

Abstract
Spinal cord ischemia/reperfusion injury (SCII) is a devastating complication of spinal or thoracic surgical procedures and can lead to paraplegia or quadriplegia. Neuronal cell damage involving mitochondrial dysfunction plays an important role in the pathogenesis of SCII. Despite the availability of various treatment options, there are currently no mitochondria-targeting drugs that have proven effective against SCII. Polydatin (PD), a glucoside of resveratrol, is known to preserve mitochondrial function in central nervous system (CNS) diseases. The aim of the present study was to explore the neuro- and mito-protective functions of PD and its underlying mechanisms. An in vitro model of SCII was established by exposing spinal cord motor neurons (SMNs) to oxygen–glucose-deprivation/reperfusion (OGD/R), and the cells were treated with different dosages of PD for varying durations. PD improved neuronal viability and protected against OGD/R-induced apoptosis and mitochondrial injury in a dose-dependent manner. In addition, PD restored the activity of neuronal mitochondria in terms of mitochondrial membrane potential (MMP), intracellular calcium levels, mitochondrial permeability transition pore (mPTP) opening, generation of reactive oxygen species (ROS), and adenosine triphosphate (ATP) levels. Mechanistically, PD downregulated Keap1 and upregulated Nrf2, NQO-1, and HO-1 in the OGD/R-treated SMNs. Likewise, PD treatment also reversed the neuronal and mitochondrial damage induced by SCII in a mouse model. Furthermore, the protective effects of PD were partially blocked by the Nrf2 inhibitor. Taken together, PD relieves mitochondrial dysfunction-induced neuronal cell damage by activating the Nrf2/ARE pathway and is a suitable therapeutic option for SCII.
1. Introduction
Spinal cord ischemia/reperfusion injury (SCII) is a devastating complication of spinal or thoracic surgical procedures that often triggers a complex series of patho-physiological events [1, 2]. Although SCII can be minimized by restoring blood and oxygen supply in a timely manner, reperfusion of the ischemic tissue may lead to mitochondrial impairment, calcium overload, and energy depletion, resulting in neuronal apoptosis [3, 4]. At the cellular level, SCII is characterized by progressive degeneration or loss of spinal motor neurons (SMNs), which directly or indirectly control muscle movement [5]. Therefore, the ischemia/reperfusion (I/R) injury in spinal cord and the ensuing pathological responses can be mitigated by preserving SMNs.
Nuclear factor-E2-related factor 2 (Nrf2), a common leucine zipper transcription factor, plays a crucial role in maintaining cellular redox homeostasis. Under physiological conditions, Nrf2 is bound to its inhibitor Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm and remains in an inactive state [6]. In the presence of a stressful stimulus, Nrf2 dissociates from Keap1 and translocates to the nucleus, where it binds to the antioxidant response element (ARE) in the promoters of its target genes and transcriptionally activates the downstream detoxifying and antioxidant enzymes [7]. In addition, the Nrf2/ARE signaling pathway maintains physiological processes in the central nervous system (CNS) [8] and also plays a crucial role after CNS I/R injury. Enhanced Nrf2 activity protects against ischemic stroke injury in vivo by mitigating oxidative stress, apoptosis, and inflammatory response after SCII [9–11]. Thus, the Nrf2/ARE axis is a promising therapeutic target for alleviating I/R-induced neuronal damage following SCII.
Polydatin (PD, structure shown in Figure 1(a)) is a natural compound isolated from the rhizome of Polygonum cuspidatum and is a known activator of the Nrf2/ARE pathway [12]. In addition, PD has well-documented anti-inflammatory, antioxidant, antiapoptotic, and immunomodulatory activities [13]. Studies increasingly show that PD has neuroprotective effects as well and can protect against I/R injury of the CNS [14, 15]. Mechanistically, PD reversed the oxidative stress-induced mitochondrial dysfunction and restored the adenosine triphosphate (ATP) content in SH-SY5Y human neuroblastoma cells [16]. Consistent with this, PD also acts as a reactive oxygen species (ROS) scavenger, which is the key to inhibiting mitochondrial permeability transition pore (mPTP) opening and membrane depolarization [17]. Therefore, it is possible that PD exerts its neuroprotective effects by restoring mitochondrial function and is a promising therapeutic option for SCII.
Figure 1.
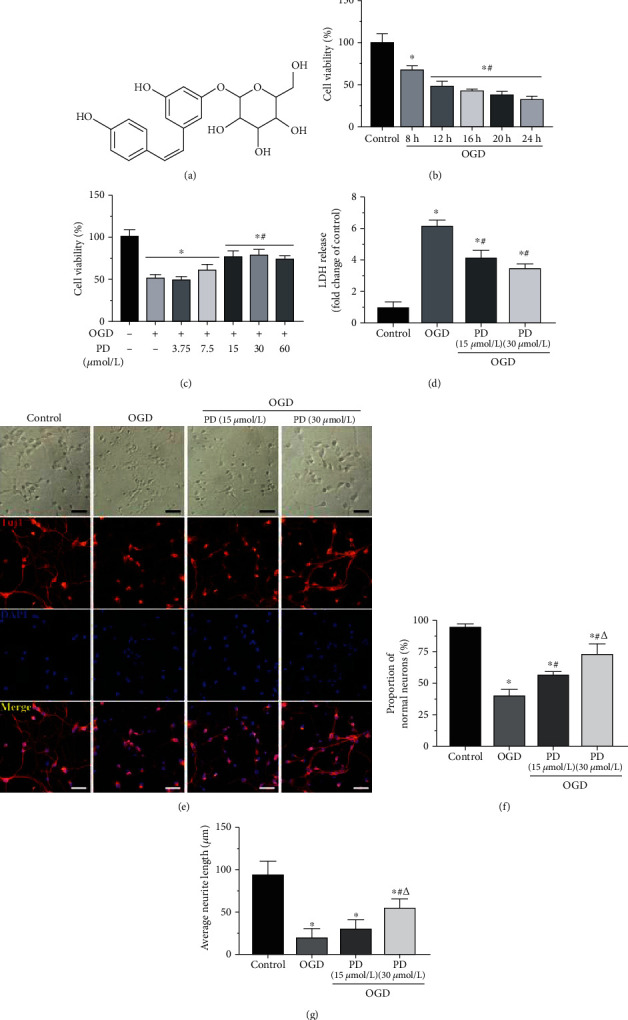
Effects of PD on the viability and morphology of SMNs exposed to OGD/R. (a) The structure of PD. (b) Percentage of viable SMNs exposed to OGD/R conditions for varying durations (8-24 h). (c) Percentage of viable SMNs with/out PD (3.75, 7.5, 15, 30, or 60 μM) treatment during hypoxic injury. (d) Extracellular LDH levels in the indicated groups. (e) Representative images showing the morphology changes in the primary neurons of indicated group and the (f) number of normal neurons and (g) average neurite length. Scale bars = 50 μm. ∗P < 0.05 vs. Control; #P < 0.05 vs. OGD; ΔP < 0.05 vs. OGD + PD (15 μM).
In this study, we analyzed the protective effects of PD on SMNs using both in vitro and in vivo I/R injury models and explored the potential regulating mechanism of the Nrf2/ARE signaling pathway.
2. Materials and Methods
2.1. Usage of Animals and Ethics Statement
Healthy pregnant and male C57BL/6J mice were purchased from the Guangdong Medical Experimental Animal Center (Foshan, China). All animal experiments were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and USE Committee of the Guangzhou University of Chinese Medicine.
2.2. Primary SMN Cultures
SMNs were isolated and cultured as previously described [18]. Once the cells attached, the medium was replaced with neurobasal medium (Gibco, Carlsbad, CA, USA) supplemented with 2% B27 (Gibco) and 0.5 mM L-glutamine (Sigma-Aldrich, St. Louis, MO, USA). Thereafter, half of the culture medium was replaced with fresh medium every other day. The cells were identified by immunostaining for β-III tubulin (Tuj1), microtubule associated protein-2 (MAP-2), choline acetyltransferase (ChAT), and glial fibrillary acidic portein (GFAP). Cells on days 5-7 were used for further experiments.
