

Abstract
Objective:
SSc-ILD is characterized by variable clinical outcomes, activation of innate immune pattern recognition receptors (PRRs), and accumulation of αSMA expressing myofibroblasts. The association of these entities with mitochondrial DNA (mtDNA), an endogenous ligand for the intracellular DNA-sensing PRRs TLR9 and cGAS-STING, has yet to be determined.
Methods:
Human lung fibroblasts from normal donors (NHLFs) and SSc-ILD explants were treated with synthetic CpG DNA and assayed for αSMA expression and extracellular mtDNA using qPCR for the human MT-ATP6 gene. Plasma MT-ATP6 concentrations were evaluated in two independent SSc-ILD cohorts and demographically matched controls. The ability of control and SSc-ILD plasma to induce TLR9 and cGAS-STING activation was evaluated with commercially available HEK 293 reporter cells. Plasma concentrations of type I interferons, IL-6, and oxidized DNA were measured with using electrochemiluminescence and ELISA-based methods, respectively. Extracellular vesicles (EVs) precipitated from plasma were evaluated for MT-ATP6 concentrations and proteomics via liquid chromatography–mass spectrometry.
Results:
NHLFs and SSc-ILD fibroblasts develop increased αSMA expression and MT-ATP6 release following CpG stimulation. Plasma mtDNA concentrations are increased in two SSc-ILD cohorts, reflective of ventilatory decline, and positively associated with both TLR9 and cGAS-STING activation as well as Type I interferon and IL-6 expression. Plasma mtDNA is not oxidized and is conveyed by EVs displaying a proteomics profile consistent with a multicellular origin.
Conclusion:
These findings demonstrate an unrecognized connection between EV-encapsulated mtDNA, clinical outcomes, and intracellular DNA-sensing PRR activation in SSc-ILD. Further study of these interactions could catalyze novel mechanistic and therapeutic insights into SSc-ILD and related disorders.
INTRODUCTION
Systemic sclerosis (SSc) is a multisystem autoimmune condition characterized by chronic inflammation and progressive fibrosis of the skin and internal organs [1]. Scleroderma-associated interstitial lung disease (SSc-ILD), a frequent complication of SSc, results in significant morbidity and mortality [2]. The clinical trajectory of SSc-ILD is highly variable as some patients experience rapid deterioration in their ventilatory function while others remain stable or improve [3, 4]. Although prognostic models with clinical parameters [5] and peripheral blood biomarkers [6] have demonstrated promise in identifying patients at risk for severe disease phenotypes, the relationship between these clinical prediction models and the molecular mechanisms driving disease remains an area of ongoing investigation.
SSc-ILD involves the pathologic remodeling of lung parenchyma and accumulation of alpha-smooth muscle actin (αSMA) expressing myofibroblasts [7]. These cells arise in response to transforming growth factor beta 1 (TGFβ1) [7, 8] and display a gene expression profile characterized in part by aberrant activation of innate immune pathways [9, 10]. The innate immune system is triggered by pattern recognition receptors (PRRs) that recognize epitopes broadly conserved across pathogens, known as pathogen-associated molecular patterns (PAMPs) derived from microbes; and endogenous ligands, known as danger-associated molecular patterns (DAMPs) that are released by injured or activated cells and tissues. [11]. Given the well-established role of autoimmunity in SSc-ILD [1], the identification and contribution of endogenous ligands to innate immune activation may provide novel mechanistic insight into the fibrotic processes affecting the lung.
Mitochondria are increasingly recognized for their ability to function as DAMPs that interact with PRRs to initiate inflammation [12]. Mitochondrial DNA (mtDNA), released either non-specifically by necrotic cells [13] or via regulated processes by stressed but viable cells [14, 15], has been shown to activate two intracellular DNA-sensing PRRs: the endosomal Toll-Like Receptor 9 (TLR9) and the cytosolic cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS). TLR9 is a receptor for DAMPs through its recognition of unmethylated, CpG-rich DNA which is prevalent in the mitochondrial genome [12]. While cGAS is widely known as a double-stranded DNA sensor [16, 17], its pleiotropic properties include its role as a receptor for exogenous mtDNA that initiates a pathway resulting in the activation of the endoplasmic reticulum membrane protein, stimulator of interferon genes (STING) [17]. TLR9 and cGAS-STING drive pro-inflammatory responses through the production of Type I interferons (IFNs) and immunogenic cytokines [12]. Although extracellular mtDNA has been associated with poor outcomes in chronic disease [18–20], its role as a DAMP for these intracellular innate immune DNA-sensors in SSc-ILD has yet to be fully understood.
To explore this role, we defined an association between mtDNA and fibroblast activation in SSc-ILD. In a direct clinical translation of our in vitro findings, we then investigated the association between circulating mtDNA, clinical outcomes, and activation of cytoplasmic DNA sensors such as TLR9 and cGAS-STING in SSc-ILD.
MATERIALS AND METHODS
Detailed methods are provided in the online supplement
Human Subjects
Discovery cohort: Plasma specimens (n=14) and corresponding clinical data were obtained from The Scleroderma Clinical Repository at Boston University. Validation cohort: Plasma specimens (n=40) and corresponding clinical data were obtained from The Yale Lung Repository (TYLR) housed within the Interstitial Lung Disease Center of Excellence at Yale School of Medicine. Control group: Plasma specimens from healthy subjects without known inflammatory or lung disease were obtained from TYLR (n=20) [26]. All human studies were performed with informed consent on protocols approved by the Institutional Review Board at each institution. SSc-ILD diagnosis was based on current consensus guidelines rendered through expert multidisciplinary discussion at each institution [21]. Demographic and clinical data were obtained at the time of the blood draw. Subjects were followed for 50 months for a relative 10% decline in forced vital capacity (FVC).
Culture and Stimulation of Human Lung Fibroblasts
Normal human lung fibroblasts (NHLFs, n=3) and SSc-ILD fibroblasts (n=3) obtained from Lonza (Lonza, Allendale, NJ) and The Scleroderma Center at the University of Pittsburgh were used between passages 3–7 and stimulated with and without 10 ng/ml of recombinant human TGFβ1 [22], 50 μg/ml CpG DNA or CpG DNA control [22, 23], and 10 μg/ml of hydroxychloroquine (HCQ) [23] as previously described.
RNA Isolation and qRT-PCR
Total cellular RNA was isolated and relative expression of αSMA was quantified using the 2-delta Ct method as previously reported [24]. Expression of αSMA was determined relative to β-actin expression and normalized to unstimulated NHLFs or SSc-ILD fibroblasts.
DNA Isolation and Quantification
Isolation of DNA and subsequent quantification of the human MT-ATP6 gene in cell culture supernatants and plasma were performed as previously described [14, 25]. MT-ATP6 concentrations in cell culture supernatants were normalized to unstimulated NHLFs or SSc-ILD fibroblasts.
TLR9 Activation
Commercially available human TLR9 expressing HEK 293 cells were cultured, passaged, plated with detection medium, stimulated with 20 μl of either healthy control or SSc-ILD plasma (either untreated or treated with 250 units/μl of Benzonase® for two hours at room temperature) for six hours, and assayed for TLR9 activation as previously described [19].
cGAS-STING Activation
Commercially available interferon stimulating gene (ISG) expressing HEK 293 cells were cultured, passaged, plated with detection medium, stimulated with 20 μl of either healthy control or SSc-ILD plasma (either untreated or treated with 250 units/μl of Benzonase® for two hours at room temperature) for six hours, and assayed for STING activation per the manufacturer’s protocol.
Quantification of Type I Interferons and Cytokines
Plasma was subject to multiplex quantification of INF-α, IFN-β, and IL-6 using the U-PLEX Biomarker Group 1 Human Assays (Meso Scale Discovery; Rockville, MD) per the manufacturer’s protocol.
Extracellular Vesicle Isolation
Extracellular vesicles (EVs) were isolated from plasma using a commercially available kit as previously described [16].
8-Oxo-2’-deoxyguanosine Quantification
Concentrations of 8-oxo-2’-deoxyguanosine were measured in the plasma and EVs using a commercially available ELISA kit as previously described [26].
Transmission Electron Microscopy
EVs were prepared and examined in a FEI Tecnai TF20 transmission electron microscope at 200 kV of accelerating voltage. Images were acquired using an AMT NanoSprint 1200 CMOS camera.
Mass Spectrometry
EVs were processed at the Keck Mass Spectrometry & Proteomics Resource of the W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University. Protein abundances were normalized to total abundances of detected spectral peaks for each sample injection. Pathway enrichment analysis was conducted via MetaCore.
