An official website of the United States government
Here's how you know
Official websites use .gov
A
.gov website belongs to an official
government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you've safely
connected to the .gov website. Share sensitive
information only on official, secure websites.
As a library, NLM provides access to scientific literature. Inclusion in an NLM database does not imply endorsement of, or agreement with,
the contents by NLM or the National Institutes of Health.
Learn more:
PMC Disclaimer
|
PMC Copyright Notice
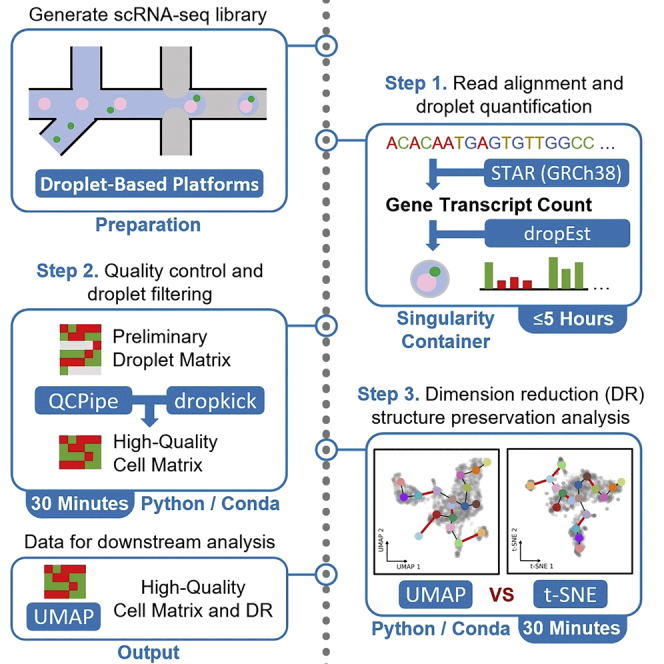
Single-cell RNA sequencing data require several processing procedures to arrive at interpretable results. While commercial platforms can serve as “one-stop shops” for data analysis, they relinquish the flexibility required for customized analyses and are often inflexible between experimental systems. For instance, there is no universal solution for the discrimination of informative or uninformative encapsulated cellular material; thus, pipeline flexibility takes priority. Here, we demonstrate a full data analysis pipeline, constructed modularly from open-source software, including tools that we have contributed.
An open-source, containerized pipeline for processing scRNA-seq data
•
The provided Singularity and Conda files permit rapid software installation
•
Multiple methods detailed for matrix quality control are provided for modular usage
•
Data entry points are highlighted for flexibility with other pipelines
Single-cell RNA sequencing data require several processing procedures to arrive at interpretable results. While commercial platforms can serve as “one-stop shops” for data analysis, they relinquish the flexibility required for customized analyses and are often inflexible between experimental systems. For instance, there is no universal solution for the discrimination of informative or uninformative encapsulated cellular material; thus, pipeline flexibility takes priority. Here, we demonstrate a full data analysis pipeline, constructed modularly from open-source software, including tools that we have contributed.
Before you begin
Single-cell, droplet-based library generation and sequencing
This pipeline was designed to take .fastq files as input generated from tag-based single-cell RNA-sequencing (scRNA-seq) libraries, such as inDrop, 10× Chromium, or Drop-Seq, according to their respective protocols (Klein et al., 2015; Macosko et al., 2015; Zheng et al., 2017). This pipeline consists of three major sections, starting from the single-cell read alignment and DropEst quantification, followed by quality control and droplet filtering, and ending with dimension reduction (DR) structure preservation analysis. Ensure that these starting libraries are generated with technologies compatible to the DropEst software, which is elaborated upon at https://github.com/hms-dbmi/dropEst (Petukhov et al., 2018). Quality thresholds at this point should depend on the guidelines set by the sequencing platform used. The single-cell read alignment and DropEst library quantification section of this pipeline utilizes a whitelist of known cell barcodes which also correspond to the sequencing platform used, ensure these are documented and accessible. We recommend encapsulating more than 1,000 single cells and sequencing them to a minimum depth of 50,000 reads per cell.
Note: For this pipeline, the necessary reference and annotation files can be found through the key resources table. Two sets of .fastq sequencing files can be found with the GEO accession numbers GSM4804820 and GSM5068493. The first is a normal, human colonic dataset and the second is a subsampled version of it, useful for the rapid testing of this pipeline’s installation on new computer systems. The expected results detailed in this protocol were generated with GSM4804820.
Singularity and R environment
The single-cell read alignment and DropEst library quantification section requires reference transcriptome and annotation files, of .fasta and .gtf format respectively, which must be locally accessible. Further information on the reference files that were as examples used (and how to download them) in this protocol can be found at https://github.com/Ken-Lau-Lab/STAR_Protocol. This modular pipeline makes use of containerized packages, which have been organized through Singularity. For Debian systems, download the latest Go libraries to a local directory with the following:
Singularity must be in installed to use our Singularity image, containing the libraries required for DropEst and STAR alignment. The image can be found at https://hub.docker.com/r/ramiremars/star_dropest and can be installed with the following code along with Singularity itself:
VERSION: This is the version of singularity that will be installed. Set to “3.7.0” in this example.
Python environment preparation
The heuristic droplet filtering, automated droplet filtering with dropkick, and post-processing and dimension reduction structure preservation analysis sections use a combination of command line tools and Jupyter Notebooks managed through a Conda Python environment. Please reference the Anaconda installation guidelines (Python >=3.8). Though this protocol is a full pipeline for scRNA-seq reads, its sections can be used modularly for the quality control (QC) and preliminary analysis of data at various stages of processing. are compatible with pre-computed droplet matrices, being a count matrix where the rows represent cell barcodes and columns represent detected, expressed genes. Along with an example dataset, the necessary python scripts and tutorial Jupyter Notebooks are available on our GitHub repository, where further Python environment setup instructions can also be found: https://github.com/Ken-Lau-Lab/STAR_Protocol.
Hardware - The full pipeline we present is computationally intensive, given the read alignment and demultiplexing steps that occur in the single-cell read alignment and DropEst library quantification section. We recommend running the first section of this pipeline on a high-performance workstation or computing cluster. The heuristic droplet filtering and automated droplet filtering with dropkick sections have lower requirements and do not need to be run on high-performance machines. Note that these requirements will be dependent on the read depth and barcode number of the library of interest.
Pause point: Throughout this protocol, each section will have multiple pause points, which are marked by the saving of files to the respective computer’s hard disk. These outputs are detailed in the expected outcomes section, as these outputs are heterogeneous in file format, size, and timing.
Step-by-step method details
Single-cell read alignment and dropEst library quantification
Timing: 5 h, for 120 million reads on a compute node with 12 cores and 48 GB of memory. 1.5 h for example subset dataset (GSM4804820) with the same specifications. Processing time decreases linearly with the number of cores available.
