

Abstract
Bruton tyrosine kinase (BTK) inhibitor ibrutinib has been validated as an effective drug to treat B cell malignancies. Combined therapies comprising ibrutinib and anti-CD20 antibodies like rituximab were designed as a backbone in many clinical trials. However, the off-target inhibition of ibrutinib on interleukin-2 (IL-2)-inducible T cell kinase (ITK) may reduce rituximab’s antibody-dependent cellular cytotoxicity (ADCC) efficacy. Orelabrutinib (Orel), a novel BTK inhibitor, was designed with high selectivity to BTK. In our study, we demonstrated in preclinical models that orelabrutinib in combination with rituximab could preserve NK-cell-mediated ADCC induced by rituximab and enhanced the apoptosis of tumor cells in vitro. The addition of orelabrutinib to rituximab had produced promising combined anti-tumor effects in B cell lymphomas in vivo. Collectively, combination therapy of orelabrutinib with rituximab would benefit patients with B cell lymphoma, especially those with relapsed or refractory disease.
Keywords: anti-CD20 antibody, BTK inhibitor, B cell lymphoma, combined effects
Graphical abstract
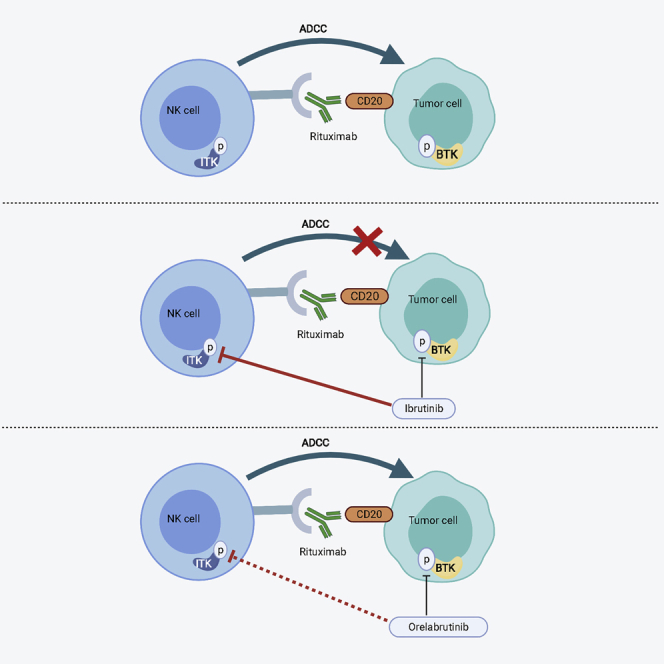
The highly selective BTK inhibitor orelabrutinib has no significant effect on IL-2-inducible T cell kinase, which preserved the ADCC effects of rituximab induced by NK cells. The combination of orelabrutinib and rituximab will likely benefit patients with B cell malignant tumors, especially those with relapsed or refractory disease.
Introduction
B cell lymphoma is a kind of non-Hodgkin lymphoma (NHL) and comprises a diverse group of malignant diseases, ranging in behavior from indolent to highly aggressive. The introduction of rituximab, a monoclonal antibody directed against CD20, ushered the era of immunotherapy for NHL and made R-CHOP (rituximab plus cyclophosphamide, hydroxydaunorubicin, oncovin [vincristine], and prednisone or prednisolone) become the standard therapy of B-NHL.1,2 However, about 1/3 patients with B-NHL developed relapsed or refractory diseases. For refractory or relapsed patients, especially for patients not suitable for autologous stem cell transplantation (ASCT), exploring specific targets and developing effective combination therapy regimens have become the primary task to improve the current treatment status of B-NHL.
Bruton tyrosine kinase (BTK) is an important regulator of B cell receptor (BCR) signaling pathway.3,4 Chronic and sustained activation of the BCR signaling pathway is related to the pathogenesis of many subtypes of B cell malignancies.5 The first-generation BTK inhibitor ibrutinib has presented with remarkable efficacy in several hematological malignancies, and it has been currently approved for the treatment of mantle cell lymphoma (MCL), chronic lymphocytic leukemia (CLL), and Waldenstrom macroglobulinemia (WM).6, 7, 8, 9, 10, 11, 12 To improve the prognosis of patients with B cell malignancies with disease progression or relapse, combined therapy like ibrutinib with anti-CD20 antibodies has been a backbone clinically. However, inhibition of interleukin-2 (IL-2)-inducible T cell kinase (ITK) by ibrutinib regulates T cell populations and suppresses NK-cell-mediated antibody-dependent cellular cytotoxicity (ADCC) in response to anti-CD20 antibodies in vitro, and thus, the combination of these two agents has not yet been shown to be more effective than each alone.13, 14, 15, 16
Orelabrutinib (ICP-022) is a newly developed BTK inhibitor with high selectivity. Preclinical data showed that orelabrutinib has significant BTK inhibition activity with an IC50 at 1.6 nM.17 When performed at 1 μM against a panel of 456 kinases, orelabrutinib only targeted BTK with >90% inhibition lacking inhibition on many additional kinases, including EGFR, TEC, and bone marrow tyrosine kinase, demonstrating orelabrutinib’s superior kinase selectivity. Its excellent safety profiles have been demonstrated in phase I study, and once daily dosing regimen with orelabrutinib could achieve sustained BTK occupancy at 24 h.17 Phase 2 study of orelabrutinib in relapsed/refractory MCL in our center has shown its promising efficacy with 85.9% ORR (overall remission rate) and 27.3% CR (complete remission).18 In the present study, we intended to determine whether the lack of ITK inhibition of orelabrutinib would make it a better candidate for the combination with rituximab and further improve the efficacy of rituximab.
Results
Orelabrutinib inhibited B cell lymphoma cell proliferation in vitro
Orelabrutinib is a novel highly selective BTK inhibitor. Compared with BTK inhibitor ibrutinib, the anti-tumor activities of orelabrutinib on the proliferation of 7 types of B cell lymphoma cell lines were first analyzed using the cell viability assay. As shown in Figure 1A, orelabrutinib exhibited anti-proliferative activity in a dose-dependent manner. Activated B-cell like-diffuse large B cell lymphoma (DLBCL) cell lines TMD8 and HBL-1 were more sensitive to novel BTK inhibitor treatment as compared to other cell lines (Figure 1B). To further assess the effect of BTK inhibitor on activation of BCR signaling pathway, the phosphorylation of BTK and downstream cascades was analyzed in HBL-1 and Z138 cells by immunoblotting analysis. As shown in Figure 1C, compared with ibrutinib, treatment with orelabrutinib dramatically reduced phosphorylation of BTK at the Tyr223 residue in a dose-dependent manner, which indicated the full activation of BTK. Meanwhile, the activation of downstream substrate of BTK phospholipase C-γ2 (PLCγ2) was also downregulated by orelabrutinib treatment. More importantly, compared with vehicle treatment, the orelabrutinib treatment decreased activation of nuclear factor κB (NF-κB) signaling pathway, which is essential for the growth and survival of various lymphoma subtypes, especially in activated B-cell like-DLBCL (Figure 1C). These results demonstrated the significant activity of orelabrutinib in cell lines.
