

Abstract
Molecular alterations that contribute to long-term (LT) and short-term (ST) survival in ovarian High-Grade Serous Carcinoma (HGSC) may be used as precision medicine biomarkers. DNA promoter methylation is an early event in tumorigenesis, which can be detected in blood and urine, making it a feasible companion biomarker to somatic mutations for early detection and targeted treatment workflows.
We compared the methylation profile in 12 high-grade serous ovarian cancer (HGSC) tissue samples to 30 fallopian tube epithelium samples, using the Infinium Human Methylation 450K Array. We also used 450K methylation arrays to compare methylation among HGSCs long-term survivors (more than 5 years) and short-term survivors (less than 3 years). We verified the array results using bisulfite sequencing and Methylation Specific PCR (qMSP). in another cohort of HGSC patient samples (n=35). Immunoblot and clonogenic assays after pharmacologic unmasking show that HIST1H2BB and MAGI2 promoter methylation down regulates mRNA expression levels in ovarian cancer cells. We then used qMSP in paired tissue, ascites, plasma/serum, vaginal swabs and urine from a third cohort of HGSC cancer patients (n=85) to test the clinical potential of HIST1H2BB and MAGI2 in precision medicine workflows. We also performed next-generation exome sequencing of 50 frequently mutated in human cancer genes, using the Ion AmpliSeqCancer Hotspot Panel, to show that the somatic mutation profile found in tissue and plasma can be quantified in paired urine samples from HGSC patients.
Our results suggest that HIST1H2BB and MAGI2 have growth-suppressing roles and can be used as HGSC precision medicine biomarkers.
Keywords: HIST1H2BB, MAGI2, HGSC, ovarian carcinoma methylation and somatic mutation markers, precision medicine
Introduction
Ovarian cancer is the fifth cause of cancer deaths among women, and the most lethal gynecological malignancy[1]. Clinical and molecular factors that contribute to long-term (LT) and short-term (ST) survival in ovarian high-grade serous cancer (HGSC) are lacking and only a few molecular alterations of response to therapy have been identified. Somatic mutations are rare in HGSC [2], BRCA1/2 germline mutations, and homologous repair deficiency in HGSC are among the few validated molecular predictors of response to platinum therapy and poly-ADP polymerase (PARP) inhibitors [3–6].
DNA promoter methylation is an early event in tumorigenesis, and can be detected in blood and other body fluids, making it a feasible biomarker for early detection of tumors.[7–9] In addition, DNA methylation has potential as a prognostic biomarker. For instance, the FDA has recently approved EpiColon, a blood-based test for diagnosis of colorectal cancer based on methylation of septin 9.[10] Detection of promoter methylation in tissues and biofluids represents a potential biomarker strategy for ovarian cancer diagnosis and therapeutic management within precision medicine workflows.
Until recently, epithelial ovarian cancers were thought to arise from the ovarian surface epithelial cells [11]. However, recent studies suggest that many of HGSCs arise from lesions in the fallopian tubes[12–17]. In this study, we sought to identify genes differentially methylated between fallopian tube tissue and HGSC and test whether ovarian cancer associated-DNA methylation and somatic mutations measured in tissue can be reproducibly measured in urine samples.
Materials and Methods
Patient samples
The study population consists of samples from three patient cohorts (n=77) and publicly available data from 1,742 patients: a retrospective cohort of HGSC Formalin Fixed and Paraffin Embedded (FFPE) samples selected from Johns Hopkins Pathology Department tumor bank (n=12); a cohort of women who were seen in the Ohio State University School of Medicine Obstetrics and Gynecology Department (n=30); a cohort of HGSC patients who were seen in Mercy Medical Center in Baltimore, Maryland (n=35); and data from the Cancer Genome Atlas Project (TCGA). The inclusion criterion was to have a clinical diagnosis of HGCS (ICD9-CM code 183), all determined by pathologists at two different institutions Hopkins and Mercy Medical Center. The Institutional Review Boards of Ohio State School of Medicine, Mercy Medical Center and Johns Hopkins School of Medicine (NA_00020633) approved the research protocols. Informed written consent was obtained from all patients included in the study.
Cancer Genome Atlas Project (TCGA) data
TCGA data was downloaded and analyzed for DNA methylation alterations using the minfi package. Somatic mutation and expression data were downloaded from the CBioPortal (http://www.cbioportal.org/).
DNA extraction
DNA was extracted from frozen normal fallopian tube epithelium, FFPE HGSC tissue samples, as well as from normal and ovarian cancer cell lines. Biofluids DNA was extracted as previously described.[11, 18] The protocol for trans-renal DNA extraction reduces the possibility of contamination from urinary tract DNA, by selecting for small and extra cellular DNA. Samples were digested using 1% SDS and 20 ug/ml proteinase K for 48–72 hours at 48°C, followed by phenol/chloroform extraction and ethanol precipitation.
Discovery with Next Generation Sequencing
We examined the Ion AmpliSeq™Cancer Hotspot Panel v2 to profile 50 frequently mutated in human cancer genes in FPPE tissue DNA from four patients with short-term survival and four patients with long-term survival, the eight of which looked identical under the microscope in a pathology laboratory. The cancer Hotspot Panel was also examined paired tissue, plasma and urine samples from two HGSC patients. Libraries for the discovery cohort were generated using the Ion AmpliSeq Library kit 2.0 according to the manufacturer’s instructions (Life Technologies, Carlsbad, USA). Included in this panel were primers for 207 amplicons covering 2800 Catalog of Somatic Mutations in Cancer of 50 genes with known cancer associations: ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAS, GNAQ, HNF1A, HRAS, IDH1, JAK2, JAK3, IDH2, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53 and VHL. (COSMIC, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic). 10ng DNA from the tumor samples was used as the template to prepare the library. Amplified libraries were quantified using the Qubit 2.0 Fluorometer and the High Sensitivity Qubit Assay Kit (Life Technologies). Amplified libraries were assessed for quality (size and concentration) using the Agilent 2100 Bioanalyzer Instrument (Agilent Technologies, Santa Clara, CA) following the Bioanalyzer standard protocol. The AmpliSeq libraries were clonally amplified on to Ion Sphere Particles (ISPs) using emulsion PCR following standard Ion Torrent protocols. ISP preparation was performed using the automated Ion Torrent OneTouch2 system following the manufacturer’s protocol (MAN0007220 Revision 4.0). The Qubit Fluorometer was used to assess ISP quality after ISP preparation but before ISP enrichment. Up to eight specimens were barcoded with Ion Xpress Barcode Adapters (Life Technologies), pooled, and run on a single Ion 318 chip. This includes multiple patient samples and one control, which we rotate among water, normal, and a mix of positive control cell lines.
Methylation 450K arrays
We sought to determine genes differentially methylated in ovarian cancer as compared to fallopian tube epithelium, therefore we compared the methylation status in 12 HGSC FFPE tissue samples to 30 fallopian tube epithelium samples using the genome-wide Infinium HumanMethylation 450K Array. The HumanMethylation450K DNA BeadChip assay was used to perform unbiased genome-wide DNA methylation analysis. Bisulfite modification of genomic DNA (2μg) was performed with EpiTect Bisulfite Kit (QIAGEN) according to the manufacturer’s protocol. We hybridized bisulfite converted DNA to the HumanMethylation450K array to identify differentially methylated regions (DMRs) in HGSC FFPE samples (n=12) and normal fallopian tube epithelium frozen tissue samples (n=30). To validate these results, we performed quantitative Methylation Specific PCR (qMSP) in another cohort of HGSC patient samples (n=35).