2.3. Establishment of OGD/R Model and Treatment
The oxygen-glucose-deprivation/reperfusion (OGD/R) model was established as described by Sun et al. [19]. After washing twice with Hank's balanced salt solution (HBSS), SMNs were cultured in glucose-free Dulbecco's modified eagle medium (DMEM; Gibco) and then incubated in a hypoxic chamber with 94% N2, 5% CO2, and 1% O2 at 37°C for varying durations. During the reoxygenation period, the glucose-free media was replaced with complete DMEM, and the cells were cultured under normoxic conditions (75% N2, 5% CO2, and 20% O2) for 24 h. Control cells were not exposed to OGD/R. PD (Sigma-Aldrich, dissolved in DMSO) and brusatol (100 nM; Chengdu Herbpurify, Chengdu, China) were added to the medium at the beginning of OGD and maintained throughout the hypoxic period.
2.4. Assessment of Cell Viability and Cytotoxicity
Cell viability was evaluated using a cell counting kit-8 (CCK-8) assay (Beyotime, Shanghai, China) according to the manufacturer's instructions, and the absorbance at 450 nm was measured with a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). The suitably treated cells were harvested, and lactate dehydrogenase (LDH) activity was analyzed using a specific kit as instructed by the manufacturer. The optical density of each well was then determined spectrophotometrically at a wavelength of 450 nm using an automatic microplate reader.
2.5. Apoptosis Assays
Neuronal cell death was quantitatively assessed by terminal deoxyribonucleotidyl transferse- (TdT-) mediated biotin-16-dUTP nick-end labeling (TUNEL) assay with a Fluorescein-FragEL DNA fragmentation detection kit and Annexin V-FITC/PI double staining as reported previously [20]. The stained cells were acquired in a flow cytometer, and the percentage of apoptotic cells was analyzed.
2.6. Intracellular Ca2+ Measurement
Free intracellular Ca2+ ([Ca2+]i) levels were measured by staining with the fluorescence Ca2+ indicator Fluo-3/AM (Beyotime). The suitably treated cells were harvested, washed with PBS, and incubated with 2 μM Fluo-3/AM at 37°C in the dark for 30 min. After washing twice with PBS to remove any unbound dye, the fluorescence intensity of Fluo-3/AM was measured using a microplate reader at 488 nm excitation and 525 nm emission wavelengths.
2.7. Detection of mPTP Opening
The opening of mPTP in the SMNs was evaluated by Calcein/AM staining in the presence of CoCl2 using an mPTP assay kit (Life Technologies, Carlsbad, CA, USA). Briefly, the cells were incubated with Calcein/AM and CoCl2 at 37°C in the dark for 40 min. The calcein that leaks out from the opened mPTP is quenched by Co2+, and the reduction in absorbance is measured. The stained neurons were rinsed thrice with HBSS, and fluorescence intensity was measured using a microplate reader with excitation and emission wavelengths of 488/525 nm.
2.8. Mitochondrial Membrane Potential (MMP/ΔΨm) Assessment
The change in MMP was measured using the lipophilic cationic probe JC-1. The suitably treated cells were incubated with an equal volume of JC-1 solution (5 μg/mL; Yeasen Biotech, Shanghai, China) at 37°C for 30 min. After rinsing thrice with PBS, the stained cells were observed under a confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany). Mitochondrial depolarization was evaluated in terms of the proportion of JC-1 monomers (green fluorescence at 530 nm emission) to aggregates (red fluorescence at 590 nm).
2.9. Measurement of ROS Levels
MitoSOX Red was used to detect the levels of mitochondrial superoxide radicals. SMNs exposed to OGD/R with/out PD treatment were incubated with 2 μM MitoSOX Red (Life Technologies, Carlsbad, CA, USA) at 37°C in the dark for 20 min and washed twice with PBS. The cells were viewed under a fluorescence microscope (Olympus, Tokyo, Japan), and the fluorescence intensity was quantified using the ImageJ software (NIH, Bethesda, MD, USA).
2.10. Measurement of ATP Levels
ATP release was measured using the luciferase-based ATP detection assay (Beyotime) according to the manufacturer's protocol. Briefly, the suitably treated neurons were lysed and centrifuged for 5 min at 12,000 × g. The supernatants were aspired, and 100 μL of each sample was mixed with 100 μL ATP detection solution. The luminescence was measured using an automatic microplate luminometer, and the ATP level was quantified from the standard curve plotted using known concentrations (1 nM–1 μM).
2.11. Establishment of SCII Model and Treatment
A total of 100 male C57BL/6 mice (8-10 weeks) weighing 20-25 g were randomly divided into the sham-operated, SCII model, PD-treated, and PD + Brusatol groups (N = 25 each). SCII was induced as previously reported with minor modifications [21]. Briefly, the aortic arch was exposed using a cervico-thoracic approach and cross-clamped between the thoracic aortic distal and left subclavian artery for 5 min to induce ischemia. After confirming ≥90% reduction in distal flow, the clamps were removed to start reperfusion. Successful induction of SCII leads to swaying of legs and slow paralysis. And animals with BBB scores higher than 2 at 24 h after I/R injury were excluded. The mice in the sham-operated group underwent the same procedure without occlusion. PD (30 mg/kg per day) was administered intragastrically for 2 days before surgery and continued until the animals were sacrificed. The Nrf2 inhibitor brusatol (2 mg/kg) was intraperitoneally (i.p.) injected once daily into mice in the PD + Brusatol group 2 h before the PD gavage. All mice were sacrificed one week postsurgery, and the spinal cords were harvested for subsequent analysis.
2.12. Determination of Malondialdehyde (MDA), Superoxide Dismutase (SOD), and Glutathione (GSH)
Following the manufacturer's instructions (Jiancheng, Nanjing, China) for the MDA, SOD, and GSH assays, the liquid supernatant of tissue homogenates was collected for measurement, as previously described [19]. The results were normalized to total protein content in spinal cords determined by BCA assay (Beyotime).
2.13. Immunofluorescence Assay
Immunofluorescence was carried out by standard protocol as previously described [22]. We used primary antibodies against Tuj1 (1 : 400; CST, Danvers, MA, USA), MAP-2 (1 : 200, CST), ChAT (1 : 200; Abcam, Cambridge, MA, USA), GFAP (1 : 500; Abcam), neuronal nuclear antigen (NeuN, 1 : 300; Abcam), Caspase-3 (1 : 400; CST), and Nrf2 (1 : 200; R&D, Minneapolis, MN, USA). Alexa Fluor® 488 donkey anti-mouse IgG (1 : 200; Invitrogen, Carlsbad, CA, USA) and Cy3-labeled goat anti-rabbit IgG (1 : 300; Invitrogen) were used as second antibodies.
2.14. Western Blotting
Western blotting was performed following standard methods [23]. Briefly, equal amounts of denatured protein from cellular/tissue homogenates were separated electrophoretically by SDS-PAGE and subsequently transferred onto PVDF membranes (Millipore, Bedford, MA, USA). The latter were blocked with 5% skim milk and incubated overnight with primary antibodies for Caspase-3, Bcl-2, Bax, Keap1, Nrf2, NQO1, HO-1, and cytochrome-c (Cyt-c) at 4°C. After washing thrice with buffer solution, the membranes were incubated with corresponding HRP-conjugated secondary antibodies at room temperature for 1 h. The protein bands on the membranes were visualized by enhanced chemiluminescence system (ECL) and densitometrically quantified with the ImageJ software. β-Tubulin served as an internal control for normalization.
2.15. Nissl Staining
Nissl staining was performed following standard methods. Briefly, the spinal cord tissues were fixed, cut into 5 μm thick transverse sections, and embedded in paraffin. The sections were stained with 0.25% cresyl violet for 5 min to label the ventral motor neurons (VMNs).
2.16. Transmission Electron Microscopy (TEM)
Spinal cord specimens were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer for 2 h at 4°C, followed by staining with 2% osmium tetroxide. After blocking with 2% uranyl acetate, the tissue samples were dehydrated through acetone and ethanol gradient and immersed in epoxy resin. The blocks were polymerized at 60°C for 2 days and cut into ultrathin sections that were mounted on 300-mesh copper grids, counter-stained with uranyl acetate and lead citrate. All sections were examined with a JEM-1200EX electron microscope (JEOL, Tokyo, Japan).
2.17. Statistical Analysis
Quantitative data are expressed as mean ± standard deviation of at least three independent experiments and were analyzed statistically by one-way ANOVA or unpaired Student's t-test using the SPSS 24.0 software (SPSS Inc., Chicago, IL, USA). Graphs were generated by the GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA, USA). P < 0.05 was defined as statistically significant.
