

Abstract
Despite the remarkable success of chimeric antigen receptor-modified T (CAR-T) cell therapy for blood malignancies, the clinical efficacy of this novel therapy in solid tumor treatment is largely limited by the immunosuppressive tumor microenvironment (TME). For instance, immune checkpoints (e.g., programmed cell death protein 1 [PD-1]/programmed death ligand 1 [PD-L1]) in TME play an important role in inhibiting T cell proliferation and functions. Transforming growth factor β (TGF)-β secreted by cancer cells in TME induces regulatory T cells (Tregs) and inhibits cytotoxic T cells. To overcome the inhibitory effect of immune checkpoints, we have previously engineered CAR-T cells to secrete anti-PD-1 to block the PD-1/PD-L1 pathway activity, a step demonstrating superior antitumor efficacy compared with conventional CAR-T cells. In this study, we engineered CAR-T cells that secrete bispecific trap protein co-targeting PD-1 and TGF-β, with the aim of further improving antitumor immunity. Compared with conventional CAR-T cells and anti-PD-1-secreting CAR-T cells, data from in vitro and in vivo experiments showed that CAR-T cells with trap protein secretion further attenuated inhibitory T cell signaling, enhanced T cell persistence and expansion, and improved effector function and resistance to exhaustion. In the xenograft mouse model, CAR-T cells with trap protein secretion exhibited significantly enhanced antitumor immunity and efficacy. With these observations, we demonstrate the potential of trap protein self-secreting CAR-T cells as a potent therapy for solid tumors.
Keywords: chimeric antigen receptor, tumor growth factor beta, immune checkpoint, PD-1, PD-L1, tumor microenvironment, amored CAR-T cells
Graphical abstract
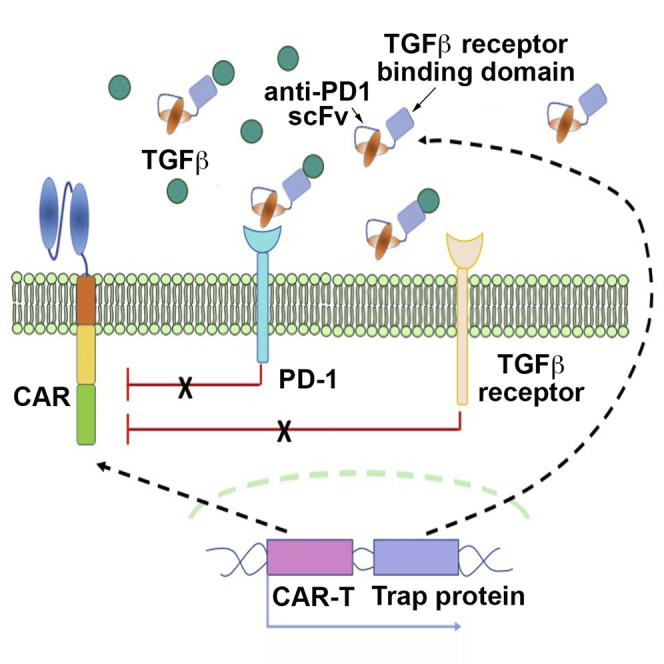
CAR-T cells, engineered to secrete bispecific trap protein co-targeting PD-1 and TGF-β, can attenuate inhibitory T cell signaling, enhance T cell persistence and expansion, and improve effector function and resistance to exhaustion. In vivo studies confirm that such engineered CAR-T cells display significantly enhanced antitumor immunity and efficacy.
Introduction
Over the last two decades, adoptive transfer of chimeric antigen receptor-engineered T (CAR-T) cells has emerged as a promising therapeutic strategy for management and treatment of cancer.1, 2, 3 In general, CARs are synthetic proteins expressed on the T cell surface. CARs consist of an extracellular antigen-binding domain, a hinge, a transmembrane domain, and both intracellular costimulatory and activation domains. Upon antigen recognition, CAR-T cells can be activated and exhibit a major histocompatibility complex (MHC)-unrestricted tumor cell killing effect.4 The potential of CAR-T therapy for hematological malignancies has been validated by clinical studies. Two anti-CD19 CAR-T products have been approved by the US Food and Drug Administration (FDA) encouraging more research aimed at developing CAR-T therapies with higher remission rates in more cancer types.5 However, despite extensive efforts, the success of CAR-T therapy is not yet extrapolated to solid tumors.
The tumor microenvironment (TME) in solid tumor imposes immunosuppression on CAR-T cells, constituting a critical challenge to the success of CAR-T therapy in solid tumors. Besides the physical and metabolic barriers of, for example, low oxygen, low nutrient, or low pH, multiple mechanisms in TME act to inhibit CAR-T cell function and expansion.6,7 For instance, tumor cells have upregulated expression of immune-checkpoint ligands, such as programmed cell death ligand 1 (PD-L1). When PD-L1 binds to its receptor programmed cell death protein 1 (PD-1) on CAR-T cells, the immune-checkpoint interaction activates immunosuppressive cell signaling that causes CAR-T cell dysfunction and exhaustion, ultimately leading to the immune tolerance of tumor cells.8,9 Recent preclinical and clinical studies have shown that knockdown or knockout of the PD-1 gene in CAR-T cells or combining immune-checkpoint blockades with CAR-T cells could significantly augment T cell immune response and enhance the antitumor efficacy of CAR-T therapy.10, 11, 12 Therefore, to provide a new approach for combinational CAR-T therapy, we previously developed CAR-T cells secreting checkpoint inhibitors to block the PD-1/PD-L1 interaction.13 Compared with systemic administration of the PD-1 antibody with CAR-T cells, anti-PD-1 self-secreting CAR-T cells have proven more functional and expandable, as well as more efficient in mediating tumor eradication.13
Another well-defined immunosuppressive mechanism in TME comes from the soluble molecules secreted by tumor cells, stromal cells, and suppressive immune cells. Among these molecules, transforming growth factor β (TGF-β) is particularly important in inhibiting T cell effector function and inducing T cell differentiation into the regulatory phenotype.14, 15, 16 Immunosuppression from TGF-β is potent and associated with immune-checkpoint signaling pathways.17,18 Studies have found that active TGF-β signaling in TME might be responsible for the poor response rates observed in clinical trials of checkpoint inhibitors, especially in the treatment for prostate cancer, ovarian cancer, and breast cancer.19,20 Seeking to boost antitumor immunity, several studies have reported on checkpoint inhibitors with anti-TGF-β monoclonal antibodies or small molecule inhibitors of the TGF-β receptor.21, 22, 23 In a recent study, Ravi et al.24 reported a bifunctional antibody-ligand trap protein comprising an anti-PD-L1 antibody fused with a TGF-βRII ectodomain sequence. Through the dual-targeting effect, the trap protein simultaneously blocks immune checkpoints and inhibits TGF-β-mediated differentiation of regulatory T cells (Tregs), thereby offering a promising strategy for cancers that fail to respond to immune-checkpoint inhibitors.
Inspired by the findings from combination therapies that co-target immune checkpoints and the TGF-β signaling pathway, we herein report the development of anti-PD-1 self-secreting CAR-T cells with the capacity to release trap proteins (Figure 1A). It consists of an anti-PD-1 single-chain fragment variant (scFv) fused with a TGF-βRII ectodomain, and it is expected to enhance CAR-T therapy for solid tumors. In our xenograft mouse model, CAR-T cells with trap protein secretion exhibited enhanced tumor infiltration, expansion, and antitumor efficacy when compared with both parental CAR-T cells and anti-PD-1 self-secreting CAR-T cells. We demonstrated that trap protein secretion empowered CAR-T cells with notable potential to eradicate solid tumors and prevent tumor relapse.