We read the data into R using the illuminaio package[19]. For data normalization we used the minfi package to apply the Noob background subtraction and dye-bias correction[20] followed by normalization and identification of DMRs between cases and controls [21]. The minfi package provides tools for analyzing Illumina’s methylation arrays and includes methods for preprocessing, quality assessment, and detection of differentially methylated regions from the kilobase to the megabase scale. We performed pre-processing with the minfi package applying a version of subset quantile normalization to the Meth and Unmeth intensities separately. The distribution of type I and type II probes was forced to be the same by first quantile normalizing the type II probes across samples and then interpolating a reference distribution to which the type I probes are normalized. The stratified quantile normalization method is implemented by the preprocessQuantile function (the function does no background correction and removes zeros using the fix2 MethOutlier function). This algorithm relies on the assumptions necessary for quantile normalization and involves both within- and between- sample normalization. We then intersected the statistically significant DMRs (FWER p < 0.05) that discriminate ovarian cancer from fallopian tube epithelium and DMRs that discriminate HGSC lesions with short- (<2 years) and long-term survival (>5 years).
Cell culture
Normal ovarian cell line Ose2a was grown in DMEM-F12. The ovarian cancer cell lines OVCAR5 and SkOV3 were cultured in McCoy’s media. Ovarian cancer cell line IGROV was cultured in RPMI-1640, and CaOV3 cells were grown in DMEM. All cells were grown in the presence of Pen/Strep (100 units/mL penicillin and 100 μg/mL streptomycin) and supplemented with 10% fetal bovine serum.
Bisulfite-sequencing
Bisulfite conversion of 1–2 μg of genomic DNA (from ovarian cancer cell lines, normal fallopian tube tissue, and HGSC FFPE tissue) was performed using the EpiTect Bisulfite kit (Qiagen) and used for amplification by qMSP. Bisulfite-converted DNA from the ovarian cancer cell lines was amplified by touchdown PCR, using Taqman primers and probes designed to a region in the promoter of HIST1H2BB or MAGI2. This amplified region is contained within the area detected in the methylation arrays and comprises 13 CpG sites in HIST1H2BB, and 23 CpG sites in MAGI2. These primers amplify the methylated and unmethylated sequences. The products of touchdown PCR were run on a 1.5% agarose gel and the corresponding DNA bands were excised from the gel and extracted using the Gel extraction kit (Qiagen). DNA was sent for bisulfite-sequencing to Genewiz. Sequences were manually analyzed to determine whether these regions of the HIST1H2BB and MAGI2 promoters were methylated; methylated cytosines remain as cytosines while unmethylated cytosines convert to uracils after bisulfite-conversion and uracils then convert to thymines during amplification.[22]
Quantitative Methylation-Specific PCR (qMSP)
Specific Taqman primers and probes were designed to amplify the bisulfite-converted promoter region of HIST1H2BB and MAGI2, primers amplify the methylated DNA. Titration of normal human methylated DNA (Zymo Research) was used to generate a standard curve for absolute quantification. The ratio between the values of HIST1H2BB or MAGI2, and the reference gene ACTB provide the relative methylation level (100 x target gene/reference gene).
Reverse-transcription RT-PCR
Total RNA was assessed for HIST1H2BB, MAGI2 and GAPDH expression levels using quantitative real-time reverse transcription (RT)-PCR (TaqMan). Reverse transcription was performed with random hexamer primers and Superscript II Reverse Transcriptase (Invitrogen Corp.) according to manufacturer’s instructions. Quantitative RT-PCR was then performed on the Applied Biosystems 7900 Sequence Detection Instrument (Applied Biosystems) using TaqMan expression assays (Life Technologies).
5-Aza-2’-deoxycytidine and Trichostatin A treatments
Ovarian cancer cells were incubated in the presence or absence of 2.5 uM 5-Aza-2-deoxycytidine for 4 days. Trichostatin A (TSA) was added on the last day and total RNA was extracted, and RT-PCR performed to measure MAGI2 mRNA expression.
siRNA transfections and clonogenic assays
CaOV3 and OVCAR5 cells were transfected with 25 nM MAGI2 siRNA (Santa Cruz Biotechnology-sc) or control siRNA (Dharmacon). Total RNA was extracted 48 hours post transfection and RT-PCR analysis of MAGI2 mRNA expression was performed. For clonogenic assays, 48 hours after transfection cells were plated in 6-well plates and allowed to grow for ~10 days. Colonies were then visualized by crystal violet (0.5% crystal violet in 50% methanol) staining.
Immunoblotting analysis
Cells were washed and cell lysates were prepared in RIPA lysis buffer (150 mM Tris-HCl, pH 6.8, 25% glycerol, and 5% SDS). Cell lysates were separated by SDS-PAGE on 4–12 or 10–20% Tris-Glycine gels and transferred to PVDF membranes. The membranes were blocked with TBS-T + 5% non-fat dry milk and incubated overnight at 4 °C with antibodies specific for the indicated proteins (H2B, MAGI2, or GAPDH). After washing, the membranes were incubated with horseradish-peroxidase conjugated secondary antibodies. Protein detection was performed by enhanced chemiluminiscence (Amersham).
Identification of DNA methylation and somatic mutation alterations in FFPE samples, cervical swabs, plasma and urine
Spearman’s rank correlation coefficient calculation was conducted on all pairwise comparisons of normalized DNA methylation ratios measurements in the biological specimen types (tumor, cervical swabs, serum/plasma, ascites and urine) in order to determine a non-parametric measure of correlation. For these same measurement comparisons, we conducted pairwise Wilcoxon signed-rank tests. We also plotted box plots with square root transformed y-axes of normalized DNA methylation ratio measurements to visualize differences in biological specimen type means among matched pairs from the same patients. Each gene’s pairwise tests (9 each, for which Wilcoxon signed-rank tests could be conducted) were considered independent families of tests therefore did not require adjusting alpha for the total pairwise tests. Since the results would be interpreted as exploratory rather than definite and each comparison could be considered to address a question with considerably different implications (e.g., discovering more methylated gene in urine than in serum/plasma has a considerably different practical implication than discovering more methylated gene in ascites than in tumor), we considered each pairwise comparison to belong to a family of a secondary hypothesis and therefore kept each pairwise alpha at 0.05. Supporting this choice, the false discovery rate of rejecting the null hypothesis of no difference when it is true is expected to be no more than 5% due to random chance for each gene at the unadjusted alpha of 0.05, however 22% of comparisons for each gene had significant differences at the 0.05 level.
We analyzed the concordance of somatic genomic variants across four biological specimen types: paired FFPE samples, cervical epithelium, plasma and urine in two HGSC patients.
For the FFPE samples we tested differences of proportions between survival groups for all loci mutated at least once in each survival group, for every specific locum at the Single Nucleotide Variants (SNVs) level, and by gene for all SNV loci per gene in the Hot Spot panel. We also constructed logistic regression models for each binary DNA methylation-mutation combination coded, as dependent variable, mean methylation beta value across all CpGs in a gene (where ≤0.3 was coded as non-methylated and >0.3 was coded as methylated and ordinal coded, as independent variable, each SNV loci per gene. Due to the small sample size (N=8), we calculated the p-value from the likelihood ratio comparing the model with the predictor SNV loci per gene to the null model of a simple average of the dependent variable, since this is the most accurate method for calculating p-values with such low sample sizes. Histograms were analyzed and summary statistics were calculated for the gene count data of significant associations, before adjustment. We performed FDR adjustments for each analysis with more than one test, and either reported based only on the adjusted p-values or noted where adjusted p-values became non-significant. We set our alpha at 0.05 for these analyses. We prepared a clustered heatmap of all significant associations between each SNV loci per gene and DNA methylation status per gene. In this figure, every SNV loci per gene were denoted in the rows while DNA methylation status per gene were denoted in the columns. Intersections between specific rows and columns represent the association for that mutation-methylation pair, where a green spot signifies that the pair has a significant association and a blank spot signifies that the pair does not have a significant association. Due to the large amount of DNA methylation measurements per gene, we only identified in the x-axis genes with significantly different methylated regions (DMRs) less than 6,000 bp from the promotor and with L-values ≥4 when comparing short term and long term HGSC survivors, obtained in a previous analysis.
For the paired samples we compared the inter-rater agreement of Ion AmpliSeq™ Cancer Hotspot Panel (Life Technologies Corporation) somatic genotype prediction in cancer related genes using Cohen’s kappa coefficient. Two versions of this analysis were conducted: One in which all missing predictions for Single Nucleotide Variants (SNVs) were kept in the analysis; and another one which only kept SNVs with predictions for both compared measures.
Results
HIST1H2BB and MAGI2 are differentially methylated in high-grade serous ovarian cancers
Our genome-wide analysis identified 161 gene-associated DMRs that differ between HGSC and fallopian tube epithelium samples. These genes are involved in regulation of transcription (GO:0045449 and GO:0006355); regulation of metabolic process (GO:0051252); regulation of apoptosis (GO:004298); regulation of programmed cell death (GO:0043067) and (GO:0010941). Specifically, we found HIST1H2BB and MAGI2 promoters were the most differentially methylated regions, by p-value measurement, when comparing HGSC with normal fallopian tube samples (Figure 1A) and, in subset analysis, both promoters gained methylation in HGSC patients with long-term survival (n=6) compared to patients with short-term survival (n=6) (Figure 1B). Consistent with our 450K array results, our quantitative Methylation-Specific PCR (qMSP) results confirmed higher methylation levels of HIST1H2BB and MAGI2 in patients with long-term survival compared to short-term survivors (Figure 1C). Together, our results suggest that HIST1H2BB and MAGI2 are differentially methylated in HGSC.
Figure 1.

HIST1H2BB and MAGI-2 are differentially methylated in HGSC as compared to normal fallopian tube epithelium; and in HGSCs of patients with long-term survival as compared to short term survival. DNA was extracted, bisulfite-converted and analyzed by 450 K Methylation arrays and qMSP. A) Dot plots of methylation levels (Beta-values) on promoter CpG sites of HIST1H2BB and MAGI-2 in 30 normal fallopian tube samples (red) as compared to 12 HGSC samples (black). B) Dot plots of methylation levels (Beta-values) on promoter CpG sites of HIST1H2BB and MAGI-2 in 6 HGSC samples with long-term survival (red) as compared to 6 HGSC samples with short-term survival (black). C) qMSP analysis of DNA from 6 HGSC tissue samples of long-term survivors (blue) as compared to 6 HGSC tissue samples of short-term survivors (red), using primers to promoter CpG sites of HIST1H2BB or MAGI-2. Overall survival time in months is shown. D) qMSP analysis of DNA from ovarian cancer cells and a normal ovarian cell line (Ose2a), using primers to promoter CpG sites of HIST1H2BB or MAGI-2. E) RT-PCR analysis in ovarian cancer cells and a normal ovarian cell line, using primers to measure mRNA expression of HIST1H2BB or MAGI-2.
HIST1H2BB and MAGI2 are methylated in ovarian cancer cell lines and methylation is inversely correlated with expression
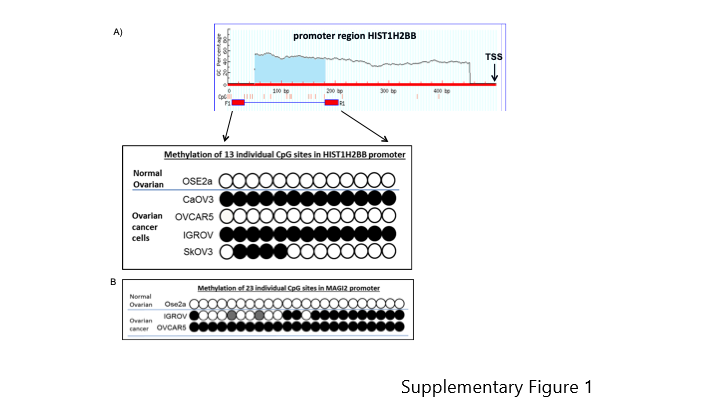
To determine whether HIST1H2BB and MAGI2 were also methylated in established ovarian cancer cell lines we performed bisulfite-sequencing and qMSP analysis. We designed primers to a region of the HIST1H2BB promoter spanning 300–500 bp upstream of the TSS. This region comprises 13 CpG sites also contained within the differentially methylated region identified by the methylation array. As shown in Supplementary Figure 1A, all CpG sites in this HIST1H2BB promoter region are methylated in two of the ovarian cancer cell lines examined, CaOV3 and IGROV as compared to normal ovarian cells Ose2a, which is completely unmethylated. For this gene, one of the cell lines, SkOV3 was partially methylated and OVCAR5 was completely unmethylated. To confirm our results, we also performed qMSP analyses using methylation specific TaqMan primers and probes to the methylated region of HIST1H2BB. CaOV3 and IGROV had higher methylation levels, whereas OVCAR5 and SkOV3 had lower methylation, consistent with our bisulfite-sequencing results.
We also analyzed methylation of the MAGI2 promoter by bisulfite-sequencing and qMSP. MAGI2 was completely methylated in 23 CpG sites in the promoter region in one of the cancer cell lines examined, OVCAR5, and partially methylated in IGROV cancer cells. In contrast, normal ovarian cells, were completely unmethylated (Supplementary Figure 1B). Results of qMSP analysis show MAGI2 methylation in all ovarian cancer cell lines, in OVCAR5 and IGROV, as well as in 2 additional cell lines examined. Therefore, our results suggest that HIST1H2BB and MAGI2 are methylated in ovarian cancer cells (Figure 1D.
We performed Real Time RT-PCR to measure mRNA expression of HIST1H2BB and MAGI2 in ovarian cancer cells to determine whether HIST1H2BB and MAGI2 promoter methylation was associated with silencing of expression. Methylation of HIST1H2BB and MAGI2 is inversely correlated with mRNA expression in most cell lines. CaOV3 and IGROV which have higher HIST1H2BB methylation levels have lower HIST1H2BB mRNA expression, and SkOV3 and OVCAR5 have lower HIST1H2BB methylation levels and higher mRNA expression levels (Figure 1E). For MAGI2 most cells have low mRNA expression levels consistent with increased methylation (Figure 1E).
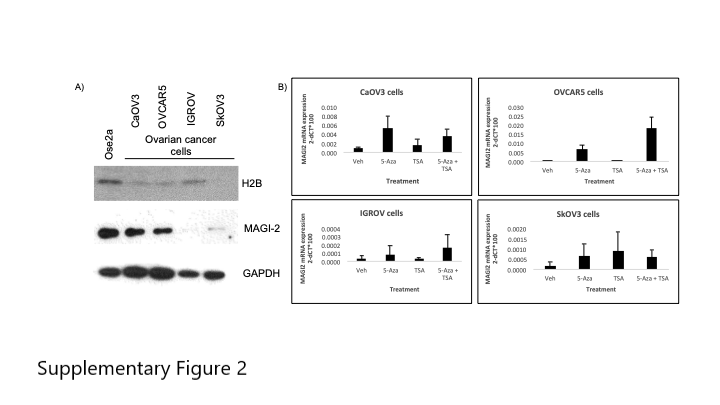
We performed immunoblots to determine whether the expression of HIST1H2BB and MAGI2 was also decreased at the protein level. H2B.F is the protein product of HIST1H2BB, and its levels were lower in the 4 ovarian cancer cell lines examined, as compared to normal ovarian cells. Similarly, MAGI2 protein levels were lower in the ovarian cancer cells, as compared to normal ovarian cells. Taken together, these results suggest that methylation of HIST1H2BB and MAGI2 could lead to their decreased mRNA and protein expression in ovarian cancer cells (Figure Supplementary Figure 2A).
MAGI2 is re-expressed upon cell treatment with demethylating agent 5-aza-2’-deoxycytidine
Inhibiting methylation should cause re-expression of MAGI2 or HIST1H2BB if methylation is contributing to their silencing of expression. Therefore, we treated ovarian cancer cells with demethylating agent 5-aza-2’-deoxycytidine to determine whether methylation might be involved in silencing of MAGI2 or HIST1H2BB. Silencing of expression by methylation is often accompanied by deacetylation, for that reason we also treated cells with a deacetylase inhibitor, Trichostatin A. In all ovarian cancer cells, treatment with 5-aza-2’-deoxycytidine with or without TSA increased expression of MAGI2 (Supplementary Figure 2B). This suggests that MAGI2 expression is regulated by methylation in ovarian cancer cells.
MAGI2 downregulation increases survival of ovarian cancer cells
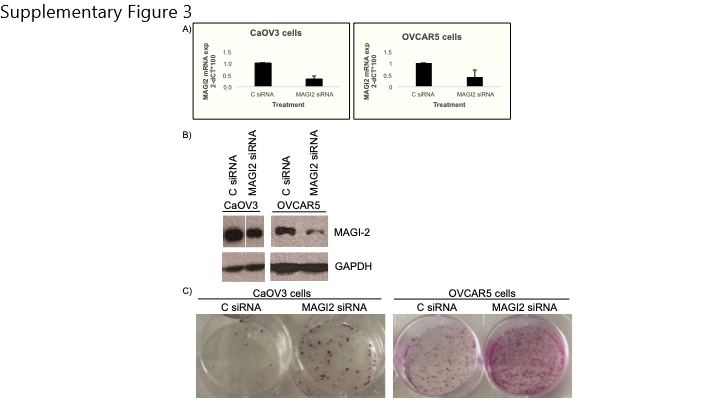
To determine whether the silencing of MAGI2 might have a role in the growth of ovarian cancer cells, we set to downregulate MAGI2 in CaOV3 and OVCAR 5 cells, which have protein expression of MAGI2 (Supplementary Figure 3A–B). We downregulated MAGI2 with siRNA and performed clonogenic assays (Supplementary Figure 3C). Downregulation of MAGI2 expression increased survival of ovarian cancer cells in CaOV3 and OVCAR5 cells, evidenced by an increased number of colonies in the MAGI2 siRNA treated cells. These results suggest that the presence of MAGI2 has a growth-inhibitory role and inhibiting its expression by methylation might confer a growth advantage to the ovarian cancer cells.
Identification of genomic and epigenomic alterations in FFPE samples from short-term and long-term survival patients
Statistically significant differentially mutated genes were usually not mutated in the short survival samples. In statistically significant differentially mutated genes, the proportion of mutations by gene was always higher in the long survival group, with the exception of the CSF1R gene, which was more mutated in the short survival group. The CSF1R (Colony Stimulating Factor 1 Receptor) gene plays an important role in innate immunity and in inflammatory processes (Supplementary Table 1).
The proportion of mutated alleles at the SNV level in the long survival group was statistically significantly larger than the proportion in the short survival group. The only significantly different mutation proportions at the SNV level were the chr17 7578395 G->A allele in the rs587780070 SNV of the TP53 gene and the chr9 21971120 G->A allele in the rs121913388 SNV of the CDKN2A gene (GRCh37), with long survival samples having more of the mutated TP53 allele and short survival samples having more of the mutated CDKN2A allele. Both of these genes are known tumor suppressors. However, neither of these two differences at the SNV level remained statistically significant after FDR adjustment, possibly due to the lower sample size of measured loci at the SNV level for each test (Supplementary Table 2).
A considerable fraction (15% ) of combinations of Differentially Methylated Regions (DMRs) associations with gene mutations were found to have unadjusted p-values under 0.05, which is three times what is expected by chance, however none of these 62272 significant associations out of 426104 tests remained significant after FDR adjustment that took into account all conducted tests probably due to small sample sizes available for these calculations. These significant, yet unadjusted associations displayed some interesting patterns, namely, in these association with p-values below 0.05, only 6154 out of 8696 DMRs measured (71% of DMRs, or 47% taking false positives into account) were represented in this group, and the 6191 DMRs were associated with ≤5 SNVs or ≥35 SNVs out of the 49 gene mutations in the Hot Spot panel, while the 49 mutated genes displayed a normal distribution, of each being associated with an average of 1271 DMRs (standard deviation of 402 DMRs) out of 8696 DMRs (15% of DMRs) (Supplementary Table 3). A heatmap was created to visualize and confirm these patterns we found in the significant associations (Supplementary Figure 4).
Identification of genomic and epigenomic alterations in cervical swabs, plasma and urine
We found somatic mutations and single nucleotide variants (SNVs) in paired tissue from cervical swabs, serum/plasma and urine, showing that liquid biopsies, which are measured in blood/plasma, can also be measured in urine with high concordance (Supplementary Tables 4–5). In order to quantify how faithfully these genotypes were identified in urine, we calculated a concordance statistic using Cohen’s kappa coefficient, which revealed that for SNVs present in each comparison pair, the correlation was 0.84, 0.77 and 0.92 for Tissue-Plasma, Tissue-Urine and Plasma-Urine in patient TG01, and 0.6, 0.6 and 1, respectively for patient TG04 (Table 1). These data represent concordance of genomic variant detection in paired tissue, plasma and urine samples, with very high concordance between plasma and urine.