Statistical Analysis
Data distribution was assessed using D’Agostino-Pearson omnibus test or Shapiro-Wilk test based on the sample size. Categorical data was compared with a chi-square test. For continuous data, parametric comparisons were made using Student’s t-test, and non-parametric data were compared using Mann-Whitney. These evaluations were performed using GraphPad Prism 8.3.1 (GraphPad Software, La Jolla, CA). Receiver operator curve (ROC) analysis of the Yale cohort determined the Youden Index for mtDNA, Kaplan-Meier analysis determined its association with disease progression, and multivariate Cox regression hazards ratio was used to model association with relevant covariates using MedCalc 12 (MedCalc Software, Belgium). Wilcoxon Ranked Sum on R statistical package (version 3.3.1) was used for multiple hypothesis testing of proteomics data. A p-value of < 0.05 and false discovery rate (FDR) < 0.10 were considered significant.
RESULTS
Exogenous CpG stimulation drives fibroblast activation in SSc-ILD
To begin to determine the role of DNA-sensing PRRs and fibroblast activation in SSc-ILD, we first subjected NHLFs to the absence or presence of 10 ng/ml of TGFβ1 and assessed for fibroblast differentiation by assaying for αSMA expression and extracellular mtDNA release using the well-validated method of quantifying the human mtDNA specific gene, MT-ATP6 [18, 19]. Consistent with our previous findings, NHLFs exposed to TGFβ1 exhibited substantially higher fold changes in αSMA expression (1.000 vs 5.565, p=0.0004, Figure 1A) and extracellular release of MT-ATP6 (1.000 vs 1.782, p=0.032, Figure 1B). We then predicted that these responses would be augmented by intracellular PRR agonism. Subsequently, we subjected TGFβ1 stimulated NHLFs to 50 μg/ml of exogenous CpG DNA, which resulted in significantly higher fold changes in both αSMA expression (1.000 vs 7.959, p=0.0003, Figure 1A) and extracellular MT-ATP6 concentrations (1.000 vs 2.520, p=0.027, Figure 1B). A role for intracellular DNA-sensing PRRs was further evaluated in cells treated with or without 10 μg/ml of HCQ, which possesses inhibitory effects on cytoplasmic DNA-sensing PRRs, including TLR9 [22] and cGAS-STING [27]. Here, TGFβ1 and CpG DNA exposed NHLFs treated with HCQ expressed less αSMA than cells subjected to both TGFβ1 and CpG DNA stimulation (3.464 vs 7.959, p=0.003, Figure 1A), or cells treated with TGFβ1 alone (3.464 vs 5.565, p=0.020, Figure 1A). Extracellular MT-ATP6 concentrations also decreased upon HCQ treatment but were not statistically significant when compared to TGFβ1 and CpG DNA stimulation (1.501 vs 2.520, p=0.020, Figure 1B) or TGFβ1 stimulation alone (1.501 vs 1.782, p=0.409, Figure 1B). Seeking a connection to scleroderma lung involvement, we then repeated the above experiments with fibroblasts obtained from explanted SSc-ILD lungs. Compared to unstimulated cells, SSc-ILD fibroblasts subjected to CpG DNA stimulation displayed higher fold changes in αSMA expression (1.000 vs 3.708, p=0.018, Figure 1C) and extracellular MT-ATP6 concentration (1.000 vs 2.591, p=0.002, Figure 1D). When these cells were subsequently treated with HCQ, they exhibited lower αSMA expression (1.261 vs 3.708, p=0.026, Figure 1C) and, unlike NHLFs, reduced extracellular MT-ATP6 release (1.225 vs 2.591, p=0.004, Figure 1D). These data provide evidence of the profound modulatory effects of intracellular DNA sensors on fibroblast activation, particularly in the SSc-ILD disease state.
Figure 1. CpG stimulation of SSc-ILD fibroblasts is opposed by hydroxychloroquine (HCQ).
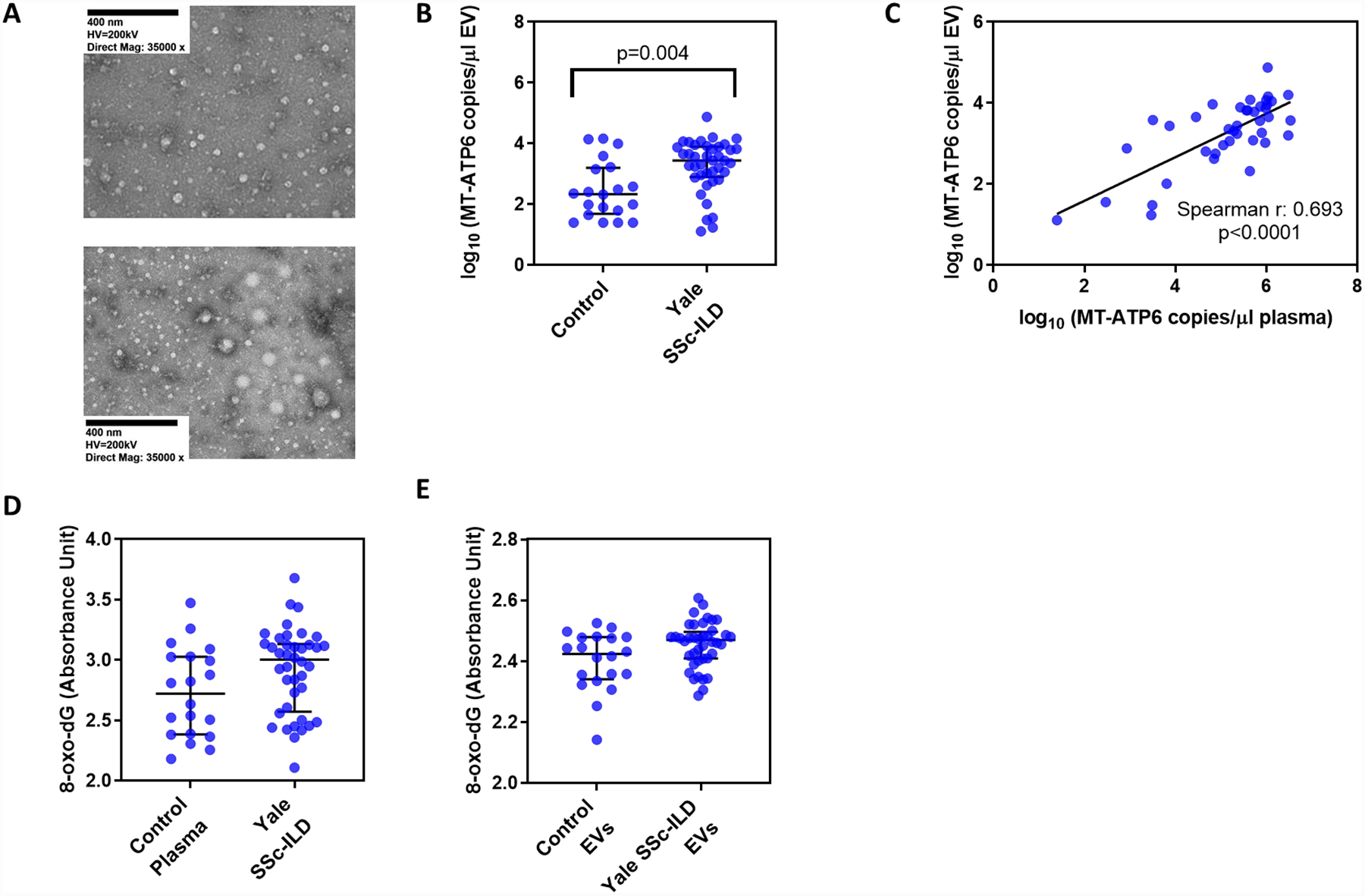
A, B. Relative to unstimulated cells (n=3), NHLFs stimulated with TGFβ1 (n=3) exhibited (A) higher fold change in αSMA expression and (B) release of extracellular MT-ATP6 copy numbers. Stimulation with exogenous CpG DNA augmented fold change in (A) αSMA expression and (B) extracellular MT-ATP6 concentrations. (A) Consequently, when these same cells stimulated with TGFβ1 + CpG DNA were then subjected to hydroxychloroquine (HCQ) to inhibit intracellular PRR activation, they expressed significantly less αSMA, even when compared to TGFβ1 alone. (B) HCQ treatment led to a reduction in MT-ATP6 concentrations that trended towards statistical significance. C, D. Fibroblasts derived from SSc-ILD lung explants (n=3) treated with CpG DNA showed elevated (C) αSMA expression and (D) extracellular MT-ATP6 concentrations. Treatment of these CpG DNA stimulated SSc-ILD fibroblasts with HCQ attenuated both (C) αSMA expression and (D) extracellular MT-ATP6 release. Expression of αSMA was determined relative to β-actin expression and normalized to unstimulated NHLFs or SSc-ILD fibroblasts. MT-ATP6 concentrations for cell culture supernatants were normalized to unstimulated NHLFs or SSc-ILD fibroblasts. **p<0.005; *p<0.05. Dotted line indicates baseline condition.