Figure 1.

Generation and characterization of CAR-T cells and target cells
(A) Schematic diagram of CAR-T cells with trap protein secretion in the tumor microenvironment. (B) Schematic representation of CAR constructs of the parental anti-CD19 CAR (CD19 CAR), anti-CD19 CAR with anti-PD-1 scFv secretion (CD19 CAR-αPD-1) and anti-CD19 CAR with trap protein secretion (CD19 CAR-Trap). (C) Expression of CARs in primary human T cells. CAR-T cells were stained with biotin-conjugated rat anti-mouse F(ab′)2 antibody, followed by APC-conjugated streptavidin, to detect CAR expression on the cell surface. NT indicates non-transduced T cells, which were used as a control. (D and E) Expression of PD-L1 and CD19 on the cell surface of target cell lines. (F) Secretion level of TGF-β by target cell lines. The concentrations of TGF-β in the conditioned cell culture medium were shown in the bar graph (n = 3, mean ± SD).
Results
Generation and characterization of CAR-T cells and target cell lines
The schematic representation of the retroviral vector constructs used in this study is shown in Figure 1B. Based on the construct of the second-generation anti-human CD19 CAR (CD19 CAR) that contains an anti-human CD19 scFv, a hinge and transmembrane domain, an intracellular CD28 costimulatory domain, and a CD3ζ activation domain, we generated the anti-CD19 CAR with anti-PD-1 scFv secretion (CD19 CAR-αPD-1) by using a P2A element as the linker between the CD19 CAR sequence and the anti-PD-1 scFv sequence. The feasibility and functionalities of CD19 CAR-αPD-1 have been demonstrated by various in vitro and in vivo experiments in our previous study.13 In the present study, we further engineered the anti-CD19 CAR with trap protein secretion (CD19 CAR-Trap) by fusing the anti-PD-1 scFv sequence with a TGF-β-binding sequence derived from TGF-βRII through a glycine-serine (GS) linker.
Human peripheral blood mononuclear cells (PBMCs) were activated and transduced with each of the three CAR constructs. As shown in Figure 1C, CARs were expressed in primary lymphocytes at a similarly high level (>60%). During the 2-week T cell expansion phase, CAR expression levels were stably maintained. After performing freeze and thaw, about 40% of CAR expression was maintained. To test the antigen-specific functionalities of CAR-T cells, we engineered three different target cell lines, SKOV3-CD19, H292-CD19, and PC3-PD-L1-CD19, also termed as PC3-CD19. The three target cell lines showed similar expression levels of antigen CD19 and TGF-β (Figures 1E and 1F), but for immune-checkpoint molecule PD-L1, PC3-CD19 had a significantly higher expression level than the other two cell lines (Figure 1D). Since the cell line with a higher expression of TGF-β and PD-L1 might serve as a better tool to test the effect of the bifunctional trap protein on CAR-T cells, PC3-CD19 was used in most of the following experiments.
Characterization of trap protein
To assess trap protein synthesized by engineered cells, 293T cells were transfected to produce trap protein labeled with a His-tag. 3 days post-transfection, the cell culture supernatant was harvested and used to incubate 293T cells engineered to express PD-1 on the cell surface. After 1 h of incubation, trap protein binding to 293T-PD-1 was detected by either anti-His-tag antibody or anti-mouse F(ab′)2 antibody (Figures 2A and 2B).
Figure 2.

Characterization of trap protein
(A and B) Detection of trap protein binding to 293T-PD-1 cells by anti-His-tag antibody or anti-mouse Fab antibody. Cell culture supernatant was collected from 293T cells transfected with the trap protein vector and used to incubate 293T cells with PD-1 expression at room temperature for 1 h. Cell culture soup collected from wild-type 293T cells was used as control. (C) Trap protein purified from transfected 293T cells was analyzed by western blot. (D and E) Binding ability of trap to PD-1 and TGF-β using sandwich ELISA, wherein purified trap protein was added to PD-1-Fc- or TGF-β-coated plates, followed by detection by anti-His-tag antibody conjugated with HRP.
Furthermore, trap protein was purified from cell culture supernatant of transfected 293T cells using a His-tag-labeled protein purification protocol. Western blotting analysis confirmed the secretion of the trap protein with a molecular weight of 56 kDa (Figure 2C). The bifunctional binding activity of the trap protein to PD-1 and TGF-β was confirmed by enzyme-linked immunosorbent assay (ELISA), wherein purified protein was added to recombinant human PD-1-Fc- or TGF-β-coated plates at different concentrations, and the binding was detected by anti-His-tag antibody (Figures 2D and 2E).
Trap protein secretion changes cytokine expression and improves proliferation of CAR-T cells in response to antigen stimulation
To study how trap protein affects the response of CAR-T cells to target cells in vitro, we co-cultured CAR-T cells with PC3-CD19 cells at a 1:1 cell-to-cell ratio for 16 h in the presence of the protein transport inhibitor. Compared with non-transduced (NT) control cells, we found that CAR-T cells all responded to the stimulation from target cells with increased expression of the proinflammatory cytokines interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and interleukin (IL)-2. However, the effects of protein drug secretion on the cytokines were quite different. For IFN-γ, CD19 CAR-αPD-1 and CD19 CAR-Trap failed to exhibit a higher percentage of IFN-γ+ T cells than CD19 CAR in either CD4+ or CD8+ T cell populations (Figure 3A). For TNF-α, CD19 CAR-Trap showed more production than CD19 CAR in both CD4+ and CD8+ T cell populations, whereas CD19 CAR-αPD-1 only had an effect similar to that of the CD4+ T cell population (Figure 3B). When IL-2 was examined, no difference was detected with the exception of the CD19 CAR-αPD-1 group, which exhibited a higher expression of the CD4+ T cell population compared with the other two groups (Figure 3C). Besides PC3-CD19, the immune response of CAR-T cells upon stimulation from H292-CD19 and SKOV3-CD19 was confirmed by intracellular IFN-γ staining (Figure S1).
Figure 3.

Reponses of CAR-T cells upon stimulation from target cells
CAR-T cells were co-cultured with PC3-PD-L1-CD19 (also termed PC3-CD19) cells for 16 h in the presence of protein transport inhibitor Brefeldin A. (A–F) IFN-γ (Α), TNF-α (Β), IL-2 (C), Ki67 (D), granzyme B (E), and CD107a (F) were measured by flow cytometry, and the percentages of marker-positive cells were shown in the bar graphs. NT cells were used as a control (n = 3, mean ± SD).
Granzyme B and CD107a expression were measured to assess the cytotoxic function of CAR-T cells upon antigen stimulation. Unlike the results of proinflammatory cytokine production, CD19 CAR-Trap showed a consistently enhanced expression of granzyme B and CD107a in its CD8+ T cell population in all four groups (Figures 3E and 3F). CD19 CAR-αPD-1 had an improved production of granzyme B over that of CD19 CAR in the CD8+ T cell population, but no significant improvement was observed in the production of CD107a (Figures 3D and 3E).
Meanwhile, the expression of Ki67 in CAR-T cells was measured to elucidate the cell-proliferative potential. Among three CAR-T groups, it was found that CD19 CAR-Trap had the highest positive rate of Ki67 in CD4+ or CD8+ T cell populations, followed by CD19 CAR-αPD-1 (Figure 3D).
Taken together, these results clearly show that trap protein secretion differentially influenced T cell functional markers. Although trap protein did not profoundly affect pro-inflammatory cytokine expression, it did significantly enhance the expression of cell proliferation marker and cytotoxicity-related molecules, indicating that CAR-T cells with trap protein secretion can proliferate faster and exert a more potent target killing effect upon short-term stimulation.
