, 3451–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Echols N; Harrison P; Balasubramanian S; Luscombe NM; Bertone P; Shang Z; Gerstein M Comprehensive Analysis of Amino Acid and Nucleotide Composition in Eukaryotic Genomes, Comparing Genes and Pseudogenes. Nucleic Acids Res 2002, 30, 2515–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Casey JR; Grinstein S; Orlowski J Sensors and Regulators of Intracellular pH. Nat. Rev. Mol. Cell Biol 2010, 11, 50–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.