Extracellular mtDNA is enriched in the plasma of two independent SSc-ILD cohorts
Because these results suggested an association between intracellular DNA sensors and fibroblast activation in SSc-ILD, we next sought to translate our findings in vivo. We first evaluated plasma from two well-characterized cohorts of SSc-ILD subjects: our discovery cohort of 14 subjects from Boston and our validation cohort of 40 subjects from Yale. A cohort of 20 demographically matched healthy individuals was also employed. As described in Table 1, both cohorts consisted of mostly Caucasian female subjects with a significant smoking history (p=0.005). Pulmonary function testing revealed relatively preserved percent predicted forced vital capacity (FVC%) and mean hemoglobin corrected diffusion capacity of the lung for carbon monoxide (DLCO-Hgb%). Because we have previously shown that the extracellular mtDNA arising in the setting of chronic lung disease can be detected in the circulation [18, 19], we first evaluated plasma samples from the relatively small Boston cohort and demographically matched controls using the above method to measure copy numbers of MT-ATP6 [18, 19]. Relative to control subjects, plasma MT-ATP6 concentrations were substantially elevated in SSc-ILD subjects (2.384 vs. 4.852 log copies/μl, p<0.0001, Figure 2A). Since these findings are limited to a small number of subjects in a cross-sectional study, it was necessary for us to validate them in the larger, longitudinal cohort at Yale, where we found similarly elevated plasma concentrations of MT-ATP6 (2.384 vs 5.491 log copies/μl, p<0.0001, Figure 2B). These data suggest the presence of a ligand for intracellular DNA-sensing PRRs, specifically mtDNA, in the circulation of subjects with SSc-ILD.
Table 1.
Clinical characteristics of the study population.
| Healthy Controls | Boston SSc-ILD Subjects | Yale SSc-ILD Subjects | p-value | |
|---|---|---|---|---|
| N | 20 | 14 | 40 | |
| Age (years) | ||||
| Mean ± SD | 51.05± 18.14 | 55.24± 10.12 | 52.37 ± 12.79 | 0.882 |
| Gender n (%) | ||||
| Male | 5 (25.00) | 4 (28.57) | 12 (30.00) | 0.769 |
| Female | 15 (75.00) | 10 (71.43) | 28 (70.00) | |
| Race n (%) | ||||
| Caucasian | 16 (80.00) | 11 (78.57) | 31 (77.50) | >0.999 |
| African American | 3 (15.00) | 3 (21.43) | 7 (17.50) | |
| Hispanic | 0 (0.00) | 0 (0.00) | 1 (2.50) | |
| Asian | 1 (5.00) | 0 (0.00) | 1 (2.50) | |
| Smoking Status n (%) | ||||
| Ever/Current | 2 (10.00) | 7(50.00) | 23 (57.50) | 0.005 |
| Never | 18 (90.00) | 7 (50.00) | 17 (42.50) | |
|
FVC% at enrollment (percent predicted mean ± SD) |
77.56± 15.19 | 73.89± 18.39 | 0.386 | |
|
DLCO-Hgb% at enrollment (percent predicted mean ± SD) |
52.23 ± 19.89 | 54.08 ± 16.25 | 0.846 |
Figure 2. Plasma mtDNA is elevated in two independent SSc-ILD cohorts and is predictive of lung function decline.
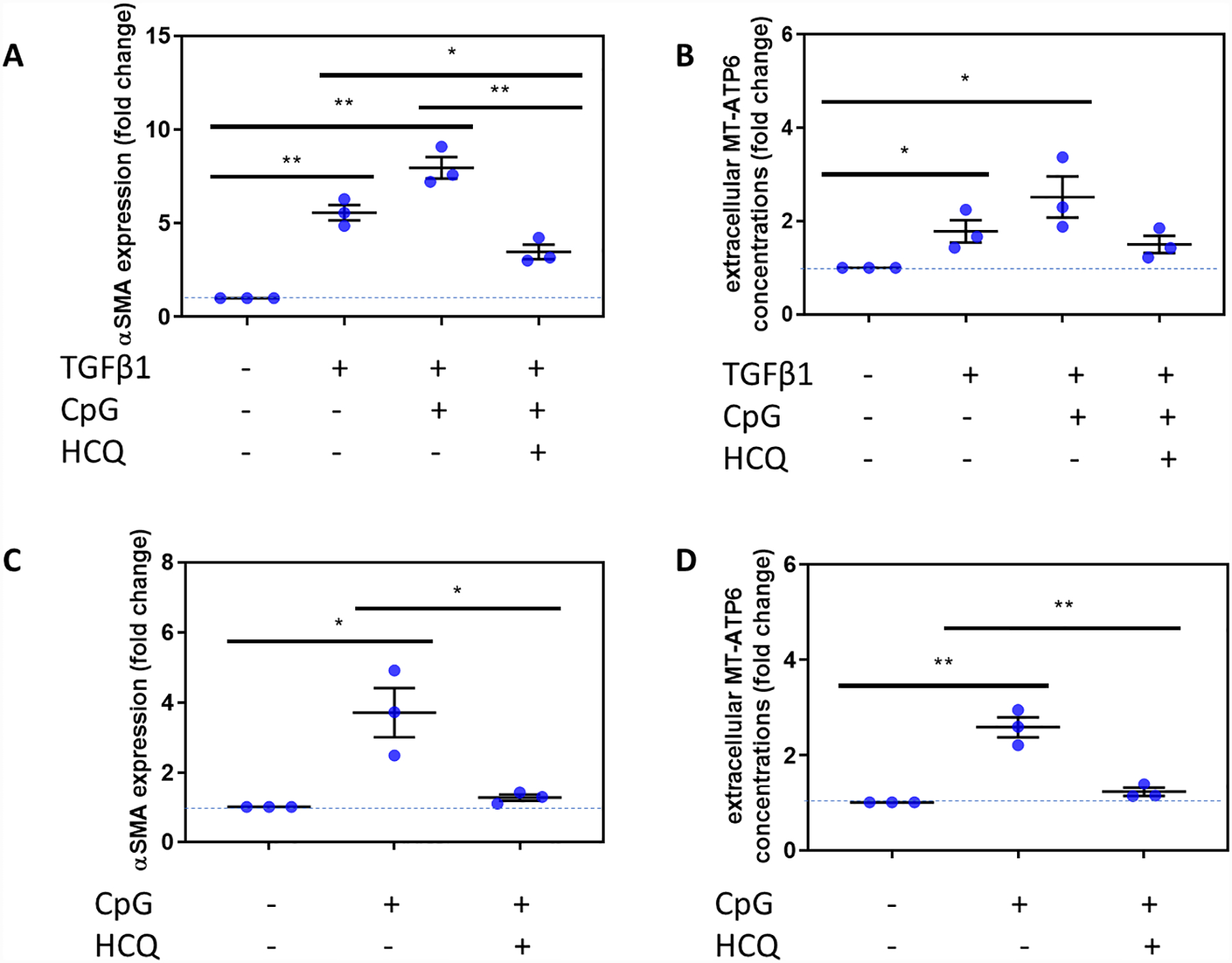
Relative to demographically matched healthy controls (n=20), plasma obtained from a SSc-ILD cohorts obtained from (A) Boston (n=14) and (B) Yale (n=40) exhibited substantially increased MT-ATP6 concentrations. Data are presented as log base 10 of the MT-ATP6 copies per microliter of plasma. (C) Receiver operator curve analysis revealed that a plasma MT-ATP6 concentration of 4.85 (log base 10) copies per microliter can be reliably used to stratify patients as high-risk (>4.85 log copies/μl) or low-risk (≤4.85 log copies/μl) for > 10% relative decline in forced vital capacity (FVC). (D) Kaplan-Meier plot for preserved FVC shows that compared to subjects whose plasma MT-ATP6 concentrations are ≤4.85 log copies/μl (top line, black), those with a plasma MT-ATP6 concentration >4.85 log copies/μl (bottom line, red) experienced significantly higher risk of a > 10% relative decline in FVC over 50 months, independent of smoking, age, gender, race, baseline FVC%, diffusion capacity for carbon monoxide corrected for hemoglobin (DLCO-Hgb%,) and modified Rodnan skin score (MRSS).