This section encompasses the library demultiplexing, read alignment, droplet count matrix estimation, and preliminary quality assessment with the DropEst library, the STAR aligner, and the scRNABatchQC R package, respectively (Dobin et al., 2013; Liu et al., 2019; Petukhov et al., 2018). First, dropTag takes paired-end, raw .fastq files and tags them in the context of unique molecular identifiers (UMIs) and cellular barcodes for the demultiplexing process. This is dependent on the scRNA-seq platform’s barcode whitelist; in this case we use the inDrop V1 and V2 barcodes. Before running the actual alignment process, a genome index must first be generated with respect to the reference and annotation files. STAR is a fast, scalable RNA-seq aligner which has splice awareness and takes the multiple tagged fastq.gz files generated by dropTag and aligns them using a reference genome index. The sorted .bam file generated by STAR alignment is used as an input to dropEst, which generates a barcode by gene count matrix, or droplet matrix, from the STAR aligned transcripts. Finally, scRNABatchQC is used to provide summary statistics and a quality assessment of the generated droplet matrix. This droplet matrix is further filtered in the heuristic droplet filtering and automated droplet filtering with dropkick section variants.
Note: This protocol serves as a reference for an order-of-operations and their parameters in our open-source pipeline; organized and executable scripts, with proper file and directory references, are available at: https://github.com/KenLauLab/STAR_Protocol/.
CRITICAL: The dropEst repository should be made locally available to explore its configurations and files by cloning from https://github.com/hms-dbmi/dropEst. This repository is also fully available within the provided Singularity container, whose configs can be displayed with the following command:
c, config filename: The file path to the .xml file containing estimation parameters within the Singularity container, which includes platform-specific information. Further parameters contained within this .xml file are described by Petukhov, et al. (Petukhov et al., 2018):
<reads_R1>, <reads_R2>: These are positional arguments which should be replaced with the paths to the fastq files representing the R1 and R2 reads respectively; set to “reads_R1.fastq” and “reads_R2.fastq” in this example. R1 corresponds to the barcode read and R2 corresponds to the gene read.
Pause point: The output of step 1 is saved as multiple tagged .fastq files, further detailed in the expected outcomes section.
2.
Create a directory for operations to be performed and generate the index file:
To create a genome index, the user must provide the reference genome (.fasta file) and the corresponding annotation file (.gtf) and run STAR with following parameters:
a.
runMode: Mode to run, example set to “genomeGenerate”
b.
runThreadN: The number of threads to generate the index file with, this step speeds up with higher values and is limited by the CPU used. Set to “16” in the example.
c.
genomeDir: Path to directory where files will be stored, set in example to “STAR_index”
d.
genomeFastaFiles: Path to genome .fasta file, set in example to “primary_assembly.fa”
e.
sjdbGTFfile: Path to annotation .gtf file, set in example to “annotation.gtf”
f.
sjdbOVerhang: The number of bases to concatenate from donor and acceptor sides of splice junctions. Set in example as “99”.
CRITICAL: Step 2 must be re-run for different reference genome and annotation file versions, as the generated genomic index will be unique to each version.
Pause point: The output of step 2 is saved as a genomic index file, further detailed in the expected outcomes section.
3.
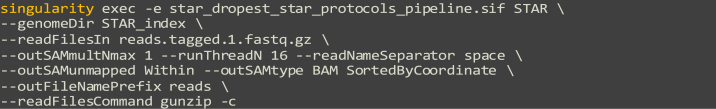
Run the single-cell alignment process with STAR, using our Singularity container:
genomeDir: The path to the directory containing the STAR index file. Set in example as “STAR_index”
b.
readFilesIn: The path to the tagged .fastq file(s), where multiple tagged .fastq files can be input, set as “reads.tagged.1.fastq.gz” in example.
c.
outSAMmultNmax: Maximum number of multiple alignments for a read that will be output to the .sam/.bam files, example set to “1”
d.
runThreadN: Number of threads, example set to “12”, increase value to speed up performance.
e.
readNameSeparator: Characters separating the part of the read names that will be trimmed in output, example set to “space”
f.
outSAMunmapped: Output unmapped reads within the main ,sam file, example set to “Within”
g.
outSAMtype: Output formatting of .bam file, example set to “BAM SortedByCoordinate”
h.
outFileNamePrefix: Output file name prefix, set here as “reads”
i.
readFilesCommand: Command to decompress fastq.gz files, example set to “gunzip –c”
CRITICAL: Ensure that there is sufficient memory overhead for this step, with a minimum of 32 GB allotted, as spikes in memory usage may prematurely end the alignment process.
Pause point: The output of step 3 is saved as a .bam, with several attributes, further detailed in the expected outcomes section.
4.
Run DropEst, configured here for an inDrop library, with the following:
F, filtered-bam: Print tagged bam file after the merge and filtration
e.
o, output-file filename: The output file name, example set to “sample_name”
f.
g, genes filename: Gene annotation file (.bed or .gtf), example set to “annotation.gtf”
g.
L: This is parameter has several options which denote the inclusion of count UMIs with reads that correspond to specific parts of the genome. Set to “eiEIBA” in the example.
i.
e: UMIs with exonic reads only
ii.
i: UMIs with intronic reads only
iii.
E: UMIs, which have both exonic and not annotated reads
iv.
I: UMIs, which have both intronic and not annotated reads
v.
B: UMIs, which have both exonic and intronic reads
vi.
A: UMIs, which have exonic, intronic and not annotated reads
<readsAligned.sortedByCoord.out.bam>: Positional argument for the input bam file. Set in example as “readsAligned.sortedByCoord.out.bam”.
CRITICAL: Like step 3, this is a memory-intensive step. Ensure that there is sufficient memory overhead for this step, with a minimum of 32 GB allotted.
Pause point: The output of step 4 is saved as a .bam and a .rds file, both with several attributes, further detailed in the expected outcomes section.
5.
From the output of DropEst, generate sparse count matrices with the code as follows:
--args: Path to the output .rds file, set in example as “sample_name.rds”.
Note: The source of the invoked R script can be viewed from within singularity container using vi. This script simply loads the .rds file, its contained data, and writes matrix files that are interoperable between different processing pipelines:
Pause point: The output of step 5 is saved as three files representing the droplet matrix, feature labels, and barcode labels, which are further detailed in the expected outcomes section.
--args: Consists of two parts, positionally, the species and target .csv file. In this example, set as “hsapiens” and “sample_name.rds_cm.csv”.
Pause point: The output of step 6 is saved as an .html file, further detailed in the expected outcomes section.
Variant 1. Heuristic droplet filtering
Timing: 15 to 30 min, depending on the size of the droplet matrix and cores available for certain parallelized functions.
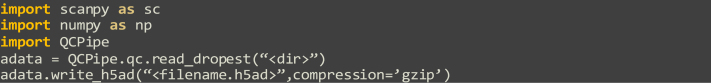
This section and its variant describe the barcode filtering of the droplet matrix, and can be used modularly if the user has a pre-computed matrix, either from the single-cell read alignment and DropEst library quantification section of this protocol or an external source, so long as the rows represent cell barcodes and columns represent genes. The output for this section will be a cell matrix, differing from a droplet matrix in that it only contains gene read counts from only high-quality, intact single cells. Primarily, this section will be performed interactively with Jupyter Notebooks running within a Conda environment, making extensive use of the AnnData Python class and scanpy library. First, a data-driven cutoff, by means of finding the inflection point in a cumulative sum curve of ranked barcode counts, is generated and used to minimize information-spars barcodes. Second, a distribution of uniquely detected genes per droplet is automatically thresholded through Otsu’s method, separating the remaining information-rich and information-sparse droplets and generating a binary metadata label. Third, tissue-specific gene expression signatures are visualized after DR to pinpoint cell populations of interest for downstream analysis. Fourth, unsupervised clustering is performed to discretize the single-cell transcriptional landscape. Finally, by heuristically integrating these metrics and expression signatures, populations of intact single-cells and their respective high-quality transcriptomes can be selected and saved to an independent file.