Figure 1.

Orelabrutinib inhibited B cell lymphoma cell proliferation in vitro
(A) B cell lymphoma cell lines (TMD8, HBL-1, SU-DHL-6, WSU-NHL, Mino, Z138, and Raji) were treated with indicated concentrations of orelabrutinib (Orel) or ibrutinib (IBR) for 72 h. Cell viability was measured by Cell Titer-Glo luminescent cell viability assay. Data are shown as mean ± SD of three biological replicates. (B) Western blot analysis for phosphorylation levels of Bruton’s tyrosine kinase (BTK) and its related signaling pathway proteins in Z138 and HBL-1 cells after IBR or Orel treatment for 2 h. (C) The relative phosphorylation levels of signaling proteins from three biologically repeated experiments were quantified by measuring the relative intensity of phosphorylated bands to the β-actin bands. ∗p < 0.05 and ∗∗p < 0.01 compared with control group.
Orelabrutinib lacked inhibition on cellular ITK compared with ibrutinib
Due to the different selectivity profile of orelabrutinib from ibrutinib, the inhibitory activity of orelabrutinib toward ITK was analyzed in YT, NK92 cell line, and primary peripheral blood mononuclear cells (PBMCs) obtained from healthy donors. As shown in Figure 2B, ibrutinib may affect the viability of YT, NK92, and PBMCs through reduction of ITK phosphorylation. Meanwhile, the novel BTK inhibitor orelabrutinib exhibited high selectivity without significant inhibitory effects on the activation of ITK and IκBα, which retain the NF-κB p65 in the cytoplasm and translocation and activation of p65 in the nucleus. Besides, EGFR inhibition of ibrutinib was thought associated with adverse effects in clinical; thus, phosphorylation of EGFR in DHL-6 and Mino cells was assessed by western blotting after indicated treatment. We observed that orelabrutinib has little EGFR inhibition compared with ibrutinib (Figure S1). Altogether, compared with ibrutinib, orelabrutinib has high selectivity on BTK inhibition.
Figure 2.

Orelabrutinib lacked off-target inhibition effect on cellular IL-2-inducible T cell kinase (ITK) compared with ibrutinib
(A and B) p-ITK and p-IκBα were assessed of proteins obtained from YT and NK-92 cells (two kinds of NK/T lymphoma cell lines expressing ITK) treated with ibrutinib or orelabrutinib for 2 h. PBMCs obtained from healthy adults were also used to verify the different effects of orelabrutinib and ibrutinib on ITK inhibition. After being pretreated with or without CD3/CD28-activating agent, PBMCs were treated with different concentrations of BTK inhibitors, orelabrutinib or ibrutinib. Proteins were extracted from pre-treated cells, and proteins of p-ITK and its associated downstream pathway were analyzed by western blot. These western blots were quantified from three biologically repeated tests. ∗p < 0.05 and ∗∗p < 0.01 compared with control group. For the western blots quantitation of PBMCs, control group activated with CD3/CD28 was regarded as control arm. (C) Cell viability was measured by Cell Titer-Glo luminescent cell viability assay in YT and NK-92 cells after being treated with indicated concentration of ibrutinib and Orel for 72 h. HBL-1, a DLBCL cell line, is highly sensitive to BTK inhibitors, and NK92 cell line has high ITK expression. Cell viability was compared in HBL-1, NK92, and YT cells treated with increasing concentration of Orel. The presentation of cell viability was normalized to no treatment control.
Orelabrutinib preserved rituximab-mediated cytotoxicity
Many reports have shown ITK inhibition by ibrutinib antagonizes the efficacy of NK-cell-mediated ADCC. Because the novel BTK inhibitor has no significant impact on ITK activity, the combined anti-tumor effect of orelabrutinib with rituximab was analyzed in cell viability assay. The primary NK cells obtained from healthy donors pretreated with BTK inhibitors were co-cultured with various subtypes of malignant B cells in presence of rituximab treatment. As shown in Figure 3A, ibrutinib treatment compromised NK-cell-mediated ADCC activities through inhibition of ITK activity. On the other hand, the combination of orelabrutinib and rituximab treatment enhanced NK-cell-mediated ADCC toward various B cell lymphoma in different the effector to target cell ratio (E:T ratio) (Figure 3B). Interestingly, when co-cultured with TMD8 and Z138 cells, the activation of NK cells was seen (Figure S2). The main mechanisms of action of rituximab depend on ADCC, but it also comprises complement-dependent cytotoxicity (CDC). We thus explore the impacts of BTK inhibitors on CDC function of rituximab. We discovered that, in Mino and TMD8 cell lines, ibrutinib, but not orelabrutinib, reduced complement-dependent cytotoxicity (CDC), slightly in line with the CD20 modulation (Figure S3). These results validated the predominance of orelabrutinib in combination with rituximab.
Figure 3.

Orelabrutinib preserved rituximab-mediated cytotoxicity
(A) NK cells pretreated with 1 μmol/L BTK inhibitor, ibrutinib, or Orel were co-cultured with B lymphoma cells at different E:T ratio in the presence of rituximab (RTX) at a concentration of 10 μg/mL or not. (B) Pretreated NK cells with 1 μmol/L ibrutinib or Orel were co-cultured with B lymphoma cells at 5:1 in the presence of 10 μg/mL rituximab or PBS. (C) Rituximab (10 μg/mL) or PBS was combined with the increasing concentration of Orel to evaluate tumor cell lysis induced by NK-cell-mediated antibody-dependent cell-mediated cytotoxicity (ADCC). Cell supernatant was collected after 4-h co-culture and then was measured for lactate dehydrogenase (LDH) with Non-Radioactive Cytotoxicity Assay. Each cell line was biologically tested for at least 3 times. ∗p < 0.05 and ∗∗p < 0.01 compared with control group.