3. Results
3.1. Verification of Spinal Cord Motor Neuronal Culture and PD Dose
The cells were isolated from the spinal cord of 14-day-old mouse embryos (Figure 2(a)) and cultures as per established protocols. The cells gradually enlarged, and their neurites grew longer to form vastly interconnected networks (Figures 2(b)–2(d)). Immunofluorescence showed that the majority of the cells were cholinergic neurons, while only a few were astrocytes (Figures 2(e)–2(h)). In agreement with the findings of Hong et al. [24], the viability of cultured neurons decreased in time-dependent manner after 8 to 24 h of OGD. Hypoxic exposure of 12 h decreased cell viability by 50% (Figure 1(b)) and was selected for the subsequent experiments. As shown in Figure 1(c), PD treatment significantly increased the proportion of viable cells at the doses of 15 and 30 μM, which were used for all experiments.
Figure 2.
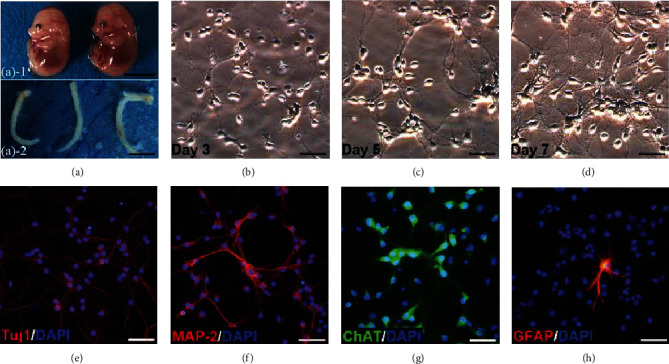
Isolation, culture, and identification of primary SMNs. (a) Spinal cords were dissected from the 14-day-old mouse embryos. Scale bars = 50 mm. Representative images of SMNs on (b) Day 3, (c) Day 5, and (d) Day 7. Primary SMNs were labeled with (e) Tuj1, (f) MAP-2, (g) ChAT, and (h) GFAP. Scale bars = 50 μm.
3.2. PD Attenuates OGD/R-Induced Apoptosis of SMNs by Preserving Mitochondrial Function
The effect of OGD/R injury and the neuroprotective effects of PD on primary cultured SMNs were evaluated in terms of secreted LDH, which is an indicator of cell membrane disintegration and cytotoxicity. OGD conditions significantly increased the LDH levels, which were restored by PD treatment in a dose-dependent manner (Figure 1(d)). Hypoxic exposure also induced morphological changes in the neurons, such as shrunken cell somas, thinner dendrites, and breakage of neuronal fibers, all of which were partially reversed by PD treatment in a dose-dependent manner (Figure 1(e)). In addition, the apoptotic ratio after OGD/R insult was as high as 43.25% ± 3.18% as indicated by Annexin V/PI staining, and significantly decreased to 20.18% ± 2.49% and 11.47% ± 2.17% in the presence of 15 μM and 30 μM PD, respectively. Similar results were obtained with the TUNEL assay as well (Figures 3(b) and 3(d)). Consistent with this, the antiapoptotic protein Bcl-2 was downregulated, and the proapoptotic proteins Bax and c-Caspase-3 were significantly upregulated in the neurons after OGD/R injury and markedly reversed by PD in a dose-dependent manner (Figures 3(e)–3(h)).
Figure 3.
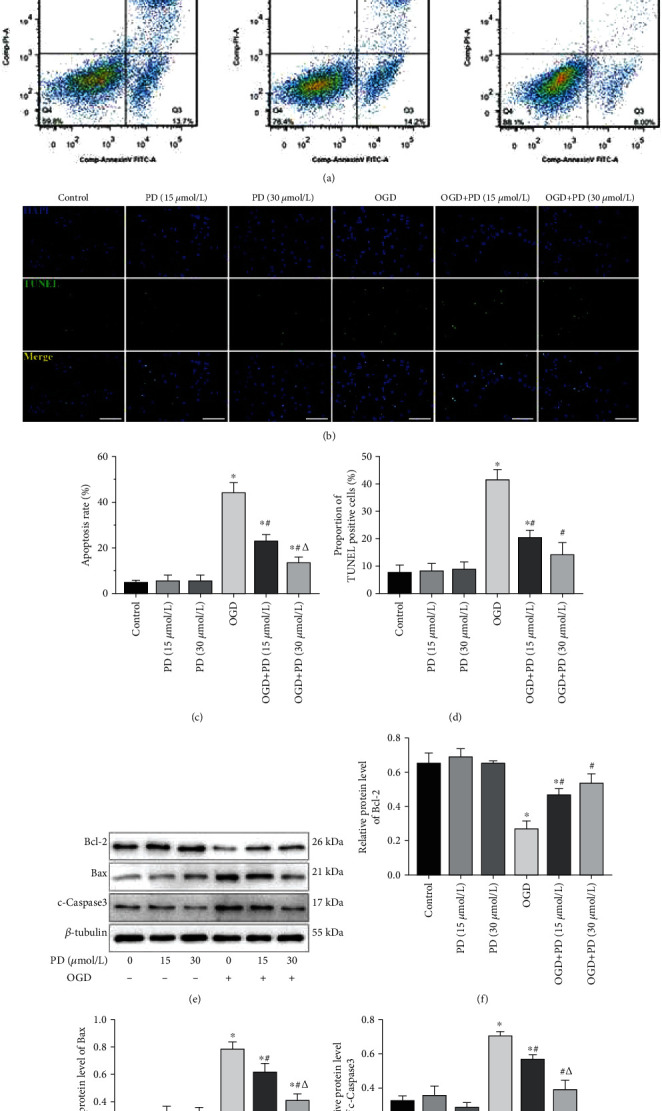
PD attenuates OGD/R-induced damage in SMNs. Cells were divided into the Control, PD (15 μM), PD (30 μM), OGD/R, OGD/R + PD (15 μM), and OGD/R + PD (30 μM) groups. (a) Representative images showing Annexin V-FITC/PI stained cells. (b) Representative images showing TUNEL-positive cells. Scale bars = 100 μm. Quantification of the apoptotic cells in (c) Annexin V-FITC/PI and (d) TUNEL assays. (e) Immunoblot showing Bcl-2, Bax, and c-Caspase-3 protein levels in the neurons of the indicated groups. (f–h) Quantification of the relative protein levels. ∗P < 0.05 vs. Control; #P < 0.05 vs. OGD; ΔP < 0.05 vs. OGD + PD (15 μM).
The mitochondria plays a critical role in hypoxia-induced neuronal apoptosis [25, 26], which is initiated by intracellular calcium dyshomeostasis and mPTP opening. The increase in Ca2+ levels during hypoxia triggers the opening of mPTPs, which in turn exacerbates mitochondrial dysfunction. To this end, we assessed [Ca2+]i levels and the extent of mPTP opening. The [Ca2+]i increased rapidly after 6 h of OGD exposure and reached its peak after 12 h of OGD exposure. However, treatment with 15 and 30 μM PD decreased the [Ca2+]i in the 12 h OGD group to 32.8% ± 3.22 and 59.52% ± 4.30, respectively (Figure 4(a)). Furthermore, hypoxic insult resulted in a gradual and significant decrease in calcein fluorescence intensity compared to the Control group, indicating increased mPTP opening in the former (Figure 4(b)). However, compared to untreated neurons (5.11 ± 1.04 U/mg protein), the normalized relative fluorescence units (NRFU) of calcein were higher in cultures treated with 15 μM and 30 μM PD during the 12 h OGD (8.26 ± 1.48 and 11.62 ± 1.17 U/mg protein). Thus, PD can restore intracellular calcium homeostasis and reduce the extent of mPTP opening in SMNs in response to hypoxia/reoxygenation.
Figure 4.
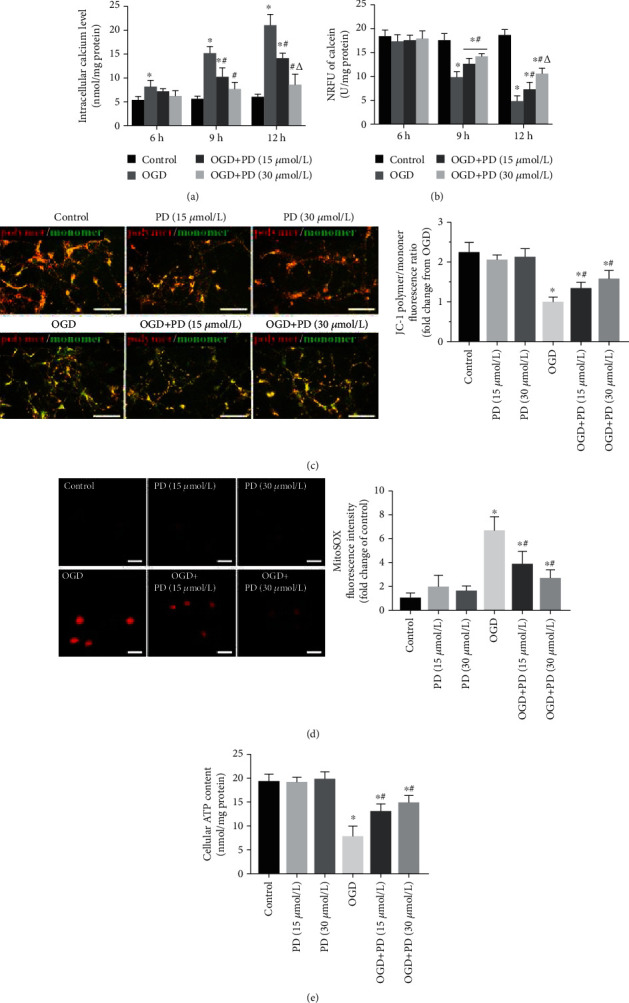
PD restores mitochondrial functions in OGD/R-injured neurons. (a) [Ca2+]i levels in cells exposed to normoxia or hypoxia-reperfusion in the indicated groups. (b) Normalized relative fluorescence units (NRFU) of calcein indicating mPTP opening in the differentially treated cells. (c) The ratios of polymeric (red) and monomeric (green) forms of JC-1 corresponding to the MMP in the indicated groups. Scale bars = 100 μm. (d) MitoSOX fluorescence intensity indicative of ROS levels. Data are presented as fold change over the Control group. Scale bars = 10 μm. (e) Neuronal ATP release (nmol/mg protein) as measured by a luciferase-based assay. ∗P < 0.05 vs. Control; #P < 0.05 vs. OGD; ΔP < 0.05 vs. OGD + PD (15 μM).
To further explore the effects of PD on mitochondrial function in OGD/R-injured SMNs, we evaluated other indicators of mitochondrial activity such as MMP, ROS levels, and cellular ATP release. As revealed in Figure 4(c), OGD/R injury caused a significant decline in the ΔΨm of SMNs compared to the controls, which was largely reversed by PD treatment in a dose-dependent manner. Furthermore, OGD/R also induced excessive ROS production and a sharp decrease in cellular ATP levels from 20.17 ± 1.02 to 8.64 ± 1.43 nmol/mg protein (Figure 4(d)). PD not only reduced the ROS levels but also alleviated hypoxia-induced ATP decline (Figure 4(e)). Taken together, PD protects SMNs from OGD/R injury by preserving healthy mitochondria. Since 30 μm PD was most effective against OGD/R injury, this dosage was used for subsequent experiments.
3.3. PD Exerts Its Mito-Protective Effects in SMNs by Activating the Nrf2/ARE Signaling Pathway
Previous studies have shown that the Nrf2/ARE signaling pathway is involved in quenching excessive ROS and reversing OGD/R-induced mitochondrial dysfunction and neuronal apoptosis [27, 28]. In addition, PD is a known Nrf2-signal activator. We found that OGD/R significantly increased Keap1 level and downregulated the Nrf2, NQO-1, and HO-1 proteins in SMNs, which was reversed by PD in a concentration-dependent manner (Figure 5(a)). Therefore, we hypothesized that the therapeutic effects of PD illustrated so far are induced by activation of the Nrf2/ARE signaling pathway. To verify this hypothesis, Nrf2 protein expression was inhibited using brusatol, which selectively regulates the level by enhances Nrf2 degradation and ubiquitination. Consistent with our hypothesis, the antiapoptotic effects of PD during OGD/R were partly suppressed by brusatol (Figures 5(b)–5(d)). Furthermore, brusatol also blocked the ameliorative effect of PD on intracellular calcium homeostasis and mPTP opening (Figures 6(a) and 6(b)). Inhibition of the Nrf2/ARE axis neutralized the ability of PD to increase MMP, scavenge ROS, and boost ATP production in the OGD-exposed SMNs (Figures 6(c)–6(e)). Taken together, PD reverses OGD/R-induced neuronal apoptosis and mitochondrial dysfunction by activating the Nrf2/ARE pathway.
Figure 5.
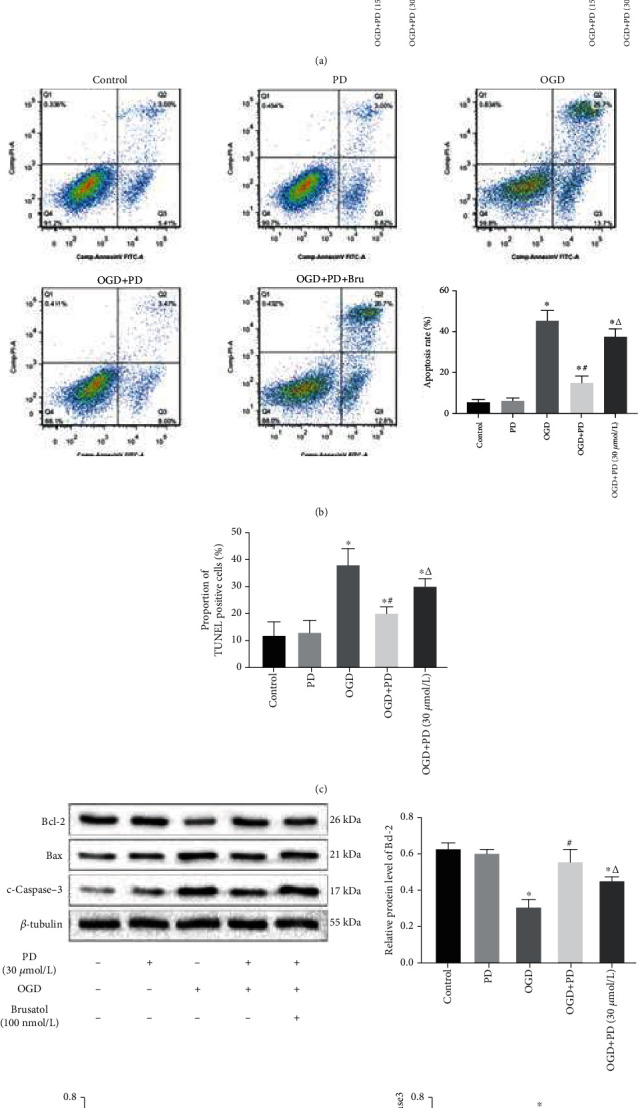
PD exerts its antiapoptotic effects by activating the Nrf2/ARE pathway. (a) Immunoblot showing levels of Keap1, Nrf2, NQO-1, and HO-1 protein in the neurons of different groups. ∗P < 0.05 vs. Control; #P < 0.05 vs. OGD; ΔP < 0.05 vs. OGD + PD (15 μM). Then, cells were divided into the Control, OGD/R, OGD/R + PD, and OGD/R + PD + Brusatol groups. (b) Percentage of apoptotic cells as detected by Annexin V-FITC/PI assay. (c) Percentage of TUNEL-positive apoptotic cells. (d) Immunoblot showing Bcl-2, Bax, and c-Caspase-3 protein levels in each group. ∗P < 0.05 vs. Control; #P < 0.05 vs. OGD; ΔP < 0.05 vs. OGD + PD.
Figure 6.
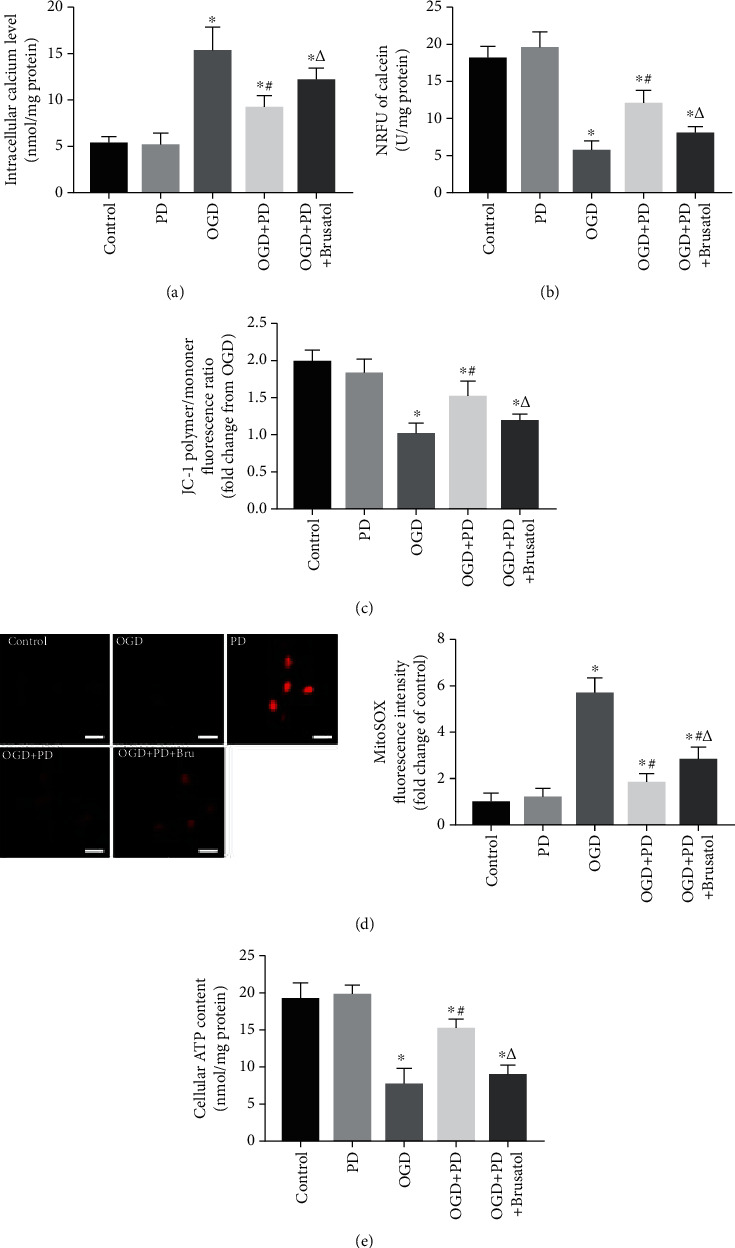
PD-mediated mito-protective depends on the Nrf2/ARE pathway. (a) [Ca2+]i levels in cells exposed to normoxia or hypoxia-reperfusion in the indicated groups. (b) NRFU of calcein indicating mPTP opening in the differentially treated cells. (c) The ratios of polymeric (red) and monomeric (green) forms of JC-1 corresponding to the MMP in the indicated groups. (d) MitoSOX fluorescence intensity indicative of ROS levels. Data are presented as fold change over the Control group. Scale bars = 10 μm. (e) Neuronal ATP release (nmol/mg protein) as measured by a luciferase-based assay. ∗P < 0.05 vs. Control; #P < 0.05 vs. OGD; ΔP < 0.05 vs. OGD + PD.
3.4. PD Protects SMNs In Vivo from SCII by Reversing Mitochondrial Dysfunction
SCII triggers extensive neuronal apoptosis and tissue damage in the spinal cords. To evaluate the potential therapeutic role of PD in vivo, we established a mouse model of SCII. Notably, SCII markedly increased the percentage of c-Caspase 3-positive neurons in the spinal cord tissues at 7 days post injury (dpi) compared to the sham-operated group (Figures 7(a) and 7(b)). PD treatment significantly reduced the number of apoptotic neurons in the SCII model, whereas coadministration of brusatol suppressed the beneficial effects of PD. Similar trends were observed in the levels of apoptosis-related proteins in the affected tissues of different groups (Figure 7(c)). Furthermore, PD-mediated reversal of SCII-induced VMNs loss was also partially blocked by brusatol (Figure 7(d)). Ultrastructural examination of spinal cords by TEM revealed peripheral edema and disintegrated neurons in the SCII group (Figure 7(e), black arrows). The neuronal damage recovered considerably with PD treatment, while coadministration of brusatol prevented further recovery.
Figure 7.
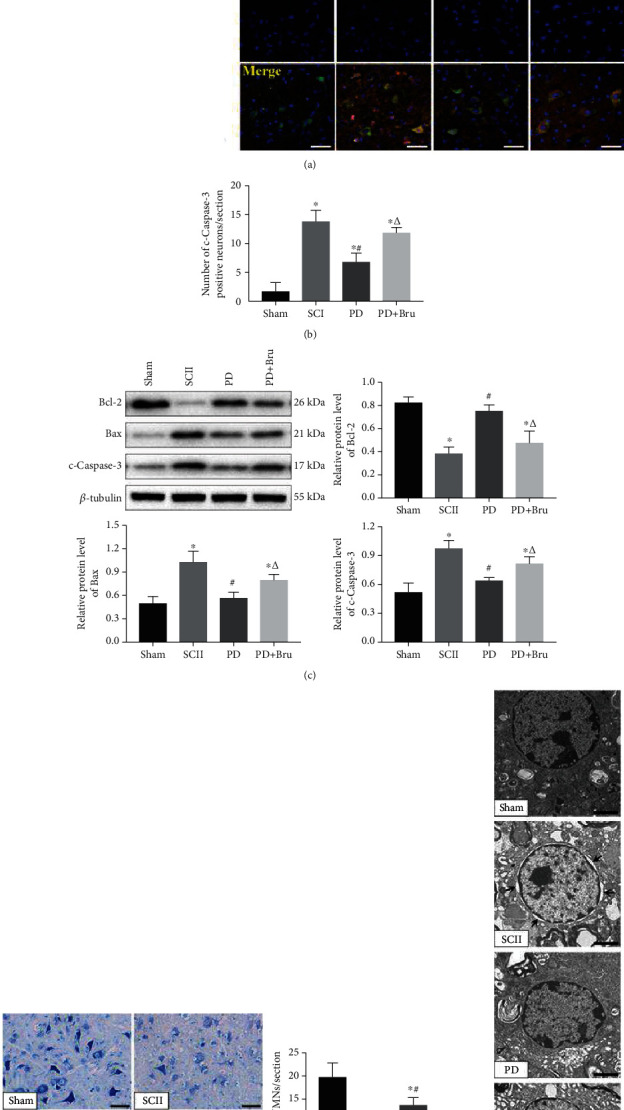
PD treatment protects SMNs from SCII-induced apoptosis. (a, b) Immunofluorescence images showing colocalization of NeuN (green) and c-Caspase-3 (red) in the spinal cord of the indicated groups. Scale bar = 50 μm. (c) Quantified expression of Bcl-2, Bax, and c-Caspase-3. (d) Representative images of Nissl staining at 7 dpi and the number of VMNs. Scale bars = 50 μm. (e) TEM images showing microstructures of neurons. Black arrows indicate peripheral edema and disintegrated organelles. Scale bars = 2 μm. ∗P < 0.05 vs. Sham; #P < 0.05 vs. SCII; ΔP < 0.05 vs. PD.
SCII was accompanied by a significant reduction in SOD activity and GSH levels and an increase in MDA content in the affected tissues. PD administration alleviated SCII-induced oxidative stress, whereas brusatol inhibited the antioxidant capacity of PD (Figures 8(a)–8(c)). Sham-operated animals had normal mitochondria with intact membranes and cristae in the spinal neurons. However, SCII resulted in swollen and irregularly shaped mitochondria with disrupted cristae (Figure 8(d), black arrows). The mito-structural aberrations were partially alleviated by PD treatment, except in the presence of brusatol. Consistent with the above, the mitochondrial apoptotic-related protein (Cyt-c) was significantly upregulated in the spinal cords at 7 dpi and decreased after PD but not PD + brusatol treatment (Figure 8(e)). Taken together, the neuroprotective role of PD in SCII depends on its ability to mitigate oxidative stress and mitochondrial dysfunction.
Figure 8.
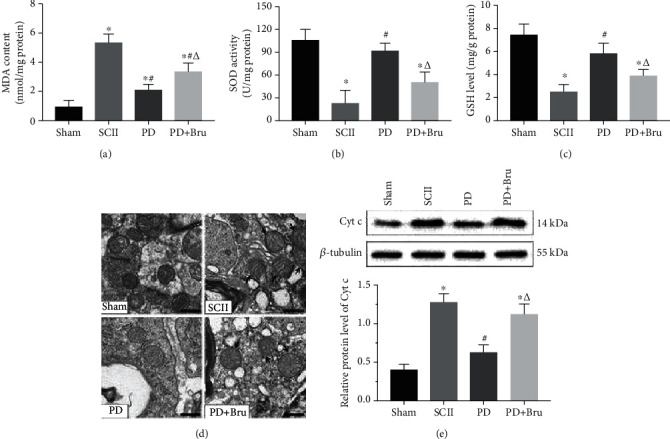
PD alleviates oxidative stress and mitochondrial dysfunction caused by SCII. (a) MDA content, (b) SOD activity, and (c) GSH levels in the spinal cords at 7 dpi. (d) TEM images showing mitochondrial ultrastructure. Arrows indicate swollen mitochondria. Scale bars = 500 nm. (e) Relative Cyt-c protein expression levels in each group. ∗P < 0.05 vs. Sham; #P < 0.05 vs. SCII; ΔP < 0.05 vs. PD.
4. Discussion
Despite recent advances in surgical techniques, patients undergoing spinal or thoracic surgery remain at high risk of postoperative complications. SCII is a life-threatening complication that can lead to permanent paraplegia due to SMN damage. Mitochondrial dysfunction is one of the key pathological events occurring early during I/R injury [29]. In addition, mitochondria play a critical role in neuronal development and maturation by modulating ATP biosynthesis, Ca2+ buffering, ROS production and sequestration, and apoptosis [30–32]. However, mitochondria are highly vulnerable to ischemic insult due to the concomitant deficiency of oxygen and glucose [19]. Therefore, mitochondria-targeted therapies that restore its function can potentially obviate the associated pathological conditions. Given the established neuroprotective effects of PD, we analyzed its potential role in regulating neuronal and mitochondrial injuries under I/R and explored the underlying regulatory mechanisms.
SCII leads to extensive neuronal apoptosis and necrosis in the spinal cord, which involves complex interactions among members of the Bcl-2 family of proteins (including Bcl-2 and Bax) [33, 34]. The proapoptotic protein Bax translocates from the cytosol to mitochondria and promotes apoptosis by inducing mitochondrial membrane depolarization and Cyt-c release [35]. On the other hand, the antiapoptotic Bcl-2 inhibits apoptosis by preventing the release of Cyt-c into the cytoplasm [36]. In addition, the Bcl-2 protein family maintains mitochondrial stability by modulating the Bcl-2/Bax balance [37]. Caspase-3 is a cysteine protease required for DNA fragmentation and morphological changes associated with apoptosis [38]. Consistent with previous studies, we found that OGD/R and SCII significantly downregulated Bcl-2 and upregulated Bax and c-Caspase-3. PD treatment restored the levels of the apoptosis-related proteins in a dose-dependent manner. In addition, PD also alleviated the morphological damage (peripheral edema, disintegrated organelles, and breakage of neurites) of motor neurons induced by OGD/R or SCII and decreased the proportion of apoptotic cells. A similar protective effect of PD was also observed against H2O2-induced apoptosis in BMSCs [39].
Studies increasingly show that the cytoprotective action of PD under hypoxia is related to its antioxidant activity, excitotoxicity, and inhibition of calcium ingress [39–41]. In addition, activation of the mitochondrial permeability transition (mPT) can lead to a bioenergetic, biosynthetic, and redox crisis in cells, resulting in mitochondrial dysfunction [42], which triggers neuronal apoptosis during hypoxia/reoxygenation [30]. Damaged mitochondria also produce excessive amounts of ROS that inhibit the respiratory complexes and exacerbate mitochondrial dysfunction [43]. Furthermore, ROS production triggered by OGD/R and other stresses causes morphological disintegration of neuronal mitochondria and initiates the apoptosis cascade [26]. Studies show that an increase in intracellular free Ca2+ levels predisposes neurons to injury, and the initiation of Ca2+-dependent mPT in particular is associated with hypoxia-induced apoptosis [44, 45]. PD modulates [Ca2+]i in ventricular myocytes and mast cells [46, 47] and also attenuated [Ca2+]i elevation in SMNs undergoing hypoxia/reoxygenation. Furthermore, PD protected SMNs from OGD/R-induced apoptosis by suppressing Ca2+-dependent mPT. It also alleviated loss of MMP in the cells exposed to OGD/R, as well as SCII-induced mitochondrial injury (apparently swollen with poorly defined cristae) in vivo and blocked the Bcl-2/Cyt-c apoptotic pathway.
The mPT depends on the opening of mPTPs located at the junction between the outer and inner mitochondria membranes [48]. The mPTPs of the inner mitochondrial membranes are normally closed but can open following I/R injury, allowing medium-sized molecules to move freely across the membrane [49, 50], which eventually leads to mitochondrial swelling and relocation of apoptogenic substances [51]. Therefore, mPTP opening is regarded as a critical determinant in the genesis of mitochondrial dysfunction and apoptosis. The stilbenoid compound resveratrol protects mitochondria and prevents I/R injury by targeting mPTP through translocation of GSK-3β, which ultimately interacts with cyclophilin D to regulate membrane permeability [52]. Likewise, its glucoside PD also significantly inhibited mPTP opening in the injured SMNs. A major consequence of hypoxia/reoxygenation injury-triggered mitochondrial dysfunction is ATP depletion [19]. PD effectively scavenged mitochondrial ROS and restored ATP production in the OGD/R neurons. Thus, PD-mediated neuroprotection of PD is mainly due to prevention of mitochondrial injury and renewal of its functions. Mitochondrial apoptosis is regulated by multiple factors and signaling pathways. The ubiquitously expressed Nrf2 is a key mediator of the antioxidant response via its interaction with ARE [53]. The Nrf2/ARE transcriptional pathway is the master regulator of multiple patho-physiological processes, including oxidative stress, inflammation, and mitochondrial dysfunction. In addition, coordinated upregulation of ARE-driven genes protects organs from I/R injury [54, 55]. A recent study showed that the antioxidant MitoQ alleviated renal I/R injury by promoting Nrf2-mediated mitophagy [56]. Furthermore, Nrf2/ARE activation has been shown to reduce mitochondrial dysfunction after traumatic brain injury in mice [57]. However, few studies have focused on the links between SMNs apoptosis, SCII, mitochondrial dysfunction, and Nrf2/ARE signaling. To the best of our knowledge, this is the first to report that activation of the Nrf2/ARE pathway attenuates I/R injury-induced SMNs apoptosis by preserving functional mitochondria (Figure 9). In addition, the protective effects of PD were neutralized by the Nrf2 inhibitor brusatol, indicating that the therapeutic action of PD is dependent on the Nrf2/ARE axis.
Figure 9.
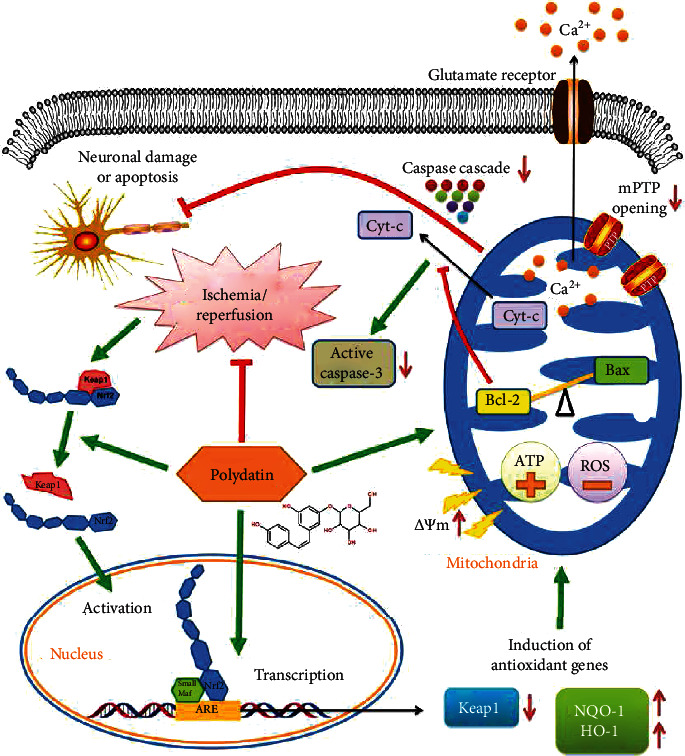
Mechanistic basis of the neuroprotective action of PD in SCII. PD promotes the nuclear translocation of Nrf2 and activates Nrf2/ARE singling, thereby inducing the ARE-driven genes NQO-1 and HO-1 that attenuate mitochondrial dysfunction. It also restores the Bcl-2/Bax balance and blocks Cyt-c-initiated Caspase cascade, which prevents neuronal damage and apoptosis.