<filename.h5ad>: The filename of the .h5ad file generated in step 7
b.
<mitochondrial nomenclature>: The mitochondrial nomenclature of the dataset, given the gene symbol. This will vary depending on the gene nomenclature and species. For example, human mitochondrial gene symbols are designated with “MT-”, whereas mouse symbols are preceded by “mt-”.
Note: The following steps assume that the notebook kernel activated in step 8 is not subsequently deactivated or restarted; thus, library import statements are not detailed further.
9.
Perform the first-pass inflection point-based filtering:
<estimated number of cells encapsulated>: This number represents the estimated number of cells encapsulated during the library generation process and is based on the flow time and rate of the process.
CRITICAL: If the droplet matrix to be used in this step was generated through an external pipeline, ensure that it is ordered, starting with barcodes associated with the most to the least detected reads. Step 9 will fail if the data are not ordered as such. This ordering, however, is automatically performed in the DropEst output preparation in step 7.
10.
Automatically identify cells with relatively high transcriptional diversity:
Optional: Perform feature selection with highly_variable_genes or nvr after installing the required packages. These methods can be run in a Jupyter Notebook:
Note: Only one of these feature selection methods should be used at a time. Also, ensure that the data’s stage of normalization and transformation complies with the requirements of these feature selection methods.
13.
Perform the initial dimensionality reduction with PCA:
highly_variable_genes: This parameter is used to indicate whether to use feature selected variables, set in this example as “False”.
14.
Generate a K-nearest neighbors graph (KNN) from the PCA-based distance matrix. This is run with a K of approximately the square root of the total number of barcodes, balancing the influence of local and global distances:
The parameter for running this Leiden clustering is as follows:
a.
resolution: The clustering resolution, where a higher number leads to more, smaller clusters, and a lower number leads to fewer, larger clusters. The example is set to “1”.
The parameter for running this UMAP is as follows:
a.
min_dist: The minimum distance allowed for each cell or data point in the 2-dimensional projection. The example is set as “0.25”. Lower min_dist values cause the data points to be more compact in 2D space, and vice versa for higher values.
17.
Visualize factors useful in the heuristic determination of high-quality cell barcodes:
The parameter for running this UMAP is as follows:
a.
color: The values stored in the AnnData object which are to be visualized in on a 2-dimensional projection, in this example we visualize some “gene”, “leiden_labels”, “pct_counts_Mitochondrial”, “pct_counts_in_top_200_genes”, and “relative_transcript_diversity_threshold”. These factors are used in the heuristic selection of clusters.
b.
use_raw: Whether to visualize normalized and scaled values or the raw count values within the AnnData object droplet matrix. Set to “False” in this example.
CRITICAL: By priority, clusters of droplet barcodes should be selected based on these criteria in step 18:
c.
Marker gene expression and specificity: These genes will vary between the biological system of interest as well as the heterogeneity of cell input. Colorectal tumors, for example, will have a mixture of epithelial and immune cells, and markers would be used accordingly.
d.
The number of uniquely expressed genes: This is a strong predictor of encapsulated cells, and empty droplets are unlikely to contain a biologically relevant diversity of gene transcripts. This should be maximized unless there is a particular cell type that is known to express very few unique transcripts.
e.
Mitochondrial gene count percentage: This is important because encapsulated cells undergoing lysis will contain a high percentage of mitochondrial reads, effectively adding noise to a droplet due to the removal of more informative genes from a limited read count pool.
f.
Ambient gene expression: This is akin to the mitochondrial gene count percentage, as the encapsulation substrate may contain the remnants of lysed cells, often consisting of mitochondrial genes, but may vary per cell type. This should be minimized.
g.
Total counts: As the number of transcripts detected represents the amount of raw transcriptional information contained within a droplet. This should also be maximized unless there is a particular cell type that is known to express very few unique transcripts.
18.
Select and visualize the cells based off the heuristic criteria using discretized Leiden clusters:
<cluster selection>: The set of Leiden clusters to be selected and passed as a list of characters such as [‘1’,’2’, … ‘n’].
b.
legend_loc: This parameter indicates where the cluster legends will be displayed, set as “on data” in this example.
c.
legend_fontoutline: This parameter is used to render an outline on the cluster legends for readability, set as “True” in this example.
d.
legend_fontsize: This parameter designates the size of the font, set to 10 in this example.
19.
Ensure that the selected cells comply with the heuristic criteria by reviewing the outputs of steps 17 and 18; then save this selection to a .h5ad file.
selection: The observation attribute used to subset the data, as defined earlier, this example is set as “Cell_Selection”
b.
<Filtered_Data.h5ad>: The filename to save the compressed .h5ad as, set in this example as “<Filtered_Data.h5ad>”.
Pause point: The output of step 19, a filtered cell matrix, is saved as an .h5ad file, further detailed in the expected outcomes section.
Variant 2. Automated droplet filtering with dropkick
Timing: 5 to 10 min, depending on the size of the droplet matrix.
This variant serves the same function as the heuristic droplet filtering section. For automated droplet filtering in Python, dropkick is a machine learning tool that builds a probabilistic model of single-cell barcode transcriptome quality and returns a score for all barcodes in the input scRNA-seq droplet matrix (see step 7 for generating .h5ad from DropEst files) (Heiser et al., 2020). dropkick can be run from the command line or interactively in a Jupyter Notebook. A command line interface exists for its two primary modules designed for QC reporting and filtering, whose usages are outlined as follows.
<path/to/counts[.h5ad|.csv]>: The file path to the droplet matrix file of interest, which can be either .h5ad or .csv file.
Note: If the input counts are in .csv format, ensure that the file is in cells by genes configuration with labels for gene identities as column headers. The output from the step 7 can be used here directly.
22.
Run the dropkick filtering algorithm with the run function:
<path/to/counts[.h5ad|.csv]>: The file path to the droplet matrix file of interest, which can be either .h5ad or .csv file.
b.
j: The number of jobs used to parallelize the training and cross-validation of the dropkick model. We recommend adjusting the `-j` flag according to the number of available CPUs. If using a machine with more than five cores, `-j 5` is optimal for the five-fold cross validation performed by dropkick, and model training is usually completed in less than two minutes.
Note: All available user parameters can be found by running `dropkick run -h`. Default parameters are typically fast and robust for most datasets across encapsulation platforms, tissues, and levels of ambient background, see the troubleshooting section for further points of optimization.
Pause point: The output of step 22 is a .h5ad file, saved to disk, containing the input droplet matrix with additional metadata consisting of cell quality scores and binary labels. This is further detailed in the expected outcomes section.
23.
In a Jupyter Notebook, as described in step 19, load the dropkick-generated .h5ad file with the appropriate libraries and generate the filtered cell matrix:
selection: The observation attribute to use to subset the data, as defined earlier, this example is set as “dropkick_label”.
b.