Synergistic effects of orelabrutinib and rituximab on inducing apoptosis of tumor cells
To determine tumor cell apoptosis post-treatment, tumor cells (TMD8 and Z138) were treated with indicated concentrations of orelabrutinib and ibrutinib for 48 h. Flow cytometry analysis of TMD8 cells showed a significant increase in the percentage of apoptotic cells (ibrutinib: from 30.5% to 61.6%, p < 0.01; Orel: from 26.51% to 68.0%, p < 0.01). Similar results were observed in Z138 cells (ibrutinib: from 7.65% to 13.14%, p < 0.05; Orel: from 9.16% to 18.34%, p < 0.05). Orelabrutinib and ibrutinib had similar effects on the induction of tumor cells apoptosis (Figures 4A and 4B). Next, the combined effects in inducing cells apoptosis were further analyzed after the combination of orelabrutinib or ibrutinib with rituximab treatment. Flow cytometry analysis of TMD8 cells showed that the apoptotic number of cells significantly increased from 31.4% in the rituximab group to 68.8% in combination treatment group (Figure 4C; p < 0.01). However, combination of ibrutinib and rituximab has not shown synergistic effects in promoting apoptosis. Similar results were observed in Z138 cells (Figure 4C). These data demonstrated that the addition of orelabrutinib to rituximab could increase the apoptosis of tumor cells in vitro significantly.
Figure 4.

Orelabrutinib enhanced apoptosis of tumor cells induced by rituximab
(A and B) B cell lymphoma cells (TMD8 and Z138) were treated with increasing concentration of ibrutinib or Orel for 48 h; after then, cells were stained with PI and Annexin V-FITC. Statistical analysis was performed using one-way ANOVA. Values from three independent experiments are presented as percentages of vehicle in mean ± SD. ∗p < 0.05 and ∗∗p < 0.01 compared among these groups. (C) NK cells isolated from healthy volunteers were pretreated with Orel or ibrutinib at 1 μM for 1 h, TMD8 and Z138 were transduced with firefly GFP, and then rituximab or PBS was added to wells and co-incubated with pretreated NK cells. After incubation at 37°C for 48 h, cells were stained with PI and Annexin-633. GFP+ cells were gated to analyze tumor apoptosis by flow cytometer. The column graph represents the percent of cell apoptosis for each group. Statistical analysis was performed using one-way ANOVA. Values of three biologically independent tests are presented as percentages of vehicle in mean ± SD. ∗p < 0.05 compared with vehicle group; ∗∗p < 0.01 compared with control group. #p < 0.05 compared with rituximab group; ##p < 0.01 compared with rituximab group.
Orelabrutinib combined with rituximab effectively inhibited tumor growth in animal models
Because co-treatment of orelabrutinib with rituximab induced proliferation inhibition in B cell lymphoma cell lines in vitro, the TMD8 cell-line-derived xenograft (CDX) and patient-derived xenograft (PDX) tumor-bearing models were used to assess the anti-tumor activity of drug combination in vivo. In both the TMD8 mouse model and the PDX model, 10 mg/kg orelabrutinib daily or 200 μg/dose rituximab weekly produced a slight reduction in tumor volume; however, when they were combined, the tumor volume was significantly reduced, demonstrating the promising combined effect of the two drugs. Moreover, treatment was well tolerated and no evident weight loss or mice death was found (Figures 5A and 5B). At the end of 3-week treatment, exposure to rituximab or orelabrutinib alone produced different degrees inhibition of tumor growth (61.5% and 30.2%, respectively) in TMD8 xenograft models. However, when treated with the combination of rituximab with orelabrutinib, tumor growth was restrained by 99.2%, demonstrating the evident combined effects of rituximab and orelabrutinib. (Figure 5A). Moreover, in the PDX model (Figure 5B), a similar inhibitory effect was observed in the combined group to that in TMD8 model, though this PDX model was insensitive to rituximab. To further detect the post-treatment apoptosis status of tumor tissues, terminal deoxynucleotidyl transferase nick-end-labeling (TUNEL) staining was conducted on paraffin-embedded samples collected from the TMD8 tumor mouse model. Compared with that of vehicle, orelabrutinib, and rituximab groups, the combination group showed increased apoptosis of tumor cells in TMD8 animal model (Figures 6A and 6B). Western blotting was then applied to assess the apoptotic proteins from PDX tumor tissues (Figure S5C). And NK-cell assay targeting Z138 cells also verified the combination effects of orelabrutinib and rituximab in these two animal models (Figure 5C). Ki-67 and granzyme B staining of tumor sections from TMD8 model suggested the significant combined effects of orelabrutinib and rituximab not only on tumor inhibition but on the regulation of immunity (Figures 6C and 6D). Collectively, the combination therapy of orelabrutinib with rituximab would produce promising effects in the treatment of B cell lymphomas, even in those refractory to rituximab.
Figure 5.

Orelabrutinib combined with rituximab effectively inhibited tumor growth in animal models
(A) CB.17/SCID mice were inoculated subcutaneously with 1 × 107 TMD8 cells in 0.1 mL PBS with Matrigel (1:1 ratio); n = 5 per group. (B) 1 mm3 patient tumor tissue was inoculated subcutaneously into the right flank of per mouse. n = 6 per group. Orelabrutinib (10 mg/kg, bid) and rituximab (200 μg/dose, weekly) were administered to the tumor-bearing mice when the tumor volume achieved 150 mm3. Body weight and tumor volume of mice were measured every other day during treatment. (C) Splenocytes of mice from different group were co-cultured with MCL cell line Z138 for 4 h. NK cell cytotoxic activity was then measured using the Nonradioactive Cytotoxicity Assay Kit. Each column represents the mean value of the triplicate experiments. (D) Hematoxylin and eosin (H&E) staining of tumor tissues from TMD8 cell-line-derived xenograft (CDX) tumor model and patient-derived xenograft (PDX) model. Scale bar: 70 μm. ∗p < 0.05 compared with control group, ∗∗p < 0.01 compared with control group, and ∗∗∗p < 0.001 compared with control group; #p < 0.05 compared with rituximab group and ###p < 0.001 compared with rituximab group.
Figure 6.