In conclusion, PD can attenuate hypoxia/reoxygenation-induced neuronal injury in vitro and in vivo by restoring mitochondrial function by activating the Nrf2/ARE signaling pathway and is a promising therapeutic agent against SCII.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 81673992 and 82074451), the National Natural Science Foundation of China Youth Fund (No. 81704095), the Natural Science Foundation of Guangdong Province of China (No. 2019A1515010323), and the Science and Technology Project of Guangdong Province (No. 2016A020226008).
Abbreviations
- CNS:
Central nervous system
- SCII:
Spinal cord ischemia/reperfusion injury
- PD:
Polydatin
- OGD/R:
Oxygen–glucose-deprivation/reperfusion
- SMNs:
Spinal cord motor neurons
- TUNEL:
Terminal deoxyribonucleotidyl transferse- (TdT-) mediated biotin-16-dUTP nick-end labeling
- mPTP:
Mitochondrial permeability transition pore
- MMP:
Mitochondrial membrane potential
- ROS:
Reactive oxygen species
- ATP:
Adenosine triphosphate
- LDH:
Lactate dehydrogenase
- MDA:
Malondialdehyde
- SOD:
Superoxide dismutase
- GSH:
Glutathione
- TEM:
Transmission electron microscopy
- Keap1:
Kelch-like ECH-associated protein 1
- Nrf2:
Nuclear factor E2-related factor 2
- ARE:
Antioxidant response element
- NQO-1:
NAD(P)H: quinine oxidoreductase-1
- HO-1:
Heme oxygenase-1.
Contributor Information
Qing Zhang, Email: zhangqinggys@163.com.
Dingkun Lin, Email: lindingkun@126.com.
Data Availability
The datasets used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare no conflicts of interest regarding this manuscript.
Authors' Contributions
L.D.K. and H.Y.H. did the conception and design of the research. Z.J.H., L.X., and L.D. performed the experiments. H.Y. and C.S.D. analyzed the data. X.Z.F. and L.J.Y. prepared the figures. Z.J.H., L.X., and Y.W.Y. performed the drafting of the article. All authors read and approved the final manuscript. Jiheng Zhan and Xing Li contributed equally to this work.
References
- 1.Zhang Q., Huang C., Meng B., Tang T., Shi Q., Yang H. Acute effect of ghrelin on ischemia/reperfusion injury in the rat spinal cord. International Journal of Molecular Sciences. 2012;13(8):9864–9876. doi: 10.3390/ijms13089864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li X. Q., Chen F. S., Tan W. F., Fang B., Zhang Z. L., Ma H. Elevated microRNA-129-5p level ameliorates neuroinflammation and blood-spinal cord barrier damage after ischemia-reperfusion by inhibiting HMGB1 and the TLR3-cytokine pathway. Journal of Neuroinflammation. 2017;14(1):p. 205. doi: 10.1186/s12974-017-0977-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sims N. R., Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta. 2010;1802(1):80–91. doi: 10.1016/j.bbadis.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Xue B., Huang J., Ma B., Yang B., Chang D., Liu J. Astragaloside IV protects primary cerebral cortical neurons from oxygen and glucose deprivation/reoxygenation by activating the PKA/CREB pathway. NEUROSCIENCE. 2019;404:326–337. doi: 10.1016/j.neuroscience.2019.01.040. [DOI] [PubMed] [Google Scholar]
- 5.Wang L., Li S., Liu Y., et al. Motor neuron degeneration following glycine-mediated excitotoxicity induces spastic paralysis after spinal cord ischemia/reperfusion injury in rabbit. American Journal of Translational Research. 2017;9(7):3411–3421. [PMC free article] [PubMed] [Google Scholar]
- 6.Itoh K., Wakabayashi N., Katoh Y., et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annual Review of Pharmacology and Toxicology. 2013;53(1):401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee C. S., Lee C., Hu T., et al. Loss of nuclear factor E2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(20):8408–8413. doi: 10.1073/pnas.1019209108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satoh T., Okamoto S.-I., Cui J., et al. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophillic phase II inducers. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(3):768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L., Yao Y., He R., et al. Methane ameliorates spinal cord ischemia-reperfusion injury in rats: antioxidant, anti-inflammatory and anti-apoptotic activity mediated by Nrf2 activation. Free Radical Biology & Medicine. 2017;103:69–86. doi: 10.1016/j.freeradbiomed.2016.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Fu J., Sun H., Zhang Y., et al. Neuroprotective effects of luteolin against spinal cord ischemia-reperfusion injury by attenuation of oxidative stress, inflammation, and apoptosis. Journal of Medicinal Food. 2018;21(1):13–20. doi: 10.1089/jmf.2017.4021. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y., Li Y., Feng Q., Shao M., Yuan F., Liu F. Polydatin attenuates cadmium-induced oxidative stress via stimulating SOD activity and regulating mitochondrial function in _Musca domestica_ larvae. CHEMOSPHERE. 2020;248:p. 126009. doi: 10.1016/j.chemosphere.2020.126009. [DOI] [PubMed] [Google Scholar]
- 13.Du Q.-H., Peng C., Zhang H. Polydatin: a review of pharmacology and pharmacokinetics. Pharmaceutical Biology. 2013;51(11):1347–1354. doi: 10.3109/13880209.2013.792849. [DOI] [PubMed] [Google Scholar]
- 14.Sun Z., Wang X. Protective effects of polydatin on multiple organ ischemia-reperfusion injury. Bioorganic Chemistry. 2020;94:p. 103485. doi: 10.1016/j.bioorg.2019.103485. [DOI] [PubMed] [Google Scholar]
- 15.Tang K. S., Tan J. S. The protective mechanisms of polydatin in cerebral ischemia. EUROPEAN JOURNAL OF PHARMACOLOGY. 2019;842:133–138. doi: 10.1016/j.ejphar.2018.10.039. [DOI] [PubMed] [Google Scholar]
- 16.Bai H., Ding Y., Li X., et al. Polydatin protects SH-SY5Y in models of Parkinson’s disease by promoting Atg5-mediated but parkin-independent autophagy. Neurochemistry International. 2020;134:p. 104671. doi: 10.1016/j.neuint.2020.104671. [DOI] [PubMed] [Google Scholar]
- 17.Schieber M., Chandel N. S. ROS function in redox signaling and oxidative stress. Current Biology. 2014;24(10):R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S., Tian R., Li H., Chen M., Zhang H., Lin D. Optimized methods for rapidly dissecting spinal cords and harvesting spinal motor neurons with high survival and purity from rats at different embryonic stages. Journal of Spinal Cord Medicine. 2018;41(3):281–291. doi: 10.1080/10790268.2017.1329075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun S., Hu F., Wu J., Zhang S. Cannabidiol attenuates OGD/R-induced damage by enhancing mitochondrial bioenergetics and modulating glucose metabolism via pentose-phosphate pathway in hippocampal neurons. Redox Biology. 2017;11:577–585. doi: 10.1016/j.redox.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X., Zhan J., Hou Y., et al. Coenzyme Q10 regulation of apoptosis and oxidative stress in H2O2 induced BMSC death by modulating the Nrf-2/NQO-1 signaling pathway and its application in a model of spinal cord injury. Oxidative Medicine and Cellular Longevity. 2019;2019:15. doi: 10.1155/2019/6493081.6493081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bell M. T., Puskas F., Agoston V. A., et al. Toll-like receptor 4-dependent microglial activation mediates spinal cord ischemia-reperfusion injury. Circulation. 2013;128(11_suppl_1):S152–S156. doi: 10.1161/CIRCULATIONAHA.112.000024. [DOI] [PubMed] [Google Scholar]
- 22.Zhan J., Li X., Luo D., et al. Polydatin promotes the neuronal differentiation of bone marrow mesenchymal stem cells in vitro and in vivo: involvement of Nrf2 signalling pathway. Journal of Cellular and Molecular Medicine. 2020;24(9):5317–5329. doi: 10.1111/jcmm.15187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhan J., He J., Chen M., Luo D., Lin D. Fasudil promotes BMSC migration via activating the MAPK signaling pathway and application in a model of spinal cord injury. Stem Cells International. 2018;2018:12. doi: 10.1155/2018/9793845.9793845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong S., Lee J. E., Kim C. Y., Seong G. J. Agmatine protects retinal ganglion cells from hypoxia-induced apoptosis in transformed rat retinal ganglion cell line. BMC NEUROSCIENCE. 2007;8(1):p. 81. doi: 10.1186/1471-2202-8-81. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Wang X. The expanding role of mitochondria in apoptosis. Genes & Development. 2001;15(22):2922–2933. [PubMed] [Google Scholar]