<Filtered_Data.h5ad>: The filename to save the compressed .h5ad as, set in this example as “<Dropkick_Filtered_Data.h5ad>”.
Note: It is good practice to ensure that the selected cells also comply with the heuristic cell selection criteria as discussed in step 17. The entirety of the heuristic droplet filtering section can also be performed with a dropkick-labeled droplet matrix (the output from step 22), further augmenting cluster selection heuristics with learned metadata.
Pause point: The output of step 23, a filtered cell matrix, is saved as an .h5ad file, further detailed in the expected outcomes section.
Post-processing and dimension reduction structure preservation analysis
Timing: 15–30 min depending on the complexity and heterogeneity of the data at hand.
The final phase of this pipeline is centered around generating a representative two-dimensional projection of a filtered cell matrix to accurately visualize the global and local populational heterogeneity within dataset. Using scRNA-seq data to address hypotheses necessitates robust visualizations to counteract stochasticity inherent to several popular non-linear dimensionality algorithms. This stochasticity is often unaccounted for during downstream and may interfere with the representation of cellular relationships along the transcriptomic landscape, warping the perceived distances between cell types in 2D space. This section walks through the quantitative evaluation of two popular embedding techniques, t-SNE (van der Maaten and Hinton, 2008) and UMAP (McInnes et al., 2018), to determine the more reliable visualization strategy for a particular dataset. First, each latent space, or non-linearly projected, representation of the cell matrix is generated. Second, after identifying putative cell types in the data, discrepancies between these latent and native, or linearly transformed, spaces are calculated on global and local scales. Finally, rearrangements in subpopulation adjacencies are calculated on a graphical basis, allowing for users to choose the latent representation which minimizes discrepancies in latent-native space distances as well as in subpopulation adjacencies; both factors may greatly influence the biological interpretation of the data.
24.
Refer to the normalization, transformation, scaling, and DR guidelines in steps 11–15, as this section uses the same processes. Ensure that the cell count matrix has been processed, up to the Leiden clustering calculation, before proceeding.
25.
Using a calculated 50-component PCA, calculate a t-SNE representation of the cell count matrix.
use_rep: The representation of the single-cell data to use to initialize t-SNE nonlinear embedding, set as “X_pca”, or the 50-dimensional PCA representation in this example.
b.
perplexity: This is the effective nearest neighbors that are utilized in the t-SNE embedding, set to the square-root (rounded-down) of the total number of cells being examined.
26.
Next, generate a coarse-grained similarity graph using communities detected through the Leiden algorithm:
init_pos: The representation of the data that is used for the initialization of the UMAP visualization, the example is set as “paga”, as calculated in step 26.
28.
Run the structure_preservation_sc function to calculate global latent-native space discrepancies:
latent: The target latent representation of the data to be evaluated, in this example we start with “X_tsne”, this parameter can be replaced with “X_umap” to evaluate UMAP representations (Figures 6C and 6D)
b.
native: The native space representation of the data to compare the latent representation with, set as “X_pca” in the example due to the linear nature of its decomposition.
c.
k: The k number of nearest neighbors for use in structure preservation analysis, set to the square-root (rounded-down) of the total number of cells being examined calculated in step 25.
29.
Perform differential gene expression (DE) testing to derive transcriptional signatures from the detected subpopulations of cells, whose local latent-native distance discrepancies should be quantified:
groupby: The dataset labels between which to perform DE testing, in this case we use the “leiden” clusters.
CRITICAL: Ensure that all detected Leiden clusters can be reasonably identified through their gene expression signatures as described in literature. Unless a particular subpopulation is expected to be novel, cluster-to-cluster comparisons will not be biologically meaningful unless properly annotated. Note that the annotation of gene expression signatures is out of the scope of this protocol and will vary for each tissue of interest.
30.
Subset single-cell cluster(s) of interest to perform latent-native discrepancy evaluation on a cluster-by-cluster basis (defined through the detection of known marker genes and the signature detection of step 29):
uns_keys: The distances of interest to be subset, which are stored in the unstructured (.uns) attribute of the AnnData object. Set in the example as “X_pca_distances”, “X_tsne_distances”, and “X_umap_distances”
b.
obs_col: This parameter indicates which observation attribute to use to subset the data, as defined earlier, this example is set as “leiden”
c.
IDs: The observational IDs in which subsets of cells, Leiden cluster IDs in this example, are selected.
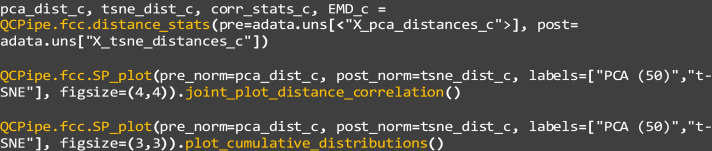
31.
Perform the latent-native discrepancy calculations and visualize them using the distance_stats, SP_plot, joint_plot_distance_correlation, and plot_cumulative_distributions functions:
pre: The calculated distances before generating the latent space representation of the data, which is are the “X_pca” distances in this example.
b.
post: The calculated distances after generating the latent space representation of the data, which is are the “X_tsne” distances in this example. For comparisons between these latent space representations, users can replace “tsne” with “umap” to test the latter embedding (Figures 7A, 7B, 7F, and 7G).
c.
pre_norm: A flattened vector of normalized, unique cell-cell distances before transformation, as output by “distance_stats”. This is calculated for the PCA representation in the example.
d.
post_norm: A flattened vector of normalized, unique cell-cell distances after transformation, as output by “distance_stats”. This is calculated for the t-SNE representation in the example.
e.
labels: The labels for the pre- and post- transformation data, set as “PCA (50)” and “t-SNE” in this example.
f.
figsize: The size of the figure, in terms of width and height. Set as “(4,4)” and “(3,3)” respectively in this example.
Note: Step 32 should be repeated as necessary with each latent space representation of interest. Here we recommend also running it with the UMAP representation calculated in step 27.
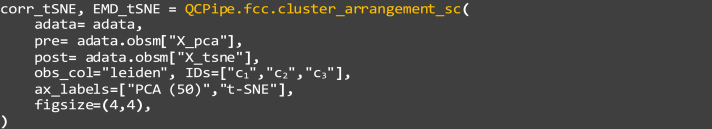
32.
Compare these distances between subpopulations of cells, being clusters c1, c2, and c3 in this example as defined in step 30:
pre: The coordinates of each single-cell before transformation, “X_pca”, or the 50-dimensional PCA are used in this case.
b.
post: The coordinates of each single-cell after transformation, “X_tsne”, or the 50-dimensional PCA are used in this case. For comparisons between these latent space representations, users can replace “tsne” with “umap” to test the latter embedding (Figures 7C, 7D, 7H, and 7I).
c.
obs_col: This parameter indicates which observation attribute to highlight by color, this example is set as “leiden”
d.
IDs: The observational IDs in which subsets of cells, Leiden cluster IDs in this example, are selected.
e.
ax_labels: The labels for the pre- and post- transformation data, set as “PCA (50)” and “t-SNE” in this example, to be plotted as axis labels.
f.
figsize: The size of the figure, in terms of width and height. Set as “(4,4)” and “(3,3)” respectively in this example.
