

Abstract
Prof. Geoffrey Burnstock originated the concept of purinergic signaling. He demonstrated the interactions and biological roles of ionotropic P2X and metabotropic P2Y receptors. This review paper traces the historical origins of many currently used antagonists and agonists for P2 receptors, as well as adenosine receptors, in early attempts to identify ligands for these receptors – prior to the use of chemical libraries for screening. Rather than presenting a general review of current purinergic ligands, we focus on common chemical scaffolds (privileged scaffolds) that can be adapted for multiple receptor targets. By carefully analyzing the structure activity relationships, one can direct the selectivity of these scaffolds toward different receptor subtypes. For example, the weak and non-selective P2 antagonist reactive blue 2 (RB-2) was derivatized using combinatorial synthetic approaches, leading to the identification of selective P2Y2, P2Y4, P2Y12 or P2X2 receptor antagonists. A P2X4 antagonist NC-2600 is in a clinical trial, and A3 adenosine agonists, show promise for chronic pain. P2X7 antagonists have been in clinical trials for depression (JNJ-54175446), inflammatory bowel disease (IBD), Crohn’s disease, rheumatoid arthritis, inflammatory pain and chronic obstructive pulmonary disease (COPD). P2X3 antagonists are in clinical trials for chronic cough, and an antagonist named after Burnstock, gefapixant, is expected to be the first P2X3 antagonist filed for approval. We are seeing that the vision of Prof. Burnstock to use purinergic signaling modulators, most recently at P2XRs, for treating disease is coming to fruition.
Keywords: P2X receptor, P2Y receptor, adenosine receptor, purinergic agonist, purinergic antagonist, scaffold
Graphical Abstract
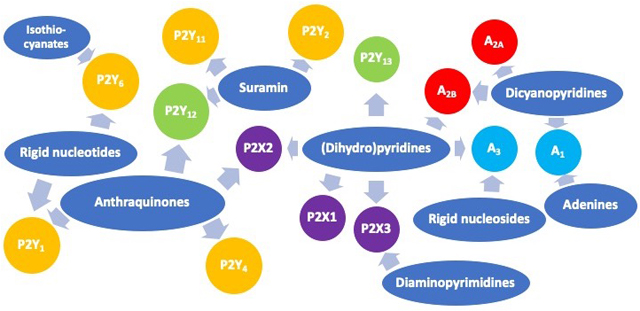
Decades-long exploration of structure-activity relationships of known P2/adenosine ligand scaffolds and eventually novel scaffolds has directed the selectivity toward various receptor subtypes, leading to receptor probes and clinical drugs.
1. Introduction
This paper is dedicated to the memory of our inspirational friend and colleague Prof. Geoffrey Burnstock (1929–2020), who began the field of purinergic signaling and guided it to ever greater achievements throughout his long, active research career of roughly six decades. He discovered non-adrenergic, non-cholinergic nerves and found their neurotransmitter to be ATP [1]. Although not a medicinal chemist himself, he recognized the need for tool compounds in order to adequately characterize a receptor, and especially because his own proposal of purinergic receptors [1,2] was initially met with skepticism and even hostility. In 1976, Burnstock addressed the reluctance of biologists to accept his concepts of co-transmission (ATP coreleased with other transmitters), and purinergic signaling in general, by stating: “Gifted and meticulous workers will perform remarkable contortions to fit their data into accepted dogma, especially if established by powerful and brilliant personalities at the forefront of the field. They will often dismiss or ignore data that fall outside interpretation by current theory, searching hard for technical or artefactual explanations. Once a new attitude becomes acceptable, then the same data can be miraculously redeployed to support it.” [3]. Burnstock’s concepts proposed in the early 1970s were eventually fully accepted, but only after two decades.
Thus, selective agonists and antagonists that could distinguish effects of extracellular ATP (Figure 1A) in different tissues, e.g. inhibitory vs. stimulatory effects on smooth muscle, were desperately needed to validate the concept of purinergic receptors prior to their cloning in the 1990s. Eventually it became clear that there were two families of purinergic P2 receptors, eight P2Y (GPCRs, numbered 1, 2, 4, 6, 11, 12, 13 and 14) and seven P2X (ligand-gated ion channels, numbered 1–7) receptors [4–6]. Burnstock’s early studies with ATP as a neurotransmitter included a search for P2 receptor antagonists, by analogy to competitive antagonists, theophylline and other alkylxanthines, at the adenosine (P1) receptors. He was limited to available chemicals, and the first compounds he and others discovered to block ATP effects were polyanionic compounds, including organic dyes, or a peptide (apamin, later found to inhibit P2Y-downstream small conductance calcium-activated potassium channels instead of P2 receptors) that only weakly antagonized. Although initially considered difficult scaffolds for determining the structure-activity relationship (SAR) at P2 receptors, much later some of the chemical scaffolds, so-called “privileged structures” explored by Geoff Burnstock as P2 antagonists were re-engineered chemically for higher affinity and selectivity. Thus, we have chosen to focus in this review paper on common chemical scaffolds that can often be adapted for multiple purinergic receptor targets. We wished to provide a different perspective in this review on the topic of purine medicinal chemistry, which has been written about in elaborate detail many times in the past few years [6–14]. We provide a sampling of the more important structures in each class, focusing on both history and the cutting edge, especially for the P2X receptors.
Figure 1.
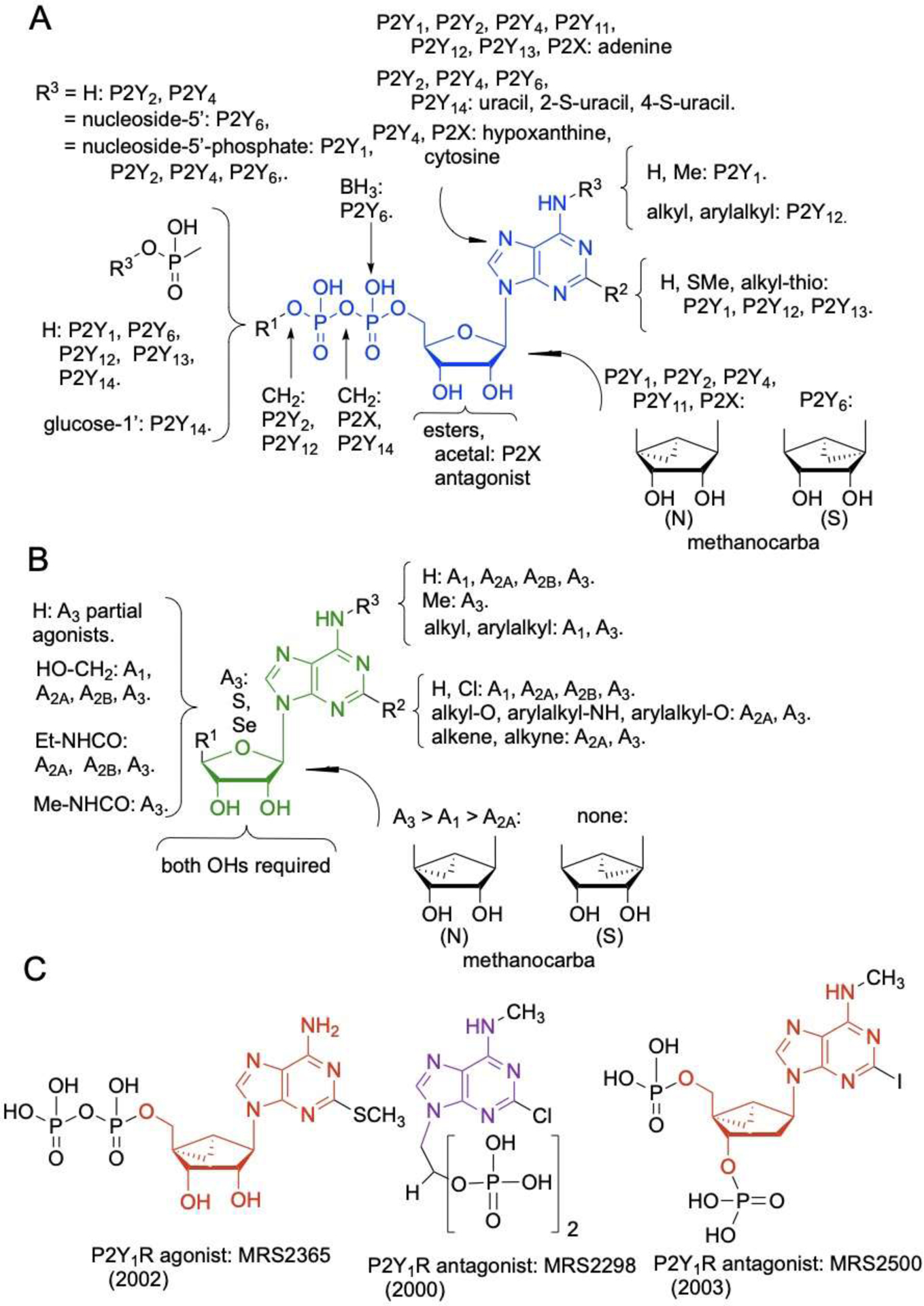
Generalized SAR of the native scaffolds of the P2 (A, C) and adenosine (B) receptors. Note that there is often a complex relationship between multiple modifications. It is necessary to consult detailed original publications and reviews for individual structures [5–6–8,10–12,51,58,60,66,93–96]. The notations indicate general effects of the chemical substitution shown (although not applicable in all cases of the same substitution). For example, in part A: “R3=H: P2Y2, P2Y4” means a H at this position is recognized by P2Y2 and P2Y4 receptors. Scaffold backbone structures are colored as follows: blue, ADP; green, 4′-truncated adenosine; red, (N)-methanocarba adenosine or deoxyadenosine; violet, 9-alkyladenine. The effects, on receptor affinity and selectivity, of substitution at different positions of the same scaffold are often interdependent. In some cases, agonists were converted into antagonists by specific structural changes. Years shown for each compound indicate an early, major report in the literature.
Molecular scaffolds are used for multiple applications in medicinal chemistry [15], including establishing molecular hierarchies, their association with diverse biological activities, transferring biological activity at a given target to a similar scaffold (scaffold hopping) and their use as fragments in higher order ligand structures. Such scaffold databases now can serve the purposes of artificial intelligence (AI) approaches to drug discovery and prediction of absorption, distribution, metabolism, excretion, toxicity (ADMET) properties [16]. Even by empirical trial-and-error probing, a relatively nonselective lead compound that binds to multiple subtypes within a receptor family can be modified to be more selective for a subset within that family.
In addition to optimizing the affinity of common scaffolds and other receptor ligands by studying their SAR, medicinal chemists often have as a goal to discover the most selective compounds achievable. However, for some applications drugs of dual or multiple activities would be desirable, especially when there is additivity or synergism of the two or more pharmacological effects (so-called polypharmacology). Thus, by comparing the SAR of a given scaffold at multiple receptors, one can in some cases combine pharmacological activities in a therapeutically useful manner by fusing or tethering pharmacophores [17,18].
2. Early attempts to identify P2R agonists and antagonists
The P2 receptor selectivity of nucleotide agonists was explored on an ATP/ADP scaffold (Figure 1A), and the difference in order of potency at each of the tissues (and later defined subtypes) provided an initial pharmacological toolset for defining the P2 receptors prior to their complete cloning [19]. For example, there were different agonist potency orders at the inhibitory taenia coli P2 receptor, later shown to correspond to the P2Y1R, compared to the excitatory guinea pig ileum receptor, which corresponds to the P2X1R [20]. However, the in situ instability of ATP analogues to rapid phospho-ester hydrolysis and the potentially opposing effect of the breakdown product adenosine (Figure 1B) that is produced, along with its selective uptake, are all factors leading to Burnstock’s statement that the “true order of potency may be obscured in isolated organ experiments.” [20].
Nevertheless, P2 agonist SAR analysis begun in the 1970s was largely confirmed in later studies. It was clear that substitution of the adenine 2-position of ATP/ADP with a methylthio or chloro group increased potency selectively at the taenia coli (P2Y) receptor [19,20]. On the other hand, α,β-methylene-ATP and β,γ-methylene-ATP, containing more stable phosphonate bridges, were selective activators of the vas deferens and urinary bladder (P2X) receptor of guinea-pig and rat [4,19]. Also, modest stereoselectivity in agonist SAR, a hallmark of receptor action, was observed, especially at P2Y receptors [21]. Stereoisomers of several ATP analogues were used to further classify the P2 purinoceptors [4]. The chirality was compared either in the form of a non-natural (mirror image) L-ribose moiety or introduced as a non-bridging sulfur atom on the α-phosphate group of ATP or ADP. Of the latter, the Rp diastereoisomers were more potent than the corresponding Sp isomer at the taenia coli P2Y but not bladder P2X receptor [21,22]. The L-nucleotides were weaker as P2 agonists than the D-ribose-containing native nucleotides at the taenia coli and vas deferens, but not bladder P2 receptor (all guinea pig) [4]. Early, validated P2 receptor antagonists were an important addition to the P2 receptor ligand toolset beginning in the late 1980s. Burnstock not only studied compounds that activate or inhibit purinergic signaling, but also compounds that potentiate the actions of purinergic agents by inhibiting their enzymatic breakdown or uptake [1], which now relates to the emerging area of ecto-nucleotidase inhibitors for cancer and other diseases [23,24]. Inhibitors of adenosine kinase, adenosine deaminase and nucleoside transporters raise endogenous adenosine levels [25].
The first P2 receptor antagonists were mainly of high molecular weight and not typical drug-like molecules. Suramin (Figure 2A) is a trypanocidal drug developed during World War I from colorless intermediates used in dye synthesis [26]. Based on a hint from its inhibition of intracellular Na-K-ATPase, suramin was tested against another activity involving ATP and was found to antagonize P2X (later shown to be P2X1) receptor-induced contraction of the vas deferens in 1988 [27]. Later, suramin was shown to be an ATP antagonist in neuronal-derived cells (PC12 cells, from which the P2X2 receptor was later cloned [28]) as well as other smooth muscle cells, including the guinea pig taenia coli [29, 30]. Suramin was a more potent antagonist at the bladder smooth muscle P2X receptor (i.e. P2X1) compared to the PC12 cell P2X receptor (i.e. P2X2) [31].
Figure 2.