Excessive plasma mtDNA is predictive of ventilatory decline
Our group [18, 19] and others [20, 25] have demonstrated the prognostic significance of elevated plasma mtDNA in several clinical contexts. To date, however, such an evaluation has not been performed in the SSc-ILD setting. Disease progression, commonly defined as a >10% relative decline in FVC in clinical trials, is an important measure of clinical phenotypes, treatment response, and drug efficacy [4]. Using longitudinal pulmonary function testing data available from the Yale cohort, we examined the value of plasma mtDNA in predicting ventilatory decline during a 50-month longitudinal follow-up period. ROC analysis revealed that a plasma MT-ATP6 copy number of 4.85 log copies/μl can stratify subjects as low-risk (≤4.85 log copies/μl) or high-risk (>4.85 log copies/μl) for >10% relative FVC decline (area under the curve [AUC]: 0.680, p=0.032, Figure 2C). Clinical characteristics of subjects with low and high MT-ATP6 concentrations, based on this threshold value, are shown in Table S1. There were no differences in the type of SSc, antibodies involved, rates of gastroesophageal reflux, modified Rodnan skin scores (MRSS), FVC%, or DLCO-Hgb% between the two groups. Kaplan-Meier analysis revealed that values exceeding the 4.85 log copies/μl cutoff were significantly associated with a >10% relative decline in FVC (hazard ratio [HR] = 3.46, 95% confidence interval [CI]: 1.38–8.96, p=0.010, Figure 2D), independent of smoking, age, gender, race, MRSS, and baseline FVC% or DLCO-Hgb%. These data demonstrate that the level of circulating extracellular mtDNA displays outcome prediction properties in SSc-ILD subjects.
Plasma mtDNA is associated with TLR9 and cGAS-STING activation
Given the clinical significance of this circulating mtDNA, we then explored its biologic relevance. Because extracellular mtDNA can be recognized by cytosolic DNA sensors such as TLR9 and cGAS in various inflammatory diseases [12], we aimed to determine if this is the case in SSc-ILD. We first stimulated a commercially available TLR9 reporter cell line with plasma obtained from healthy controls and the Yale SSc-ILD cohort (limited sample quantities precluded this analysis in the Boston cohort). Relative to plasma from healthy controls, SSc-ILD plasma robustly activated TLR9 (0.182 vs 0.320 absorbance units [AU], p<0.0001, Figure 3A) in a manner that showed a positive correlation with plasma MT-ATP6 concentrations (Spearman r=0.358, p=0.023, Figure 3B). To determine whether the extracellular mtDNA intrinsic to the plasma activates TLR9, we subjected the same plasma to Benzonase®, an endonuclease that attacks and degrades all forms of DNA, prior to stimulating cells. This resulted in a significant reduction of TLR9 activation (0.291 vs 0.320 AU, p=0.027, Figure 3C). Because the cGAS-STING pathway can be activated in parallel to the TLR9 pathway [12], we then repeated the above experiments with a cGAS-STING ISG reporter cell line. Similar to TLR9, we found that the Yale SSc-ILD plasma samples vigorously activated cGAS-STING (0.335 vs 0.858 AU, p<0.0001, Figure 3D) in a manner that also correlated with their respective plasma MT-ATP6 concentrations (Spearman r=0.426, p=0.006, Figure 3E). Benzonase® digestion subsequently led to a substantial reduction in cGAS-STING activation (0.742 vs 0.858 AU, p=0.002, Figure 3F). These data show that extracellular mtDNA present in the circulation has agonistic effects on innate immune cytoplasmic DNA sensors in SSc-ILD.
Figure 3. Plasma mtDNA is associated with TLR9 and cGAS-STING activation.
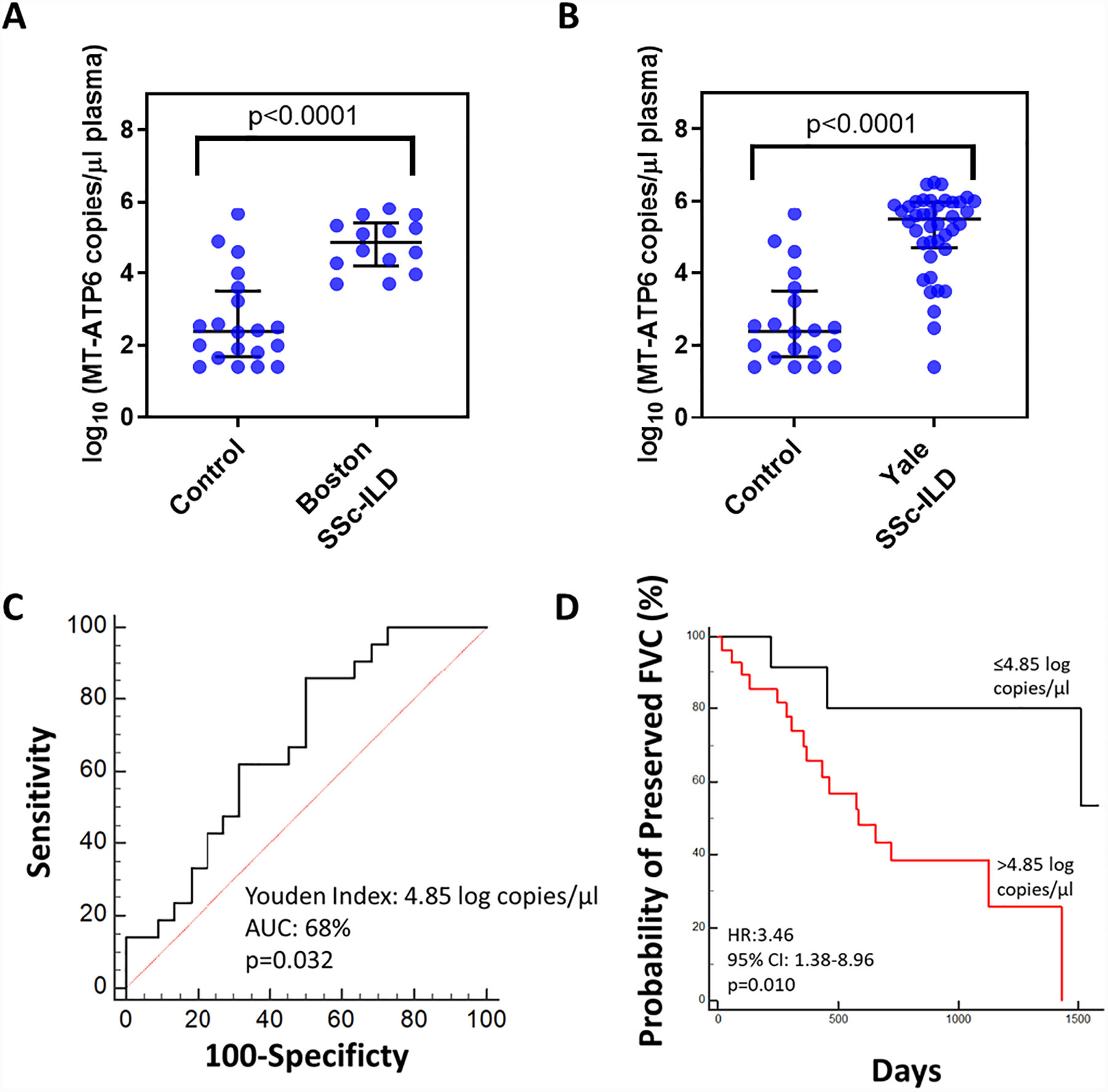
A-C. A commercially available TLR9 reporter cell line was stimulated with plasma obtained from healthy controls and the Yale SSc-ILD cohort. Relative to control plasma, SSc-ILD plasma (A) robustly activated TLR9 in a manner that (B) showed a positive correlation with plasma MT-ATP6 concentrations and (C) was abrogated by treatment with Benzonase®. D-F. Because the cGAS-STING pathway can be activated in parallel with TLR9, the above experiment was repeated with the cGAS-STING ISG reporter cell line. Here, SSc-ILD plasma (D) vigorously activated cGAS-STING in a manner that also (E) correlated with their respective plasma MT-ATP6 concentrations and (F) was opposed by Benzonase® digestion.