- 26.Kristian T. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. CELL CALCIUM. 2004;36(3-4):221–233. doi: 10.1016/j.ceca.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 27.Xu P., Liu Q., Xie Y., et al. Breast cancer susceptibility protein 1 (BRCA1) rescues neurons from cerebral ischemia/reperfusion injury through NRF2-mediated antioxidant pathway. Redox Biology. 2018;18:158–172. doi: 10.1016/j.redox.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denzer I., Munch G., Friedland K. Modulation of mitochondrial dysfunction in neurodegenerative diseases via activation of nuclear factor erythroid-2-related factor 2 by food-derived compounds. Pharmacological Research. 2016;103:80–94. doi: 10.1016/j.phrs.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 29.Kahl A., Stepanova A., Konrad C., et al. Critical role of flavin and glutathione in complex I-mediated bioenergetic failure in brain ischemia/reperfusion injury. Stroke. 2018;49(5):1223–1231. doi: 10.1161/STROKEAHA.117.019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rugarli E. I., Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO Journal. 2012;31(6):1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flippo K. H., Strack S. Mitochondrial dynamics in neuronal injury, development and plasticity. Journal of Cell Science. 2017;130(4):671–681. doi: 10.1242/jcs.171017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tait S. W., Green D. R. Mitochondrial regulation of cell death. Cold Spring Harbor Perspectives in Biology. 2013;5(9) doi: 10.1101/cshperspect.a008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gong G., Yuan L. B., Hu L., et al. Glycyrrhizin attenuates rat ischemic spinal cord injury by suppressing inflammatory cytokines and HMGB1. Acta Pharmacologica Sinica. 2012;33(1):11–18. doi: 10.1038/aps.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chao D. T., Korsmeyer S. J. BCL-2 family: regulators of cell death. Annual Review of Immunology. 1998;16(1):395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 35.Heiskanen K. M., Bhat M. B., Wang H. W., Ma J., Nieminen A. L. Mitochondrial depolarization accompanies cytochrome cRelease during apoptosis in PC6 cells. Journal of Biological Chemistry. 1999;274(9):5654–5658. doi: 10.1074/jbc.274.9.5654. [DOI] [PubMed] [Google Scholar]
- 36.Rossé T., Olivier R., Monney L., et al. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome _c_. NATURE. 1998;391(6666):496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- 37.Brunelle J. K., Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. Journal of Cell Science. 2009;122(4):437–441. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janicke R. U., Sprengart M. L., Wati M. R., Porter A. G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. Journal of Biological Chemistry. 1998;273(16):9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 39.Chen M., Hou Y., Lin D. Polydatin protects bone marrow stem cells against oxidative injury: involvement of Nrf 2/ARE pathways. Stem Cells International. 2016;2016:10. doi: 10.1155/2016/9394150.9394150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah F. A., Kury L. A., Li T., et al. Polydatin attenuates neuronal loss via reducing neuroinflammation and oxidative stress in rat MCAO models. Frontiers in Pharmacology. 2019;10:p. 663. doi: 10.3389/fphar.2019.00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang X., Liu W., Deng J., et al. Polydatin protects cardiac function against burn injury by inhibiting sarcoplasmic reticulum Ca2+ leak by reducing oxidative modification of ryanodine receptors. Free Radical Biology & Medicine. 2013;60:292–299. doi: 10.1016/j.freeradbiomed.2013.02.030. [DOI] [PubMed] [Google Scholar]
- 42.Tsujimoto Y., Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. APOPTOSIS. 2007;12(5):835–840. doi: 10.1007/s10495-006-0525-7. [DOI] [PubMed] [Google Scholar]
- 43.Whiteman M., Armstrong J. S., Cheung N. S., et al. Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. FASEB Journal. 2004;18(12):1395–1397. doi: 10.1096/fj.03-1096fje. [DOI] [PubMed] [Google Scholar]
- 44.Shimizu S., Bowman P. S., Thorne G., Paul R. J. Effects of hypoxia on isometric force, intracellular Ca2+, pH, and energetics in porcine coronary artery. Circulation Research. 2000;86(8):862–870. doi: 10.1161/01.RES.86.8.862. [DOI] [PubMed] [Google Scholar]
- 45.Choi D. W. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. TRENDS IN NEUROSCIENCES. 1988;11(10):465–469. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- 46.Deng J., Liu W., Wang Y., Dong M., Zheng M., Liu J. Polydatin modulates Ca2+ handling, excitation-contraction coupling and β-adrenergic signaling in rat ventricular myocytes. Journal of Molecular and Cellular Cardiology. 2012;53(5):646–656. doi: 10.1016/j.yjmcc.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 47.Yang B. Polydatin attenuated food allergy via store-operated calcium channels in mast cell. World Journal of Gastroenterology. 2013;19(25):3980–3989. doi: 10.3748/wjg.v19.i25.3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonora M., Morganti C., Morciano G., Giorgi C., Wieckowski M. R., Pinton P. Comprehensive analysis of mitochondrial permeability transition pore activity in living cells using fluorescence-imaging-based techniques. Nature Protocols. 2016;11(6):1067–1080. doi: 10.1038/nprot.2016.064. [DOI] [PubMed] [Google Scholar]
- 49.Wang X., Song R., Bian H. N., Brunk U. T., Zhao M., Zhao K. S. Polydatin, a natural polyphenol, protects arterial smooth muscle cells against mitochondrial dysfunction and lysosomal destabilization following hemorrhagic shock. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2012;302(7):R805–R814. doi: 10.1152/ajpregu.00350.2011. [DOI] [PubMed] [Google Scholar]
- 50.Kerr P. M., Suleiman M. S., Halestrap A. P. Reversal of permeability transition during recovery of hearts from ischemia and its enhancement by pyruvate. American Journal of Physiology-Heart and Circulatory Physiology. 1999;276(2):H496–H502. doi: 10.1152/ajpheart.1999.276.2.H496. [DOI] [PubMed] [Google Scholar]
- 51.Baines C. P. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Research in Cardiology. 2009;104(2):181–188. doi: 10.1007/s00395-009-0004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xi J., Wang H., Mueller R. A., Norfleet E. A., Xu Z. Mechanism for resveratrol-induced cardioprotection against reperfusion injury involves glycogen synthase kinase 3β and mitochondrial permeability transition pore. European Journal Of Pharmacology. 2009;604(1-3):111–116. doi: 10.1016/j.ejphar.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moi P., Chan K., Asunis I., Cao A., Kan Y. W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(21):9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ke B., Shen X. D., Zhang Y., et al. KEAP1-NRF2 complex in ischemia-induced hepatocellular damage of mouse liver transplants. Journal of Hepatology. 2013;59(6):1200–1207. doi: 10.1016/j.jhep.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peake B. F., Nicholson C. K., Lambert J. P., et al. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. American Journal of Physiology-Heart and Circulatory Physiology. 2013;304(9):H1215–H1224. doi: 10.1152/ajpheart.00796.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiao L., Xu X., Zhang F., et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biology. 2017;11:297–311. doi: 10.1016/j.redox.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller D. M., Singh I. N., Wang J. A., Hall E. D. Nrf2-ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Experimental Neurology. 2015;264:103–110. doi: 10.1016/j.expneurol.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used to support the findings of this study are available from the corresponding author upon request.