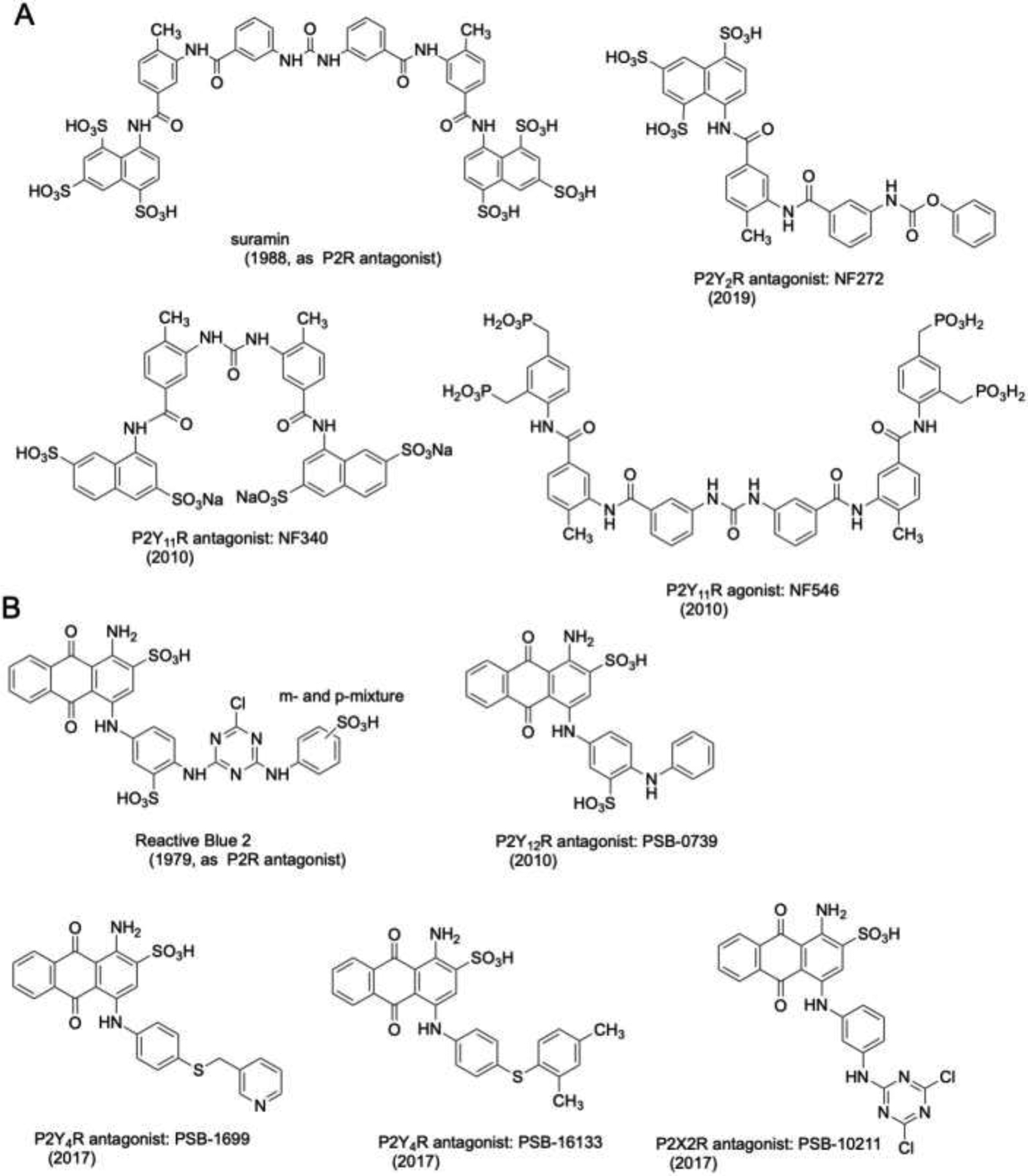
Early-identified large scaffolds for P2R antagonists, suramin (A) and Reactive Blue 2, an anthraquinone (B). Affinities of many these compounds and their original references are collected [6,66–80]. Years shown for each compound indicate an early, major report in the literature.
Following a proceedings abstract in 1979, Reactive Blue 2 (RB2) (Figure 2B) was shown to be a moderately potent antagonist of P2YRs [32,33] and P2XRs [34]. There was ambiguity about the structure of the RB2 family of dyes, which was finally analyzed by Glänzel et al. [31] to show that the para-substituted isomer held the desired P2Y receptor activity. High concentrations of the early P2 receptor antagonists were often used, for example 10–100 μM of RB2 and suramin [27,35,36]. In addition to some of the dyes coloring tissue preparations, most of these early P2 receptor antagonists were promiscuous in their interaction with diverse proteins [37] and, thus, were of limited use in studying P2 receptors pharmacologically. For example, other actions of suramin include inhibition of protein-tyrosine phosphatases, sirtuins and reverse transcriptase [27,38]. RB2 is also an inhibitor of glutathione S-transferase and protein kinases [39,40]. Therefore, more definitive P2 receptor ligands were evidently needed - but it took decades after the early phases of Burnstock’s work to achieve this goal, and many of the P2X and P2Y receptors still lack potent and selective agonists and antagonists.
PPADS (pyridoxal phosphate 6-azophenyl-2′,4′-disulfonic acid, Figure 3A) was first reported in 1992 by Lambrecht and coworkers as a P2X receptor antagonist [41] and soon thereafter used by Burnstock’s group to characterize P2X receptors in the rabbit urinary bladder [42]. PPADS was also shown to block P2Y1 but not P2Y12 receptors [28]. However, PPADS and some derivatives were later found to inhibit NTPDase1 (ecto-apyrase, CD39) and NTPDase2 (ecto-ATPase, CD39L1) [43] that convert ATP/UTP to AMP/UMP, which complicates their use as tool compounds at P2 receptors.
Figure 3.
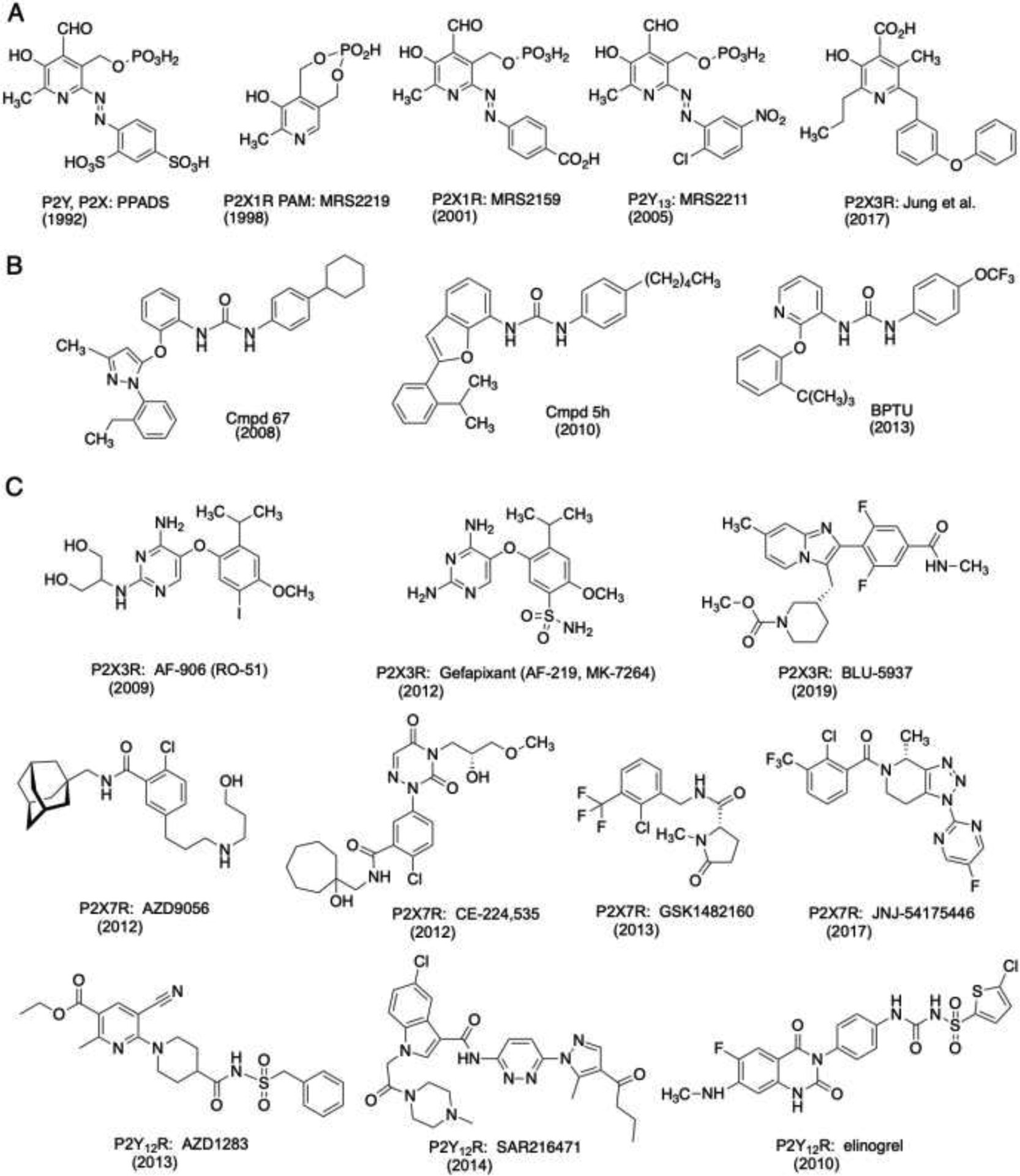
Small scaffolds for P2R antagonists, including PAMs and NAMs related to pyridoxal phosphate (A), N,N′-diarylureas that allosterically antagonize P2Y1R (B) and various P2X3R, P2X7R and P2Y12R (C) antagonists. Gefapixant, BLU-5937, AZD9056, CE-224,535, GSK1482160 and JNJ-54175446 have been in clinical trials [66–68,116–120]. The pyridoxal phosphates are known to interact with various P2X and P2Y receptors as antagonists or PAMs, while the diarylureas are specific for P2Y1R. This specificity is a function of their unusual binding site on the outer surface of the receptor where the urea forms a bidentate H-bond with a backbone carbonyl group. Affinities of many these compounds and their original references are collected [6, 66–68,116–120]. Years shown for each compound indicate an early, major report in the literature.
3. Subsequently analyzed SAR of early P2 ligand scaffolds
3.1. P2 Receptor Agonists.
ATP was first noted by Burnstock and colleagues to contract vas deferens and bladder smooth muscles by activating what were later categorized as P2X receptors [4]. However, P2Y receptors were initially described by Burnstock as “inhibitory” due to ATP’s inducing relaxation of the taenia coli (gastrointestinal inhibitory junction potential [44]) and the vasculature through endothelial cells, later designated as P2Y1 receptor effects [4]. Now it is evident that five of the P2Y receptor subtypes (1, 2, 4, 6 and 11) couple to Gq protein, the P2Y11 receptor in addition to Gs, while the remaining three subtypes couple to Gi. Although Gq-coupled receptors may be stimulatory in neurons and astrocytes in the brain, e.g. by inducing Ca2+ waves, they can also be inhibitory [45]. For example, various presynaptic Gq-coupled P2Y receptors inhibit the release of neurotransmitters such as glutamate [46].
3.1.1. Mononucleotides.
The high P2 receptor potency when a 2-alkylthio group was installed on the ATP scaffold (Figure 1A) was first discovered in 1972 by Gough et al. [22,47] and later elaborated chemically by Cusack and Hourani for activation of P2Y1 and P2Y12 receptors [21, 22] and by Jacobson and coworkers [48]. The series of extended 2-alkylthio derivatives of ATP, ADP and AMP, based originally on 2-MeS-ATP, showed high affinity at P2 receptors, particularly P2Y1R and P2Y12R [49]. The substitution of a ring-constrained bicyclic system for ribose, known as (N)-methanocarba (bicyclo[3.1.0]hexane), introduced P2Y1R selectivity in potent agonist MRS2365 (Figure 1C) [50]. The SAR progression of 2-alkylthio-ATP analogues at the P2Y12R led eventually to a 5′-triphosphate analogue having a β,γ-dichloromethylene bridge for stability, i.e. cangrelor (AR-C69931MX) [51], as an approved antithrombotic drug, and even to an uncharged, potent P2Y12R antagonist, as the approved drug ticagrelor (AZD6140, a nucleoside analogue) [51].
Novel uracil mononucleotides as P2Y2,4,6 receptor agonists have been introduced [52,53]. This followed the discovery that UTP (Figure 1A) and its analogues induced P2Y-like activities, which was later confirmed with the cloning of these receptors beginning with the mouse P2Y2 receptor from neuroblastoma cells [54]. Nucleotides with alternative nucleobases have been shown to bind to P2Y and P2X receptors [55,56]. Efforts to chemically stabilize the di- or triphosphate moieties of P2R agonists include the introduction of a chiral boranophosphate moiety, for example in an analogue of P2Y6R agonist UDP [57].
3.1.2. Dinucleotides.
The P2 receptors can accommodate dinucleotides as well as mononucleotides [58], and an extracellular dinucleoside tetraphosphate (Up4A) is an endothelial-derived transmitter substance [59]. Each P2R subtype has a different preferred number of phosphate units in the chain [58], e.g. Np3N (where N is a nucleoside) for P2Y6R [52] and Np4N for P2XRs, P2Y2R and P2Y4R [10,58,60]. Other examples of dinucleotides that are useful P2R pharmacological probes are: Ip5I (P2X1 [61]), Ap4A (P2Y12), Up4U (P2Y2,4) and MRS2957 (P2Y6) [53]. Ap4A analogues with defined stereochemistry have also been studied as dual P2Y1R/P2Y12R antagonists [17]. Dinucleotide Up4U (diquafosol, a long-acting P2Y2R agonist) is approved for treating dry eye syndrome in Japan and Korea, while its dinucleotide analogue denufosol failed to receive FDA approval after multiple clinical trials for cystic fibrosis [62]. Diquafosol, used as a 3% ophthalmic solution for treating dry eye, increases secretion from the meibomian glands in the eyelid [63].
3.1.3. Nucleotide sugars.
UDP-glucose potently activates the P2Y14R and acts as a damage associated molecular pattern (DAMP) in various inflammatory conditions [64]. Thus, a series of P2Y14R antagonists based on well-explored naphthalene and phenyl-triazole scaffolds have shown efficacy in models of inflammation, chronic neuropathic pain and asthma [64,65]. Other UDP-sugars are less potent than UDP-glucose, and UDP also acts as a P2Y14R agonist or partial agonist [11,58].
3.2. P2 Antagonists.
In recent years, the discovery of subtype-selective P2YR and P2XR antagonists has accelerated [6,9,10]. Novel scaffolds have been discovered that can be directed toward multiple P2R subtypes by appropriate derivatization [6,66]. Nevertheless, as described here, earlier scaffolds have also been useful in the discovery of P2R antagonists (Figure 2A). Multiple studies have shown that weak, non-drug-like, nonselective P2 receptor antagonists, such as RB-2, can still be utilized as a starting point leading to potent receptor subtype-selective compounds.
3.2.1. Suramin analogues.
A tetraphosphonate analogue related to antagonist suramin (Figure 2A), NF546, acts as a selective P2Y11R agonist, which is an example of using a common scaffold both to shift to a different receptor subtype and to change the pharmacological efficacy [67]. A suramin derivative, NF272, was identified as a competitive P2Y2 receptor antagonist, although it was <10-fold selective compared to P2Y2 and P2Y6 and at least as potent at P2Y1, P2Y11 and P2Y12 [68]. NF272 also blocks P2X1R. The suramin derivative NF770 is a relatively potent and selective P2X2R antagonist, which was proposed to display a competitive mechanism of action [69].
Recently, suramin-derived benzenesulfonates, benzenesulfonamides and related derivatives were found to act as P2Y2R antagonists with ancillary inhibitory activity at the related orphan GPCR GPR17. Dual inhibitors of these proinflammatory receptors may be advantageous for the therapy of inflammatory diseases [70].
3.2.2. RB2 analogues.
The group of August W. Frahm was the first to synthesize RB-2 analogs (anthraquinone derivatives) with the aim of optimizing their interactions with P2Y receptor subtypes. MG 50–3-1 (structure not shown) was reported to be a nanomolar antagonist at the P2Y1-like receptor of guinea-pig taenia coli, showing only weak activity at the P2X1 receptor in rat vas deferens [71]. Subsequently, an improved synthetic procedure was developed for MG 50–3-1 to provide sufficient amounts of the compound for additional experiments [72]. At the human P2Y1R it was weaker (IC50 350 nM) than at the guinea pig preparation and also blocked the human P2Y4R (153 nM).
Broad, systematic SAR analysis of truncated RB-2 derivatives became possible by the development of combinatorial synthetic approaches [73] and, later on, by an improved Ullmann coupling reaction of bromo-substituted anthraquinone derivatives and amines/anilines [74,75]. This led to the identification of antagonists with selectivity for P2Y2, P2Y4, P2Y12 or P2X2 receptors. The highly potent and selective P2Y12R antagonist PSB-0739 (Figure 2B) showing low nano- to subnanomolar potency has become a useful pharmacological tool [76,77]. For example, this antagonist was used to demonstrate that blocking the P2Y12 receptor in the spinal cord reduces chronic pain and cytokine production [78]. Mutagenesis and docking studies confirmed that it acts as a competitive antagonist [77,79]. Recently, the first P2Y4-selective antagonists, namely the anthraquinone derivatives PSB-1699 and PSB-16133 (Figure 2B), were reported [80]. Mutagenesis and molecular modeling studies, based on X-ray structures of related GPCRs, defined their P2Y4R binding sites and interactions [81–83]. Optimization of anthraquinone derivatives related to RB-2 also led to potent, selective P2X2R antagonists (PSB-10211 and PSB-1011) and positive allosteric modulators (e.g. PSB-10129) [84].
3.2.3. PPADS and other pyridine analogues.
Aryl-diazo-bridged pyridoxal phosphate derivative PPADS serves as a versatile P2 receptor antagonist scaffold, and its binding region on a P2X1 receptor homology model was recently characterized [85]. PPADS analogues (Figure 3) have been developed as useful research tools with selectivity for various P2 receptor subtypes, although they contain several non-druglike structural features (aryl-azo, aldehyde and phosphate ester groups) [41,42,86–89]. Analogues of PPADS have been applied to antagonize both P2YRs and P2XRs. MRS2211 is a selective P2Y13R antagonist. PPADS analogues were found by mutagenesis to occupy the same binding region of the P2Y1R as nucleotide antagonists, and the extracellular loops participate in the ligand selectivity in comparison to the P2Y6R [82]. A P2X1 receptor positive allosteric modulator (PAM) MRS2219 was derived from a PPADS scaffold [90]. High affinity at P2X3R was also found for the PPADS scaffold [89]. Recently, a PPADS analogue lacking azo, aldehyde and phosphate groups was found to relieve pain in the mouse by blocking the P2X3 receptor (Figure 3A) [84]. Dihydropyridines (including nicardipine) and related anionic 5-phosphonate analogues were found to antagonize the rat P2X2R and P2X4R [91].
3.2.4. DIDS analogues.
The P2Y receptor antagonism by the irreversible chloride channel blocker DIDS (4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid) was used as a lead compound for several reactive and sparingly water soluble diisothiocyanate derivatives as P2Y antagonists, including P2Y6-selective MRS2578 (structure not shown) [92]. Other diisothiocyanate analogues favored various P2Y receptor combinations. DIDS was earlier found to antagonize the rat P2Y6 receptor [92], as well as the bladder smooth muscle P2X1 receptor (IC50 3 μM), but not the PC12 cell P2X2 receptor [31].
3.2.5. P2 agonists that became P2X antagonists by structural modification.
Some of the first P2 nucleotide ligands derived from ATP could not readily be examined by analogue synthesis, due to difficulty of synthesis and uncertainty about the precise structure. For example, the potent P2X1/P2X7 receptor agonist Bz-ATP [13,97] appears to be a mixture of 2′- and 3′-monoesters. Some of the 2′- and 3′-derivatives of ATP have been found to antagonize P2 receptors [98]. Notably, an early antagonist photoaffinity probe arylazidoaminopropionyl-ATP (ANAPP3, structure not shown) also had an ambiguous structure and its use was curtailed after the 1980s. Nevertheless, ANAPP3 was the only P2R antagonist described when the distinction between P2X and P2Y receptors was proposed by Burnstock and Kennedy in 1985 [4], which illustrated the pressing need at that time to identify new ligands. In the absence of potent antagonists, pre-exposure to α,β-methylene-ATP was used to block the subsequently termed P2X1 (but not P2X2) receptor effects by desensitization. Oxidized ATP (o-ATP, structure not shown) was originally used to block and affinity-label nucleotide binding sites in mast cells [99], later cloned as the P2X7 (originally termed P2Z) receptor [100]. It was shown to be an irreversible antagonist at recombinant P2X1, P2X2 and P2X7 receptors [31,100]. However, o-ATP is also not suitable for analogue synthesis, because it is both unstable and chemically reactive towards amines. Furthermore, the possibility of off-target interactions, for example with other ATP-binding proteins is enormous.
The first ATP analogue generally useful as a P2X antagonist was 2′,3′-O-(2,4,6-trinitrophenyl)adenosine 5′-triphosphate (TNP-ATP, structure not shown), which has some selectivity for P2X2 and P2X2/3 and P2X4 receptors [101–103] but also binds to diverse ATP-recognizing proteins such as protein kinases and ATPases [104]. Dal Ben et al. [98] recently explored the SAR of ATP analogues with large acetal groups at the 2′- and 3′-hydroxyls as P2X receptor antagonists.
3.2.6. P2 agonists that became P2Y antagonists by structural modification.
In 1996, Boyer et al. discovered that naturally occurring bis-phosphate derivatives of adenosine block the P2Y1 receptor [110]. Based on this finding, Jacobson, Harden and coworkers explored the SAR in great detail [50,94–96]. Bioisosteric substitutes for the ribose moiety, i.e. cyclic alkyl and acyclic groups have been discovered (Figure 1C), including acyclic phosphonate MRS2298 [96]. Introduction of a rigid (N)-methanocarba ring system as in place of ribose resulted in MRS2500 as a sub-nM antagonist that is highly selective for the P2Y1R [97]. The SAR for nucleotide antagonists at the P2Y1 receptor largely resembles agonist SAR, except that a 3′,5′-bisphosphate is required, instead of a 5′-diphosphate, and a 2′-deoxy modification is favored [111]. Nevertheless, the bicyclic (N)-methanocarba modification was highly affinity-enhancing for both P2Y1R nucleotide agonists and antagonists. Attempts to apply the same strategy of separating the diphosphate moiety into bisphosphates to another 5′-diphosphate activated receptor, P2Y6, failed to produce antagonists, indicating a major structural difference between these two Gq-coupled receptors. In fact, P2Y6 agonists require the opposite ring twist of ribose, and a South (S)-methanocarba modification, but not (N) is tolerated in UDP analogues [53,58]. Among non-nucleotide P2Y2R antagonists, such as AR-C118925XX (structure not shown), a uracil core is shared with P2Y2R agonists [9].
4. Recently identified scaffolds for P2X, P2Y and adenosine receptors
4.1. Novel P2X receptor ligand scaffolds and antagonist clinical trials.
As reviewed recently by Burnstock [112], there are numerous potential clinical applications of P2 receptor-based drugs. Geoff Burnstock and colleagues reported that the P2X3 receptor is localized in sensory neurons of the dorsal root ganglia [113], and perhaps in the future P2X3R antagonists may be applied to inflammatory or neuropathic pain control [66,114,115]. P2X3 antagonists have also been considered for further indications including control of neuropathic pain, chronic pain, e.g. associated with endometriosis, sleep apnea, and overactive bladder [116].
A large pharmaceutical effort identified the class of diaminopyrimidines (DAPs) as a scaffold for P2X2/3 or P2X3R antagonists (Figure 3C) [63,103]. One the P2X3 receptor antagonists in this series, gefapixant [100,104], is a hopeful drug for chronic cough that has recently completed two Phase 3 clinical trials (COUGH-1 and COUGH-2) [117]. The higher dose of 45 mg twice per day significantly improved symptoms of chronic cough and thus met the primary clinical endpoint. As an adverse event, disturbance of taste perception was observed. Geoff participated in the discovery of Gefapixant, and the compound is named after him (Gef = Geoff; pixant = P2X receptor antagonist). Gefapixant is expected to be the first P2X3 antagonist that will be filed for approval. The compound acts as a negative allosteric modulator (NAM) at a novel P2XR site, bridging the left flipper, lower body, and dorsal fin regions of the P2X3R [118].
Further P2X3 receptor antagonists containing diverse scaffolds have been developed that are clinically evaluated, including BLU-5937 [66,119] (Figure 3C), BAY 1817080 [120] and S-600918 (structures undisclosed). All of these antagonists are allosteric modulators, which may bind to different sites on the ion channel receptor. BLU-5937 displays selectivity for P2X3R homomers over P2X2/3 heteromers, which is expected to reduce or abolish taste sensitivity (dysgeusia) [119]. In a Phase 2 clinical study in chronic cough (RELIEF), BLU-5937 had low impact on taste perception, but also failed to reach its primary endpoint [121]. BAY 1817080 was evaluated in a Phase 1/2A clinical trial in chronic cough and, like gefapixant, the antagonist did show efficacy. S-600918, another P2X3 antagonist, probably belonging to a previously disclosed series of pyrrolinones [122] that was evaluated in a phase 2a study, also showed a potential to reduce cough with moderate effects on taste perception [123].
One P2X4R antagonist, NC-2600 (structure not disclosed), is in clinical trials for chronic neuropathic pain [124], and further P2X4R antagonists for the treatment of peripheral inflammatory pain and for chronic neuropathic pain are in the pipeline. Microglial P2X4R is upregulated in pain and injury, leading to release of brain-derived neurotrophic factor (BDNF), a short-term promoter of pain. P2X4R antagonists are also a potential treatment during the early stages after a stroke [125]. Putative mechanisms of P2X4 anti-ischemic neuroprotection include fewer pro-inflammatory myeloid cells entering the brain, reduced plasma membrane P2X4R expression, and increased blood brain barrier integrity [125].
Chemically diverse heterocyclic P2X7 receptor antagonists (Figure 3D) have been developed, and several have been in multiple clinical trials for inflammatory bowel disease (IBD), Crohn’s disease, rheumatoid arthritis, inflammatory pain, chronic obstructive pulmonary disease (COPD) and more recently for CNS disorders, in particular depression [66,115,126–129]. P2X7 activation induces the production of pro-inflammatory IL-1β and reactive oxygen species (ROS) by macrophages, and mast cells may also be involved in the protective effects of P2X7 antagonists in IBD [129,130]. P2X7 antagonist JNJ-54175446 is currently in a Phase II clinical trial for major depressive disorder [127]. Brain-penetrant P2X7 receptor antagonist PET imaging agents have been designed [66,127].
4.2. Novel P2Y receptor ligand scaffolds, clinical trials and approved drugs.
In addition to orthosteric antagonists cangrelor and ticagrelor (mentioned above), there are two allosteric P2Y12R antagonists, approved as antithrombotic agents, clopidogrel and prasugrel, that act as prodrugs that require conversion in vivo; further compounds are in development [131]. The thienopyridine class of P2Y12R prodrugs was first discovered empirically as antithrombotic agents, and only later associated with this receptor subtype [115]. Various other non-nucleotide heterocyclic classes of high affinity P2Y12R antagonists have been reported (Figure 3C) [11,132], but none have been approved as antithrombotic drugs. The X-ray structure of the P2Y12R complex of one such orthosteric antagonist AZD1283 (a cyanopyridine derivative containing a polar sulfonylcarbamoyl group as a diphosphate bioisostere), was reported [133]. Similarly, elinogrel contains a polar sulfonylurea group, but its Phase 2 clinical trial was discontinued in 2012.
Activation of both P2Y1R and P2Y12R is needed for stable thrombus formation; thus, blocking each individually has an antithrombotic effect [11]. A class of N,N′-diarylureas was discovered (Figure 3B), initially from screening of chemical libraries, to be orally active P2Y1R antagonists, and their antithrombotic activity was established [134]. The allosteric antagonist BPTU binds to the lipid-exposed outer surface of the P2Y1R and its SAR has been analyzed in light of the determination of the X-ray structure of its P2Y1R complex [50]. An in silico screen using the X-ray structure identified 1-indolinoalkyl-2-phenolic derivatives as P2Y1R agonists with anticancer potential [135]. However, no P2Y1R ligands are yet approved for clinical use.
4.3. Adenosine receptor ligand scaffolds.
The physiological effects of adenosine were discovered decades before Burnstock’s pioneering studies on ATP [4], and the search for new ligands for adenosine receptors developed earlier than for the P2 receptors. Since adenosine derivatives (Figure 1B) and the prototypical receptor antagonists, xanthines (Figure 4), were already commonly found in the compound collections of academic and pharmaceutical laboratories by the 1970s [136,137], it was relatively straightforward to begin the SAR process using compounds off the shelf as each of the four adenosine receptor subtypes (A1, A2A, A2B and A3) was discovered [6–8,138]. Purine nucleobases, such as adenine derivatives were also plentiful, and their SAR as antagonists was explored early (Figure 5). One of the first entries into adenosine receptor SAR was by Bruns [139], who wrote to numerous laboratories around the world requesting samples of adenine nucleosides and other purines to test for activation or inhibition in fibroblasts of what was later termed the A2B receptor. The subsequent process of structural optimization of adenosine receptor agonists and antagonists through de novo chemical synthesis was not as challenging as for the P2 receptors, as there was already a large literature surrounding preparation of xanthines and nucleoside analogues [137]. Unlike P2 receptors, each of the adenosine receptor subtypes already has definitive agonists and antagonists. Numerous clinical trials of adenosine receptor agonists and antagonists have occurred. The only synthetic ligands thus far approved for human use are an A2A agonist and A2A antagonist [140,141]. Nevertheless, interest in developing adenosine receptor ligands therapeutically has accelerated recently, for example, with A2A antagonists for boosting the innate immune response in the tumor microenvironment [142,143].
Figure 4.
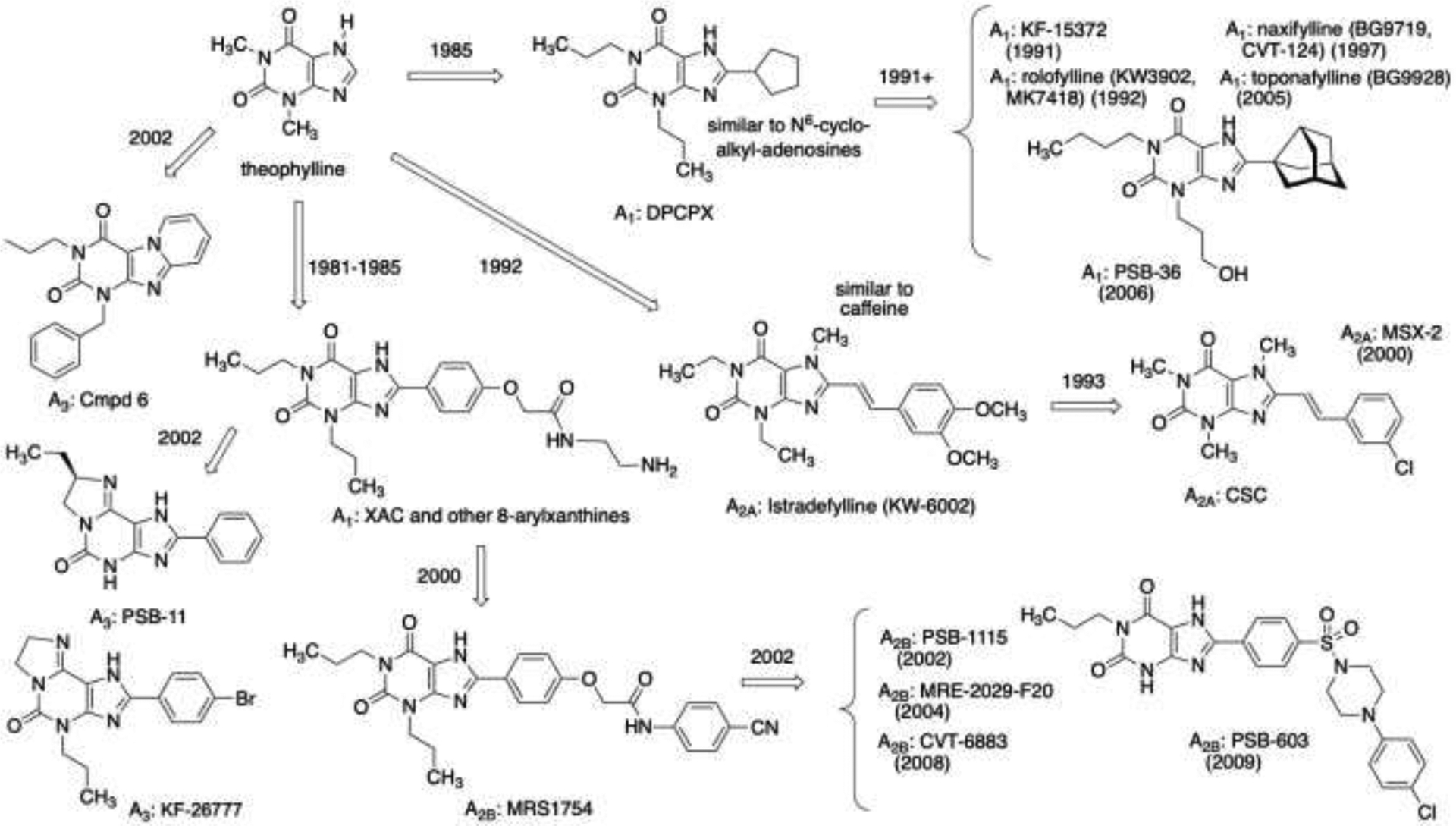
Adenosine receptor antagonists derived from a xanthine scaffold. The prototypical xanthine is theophylline, which is slightly more potent than caffeine at the adenosine receptors. The receptor affinities of many of the compounds cited can be found in the original references [6–8,12,136–140,156–159]. Years shown for each compound indicate an early, major report in the literature.
Figure 5.
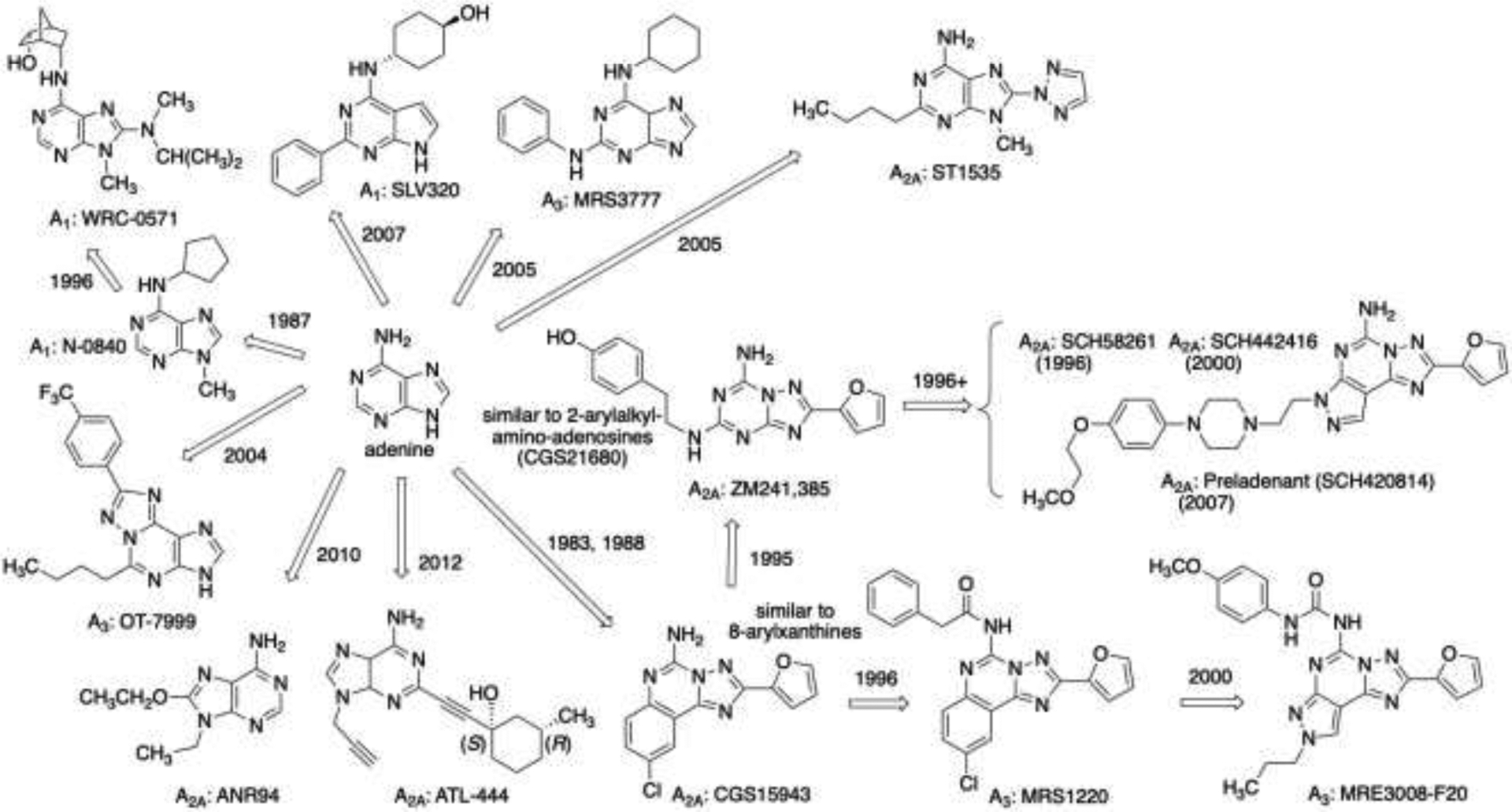
Adenosine receptor antagonists derived from an adenine scaffold, stemming from early key reports [105,136,139]. Without the ribose moiety of adenosine, adenine derivatives are consistently antagonists or inverse agonists at the adenosine receptors. Diverse A2A antagonists have been in clinical trials for Parkinson’s disease and cancer immunotherapy. The receptor affinities of many of the compounds cited can be found in the original references [6–8,12,105,146,161]. Years shown for each compound indicate an early, major report in the literature.
4.3.1. Adenosine agonist scaffolds.
Most adenosine receptor agonists are derivatives of adenosine (Figure 1B), but several classes of nonnucleoside agonists have been found (Figure 6C) [144]. Although the generalized SAR patterns of adenosine derivatives as agonists are shown in Figure 1B, specific agonist structures are not depicted here but rather are found in references [6–8]. Adenine is the address portion of the adenosine scaffold, while the ribose is the message portion. Thus, modification of the ribose ring has been shown to decrease efficacy at the A1, A2A and A3 receptor, leading to partial agonists and antagonists [145]. Adenines lacking the ribose moiety entirely are often adenosine receptor antagonists (Figure 5), and this is one of the widely explored scaffolds for such antagonists besides xanthines [105,146].
Figure 6.
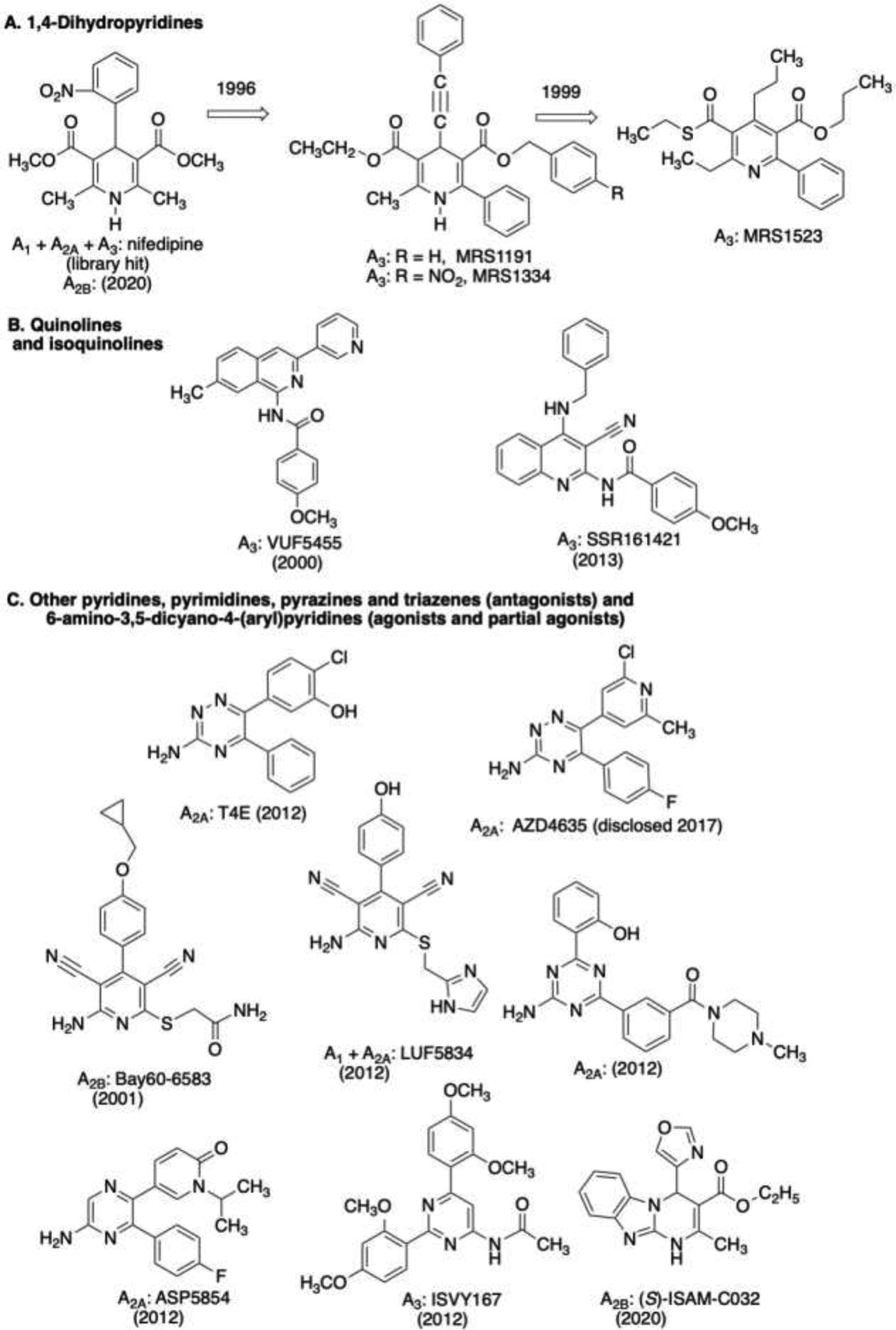
Adenosine receptor ligands based on other selected heterocyclic scaffolds, particularly those that were modified from library screening hits and can be assembled by multicomponent condensation. Numerous non-purine heterocycles have been found to bind to adenosine receptors (particularly A2A and A3 receptors [7,8]), and these compounds represent a small fraction of those diverse structures. 3,5-Dicyanopyridines (C) are agonists or partial agonists [106,144], while the other heterocycles shown are antagonists. The receptor affinities of many of the compounds cited can be found in the original references [6–8,12, 106,107,144,154,169,170,172]. Years shown for each compound indicate an early, major report in the literature.
Adenine nucleosides are well explored, either as single receptor subtype-selective agonists or agonists of mixed selectivity. For example, modification at both C2 and 5′ positions resulted in the first A2A-selective agonist, CGS21680 [147]. 2-Arylalkylether MRS3997 (structure not shown) is a potent, full agonist at both A2A and A2B receptors, but not at the other two subtypes [148]. 2-Alkynyladenosine derivatives have been developed to act at A2A and A3 receptors [7,8,149]. The effects on selectivity of substitution at the C2, N6 and 5′ positions of adenosine (Figure 1B) are often interdependent [126]. Certain novel nucleobases in place of adenine in nucleoside derivatives are also known to act as adenosine receptor agonists [150]. Rigid pseudoribose moieties, such as the (N)-methanocarba ring system, have been used to gain selectivity of adenosine agonists for the A3 receptor [7].
Most of the adenosine agonists in clinical trials so far have failed in development [140], either for problems of efficacy or side effects, or for commercial reasons. One example of the latter is O-methyl-isoguanosine (BVT.115959), which was terminated although it showed efficacy in a randomized, placebo-controlled Phase II trial for diabetic neuropathic pain. Although the ascribed mechanism was activation of the A2A receptor, it proved to be 6-fold more potent in hA1 and hA3 receptor binding than at the hA2A receptor [151]. This is a hopeful indicator of the potential future use of A1/A3 receptor agonists for chronic pain treatment. Both A1 and A3 agonists demonstrate efficacy for treating neuropathic pain [124]. For example, highly selective (N)-methanocarba A3 agonist MRS5980, potently prevents or reverses mechano-hypersensitivity in chronic pain models and, when combined with morphine, makes the opioid effective upon chronic use with no tolerance or withdrawal behaviors [152]. Of note, the A3-induced mast cell degranulation that occurs in rodent species is absent in higher species including human [140]. A3 agonists have progressed to advanced clinical trials for autoimmune inflammatory diseases (psoriasis and rheumatoid arthritis) and for liver diseases (cancer and non-alcoholic steatoheaptitis) [8].
There are two classes of non-nucleoside adenosine agonists, dicyanopyridines and (cyano)pyrimidines (Figure 6C) [106,144]. With appropriate functionalization, this scaffold can be directed to bind with A1, A2A and A2B receptors, either individually or as mixed agonists. For example, the commonly used A2B receptor agonist BAY 60–6583, which is actually a partial agonist at the A2BAR [107] and an antagonist at A1 and A3 receptors at higher concentrations [108], several A1 receptor agonists (Capadenoson and Neladenoson) that were in clinical trials for cardioprotection/heart failure [109], and a mixed A1 /A2A tool compound (agonist LUF5834) all contain a common 2-amino-4-phenyl-pyridine-3,5-dicarbonitrile scaffold. Not only is this scaffold variable in its target adenosine receptor subtype(s), small changes in functional groups dramatically alter the relative efficacy in this series, even inducing receptor antagonism/inverse agonism in selected cases [153]. Molecular modeling combined with site-directed mutagenesis provided evidence for the accommodation of this scaffold in the adenosine receptor orthosteric binding site [154, 155].
4.3.1. Adenosine antagonist scaffolds
We have selected a few illustrative examples from the myriad of adenosine receptor antagonist scaffolds [6–8]. The alkylxanthines are the prototypical adenosine receptor antagonists, and countless analogues have been reported, leading to selectivity for each of the four adenosine receptor subtypes (Figure 4) [12]. The inclusion of an 8-phenyl or 8-cycloalkyl moiety on the xanthine scaffold provided antagonist selectivity for the A1 receptor, in parallel to similar affinity-enhancing derivatization of adenosine at the N6 position. The 8-cycloalkyl group could be bridged to further enhance the A1 receptor affinity as with PSB-36 [108]. An 8-phenylxanthine with an amine-functionalized chain (XAC) has provided many fluorescent probes and other conjugates for detection and characterization of adenosine receptors [156], as this alkylamino group extends out of the orthosteric binding site and is not constrained. 8-Phenylxanthines later were found to be more versatile in their applicability to the A2B receptor, for which the first selective antagonist was MRS1754 [157,158]. The xanthine scaffold later showed a preference for 3-H at the A2B receptor (PSB-603), compared to 3-propyl substitution in A1 receptor antagonists [159]. 8-Styryl groups on the xanthine scaffold provide A2A receptor antagonists, including the approved drug istradefylline (KW-6002) for Parkinson’s disease treatment [160]. Cyclized xanthines have provided antagonists of the A3 receptor [8]. Examples are PSB-11, KF-26777 and Cmpd 6 (Figure 4).
Many adenine derivatives, without the ribose moiety, have been reported as mentioned above as subtype-selective adenosine receptor antagonists (Figure 5) [161]. The original discovery that adenine derivatives, per se, antagonize the adenosine receptors was reported by John Daly and coworkers, e.g. with A1 antagonist N-0840 [105]. A survey of non-xanthine heterocylic antagonists of the adenosine receptors was published in 1988 [162]. During the same time period, basing alternative heterocycles on the adenine/purine scaffold lacking ribose, i.e. fused 5- and 6-membered nitrogen heterocycles, led to early antagonists of the A2A receptor from pharmaceutical labs, i.e. CGS15943 [163, first disclosed in an abstract in 1983] and ZM241,385 [164]. Further elaboration of the heterocyclic core resulted in more selective A2AAR antagonists, such as pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine derivative SCH442416 [165]. This tricyclic series has progressed to an A2AAR antagonist, preladenant (SCH420814, MK-3814) [166], that was in clinical trials for Parkinson’s disease and is currently being repurposed to cancer immunotherapy.
Flavones, of the flavonoid family were discovered in early library screening to bind as antagonists to various adenosine receptors, although flavonoids notoriously have many off-target actions [167]. Another class of phytochemicals, coumarins, which have a rearranged backbone relative to flavonoids, have provided selective A3AR antagonists [168].
1,4-Dihydropyridines (DHP) have long been used clinically as calcium channel blockers. Jacobson and colleagues detected A3 adenosine receptor interactions of some common DHPs (Figure 6A) by screening several thousand members of a small, manually collected chemical library, prior to the widespread use of commercial libraries [8,169]. Subsequently, the calcium channel interactions were deselected by chemical modification of this scaffold, and at the same time A3 receptor affinity was greatly increased, leading to the identification of often-used A3-selective 1,4-dihydropyridine antagonists MRS1191 and its 4-nitrobenzyl derivative MRS1334. The same scaffold was oxidized systematically to direct the A3 SAR to pyridine derivative MRS1523, an effective antagonist at rodent homologues as well as the human A3 receptor. Recently, the DHP nifedipine was found to block the A2B receptor at clinically relevant concentrations, suggesting its possible use in treating diarrhoea [170]. To illustrate the versatility of DHPs as a privileged scaffold, they were also directed toward modulation of the P2X2 receptor [91]. Sotelo and coworkers discovered novel, selective AR antagonist scaffolds containing a pyrimidine core that can be assembled from condensation of multiple prefunctionalized components [171]. These antagonists can bind to different AR subtypes depending on the functionalization, but recently high affinity and selectivity at the A2B receptor was reported for (S)-ICAM-C032 [171]. Quinoline and isoquinoline scaffolds (Figure 6B) have also been explored as A3 antagonist scaffolds [172].
5. Structure-based approaches for purinergic agonists and antagonists
Since the first X-ray structure of an A2A adenosine receptor was reported in 2008 [173], ligand design at purinergic receptors has become increasingly structure-based. A recent count yielded fifty-seven purinergic GPCR structures (52 AR structures and 5 P2YR structures) deposited in the Protein Data Bank (PDB), comprising an impressive ~14% of all GPCR structures known [174]. Thus, the structures of P2Y1R, P2Y12R, A1AR and especially the A2AAR have been leading examples to help guide the application of structure-based design to medicinal chemistry [50,173–177]. P2XR structures are beginning to be useful in medicinal chemistry, with their more complex structure of three interwound subunits, each approximating the form of a dolphin [178]. For example, the interaction of antagonists having a pyroglutamide scaffold with the P2X7R has been modeled [130].
Each of these receptor classes presented unanticipated features in their 3D structures. For example, an allosteric antagonist BPTU was the first GPCR ligand to be discovered to bind entirely outside the heptahelical bundle and in close contact with the membrane phospholipids [175]. The same study found that the binding site for nucleotide antagonist MRS2500 did not share any contact points in common with BPTU. The orientation of typical adenosine receptor agonists and antagonists in the A2A receptor showed a portion that is facing the extracellular medium, rather than being localized in the canonical binding site at ~1/3 the total depth through the transmembrane region [173,176].
An A2A antagonist that was designed completely by structure-based methods, a 1,2,4-triazin-3-amine derivative AZD-4365 (HTL-1071), is in clinical trials for cancer immunotherapy [142]. The design of this antagonist series was guided by biophysical mapping of the binding site, based on multiple A2A receptor X-ray structures. Its effects on multiple cell types contribute to its in vivo efficacy, including improved function of CD103+ dendritic cells and T cells in the tumor microenvironment [143].
6. Conclusions
Ligand development at P2 and at P1 (adenosine) receptors followed different paths. The adenosine receptors are currently most advanced in ligand discovery, and the discovery of potent ligands of P2Y and then P2X receptors followed chronologically. The search for ligands of the purinergic receptors progressed through phases from the early need to prove the existence of P2 receptors until its present relevance for disease treatment was established. Initially, the P2 agonists and antagonists were poorly suited for pharmacological studies. However, intense efforts to modify known scaffolds and eventually to discover novel scaffolds have been sustained since the 1990s. Diverse scaffolds were chemically modified to direct the selectivity toward different subtypes of P2X, P2Y and adenosine receptors. Some of the interactions of the versatile scaffolds presented here are summarized in Figure 7. The medicinal chemistry of purinergic receptors has progressed from useful tool compounds to, in some cases, promising experimental drugs. With Geoffrey Burnstock’s inspiration and encouragement, numerous clinical trials have ensued using various P2XR antagonists. Thus, the vision of Prof. Burnstock to use purinergic signaling modulators, most recently at P2Rs, for treating disease is coming to fruition.
Figure 7.
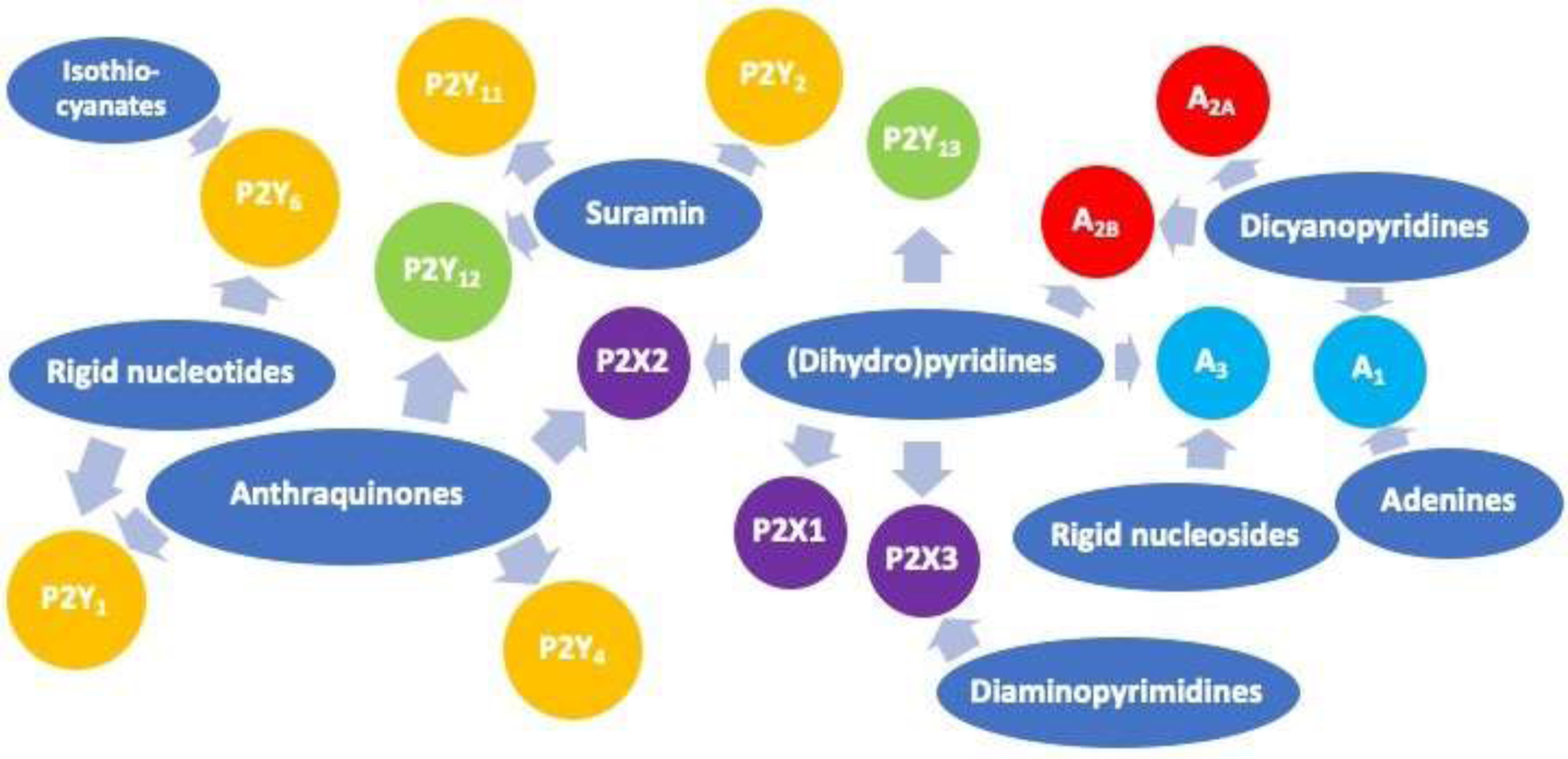
Summary of some of the P2X, P2Y and adenosine receptor interactions of the versatile scaffold classes (blue) presented here. The receptor color coding is: P2X ion channels (violet), P2Y (Gq-coupled in orange, Gi-coupled in green) and adenosine (Gi-coupled in cyan, Gs-coupled in red). Agonist/antagonist ligands are not distinguished. The large class of xanthine antagonists (Figure 4) is not shown.
Acknowledgments
We acknowledge funding from the NIDDK Intramural Research Program (ZIADK31116, ZIADK31117, ZIADK31126). C.E.M. is grateful by support of the Deutsche Forschungsgemeinschaft (SFB1328, FOR2372, GRK1873).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no conflict of interest.
References
- [1].Burnstock G, Purinergic nerves, Pharmacol. Rev 24 (1972) 509–581. [PubMed] [Google Scholar]
- [2].Burnstock G, Fredholm BB, North RA and Verkhratsky A (2010), The birth and postnatal development of purinergic signalling. Acta Physiologica, 199: 93–147. [DOI] [PubMed] [Google Scholar]
- [3].Burnstock G, Do some nerve cells release more than one transmitter?, Neuroscience. 1 (1976) 239–248. 10.1016/0306-4522(76)90054-3. [DOI] [PubMed] [Google Scholar]
- [4].Burnstock G, Kennedy C, Is there a basis for distinguishing two types of P2-purinoceptor?, Gen. Pharmacol 16 (1985) 433–440. 10.1016/0306-3623(85)90001-1. [DOI] [PubMed] [Google Scholar]
- [5].Abbracchio MP, Burnstock G, Purinoceptors: are there families of P2X and P2Y purinoceptors?, Pharmacol. Ther 64 (1994) 445–475. 10.1016/0163-7258(94)00048-4. [DOI] [PubMed] [Google Scholar]
- [6].Jacobson KA, Müller CE, Medicinal chemistry of adenosine, P2Y and P2X receptors, Neuropharmacology. 104 (2016) 31–49. 10.1016/j.neuropharm.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Baraldi S, Baraldi PG, Oliva P, Toti KS, Ciancetta A, Jacobson KA, A2A Adenosine Receptor: Structures, Modeling, and Medicinal Chemistry, in: Borea PA, Varani K, Gessi S, Merighi S, Vincenzi F (Eds.), The Adenosine Receptors, Springer International Publishing, Cham, 2018: pp. 91–136. 10.1007/978-3-319-90808-3_5. [DOI] [Google Scholar]
- [8].Jacobson KA, Tosh DK, Gao ZG, Yu J, Suresh RR, Rao H, Romagnoli R, Baraldi PG, Tabrizi MA, Medicinal chemistry of the A3 adenosine receptor. In: The Adenosine Receptors, The Receptors, Varani K (ed.). Springer, 2018, 34:169–198. [Google Scholar]
- [9].Conroy S, Kindon N, Kellam B, Stocks MJ, Drug-like Antagonists of P2Y Receptors—From Lead Identification to Drug Development, J. Med. Chem 59 (2016) 9981–10005. 10.1021/acs.jmedchem.5b01972. [DOI] [PubMed] [Google Scholar]
- [10].Lambertucci C, Dal Ben D, Buccioni M, Marucci G, Thomas A, Volpini R, Medicinal chemistry of P2X receptors: agonists and orthosteric antagonists, Curr. Med. Chem 22 (2015) 915–928. 10.2174/0929867321666141215093513. [DOI] [PubMed] [Google Scholar]
- [11].Jacobson KA, Delicado EG, Gachet C, Kennedy C, von Kügelgen I, Li B, Miras-Portugal MT, Novak I, Schöneberg T, Perez-Sen R, Thor D, Wu B, Yang Z, Müller CE, Update of P2Y receptor pharmacology: IUPHAR Review 27, Br. J. Pharmacol 177 (2020) 2413–2433. 10.1111/bph.15005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Müller CE, Jacobson KA, Xanthines as adenosine receptor antagonists, Handb Exp Pharmacol. (2011) 151–199. 10.1007/978-3-642-13443-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Khakh BS, Burnstock G, Kennedy C, King BF, North RA, Séguéla P, Voigt M, Humphrey PP, International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits, Pharmacol. Rev 53 (2001) 107–118. [PubMed] [Google Scholar]
- [14].Müller CE, Baqi Y, Namasivayam V. Agonists and antagonists for purinergic receptors. Methods Mol Biol 2020;2041:45–64. doi: 10.1007/978-1-4939-9717-6_3. [DOI] [PubMed] [Google Scholar]
- [15].Hu Y, Stumpfe D, Bajorath J, Computational exploration of molecular scaffolds in medicinal chemistry, J. Med. Chem 59 (2016) 4062–4076. 10.1021/acs.jmedchem.5b01746. [DOI] [PubMed] [Google Scholar]
- [16].Yang H, Sun L, Wang Z, Li W, Liu G, Tang Y, ADMETopt: A Web Server for ADMET Optimization in Drug Design via Scaffold Hopping, J. Chem. Inf. Model 58 (2018) 2051–2056. 10.1021/acs.jcim.8b00532. [DOI] [PubMed] [Google Scholar]
- [17].Chang H, Yanachkov IB, Dix EJ, Yanachkova M, Li Y, Barnard MR, Wright GE, Michelson AD, Frelinger AL, Antiplatelet activity, P2Y1 and P2Y12 inhibition, and metabolism in plasma of stereoisomers of diadenosine 5′,5″-P1,P4-dithio-P2,P3-chloromethylenetetraphosphate, PLoS ONE. 9 (2014) e94780. 10.1371/journal.pone.0094780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jacobson KA, Xie R, Young L, Chang L, Liang BT A novel pharmacological approach to treating cardiac ischemia: binary conjugates of A1 and A3 adenosine receptor agonists. J. Biol. Chem, 2000, 275:30272–30279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Burnstock G, Fischer B, Hoyle CHV, Maillard M, Ziganshin AU, Brizzolara AL, von Isakovics A, Boyer JL, Harden TK, Jacobson KA, Structure activity relationships for derivatives of adenosine-5′-triphosphate as agonists at P(2) purinoceptors: Heterogeneity Within P2X and P2Y Subtypes, Drug Dev. Res 31 (1994) 206–219. 10.1002/ddr.430310308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Burnstock G, Purinergic receptors, J. Theor. Biol 62 (1976) 491–503. 10.1016/0022-5193(76)90133-8. [DOI] [PubMed] [Google Scholar]
- [21].Cusack NJ, Hourani SM, Platelet P2 receptors: from curiosity to clinical targets, J. Auton. Nerv. Syst 81 (2000) 37–43. 10.1016/s0165-1838(00)00151-x. [DOI] [PubMed] [Google Scholar]
- [22].Jacobson KA, Adenosine (P1) and ATP (P2) receptors, Comprehensive Medicinal Chemistry, Pergamon Press, London, 1990: pp. 601–642. [Google Scholar]
- [23].Vaisitti T, Arruga F, Guerra G, Deaglio S, Ectonucleotidases in Blood Malignancies: A Tale of Surface Markers and Therapeutic Targets, Front Immunol. 10 (2019) 2301. 10.3389/fimmu.2019.02301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zimmermann H History of ectonucleotidases and their role in purinergic signaling. Biochem Pharmacol. [this issue] [DOI] [PubMed] [Google Scholar]
- [25].Boison D, Yegutkin GG (2019). Adenosine Metabolism: Emerging Concepts for Cancer Therapy. Cancer Cell, 36(6), 582–596. 10.1016/j.ccell.2019.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dressel J, (Oesper RE, translator), The discovery of Germanin by Oskar Dressel and Richard Kothe, J. Chem. Educ 38 (1961) 620. 10.1021/ed038p620. [DOI] [Google Scholar]
- [27].Dunn PM, Blakeley AG, Suramin: a reversible P2-purinoceptor antagonist in the mouse vas deferens, Br. J. Pharmacol 93 (1988) 243–245. 10.1111/j.1476-5381.1988.tb11427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Brake AJ, Wagenbach MJ, Julius D, New structural motif for ligand-gated ion channels defined by an ionotropic ATP receptor, Nature. 371 (1994) 519–523. 10.1038/371519a0. [DOI] [PubMed] [Google Scholar]
- [29].Den Hertog A, Nelemans A, Van Den Akker J The inhibitory action of suramin on the P2-purinoceptor response in smooth muscle cells of guinea-pig taenia caeci. Eur. J. Pharmacol 166 (1989) 531–534. 10.1016/0014-2999(89)90370-1 [DOI] [PubMed] [Google Scholar]
- [30].Nakazawa K, Fujimori K, Takanaka A, Inoue K, Reversible and selective antagonism by suramin of ATP-activated inward current in PC12 phaeochromocytoma cells, British Journal of Pharmacology. 101 (1990) 224–226. 10.1111/j.1476-5381.1990.tb12117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Evans RJ, Lewis C, Buell G, Valera S, North RA, Surprenant A, Pharmacological characterization of heterologously expressed ATP-gated cation channels (P2 purinoceptors), Molecular Pharmacology 48 (1995) 178–183. http://molpharm.aspetjournals.org/content/48/2/178 [PubMed] [Google Scholar]
- [32].Burnstock G, Warland JII, P2-purinoceptors of two subtypes in the rabbit mesenteric artery: reactive blue 2 selectively inhibits responses mediated via the P2y-but not the P2X-purinoceptor Br. J. Pharmacol (1987), 90, 383–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Boyer JL, Zohn IE, Jacobson KA, Harden TK, Differential effects of P2-purinoceptor antagonists on phospholipase C- and adenylyl cyclase-coupled P2Y-purinoceptors, British Journal of Pharmacology. 113 (1994) 614–620. 10.1111/j.1476-5381.1994.tb17034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Inoue K, Nakazawa K, Ohara-Imaizumi M, Obama T, Fujimori K, Takanaka A, Antagonism by reactive blue 2 but not by brilliant blue G of extracellular ATP-evoked responses in PC12 phaeochromocytoma cells, Br. J. Pharmacol 102 (1991) 851–854. 10.1111/j.1476-5381.1991.tb12265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Glänzel M, Bültmann R, Starke K, Frahm AW, Constitutional isomers of Reactive Blue 2 – selective P2Y-receptor antagonists?, Eur J Med Chem. 38 (2003) 303–312. 10.1016/S0223-5234(02)01449-6. [DOI] [PubMed] [Google Scholar]
- [36].Brown J, Brown CA, Evaluation of reactive blue 2 derivatives as selective antagonists for P2Y receptors, Vascular Pharmacology. 39 (2002) 309–315. 10.1016/S1537-1891(03)00030-2. [DOI] [PubMed] [Google Scholar]
- [37].Malik EM, Müller CE. Anthraquinones as pharmacological tools and drugs. Med Res Rev. 2016;36(4):705–748. [DOI] [PubMed] [Google Scholar]
- [38].Wiedemar N, Hauser DA, Mäser P, 100 Years of Suramin, Antimicrob. Agents Chemother 64 (2020). 10.1128/AAC.01168-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yasgar A, Shultz J, Zhou W, Wang H, Huang F, Murphy N, Abel EL, DiGiovanni J, Inglese J, Simeonov A, A high-throughput 1,536-well luminescence assay for glutathione s-transferase activity, Assay Drug Dev Technol. 8 (2010) 200–211. 10.1089/adt.2009.0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Coughlan SJ, Davenport JW, Hind G, Reactive blue 2 is a potent inhibitor of a thylakoid protein kinase, European Journal of Biochemistry. 197 (1991) 467–471. 10.1111/j.1432-1033.1991.tb15933.x. [DOI] [PubMed] [Google Scholar]
- [41].Lambrecht G, Friebe T, Grimm U, Windscheif U, Bungardt E, Hildebrandt C, Bäumert HG, Spatz-Kümbel G, Mutschler E, PPADS, a novel functionally selective antagonist of P2 purinoceptor-mediated responses, Eur. J. Pharmacol 217 (1992) 217–219. 10.1016/0014-2999(92)90877-7. [DOI] [PubMed] [Google Scholar]
- [42].Ziganshin AU, Hoyle CH, Bo X, Lambrecht G, Mutschler E, Bäumert HG, Burnstock G, PPADS selectively antagonizes P2X-purinoceptor-mediated responses in the rabbit urinary bladder., Br J Pharmacol. 110 (1993) 1491–1495. 10.1111/j.1476-5381.1993.tb13990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hoffmann C, Heine P, Pradel G, Kim Y-C, Jacobson KA, Zimmermann H, Inhibition of ecto-apyrase and ecto-ATPase by pyridoxal phosphate-related compounds, Drug Dev. Res 51 (2000) 151–158. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].King BF, Burnstock and the legacy of the inhibitory junction potential and P2Y1 receptors. Purinergic Signalling, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Betke KM, Wells CA, Hamm HE. GPCR mediated regulation of synaptic transmission. Prog Neurobiol. 2012. March;96(3):304–321. doi: 10.1016/j.pneurobio.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Guzman SJ, Gerevich Z (2016). P2Y receptors in synaptic transmission and plasticity: Therapeutic potential in cognitive dysfunction. Neural Plasticity, 2016, 1207393. 10.1155/2016/1207393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gough GR, Maguire MH, Satchell DG, Three new adenosine triphosphate analogs. Synthesis and effects on isolated gut, J. Med. Chem 16 (1973) 1188–1190. 10.1021/jm00268a028. [DOI] [PubMed] [Google Scholar]
- [48].Fischer B, Boyer JL, Hoyle CHV, Ziganshin AU, Brizzolara AL, Knight GE, Zimmet J, Burnstock G, Harden TK, Jacobson KA, Identification of potent, selective P2Y-purinoceptor agonists: structure-activity relationships for 2-thioether derivatives of adenosine 5′-triphosphate, J. Med. Chem 36 (1993) 3937–3946. 10.1021/jm00076a023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Boyer JL, O’Tuel JW, Fischer B, Jacobson KA, Harden TK, Potent agonist action of 2-thioether derivatives of adenine nucleotides at adenylyl cyclase-linked P2y-purinoceptors, Br. J. Pharmacol 116 (1995) 2611–2616. 10.1111/j.1476-5381.1995.tb17215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ciancetta A, O’Connor RD, Paoletta S, Jacobson KA, Demystifying P2Y1 Receptor Ligand Recognition through Docking and Molecular Dynamics Analyses, J. Chem. Inf. Model 57 (2017) 3104–3123. 10.1021/acs.jcim.7b00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Springthorpe B, Bailey A, Barton P, Birkinshaw TN, Bonnert RV, Brown RC, Chapman D, Dixon J, Guile SD, Humphries RG, Hunt SF, Ince F, Ingall AH, Kirk IP, Leeson PD, Leff P, Lewis RJ, Martin BP, McGinnity DF, Mortimore MP, Paine SW, Pairaudeau G, Patel A, Rigby AJ, Riley RJ, Teobald BJ, Tomlinson W, Webborn PJH, Willis PA, From ATP to AZD6140: The discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis, Bioorg. Med. Chem. Lett 17 (2007) 6013–6018. 10.1016/j.bmcl.2007.07.057. [DOI] [PubMed] [Google Scholar]
- [52].Attah IY, Neumann A, Al-Hroub H, Rafehi M, Baqi Y, Namasivayam V, Müller CE, Ligand binding and activation of UTP-activated G protein-coupled P2Y2 and P2Y4 receptors elucidated by mutagenesis, pharmacological and computational studies, Biochimica et Biophysica Acta (BBA) - General Subjects. 1864 (2020) 129501. 10.1016/j.bbagen.2019.129501. [DOI] [PubMed] [Google Scholar]
- [53].Toti KS, Jain S, Ciancetta A, Balasubramanian R, Chakraborty S, Surujdin R, Shi Z-D, Jacobson KA, Pyrimidine nucleotides containing a (S)-methanocarba ring as P2Y6 receptor agonists. MedChemComm 8 (2017) 1897–1908. 10.1039/c7md00397h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lustig KD, Shiau AK, Brake AJ, et al. (1993) Expression cloning of an ATP receptor from mouse neuroblastoma cells. Proc. Natl. Acad. Sci. U.S.A 90, 5113–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Patel K, Barnes A, Camacho J, Paterson C, Boughtflower R, Cousens D, Marshall F, Activity of diadenosine polyphosphates at P2Y receptors stably expressed in 1321N1 cells Eur. J. Pharmacol 2001, 430; 203–210 [DOI] [PubMed] [Google Scholar]
- [56].Cinkilic O, King BF, Van Der Giet M, Schlüter H, Zidek W, Burnstock G. Selective agonism of group I P2X receptors by dinucleotides dependent on a single adenine moiety. J. Pharmacol. Exp. Ther 2001, 299:131–136. [PubMed] [Google Scholar]
- [57].Ginsburg-Shmuel T, Haas M, Grbic D, Arguin G, Nadel Y, Gendron F-P, Reiser G, Fischer B, UDP made a highly promising stable, potent, and selective P2Y6-receptor agonist upon introduction of a boranophosphate moiety, Bioorg. Med. Chem 20 (2012) 5483–5495. 10.1016/j.bmc.2012.07.042. [DOI] [PubMed] [Google Scholar]
- [58].Jacobson KA, Paoletta S, Katritch V, Wu B, Gao Z-G, Zhao Q, Stevens RC, Kiselev E, Nucleotides acting at P2Y receptors: Connecting structure and function, Mol. Pharmacol 88 (2015) 220–230. 10.1124/mol.114.095711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhou Z, Matsumoto T, A 15-Year Study on Up4A in Cardiovascular Disease, Front. Pharmacol 11 (2020). 10.3389/fphar.2020.01200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Shaver SR, Rideout JL, Pendergast W, Douglass JG, Brown EG, Boyer JL, Patel RI, Redick CC, Jones AC, Picher M et al. (2005) Structure-activity relationships of dinucleotides: Potent and selective agonists of P2Y receptors. Purinergic Signalling 1: 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dunn PM, Liu M, Zhong Y, King BF, Burnstock G, Diinosine pentaphosphate: an antagonist which discriminates between recombinant P2X3 and P2X2/3 receptors and between two P2X receptors in rat sensory neurones, Br J Pharmacol. 130 (2000) 1378–1384. 10.1038/sj.bjp.0703404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Khalafalla MG, Woods LT, Jasmer KJ, Forti KM, Camden JM, Jensen JL, Limesand KH, Galtung HK, Weisman GA, P2 receptors as therapeutic targets in the salivary gland: from physiology to dysfunction, Front Pharmacol. 11 (2020). 10.3389/fphar.2020.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kang DH, Lee YW, Hwang KY, Koh KM, Kwon YA, Kim BY, Song SW, Kim KY. Changes of tear film lipid layer thickness by 3% diquafosol ophthalmic solutions in patients with dry eye syndrome. Int J Ophthalmol. 2019. October 18;12(10):1555–1560. doi: 10.18240/ijo.2019.10.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Battistone MA, Mendelsohn AC, Spallanzani RG, Allegretti AS, Liberman RN, Sesma J, Kalim S, Wall SM, Bonventre JV, Lazarowski ER, Brown D, Breton S, Proinflammatory P2Y14 receptor inhibition protects against ischemic acute kidney injury in mice, J. Clin. Invest 130 (2020) 3734–3749. 10.1172/JCI134791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jung YH, Yu J, Wen Z, Salmaso V, Karcz TP, Phung NB, Chen Z, Duca S, Bennett JM, Dudas S, Cook DN, Salvemini D, Gao ZG, Jacobson KA Exploration of alternative scaffolds for P2Y14 receptor antagonists containing a biaryl core. J. Med. Chem, 2020, 63:9563–9589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Illes P, Müller CE, Jacobson KA, Grutter T, Nicke A, Fountain SJ, Kennedy C, Schmalzing G, Jarvis MF, Stojilkovic SS, King BF, Di Virgilio F Update of P2X receptor properties and their pharmacology: IUPHAR Review: x, Br. J. Pharmacol, 2020, submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Meis S, Hamacher A, Hongwiset D, Marzian C, Wiese M, Eckstein N, Royer H-D, Communi D, Boeynaems J-M, Hausmann R, Schmalzing G, Kassack MU, NF546 [4,4’-(carbonylbis(imino-3,1-phenylene-carbonylimino-3,1-(4-methyl-phenylene)-carbonylimino))-bis(1,3-xylene-alpha,alpha’-diphosphonic acid) tetrasodium salt] is a non-nucleotide P2Y11 agonist and stimulates release of interleukin-8 from human monocyte-derived dendritic cells, J. Pharmacol. Exp. Ther 332 (2010) 238–247. 10.1124/jpet.109.157750. [DOI] [PubMed] [Google Scholar]
- [68].Brockmann N, Sureechatchaiyan P, Müller D et al. Profiling of a suramin-derived compound library at recombinant human P2Y receptors identifies NF272 as a competitive but non-selective P2Y2 receptor antagonist. Purinergic Signalling 15, 287–298 (2019). 10.1007/s11302-019-09663-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wolf C, Rosefort C, Fallah G, Kassack MU, Hamacher A, Bodnar M, Wang H, Illes P, Kless A, Bahrenberg G, Schmalzing G, Hausmann R (2010). Molecular determinants of potent P2X2 antagonism identified by functional analysis, mutagenesis and homology docking. Mol. Pharmacol, mol.110.068700. 10.1124/mol.110.068700 [DOI] [PubMed] [Google Scholar]
- [70].Pillaiyar T, Funke M, Al-Hroub H, Weyler S, Ivanova S, Schlegel J, Abdelrahman A, Müller CE. Design, synthesis and biological evaluation of suramin-derived dual antagonists of the proinflammatory G protein-coupled receptors P2Y2 and GPR17. Eur J Med Chem. 2020, 111789. doi: 10.1016/j.ejmech.2019.111789. [DOI] [PubMed] [Google Scholar]
- [71].Glänzel M, Bültmann R, Starke K, Frahm AW, Structure-activity relationship of novel P2-receptor antagonists structurally related to Reactive Blue 2, Eur. J. Med. Chem 40 (2005) 1262–1276.doi: 10.1016/j.ejmech.2005.07.007. [DOI] [PubMed] [Google Scholar]
- [72].Baqi Y, Müller CE, Convergent synthesis of the potent P2Y receptor antagonist MG 50–3-1 based on a regioselective Ullmann coupling reaction, Molecules 17 (2012) 2599–2615. Doi: 10.3390/molecules17032599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Weyler S, Baqi Y, Hillmann P, Kaulich M, Hunder AM, Müller IA, Müller CE. Combinatorial synthesis of anilinoanthraquinone derivatives and evaluation as non-nucleotide-derived P2Y2 receptor antagonists. Bioorg Med Chem Lett. 2008. January 1;18(1):223–227. doi: 10.1016/j.bmcl.2007.10.082. [DOI] [PubMed] [Google Scholar]
- [74].Baqi Y, Müller CE. Rapid and efficient microwave-assisted copper(0)-catalyzed Ullmann coupling reaction: general access to anilinoanthraquinone derivatives. Org Lett. 2007, 9(7):1271–1274. doi: 10.1021/ol070102v. [DOI] [PubMed] [Google Scholar]
- [75].Baqi Y, Müller CE. Synthesis of alkyl- and aryl-amino-substituted anthraquinone derivatives by microwave-assisted copper(0)-catalyzed Ullmann coupling reactions. Nat Protoc. 2010, 5(5):945–953. doi: 10.1038/nprot.2010.63. Epub 2010 Apr 29. [DOI] [PubMed] [Google Scholar]
- [76].Baqi Y, Atzler K, Köse M, Glänzel M, Müller CE, High-affinity, non-nucleotide-derived competitive antagonists of platelet P2Y12 receptors, J. Med. Chem 52 (2009) 3784–3793. [DOI] [PubMed] [Google Scholar]
- [77].Hoffmann K, Baqi Y, Morena MS, Glänzel M, Müller CE, von Kügelgen I, Interaction of new, very potent non-nucleotide antagonists with Arg256 of the human platelet P2Y12 receptor, J. Pharmacol. Exp. Ther 331 (2009) 648–655. doi: 10.1124/jpet.109.156687. [DOI] [PubMed] [Google Scholar]
- [78].Horváth G, Gölöncsér F, Csölle C, Király K, Andó RD, Baranyi M, Koványi B, Máté Z, Hoffmann K, Algaier I, Baqi Y, Müller CE, Von Kügelgen I, Sperlágh B. Central P2Y12 receptor blockade alleviates inflammatory and neuropathic pain and cytokine production in rodents. Neurobiol Dis. 2014. October;70(100):162–178. doi: 10.1016/j.nbd.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Paoletta S, Sabbadin D, von Kügelgen I, Hinz S, Katritch V, Hoffmann K, Abdelrahman A, Straßburger J, Baqi Y, Zhao Q, Stevens RC, Moro S, Müller CE, Jacobson KA. Modeling ligand recognition at the P2Y12 receptor in light of X-ray structural information. J Comput Aided Mol Des. 2015, 29(8):737–756. doi: 10.1007/s10822-015-9858-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Rafehi M, Malik EM, Neumann A, Abdelrahman A, Hanck T, Namasivayam V, Müller CE, Baqi Y (2017). Development of potent and selective antagonists for the UTP-activated P2Y4 receptor. J Med Chem. 60(7), 3020–3038. 10.1021/acs.jmedchem.7b00030 [DOI] [PubMed] [Google Scholar]
- [81].Hillmann P, Ko G-Y, Spinrath A, Raulf A, von Kügelgen I, Wolff SC, Nicholas RA, Kostenis E, Höltje H-D, Müller CE (2009). Key determinants of nucleotide-activated G protein-coupled P2Y2 receptor function revealed by chemical and pharmacological experiments, mutagenesis and homology modeling. J Med Chem. 52(9), 2762–2775. 10.1021/jm801442p [DOI] [PubMed] [Google Scholar]
- [82].Rafehi M, Neumann A, Baqi Y, Malik EM, Wiese M, Namasivayam V, Müller CE (2017). Molecular recognition of agonists and antagonists by the nucleotide-activated G protein-coupled P2Y2 receptor. J Med Chem. 60(20), 8425–8440. 10.1021/acs.jmedchem.7b00854 [DOI] [PubMed] [Google Scholar]
- [83].Attah IY, Neumann A, Al-Hroub H, Rafehi M, Baqi Y, Namasivayam V, Müller CE. Ligand binding and activation of UTP-activated G protein-coupled P2Y2 and P2Y4 receptors elucidated by mutagenesis, pharmacological and computational studies. Biochim Biophys Acta Gen Subj. 2020, 1864(3):129501. doi: 10.1016/j.bbagen.2019.129501. [DOI] [PubMed] [Google Scholar]
- [84].Baqi Y, Hausmann R, Rosefort C, Rettinger J, Schmalzing G, Müller CE, Discovery of potent competitive antagonists and positive modulators of the P2X2 receptor, J. Med. Chem 54 (2011) 817–830. [DOI] [PubMed] [Google Scholar]
- [85].Huo H, Fryatt AG, Farmer LK, Schmid R, Evans RJ. Mapping the binding site of the P2X receptor antagonist PPADS reveals the importance of orthosteric site charge and the cysteine-rich head region. J Biol Chem. 2018, 293(33):12820–12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Guo D, von Kügelgen I, Moro S, Kim YC, Jacobson KA Evidence for the recognition of non-nucleotide antagonists within the transmembrane domains of the human P2Y1 receptor. Drug Devel. Res, 2002, 57:173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kim YC, Brown SG, Harden TK, Boyer JL, Dubyak G, King BF, Burnstock G, Jacobson KA, Structure-activity relationships of pyridoxal phosphate derivatives as potent and selective antagonists of P2X1 receptors, J. Med. Chem 44 (2001) 340–349. 10.1021/jm9904203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Jung Y-H, Kim YO, Lin H, Cho J-H, Park J-H, Lee S-D, Bae J, Kang KM, Kim Y-G, Pae AN, Ko H, Park C-S, Yoon MH, Kim Y-C, Discovery of Potent Antiallodynic Agents for Neuropathic Pain Targeting P2X3 Receptors, ACS Chem Neurosci. 8 (2017) 1465–1478. 10.1021/acschemneuro.6b00401. [DOI] [PubMed] [Google Scholar]
- [89].Brown SG, Kim Y-C, Kim S-A, Jacobson KA, Burnstock G, King BF, Actions of a Series of PPADS Analogs at P2X1 and P2X3 Receptors, Drug Dev Res. 53 (2001) 281–291. 10.1002/ddr.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Jacobson KA, Kim YC, Wildman SS, Mohanram A, Harden TK, Boyer JL, King BF, Burnstock G A pyridoxine cyclic-phosphate and its 6-arylazo-derivative selectively potentiate and antagonize activation of P2X1 receptors. J. Med. Chem, 1998, 41:2201–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Jacobson KA, Kim YC, King BF In search of selective P2 receptor ligands: interaction of dihydropyridine derivatives at recombinant rat P2X2 receptors. J. Auton. Nerv. System, 2000, 81:152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Mamedova LK, Joshi BV, Gao Z-G, von Kügelgen I, Jacobson KA, Diisothiocyanate derivatives as potent, insurmountable antagonists of P2Y6 nucleotide receptors, Biochem. Pharmacol 67 (2004) 1763–1770. 10.1016/j.bcp.2004.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Herold CL, Qi AD, Harden TK, Nicholas RA. Agonist versus antagonist action of ATP at the P2Y4 receptor is determined by the second extracellular loop. J Biol Chem. 2004, 279(12):11456–11464. doi: 10.1074/jbc.M301734200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Ravi RG, Kim HS, Servos J, Zimmermann H, Lee K, Maddileti S, Boyer JL, Harden TK, Jacobson KA Adenine nucleotides analogues locked in a Northern methanocarba conformation: Enhanced stability and potency as P2Y1 receptor agonists. J. Med. Chem, 2002, 45:2090–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kim HS, Ohno M, Xu B, Kim HO, Choi Y, Ji XD, Maddileti S, Marquez VE, Harden TK, Jacobson KA 2-Substitution of adenine nucleotide analogues containing a bicyclo[3.1.0]hexane ring system locked in a Northern conformation: Enhanced potency as P2Y1 receptor antagonists. J. Med. Chem, 2003, 46:4974–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kim HS, Barak D, Harden TK, Boyer JL, Jacobson KA, Acyclic and cyclopropyl analogues of adenosine bisphosphate antagonists of the P2Y1 receptor: Structure activity relationships and receptor docking. J. Med. Chem, 2001, 44:3092–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kopp R, Krautloher A, Ramírez-Fernández A, Nicke A (2019). P2X7 interactions and signaling - making head or tail of it. Frontiers in Molecular Neuroscience, 12, 183. 10.3389/fnmol.2019.00183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Dal Ben D, Buccioni M, Lambertucci C, Marucci G, Spinaci A, Marchenkova A, Abdelrahman A, Nistri A, Müller CE, Volpini R, Investigation on 2′,3′-O-substituted ATP derivatives and analogs as novel P2X3 receptor antagonists, ACS Med Chem Lett. 10 (2019) 493–498. 10.1021/acsmedchemlett.8b00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Murgia M, Hanau S, Pizzo P, Rippa M, Di Virgilio F, Oxidized ATP. An irreversible inhibitor of the macrophage purinergic P2Z receptor, J. Biol. Chem 268 (1993) 8199–8203. [PubMed] [Google Scholar]
- [100].Surprenant A, Rassendren F, Kawashima E, North RA, and Buell G (1996) The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science (Wash DC) 272:735–738. [DOI] [PubMed] [Google Scholar]
- [101].Virginio C; Robertson G; Surprenant A; North RA Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Mol. Pharmacol, 1998, 53(6), 969–973. [PubMed] [Google Scholar]
- [102].Pasqualetto G, Brancale A, Young MT, The molecular determinants of small-molecule ligand binding at P2X receptors, Front Pharmacol. 9 (2018). 10.3389/fphar.2018.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Abdelrahman A, Namasivayam V, Hinz S, Schiedel AC, Köse M, Burton M, El-Tayeb A, Gillard M, Bajorath J, de Ryck M, Müller CE (2017). Characterization of P2X4 receptor agonists and antagonists by calcium influx and radioligand binding studies. Biochem. Pharmacol, 125, 41–54. 10.1016/j.bcp.2016.11.016 [DOI] [PubMed] [Google Scholar]
- [104].Hiratsuka T (2003), Fluorescent and colored trinitrophenylated analogs of ATP and GTP. Eur. J. Biochem, 270: 3479–3485. doi: 10.1046/j.1432-1033.2003.03748.x [DOI] [PubMed] [Google Scholar]
- [105].Ukena D, Padgett WL, Hong O, Daly JW, Daly DT, Olsson RA (1987). N 6-Substituted 9-methyladenines: A new class of adenosine receptor antagonists. FEBS Letters, 215(2), 203–208. 10.1016/0014-5793(87)80146-1 [DOI] [PubMed] [Google Scholar]
- [106].Rosentreter U, Henning R, Bauser M, Krämer T, Vaupel A, Hübsch W, Dembowsky K, Salcher-Schrauf-Stätter O, Stasch JP, Krahn T and Perzborn E (2001) inventors, Bayer, assignee. Substituted 2-thio-3,5-dicyano-4-aryl-6-aminopyridines and the use thereof as adenosine receptor ligands. WO Patent 2001/025210. [Google Scholar]
- [107].Hinz Sonja, Svenja K Lacher, Benjamin F Seibt, Christa E Müller, BAY60–6583 acts as a partial agonist at adenosine A2B receptors. J. Pharmacol. Exp. Ther 2014, 349(3):427–436. doi: 10.1124/jpet.113.210849. [DOI] [PubMed] [Google Scholar]
- [108].Alnouri MW; Jepards S; Casari A; Schiedel AC; Hinz S; Müller CE Selectivity is species-dependent: Characterization of standard agonists and antagonists at human, rat, and mouse adenosine receptors. Purinergic Signal. 2015, 11, 389–407. DOI: 10.1007/s11302-015-9460-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Meibom D, Albrecht-Küpper B, Diedrichs N, Hübsch W, Kast R, Krämer T, Krenz U, Lerchen H-G, Mittendorf J, Nell PG, Süssmeier F, Vakalopoulos A, Zimmermann K, Neladenoson bialanate hydrochloride: A prodrug of a partial adenosine A1 receptor agonist for the chronic treatment of heart diseases. ChemMedChem 2017, 12, 728. DOI: 10.1002/cmdc.201700151 [DOI] [PubMed] [Google Scholar]
- [110].Boyer JL, Romero-Avila T, Schachter JB, Harden TK, Identification of competitive antagonists of the P2Y1 receptor, Mol. Pharmacol 50 (1996) 1323–1329. [PubMed] [Google Scholar]
- [111].Camaioni E, Boyer JL, Mohanram A, Harden TK, Jacobson KA Deoxyadenosine-bisphosphate derivatives as potent antagonists at P2Y1 receptors. J. Med. Chem, 1998, 41:183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Burnstock G Purinergic Signalling: Therapeutic Developments. Front Pharmacol. 2017, 8:661. doi: 10.3389/fphar.2017.00661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Chen CC, Akopian AN, Sivilottit L, Colquhoun D, Burnstock G, Wood JN (1995) A P2X purinoceptor expressed by a subset of sensory neurons. Nature 377, 428–431. [DOI] [PubMed] [Google Scholar]
- [114].Jarvis MF and Khakh BS (2009) ATP-gated P2X cation-channels. Neuropharmacology 56, 208–215. [DOI] [PubMed] [Google Scholar]
- [115].di Virgilio F, Jacobson KA, Williams M Geoffrey Burnstock - An accidental pharmacologist. Biochem. Pharmacol, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Ford AP. In pursuit of P2X3 antagonists: novel therapeutics for chronic pain and afferent sensitization. Purinergic Signal. 2012, 8(Suppl 1):3–26. doi: 10.1007/s11302-011-9271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Morice AH, Kitt MM, Ford AP, Tershakovec AM, Wu W-C, Brindle K, Thompson R, Thackray-Nocera S, Wright C, The effect of gefapixant, a P2X3 antagonist, on cough reflex sensitivity: a randomised placebo-controlled study, Eur. Respir. J 54 (2019). 10.1183/13993003.00439-2019. [DOI] [PubMed] [Google Scholar]
- [118].Wang J, Wang Y, Cui W-W, Huang Y, Yang Y, Liu Y, W-S Zhao, Cheng X-Y, Sun W-S, Cao P, Zhu MX, Wang R, Hattori M, Yu Y. Druggable negative allosteric site of P2X3 receptors. Proc Natl Acad Sci 2018, 115 (19), 4939–4944; DOI: 10.1073/pnas.1800907115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Garceau D, Chauret N (2019) BLU-5937: A selective P2X3 antagonist with potent anti-tussive effect and no taste alteration. Pulmon. Pharmcol. Ther 56: 56–62. [DOI] [PubMed] [Google Scholar]
- [120].Morice A, Smith J, McGarvey L, Birring S, Parker SM, Turner A, Gashaw I, Fels L, Klein S, Francke K, Friedrich C. Safety and efficacy of BAY 1817080, a P2X3 receptor antagonist, in patients with refractory chronic cough (RCC), American Thoracic Society 2020 International Conference, May 15–20, 2020, Philadelphia, PA, DOI: 10.1164/ajrccm-conference.2020.201.1_MeetingAbstracts.A7648 [DOI] [Google Scholar]
- [121].Bellus chokes in chronic cough, https://www.evaluate.com/vantage/articles/news/trial-results/bellus-chokes-chronic-cough, accessed Oct. 5, 2020
- [122].Tobinaga H, Kameyama T, Asahi K, Horiguchi T, Oohara M, Tada Y, Fuchino K, Jikihara S, Endoh T, Kurihara N, Kanda Y, Ogawa M, Tamura N, Yagi S, Taniguchi E, Takahara Y, Shimada S, Takeyama C, Yamamoto S, Shinohara S, Kai H (2019). Pyrrolinone derivatives as a new class of P2X3 receptor antagonists Part 2: Discovery of orally bioavailable compounds. Bioorg Med Chem Lett 29(5), 688–693. 10.1016/j.bmcl.2019.01.039 [DOI] [PubMed] [Google Scholar]
- [123].Niimi A, Ishihara H, Hida H, Miyazaki S, Late Breaking Abstract - Phase 2a randomised, double-blind, placebo-controlled, crossover study of a novel P2X3 receptor antagonist S-600918 in patients with refractory chronic cough. Eur. Respir. J 2019, 54: RCT452. DOI: 10.1183/13993003.congress-2019.RCT452 [DOI] [Google Scholar]
- [124].Jacobson KA, Giancotti LA, Lauro F, Mufti F, Salvemini D, Treatment of chronic neuropathic pain: purine receptor modulation, Pain. 161 (2020) 1425–1441. 10.1097/j.pain.0000000000001857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Srivastava P, Cronin CG, Scranton VL, Jacobson KA, Liang BT, Verma R Neuroprotective and neuro-rehabilitative effects of acute purinergic receptor P2X4 (P2X4R) blockade after ischemic stroke. Exp. Neurol, 2020, 329:113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Gelin CF, Bhattacharya A, Letavic MA, P2X7 receptor antagonists for the treatment of systemic inflammatory disorders, Prog. Med. Chem 59 (2020) 63–99. 10.1016/bs.pmch.2019.11.002. [DOI] [PubMed] [Google Scholar]
- [127].Bhattacharya A, Ceusters M, Targeting neuroinflammation with brain penetrant P2X7 antagonists as novel therapeutics for neuropsychiatric disorders, Neuropsychopharmacology. 45 (2020) 234–235. 10.1038/s41386-019-0502-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Jarvis MF, Geoffery Burnstock’s influence on the evolution of P2X3 receptor pharmacology. Purinergic Signal, submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Ochoa-Cortes F, Liñán-Rico A, Jacobson KA, Christofi FL Potential for developing purinergic drugs for gastrointestinal diseases. Inflamm. Bowel Dis, 2014, 20:1259–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Homerin G, Jawhara S, Dezitter X, Baudelet D, Dufrénoy P, Rigo B, Millet R, Furman C, Ragé G, Lipka E, Farce A, Renault N, Sendid B, Charlet R, Leroy J, Phanithavong M, Richeval C, Wiart J-F, Allorge D, … Ghinet A (2020). Pyroglutamide-based P2X7 receptor antagonists targeting inflammatory bowel disease. J. Med. Chem 63(5), 2074–2094. 10.1021/acs.jmedchem.9b00584 [DOI] [PubMed] [Google Scholar]
- [131].Baqi Y, Müller CE. Antithrombotic P2Y12 receptor antagonists: recent developments in drug discovery. Drug Discov Today. 2019. January;24(1):325–333. doi: 10.1016/j.drudis.2018.09.021. [DOI] [PubMed] [Google Scholar]
- [132].Paoletta S, Sabbadin D, von Kügelgen I, Hinz S, Katritch V, Hoffmann K, Abdelrahman A, Straßburger J, Baqi Y, Zhao Q, Stevens RC, Moro S, Müller CE, Jacobson KA Modeling ligand recognition at the P2Y12 receptor in light of X-ray structural information. J. Comput. Aided Mol. Des, 2015, 29:737–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Zhang K, Zhang J, Gao ZG, Zhang D, Zhu L, Han GW, Moss SM, Paoletta S, Kiselev E, Lu W, Fenalti G, Zhang W, Müller CE, Yang H, Cherezov V, Katritch V, Han GW, Jacobson KA, Stevens RC, Wu B, Zhao Q Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature, 2014, 509:115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Yang W, Wang Y, Lai A, Qiao J, Wang T, Hua J, … Lam P (2014). Discovery of 4-aryl-7-hydroxyindoline based P2Y1 antagonists as novel antiplatelet agents. J. Med. Chem, 57, 14, 6150–6164. 10.1021/jm5006226 [DOI] [PubMed] [Google Scholar]
- [135].Le HTT, Rimpilainen T, Konda Mani S et al. Synthesis and preclinical validation of novel P2Y1 receptor ligands as a potent anti-prostate cancer agent. Sci. Rep 9, 18938 (2019). 10.1038/s41598-019-55194-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Daly JW Adenosine receptors: Targets for future drugs. J. Med. Chem 1982, 25, 197–207. [DOI] [PubMed] [Google Scholar]
- [137].Olsson RA, Robert Berne: His Place in the History of Purine Research. Drug Devel. Res 58:296–301 (2003) [Google Scholar]
- [138].Müller CE, Jacobson KA Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta - Biomembranes, 2011, 1808:1290–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Bruns RF, Adenosine receptor activation in human fibroblasts: nucleoside agonists and antagonists. Canadian J. Physiol. Pharmacol, 1980, 58(6): 673–691, 10.1139/y80-110 [DOI] [PubMed] [Google Scholar]
- [140].Jacobson KA, Tosh DK, Jain S, Gao ZG Historical and current adenosine receptor agonists in preclinical and clinical development. Frontiers Cell. Neurosci, 2019, 13:124, DOI: 10.3389/fncel.2019.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Jacobson KA, Gao ZG, Matricon P, Eddy MT, Carlsson J A2A adenosine receptor antagonists: From caffeine to selective non-xanthines. Br. J. Pharmacol, in press, DOI: 10.1111/bph.15103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Congreve M, Andrews SP, Doré AS, Hollenstein K, Hurrell E, Langmead CJ, … Marshall FH (2012). Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem 55(5), 1898–1903. 10.1021/jm201376w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Borodovsky A, Barbon CM, Wang Y, et al. Small molecule AZD4635 inhibitor of A2AR signaling rescues immune cell function including CD103+ dendritic cells enhancing anti-tumor immunity. J. Immunother. Cancer 2020;8:e000417. doi: 10.1136/jitc-2019-000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Dal Ben D, Lambertucci C, Buccioni M, Martí Navia A, Marucci G, Spinaci A, Volpini R, Non-nucleoside agonists of the adenosine receptors: An overview, Pharmaceuticals (Basel). 12(4), (2019), 150. 10.3390/ph12040150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Tosh DK, Salmaso V, Rao H, Bitant A, Fisher CL, Lieberman DI, Vorbrüggen H, Reitman ML, Gavrilova O, Gao Z-G, Auchampach JA, Jacobson KA, Truncated (N)-methanocarba nucleosides as partial agonists at mouse and human A3 adenosine receptors: Affinity enhancement by N6-(2-phenylethyl) substitution, J. Med. Chem 63 (2020) 4334–4348. 10.1021/acs.jmedchem.0c00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Lambertucci C, Marucci G, Dal Ben D, Buccioni M, Spinaci A, Kachler S, Klotz K-N, Volpini R (2018). New potent and selective A1 adenosine receptor antagonists as potential tools for the treatment of gastrointestinal diseases. Eur. J. Med. Chem 151, 199–213. 10.1016/j.ejmech.2018.03.067 [DOI] [PubMed] [Google Scholar]
- [147].Hutchison AJ, Williams M, de Jesus R, Yokoyama R, Oei HH, Ghai GR, Webb RL, Zoganas HC, Stone GA, Jarvis MF. 2-(Arylalkylamino)adenosin-5’-uronamides: A New Class of Highly Selective Adenosine A2 Receptor Ligands. J. Med. Chem 1990;33(7):1919–1924. [DOI] [PubMed] [Google Scholar]
- [148].Adachi H, Palaniappan KK, Ivanov AA, Bergman N, Gao ZG, Jacobson KA Structure-activity relationships of 2,N6,5′-substituted adenosine derivatives with potent activity at the A2B adenosine receptor. J. Med. Chem, 2007, 50:1810–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Dal Ben D, Buccioni M, Lambertucci C, Marucci G, Volpini R, Cristalli G. The importance of alkynyl chain presence for the activity of adenine nucleosides/nucleotides on purinergic receptors. Curr. Med. Chem, 2011, 18, DOI: 10.2174/092986711795328391. [DOI] [PubMed] [Google Scholar]
- [150].Rodríguez D, Chakraborty S, Warnick E, Crane S, Gao ZG, O’Connor RO, Jacobson KA, Carlsson J Structure-based screening of uncharted chemical space for atypical adenosine receptor agonists. ACS Chem. Biol, 2016, 11:2763–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Gao ZG, Mamedova LK, Chen P, Jacobson KA 2-Substituted adenosine derivatives: Affinity and efficacy at four subtypes of human adenosine receptors. Biochem. Pharmacol, 2004, 68:1985–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Doyle TM, Largent-Milnes TM, Chen Z, Staikopoulos V, Esposito E, Dalgarno R, Fan C, Tosh DK, Cuzzocrea S, Jacobson KA, Trang T, Hutchinson MR, Bennett GJ, Vanderah TW, Salvemini D Chronic morphine-induced changes in signaling at the A3 adenosine receptor contribute to morphine-induced hyperalgesia, tolerance and withdrawal. J. Pharmacol. Exp. Ther, 2020, 374, 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Chang LC, von Frijtag Drabbe Künzel JK, Mulder-Krieger T, et al. A series of ligands displaying a remarkable agonistic-antagonistic profile at the adenosine A1 receptor. J Med Chem. 2005;48(6):2045–2053. doi: 10.1021/jm049597+ [DOI] [PubMed] [Google Scholar]
- [154].Lane R, Klein Herenbrink C, van Western GJP, Spoorendonk JA, Hoffmann C, IJzerman AP (2012). A novel non-ribose agonist, LUF5834, engages distinct residues from that of adenosine-like ligands to activate the adenosine A2A receptor. Mol. Pharmacol 81(3), 475–487. \ 10.1124/mol.111.075937 [DOI] [PubMed] [Google Scholar]
- [155].Thimm D, Schiedel AC, Sherbiny FF, Hinz S, Hochheiser K, Bertarelli DCG, Maaß A, Müller CE (2013). Ligand-specific binding and activation of the human adenosine A2B receptor. Biochemistry, 52(4), 726–740. 10.1021/bi3012065 [DOI] [PubMed] [Google Scholar]
- [156].Jacobson KA, Ukena D, Padgett W, Kirk KL, Daly JW Molecular probes for extracellular adenosine receptors. Biochem. Pharmacol, 1987, 36:1697–1707. DOI: 10.1016/0006-2952(87)90056-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Kim YC, Ji X. d., Melman N, Linden J, Jacobson KA Anilide derivatives of an 8-phenylxanthine carboxylic congener are highly potent and selective antagonists at human A2B adenosine receptors. J. Med. Chem, 2000, 43:1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Hayallah AM, Sandoval-Ramírez J, Reith U, Schobert U, Preiss B, Schumacher B, Daly JW, Müller CE. 1,8-disubstituted xanthine derivatives: synthesis of potent A2B-selective adenosine receptor antagonists. J Med Chem. 2002. March 28;45(7):1500–1510. doi: 10.1021/jm011049y. [DOI] [PubMed] [Google Scholar]
- [159].Borrmann T, Hinz S, Bertarelli DC, et al. 1-alkyl-8-(piperazine-1-sulfonyl)phenylxanthines: development and characterization of adenosine A2B receptor antagonists and a new radioligand with subnanomolar affinity and subtype specificity. J Med Chem. 2009;52(13):3994–4006. doi: 10.1021/jm900413e [DOI] [PubMed] [Google Scholar]
- [160].Chen JF, Cunha RA. The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020;16(2):167–174. doi: 10.1007/s11302-020-09694-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Volpini R, Dal Ben D, Lambertucci C, et al. (2010) Adenosine A2A receptor antagonists: New 8-substituted 9-ethyladenines as tools for in vivo rat models of Parkinson’s disease. ChemMedChem 4, 1010–1019. DOI: 10.1002/cmdc.200800434 [DOI] [PubMed] [Google Scholar]
- [162].Daly JW, Hong O, Padgett WL, Shamim MT, Jacobson KA, Ukena D Non-xanthine heterocycles: activity as antagonists of A1- and A2-adenosine receptors. Biochem Pharmacol, 1988, 37:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Francis JE, Cash WD, Psychoyos S, Ghai G, Wenk P, Friedmann RC, … Furness P (1988). Structure-activity profile of a series of novel triazoloquinazoline adenosine antagonists. J. Med. Chem 31(5), 1014–1020. 10.1021/jm00400a022 [DOI] [PubMed] [Google Scholar]
- [164].Poucher SM, Keddie JR, Singh P, Stoggall SM, Caulkett P, Jones G and Collis M (1995), The in vitro pharmacology of ZM 241385, a potent, non-xanthine, A2a selective adenosine receptor antagonist. Br. J. Pharmacol 115: 1096–1102. doi: 10.1111/j.1476-5381.1995.tb15923.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Todde S, Moresco RM, Simonelli P, Baraldi PG, Cacciari B, Spalluto G, Varani K, Monopoli A, Matarrese M, Carpinelli A, Magni F, Kienle MG, Fazio F (2000) Design, radiosynthesis, and biodistribution of a new potent and selective ligand for in vivo imaging of the adenosine A2A receptor system using positron emission tomography [2]. J. Med. Chem 43:4359–4362. doi: 10.1021/jm0009843 [DOI] [PubMed] [Google Scholar]
- [166].Neustadt BR, Hao J, Lindo N, Greenlee WJ, Stamford AW, Tulshian D, Ongini E, Hunter J, Monopoli A, Bertorelli R, et al. (2007) Potent, selective, and orally active adenosine A2A receptor antagonists: arylpiperazine derivatives of pyrazolo[4,3-e-]-1,2,4-triazolo[1,5-c]pyrimidines. Bioorg. Med. Chem. Lett 17: 1376–1380. [DOI] [PubMed] [Google Scholar]
- [167].Jacobson KA, Moro S, Manthey JA, West PL, Ji X-D, Interactions of flavones and other phytochemicals with adenosine receptors, Adv. Exp. Med. Biol 505 (2002) 163–171. 10.1007/978-1-4757-5235-9_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Matos MJ, Vilar S, Vazquez-Rodriguez S, Kachler S, Klotz K-N, Buccioni M, Delogu G, Santana L, Uriarte E, Borges F, Structure-based optimization of coumarin hA3 adenosine receptor antagonists, J. Med. Chem 63 (2020) 2577–2587. 10.1021/acs.jmedchem.9b01572. [DOI] [PubMed] [Google Scholar]
- [169].Jiang J, Li AH, Jang SY, Chang L, Melman N, Moro S, Ji X, Lobkovsky EB, Clardy JC, Jacobson KA, Chiral resolution and stereospecificity of 6-phenyl-4-phenylethynyl- 1,4-dihydropyridines as selective A3 adenosine receptor antagonists, J. Med. Chem 42 (1999) 3055–3065. 10.1021/jm980688e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Asano T, Noda Y, Tanaka K-I, Yamakawa N, Wada M, Mashimo T, Fukunishi Y, Mizushima T, Takenaga M, A2B adenosine receptor inhibition by the dihydropyridine calcium channel blocker nifedipine involves colonic fluid secretion, Sci. Rep 10 (2020) 3555. 10.1038/s41598-020-60147-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Mallo-Abreu A, Prieto-Díaz R, Jespers W, Azuaje J, Majellaro M, Velando C, García-Mera X, Caamaño O, Brea J, Loza MI, Gutiérrez-de-Terán H, Sotelo E, Nitrogen-walk approach to explore bioisosteric replacements in a series of potent A2B adenosine receptor antagonists, J. Med. Chem 63 (2020) 7721–7739. 10.1021/acs.jmedchem.0c00564. [DOI] [PubMed] [Google Scholar]
- [172].van Muijlwijk-Koezen JE, Timmerman H, IJzerman AP. The adenosine A3 receptor and its ligands. Prog Med Chem. 2001;38:61–113. doi: 10.1016/s0079-6468(08)70092-4 DOI: 10.1016/s0079-6468(08)70092-4 [DOI] [PubMed] [Google Scholar]
- [173].Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC, The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist, Science 322 (2008) 1211–1217. 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Jacobson KA, Salmaso V Purinergic signaling: Impact of GPCR structures on rational drug design. ChemMedChem, in press, 2020, doi: 10.1002/cmdc.202000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Zhang D, Gao Z-G, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, Han GW, Liu H, Cherezov V, Katritch V, Jiang H, Stevens RC, Jacobson KA, Zhao Q, Wu B, Two disparate ligand-binding sites in the human P2Y1 receptor, Nature. 520 (2015) 317–321. 10.1038/nature14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao Z-G, Cherezov V, Stevens RC, Structure of an agonist-bound human A2A adenosine receptor, Science 332 (2011) 322–327. 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Zhang K, Zhang J, Gao ZG, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Müller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q Agonist-bound structure of the human P2Y12R receptor. Nature, 2014, 509:119–122. DOI: 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Schmid R, Evans RJ, ATP-gated P2X receptor channels: Molecular insights into functional roles, Annu. Rev. Physiol 81 (2019) 43–62. 10.1146/annurev-physiol-020518-114259. [DOI] [PubMed] [Google Scholar]