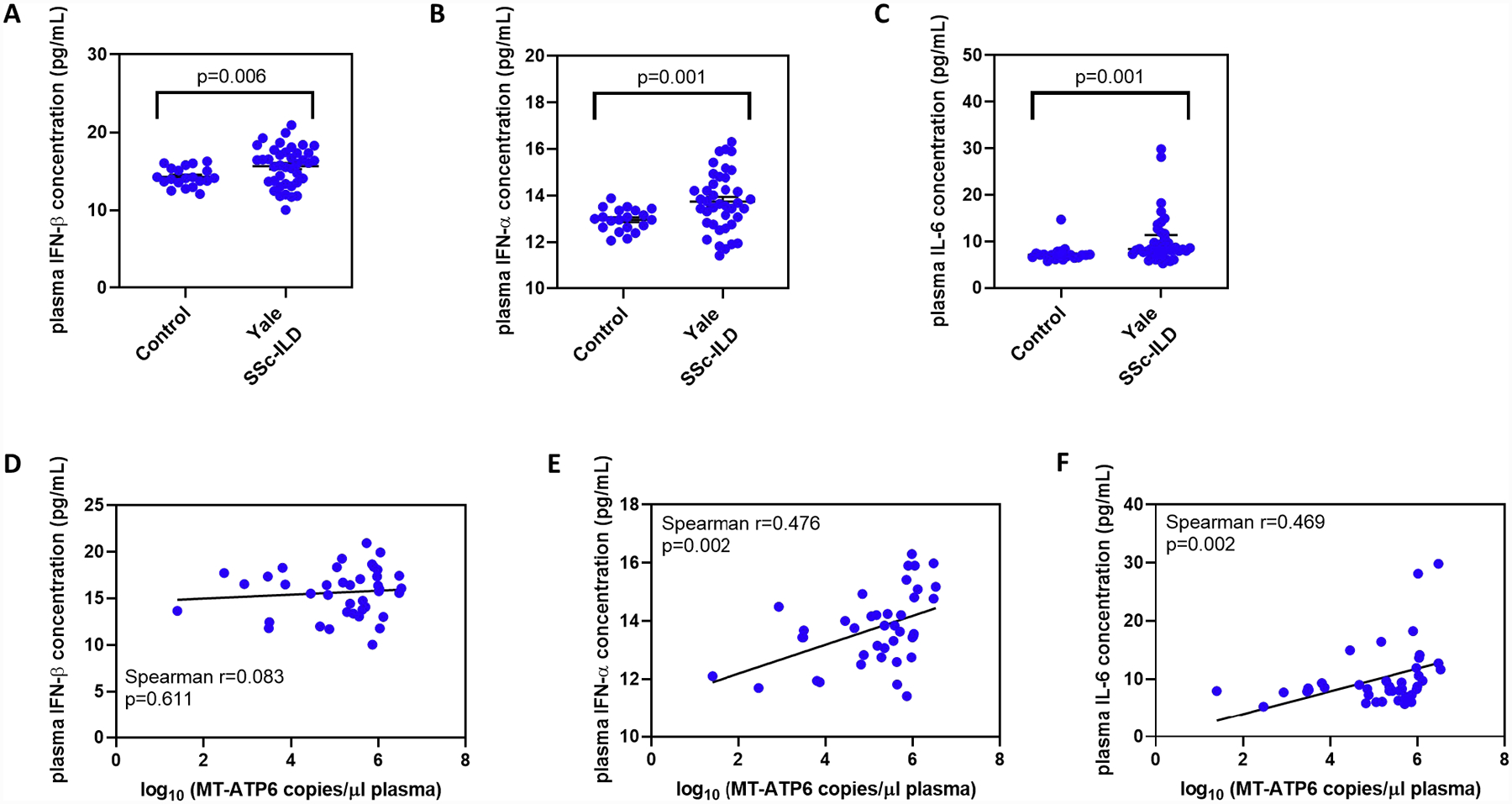
Plasma mtDNA is associated with increased expression of Type I IFNs and IL-6
The TLR9 and cGAS-STING signaling pathways result in an intracellular DNA-driven immune response characterized by the excessive production of Type I IFNs and immunogenic cytokines such as IL-6 [12, 28]. To determine if extracellular mtDNA elicited a similar response in SSc-ILD, we assayed IFN-β, IFN-α, and IL-6 concentrations in the same plasma samples analyzed above. Relative to controls, Yale SSc-ILD subjects demonstrated elevated plasma concentrations of IFN-β (14.26 vs 15.64 pg/ml, p=0.006, Figure 4A), IFN-α (12.96 vs 13.74 pg/ml, p=0.001, Figure 4B), and IL-6 (7.091 vs 8.368 pg/ml, p=0.001, Figure 4C). Next, we explored an association between circulating mtDNA and these inflammatory mediators. Although plasma MT-ATP6 concentrations did not correlate with IFN-β (Spearman r=0.083, p=0.611, Figure 4D), a significant association was found with IFN-α (r=0.476, p=0.002, Figure 4E) and IL-6 (r=0.469, p=0.002, Figure 4F). These findings reveal an association between circulating mtDNA and Type I IFNs and IL-6, further supporting its potential to activate DNA-sensing PRRs in SSc-ILD.
Figure 4. Plasma mtDNA is associated with increased expression of Type 1 interferons and IL-6.
A commercially available multiplex assay was used to quantify IFN-β, IFN-α, and IL-6 concentrations in plasma samples obtained from healthy controls and the Yale SSc-ILD cohort. Relative to controls, Yale SSc-ILD subjects expressed elevated plasma concentrations of (A) IFN-β, (B) IFN-α, and (C) IL-6. Although plasma MT-ATP6 concentrations did not correlate with (D) IFN-β, a significantly positive correlation was detected with both (E) IFN-α and (F) IL-6.
Extracellular mtDNA is conveyed in extracellular vesicles in the plasma
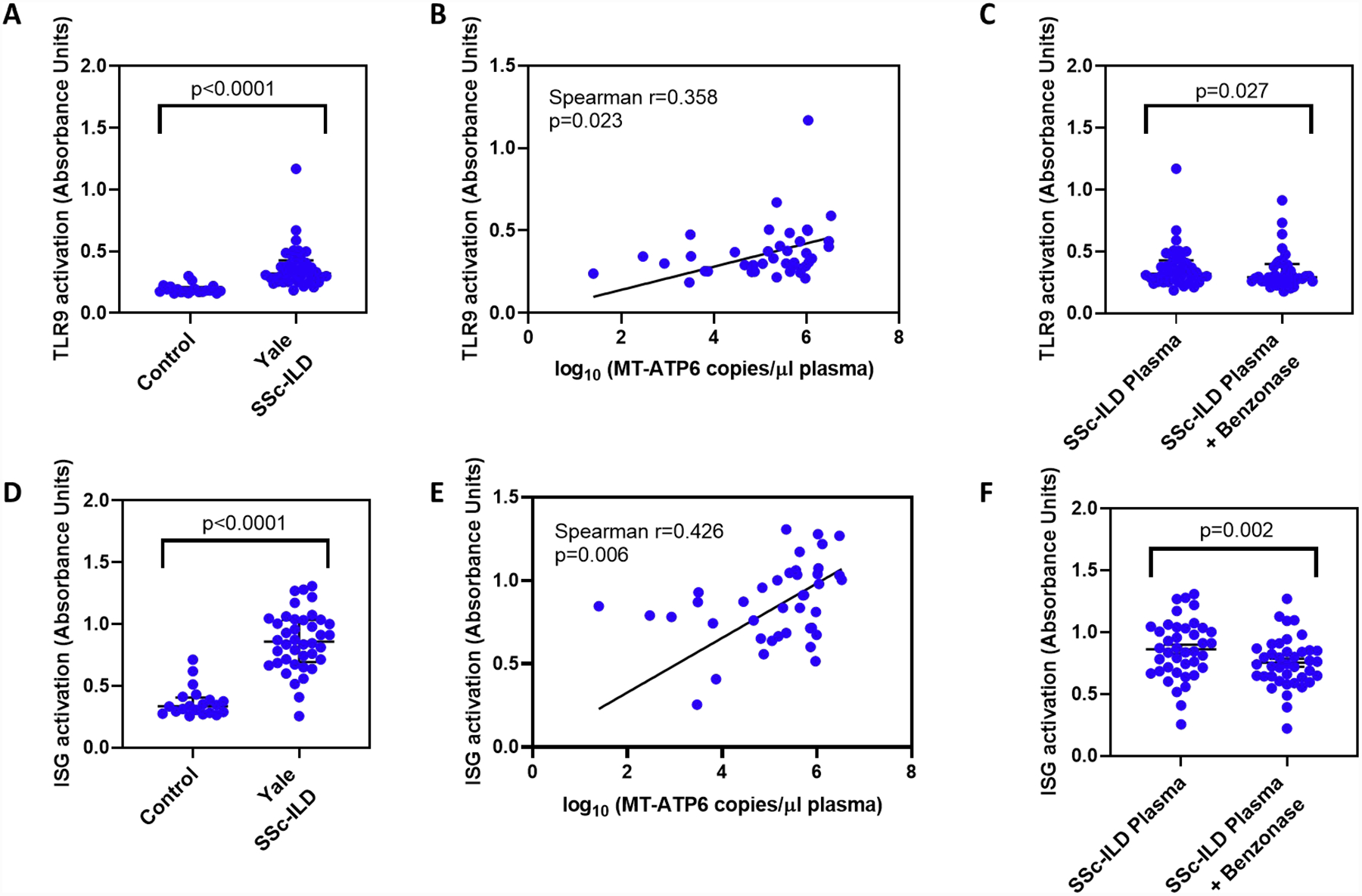
Given the clinical and biological significance of extracellular mtDNA in SSc-ILD, we then aimed to explore its nature, starting with how it is shuttled in the circulation. Cells extrude their mtDNA into the extracellular space as either free nucleic acids during events like necroptosis, or via an active process involving encapsulation in extracellular vesicles (EVs) [29]. EVs are increasingly recognized for their pathogenic role in lung disease [30] but have been only minimally studied in SSc-ILD. Therefore, we isolated and analyzed EVs from the plasma of healthy controls and Yale SSc-ILD subjects. Transmission electron microscopy revealed the presence of EVs in the plasma samples (Figure 5A). Relative to EVs isolated from control subjects, EVs from SSc-ILD subjects contained substantially higher concentrations of MT-ATP6 (2.338 vs 3.441 log copies/μl, p=0.004, Figure 5B) that strongly correlated with their respective plasma MT-ATP6 copy numbers (Spearman r=0.693, p<0.0001, Figure 5C). These data show that extracellular mtDNA is conveyed in the plasma within EVs in SSc-ILD.
Figure 5. Extracellular vesicles convey extracellular mtDNA that is not heavily oxidized.
(A) Transmission electron microscopy reveals the presence of extracellular vesicles (EVs) in plasma of healthy controls (top) and Yale SSc-ILD subjects (bottom) that were isolated using a commercially available precipitation method. As compared to healthy controls, (B) EVs isolated from SSc-ILD subjects contained substantially higher MT-ATP6 concentrations that (C) showed a strong positive correlation with their respective plasma MT-ATP6 concentrations. Concentrations of 8-oxo-2’-deoxyguanosine (8-oxo-dG), the most frequent oxidative DNA damage present in mtDNA, were similar between healthy controls and SSc-ILD subjects when assayed in the (D) plasma and (E) EVs.
Circulating mtDNA is not excessively oxidized in SSc-ILD
It has been reported that extracellular mtDNA exists in oxidized and non-oxidized states, with the former reflecting the presence of reactive oxygen species associated with severe cellular stress [14]. Believing that this was the state of the extracellular mtDNA found in SSc-ILD, we assayed for the presence of 8-oxo-2’-deoxyguanosine (8-oxo-dG), the most frequent derivative of oxidative DNA damage present in mtDNA [31], using a commercially available ELISA. We first examined the levels of 8-oxo-dG in the plasma, finding that relative to controls, SSc-ILD subjects contained mildly, yet not statistically significantly, higher plasma 8-oxo-dG concentrations (2.723 vs 3.004 AU, p=0.061, Figure 5D). We then compared the 8-oxo-dG levels in the EVs of controls and SSc-ILD subjects, where EVs from both groups displayed similar concentrations of 8-oxo-dG (2.424 vs 2.470 AU, p=0.062, Figure 5E). These findings demonstrate that the capacity for extracellular mtDNA to activate intracellular DNA-sensing PRRs in SSc-ILD does not appear to be dependent upon its degree of oxidation.
Proteomics of extracellular vesicles
The above analysis was predicated upon identifying the nature of the circulating mtDNA encapsulated in EVs. However, because of the well-described pleotropic effects of EVs [30], we then characterized their role in SSc-ILD by identifying their proteomic profile through mass spectrometry. As shown in Table 2, we identified 38 proteins whose content differed between samples from SSc-ILD subjects and healthy controls. Unbiased pathway enrichment analysis revealed that EVs derived from SSc-ILD subjects possess a proteomic profile associated with platelet activation, cell adhesion, and immune responses (Table S2). While this work is exploratory in nature, our proteomics data suggest that extracellular mtDNA is cargoed in EVs that are derived from multiple cellular sources and contain potential mediators of the SSc-ILD disease state.
Table 2.
Differentially expressed proteins by extracellular vesicles between healthy controls and SSc-ILD subjects.
| Protein symbol | Protein name | Healthy control EVs (normalized mean protein abundance) | SSc-ILD EVs (normalized mean protein abundance) | Fold change | Log 2 (fold chanqe) | p-value | False discovery rate (FDR) |
|---|---|---|---|---|---|---|---|
| MBP | Myelin basic protein | 298,699.491 | 18,018.435 | 0.060 | −4.051 | 0.013 | 0.084 |
| S352B | Solute carrier family 35-member E2B | 413,406.705 | 91,711.178 | 0.222 | −2.172 | 0.001 | 0.034 |
| CBPB2 | Carboxypeptidase B2 | 83,389.229 | 23,457.862 | 0.281 | −1.830 | 0.001 | 0.024 |
| IPSP | Plasma serine protease inhibitor | 46,606.330 | 17,172.169 | 0.368 | −1.440 | 0.001 | 0.034 |
| ODF2L | Protein BCAP | 807,909.988 | 369,846.311 | 0.458 | −1.127 | 0.013 | 0.084 |
| TLN1 | Talin-1 | 1,170,682.528 | 620,758.318 | 0.530 | −0.915 | 0.005 | 0.056 |
| C02 | Complement C2 | 544,273.948 | 868,274.262 | 1.595 | 0.674 | 0.013 | 0.084 |
| FIBG | Fibrinogen gamma chain | 447,110,849.500 | 743,745,646.500 | 1.663 | 0.734 | 0.001 | 0.024 |
| FIBB | Fibrinogen beta chain | 584,567,294.000 | 973,307,939.000 | 1.665 | 0.736 | 0.008 | 0.072 |
| FIBA | Fibrinogen alpha chain | 405,289,365.800 | 712,152,418.600 | 1.757 | 0.813 | 0.005 | 0.056 |
| AACT | Alpha-1-antichymotrypsin | 4,417,985.251 | 7,898,641.799 | 1.788 | 0.838 | 0.008 | 0.072 |
| CFAB | Complement factor B | 9,133,598.347 | 18,292,154.820 | 2.003 | 1.002 | 0.003 | 0.052 |
| HV349 | Immunoglobulin heavy variable 3–49 | 317,699.642 | 650,526.223 | 2.048 | 1.034 | 0.013 | 0.084 |
| PROS | Vitamin K-dependent protein S | 6,703,106.172 | 13,836,417.230 | 2.064 | 1.046 | 0.013 | 0.084 |
| IGLC2 | Immunoglobulin lambda constant 2 | 12,194,537.070 | 25,513,033.560 | 2.092 | 1.065 | 0.005 | 0.056 |
| ALBU | Serum albumin | 359,038,924.700 | 752,501,305.100 | 2.096 | 1.068 | 0.005 | 0.056 |
| HV428 | Immunoglobulin heavy variable 4–28 | 231,497.682 | 489,189.708 | 2.113 | 1.079 | 0.003 | 0.052 |
| A1AT | Alpha-1-antitrypsin | 30,200,706.180 | 64,099,387.800 | 2.122 | 1.086 | 0.005 | 0.056 |
| ITIH3 | Inter-alpha-trypsin inhibitor heavy chain H3 | 2,361,005.423 | 5,162,651.549 | 2.187 | 1.129 | 0.001 | 0.024 |
| IGK | Immunoglobulin kappa light chain | 10,529,033.790 | 23,042,556.970 | 2.188 | 1.130 | 0.013 | 0.084 |
| KVD08 | Immunoglobulin kappa variable 1D-8 | 207,857.199 | 456,720.364 | 2.197 | 1.136 | 0.008 | 0.072 |
| IGLC7 | Immunoglobulin lambda constant 7 | 2,439,393.001 | 5,547,344.019 | 2.274 | 1.185 | 0.008 | 0.072 |
| KV401 | Immunoglobulin kappa variable 4–1 | 4,116,927.954 | 9,616,626.861 | 2.336 | 1.224 | 0.008 | 0.072 |
| A1AG1 | Alpha-1-acid glycoprotein 1 | 5,086,722.212 | 12,119,094.780 | 2.382 | 1.252 | 0.008 | 0.072 |
| TRFE | Serotransferrin | 31,797,115.900 | 76,343,237.290 | 2.401 | 1.264 | 0.008 | 0.072 |
| KV108 | Immunoglobulin kappa variable 1–8 | 157,097.465 | 379,683.374 | 2.417 | 1.273 | 0.003 | 0.052 |
| A2MG | Alpha-2-macroglobulin | 994,845,834.600 | 2,474,255,750.000 | 2.487 | 1.314 | 0.013 | 0.084 |
| FINC | Fibronectin | 82,092,581.830 | 253,510,123.300 | 3.088 | 1.627 | 0.001 | 0.024 |
| A1BG | Alpha-1 B-glycoprotein | 1,036,138.623 | 3,210,831.597 | 3.099 | 1.632 | 0.001 | 0.024 |
| A2GL | Leucine-rich alpha-2-glycoprotein | 431,945.295 | 1,441,197.882 | 3.337 | 1.738 | 0.005 | 0.056 |
| HV432 | Immunoglobulin heavy variable 4-30-2 | 40,640.500 | 136,888.997 | 3.368 | 1.752 | 0.013 | 0.084 |
| LV319 | Immunoglobulin lambda variable 3–19 | 366,878.884 | 1,371,913.202 | 3.739 | 1.903 | 0.001 | 0.024 |
| IGHA1 | Immunoglobulin heavy constant alpha 1 | 142,894,367.700 | 591,828,356.700 | 4.142 | 2.050 | 0.013 | 0.084 |
| HV124 | Immunoglobulin heavy variable 1–24 | 161,635.304 | 896,574.250 | 5.547 | 2.472 | 0.001 | 0.034 |
| KVD33 | Immunoglobulin kappa variable 1D-33 | 48,787.643 | 283,167.962 | 5.804 | 2.537 | 0.013 | 0.084 |
| HV364 | Immunoglobulin heavy variable 3–64 | 6,133.476 | 37,883.670 | 6.177 | 2.627 | 0.005 | 0.056 |
| SHBG | Sex hormone-binding globulin | 12,184.601 | 88,977.382 | 7.302 | 2.868 | 0.005 | 0.056 |
| PSPB | Pulmonary surfactant-associated protein B | 1,290.473 | 38,385.290 | 29.745 | 4.895 | 0.001 | 0.024 |
DISCUSSION