Note: Step 3.9 should also be repeated as necessary with each latent space representation of interest. Here we recommend also running it with the UMAP representation calculated in step 27. Further, additional comparisons between other Leiden clusters should be performed to evaluate all potential subpopulations of interest, and these clusters should incorporate signatures highlighted in step 29.
33.
Generate a minimum-spanning tree (MST) to investigate global subpopulation arrangements and structure:
dim_name: The name of the latent representation to be plotted, being “X_tsne” and “X_umap”.
b.
obs_col: This parameter indicates which observation attribute to highlight by color, this example is set as “leiden”
c.
use_rep: The representation of the single-cell data to plot, set as “X_tsne” and “X_umap” in this example.
d.
highlight_edges: Which differing edges to highlight, representing a rearrangement of coarse cluster neighbors. “tsne_set” and “umap_set” in this example, calculated earlier.
Global comparison of two-dimensional embeddings of human colonic mucosa dataset
(A) t-SNE embedding seeded with 50-component PCA, plotted with overlay of Leiden clustering.
(B) UMAP embedding seeded with 50-component PCA and initialized with PAGA coordinates, plotted with overlay of Leiden clustering.
(C) Global structural preservation correlation plot comparing t-SNE coordinates (latent space) to 50-component PCA (native space).
(D) Same as in (C), comparing UMAP coordinates (latent space) to 50-component PCA (native space). Indicated with red arrows in (C) and (D) are latent space distance distributions which differ between t-SNE and UMAP.
(E) Top four differentially expressed genes for each Leiden cluster, with signatures for clusters 3, 4, 8, and 11 highlighted.
(B) Local structure preservation correlation plot for tuft cell cluster, comparing t-SNE coordinates (latent space) to 50-component PCA (native space).
(C) t-SNE embedding highlighting secretory lineage from stem cells (cluster 3) to goblet cells (cluster 8) and mature goblet cells (clusters 4 and 12).
(D) Structure preservation correlation plot showing distances between stem and mature cell lineage clusters.
Indicated with red arrows in (B and G) and (D and I) are latent space distance distributions which differ between t-SNE and UMAP.
(E) t-SNE embedding with minimum spanning tree (MST) drawn between Leiden cluster centroids. Red edges represent those not present in native (PCA) space.
(F–J) Same as in (A–E), for UMAP embedding.
Expected outcomes
These expected outcomes are also described in this pipeline’s repository, which can be found at https://github.com/Ken-Lau-Lab/STAR_Protocol. Examples in the repository also contain proper file and directory references for the provided files.
Note: The entirety of this pipeline and the expected outcomes detailed here are based on the GSM5068493 dataset, with the reference genome and annotation files detailed in the key resources table. The droplet and cell matrices of this dataset are also included as .h5ad files on our GitHub repository. For the sake of saving time and computational resources users may want to test the single-cell read alignment and DropEst library quantification section with our reduced dataset, GSM4804820.
Single-cell read alignment and dropEst library quantification results
Step 1: The output of the DropTag step will be tagged and demultiplexed fastq.gz files that will be aligned by STAR in subsequent steps. Note that the DropTag step can output around 20 tagged .fastq files, depending on the read depth of the input files. These outputs are also paired with a log, named tag_main.log, containing the number of reads processed, number of reads that passed the minimum quality threshold, number of reads with expected structure for the library type, and trimming statistics, given arguments specified in configs/indrop_v1_2.xml file. The subsampled dataset we provide will only produce 1 tagged .fastq file due to its size.
Step 2: The output genomic index file is necessary for STAR alignment and allows for the query time of each read alignment to be minimized. Only one of these genomic indices needs to be generated per reference and annotation pair.
Step 3: The .bam produced through STAR alignment contains several attributes which are detailed in Table 1. Additionally, a file named “Log.final.out” contains useful metrics to evaluate the quality of the mapping.
Table 1.
Expected outputs for single-cell read alignment and DropEst library quantification
Object attribute (within .bam or .rds)
Definition
Step output
NH
Number of reported alignments that contain the query in the current record.
Step 3 .bam, Step 4 .bam
HI
Query hit index, indicating the alignment record is the i-th one stored in SAM.
Step 3 .bam, Step 4 .bam
AS
Alignment score generated by aligner
Step 3 .bam, Step 4 .bam
nM
Number of mismatches per (paired) alignment
Step 3 .bam, Step 4 .bam
GX
Gene id
Step 4 .bam
CR
Cell barcode raw
Step 4 .bam
UR
UMI raw
Step 4 .bam
CB
Cell barcode
Step 4 .bam
UB
UMI
Step 4 .bam
Cm
Count matrix in sparse format
Step 4 .rds
cm_raw
Count matrix in sparse format without filtration by minimal number of UMI and by the required type of reads (-L option)
Step 4 .rds
saturation_info
Data for estimating saturation using preseqR package
Step 4 .rds
merge_targets
Vector of corrected barcodes, named with raw barcodes
Step 4: DropEst outputs multiple files, including a sorted .bam file and a .rds file. The .bam file contains read-level information, like the output of step 3, and is also outlined in Table 1. The .rds file contains information regarding the droplet matrix itself, with corrected barcode information. Both files are used to generate the final droplet matrix. Additionally, this step outputs a est_main.log file, detailing the progress of dropEst, the number of reads mapped to intergenic, exonic, and intronic regions, and further cell barcode and UMI merging statistics. This log also contains the used arguments specified in .xml configuration file.
Step 5: Droplet matrix generation utilizes both the sorted .bam and .rds file of step 4. Two directories are generated, containing the respective matrices derived from the cm and cm_raw (used for subsequent steps) fields of the .rds file. These directories contain corresponding files as follows:
a.
Counts.mtx - This file represents the demultiplexed count matrix containing the number of times a transcript of a gene was observed with a specific barcode (vars × obs). As this matrix contains all barcodes detected in the alignment of sequenced reads, it includes both barcodes associated with real, encapsulated cells as well as empty droplets and false positives. Thus, the number of barcodes within this matrix is far greater than should exist per encapsulated cell.
b.
Features.txt - This file contains the genes detected through the alignment process. Generally, this number should be between 20,000 and 30,000. Depending on the reference transcriptome and GTF annotation file used, these will be gene symbols, ENSEMBL IDs, and other associated nomenclatures.
c.
Barcodes.txt - This file contains the detected barcodes, generally these will be a series of unique nucleotide strings that represent a detected barcode. This will vary depending on the read depth of the alignment process.
Step 6: The scRNABatchQC report generates a .html report with several QC diagnostic metrics such as number of cells per sample. The output is an HTML report with metrics and diagnostic plots evaluating the quality of the experiment. The report includes three parts: QC Summary, Technical View, and Biological View. The QC summary table provides metrics including the total number of counts, cells, and genes, percentage of mitochondrial and rRNA reads, and the number of cells removed due to low quality. The Technical View diagnostic plots show different features, including distributions of counts, genes, mtRNA and rRNA percentages, expression cumulative plots, and variance explained by features. The Biological View shows the highly variable genes, differentially expressed genes according to PCA, enriched pathways according to both gene lists, PCA and t-SNE plots.