Table 1:
Cohen’s Kappa, 95% confidence interval and percent agreement for somatic mutations measured in tumor, plasma and urine.
| Specimen Comparisons | Patient 1 | Patient 1 (Without Missing Predictions) | Patient 2 | Patient 2 (Without Missing Predictions) |
|---|---|---|---|---|
| Tissue-Plasma | 0.68 (0.42–0.93); 71% | 0.84 (0.62–1.06); 86% | 0.18 (−0.06–0.42); 27% | 0.6 (0.19–1.01); 67% |
| Tissue-Urine | 0.61 (0.34–0.89); 88% | 0.77 (0.51–1.02);79% | 0.23 (−0.02–0.47); 32% | 0.6 (0.19–1.01); 67% |
| Plasma-Urine | 0.87 (0.69–1.05); 88% | 0.92 (0.76–1.08); 93% | 0.82 (0.63–1.01); 86% | 1 (1,1); 100% |
We also found HIST1H2BB and MAGI2 promoter methylation in cervical swabs, plasma/serum and urine in a subset of the samples tested, suggesting that these could be used as non-invasive early detection biomarkers. Our data suggest vaginal swab DNA has lower DNA methylation levels when compared to trans-renal (tr-DNA) DNA from urine (HIST1H2BB p=0.03; MAGI2 p=0.05) and serum/plasma DNA (HIST1H2BB p=0.03; MAGI2 p=0.02) for the genes measured. The data also indicates TrDNA is a better indicator of DNA methylation status than serum/plasma in the genes measured (Figure 2). The data also suggest the different biological specimen measures are correlated. The correlation between DNA methylation measures between tumor and TrDNA were 1 and 0.5 for HIST1H2BB and MAGI2, respectively. The correlation between DNA methylation measures between tumor and ascites were 1 for both HIST1H2BB and MAGI2. The correlation between tumor and vaginal swab DNA methylation was 0.5 in both HIST1H2BB and MAGI2.
Figure 2. Magnitude of methylation differs in DNA from paired tumor, urine (trans-renal DNA -TrDNA), plasma/serum, vaginal swabs and ascites specimens.

HIST1H2BB and MAGI2 methylation was measured by quantitative Methylation Specific PCR (qMSP) and compared in DNA from paired tumor, urine (TrDNA), plasma/serum, vaginal swab and ascites specimens. A) Box-plots show that qMSP levels of HIST1H2BB were significantly higher in TrDNA compared to vaginal swabs (p=0.03), and higher in serum/plasma compared to vaginal swabs (p=0.03). B) Box-plots show that qMSP levels of MAGI2 methylation were higher in TrDNA compared to serum/plasma (p=0.02), and higher in TrDNA compared to vaginal swabs (p=0.05).
Discussion
DNA methylation in HGSC
Epigenetic alterations are found in primary human cancers, and such aberrations are composed of DNA methylation and its linked histone modification. DNA methylation occurs in cytosine residues among the CpG islands of the promoter regions of individual genes[23]. Methylated cytosines can be bound by methyl-CpG-binding protein 2 (MeCP2), and the resulting protein-nucleotide can be incorporated into protein complexes that include histone modification enzymes leading to dynamic changes in chromatin structure[24]. As a result, DNA methylation can result in gene silencing due to impaired access of transcription factors through condensed and closed chromatin.
Promoter DNA methylation, which occurs on cytosine nucleotides across CpG islands, results in gene silencing and represents a major epigenetic alteration in human cancer. Methylation-specific PCR can amplify these modifications as candidate biomarkers in cancer cells. Such candidate biomarkers are widely scattered across the entire genome, amounting to a total of 200-300 promoter methylated genes[25]. After rigorous validation, superior biomarker candidates representing cancer-specific methylation are now considered ready for use in clinical decision-making in the development of therapeutic strategies, with possible use extending even to cases where cancer surgery is indicated[26–29].
In this study, we have used epigenome wide and molecular biology tools to show that HIST1H2BB and MAGI2 are newly described tumor suppressor genes (TSG) differentially methylated in HGSC tissue samples when compared to normal fallopian tube epithelium. We show that the expression of HIST1H2BB and MAGI2 is reduced in ovarian cancer cells with promoter methylation of these genes, a hallmark of TSGs. We also show that HIST1H2BB and MAGI2 promoter methylation discriminates between long-term and short-term HGSC survivors. Finally, we use massively parallel exome sequencing and molecular biology tools to show that HIST1H2BB and MAGI2 promoter methylation and HGSC-associated somatic mutations, can be measured in tissue, plasma and urine samples. Together we identify molecular markers that can be added to precision medicine workflows for HGSC diagnosis and treatment.
The MAGUK Inverted 2 (MAGI2) gene codes a PTEN-interacting scaffold protein implicated in cancer on the basis of rare, recurrent genomic translocations and deletions in various tumors. In the renal glomerulus, MAGI2 is exclusively expressed in podocytes, specialized cells forming part of the glomerular filter, where it interacts with the slit diaphragm protein nephrin [30]. This encoded protein is characterized by two WW domains, a guanylate kinase-like domain and multiple PDZ domains, the structural similarity of the membrane-associated guanylate kinase homologue (MAGUK) family. MAGI2 is a tight junction protein in epithelial tissues [31].
In our study, MAGI2 exhibited a growth inhibitory role in ovarian cancer cells since survival of ovarian cancer cells OVCAR5 and CaOV3 increased upon silencing of MAGI2 with siRNA. MAGI2 promoter methylation has been observed in cervical scrapings from patients with endometrial and ovarian cancer[32]. MAGI2 is significantly methylated in Cervical Intraepithelial Neoplastic grade 3+ lesions. MAGI2 is potentially implicated in β-catenin signaling, suggesting the epigenetic dysregulation of this signaling pathway during cervical cancer development[33]. MAGI2 methylation, as well as inactivating mutations of MAGI2, have also been reported in prostate cancer,[34, 35] suggesting importance of MAGI2 silencing across cancers. MAGI2 mRNA expression is decreased in prostate cancer cells and patient samples, and inclusion of MAGI2 in a biomarker gene panel improved the ability of the panel to discriminate between benign hyperplasia samples and prostate cancer. [36]
MAGI2 seems to have tumor suppressor roles in cancer, among its roles it has been reported that MAGI2 interacts with tumor suppressor protein PTEN and inhibits AKT signaling,[37] and increased PTEN protein stability and decreased AKT activation induced by MAGI2 also inhibits proliferation and migration of hepatocellular carcinoma cells.[38] Altogether, these reports suggest a growth-inhibitory role for MAGI2 and that methylation and silencing could confer a growth advantage to cancer cells.
HIST1H2BB (Histone Cluster 1 H2B Family Member B) is a Protein Coding gene. Among HIST1H2BB related pathways are Meiosis and Signaling by Rho GTPases. Gene Ontology (GO) annotations related to HIST1H2BB include sequence-specific DNA binding and protein heterodimerization activity. An important paralog of HIST1H2BB is HIST1H2BN.
Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Nucleosomes consist of approximately 146 bp of DNA wrapped around a histone octamer composed of pairs of each of the four core histones (H2A, H2B, H3, and H4). The chromatin fiber is further compacted through the interaction of a linker histone, H1, with the DNA between the nucleosomes to form higher order chromatin structures. This gene is intronless and encodes a replication-dependent histone that is a member of the histone H2B family. Transcripts from this gene lack polyA tails; instead, they contain a palindromic termination element. This gene is found in the large histone gene cluster on chromosome 6p22-p21.3
Covalent modifications of histones play a crucial role in the regulation of gene expression. HIST1H2BB impacts transcriptional regulation and elongation, mostly by ubiquitination. Histone H2B monoubiquitination (H2Bub1) has mainly been described as a regulator of transcription elongation. Genome-wide profiles show that H2Bub1 levels are negatively correlated with the accessibility of enhancers to transcriptional activators. The chromatin association of histone variant H2A.Z, which is evicted from enhancers for transcriptional activation, is stabilized by H2Bub1 by impairing access of the chromatin remodeler INO80. Thus, H2Bub1 acts as a gatekeeper of H2A.Z eviction and activation of inducible enhancers [39]. H2B ubiquitination (uH2B) also promotes histone eviction at Double Stranded Breaks independent of resection or ATP-dependent chromatin remodelers. Cells lacking uH2B, or its E3 ubiquitin ligase Bre1, exhibit hyper-resection due to the loss of H3K79 methylation[40]. H3K79 methylation by the histone methyltransferase Dot1 affects gene expression and the response to DNA damage and is enhanced by monoubiquitination of the C-terminus of histone H2B (H2Bub1). However, Dot1 and H2Bub1 are subject to bi-directional crosstalk and Dot1 possesses chromatin regulatory functions that are independent of its methyltransferase activity[41]. Regulation of Dot1-mediated H3K79 methylation and Set1-COMPASS-mediated H3K4 methylation by H2BK123 ubiquitination (H2Bub1) are evolutionarily conserved trans-histone crosstalk mechanisms. Ubiquitin acts as a “glue” to bind the nucleosome together for supporting Dot1/Set1-COMPASS functions[42, 43]. Ubiquitylation of histone H2B at lysine residue 120 (H2BK120ub) is a prominent histone posttranslational modification (PTM) associated with the actively transcribed genome. De novo ubiquitylation of H2BK120 is found to be highly sensitive to PTMs on the N-terminal tail of histone H2A, a crosstalk that extends to the common histone variant H2A.Z[44].
The impact of HIST1H2BB promoter methylation is not described in the literature. Silencing of HIST1H2BB by methylation might be expected to lead to alterations in transcriptional regulation and elongation. Due to the role of H2B in the maintenance of nucleosome structure, silencing of HIST1H2BB could lead to chromatin remodeling and disruptions in the general nucleosomal structure, impacting gene expression. In this study, we found HIST1H2BB methylated in ovarian cancer tissue and its expression reduced at the mRNA and protein levels in ovarian cancer cell lines.
In sum, we identified the promoter regions of HIST1H2BB and MAGI2 to be differentially methylated in tumor when compared to fallopian tube epithelium. We also found that HIST1H2BB and MAGI2 promoter methylation discriminates between HGSC patients with long-term survival compared to short-term survivors. QMSP results and reverse transcription RT-PCR analyses show that promoter methylation of HIST1H2BB and MAGI2 in ovarian cancer cell lines is inversely correlated with their mRNA and protein expression, when compared to normal ovarian cell lines. Treatment of ovarian cancer cell lines with demethylating agent 5-aza-2’-deoxycytidine and deacetylase inhibitor TSA induce re-expression of MAGI2. Downregulation of MAGI2 using siRNAs was shown to increase the growth of ovarian cancer cells, suggesting a growth-inhibitory role for MAGI2 in ovarian cancer. The survival results of our clonogenic assay suggest that MAGI2 expression could be a strong potential target for ovarian cancer treatment interventions. This finding must be confirmed in larger projects. QMSP also confirmed higher methylation levels of HIST1H2BB and MAGI2 in patients with long-term survival compared to short-term survivors and in paired tissue, ascites, cervical swabs, plasma/serum, and urine samples. Together these exploratory data suggest that promoter methylation of HIST1H2BB and MAGI2 can detect HGSC and identify HGSC patients with poor survival. We surmise they can be used as early detection, as well as diagnostic and prognostic biomarkers.
Early detection and intervention are likely to be the most effective means for reducing morbidity and mortality of human cancer. A noninvasive assay for detection of early-stage tumors, using massively parallel sequencing to evaluate sequence changes in circulating cell-free DNA, detected somatic mutations in 68% of ovarian cancer patients with stage I or stage II disease [45]. HGSC patients with short-term survival (less than 2 years) are characterized by focal copy number gain of CCNE1, lack of BRCA mutation signature, low homologous recombination deficiency scores, and the presence of ESR1-CCDC170 gene fusion [46]. A BRCA genetic testing assay is now being recommended to identify the estimated 3–9% of ovarian cancer patients with somatic BRCA1/2 mutations who, in addition to germline carriers, could benefit from PARP inhibitors therapy [47]. These data suggests that somatic mutation and DNA methylation testing in biofluids can be a broadly applicable approach for noninvasive detection of tumors, useful for personalized screening and therapeutic management of patients with HGSC [48].
DNA methylation and somatic mutations associations in short-term and long-term survival in HGSC
We used a Cancer Hotspot sequencing panel to compare the frequency of somatic mutations in HGSC patients with short-term and long-term survival, whose FFPE tissue slides were identical under the microscope. We found statistically significant differentially mutated genes and alleles when comparing short-term and long-term survival samples. We also found that 71% of DMRs had significant associations with mutations in HGSC. Most DMRs were usually associated either with ≤10% or with ≥71% of gene mutations in DMRs, and the average gene mutation was associated with 15% of DMRs. It is also likely that many of the gene methylations were highly correlated due to varying factors (Supplementary Figure 4). If we assume that promoter DNA methylation events occur before mutations, we can therefore hypothesize that in ovarian HGSC clusters of approximately 15% of promoter DNA methylation events can each lead to large amount of mutations (≥71%) or a small (≤10%) amount of mutations. These DNA methylation clusters together amount to 47% of promoter DNA methylation events, which explain from 71% to 100% of mutated genes.
We also quantified epigenomic alterations and somatic mutations in paired tissue, plasma/serum and urine of ovarian cancer patients, to show their potential usefulness in ovarian cancer precision medicine workflows. We found that HIST1H2BB and MAGI2 promoter DNA methylation and somatic mutations in HGSC-related genes are reproducible in plasma/serum and urine.
Detection of promoter DNA methylation and somatic mutations in various biofluids permits early detection of cancer cells during perioperative courses of clinical treatment as is illustrated in Figure 3. We performed preliminary analyses to identify the biofluids in which genomic and promoter DNA methylation measurements best correlate with paired tissue levels. Although tissue and biofluids data indicates TrDNA is a better indicator of DNA methylation status than serum/plasma, this finding might not be generalizable to other genes or cancer types, such as non-urinary tract adjacent malignancies. We controlled for direct shedding of tumor cells into the urinary tract, a way to bypass renal filtration, by only isolating cell-free DNA in urine 150 bp long or smaller, a size that allows filtration through the kidneys. Therefore, these results are possibly generalizable to non-urinary tract adjacent malignancies. Finally, to control for varying concentrations, we normalized to a housekeeping gene. We compared ratio measures of methylation, which are independent of the varying concentration between biological specimen types, making our comparisons directly valid. Although our samples sizes for the comparisons were small, the paired design leveraged sample size to achieve statistical power high enough to detect significant difference in several comparisons. Our exploratory results suggest that trans-renal DNA from urine has the potential to be a more sensitive non-invasive measure of circulating tumor DNA methylation. We hypothesize this conclusion can be broadened to DNA in general, such as circulating tumor DNA. Circulating tumor DNA is a very promising powerful biomarker of cancer for diagnosis and prognosis.[48–50]
Figure 3.

Diagram representing the use of precision methylation for personalized medicine workflows focused on ovarian cancer detection.
The main limitation of this project is sample size. We are showing associations, not cause and effect relationships in our patient data. We did not set out to prove the DNA methylation drivers of HGSC. This would require another study design with a much larger sample size to confirm the observed associations in this initial study, followed by work on patient derived tumorgrafts[51, 52]. Ultimately, molecular analyses in precision medicine may be helpful to better elucidate whether observed genomic and epigenomic alterations represent a distinct entity with clinical, immunophenotypic, and molecular characteristics or an incidental phenomenon during malignant transformation. Larger samples sizes will also provide the opportunity to systematically close the existing gap between diagnostic and prognostic data. Data science tools can be set in place to integrate molecular features, clinical information and health services organization data.