Our study lends novel insight into the molecular factors linking fibroblast activation, extracellular mtDNA, and clinical outcomes in SSc-ILD. Specifically, we observed that a ligand for intracellular DNA-sensing PRRs modulates fibroblast activation through αSMA expression and the release of their mtDNA, findings that were particularly pronounced in SSc-ILD-derived fibroblasts. In a clinical correlate of these findings, we found that elevated concentrations of extracellular mtDNA are present in the circulation of SSc-ILD subjects. Its levels are associated with ventilatory decline, potentially mediated through the activation of TLR9 and cGAS-STING pathways resulting in the downstream production of Type I interferons and IL-6. The extracellular mtDNA appears to be shuttled in EVs and not heavily oxidized. These findings demonstrate an unrecognized connection between mtDNA, extracellular vesicles, intracellular DNA-sensing PRR activation, and clinical outcomes in SSc-ILD. These findings have the potential to be relevant to the presently studied scleroderma-related lung disease, but also in other TLR9 or cGAS sting related conditions including but not limited to acute respiratory distress syndrome [32], tuberculosis [33], and systemic lupus erythematosus [34].
Through our robust translational pipeline, we found that stimulation with a ligand for cytoplasmic DNA-sensing PRRs modulates fibroblast activation and accumulation of extracellular mtDNA. One potential explanation for the latter observation may be a shift towards aerobic glycolysis and mitochondrial dysfunction, which has been reported in other forms of pulmonary fibrosis [18, 35–37] but not yet in SSc-ILD. Another explanation could be nonspecific release during necroptosis [13]. Whatever the mechanism, our findings indicate that excessive local concentrations of mtDNA in the lung may act upon fibroblasts and is associated with the production of αSMA. In a direct clinical translation of our in vitro studies, we found that enhanced concentrations of circulating mtDNA are outcome-predictive in a moderately sized SSc-ILD cohort, indicating our work’s clinical impact. Specifically, most currently available prognostic algorithms employ clinical factors to predict outcomes [38], and the use of circulating biomarkers to predict FVC decline is less well understood. Therefore, the identification of an easily measurable and reproducible mechanistic biomarker would be of great benefit to SSc-ILD patients and the clinicians who care for them.
Another novel finding of our study is that plasma mtDNA is shuttled in EVs. A growing body of work has described the significance of EVs in various ILDs, including rheumatoid arthritis-associated ILD [39], sarcoidosis [40], and idiopathic pulmonary fibrosis [16]. Moreover, recent work has demonstrated that EVs, specifically platelet-derived EVs, can convey mitochondrial DAMPS in the circulation [41]. While we were unable to precisely determine the cell of origin of the EVs found in SSc-ILD plasma due to technical limitations [42], our proteomics data suggest a significant contribution from platelets. Further investigation into the cellular source of these circulating EVs and how their extracellular mtDNA cargo mediates innate immunity in SSc-ILD could reveal a convergent molecular pathway linking divergent clinical states.
Perhaps the most exciting aspect of our work is the finding that plasma mtDNA is associated with the activation of both TLR9 and cGAS, two potent PRRs of the innate immune system. TLR9 signaling proceeds through the adaptor myeloid differentiation primary response protein 88 (MYD88), which then activates interferon regulatory factor 7 (IRF7) to enhance Type I IFN and cytokine responses [12]. Moreover, crosstalk signaling within the TLR family of PRRs has been shown to augment these responses [43]. Recent studies demonstrated the sequential activation of TLR9 and TLR3, which recognizes double-stranded RNA [44], as well as TLR4, which is activated by lipopolysaccharide components of gram-negative bacteria [45]. Both TLR3 [46] and TLR4 [47] have been implicated in fibroblast activation in SSc. In addition, we observed parallel activation of the cGAS-STING pathway in which the DNA-sensor cGAS catalyzes the synthesis of the cyclic dinucleotide cGAMP, which binds and activates STING [17]. The interaction between the TLR9 and cGAS-STING pathways has yet to be fully understood. Some studies have demonstrated their synergistic relationship, as parallel in vitro activation of TLR9 and cGAS-STING resulted in the augmented production of IFNs [48]. However, other studies have shown a sequential interaction between the two pathways, in which TLR9 signaling subsequently induces cGAS-STING activation [49]. Studies into host defenses against cutaneous Staphylococcus aureus infection additionally revealed an antagonist relationship between these two PRRs [50]. Additional work is required to characterize whether TLR9 works independently of or in concert with cGAS-STING in SSc-ILD.
While our results are promising, they are by no means definitive. Understanding whether extracellular mtDNA mediates fibroblast activation in SSc-ILD will require additional work with matched bronchoalveolar lavage and plasma samples in multiple cohorts. Our work is also limited by not having fully characterized the features of extracellular mtDNA, such as the presence of damage to its genome. While our data suggests that it is not heavily oxidized, it may possess other types of DNA damage that influence its biological importance in the chronic disease state. We have also not explored whether a connection exists between mtDNA and other PRR-associated [51] predictors of physiologic progression in SSc-ILD, such as miR155 [52] and CXCL4 [53]. Lastly, while our work implicates extracellular mtDNA present in the SSc-ILD plasma as an agonist for TLR9 and cGAS-STING, studies involving knockdown and overexpression strategies are required to fully characterize these interactions. However, despite these relatively minor limitations, our study yields novel insight into the relationship between intracellular DNA-sensing PRRs and their circulating endogenous ligand mtDNA in SSc-ILD. Additional studies in this realm may further our understanding of this difficult to treat disease.
Supplementary Material
ACKNOWLEDGEMENTS
We kindly thank Dr. Glenda Trujillo (Bristol-Myers-Squibb) for her intellectual contributions to this manuscript, and Dr. Carol Feghali-Bostwick (Medical University of South Carolina) for providing us with scleroderma fibroblasts. We highly appreciate the efforts of Donna Carrano (Yale University School of Medicine) in recruiting study subjects. Most of all, we are very grateful to all study participants for generously providing their time and plasma to this study.
Funding:
CR was supported by grants from the Parker B. Francis Foundation and the Foundation for Sarcoidosis Research. HS was supported by grants from the Scleroderma Foundation and the American Thoracic Society. JLG was supported by K01HL125474-03 grant. ELH was supported by R01HL15267701, R01HL109233, R01HL125850, and U01HL112702 grants, and grants from the Gabriel and Alma Elias Research Fund and the Greenfield Foundation. Proteomics data were collected using mass spectrometry supported by NIH SIG S10ODOD018034-01. The authors have no relevant financial conflicts of interest to disclose.