Variant 1. Heuristic droplet filtering results
Step 9: Inflection curve detection is a fast way to calculate a first pass quality cutoff for droplets and potential cells detected. The resulting curve of a dataset containing a mixed distribution of high-quality cells and empty droplets also contains information about the overall quality of the dataset. Ideally, the produced inflection curve should look similar to a logarithmic function with nonzero values. The sharpness of the angle of inflection is related to the separability of encapsulated, intact cells and empty droplets, with a sharper angle indicating a more significant distinction between the two (Figure 1A, robust inflection point indicated). In some cases, the overall library quality may be low enough to affect the generation of this curve (Figure 1C, weak inflection point indicated). Additionally, in the case of lower quality libraries, the normalized transcriptional diversity curve will contain a wider plateau when plotted by cell rank. (Figures 1B and 1D, ‘plateaus’ indicated).
(A and B) Inflection curve thresholding (A) for a high quality dataset with corresponding Total Counts to N Genes By Counts ratio plot on log scales (B).
(C and D) Inflection curve thresholding (C) for a low quality dataset with corresponding Total Counts to N Genes By Counts ratio plot on log scales (D).
Red arrows indicate inflection points (A and B), and red brackets indicate the ‘plateau’ motif in the Total Counts/N Genes By Counts plot.
Step 10: Relative transcript diversity thresholding uses the distribution of transcript diversity as both an indicator of overall library quality and a means to automatically label potential encapsulated cells. Higher quality libraries, after running the relative_diversity function, will have distinct bimodal or multimodal distributions of transcript diversity. The labeled, higher quality barcodes within the green highlight are also labeled on a corresponding UMAP (Figures 2A and 2B, bimodal distribution indicated with highlighted UMAP counterpart). Lower quality libraries will be unimodal, with few barcodes labeled as high quality (Figures 2C and 2D unimodal distribution indicated with highlighted UMAP counterpart). In addition, the visualization of these lower quality libraries will, qualitatively, appear more amorphous in a UMAP visualization, with a single observable cluster.
Relative transcript diversity distribution analysis
(Aa nd B) Relative transcript diversity histogram plot (A) for a high quality dataset with corresponding UMAP with highlighted selection (B).
(C and D) Relative transcript diversity histogram plot (C) for a low quality dataset with corresponding UMAP with highlighted selection (D).
Red arrows indicate bimodality and unimodality in (A and C), respectively, for the distributions of transcript diversity.
Steps 17–19: Leiden clustering and cell determination uses a combination of unsupervised machine learning and heuristic approaches from publicly available, open-source software. Because of the heuristic selection criteria for subpopulations of high-quality cells, the quality of subpopulation selection is augmented with prior knowledge, though this is dependent on the transcriptional nature of the cells of interest. In this example, we examine the heterogeneity of cells found in a normal, human colonic epithelium. Going in with prior knowledge of intestinal stem, transit amplifying, hematopoietic, absorptive, secretory, tuft, enteroendocrine, and intestinal epithelial cells, we examine LGR5, PCNA, PTPRC, KRT20, MUC2, POU2F3, CHGA, and EPCAM, respectively (Figure 3A). The expression of these marker genes is of the highest priority in our heuristic process, followed by transcript diversity and so on. Based on these criteria, Leiden clusters 9, 11, 16, 21, and 22 are selected (Figures 3B and 3C, selected cells indicated). The output from step 19 will be a single, compressed .h5ad file comprising a filtered cell count matrix with barcode and gene names annotated.
(A) Marker gene UMAP overlays, with scale bars denoting the normalized and transformed values.
(B) Leiden cluster labels derived through the Leiden community detection algorithm at a resolution of 2.
(C) UMAP visualization of the selected clusters, being 9, 11, 16, 21, and 22.
Variant 2. Automated droplet filtering with dropkick results
Step 21: Automated droplet filtering with the dropkick Python package provides quality control metrics and cell probabilities for each barcode that can be used to subset an unfiltered counts matrix using a gene-based logistic regression model (Heiser, et al. 2020). The dropkick QC module returns a report that quantifies ambient RNA and estimates global data quality by the profile of total counts and genes per barcode (Figures 4A and 4B). The human colonic mucosa sample contains ambient RNA that consists primarily of mitochondrial genes (Figure 4B) and accounts for nearly 90% of all reads in some empty droplets (Figure 4A), indicating that cell death and lysis contributed highly to background noise during this encapsulation.
(A) Profile of total counts (black trace) and genes (green points) detected per ranked barcode for human colonic mucosa sample. Percentage of mitochondrial (red) and ambient (blue) reads for each barcode included to denote quality along total counts profile.
(B) Ranked gene dropout rates. Ambient genes identified by dropkick are used to calculate percent ambient counts in A.
(C) Plot of coefficient values for 2,000 highly variable genes (top) and mean binomial deviance ± SEM (bottom) for model cross-validation along the lambda regularization path defined by dropkick. Top and bottom three coefficients are shown, in axis order, along with total model sparsity (top). Chosen lambda value shown as dashed vertical line.
(D) Plot of percent ambient counts versus arcsinh-transformed genes detected per barcode, with histogram distributions plotted on margins. Initial dropkick training thresholds shown as dashed vertical lines. Each point (barcode) is colored by its final cell probability after model fitting.
Step 22: After training a logistic regression model using automated heuristic thresholds, the resulting dropkick coefficient values, and deviance scores along the path of tested lambda (regularization strength) are shown (Figure 4C and saved as a “_coef.png” file) as well as final cell probabilities (Figure 4D also saved as a “_score.png” file). Further outputs from this step include a single, compressed .h5ad file comprising a filtered cell count matrix with barcode and gene names annotated. This file will retain the original input counts file name, appending “_dropkick.h5ad”. “dropkick_score” and “dropkick_label” columns in the .obs dataframe of this file provide the user with cell probabilities and default thresholded labels (dropkick_score ≥ 0.5 indicates real cell), respectively.
Post-processing and dimension reduction structure preservation analysis results
Step 28: To examine DR structure preservation on a global scale, our filtered cell counts matrix is normalized, transformed, and scaled prior to PCA for initial DR. We can explore our PCA outputs for top gene loadings in the highest PCs that indicate importance for distinguishing differences in cell populations (Figure 5A) and confirm the appropriate number of PCs for our analysis (for our human colon data: 50 PCs; Figure 5B). This PCA becomes the “native space” for downstream evaluation of data structure preservation in our two-dimensional embeddings (Figure 5C). Embeddings from t-SNE and UMAP display similar global structure and compare favorably to the native PCA space (Figures 6A–6D), with global correlation values of 0.6112 and 0.6632, Earth Mover’s Distances (EMDs) of 0.1649 and 0.1444, and K-nearest neighbor preservation values of 97.60% and 97.53% for t-SNE and UMAP, respectively. In 7C and 7D, the differing means of the cell-to-cell distance distributions are indicated. With the advantage in two out of three global metrics, UMAP outperforms t-SNE on average across all cells in our dataset. However, we can perform more in-depth analyses on local and organizational data structures to evaluate strengths and weaknesses of both embeddings.
PCA of human colonic mucosa dataset with PAGA graph
(A) Top and bottom 15 gene loadings for the first three PCs.