This challenge can be addressed with big data analytic strategies, which may include machine learning and computer algorithms to integrate patient demographic, psychosocial, clinical, pathology and molecular profiles with treatment recommendations, health insurance coverage, clinical and health information data. This integration can become the foundation for Precision Medicine platforms. In sum, we now have the understanding and capabilities of combining multiple big data streams into next generation precision medicine tools with machine learning and quantum entanglement capabilities, which will allow us to obtain precise depictions of molecular, clinical, psychosocial and contextual portraits to track health trajectories across the life-span. The markers described in this manuscript may be useful to improve our understanding of HGSC survival, guide treatment options, and inform public health strategies designed to improve HGSC survival rates.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Financial support: This research was supported by HERA Ovarian Cancer Foundation Outside the Box Grant (BV); Ovarian Cancer Research Alliance (IMS); National Cancer Institute U01CA84986 (DS) and K01CA164092 (RGP); P50CA228991 (IMS) and National Institute on Minority Health and Health Disparities R44MD014911 (RGP).
Footnotes
Conflicts of Interests: There are no conflicts of interests
References
- 1.Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC Jr., Beral V, Berek JS, Birrer MJ, Blagden S, Bookman MA, Brenton JD, Chiappinelli KB, Martins FC, Coukos G, Drapkin R, Edmondson R, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015; 15(11):668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Despierre E, Yesilyurt BT, Lambrechts S, Johnson N, Verheijen R, van der Burg M, Casado A, Rustin G, Berns E, Leunen K, Amant F, Moerman P, Lambrechts D, Vergote I, Eortc GCG and Group EGTR. Epithelial ovarian cancer: rationale for changing the one-fits-all standard treatment regimen to subtype-specific treatment. Int J Gynecol Cancer. 2014; 24(3):468–477. [DOI] [PubMed] [Google Scholar]
- 3.McLaughlin JR, Rosen B, Moody J, Pal T, Fan I, Shaw PA, Risch HA, Sellers TA, Sun P and Narod SA. Long-term ovarian cancer survival associated with mutation in BRCA1 or BRCA2. J Natl Cancer Inst. 2013; 105(2):141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Spencer S, Dougherty B, Orr M, Hodgson D, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014; 15(8):852–861. [DOI] [PubMed] [Google Scholar]
- 5.De Picciotto N, Cacheux W, Roth A, Chappuis PO and Labidi-Galy SI. Ovarian cancer: Status of homologous recombination pathway as a predictor of drug response. Crit Rev Oncol Hematol. 2016; 101:50–59. [DOI] [PubMed] [Google Scholar]
- 6.Sokolenko AP, Suspitsin EN, Kuligina E, Bizin IV, Frishman D and Imyanitov EN. Identification of novel hereditary cancer genes by whole exome sequencing. Cancer Lett. 2015; 369(2):274–288. [DOI] [PubMed] [Google Scholar]
- 7.Laird PW. The power and the promise of DNA methylation markers. Nature Reviews Cancer. 2003; 3(4):253–266. [DOI] [PubMed] [Google Scholar]
- 8.Heyn H and Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nature Reviews Genetics. 2012; 13(10):679–692. [DOI] [PubMed] [Google Scholar]
- 9.Guerrero-Preston R, Valle BL, Jedlicka A, Turaga N, Folawiyo O, Pirini F, Lawson F, Vergura A, Noordhuis M and Dziedzic A. Molecular triage of premalignant lesions in liquid-based cervical cytology and circulating cell-free dna from urine, using a panel of methylated human papilloma virus and host genes. Cancer Prevention Research. 2016; 9(12):915–924. [DOI] [PubMed] [Google Scholar]
- 10.Nian J, Sun X, Ming S, Yan C, Ma Y, Feng Y, Yang L, Yu M, Zhang G and Wang X. Diagnostic Accuracy of Methylated SEPT9 for Blood-based Colorectal Cancer Detection: A Systematic Review and Meta-Analysis. Clinical and translational gastroenterology. 2017; 8(1):e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esteller M, Sanchez-Cespedes M, Rosell R, Sidransky D, Baylin SB and Herman JG. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer research. 1999; 59(1):67–70. [PubMed] [Google Scholar]
- 12.Vang R, Shih Ie M and Kurman RJ. Fallopian tube precursors of ovarian low- and high-grade serous neoplasms. Histopathology. 62(1):44–58. [DOI] [PubMed] [Google Scholar]
- 13.Kim J, Park EY, Kim O, Schilder JM, Coffey DM, Cho CH and Bast RC Jr. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers (Basel). 2018; 10(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ducie J, Dao F, Considine M, Olvera N, Shaw PA, Kurman RJ, Shih IM, Soslow RA, Cope L and Levine DA. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat Commun. 2017; 8(1):990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang S, Onuma K, Deb P, Wang E, Lytwyn A, Sur M and Daya D. Frequency of serous tubal intraepithelial carcinoma in various gynecologic malignancies: a study of 300 consecutive cases. Int J Gynecol Pathol. 2012; 31(2):103–110. [DOI] [PubMed] [Google Scholar]
- 16.Samimi G, Trabert B, Geczik AM, Duggan MA and Sherman ME. Population Frequency of Serous Tubal Intraepithelial Carcinoma (STIC) in Clinical Practice Using SEE-Fim Protocol. JNCI Cancer Spectr. 2018; 2(4):pky061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samimi G, Trabert B, Duggan MA, Robinson JL, Coa KI, Waibel E, Garcia E, Minasian LM and Sherman ME. Processing of fallopian tube, ovary, and endometrial surgical pathology specimens: A survey of U.S. laboratory practices. Gynecol Oncol. 2018; 148(3):515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adelson ME, Feola M, Trama J, Tilton RC and Mordechai E. Simultaneous detection of herpes simplex virus types 1 and 2 by real-time PCR and Pyrosequencing. Journal of clinical virology. 2005; 33(1):25–34. [DOI] [PubMed] [Google Scholar]
- 19.Smith ML, Baggerly KA, Bengtsson H, Ritchie ME and Hansen KD. illuminaio: An open source IDAT parsing tool for Illumina microarrays. F1000Research. 2013; 2:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Triche TJ Jr., Weisenberger DJ, Van Den Berg D, Laird PW and Siegmund KD. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic acids research. 2013; 41(7):e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD and Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li L-C and Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002; 18(11):1427–1431. [DOI] [PubMed] [Google Scholar]
- 23.Du J, Johnson LM, Jacobsen SE and Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015; 16(9):519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harikrishnan KN, Chow MZ, Baker EK, Pal S, Bassal S, Brasacchio D, Wang L, Craig JM, Jones PL, Sif S and El-Osta A. Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat Genet. 2005; 37(3):254–264. [DOI] [PubMed] [Google Scholar]
- 25.Hoque MO, Kim MS, Ostrow KL, Liu J, Wisman GB, Park HL, Poeta ML, Jeronimo C, Henrique R, Lendvai A, Schuuring E, Begum S, Rosenbaum E, Ongenaert M, Yamashita K, Califano J, et al. Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res. 2008; 68(8):2661–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yokoi K, Yamashita K and Watanabe M. Analysis of DNA Methylation Status in Bodily Fluids for Early Detection of Cancer. Int J Mol Sci. 2017; 18(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yokoi K, Yamashita K, Ishii S, Tanaka T, Nishizawa N, Tsutsui A, Miura H, Katoh H, Yamanashi T, Naito M, Sato T, Nakamura T and Watanabe M. Comprehensive molecular exploration identified promoter DNA methylation of the CRBP1 gene as a determinant of radiation sensitivity in rectal cancer. Br J Cancer. 2017; 116(8):1046–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ushiku H, Yamashita K, Katoh H, Ema A, Minatani N, Kikuchi M, Kojo K, Yokoi K, Tanaka T, Nishizawa N, Ishii S, Hosoda K, Moriya H, Mieno H, Katada N, Kikuchi S, et al. Promoter DNA methylation of CDO1 gene and its clinical significance in esophageal squamous cell carcinoma. Dis Esophagus. 2017; 30(2):1–9. [DOI] [PubMed] [Google Scholar]
- 29.Ushiku H, Yamashita K, Ema A, Minatani N, Kikuchi M, Kojo K, Yokoi K, Tanaka T, Nishizawa N, Ishii S, Hosoda K, Moriya H, Mieno H, Katada N, Kikuchi S, Katoh H, et al. DNA diagnosis of peritoneal fluid cytology test by CDO1 promoter DNA hypermethylation in gastric cancer. Gastric Cancer. 2017; 20(5):784–792. [DOI] [PubMed] [Google Scholar]
- 30.Balbas MD, Burgess MR, Murali R, Wongvipat J, Skaggs BJ, Mundel P, Weins A and Sawyers CL. MAGI-2 scaffold protein is critical for kidney barrier function. Proc Natl Acad Sci U S A. 2014; 111(41):14876–14881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ihara K, Asanuma K, Fukuda T, Ohwada S, Yoshida M and Nishimori K. MAGI-2 is critical for the formation and maintenance of the glomerular filtration barrier in mouse kidney. Am J Pathol. 2014; 184(10):2699–2708. [DOI] [PubMed] [Google Scholar]
- 32.Chang CC, Wang HC, Liao YP, Chen YC, Weng YC, Yu MH and Lai HC. The feasibility of detecting endometrial and ovarian cancer using DNA methylation biomarkers in cervical scrapings. J Gynecol Oncol. 2018; 29(1):e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen YC, Huang RL, Huang YK, Liao YP, Su PH, Wang HC, Chang CC, Lin YW, Yu MH, Chu TY and Lai HC. Methylomics analysis identifies epigenetically silenced genes and implies an activation of beta-catenin signaling in cervical cancer. Int J Cancer. 2014; 135(1):117–127. [DOI] [PubMed] [Google Scholar]
- 34.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D and Sougnez C. The genomic complexity of primary human prostate cancer. Nature. 2011; 470(7333):214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JH, Dhanasekaran SM, Prensner JR, Cao X, Robinson D, Kalyana-Sundaram S, Huang C, Shankar S, Jing X and Iyer M. Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome research. 2011; 21(7):1028–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahdian R, Nodouzi V, Asgari M, Rezaie M, Alizadeh J, Yousefi B, Shahrokh H, Abolhasani M and Nowroozi M. Expression profile of MAGI2 gene as a novel biomarker in combination with major deregulated genes in prostate cancer. Mol Biol Rep. 2014; 41(9):6125–6131. [DOI] [PubMed] [Google Scholar]
- 37.Matsuda S, Nakanishi A, Wada Y and Kitagishi Y. Roles of PI3K/AKT/PTEN pathway as a target for pharmaceutical therapy. The open medicinal chemistry journal. 2013; 7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu Y, Li Z, Guo L, Wang L, Zhang L, Cai X, Zhao H and Zha X. MAGI-2 Inhibits cell migration and proliferation via PTEN in human hepatocarcinoma cells. Archives of biochemistry and biophysics. 2007; 467(1):1–9. [DOI] [PubMed] [Google Scholar]
- 39.Segala G, Bennesch MA, Pandey DP, Hulo N and Picard D. Monoubiquitination of Histone H2B Blocks Eviction of Histone Variant H2A.Z from Inducible Enhancers. Mol Cell. 2016; 64(2):334–346. [DOI] [PubMed] [Google Scholar]
- 40.Zheng S, Li D, Lu Z, Liu G, Wang M, Xing P, Wang M, Dong Y, Wang X, Li J, Zhang S, Peng H, Ira G, Li G and Chen X. Bre1-dependent H2B ubiquitination promotes homologous recombination by stimulating histone eviction at DNA breaks. Nucleic acids research. 2018; 46(21):11326–11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Welsem T, Korthout T, Ekkebus R, Morais D, Molenaar TM, van Harten K, Poramba-Liyanage DW, Sun SM, Lenstra TL, Srivas R, Ideker T, Holstege FCP, van Attikum H, El Oualid F, Ovaa H, Stulemeijer IJE, et al. Dot1 promotes H2B ubiquitination by a methyltransferase-independent mechanism. Nucleic acids research. 2018; 46(21):11251–11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chandrasekharan MB, Huang F, Chen YC and Sun ZW. Histone H2B C-terminal helix mediates trans-histone H3K4 methylation independent of H2B ubiquitination. Mol Cell Biol. 2010; 30(13):3216–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chandrasekharan MB, Huang F and Sun ZW. Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation. Epigenetics. 2010; 5(6):460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wojcik F, Dann GP, Beh LY, Debelouchina GT, Hofmann R and Muir TW. Functional crosstalk between histone H2B ubiquitylation and H2A modifications and variants. Nat Commun. 2018; 9(1):1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, Anagnostou V, Fiksel J, Cristiano S, Papp E, Speir S, Reinert T, Orntoft MW, Woodward BD, Murphy D, Parpart-Li S, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med. 2017; 9(403). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang SYC, Lheureux S, Karakasis K, Burnier JV, Bruce JP, Clouthier DL, Danesh A, Quevedo R, Dowar M, Hanna Y, Li T, Lu L, Xu W, Clarke BA, Ohashi PS, Shaw PA, et al. Landscape of genomic alterations in high-grade serous ovarian cancer from exceptional long- and short-term survivors. Genome Med. 2018; 10(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Capoluongo E, Scambia G and Nabholtz JM. Main implications related to the switch to BRCA1/2 tumor testing in ovarian cancer patients: a proposal of a consensus. Oncotarget. 2018; 9(28):19463–19468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B and Alani RM. Detection of circulating tumor DNA in early-and late-stage human malignancies. Science translational medicine. 2014; 6(224):224ra224–224ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwarzenbach H, Hoon DS and Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nature Reviews Cancer. 2011; 11(6):426–437. [DOI] [PubMed] [Google Scholar]
- 50.Dawson S-J, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin S-F, Dunning MJ, Gale D, Forshew T and Mahler-Araujo B. Analysis of circulating tumor DNA to monitor metastatic breast cancer. New England Journal of Medicine. 2013; 368(13):1199–1209. [DOI] [PubMed] [Google Scholar]
- 51.Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D, Ben-Zvi I, Stebbing J, McGuire W, Harris W, Maki R, Gaya A, Bedi A, Zacharoulis S, Ravi R, Wexler LH, et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol. 2017; 28(10):2595–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, McFarland JM, Wong B, Boehm JS, Beroukhim R and Golub TR. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017; 49(11):1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.