REFERENCES
- 1.Denton CP and Khanna D, Systemic sclerosis. The Lancet, 2017. 390(10103): p. 1685–1699. [DOI] [PubMed] [Google Scholar]
- 2.Solomon JJ, et al. , Scleroderma lung disease. European Respiratory Review, 2013. 22(127): p. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackintosh JA, et al. , Systemic Sclerosis Associated Interstitial Lung Disease: A Comprehensive Overview. Semin Respir Crit Care Med, 2019. 40(02): p. 208–226. [DOI] [PubMed] [Google Scholar]
- 4.Herzog EL, et al. , Interstitial lung disease associated with systemic sclerosis and idiopathic pulmonary fibrosis: How similar and distinct? Arthritis Rheumatol, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goh NS, et al. , Short-Term Pulmonary Function Trends Are Predictive of Mortality in Interstitial Lung Disease Associated With Systemic Sclerosis. Arthritis & Rheumatology, 2017. 69(8): p. 1670–1678. [DOI] [PubMed] [Google Scholar]
- 6.Elhai M, et al. , Performance of Candidate Serum Biomarkers for Systemic Sclerosis–Associated Interstitial Lung Disease. Arthritis & Rheumatology, 2019. 71(6): p. 972–982. [DOI] [PubMed] [Google Scholar]
- 7.Gilbane AJ, Denton CP, and Holmes AM, Scleroderma pathogenesis: a pivotal role for fibroblasts as effector cells. Arthritis research & therapy, 2013. 15(3): p. 215–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang F, et al. , Toll-like Receptor 9 Signaling Is Augmented in Systemic Sclerosis and Elicits Transforming Growth Factor beta-Dependent Fibroblast Activation. Arthritis Rheumatol, 2016. 68(8): p. 1989–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Artlett CM, et al. , The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum, 2011. 63(11): p. 3563–74. [DOI] [PubMed] [Google Scholar]
- 10.Christmann RB, et al. , Association of Interferon- and transforming growth factor β-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis & rheumatology (Hoboken, N.J.), 2014. 66(3): p. 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellson CD, et al. , Danger-associated molecular patterns and danger signals in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol, 2014. 51(2): p. 163–8. [DOI] [PubMed] [Google Scholar]
- 12.West AP and Shadel GS, Mitochondrial DNA in innate immune responses and inflammatory pathology. Nature Reviews Immunology, 2017. 17: p. 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeda A and Fadeel B, Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis, 2014. 5: p. e1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Martinez I, et al. , Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. The Journal of Clinical Investigation, 2016. 126(3): p. 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McArthur K, et al. , BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science, 2018. 359(6378). [DOI] [PubMed] [Google Scholar]
- 16.Martin-Medina A, et al. , Increased Extracellular Vesicles Mediate WNT5A Signaling in Idiopathic Pulmonary Fibrosis. American Journal of Respiratory and Critical Care Medicine, 2018. 198(12): p. 1527–1538. [DOI] [PubMed] [Google Scholar]
- 17.Roers A, Hiller B, and Hornung V, Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity, 2016. 44(4): p. 739–754. [DOI] [PubMed] [Google Scholar]
- 18.Ryu C, et al. , Extracellular Mitochondrial DNA is Generated by Fibroblasts and Predicts Death in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryu C, et al. , Plasma mitochondrial DNA is associated with extrapulmonary sarcoidosis. European Respiratory Journal, 2019. 54(2): p. 1801762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krychtiuk KA, et al. , Mitochondrial DNA and Toll-Like Receptor-9 Are Associated With Mortality in Critically Ill Patients. Crit Care Med, 2015. 43(12): p. 2633–41. [DOI] [PubMed] [Google Scholar]
- 21.van den Hoogen F, et al. , 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum, 2013. 65(11): p. 2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirillov V, et al. , Sustained activation of toll-like receptor 9 induces an invasive phenotype in lung fibroblasts: possible implications in idiopathic pulmonary fibrosis. Am J Pathol, 2015. 185(4): p. 943–57. [DOI] [PubMed] [Google Scholar]
- 23.Trujillo G, et al. , TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci Transl Med, 2010. 2(57): p. 57ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thannickal VJ, et al. , Matrix biology of idiopathic pulmonary fibrosis: a workshop report of the national heart, lung, and blood institute. Am J Pathol, 2014. 184(6): p. 1643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakahira K, et al. , Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med, 2013. 10(12): p. e1001577; discussion e1001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du Y, et al. , Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nature medicine, 2016. 22(2): p. 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.An J, et al. , Cutting edge: Antimalarial drugs inhibit IFN-beta production through blockade of cyclic GMP-AMP synthase-DNA interaction. J Immunol, 2015. 194(9): p. 4089–93. [DOI] [PubMed] [Google Scholar]
- 28.Motwani M, Pesiridis S, and Fitzgerald KA, DNA sensing by the cGAS–STING pathway in health and disease. Nature Reviews Genetics, 2019. 20(11): p. 657–674. [DOI] [PubMed] [Google Scholar]
- 29.Boudreau LH, et al. , Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood, 2014. 124(14): p. 2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McVey MJ, et al. , Extracellular Vesicles in Lung Health, Disease, and Therapy: Mini-Review and Special Call for papers. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2019. [DOI] [PubMed] [Google Scholar]
- 31.Hamilton ML, et al. , A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA. Nucleic acids research, 2001. 29(10): p. 2117–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Q, et al. , Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature, 2010. 464(7285): p. 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pahari S, et al. , Bolstering Immunity through Pattern Recognition Receptors: A Unique Approach to Control Tuberculosis. Frontiers in Immunology, 2017. 8: p. 906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caielli S, et al. , Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. The Journal of experimental medicine, 2016. 213(5): p. 697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bueno M, et al. , PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest, 2015. 125(2): p. 521–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kottmann RM, et al. , Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am J Respir Crit Care Med, 2012. 186(8): p. 740–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie N, et al. , Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am J Respir Crit Care Med, 2015. 192(12): p. 1462–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volkmann ER, et al. , Development of a Composite Outcome Measure for Systemic Sclerosis Related Interstitial Lung Disease. Rheumatology (Sunnyvale), 2015. 5(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Withrow J, et al. , Extracellular vesicles in the pathogenesis of rheumatoid arthritis and osteoarthritis. Arthritis research & therapy, 2016. 18(1): p. 286–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qazi KR, et al. , Proinflammatory exosomes in bronchoalveolar lavage fluid of patients with sarcoidosis. Thorax, 2010. 65(11): p. 1016. [DOI] [PubMed] [Google Scholar]
- 41.Marcoux G, et al. , Platelet-derived extracellular vesicles convey mitochondrial DAMPs in platelet concentrates and their levels are associated with adverse reactions. Transfusion, 2019. 59(7): p. 2403–2414. [DOI] [PubMed] [Google Scholar]
- 42.Nolan JP and Duggan E, Analysis of Individual Extracellular Vesicles by Flow Cytometry, in Flow Cytometry Protocols, Hawley TS and Hawley RG, Editors. 2018, Springer New York: New York, NY. p. 79–92. [DOI] [PubMed] [Google Scholar]
- 43.Tan RST, et al. , TLR cross-talk confers specificity to innate immunity. International reviews of immunology, 2014. 33(6): p. 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitmore MM, et al. , Synergistic Activation of Innate Immunity by Double-Stranded RNA and CpG DNA Promotes Enhanced Antitumor Activity. Cancer Research, 2004. 64(16): p. 5850. [DOI] [PubMed] [Google Scholar]
- 45.De Nardo D, et al. , Signaling Crosstalk during Sequential TLR4 and TLR9 Activation Amplifies the Inflammatory Response of Mouse Macrophages. The Journal of Immunology, 2009. 183(12): p. 8110. [DOI] [PubMed] [Google Scholar]
- 46.Agarwal SK, et al. , Toll-like receptor 3 upregulation by type I interferon in healthy and scleroderma dermal fibroblasts. Arthritis research & therapy, 2011. 13(1): p. R3–R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhattacharyya S, et al. , TLR4-dependent fibroblast activation drives persistent organ fibrosis in skin and lung. JCI insight, 2018. 3(13): p. e98850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Temizoz B, et al. , TLR9 and STING agonists synergistically induce innate and adaptive type-II IFN. European journal of immunology, 2015. 45(4): p. 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu R-H, et al. , Sequential Activation of Two Pathogen-Sensing Pathways Required for Type I Interferon Expression and Resistance to an Acute DNA Virus Infection. Immunity, 2015. 43(6): p. 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scumpia PO, et al. , Opposing roles of Toll-like receptor and cytosolic DNA-STING signaling pathways for Staphylococcus aureus cutaneous host defense. PLoS pathogens, 2017. 13(7): p. e1006496–e1006496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu F, et al. , TLR9-induced miR-155 and Ets-1 decrease expression of CD1d on B cells in SLE. Eur J Immunol, 2015. 45(7): p. 1934–45. [DOI] [PubMed] [Google Scholar]
- 52.Christmann RB, et al. , miR-155 in the progression of lung fibrosis in systemic sclerosis. Arthritis Res Ther, 2016. 18(1): p. 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Bon L, et al. , Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med, 2014. 370(5): p. 433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.