(B) Proportion of total explained variance for each of the top 30 PCs.
(C) First two PCs plotted with Leiden cluster overlay.
(D) PAGA graph constructed from k-nearest neighbors (kNN) in 50-component PCA space (k = 46), describing relationships between Leiden clusters.
Step 29: DE analysis allows us to identify cell types associated with each Leiden cluster (Figure 6E). We can use this information to dive more deeply into local structure preservation by t-SNE and UMAP embeddings; four clusters are highlighted in 7E, corresponding to example subpopulations for structure preservation analysis.
Step 30: To examine DR structure preservation for a single subpopulation, we focus on our tuft cell cluster and calculate correlation and EMD values of 0.3014 and 0.1587 for t-SNE and 0.4025 and 0.0851 for UMAP. This indicates that UMAP does a significantly better job of preserving cell-cell distances within the tuft cell population from their native PCA space (Figures 7A, 7B, 7F, and 7G).
Step 31: To examine DR structure preservation for multiple subpopulations, we investigate the distance distributions along the secretory epithelial cell lineage (Figures 7C, 7D, 7H, and 7I); we note that t-SNE slightly exaggerates distances between stem cells (cluster 3) and early goblet cells (cluster 4 and 8), as indicated by a greater shift of distances in the correlation plot above the identity line (Figures 7D and 7I). This shift is further quantified by correlation and EMD values, with a mean increase in correlation by 0.083 and a mean decrease in EMD by 0.029 comparing UMAP to t-SNE.
Step 35: For global subpopulation arrangement and structure analysis, we perform a coarse-grained global analysis of cluster arrangement by building a minimum spanning tree (MST) graph between Leiden cluster centroids in native and latent spaces. We observe five edges in the t-SNE MST that are not present in PCA space, indicating relative cluster rearrangements. UMAP, however, displays four of these edge permutations, suggesting that this embedding is more precise in maintaining populational organization than t-SNE (Figures 7E and 7J). These results indicate that further analyses and visualizations should be performed with UMAP.
Limitations
This pipeline is designed to integrate open-source software in a modular fashion, and each section has its own limitations. Primarily, dataset-to-dataset variation may lead to differing performance. Relatively high hardware requirements to run the single-cell read alignment and DropEst library quantification section may prevent some users from fully utilizing this pipeline. The heuristic droplet filtering, automated droplet filtering with dropkick, and post-processing and dimension reduction structure preservation analysis sections involve the highest likelihood of variable parameters, though the suggested guidelines will minimize unwanted variation. Currently, this pipeline is not implemented with batch-aware functions and is unable to incorporate information across replicates into the QC process. Further limitations include a set of assumptions each of these steps makes, which should be satisfied regardless of dataset-to-dataset variation that may exist. In the heuristic droplet filtering and automated droplet filtering with dropkick section variants, limitations include:
•
In step 9, a distinct inflection point will only arise given some subset of more informative, information-rich droplets. The properties of the utilized cumulative sum curve have been observed to be dependent on the encapsulation process and read depth. If all droplets within a single sequencing library contain a homogenous amount of information, an identifiable inflection may not be detected, preventing a first pass filtering of droplets.
•
For step 10, this step assumes a multimodal distribution of the number of unique genes detected per droplet. It has been demonstrated before that single-cell libraries generated from encapsulated cells, as opposed to empty droplets, present a higher diversity of detected genes (Lun et al., 2019). This step assumes that both high and low information, with respect to read counts, are represented in each dataset. Like step 9, if this diversity in droplet quality is not detected, the identification of relatively high-quality cells is not possible. Though, given the Poissonian or super-Poissonian nature of tag-based scRNA-seq cell encapsulation, it is highly unlikely a dataset will be devoid of low-information empty droplets (Zhang et al., 2019).
•
For step 14, the highest priority heuristic criterion assumes that a defined set of marker genes exists for the dataset of interest, which may not be the case for all biological specimens of interest. The likelihood of Type 2 error increases with fewer marker-defined subpopulations, as these subpopulations may simply express fewer genes. Similarly, Type 1 errors are also possible with the overreliance on agnostic metrics such as transcriptional diversity.
•
In step 21, dropkick trains a cross-validated logistic regression model of genes associated with subsets of high and low-quality cells. The accuracy of this model will be dependent on the representation and detection of these bins of cells. Similarly, the default cutoff of 0.5 for binary barcode labeling may vary depending on the properties of the cell type of interest; thus, these results should be further validated with prior knowledge about target gene transcription.
Steps 29 and 30, like step 14, are dependent on the detected or known heterogeneity of single cell subpopulations within the dataset. The assumption is made that there exist multiple subpopulations of cells that present detectable marker gene expression, and that these marker genes are known. In the case that these markers are unknown, the DE testing between clusters may draw some insight on distinct gene expression profiles. Still, statistically identified gene sets may need validation, whether through user review or gene set enrichment analysis. The analysis of structural preservation necessitates the capture of high-quality clusters, as identified through previous steps.
Troubleshooting
Problem 1
An error code arises that claims that the open file limit has been reached. This can arise in step 3 when setting a high –runThreadN parameter.
Potential solution
This occurs for certain steps involving parallelization due to the number of files written to the disk which are read simultaneously and the default Linux system variable, ulimit, is set too low. This can be amended by setting this to a higher value based on the hardware available.
Problem 2
Droplet estimation following read alignment yields fewer than expected demultiplexed droplets. This can arise after assessing the logs alignment logs of step 3.
Potential solution
This can occur because a library of insufficient quality or sequenced at insufficient depth may have led to a lack of detected high quality reads. The “Uniquely mapped reads %” from the alignment logs indicate the mapping rate of the library. Generally, high-quality libraries should have above 80% mapping rate, lower than 50% may indicate a problem with the library preparation or sample quality.
Problem 3
An error may arise during the Dropkick installation due to pip being unable to find a FORTRAN compiler, during step 7.
Potential solution
For Debian-based systems, gfortran can be installed with:
You are not detecting any mitochondrial reads in the droplet matrix, as indicated by the average pct_counts_Mitochondrial being NaN or 0, which can arise during step 17.
Potential solution
This error can occur because the calculation and visualization of mitochondrial read count percentage per single-cell transcriptome is dependent on the nomenclature of the genes themselves. Ensure that the gene nomenclature in the AnnData object is consistent with that used in setting the variable annotation “Mitochondrial”. The gene names and annotations can be checked and set with the following two lines of code, respectively:
Visually, a small population of cells is highly and specifically expressing verified marker genes, but there is no way to select this population given the Leiden cluster labels, which can arise during step 17 or step 24.
Potential solution
This can occur due to the default Leiden clustering resolution and the simple underrepresentation of that cell population of interest in the dataset. Computationally, these cells can be singled out by further increasing the Leiden clustering resolution, at the cost of generating more clusters in general. Experimentally, more cells may have to be encapsulated and sequenced to a higher read depth to capture rare cells.
Problem 6
The cumulative sum curve is too shallow to visually detect a meaningful inflection point, or the automatically detected inflection point is throwing out target cells; this can arise in step 9.