Trap protein secretion attenuates TGF-β signaling, reduces the proportion of Treg, and improves the effector cytokine secretion of CAR-T cells
We next sought to study the capability of CAR-T cells with trap protein secretion to lyse target cells by co-culturing CAR-T cells with a series of target cells at various effector-to-target ratios for 24 h. In the co-culture with H292-CD19, SKOV3-CD19, and PC3-CD19 cells, at different effector-to-target ratios, CD19 CAR-Trap exhibited a cell killing capability comparable to that of CD19 CAR and CD19 CAR-αPD-1 (Figure 4A). The antigen-specific cell lysis of CAR-T cells was so potent that saturation was observed at the effector-to-target ratio of 3:1 for H292-CD19 and PC3-CD19 cells and at 1:1 for SKOV3-CD19 cells (Figure 4A). The effect of trap protein secretion on cell killing capability was, however, not significant (ns) within 24 h of co-culture.
Figure 4.

In vitro functionality of CAR-T cells
(A) Cytotoxicity of CAR-T cells against target cells. Three groups of CAR-T cells were co-cultured for 24 h with H292-CD19, SKOV3-CD19, and PC3-CD19 cells at 1:1, 3:1, and 5:1 effector-to-target ratios. NT cells were used as a control (n = 3, mean ± SD). (B) Expression of phosphorylated Smad2/3 (pSmad2/3) in T cells after incubation in different concentrations of recombinant TGF-β for 24 h. The percentages of pSmad2/3+ T cells were shown in bar graphs (n = 3, mean ± SD; ∗∗∗p < 0.001). (C) Representative FACS gating for pSmad2/3+ T cells using NT cells without incubation as a negative control. (D) Percentages of Tregs (CD4+CD25+Foxp3+) in different CAR-T cell groups with or without co-culture with PC3-CD19 cells for 24 h. The data from FACS were shown in bar graphs. NT cells were used as a control (n = 3, mean ± SD; ∗∗∗p < 0.001). (E) CAR-T cells were co-cultured with PC3-CD19 cells for various durations. IFN-γ secretion in supernatants was quantified by ELISA (n = 3, mean ± SD; ns, not significant; ∗∗p < 0.01).
In human CD4+ T cells, antigen stimulation concomitant with TGF-β induces the expression of Foxp3 in naive CD4+ T cells and converts them to CD4+ CD25+ Foxp3+ Tregs.25 To confirm the ability of the trap protein to block the TGF-β signaling pathway, TGF-β-induced phosphorylation of Smad2/3 and Foxp3 expression in human T cells was measured. When CAR-T cells were incubated with TGF-β at various concentrations for 24 h, the phosphorylation of Smad2/3 was elevated in the NT, CD19 CAR, and CD19 CAR-αPD-1 groups in a dose-dependent manner, whereas that in the CD19 CAR-Trap group remained at the basal level (Figures 4B and 4C). The expression of Foxp3 in the CD4+ T cell population, representing the differentiation of Tregs, was measured with or without co-culture with PC3-CD19 cells. We found that CD19 CAR and CD19 CAR-αPD-1 exhibited a greater proportion of Tregs compared with NT, especially after being co-cultured with target cells. However, CD19 CAR-Trap contained fewer Tregs than CD19 CAR and CD19 CAR-αPD-1, irrespective of co-culture with target cells (Figure 4D). Notably, the proportion of Tregs in CD19 CAR-Trap T cells was lower than that in NT cells.
To evaluate the effect of trap protein on effector function, CAR-T cells were co-cultured with PC3-CD19 for different durations. The conditioned supernatants were collected and processed for IFN-γ quantification by ELISA. Upon stimulation by PC3-CD19 for 24 h and 48 h, CD19 CAR-αPD-1 and CD19 CAR-Trap secreted a similar amount of IFN-γ, which was higher than the amount secreted by CD19 CAR (Figure 4E). After 72 h of antigen stimulation, CD19 CAR-Trap had a significantly higher secretion of IFN-γ than either CD19 CAR or CD19 CAR-αPD-1 (Figure 4E).
Trap protein secretion rescues the CAR-T cell from exhaustion by limiting the upregulation of immune-checkpoint molecules
Importantly, trap protein blocks the binding between PD-1 and PD-L1, concomitant with the blocking of TGF-β signaling, as a step to remediate T cell exhaustion. Since PD-1/PD-L1 binding induces increased expression of PD-1 on the T cell surface, we investigated the effect of trap protein on PD-1 expression by co-culturing CAR-T cells with PC3-CD19 cells. After 24 h, all CAR-T groups exhibited upregulated PD-1 expression compared with NT. In contrast, PD-1 expression was significantly lower in the CD19 CAR-αPD-1 and CD19 CAR-Trap groups compared with that in the CD19 CAR group, indicating that the secreted protein drugs, anti-PD-1 scFv and trap protein, played a role in limiting the upregulation of PD-1 expression. No significant difference was observed between CD19 CAR-αPD-1 and CD19 CAR-Trap (Figure 5A). When different T cell subtypes were examined, similar results were observed in both CD4+ T cells and CD8+ T cells (Figure 5B).
Figure 5.

Expression of exhaustion markers in CAR-T cells
(A) CD3+ T cells were shown in each panel. PD-1+CD8+ T cells were gated, and their percentage over total CD3+ T cells was shown in each scatterplot. (B) The percentages of PD-1+CD4+ and PD-1+CD8+ T cells over total CD4+ and CD8+ T cells were shown in bar graphs (n = 3, mean ± SD; ∗∗∗p < 0.001). (C and D) LAG3 expression and TIM3 expression were measured by flow cytometry. The percentages of LAG3+CD8+ and TIM3+CD8+ T cells over total CD8+ T cells were shown in bar graphs (n = 3, mean ± SD; ∗∗p < 0.01). (E and F) PD-L1 expression was measured by flow cytometry. The percentages of PD-L1+CD4+ and PD-L1+CD8+ over total CD4+ and CD8+ T cells were shown in bar graphs (n = 3, mean ± SD; ∗p < 0.05; ∗∗p < 0.01).
In addition to PD-1, other immune-checkpoint molecules are expressed on the T cell surface, such as lymphocyte-activation gene 3 (LAG3), T cell immunoglobulin domain and mucin domain-containing protein 3 (TIM3), and PD-L1, contributing to T cell exhaustion upon antigen stimulation as a complex interaction network.26, 27, 28 To further evaluate the capability of trap protein to rescue CAR-T cells under these conditions, we tested the expression of LAG3, TIM3, and PD-L1 in CAR-T cells after co-culture with PC3-CD19 cells for 24 h. Again, CAR-αPD-1 and CD19 CAR-Trap had less LAG3 and PD-L1 upregulation than that exhibited by CD19 CAR (Figures 5C, 5E, and 5F). In the analysis for TIM3, CAR-αPD-1 and CD19 CAR-Trap showed a trend toward the inhibition of TIM3 upregulation, but it did not reach statistical significance (Figure 5D). The effect of trap protein to alleviate CAR-T cell exhaustion was also confirmed by the data collected from co-culture with H292-CD19 and SKOV3-CD19 (Figures S2–S5).
Trap protein-secreting CAR-T cells exhibit enhanced expansion, infiltration, and antitumor efficacy in vivo
Having confirmed the in vitro specificity and functionality of CD19 CAR-Trap in response to CD19+PD-1+ target cells, we continued to evaluate the antitumor potential of CD19 CAR-Trap cells in vivo utilizing a subcutaneous human prostate cancer xenograft model in NOD.Cg-PrkdcscidIL2Rgtm1Wjl/Sz (NSG) mice. The experimental procedure for animal study is shown in Figure 6A.
Figure 6.

Antitumor efficacy, infiltration, and functionality of CAR-T cells in vivo
(A) Schematic representation of the in vivo experimental procedure. NSG mice were subcutaneously (s.c.) injected with 3 × 106 of PC3-CD19 tumor cells into the right flank. After 16 days, when the tumors grew to 75−100 mm3, 2 × 106 of CD19 CAR, CD19 CAR-αPD-1, or CAR19 CAR-Trap T cells were adoptively transferred through intravenous (i.v.) injection. Tumor volume was measured every other day. The mice were euthanized for analysis on day 12 post-treatment. (B) Tumor growth curve for mice treated with NT, CD19 CAR, CD19 CAR-αPD-1, or CAR19 CAR-Trap T cells (n = 5, mean ± SD; ∗∗p < 0.01; ∗∗∗p < 0.001). (C) The ratio of CD8+ versus CD4+ T cells in the tumor (n = 5, mean ± SD; ∗∗∗p < 0.001). (D) The percentage of PD-1+ TILs over total TILs (n = 5, mean ± SD; ∗p < 0.05). (E) The percentage of Tregs (CD45+CD4+CD25+Foxp3+ cells) over total TILs (n = 5, mean ± SD; ∗∗p < 0.01; ∗∗∗p < 0.001). (F) The percentage of pSmad2/3+ TILs over total TILs (n = 5, mean ± SD; ∗∗p < 0.01). (G) The percentage of CD45+ T cells in tumor, blood, spleen, and bone marrow tissues of PC3-CD19 tumor-bearing mice of different groups (n = 5, mean ± SD; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001). (H) A representative FACS scatterplot of the percentage of CD45+ T cells in tumor, blood, spleen, and bone marrow tissues of different groups.
Briefly, PC3-CD19 cells (3 × 106) were injected into the right flank of NSG mice. When the average tumor size reached 75−100 mm3 on day 16 post-tumor inoculation, the tumor-bearing mice were randomized into four groups (5 mice per group), and they received 2 × 106 CAR-T cells through intravenous injection. During the 12-day tumor-growth monitoring period, groups receiving CAR-T treatment groups showed more efficacy in inhibiting tumor progression compared to the NT group. Among CAR-T treatment groups, CD19 CAR-Trap exhibited significantly slower tumor growth than CD19 CAR and CD19 CAR-αPD-1, suggesting enhanced antitumor efficacy (Figure 6B). In comparison between CD19 CAR and CD19 CAR-αPD-1, no significant difference was observed.
On day 12 post-treatment, mice were all euthanized, and T cells in tumors were analyzed. In the analysis of tumor samples, T cell infiltration of the NT group was negligible; therefore, we only focused on the three CAR-T treatment groups. In terms of tumor-infiltrating lymphocytes (TILs), the CD19 CAR-Trap groups had a higher CD8 T:CD4 T ratio than either the CD19 CAR group or CD19 CAR-αPD-1 group (Figure 6C). Consistent with the data from in vitro studies, treatment with CD19 CAR-Trap resulted in inhibition of PD-1 expression on TILs, differentiation of Foxp3+ Tregs, and TGF-β signaling (Figures 6D–6F). Furthermore, TILs of the CD19 CAR-Trap treatment group displayed a slightly higher proportion of effector memory T cells than either the CD19 CAR group or CD19 CAR-αPD-1 group (Figure S6).
To compare the CAR-T infiltration and expansion in vivo, tumor, blood, spleen, and bone marrow of each group were harvested and examined. The T cell population was identified using tissue samples from a nontreated mouse as a control (Figure 6H). We found that CD19 CAR-Trap maintained a high proportion of T cells (14%) in tumors on day 12 post-treatment, which was more than three times the T cell percentages shown in the CD19 CAR group (1%) and CD19 CAR-αPD-1 group (4%) (Figure 6G). Similar results were shown in the analysis for blood, spleen, and bone marrow. Compared with NT, all CAR-T treatment groups had more T cells present, but CD19 CAR-Trap exhibited a significantly higher level than other CAR-T groups (Figure 6G). In a nutshell, within 12 days, CD19 CAR-Trap showed a superior expansion in tumor, blood, spleen, and bone marrow.
Trap protein-secreting CAR-T cells achieve long-term remission and prevent tumor relapse
To further explore the antitumor potential of CD19 CAR-Trap, we conducted a long-term in vivo efficacy study with the dose of CAR-T cells doubled (4 × 106 cells). The establishment of the tumor model was the same as previously described. As shown in Figure 7A, tumor-bearing mice were randomized into four groups (8 mice per group) with an average tumor size of ~100 mm3. Most mice of the NT group reached the endpoint tumor size of 1000 mm3 within the 12 days. Although CD19 CAR and CD19 CAR-αPD-1 indistinguishably slowed the tumor growth rate, CD19 CAR-Trap decreased the tumor size by more than 50% (Figure 7B). Notably, we found that tumors of mice in the CD19 CAR-Trap group shrank faster when the tumor volume got smaller, and they could be detected at 16 days post-treatment. In the meantime, no significant body weight loss was observed in any treatment group (Figure 7C).
Figure 7.

Long-term antitumor efficacy and immune surveillance of CAR-T cells in vivo
(A) Schematic representation of the in vivo experimental procedure. NSG mice were s.c. injected with 3 × 106 of PC3-CD19 tumor cells into the right flank. After 16 days, when the tumors grew to ~100 mm3, 4 × 106 of CD19 CAR, CD19 CAR-αPD-1, or CAR19 CAR-Trap T cells were adoptively transferred through i.v. injection. Tumor volume was measured every other day. The mice were monitored until they met the endpoint. On day 60 post-treatment, mice in the CD19 CAR-Trap group were rechallenged with 1 × 106 PC3-CD19 tumor cells and monitored for another 2 weeks. (B) Tumor growth curve for mice treated with NT, CD19 CAR, CD19 CAR-αPD-1, or CD19 CAR-Trap T cells (n = 8, mean ± SD; ∗∗∗p < 0.001). (C) Waterfall plot analysis of tumor reduction on day 12 post-treatment for various treatment groups. (D) Body weight curve for mice in various treatment groups (n = 8, mean ± SD). (E) Survival of PC3-CD19 tumor-bearing NSG mice after indicated treatment. Overall survival curves were plotted using the Kaplan-Meier method.
After taking out the mice that met the endpoint, we continued to monitor the survival of mice for 60 days. The 8 mice of the CD19 CAR-Trap group kept complete remission until the end. In the CD19 CAR and CD19 CAR-αPD-1 groups, only 1 mouse achieved complete remission. Compared with either CD19 CAR (12.5%) or CD19 CAR-αPD-1 (12.5%), CD19 CAR-Trap significantly improved the survival rate (100%) (Figure 7D). The 8 mice of CD19 CAR-Trap group were then bled on day 61 for T cell analysis. Except for 2 mice found to develop graft-versus-host disease (GvHD) with an abnormally high percentage of T cells in blood and GvHD symptoms, 6 mice had percentages of T cells that ranged from 3% to 8% (Figure S7). We injected those 6 mice with 1 × 106 PC3-CD19 cells into the left flank subcutaneously and monitored them for another 2 weeks. None of the 6 mice developed detectable tumors.
Discussion
Our strategy to overcome the current hurdles faced by CAR-T therapy in solid tumor treatment involved the engineering of CAR-T cells to secrete bifunctional trap proteins into the TME, simultaneously targeting checkpoint-molecule PD-1 and soluble immunosuppressive molecule TGF-β. The in vitro studies showed that the secreted trap protein significantly enhanced CAR-T cell proliferation and the production of cytotoxicity-related molecules upon antigen stimulation, whereas inhibiting the activation of Tregs and upregulation of immune-checkpoint molecules in vitro. In the xenograft mouse model, we established that CAR-T cells with trap protein secretion exhibited not only superior antitumor efficacy but also enhanced tumor infiltration and expansion when compared with conventional CAR-T cells and CAR-T cells with anti-PD-1 scFv secretion.
Over the years, despite the unprecedented success that CAR-T therapy has achieved in hematological malignancies, the clinical results of it in solid tumor treatment are still far from satisfactory.29, 30, 31, 32, 33 Unlike hematological malignancies, the unique TME poses significant challenges for CAR-T cells to mount their effector function and expand at the tumor sites. PD-1/PD-L1 pathway and TGF-β signaling pathway have been demonstrated as the two major resistance mechanisms exerted by solid tumors in response to T cell antitumor activity.8,34 In clinical studies, single agents targeting only one signaling pathway are unable to induce effective immune responses in most patients.35,36 To assess the potential of CAR-T cells with trap protein secretion to solve these issues, an ideal tumor model should have three characteristics: (1) overexpression of tumor-associated antigen; (2) high expression level of PD-L1; and (3) high secretion level of TGF-β. Thus, we chose the prostate cancer cell line PC3 known to have high endogenous TGF-β secretion, and we engineered it to overexpress PD-L1 and CD19, the well-documented antigen in CAR-T research.37 Engineered PC3-CD19, together with engineered ovarian cancer cell line SKOV3-CD19 and lung cancer cell line H292-CD19, as target cell lines, provided the basis for our in vitro and in vivo analyses.
Numerous studies have shown that activation of the PD-1/PD-L1 pathway in T cells inhibits cytokine production, T cell proliferation, and effector function.8 In our previous study, the secretion of anti-PD-1 scFv was demonstrated to help CAR-T cells maintain effector function and rescue CAR-T cells from exhaustion in vitro.13 In this study, to investigate the effect of a secreted trap protein, we conducted similar co-cultures between CAR-T cells and the target cell lines, and we found that CAR-T cells with trap protein secretion showed significantly improved cytotoxic function by elevated granzyme B and CD107a expression and higher proliferation potential by higher Ki67 expression in comparison with the parental CAR-T cells. For effector cytokine IFN-γ, a higher secretion level from the trap protein self-secreting CAR-T cells was observed, especially when co-cultured with target cells for a long period. In these analyses, anti-PD-1 self-secreting CAR-T cells showed a similar, but weaker, enhancement. The different results between anti-PD-1 self-secreting CAR-T and trap protein self-secreting CAR-T might result from the blocking effect of trap protein against the TGF-β signaling pathway. Dimeloe et al.38 reported that tumor-derived TGF-β inhibits IFN-γ production by human CD4+ T cells but that the production capacity can be restored by applying TGF-β-neutralizing antibodies. In addition to inhibition of T cell proliferation, TGF-β also inhibits the expression of perforin, granzyme A, granzyme B, and IFN-γ by CD8+ T cells, as described by Thomas and Massagué.16 In the staining for CAR expression, we noticed that the self-secreting CAR-T cells had a lower CAR density than the parental CAR-T cells, probably because the insertion of additional DNA sequences, which encode anti-PD-1 scFv or trap protein, impairs vector production and transduction efficiency. However, the cytotoxic effector functions of self-secreting CAR-T cells were not comprised, as indicated by our in vitro and in vivo results.
Upon persistent antigen stimulation, the expression level of PD-1 in T cells is upregulated.39 To study the ability of the trap protein to overcome suppression posed by immune checkpoints, we analyzed the expression of PD-1 and its co-expressing immune-checkpoint molecules, such as LAG3, TIM3, and PD-L1, in CAR-T cells post-antigen stimulation. Consistent with the findings from our previous study, the self-secreting CAR-T cells alleviated upregulation of PD-1, LAG3, TIM3, and PD-L1 after co-culture with various target cell lines. Positive correlations among the expressions of PD-1, TIM3, and LAG3 have been reported by Zhou et al.40. Our data indicate that the secreted trap protein alters the signaling network of immune-checkpoint molecules, rather than merely targeting the PD-1/PD-L1 pathway. The inhibitory effect of trap protein on the expression of LAG3, TIM3, and PD-L1 might then contribute to the enhanced functions of trap protein self-secreting CAR-T cells, as well.
On the other hand, both autocrine and paracrine TGF-β in the TME promote differentiation of Tregs and attenuate the activation of T cells through the induction of Smad2/3 phosphorylation and Foxp3 expression.15 In the incubation with recombinant TGF-β, we found that the phosphorylation of Smad2/3 in conventional CAR-T cells and anti-PD-1 self-secreting CAR-T cells increased with TGF-β concentration, whereas trap protein self-secreting CAR-T cells remained at the basal level, indicating that trap protein enables CAR-T cells to counteract TGF-β-mediated signaling. Apart from tumor cell-derived TGF-β, it has been reported that T cell-derived TGF-β also regulates T cell differentiation and tolerance.41 However, it is not known if T cell-derived TGF-β will be increased in genetically modified CAR-T cells. Even without antigen stimulation, we noticed that conventional CAR-T cells and anti-PD-1 self-secreting CAR-T cells had an elevated percentage of Tregs compared with NT T cells. Protected by the inhibition of TGF-β signaling pathway, trap protein self-secreting CAR-T cells kept a small Treg proportion with or without antigen stimulation.
The superior antitumor efficacy of trap protein self-secreting CAR-T cells was demonstrated in the PC3-CD19 xenograft mouse model. At a low dose of 2 million CAR-T cells, trap protein self-secreting CAR-T cells achieved stability of tumor growth in the treated mice, whereas other treatments failed to control tumor progression. The undifferentiated efficacy of conventional CAR-T and anti-PD-1 self-secreting CAR-T cells suggests that targeting only the PD-1/PD-L1 pathway is not enough to enhance CAR-T cells in this progressive prostate tumor model. CD8+/CD4+ T cell ratio at the tumor site is another important prognostic marker in immunotherapy. In solid tumors, research has found that a high CD8+/CD4+ T cell ratio is often correlated with absence of metastasis, slow tumor progression, and improved survival.42, 43, 44 In our in vivo results, the CD8+/CD4+ T cell ratio of the trap protein self-secreting CAR-T group was twice that of the conventional CAR-T group and anti-PD-1 self-secreting CAR-T group, indicating that trap protein self-secreting CAR-T cells are skewed toward an effector phenotype, thereby possibly contributing to the enhanced inhibition of tumor progression. Although CD4+ T cells are not the major T cell population within the tumor-infiltrating T cells, they contribute to overall antitumor efficacy via multiple mechanisms in the presence of trap protein that can block TGF-β signaling. Recent studies have shown that TGF-β blockade not only increases IL-2 and IFN-γ secretion of CD4+ T helper type 1 (Th1) cells but also improves IL-4 expression of CD4+ Th2 cell, causing reorganization of tumor vasculature, tumor tissue hypoxia, and the consequent tumor cell death.45,46 For the expressions of biomarkers related to PD-1/PD-L1 and TGF-β pathways, we found consistency between in vivo and in vitro data. The trap protein self-secreting CAR-T group exhibited lower expression of PD-1, attenuated phosphorylation of Smad2/3, and a smaller proportion of Foxp3+ Tregs. Taken together, these findings can help to explain why the trap protein self-secreting CAR-T group exhibits dramatically improved antitumor efficacy and T cell expansion in tumor, spleen, and bone marrow.
In the subsequent in vivo study, with the treatment protocol slightly modified by doubling the dose of CAR-T cells, we focused on the capability of trap protein self-secreting CAR-T cells to eradicate solid tumors and to improve survival. Within 12 days, the change in dose showed no significant impact on the tumor growth of the conventional CAR-T group and anti-PD-1 self-secreting CAR-T group; it did lead to a remarkable tumor shrinkage in the trap protein self-secreting CAR-T group. Trap protein self-secreting CAR-T cells successfully eradicated tumors on day 16 post-treatment and improved survival rate compared with the conventional CAR-T group and anti-PD-1 self-secreting CAR-T group. For 6 out of the 8 mice in the trap protein self-secreting CAR-T group, it is worth noting that CAR-T cells can persist in the body for 60 days without causing GvHD. When tumor rechallenge was given by injecting tumor cells into the cured mice, no new tumor was developed, suggesting that the remaining trap protein self-secreting CAR-T cells in the mouse body prevent tumor recurrence.
In conclusion, this proof-of-concept study provides support for the strategy of utilizing self-secreting CAR-T cells for co-targeting two immunosuppressive mechanisms in the TME. This three-in-one approach could potentially improve the clinical outcome of CAR-T therapy by rescuing CAR-T cells from immunosuppression and enhancing the expansion and functionalities of CAR-T cells. Moreover, the protein drug secreted by CAR-T cells could achieve a localized accumulation at the tumor site, which may improve the safety profile of combinational CAR-T therapy by avoiding toxicities associated with systemic administration of protein drugs. Although the trap protein self-secretion strategy has shown promising results, further modifications and improvements could be made to expand CAR-T therapy in different clinical settings. First, the antigen CD19 used in this proof-of-concept study is not a solid tumor antigen. To improve the clinical relevance of this therapeutic strategy, solid tumor antigens, such as prostate-specific membrane antigen (PSMA) or human epidermal growth factor receptor 2 (HER2), could be exploited in the future. Second, given the numerous dynamic interplays between cells and molecules in the TME, the simultaneous inhibition of PD-1/PD-L1 and TGF-β signaling pathways may even have a more profound effect than the results of our in vivo study might suggest. Therefore, it would be beneficial to unveil the antitumor potential of the trap protein self-secreting CAR-T cells in syngeneic mouse models. Finally, this self-secretion platform is modular and flexible, and it can be applied to combinations of different antibodies and binding sequences for receptors or ligands. For example, the combinations of anti-PD-L1 antibody or anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibody with TGF-β, already shown to have antitumor potential in previous studies, can also be explored.24
Materials and methods
Cell lines
SKOV3 ovarian cancer cell line, PC3 prostate cancer cell line, and 293T were obtained from ATCC. NCI-H292 lung cancer cell line was kindly provided by Dr. Ite Laird-Offringa. The PC3 cell line was transduced with viral vector to express a high level of PD-L1 on the cell surface as PC3-PD-L1 and sorted to 99% purity. The SKOV3, NCI-H292, and PC3-PD-L1 cell lines were transduced with a lentiviral vector FUW-CD19. The transduced cells were then stained with anti-human CD19 antibody and sorted to 99% purity. The cell culture methods for 293T, engineered SKOV3-CD19, and H292-CD19 were described in our previous publications.13,47 PC3 and PC3-CD19 were maintained in D10 medium consisting of DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin.
Plasmid construction
The retroviral vectors encoding anti-CD19 CAR (CD19 CAR) and CD19 CAR-αPD-1 were constructed based on the MP71 retroviral vector kindly provided by Prof. Wolfgang Uckert, as described previously.13 The vector encoding CD19 CAR-Trap was generated based on CD19 CAR-αPD-1. The RV-CD19 CAR-Trap vector consisted of the following components in frame from 5′ end to 3′ end: the MP71 retroviral backbone, a NotI site, the anti-CD19 CAR, a T2A sequence, a human IL-2 leading sequence, the anti-PD-1 scFv light-chain variable region (GGGGS)3, the anti-PD-1 scFv heavy-chain variable region (GGGGS)2, a TGF-β-binding sequence, and an EcoRI site. Another trap sequence with the same components followed by a His-tag sequence was inserted into the pcDNA3.1 vector for trap protein purification and binding assessment.
The anti-PD-1 scFv in the CD19 CAR-Trap vector was derived from the amino acid sequence of monoclonal antibody 5C4-specific against human PD-1. The TGF-β-binding sequence was derived from the amino acid sequence of the ligand-binding region in the human TGF-βRII extracellular domain. The amino acid sequences were codon optimized using an online codon optimization tool, and corresponding DNA sequences were synthesized by Integrated DNA Technologies. The DNA sequence encoding trap protein was ligated into the CD19 CAR vector through the Gibson assembly method to generate the CD19 CAR-Trap vector.
Trap protein purification and western blotting analysis
Using PEIpro (Polyplus) as the transfection reagents, 293T cells were transfected with the pcDNA3.1 encoding trap protein labeled with His-tag. 3 days after transfection, cell culture supernatant was harvested, and trap protein was purified with HisPur Ni-NTA Resin (Thermo Scientific), following the manufacturer’s instruction. The purified trap protein was then quantified using a Micro BCA (bicinchoninic acid) Protein Assay Kit (Thermo Scientific).
20 ng and 200 ng of purified protein were resolved by SDS polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinylidene fluoride (PVDF) membrane (Bio-Rad). The PVDF membrane was blocked with 5% bovine serum albumin (BSA) for 2 h and then incubated with anti-6× His-tag antibody-horseradish peroxidase (HRP; 1:10,000 dilution; Abcam) at 4°C overnight. After incubation, the membrane was washed and developed with western blotting substrate (Thermo Scientific) and visualized with the enhanced chemiluminescence (ECL) machine (Bio-Rad).
Retroviral vector production
Retroviral vectors were prepared by transient transfection of 293T cells using a standard calcium phosphate precipitation method. Briefly, 293T cells were seeded in a 15-cm tissue-culture dish at a density of 0.8 × 106 cells/mL. When confluency reached 70%–80%, 293T cells were transfected with 37.5 μg of the retroviral backbone plasmid, together with 18.75 μg of the envelope plasmid RD114 and 30 μg of the packaging plasmid encoding gag-pol. The supernatants were harvested at 48 h after transfection and filtered through a 0.45-μm filter prior to use.
CAR-T cell production and expansion
Frozen human PBMCs were obtained from AllCells. The T cell culture medium (TCM) was composed of AIM-V medium with 5% (v/v) human AB serum, 10 mM HEPES, 1% (v/v) GlutaMax-100×, 12.25 mM N-acetylcysteine (NAC), 100 U/mL penicillin, and 100 μg/mL streptomycin. The cell culture was supplemented with 10 ng/mL human IL-2. After the activation by Dynabeads Human T-Expander CD3/CD28 (Invitrogen) at a bead:PBMC ratio of 3:1 for 48 h, PBMCs were transduced with retroviral vectors using the method previously described. Transduced T cells were expanded for 2 weeks in TCM, during which, time-culture medium was replenished every 2 days, and T cell density was maintained between 0.5 and 1 × 106 cells/mL.
Flow cytometry
A MACSQuant Analyzer was used for flow cytometry analysis. Before staining, cells were harvested and washed with fluorescence-activated cell sorting (FACS) buffer (PBS containing 4% BSA fraction V). To detect CAR expression on the T cell surface, we used rat anti-mouse biotin-conjugated F(ab′)2 antibody (Abcam), followed by allophycocyanin (APC)-conjugated streptavidin (BioLegend). Anti-human PD-L1 antibody and anti-human CD19 antibody (BioLegend) were used to detect the expression of PD-L1 and CD19 on the target cell surface, respectively. The following antibodies were used for T cell phenotyping: CD3-fluorescein isothiocyanate (FITC), CD45-phycoerythrin (PE), CD8-Pacific Blue, CD4-PerCP/Cy5.5, PD-1-Brilliant Violet 421, PD-L1-PE, TIM3-PE, LAG3-PerCP/Cy5.5, CD107a-APC, CD45RO-PE, CD62L-APC, and CD25-Pacific Blue (BioLegend). For cell-surface staining, cells were stained with appropriate antibodies in FACS buffer at 4°C for 15 min. For intracellular staining, cells were permeabilized by Cytofix/Cytoperm Fixation and Permeabilization Solution Kit (BD Biosciences) before being stained with appropriate antibodies in FACS buffer at 4°C for 30 min. The following antibodies were used for T cell intracellular staining: IFN-γ-PE, phospho-Smad2/3-APC, FoxP3-APC, Ki67-Brilliant Violet 421, granzyme B-PE, IL-2-APC, and TNF-α-PE. Prior to staining for cells from mouse organs (tumor, spleen, bone marrow, blood), 1× lysing solution (BD Biosciences) was used to lyse the red blood cells on ice for 10 min, as recommended by the manufacturer.
ELISA
The TGF-β secretion levels of target cells were evaluated by the Human TGF-β1 ELISA Kit (BD Biosciences). Cancer cell lines were seeded at 1 × 106 cells per well in a 6-well tissue-culture plate (Corning) for 24 h, and the supernatants were harvested. The concentration of TGF-β1 in the supernatants was determined by following the manufacturer’s protocol.
The binding ability of trap protein to PD-1 and TGF-β protein was evaluated by sandwich ELISA, wherein recombinant human PD-1-Fc (GenScript) or TGF-β protein (R&D Systems) (200 ng/mL) was coated on the plates, followed by purified trap protein (0−40 μg/mL) detected by anti-6 × His-tag antibody-HRP (1:500 diluted; Abcam).
IFN-γ secreted by T cells was measured by the Human IFN-γ ELISA Set (BD Biosciences). Briefly, 2 × 105 CAR-T cells were co-cultured with 2 × 105 PC3-CD19 cells per well in a 96-well round-bottom plate in a 200-μL vol of complete media. Supernatants were harvested at 24 h, 48 h, and 72 h after co-culture. The concentration of IFN-γ in the supernatants was determined by following the manufacturer’s protocol.
Cytotoxicity assay
The cytotoxic activity of CAR-T cells against target cells was evaluated by a 24-h co-culture assay using target cells pre-labeled with CFSE (Invitrogen), as described previously. We co-cultured NT cells and CAR-T cells with target cells (5 × 104 cells/well) at effector:target ratios of 1:1, 3:1, and 5:1. After co-culture, 7AAD (BD Biosciences) was added, as recommended by the manufacturer. Flow cytometric analysis was performed to quantify the dead target cells (CFSE+ and 7-AAD+). Cell cytotoxicity was calculated as CFSE+7-AAD+ cells/(CFSE+7-AAD− + CFSE+7-AAD+) cells.
Tumor model and adoptive transfer
The animal experiments were conducted following the animal protocol approved by the University of Southern California (USC) Institutional Animal Care and Use Committee (IACUC). 6- to 8-week-old female NSG mice (Jackson Laboratory) were inoculated with 3 × 106 PC3-CD19 cells subcutaneously. When the tumor size reached 75−100 mm3, the mice were randomized into 4 groups and were treated with 2 × 106 (in the short-term study) or 4 × 106 (in the long-term study) CAR-T-positive cells in 100 μL of PBS intravenously via tail-vein injection. An equal number of donor-matched NT T cells were used as a control. CAR expression level was normalized to 20% in all of the CAR-T groups by adding NT cells prior to injection. Tumor growth was monitored every 2 days. Tumor size was calculated as (width2 × length)/2. For the short-term study, mice were euthanized on day 12 post-treatment. Then ex vivo analysis was performed on harvested tumors, spleens, bone marrows, and blood. For the long-term study, mice were euthanized when they displayed obvious weight loss, ulceration of tumors, or tumor size larger than 1,000 mm3.
Statistical analysis
Statistical analysis was performed in GraphPad Prism, version 7.0a. The differences among groups were determined with two-way analysis of variance (ANOVA) with Tukey’s multiple comparison. Tumor growth curve was analyzed using one-way ANOVA with repeated measures (Tukey’s multiple comparison method). Mouse survival curve was evaluated by the Kaplan-Meier method. A p value <0.05 was considered statistically significant. Significance of findings was defined as follows: ns, p > 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01AI068978, R01CA170820, R01EB017206, and P01CA132681) and a grant from the Ming Hsieh Institute for Research on Engineering-Medicine for Cancer.
Author contributions
Conception and design, X.C., S.L., and P.W.; development of methodology, X.C., S.Y., S.L., and P.W.; acquisition of data, X.C., S.Y., Y.Q., H.-Y.W., J.L., Z.S.D., G.E.C., F.H., and X.Z.; analysis and interpretation of data, X.C., S.Y., H.-Y.W., and P.W.; writing, review, and/or revision, X.C., H.-Y.W., M.A.M., and P.W.; study supervision, P.W.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.03.014.
Supplemental information
References
- 1.Jackson H.J., Rafiq S., Brentjens R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016;13:370–383. doi: 10.1038/nrclinonc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sadelain M. T-cell engineering for cancer immunotherapy. Cancer J. 2009;15:451–455. doi: 10.1097/PPO.0b013e3181c51f37. [DOI] [PubMed] [Google Scholar]
- 3.Almåsbak H., Aarvak T., Vemuri M.C. CAR T Cell Therapy: A Game Changer in Cancer Treatment. J. Immunol. Res. 2016;2016:5474602. doi: 10.1155/2016/5474602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadelain M., Brentjens R., Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schultz L., Mackall C. Driving CAR T cell translation forward. Sci. Transl. Med. 2019;11:eaaw2127. doi: 10.1126/scitranslmed.aaw2127. [DOI] [PubMed] [Google Scholar]
- 6.Junttila M.R., de Sauvage F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 7.Rafiq S., Hackett C.S., Brentjens R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020;17:147–167. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez M., Moon E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon E.K., Wang L.C., Dolfi D.V., Wilson C.B., Ranganathan R., Sun J., Kapoor V., Scholler J., Puré E., Milone M.C. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014;20:4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGowan E., Lin Q., Ma G., Yin H., Chen S., Lin Y. PD-1 disrupted CAR-T cells in the treatment of solid tumors: Promises and challenges. Biomed. Pharmacother. 2020;121:109625. doi: 10.1016/j.biopha.2019.109625. [DOI] [PubMed] [Google Scholar]
- 11.John L.B., Devaud C., Duong C.P., Yong C.S., Beavis P.A., Haynes N.M., Chow M.T., Smyth M.J., Kershaw M.H., Darcy P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013;19:5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 12.Heczey A., Louis C.U., Savoldo B., Dakhova O., Durett A., Grilley B., Liu H., Wu M.F., Mei Z., Gee A. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017;25:2214–2224. doi: 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li S., Siriwon N., Zhang X., Yang S., Jin T., He F., Kim Y.J., Mac J., Lu Z., Wang S. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017;23:6982–6992. doi: 10.1158/1078-0432.CCR-17-0867. [DOI] [PubMed] [Google Scholar]
- 14.Massagué J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li M.O., Flavell R.A. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas D.A., Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 17.Oida T., Xu L., Weiner H.L., Kitani A., Strober W. TGF-beta-mediated suppression by CD4+CD25+ T cells is facilitated by CTLA-4 signaling. J. Immunol. 2006;177:2331–2339. doi: 10.4049/jimmunol.177.4.2331. [DOI] [PubMed] [Google Scholar]
- 18.Shevach E.M. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 19.Ganesh K., Massagué J. TGF-β Inhibition and Immunotherapy: Checkmate. Immunity. 2018;48:626–628. doi: 10.1016/j.immuni.2018.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Löffek S. Transforming of the Tumor Microenvironment: Implications for TGF-β Inhibition in the Context of Immune-Checkpoint Therapy. J. Oncol. 2018;2018:9732939. doi: 10.1155/2018/9732939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Principe D.R., Park A., Dorman M.J., Kumar S., Viswakarma N., Rubin J., Torres C., McKinney R., Munshi H.G., Grippo P.J., Rana A. TGFβ Blockade Augments PD-1 Inhibition to Promote T-Cell-Mediated Regression of Pancreatic Cancer. Mol. Cancer Ther. 2019;18:613–620. doi: 10.1158/1535-7163.MCT-18-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X., Wang L., Li P., Song M., Qin G., Gao Q., Zhang Z., Yue D., Wang D., Nan S. Dual TGF-β and PD-1 blockade synergistically enhances MAGE-A3-specific CD8+ T cell response in esophageal squamous cell carcinoma. Int. J. Cancer. 2018;143:2561–2574. doi: 10.1002/ijc.31730. [DOI] [PubMed] [Google Scholar]
- 23.Holmgaard R.B., Schaer D.A., Li Y., Castaneda S.P., Murphy M.Y., Xu X., Inigo I., Dobkin J., Manro J.R., Iversen P.W. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer. 2018;6:47. doi: 10.1186/s40425-018-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ravi R., Noonan K.A., Pham V., Bedi R., Zhavoronkov A., Ozerov I.V., Makarev E., V Artemov A., Wysocki P.T., Mehra R. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat. Commun. 2018;9:741. doi: 10.1038/s41467-017-02696-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen W., Jin W., Hardegen N., Lei K.J., Li L., Marinos N., McGrady G., Wahl S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baumeister S.H., Freeman G.J., Dranoff G., Sharpe A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016;34:539–573. doi: 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 27.Huang R.Y., Francois A., McGray A.R., Miliotto A., Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. OncoImmunology. 2016;6:e1249561. doi: 10.1080/2162402X.2016.1249561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koyama S., Akbay E.A., Li Y.Y., Herter-Sprie G.S., Buczkowski K.A., Richards W.G., Gandhi L., Redig A.J., Rodig S.J., Asahina H. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017;9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmed N., Brawley V., Hegde M., Bielamowicz K., Kalra M., Landi D., Robertson C., Gray T.L., Diouf O., Wakefield A. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thistlethwaite F.C., Gilham D.E., Guest R.D., Rothwell D.G., Pillai M., Burt D.J., Byatte A.J., Kirillova N., Valle J.W., Sharma S.K. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017;66:1425–1436. doi: 10.1007/s00262-017-2034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang C., Wang Z., Yang Z., Wang M., Li S., Li Y., Zhang R., Xiong Z., Wei Z., Shen J. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA+ Metastatic Colorectal Cancers. Mol. Ther. 2017;25:1248–1258. doi: 10.1016/j.ymthe.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y., Chen M., Wu Z., Tong C., Dai H., Guo Y., Liu Y., Huang J., Lv H., Luo C. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. OncoImmunology. 2018;7:e1440169. doi: 10.1080/2162402X.2018.1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newick K., O’Brien S., Moon E., Albelda S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017;68:139–152. doi: 10.1146/annurev-med-062315-120245. [DOI] [PubMed] [Google Scholar]
- 35.Nowicki T.S., Hu-Lieskovan S., Ribas A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. 2018;24:47–53. doi: 10.1097/PPO.0000000000000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohn A., Lahn M.M., Williams K.E., Cleverly A.L., Pitou C., Kadam S.K., Farmen M.W., Desaiah D., Raju R., Conkling P., Richards D. A phase I dose-escalation study to a predefined dose of a transforming growth factor-β1 monoclonal antibody (TβM1) in patients with metastatic cancer. Int. J. Oncol. 2014;45:2221–2231. doi: 10.3892/ijo.2014.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kloss C.C., Lee J., Zhang A., Chen F., Melenhorst J.J., Lacey S.F., Maus M.V., Fraietta J.A., Zhao Y., June C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018;26:1855–1866. doi: 10.1016/j.ymthe.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dimeloe S., Gubser P., Loeliger J., Frick C., Develioglu L., Fischer M., Marquardsen F., Bantug G.R., Thommen D., Lecoultre Y. Tumor-derived TGF-β inhibits mitochondrial respiration to suppress IFN-γ production by human CD4+ T cells. Sci. Signal. 2019;12:eaav3334. doi: 10.1126/scisignal.aav3334. [DOI] [PubMed] [Google Scholar]
- 39.Simon S., Labarriere N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? OncoImmunology. 2017;7:e1364828. doi: 10.1080/2162402X.2017.1364828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou G., Sprengers D., Boor P.P.C., Doukas M., Schutz H., Mancham S., Pedroza-Gonzalez A., Polak W.G., de Jonge J., Gaspersz M. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology. 2017;153:1107–1119.e10. doi: 10.1053/j.gastro.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 41.Oh S.A., Li M.O. TGF-β: guardian of T cell function. J. Immunol. 2013;191:3973–3979. doi: 10.4049/jimmunol.1301843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eerola A.K., Soini Y., Pääkkö P. A high number of tumor-infiltrating lymphocytes are associated with a small tumor size, low tumor stage, and a favorable prognosis in operated small cell lung carcinoma. Clin. Cancer Res. 2000;6:1875–1881. [PubMed] [Google Scholar]
- 43.Piersma S.J., Jordanova E.S., van Poelgeest M.I., Kwappenberg K.M., van der Hulst J.M., Drijfhout J.W., Melief C.J., Kenter G.G., Fleuren G.J., Offringa R., van der Burg S.H. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res. 2007;67:354–361. doi: 10.1158/0008-5472.CAN-06-3388. [DOI] [PubMed] [Google Scholar]
- 44.Nakano O., Sato M., Naito Y., Suzuki K., Orikasa S., Aizawa M., Suzuki Y., Shintaku I., Nagura H., Ohtani H. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001;61:5132–5136. [PubMed] [Google Scholar]
- 45.Li S., Liu M., Do M.H., Chou C., Stamatiades E.G., Nixon B.G., Shi W., Zhang X., Li P., Gao S. Cancer immunotherapy via targeted TGF-β signalling blockade in TH cells. Nature. 2020;587:121–125. doi: 10.1038/s41586-020-2850-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu M., Kuo F., Capistrano K.J., Kang D., Nixon B.G., Shi W., Chou C., Do M.H., Stamatiades E.G., Gao S. TGF-β suppresses type 2 immunity to cancer. Nature. 2020;587:115–120. doi: 10.1038/s41586-020-2836-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siriwon N., Kim Y.J., Siegler E., Chen X., Rohrs J.A., Liu Y., Wang P. CAR-T Cells Surface-Engineered with Drug-Encapsulated Nanoparticles Can Ameliorate Intratumoral T-cell Hypofunction. Cancer Immunol. Res. 2018;6:812–824. doi: 10.1158/2326-6066.CIR-17-0502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.