Combined effects of orelabrutinib and rituximab in vivo
(A and B) Apoptosis of tumor tissues from TMD8 tumor model was assessed by the terminal deoxynucleotidyl transferase nick-end-labeling (TUNEL) assay, and the nuclei were counterstained with DAPI. Representative images show apoptotic (fragmented) DNA (green staining) and the corresponding cell nuclei (blue) staining. Scale bar: 50 μm. Results are expressed as mean ± SD (n = 3 biologically independent samples). (C and D) Ki67, granzyme B, and NKp44 were assessed by immunohistochemistry of TMD8 tumor tissue. Scale bar: 60 μm. The data show the density of positivity cells for each section. Student’s t test was performed for statistical analysis. Values present as percentages of control group in mean ± SD (n = 3 biologically independent samples), ∗∗p < 0.01 compared with control group, and ∗∗∗p < 0.001 compared with control group; ##p < 0.01 compared with rituximab group and ###p < 0.001 compared with rituximab group.
Discussion
ADCC is suggested as main mechanism of action of rituximab, although the exact pathways involved are not fully elucidated. FcR stimulation of immune cells is requisite for ADCC, which determined the efficacy of anti-CD20 antibodies. Studies in animals had shown that NK cells were an indispensable part in anti-tumor effects of anti-CD20 antibodies for NK cells expressing CD16 (Fc g receptor III).19,20 Therapeutic measures potentiating NK cells’ function to enhance ADCC may produce better tumor eradication. In the present study, we showed that orelabrutinib preserved or even promoted the activity of NK cells, which to some extent exactly made it reasonable to explore the combined effects of orelabrutinib with rituximab.
BTK, as a member of TEC tyrosine kinase family, is an important enzyme in BCR signaling pathway and plays a key role in the activation and survival of B cells. Targeting BTK in malignant B cells has been shown to inhibit the activation of extracellular regulated protein kinase and PLCγ2 as well as NF-κB signal transduction.21 And BTK inhibitor ibrutinib has been approved for the treatment of refractory or relapsed hematologic malignancies.5,8,9 For refractory or relapsed patients, combined regimes of BTK inhibitors and rituximab are a backbone in many clinical settings. However, it has been reported that ibrutinib attenuates the effect of rituximab,15 and ibrutinib interfering on rituximab’s ADCC has been considered partly due to ITK off-target inhibition.22, 23, 24 Orelabrutinib is a novel high-selective inhibitor of BTK, which had demonstrated high efficacy in R/R CLL and MCL.17 In our study, we confirmed that, compared with ibrutinib, orelabrutinib has no significant inhibitory effects on NK-cell-mediated ADCC of rituximab, and the combination of these two agents produced higher anti-tumor effects than each alone in vivo or in vitro. These results indicated combined therapy of selective BTK inhibitor orelabrutinib with anti-CD20 antibody is a promising choice.
In vitro, the proliferation assay and western blotting showed that orelabrutinib has similar inhibitory effects in B cell lymphoma cell lines. Although orelabrutinib significantly inhibits the phosphorylation level of BTK at 1 μmol/L, it does not affect ITK activity compared with ibrutinib. The lack of ITK inhibition makes it to have no impact on ADCC of NK cells induced by rituximab. More importantly, our data show that orelabrutinib could promote the activity of NK cells slightly in vitro. The Fc receptor terminus of antibodies harbors a binding site for the serum protein C1q activating the complement pathway. Then, it was followed by membrane attack complexes (MACs) formation on target cells, which resulted in cell lysis. This process, termed as complement-dependent cytotoxicity (CDC), is another main function of anti-CD20 antibody.25,26 It would be worthy of noting that BTK inhibitors, in addition to the inhibition of BCR signaling, could induce early lymphocytosis in vivo by making egress of neoplastic B cells into the peripheral blood.27 This property may provide chances for subsequent CDC induction by co-administered anti-CD20 monoclonal antibodies. However, the efficiency of CDC may be attuned by the reduced CD20 expression. Although published reports differ in that whether ibrutinib has impact on CDC in vitro,28,29 our data showed that CDC was reduced slightly after 1-h pre-exposure to ibrutinib at 1 μmol/L in Mino and TMD8 cell lines, although after pretreatment with orelabrutinib, the CDC remained unchanged or even increased assessed by flow cytometry. And we also find the difference of the modulation of CD20 in these two agents, which agrees with previous reports and probably explains the different impact of CDC.28 All these results in vitro make it rational to combine orelabrutinib with rituximab. But research should be done to investigate whether the modulation of CD20 is the main reason for the CDC difference, because we find, in other B cell lymphoma cell lines, ibrutinib and orelabrutinib neither antagonize CDC of rituximab. However, it has been reported that rituximab encompasses low levels of direct B cell killing. Novel anti-CD20 antibodies, such as obinutuzumab (OBI), have been shown with stronger capacity for direct B cell killing compared with rituximab.30,31 Considering OBI had stronger direct B cell killing than rituximab, we assumed the addition of orelabrutinib to OBI could probably produce different results, although the mechanism of action of OBI also comprises ADCC. In our present study, we had verified that novel BTK inhibitor orelabrutinib showed no inhibitory effects on ADCC of rituximab. Orelabrutinib may not inhibit these functions of OBI as well. Therefore, it would also be rational to combine OBI with orelabrutinib in the treatment of B cell malignancies. Further investigation could be conducted in future research to validate the combined effects of OBI and orelabrutinib.
In vivo, we verify that Orel can enhance the anti-tumor effect of rituximab in PDX or TMD8 tumor-bearing animal model. And NK cells isolated from combined group showed higher activity, producing significant tumor cell lysis in vitro. It was widely known that NK cells play important roles in humans, and this subpopulation of lymphocytes are cytotoxic lymphocytes in the destruction of abnormal cells, like tumor cells or virus-infected cells. NK cells also regulate the immune response through cytokine and chemokine production, which activates other innate and adaptive immunity.32,33 Considering the positive effect of orelabrutinib on NK cell activity, the combination of Orel and rituximab seems to be more rational, which also partly explains the reason of the evident combined effects of these two agents in our studies. And the PDX results show that orelabrutinib has significant anti-tumor effects, even in rituximab non-sensitive patients, which were presented in tumor inhibition and the promotion of apoptosis. And in vitro, we also found that orelabrutinib but ibrutinib could promote the apoptosis of tumor cells when combined with rituximab. We assumed that the combined effect is probably due to the activation of NK cell activity and the enhanced apoptosis of tumor cells.
Immunohistochemistry assay of granzyme B and Ki67 from TMD8 model shows that the co-treatment promotes the cytotoxicity of effector cells and inhibits the activity of tumor cells. However, programmed cell death 1 ligand 1 (PD-L1) expression on tumor cells from PDX model (Figures S5A and S5B) was increased in combination and rituximab groups; probably, this is the protection of tumor cells for themselves to escape the enhanced immunity induced by rituximab or this result was associated with the resistance to rituximab of this PDX model. It has been reported that ibrutinib could promote the anti-tumor effects of anti-programmed cell death 1/PDL-1 antibodies due to ITK inhibition of T cells.34 Because the mice model (CB-17 severe combined immunodeficiency [SCID]) we used in this study only has NK cells, we could hardly explain whether the lack of off-target inhibition on ITK of orelabrutinib could impact the T-cell-associated immune conditions in vivo. But it has been reported recently that the activation of BTK-associated pathway could upregulate the expression of PD-L1.35 Our immunohistochemistry (IHC) results show that, compared with rituximab treatment, Orel and combined group had lower PD-L1-expressed cells, which means that the addition of orelabrutinib to rituximab is also a rational choice to block the immune escape of tumor cells. Moreover, it has been reported that TEC, EGFR, and bone marrow tyrosine kinase off-target inhibitions were considered basic mechanisms of adverse effects of ibrutinib,36 although orelabrutinib as a high-selective BTK inhibitor lacking these off-target inhibition has shown its safety and efficacy both in animal experiments and clinical trials.
It also has been reported that ibrutinib other than selective BTK inhibitors has unique immune-regulating capability in enhancing expansion of chronically activated T cells by reducing activation-induced cell death.37,38 And it has been reported that BTK inhibitors can also have the inhibitory effects on STAT3 pathways and then regulate the immunosuppressive microenvironment of CLL;39 we did not find these changes induced by orelabrutinib through western blotting of tumor samples from mice (data not shown), but we found that orelabrutinib had positive effects on NK-cell activity, and specific mechanism associated with this effect should be further investigated.
Selective BTK inhibitor orelabrutinib has no off-target impact on ITK and significantly enhances the anti-tumor effect of rituximab. In our study, the combination of these two agents showed promising combined efficacy in B cell lymphoma, which demonstrated that orelabrutinib combined with rituximab may be a more reasonably backbone regimen in clinical setting.
Materials and methods
Cell lines and cell culture
Human DLBCL cell lines (HBL-1 and TMD8) and MCL cell lines (Z138 and Mino) were generously given by Dr. Fu from the University of Nebraska Medical Center (Omaha, NE, USA). DLBCL cell line SU-DHL-6, Burkitt lymphoma cell line Raji, FL (follicular lymphoma) cell line WSU-NHL, and NK/T cell line NK92 and YT were obtained from American Type Culture Collection (Manassas, VA, USA). DMEM-LOW or RPMI-1640 medium (Gibco; Life Technologies) with 10% fetal bovine serum (FBS) (Biological Industries), penicillin/streptomycin, glutamine, and beta-mercaptoethanol was used for all cell cultures. All cell lines were cultured in a humidified atmosphere at 5% CO2 incubator with 37°C and were identified by short tandem repeat DNA fingerprinting analysis (Applied Biosystems, Foster City, CA, USA).
Reagents and antibodies
Orelabrutinib was a gift from INNOCARE (Beijing, China), and ibrutinib was purchased from MedChemExpress (Monmouth Junction, NJ, USA). Orelabrutinib and ibrutinib were dissolved in dimethyl sulfoxide (DMSO) (Sigma Chemical) and stored at −80°C at a suitable stock concentration. Rituximab was preserved in 4°C conditions after being diluted within 24 h. Antibodies against phospho-Tyr42-I kappaB alpha (catalog no. AF7276) and I kappaB alpha (catalog no. AF7776) were purchased from Affinity Biosciences. Antibodies against phospho-Tyr223-BTK, BTK (no. 8547), ITK (no. 2380), phospho-Tyr759-PLCγ2 (no. 3874), PLCγ2 (no. 3872), phospho-Tyr1068-EGF Receptor (D7A5) XP rabbit monoclonal antibody (mAb) (no. 3777), EGF receptor (no. 2232), phospho-Ser536-NF-κB (no. 3039), and NF-κB (no. 8242) were obtained from Cell Signaling Technology (Danvers, MA, USA). Purified mouse anti-BTK (pY551)/ITK (pY511) (cat no. 558034) was provided by BD Biosciences, and anti-β-actin (cat no. A5441) was obtained from Sigma (St. Louis, MO, USA). Anti-APC-CD56 (cat no. 362503) and anti-FITC-CD107a (cat no. 328605) used in flow cytometry were obtained from BioLegend. Anti-EFLUOR450-CD336 (NKp44) (cat no. 48336941) was from Thermo/eBioscience.
Cell viability assay
Cell viability was measured using Cell Titer-Glo Luminescent Cell viability assay system (cat no. G7572; Promega, Madison, WI, USA). First, 90 μL cells with a concentration of 5 × 104/mL were seeded in 96-well black bottom plates and incubated with indicated concentrations of orelabrutinib or ibrutinib for 72 h (orelabrutinib given dose was selected according to the reported concentration of ibrutinib). Then, assay reagent (10 μL) was added to each well. After incubating at room temperature for 10 min on an orbital shaker, cell viability was measured according to luminescent signals by an LMax II instrument (Molecular Devices, Sunnyvale, CA, USA).
PBMCs and lymphocyte isolation
PBMCs from whole blood of healthy individuals were isolated by Ficoll density gradient centrifugation. Specific leukocyte populations were negatively selected through Rosette-sep isolation from peripheral blood (STEMCELL Technologies, Cambridge, MA USA). All donors were given written informed and the procedure was accordance with the Code of Ethics of the World Medical Association and was approved by Peking University Cancer Hospital & Institute.
Quantification of apoptosis
NK cells obtained from healthy volunteers were pretreated with kinase inhibitors for 1 h, B cell lymphoma cells were transduced with firefly GFP, and then rituximab or phosphate buffered saline (PBS) was added to wells and co-incubated with pretreated NK cells. After incubation at 37°C for 48 h, cells were stained with propidium iodide (PI) and Annexin-633. To compare the induction of apoptosis induced by ibrutinib and orelabrutinib, B cell lymphoma cell lines without GFP transduction were stained with PI and annexin V-fluorescein isothiocyanate (FITC) after 48 h of treatment, and cell death was assessed using a BD Accuri C6 flow cytometer (BD Biosciences).
Western blotting analysis
Approximately 1 × 106 cells were collected and lysed in radio immunoprecipitation assay buffer (cat no. 9806, Cell Signaling Technology, Danvers, MA, USA) with phosphatase/protease inhibitor cocktail (cat no. 04693124001, Roche, Mannhein, Germany). Signaling proteins were detected through western blotting according to standard protocols previously reported.40 ECL detection reagent (cat no. RPN225, GE Healthcare, UK) was used for visualizing immunopositive bands with the chemiluminescence detection system (Alpha Innotech, San Leandro, CA, USA).
Cytotoxicity assay
Effector NK cells were isolated from whole blood obtained from healthy adults and incubated with different target B cell lymphoma cell lines loaded at an effector to a target ratio of 1:1 to 10:1, followed by 4-h NK cell-mediated ADCC assays, which were performed according to manufacturer’s protocols.24 Isolated NK cells were treated with DMSO, orelabrutinib, or ibrutinib with specific concentration for 1 h and then tumor cells were incubated with rituximab at 10 μg/mL and co-cultured with pre-treated NK cells. After 4-h co-culture, supernatant was collected and measured for lactate dehydrogenase (LDH) lysis with CytoTox 96 Non-Radioactive Cytotoxicity Assay Technical Bulletin (cat no. CTB163, Promega, Madison, WI, USA) using a standard 96-well plate reader. Cytotoxicity (named %ADCC or target cell lysis [%] in this context) was measured according to the following equation: % relative cytotoxicity/target cell lysis (%) = (experimental release − effector cell spontaneous release − target spontaneous release)/(target maximum release − target spontaneous release) × 100%.
Analysis of NK cell activation
The activation of NK cell was assessed by the percent of CD56+CD107a+ cells after different treatments. Isolated NK cells from healthy donors were co-cultured with TMD8 or Z138 cells and plated in 24-well plates for 4 h with treatment of orelabrutinib or DMSO, and cells were collected and washed with PBS and stained with surface antibodies (anti-CD56, anti-CD107a, or anti-NKp44) at room temperature for 15 min. After washing, cells were analyzed by flow cytometry.
Complement activation and cytotoxicity
Cells were incubated with ibrutinib or orelabrutinib for 1 h and then rituximab or PBS was added, and 20% pooled human AB serum was added 10 min later (for the supplement of complement). After 4-h co-treatment at 37°C, cells were collected and then stained with PI (Sigma-Aldrich, Germany), and cell death was analyzed by flow cytometry (BD Biosciences, San Jose, CA, USA).
In vivo studies
All in vivo experiments were performed on 6- to 8-week-old female CB17/Icr-Prkdcscid/IcrlcoCrl (CB-17 SCID) mice obtained from Charles River Laboratories (Beijing, China). The mice were kept in specific pathogen free animal laboratory environment that met the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The animal experiments were all approved by Peking University Cancer Hospital & Institute. Tumor cells of TMD8 (1 × 107) suspended with the equal volume of PBS and Matrigel (cat no. 356237, Corning Life Sciences, Corning, NY, USA) were inoculated subcutaneously into the right flank of every single mouse. PDX model was built by subcutaneously inoculating 1 mm3 DLBCL patient biopsy specimen tissue into the right flank of per mouse. When tumor volume reached 100−150 mm3, treatments were initiated. According to the dose of orelabrutinib and rituximab used in preliminary experiments of our center, the mice were randomly divided into four groups (n = 6−8/group): vehicle control; orelabrutinib (10 mg/kg, dissolved in 0.5% methyl cellulose) by gavage twice daily; rituximab (200 μg/dose, diluted to 1 mg/mL with normal saline) by intraperitoneal injection weekly; and their combination. Tumor volume was assessed by caliper every 2 days. Mice were finally sacrificed when tumor volume reached to 2,000 mm3. The tumor tissue samples were collected from the different groups and maintained in formalin or grinded to cell suspension for further experiments. Tumor volume was calculated according to V = ab2/2, where a and b represented the long and short tumor diameters, respectively.
NK-cell assay
This experiment was performed according to the method from Li et al.41 Harvested splenocytes from different groups (n = 3) were effector cells, and Z138 cells were applied as target cells. The E:T ratio was 10:1. The cytotoxic activity of NK cells was measured using CytoTox 96 Non-Radioactive Cytotoxicity Assay Technical Bulletin (cat no. CTB163, Promega, Madison, WI, USA) as described above.
DNA fragmentation (TUNEL) assay
Paraffin-embedded tumor samples were cut into 5-μm sections. TUNEL assay was then carried out using an in situ cell death detection kit using the in situ cell death detection kit (cat no. 11684795910, Roche, Germany) to detect DNA fragmentation according to the manufacturer’s protocol.
Immunohistochemical analysis
Paraffin-embedded tumor samples were deparaffinized, and after high-pressure-mediated antigen retrieval, tumor slides were stained with primary antibodies with recommended concentration according to standard protocols at 4°C overnight. Slides were incubated with secondary antibody for 60 min at temperature. Then, sections were stained with diaminobenzidine and counterstained with hematoxylin. All sections were then imaged with a microscope (DMI6000B; Leica Microsystems).
Statistical analysis
All experiments were repeated biologically for at least three times, and representative data are shown in the figures. All values are expressed as mean ± SD. Results were plotted with GraphPad Prism or IBM SPSS Statistics (version 22.0; IBM, New York, NY, USA). Differences between control group and treatment groups were analyzed using paired or unpaired Student’s t test. Student’s t test was also applied when analyzing differences between rituximab group and combination group. One-way ANOVA analysis was used to compare the differences among 3 treatment arms. p <0.05 was recognized as statistically significant.
Acknowledgments
We express gratitude to Innocare company (Beijing, China) for their provision of orelabrutinib (Orel). We would like to thank Dr. Kai Fu from University of Nebraska Medical Center in USA for the kind gifts of the MCL cell lines and Dr. Dingyao Xu for the English language review. We thank Professor Shaokun Shu and Weimin Zhang for their valuable advice. We also thank Dr. Huirong Ding and Dr. Xijuan Liu from the Peking University Cancer Hospital & Institute for help with the analysis of the flow cytometry results. This work was supported by the National Science Center (grant numbers 81870154 and 81670187) and the Beijing Natural Science Foundation (grant number 7172047). The funding bodies had no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication. Data can be made available upon reasonable request.
Author contributions
J.Z. and Y.S. designed the study and approved the final manuscript. H.Y. performed and evaluated the experiment, analyzed data, and wrote the manuscript. Xing Wang, J.L., and Y.Y. helped finish the in vivo experiment. N.D., Xiaogan Wang, and W.F. helped review the manuscript. L.M. reviewed and helped analyze data. D.W. helped perform in vitro study. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.03.015.
Contributor Information
Yuqin Song, Email: songyuqin622@163.com.
Jun Zhu, Email: zhu-jun2017@outlook.com.
Supplemental information
References
- 1.Herrera A.F. Noncellular immune therapies for non-Hodgkin lymphoma. Hematol. Oncol. Clin. North Am. 2019;33:707–725. doi: 10.1016/j.hoc.2019.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Sehn L.H., Donaldson J., Chhanabhai M., Fitzgerald C., Gill K., Klasa R., MacPherson N., O’Reilly S., Spinelli J.J., Sutherland J. Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. J. Clin. Oncol. 2005;23:5027–5033. doi: 10.1200/JCO.2005.09.137. [DOI] [PubMed] [Google Scholar]
- 3.Hussain A., Yu L., Faryal R., Mohammad D.K., Mohamed A.J., Smith C.I. TEC family kinases in health and disease--loss-of-function of BTK and ITK and the gain-of-function fusions ITK-SYK and BTK-SYK. FEBS J. 2011;278:2001–2010. doi: 10.1111/j.1742-4658.2011.08134.x. [DOI] [PubMed] [Google Scholar]
- 4.Tomlinson M.G., Kane L.P., Su J., Kadlecek T.A., Mollenauer M.N., Weiss A. Expression and function of Tec, Itk, and Btk in lymphocytes: evidence for a unique role for Tec. Mol. Cell. Biol. 2004;24:2455–2466. doi: 10.1128/MCB.24.6.2455-2466.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis R.E., Ngo V.N., Lenz G., Tolar P., Young R.M., Romesser P.B., Kohlhammer H., Lamy L., Zhao H., Yang Y. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Advani R.H., Buggy J.J., Sharman J.P., Smith S.M., Boyd T.E., Grant B., Kolibaba K.S., Furman R.R., Rodriguez S., Chang B.Y. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burger J.A., Buggy J.J. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) Leuk. Lymphoma. 2013;54:2385–2391. doi: 10.3109/10428194.2013.777837. [DOI] [PubMed] [Google Scholar]
- 8.Byrd J.C., Furman R.R., Coutre S.E., Flinn I.W., Burger J.A., Blum K.A., Grant B., Sharman J.P., Coleman M., Wierda W.G. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang M.L., Rule S., Martin P., Goy A., Auer R., Kahl B.S., Jurczak W., Advani R.H., Romaguera J.E., Williams M.E. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brower V. Ibrutinib promising in subtype of DLBCL. Lancet Oncol. 2015;16:e428. doi: 10.1016/S1470-2045(15)00192-8. [DOI] [PubMed] [Google Scholar]
- 11.Burger J.A., Tedeschi A., Barr P.M., Robak T., Owen C., Ghia P., Bairey O., Hillmen P., Bartlett N.L., Li J., RESONATE-2 Investigators Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N. Engl. J. Med. 2015;373:2425–2437. doi: 10.1056/NEJMoa1509388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Treon S.P., Tripsas C.K., Meid K., Warren D., Varma G., Green R., Argyropoulos K.V., Yang G., Cao Y., Xu L. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N. Engl. J. Med. 2015;372:1430–1440. doi: 10.1056/NEJMoa1501548. [DOI] [PubMed] [Google Scholar]
- 13.Dubovsky J.A., Beckwith K.A., Natarajan G., Woyach J.A., Jaglowski S., Zhong Y., Hessler J.D., Liu T.M., Chang B.Y., Larkin K.M. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–2549. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Younes A., Thieblemont C., Morschhauser F., Flinn I., Friedberg J.W., Amorim S., Hivert B., Westin J., Vermeulen J., Bandyopadhyay N. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol. 2014;15:1019–1026. doi: 10.1016/S1470-2045(14)70311-0. [DOI] [PubMed] [Google Scholar]
- 15.Kohrt H.E., Sagiv-Barfi I., Rafiq S., Herman S.E., Butchar J.P., Cheney C., Zhang X., Buggy J.J., Muthusamy N., Levy R. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood. 2014;123:1957–1960. doi: 10.1182/blood-2014-01-547869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burger J.A., Keating M.J., Wierda W.G., Hartmann E., Hoellenriegel J., Rosin N.Y., de Weerdt I., Jeyakumar G., Ferrajoli A., Cardenas-Turanzas M. Safety and activity of ibrutinib plus rituximab for patients with high-risk chronic lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol. 2014;15:1090–1099. doi: 10.1016/S1470-2045(14)70335-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhillon S. Orelabrutinib: first approval. Drugs. 2021;81:503–507. doi: 10.1007/s40265-021-01482-5. [DOI] [PubMed] [Google Scholar]
- 18.Song Y., Song Y., Liu L., Zhang M., Li Z., Ji C., Xu W., Liu T., Xu B., Wang X. Safety and efficacy of orelabrutinib monotherapy in Chinese patients with relapsed or refractory mantle cell lymphoma: a multicenter, open-label, phase II study. Blood. 2019;134:755. [Google Scholar]
- 19.Clynes R.A., Towers T.L., Presta L.G., Ravetch J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 20.Bowles J.A., Weiner G.J. CD16 polymorphisms and NK activation induced by monoclonal antibody-coated target cells. J. Immunol. Methods. 2005;304:88–99. doi: 10.1016/j.jim.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 21.Herman S.E., Mustafa R.Z., Gyamfi J.A., Pittaluga S., Chang S., Chang B., Farooqui M., Wiestner A. Ibrutinib inhibits BCR and NF-κB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. 2014;123:3286–3295. doi: 10.1182/blood-2014-02-548610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao X., Bodo J., Sun D., Durkin L., Lin J., Smith M.R., Hsi E.D. Combination of ibrutinib with ABT-199: synergistic effects on proliferation inhibition and apoptosis in mantle cell lymphoma cells through perturbation of BTK, AKT and BCL2 pathways. Br. J. Haematol. 2015;168:765–768. doi: 10.1111/bjh.13149. [DOI] [PubMed] [Google Scholar]
- 23.Yasuhiro T., Sawada W., Klein C., Kozaki R., Hotta S., Yoshizawa T. Anti-tumor efficacy study of the Bruton’s tyrosine kinase (BTK) inhibitor, ONO/GS-4059, in combination with the glycoengineered type II anti-CD20 monoclonal antibody obinutuzumab (GA101) demonstrates superior in vivo efficacy compared to ONO/GS-4059 in combination with rituximab. Leuk. Lymphoma. 2017;58:699–707. doi: 10.1080/10428194.2016.1201567. [DOI] [PubMed] [Google Scholar]
- 24.Reiff S.D., Muhowski E.M., Guinn D., Lehman A., Fabian C.A., Cheney C., Mantel R., Smith L., Johnson A.J., Young W.B. Noncovalent inhibition of C481S Bruton tyrosine kinase by GDC-0853: a new treatment strategy for ibrutinib-resistant CLL. Blood. 2018;132:1039–1049. doi: 10.1182/blood-2017-10-809020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou X., Hu W., Qin X. The role of complement in the mechanism of action of rituximab for B-cell lymphoma: implications for therapy. Oncologist. 2008;13:954–966. doi: 10.1634/theoncologist.2008-0089. [DOI] [PubMed] [Google Scholar]
- 26.Bondza S., Marosan A., Kara S., Lösing J., Peipp M., Nimmerjahn F., Buijs J., Lux A. Complement-dependent activity of CD20-specific IgG correlates with bivalent antigen binding and C1q binding strength. Front. Immunol. 2021;11:609941. doi: 10.3389/fimmu.2020.609941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang B.Y., Francesco M., De Rooij M.F., Magadala P., Steggerda S.M., Huang M.M., Kuil A., Herman S.E., Chang S., Pals S.T. Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood. 2013;122:2412–2424. doi: 10.1182/blood-2013-02-482125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bojarczuk K., Siernicka M., Dwojak M., Bobrowicz M., Pyrzynska B., Gaj P., Karp M., Giannopoulos K., Efremov D.G., Fauriat C. B-cell receptor pathway inhibitors affect CD20 levels and impair antitumor activity of anti-CD20 monoclonal antibodies. Leukemia. 2014;28:1163–1167. doi: 10.1038/leu.2014.12. [DOI] [PubMed] [Google Scholar]
- 29.Bobrowicz M., Dwojak M., Pyrzynska B., Stachura J., Muchowicz A., Berthel E., Dalla-Venezia N., Kozikowski M., Siernicka M., Miazek N. HDAC6 inhibition upregulates CD20 levels and increases the efficacy of anti-CD20 monoclonal antibodies. Blood. 2017;130:1628–1638. doi: 10.1182/blood-2016-08-736066. [DOI] [PubMed] [Google Scholar]
- 30.Rougé L., Chiang N., Steffek M., Kugel C., Croll T.I., Tam C., Estevez A., Arthur C.P., Koth C.M., Ciferri C. Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science. 2020;367:1224–1230. doi: 10.1126/science.aaz9356. [DOI] [PubMed] [Google Scholar]
- 31.Marshall M.J.E., Stopforth R.J., Cragg M.S. Therapeutic antibodies: what have we learnt from targeting CD20 and where are we going? Front. Immunol. 2017;8:1245. doi: 10.3389/fimmu.2017.01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tarazona R., Campos C., Pera A., Sanchez-Correa B., Solana R. Flow cytometry analysis of NK cell phenotype and function in aging. Methods Mol. Biol. 2015;1343:9–18. doi: 10.1007/978-1-4939-2963-4_2. [DOI] [PubMed] [Google Scholar]
- 33.Böttcher J.P., Bonavita E., Chakravarty P., Blees H., Cabeza-Cabrerizo M., Sammicheli S., Rogers N.C., Sahai E., Zelenay S., Reis E Sousa C. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172:1022–1037.e14. doi: 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sagiv-Barfi I., Kohrt H.E., Czerwinski D.K., Ng P.P., Chang B.Y., Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA. 2015;112:E966–E972. doi: 10.1073/pnas.1500712112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vincent-Fabert C., Roland L., Zimber-Strobl U., Feuillard J., Faumont N. Pre-clinical blocking of PD-L1 molecule, which expression is down regulated by NF-κB, JAK1/JAK2 and BTK inhibitors, induces regression of activated B-cell lymphoma. Cell Commun. Signal. 2019;17:89. doi: 10.1186/s12964-019-0391-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byrd J.C., Harrington B., O’Brien S., Jones J.A., Schuh A., Devereux S., Chaves J., Wierda W.G., Awan F.T., Brown J.R. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2016;374:323–332. doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ping L., Ding N., Shi Y., Feng L., Li J., Liu Y., Lin Y., Shi C., Wang X., Pan Z. The Bruton’s tyrosine kinase inhibitor ibrutinib exerts immunomodulatory effects through regulation of tumor-infiltrating macrophages. Oncotarget. 2017;8:39218–39229. doi: 10.18632/oncotarget.16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Long M., Beckwith K., Do P., Mundy B.L., Gordon A., Lehman A.M., Maddocks K.J., Cheney C., Jones J.A., Flynn J.M. Ibrutinib treatment improves T cell number and function in CLL patients. J. Clin. Invest. 2017;127:3052–3064. doi: 10.1172/JCI89756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kondo K., Shaim H., Thompson P.A., Burger J.A., Keating M., Estrov Z., Harris D., Kim E., Ferrajoli A., Daher M. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia. 2018;32:960–970. doi: 10.1038/leu.2017.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J., Wang X., Xie Y., Ying Z., Liu W., Ping L., Zhang C., Pan Z., Ding N., Song Y., Zhu J. The mTOR kinase inhibitor everolimus synergistically enhances the anti-tumor effect of the Bruton’s tyrosine kinase (BTK) inhibitor PLS-123 on Mantle cell lymphoma. Int. J. Cancer. 2018;142:202–213. doi: 10.1002/ijc.31044. [DOI] [PubMed] [Google Scholar]
- 41.Ashizawa T., Iizuka A., Nonomura C., Kondou R., Maeda C., Miyata H., Sugino T., Mitsuya K., Hayashi N., Nakasu Y. Antitumor effect of programmed death-1 (PD-1) blockade in humanized the NOG-MHC double knockout mouse. Clin. Cancer Res. 2017;23:149–158. doi: 10.1158/1078-0432.CCR-16-0122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.