Potential solution
This error occurs primarily for the same reasons as step 1, Problem a. The lack of a detectable inflection curve, as provided in our expected outputs example, indicates low quality library inputs. Alternatively, if high quality cells expressing target marker genes are suspected to be removed as part of this inflection curve first pass, the user can manually set the initial quality cutoff with the following code, where <user_threshold> indicates the top N droplets to retain for further quality control:
dropkick is labeling cells with high and specific expression of verified marker genes as empty droplets; this can arise in step 23.
Potential solution
This error can happen due to a combination of reasons detailed in step 1, Problem a, and the dependencies on logistic regression parameters. Since the binary dropkick_label is based on a threshold at 0.5, adjustments can be made to be more or less inclusive of droplets based on the dropkick_score distribution. This can be done with the following code, where <user_threshold> is a number between 0 and 1, with 0 being the least stringent and 1 being the most permissive:
Further optimizations to the generation of this dropkick score can be done through adjustments to the logistic regression parameters such as specifying more iterations, a range of alpha values, or a longer “lambda path”.
Problem 8
Latent and native space distances or subpopulation rearrangements are inconsistent between runs and machines; this can arise in steps 28, 30, 31, 32, and 35.
Potential solution
Ensure that the random seeds are consistent between different runs. Three sources of random variation originate in the PYTHONHASHSEED, the numpy library seed, and the random library seed. Still, note there may be some minor variation due to the current implementation of UMAP used in scanpy.
Problem 9
Individual subpopulations during DR structure preservation yields lack biological heterogeneity, thus affecting the interpretation of DE testing and cluster-based DR structure analysis; this can arise in steps 29, 30, 32, and 35.
Potential solution
The clustering resolution of the Leiden algorithm will need to be adjusted based on the dataset examined. A common heuristic to selecting this resolution parameter is to increase its value until the smallest, marker-indicated subpopulation of cells is discretely identified as a cluster.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ken Lau (ken.s.lau@vanderbilt.edu).
This study was supported by US National Institutes of Health (NIH) grants U2CCA233291 and U54CA217450 (MARS, CNH, QL, KSL), R01DK103831 (KSL), and T32LM012412 (BC). Additional testing was performed by Harrison Kiang.
Author contributions
Writing, B.C., C.N.H., and M.A.R.-S.; development, B.C., C.N.H., and M.A.R.-S.; processing, B.C., C.H., and M.A.R.-S.; funding acquisition, K.S.L. and Q.L.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Bob Chen, Email: bob.chen@vanderbilt.edu.
Ken S. Lau, Email: ken.s.lau@vanderbilt.edu.
References
Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barnett D.W., Garrison E.K., Quinlan A.R., Strömberg M.P., Marth G.T. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011;27:1691–1692. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bates D., Eddelbuettel D. Fast and elegant numerical linear algebra using the rcppeigen package. J. Stat. Softw. 2013;52:1–24. [Google Scholar]
Chen B., Herring C.A., Lau K.S. pyNVR: investigating factors affecting feature selection from scRNA-seq data for lineage reconstruction. Bioinformatics. 2018;35:2335–2337. doi: 10.1093/bioinformatics/bty950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Csardi G., Nepusz T. The igraph software package for complex network research. InterJ. Comp. Syst. 2006:1695. [Google Scholar]
Eddelbuettel D., Francois R. Rcpp: Seamless R and C++ Integration. J. Stat. Softw. 2011;1 [Google Scholar]
Flamary R., Courty N. Transport library; 2017. POT Python Optimal. [Google Scholar]
Hagberg A.A., Schult D.A., Swart P.J. Exploring network structure, dynamics, and function using NetworkX. In: Varoquaux G., Vaught T., Millman J., editors. Proceedings of the 7th Python in Science Conference. 2008. pp. 11–15. [Google Scholar]
Harris C.R., Millman K.J., van der Walt S.J., Gommers R., Virtanen P., Cournapeau D., Wieser E., Taylor J., Berg S., Smith N.J. Array programming with NumPy. Nature. 2020;585:357–362. doi: 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Heiser C.N., Lau K.S. A quantitative framework for evaluating single-cell data structure preservation by dimensionality reduction techniques. Cell Rep. 2020;31:107576. doi: 10.1016/j.celrep.2020.107576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Heiser C.N., Wang V.M., Chen B., Hughey J.J., Lau K.S. Automated quality control and cell identification of droplet-based single-cell data using dropkick. BioRxiv. 2020 doi: 10.1101/2020.10.08.332288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D.A., Kirschner M.W., Zilionis R. Droplet barcoding for single-cell transcriptomics applied. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kluyver T., Ragan-Kelley B., Pérez F., Granger B., Bussonnier M., Frederic J., Kelley K., Hamrick J., Grout J., Corlay S. Jupyter Notebooks -- a publishing format for reproducible computational workflows. In: Loizides F., Schmidt B., editors. Positioning and Power in Academic Publishing: Players, Agents and Agendas. 2016. pp. 87–90. [Google Scholar]
Kurtzer, G. M. (2016, August 23). Singularity 2.1.2 - Linux application and environment containers for science. Zenodo. 10.5281/zenodo.60736 [DOI]
Liu Q., Sheng Q., Ping J., Ramirez M.A., Lau K.S., Coffey R.J., Shyr Y. scRNABatchQC: multi-samples quality control for single cell RNA-seq data. Bioinformatics. 2019;35:5306–5308. doi: 10.1093/bioinformatics/btz601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lun A.T.L., Riesenfeld S., Andrews T., Dao T.P., Gomes T., Jamboree, participants in the 1st H.C.A. Marioni J.C. EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 2019;20:63. doi: 10.1186/s13059-019-1662-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
McInnes L., Healy J., Saul N., Großberger L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018 [Google Scholar]
Petukhov V., Guo J., Baryawno N., Severe N., Scadden D.T., Samsonova M.G., Kharchenko P.V. dropEst: Pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 2018;19:78. doi: 10.1186/s13059-018-1449-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
R Core Team (2020). R: A language and environment for statistical computing. R A Lang. Environ. Stat. Comput. R Found. Stat. Comput. Vienna, Austria.
Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R. Ensembl 2020. Nucleic Acids Res. 2020;48:D682–D688. doi: 10.1093/nar/gkz966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang X., Li T., Liu F., Chen Y., Yao J., Li Z., Huang Y., Wang J. Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol. Cell. 2019;73:130–142. doi: 10.1016/j.molcel.2018.10.020. [DOI] [PubMed] [Google Scholar]
Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.




Pause point: Throughout this protocol, each section will have multiple pause points, which are marked by the saving of files to the respective computer’s hard disk. These outputs are detailed in the expected outcomes section, as these outputs are heterogeneous in file format, size, and timing.
Timing: 5 h, for 120 million reads on a compute node with 12 cores and 48 GB of memory. 1.5 h for example subset dataset (GSM4804820) with the same specifications. Processing time decreases linearly with the number of cores available.
CRITICAL: The dropEst repository should be made locally available to explore its configurations and files by cloning from https://github.com/hms-dbmi/dropEst. This repository is also fully available within the provided Singularity container, whose configs can be displayed with the following command:


























 The parameter for running the PCA is as follows:
The parameter for running the PCA is as follows:



 The parameter for running this UMAP is as follows:
The parameter for running this UMAP is as follows: