

Abstract
Background & Aims
aISCs (aISCs) are sensitive to acute insults including chemotherapy and irradiation. Regeneration after aISC depletion has primarily been explored in irradiation (IR). However, the cellular origin of epithelial regeneration after doxorubicin (DXR), a common chemotherapeutic, is poorly understood.
Methods
We monitored DXR’s effect on aISCs by enumerating Lgr5-eGFP+ and Olfm4+ crypts, cleaved caspase-3 (CASP3+) immunofluorescence, and time-lapse organoid imaging. Lineage tracing from previously identified regenerative cell populations (Bmi1+, Hopx+, Dll1+, and Defa6+) was performed with DXR damage. Lineage tracing from aISCs was compared with lineage tracing from early progeny cells (transit-amplifying cells arising from aISCs 1 day predamage) in the context of DXR and IR. We compared stem cell and DNA damage response (DDR) transcripts in isolated aISCs and early progeny cells 6 and 24 hours after DXR.
Results
Epithelial regeneration after DXR primarily arose from early progeny cells generated by aISCs. Early progeny cells upregulated stem cell gene expression and lacked apoptosis induction (6 hours DXR: 2.5% of CASP3+ cells, p<0.0001). aISCs downregulated stem cell gene expression and underwent rapid apoptosis (6 hours DXR: 63.4% of CASP3+ cells). There was minimal regenerative contribution from Bmi1+, Hopx+, Dll1+, and Defa6+-expressing populations. In homeostasis, 48.4% of early progeny cells were BrdU+, and expressed low levels of DDR transcripts.
Conclusions
We show that DXR effectively depleted aISCs in the small intestine and subsequent epithelial regeneration depended on nonquiescent early progeny cells of aISCs. The chemoresistant phenotype of the early progeny cells may rely on a dampened DDR in contrast to aISCs’ robust DDR, which facilitates expeditious apoptosis.
Keywords: Gastrointestinal, irradiation, LGR5, DNA damage response
Abbreviations used in this paper: aISC, active intestinal stem cell; BrdU, 5-bromo-2’-deoxyuridine; CASP3, cleaved caspase-3; DDR, DNA damage response; DMEM, Dulbecco’s modified Eagle medium; DXR, doxorubicin; FACS, fluorescent-activated cell sorting; FBS, fetal bovine serum; GFP, green fluorescent protein; HR, homologous recombination; IP, intraperitoneal; IR, irradiation; PBS, phosphate-buffered saline; qRT-PCR, quantitative reverse-transcription polymerase chain reaction; TA, transit amplifying; TAM, tamoxifen; TBI, total body irradiation
Graphical abstract

Summary.
Early progeny of intestinal stem cells, residing in the transit-amplifying region, are responsible for epithelial repair after doxorubicin-induced intestinal stem cell depletion. These chemoresistant, actively cycling cells demonstrate the importance of cellular plasticity in epithelial regeneration after chemotherapy.
The gastrointestinal epithelium is composed of a cell lining that renews every 4–7 days, forming a barrier between the systemic circulation of the organism and the lumen of the intestine.1 In homeostasis, the rapid renewal of the epithelium originates from active intestinal stem cells (aISCs), which exist at the base of the crypt intercalated between Paneth cells.2 These cells are marked by Lgr5, which encodes a G protein–coupled receptor for R-spondins, and Olfm4, a Notch target gene, with high fidelity within the murine and human small intestine.2, 3, 4 These crypt-base stem cells are highly susceptible to irradiation and chemotherapy-induced injury.5, 6, 7 After acute injury to aISCs by irradiation, a reserve population of quiescent stem cells, labeled by Bmi1 or Hopx near the +4 crypt position, regenerate the epithelium.8, 9, 10 Recent studies demonstrate that Bmi1 marks cells of the enteroendocrine lineage.11 Others have demonstrated the inherent plasticity of the intestinal epithelium, with varying degrees of regeneration originating from Dll1+ or Atoh1+ secretory progenitors,12, 13, 14, 15 differentiated Alpi1+ enterocyte precursors,16 and differentiated Defa4+, Lyz1+, or Bhlha15+ Paneth cells.17, 18, 19
Doxorubicin (DXR), a chemotherapy drug, and irradiation (IR) are considered to be similar injury models,20, 21, 22 although the regenerative capacity of non–stem cells with DXR has not been fully explored.6,19 Hayakawa et al19 demonstrated a reduction of aISCs within 24 hours after 15 mg/kg DXR. Despite this decrease in aISCs, lineage tracing from the Lgr5 locus performed at the time of injury did not differ from noninjured intestines, indicating that the majority of crypts retained sufficient aISCs to repopulate the epithelium at this dose. Our previously published study demonstrated that there was an expansion of putative +4 or quiescent stem cells during the regenerative phase after DXR, although no transgenic mouse models were used to identify these cells, thus the findings required further investigation.6 Secretory precursors and differentiated Paneth cells have a modest but variable (0–50 lineage trace events per 5 cm)17 contribution to epithelial regeneration that has been identified in multiple studies.15,17,19,23 This reversion phenomenon appears dependent on Notch activation after DXR and IR.17, 18, 19
In the current study, we were interested in determining what epithelial cell populations contribute to aISC regeneration in the setting of DXR-induced injury. We undertook a survey of lineage tracing from different cell populations that have previously demonstrated reversion to a stem-like state after aISC depletion. We opted to perform all lineage trace experiments in the jejunum, as it comprises the majority of the small intestinal length and has been the focus for our other studies. It has been postulated that the highest capacity for reversion to a stem-like state would exist in the early transit-amplifying (TA) zone, but the high degree of plasticity and rapid migration of the TA cells has made identifying these cells difficult.24, 25, 26 We labeled early TA cells by inducing lineage tracing of daughter cells descended from Lgr5+ or Olfm4+ aISCs 1 day prior to inducing aISC damage. We refer to these labeled cells as early progeny cells throughout to clarify that they are recently descended from aISCs and represent a distinct population separate from aISCs.
Here, we show that DXR results in depletion of aISCs. After DXR-induced loss of aISCs, early progeny cells contribute to epithelial regeneration. However, after IR-induced loss of aISCs, there was not a similar recruitment of early progeny cells for regeneration, suggesting damage-dependent differences. After DXR, early progeny cells rarely underwent apoptosis, in contrast to the highly apoptotic aISC population. In homeostasis, early progeny cells are actively cycling, similar to aISCs. Therefore, quiescence does not appear to be a mechanism by which these cells are chemoresistant. Early progeny cells in homeostasis have decreased expression of DNA damage response transcripts in comparison with aISCs. A dampened DNA damage response may underlie the enhanced survival of early progeny cells in the face of chemotherapy. These data are supportive of early progeny cells as a highly flexible population with substantial capacity for regeneration after loss of aISCs.
Results
aISCs Are Rapidly Lost From the Jejunal Crypt After DXR-Induced Damage
We first determined the impact of high-dose DXR (20 mg/kg intraperitoneal [IP]) on aISCs over time in the murine jejunum by assessing the retention of 2 stem cell markers, Lgr5 and Olfm4. The Lgr5IRES-eGFP-CreERT2 (hereafter referred to as Lgr5eGFP-CreERT2) mouse exhibits mosaicism (Figure 1A), resulting in fluorescence in about 30% of jejunal crypts.2,3 The percentage of GFP (green fluorescence protein)+ crypts declined within 24 hours after DXR administration (Figure 1A and B) and was persistently depleted out to 5 days after injury (Figure 1A and B). Additionally, the Lgr5 transcript was downregulated in jejunal tissue after DXR (Figure 1C).
Figure 1.

aISCs are rapidly lost after DXR. (A) Representative images of Lgr5eGFP+ fluorescence in the jejunum of Lgr5eGFP-CreERT2 mice over time (indicated in hours) after DXR injection. Scale bar = 20 μm. (B) Quantification of the percentage of Lgr5eGFP+ crypts of total crypts counted after DXR. Positive crypts contained at least 2 Lgr5eGFP+ aISCs. (C) Log2 fold change of Lgr5 expression normalized to Actb in jejunal tissue after DXR. (D) Representative images of OLFM4 immunofluorescence in jejunal crypts of control mice and 72 hours post-DXR. Arrows indicate faint positivity retained at the +4 position. Scale bar = 20 μm. (E) Quantification of OLFM4 immunofluorescence scoring from control mice and after DXR. See methods for staining rubric. (F) Representative images of Olfm4 in situ hybridization of jejunal crypts of control mice and 72 hours post-DXR. Scale bar = 20 μm. (G) Log2 fold change of Olfm4 expression normalized to Actb in jejunal tissue after DXR. Data are presented as mean ± SD. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001; 1-way analysis of variance followed by Dunnett’s post hoc test.
OLFM4 immunofluorescence and Olfm4 in situ hybridization were used to independently identify aISCs without utilizing a transgenic mouse model. The cell population marked by OLFM4 highly overlaps with the LGR5+ population.3 In control tissues, all crypts examined exhibited several cells expressing OLFM4 at the base (Figure 1D). Consistent with the findings in the Lgr5eGFP-CreERT2 mice, OLFM4 expression declined rapidly and persisted out to at least 72 hours (Figure 1E). Most crypts retained very faint OLFM4 immunopositivity in 1–2 cells, and subjectively this positivity was retained in the +4 cell position (Figure 1D, arrows). We also identified minimal Olfm4 messenger RNA via in situ hybridization at 72 hours after DXR (Figure 1F). Finally, the Olfm4 transcript was rapidly and persistently downregulated after DXR in jejunal tissue (Figure 1G). These findings demonstrate that crypt-base stem cells as marked by Lgr5 and Olfm4 are rapidly depleted by DXR.
aISCs Exhibit Cleaved Caspase-3 Immunopositivity and Undergo Expulsion From the Crypt Base After Exposure to DXR
We were interested in monitoring the fate of the Lgr5+ cell after DXR. Were these aISCs retained but downregulated expression of stem cell markers,27 or did they initiate cellular death? We co-localized Lgr5eGFP+ fluorescence with cleaved caspase-3 (CASP3) immunofluorescence after DXR injection in vivo (Figure 2A). CASP3 identifies cells that have initiated the caspase cascade leading to apoptosis, a common sequela to significant or irreparable DNA damage.28 Additionally, we monitored Lgr5eGFP+ enteroids via time-lapse imaging after in vitro application of DXR.
Figure 2.

aISCs undergo apoptosis and expulsion from the crypt base after exposure to DXR. (A) Representative images of CASP3 immunofluorescence co-localizing with Lgr5eGFP+ cells in a jejunal crypt at 6 hours and 24 hours after DXR as compared with control. Scale bar = 10 μm. (B) Quantification of the percentage of CASP3+ cells that are Lgr5eGFP+ or non-Lgr5eGFP+ within the crypt epithelium at 6 hours and 24 hours after DXR as a percentage of total CASP3+ cells/crypt (mean ± SD; n = 3 per time point, 10 crypts/mouse). ∗∗P < .01; 2-way analysis of variance followed by Bonferroni’s post hoc test. (C) Combined GFP fluorescence and differential inference contrast time lapse of 1 representative Lgr5eGFP+ enteroid after DXR. Scale bar = 50 μm. Within the inset image of the Lgr5eGFP+ bud, the asterisk indicates the autofluorescent lumen and the dotted line identifies Lgr5eGFP+ cells present initially in the enteroid bud, which are subsequently extruded into the lumen over time after DXR application. The yellow double-headed arrow is an example of the measurements performed in (D). Two-dimensional deconvolution was applied to the GFP channel. (D) Quantification of measured distance (μm) of maximal GFP+ intensity (representing Lgr5eGFP+ cells) from the basolateral membrane of the enteroid bud over time after DXR or vehicle (media) application to Lgr5eGFP+ enteroids. 4 ng/μL DXR (n = 7 GFP+ enteroid buds) or vehicle (n = 4 GFP+ enteroid buds) was applied in vitro and enteroids were imaged by time-lapse microscopy. Data are presented as mean ± SEM. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; repeated-measures 2-way analysis of variance followed by Holm-Sidak post hoc test.
Lgr5eGFP+ cells accounted for 69.3% of the total CASP3+ cells in jejunal GFP-expressing crypts at 6 hours after DXR, which then dropped to 45% by 24 hours (Figure 2A and B). The high percentage of Lgr5eGFP+/CASP3+ cells compared with non-Lgr5eGFP+/CASP3+ cells suggests that aISCs are highly chemosensitive, given that there is a small number of aISCs (14 cells/crypt) in relation to the number of total crypt cells (∼250 cells/crypt).29,30 Consistent with our in vivo results, DXR application in vitro to Lgr5eGFP+ enteroids resulted in rapid expulsion of GFP+ cells into the crypt lumen (Figure 2C). Expulsion from the enteroid bud was tracked by measuring the distance between the basolateral membrane of the bud and the maximal GFP fluorescence over time (Figure 2C, arrow). The GFP signal separated from the basolateral aspect of the bud around 5.5 hours after DXR application, indicating expulsion of Lgr5eGFP+ cells into the lumen (Figure 2D). Consistent with our in vitro findings, we occasionally find intraluminal GFP in Lgr5eGFP+ crypts in tissues from DXR-treated mice (an example is present in Figure 10A) (6 hours DXR). These findings demonstrate that DXR causes rapid apoptosis and removal of aISCs from the crypt epithelium.
Figure 10.

Early progeny of aISCs lack activation of CASP3 and broadly upregulate DNA damage response transcripts after DXR. (A) Representative images of CASP3 immunofluorescence in crypts of Lgr5eGFP-CreERT2 reporter mice, using the experimental design in Figure 6A and C. An early progeny cell is indicated by the white dotted line in each experimental group. Scale bar = 10 μm. (B) Quantification of the percentage of CASP3+ cells that are Double+ aISCs (GFP+/tdTomato+) or early progeny cells (tdTomato+) within the crypt epithelium at 6 hours and 24 hours after DXR as a percentage of total CASP3+ cells/crypt (mean ± SD; n = 3 per time point, 10 crypts/mouse). ∗∗P < .01; ∗∗∗∗P < .0001; 2-way analysis of variance followed by Bonferroni’s post hoc test. (C) Mean log2 fold change of DDR–associated transcripts in the indicated populations isolated from control mice 24 hours after TAM (n = 3, normalized to double negative epithelial cells). Cells were isolated via FACS as in Figure 6A. (D) Mean log2 fold change of DDR transcripts in the indicated population 6 hours (n = 2) and 24 hours (n = 3) after DXR. Each cell population was normalized to its control population (n = 3) (Figure 8D) to demonstrate DXR-specific responses within the identified cell type. Cells were isolated via FACS as in Figure 6C. Statistically different gene expression (P values in text) was calculated by 2-way analysis of variance, and the false discovery rate was controlled with the 2-stage step-up method of Benjamini, Krieger, and Yekutieli.
aISCs’ Ability to Produce New Progeny Is Severely Impaired After Exposure to DXR and IR
The primary function of aISCs is to produce progeny, with cell division occurring on average every ∼21.5 hours.31 We hypothesized that no new progeny would be produced after severe damage to aISCs. We tested this hypothesis by simultaneously inducing lineage tracing from aISCs and administering DXR or IR. We used 3 transgenic mouse models crossed to Rosa26LSL-tdTomato mice to label aISCs: Lgr5eGFP-CreERT2, Lgr5IRES-CreERT2(hereafter referred to as Lgr5CreERT2), and Olfm4IRES-eGFP-CreERT2 (hereafter referred to as Olfm4eGFP-CreERT2).2,3,32
We injected mice with tamoxifen (TAM) (50 mg/kg IP) within 5 minutes of either DXR (20 mg/kg IP) or IR (12 Gy total body irradiation) to label surviving aISCs or progeny after damage (Figure 3A). This TAM protocol delivers less drug than previously published genotoxic dosages, which impair organoid formation from treated Lgr5eGFP-CreERT2 mice.33 Full lineage tracing from the crypt base to the villus tip was observed in control Lgr5eGFP-CreERT2 (Figure 3B) and Olfm4eGFP-CreERT2 (Figure 3C) reporter mice, and approximately 80% of the total villus length in Lgr5CreERT2 reporter mice (Figure 3D). This indicates near-total epithelial turnover within 5 days, excepting long-lived Paneth cells.
Figure 3.

DXR and IR impair aISCs’ regenerative capacity. (A) Experimental design to identify lineage tracing from aISCs after DXR or IR. Control mice were administered 1 TAM injection, and received no additional treatments. Experimental mice were injected with TAM, to initiate lineage tracing from aISCs, immediately followed by DXR or IR, to injure aISCs. All tissues were collected 5 days after TAM injection. (B–D) Representative images and quantification of lineage tracing emanating from Lgr5 or Olfm4-expressing cells in control and DXR or IR-treated mice as indicated. Data are presented as mean ± SD. Lineage trace events were identified as >5 contiguous tdTomato+ cells emanating from a crypt base. ∗∗∗P < .001; ∗∗∗∗P < .0001; Student’s t test. Scale bar = 50 μm.
Lineage tracing originating from aISCs decreased after DXR (Figure 3B and C), with only 23.3% labeled DXR-treated crypts vs 99.7% of all control crypts in Olfm4eGFP-CreERT2 reporter mice (P < .001) (Figure 3C). Lineage tracing after IR was similarly impaired in Lgr5CreERT2 reporter mice (24%), as compared with control (97.2%) (P < .001) (Figure 3D). As demonstrated in Figure 2B, where Lgr5eGFP+ cells were positive for CASP3 by 6 hours, DXR has a rapid onset of toxicity to the epithelium. TAM undergoes hepatic metabolism to 4-OHT (4-hydroxy-tamoxifen), which is distributed body-wide within 4–8 hours after intraperitoneal injection in mice.34 Thus, we considered that perhaps 4-OHT–mediated recombination was impaired by DXR’s topoisomerase II alpha poisoning or by DXR-DNA adducts. Therefore, we also induced lineage tracing 6 hours pre-DXR, allowing sufficient time for initiation of Cre-mediated recombination prior to damage. However, this did not significantly alter the lineage tracing observed from aISCs (Figure 4A). Surviving aISCs, induced to lineage trace 1 day post-DXR, also had limited regenerative capacity (Figure 4B). Together, these data indicate that the majority of aISCs do not produce epithelial progeny after DXR or IR.
Figure 4.
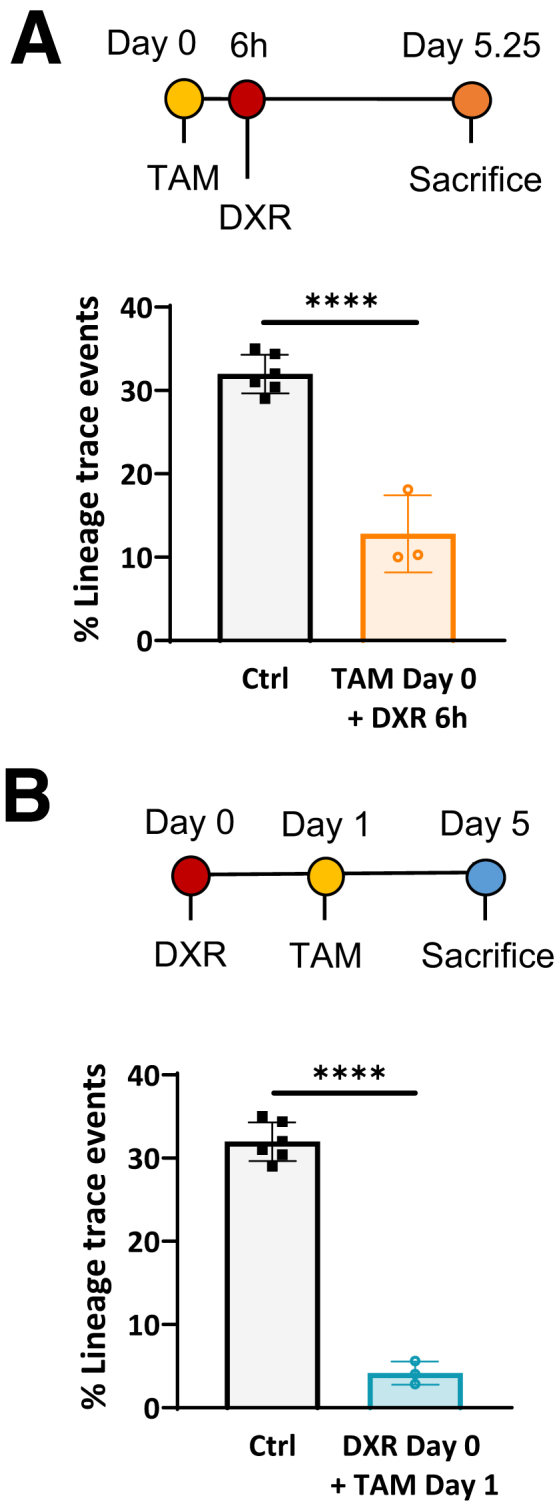
Regenerative contribution from Lgr5+aISCs is not influenced by TAM administration. (A, B) Experimental design for TAM (50 mg/kg IP) and DXR (20 mg/kg IP) injections in Lgr5eGFP-CreERT2 reporter mice, followed by quantification of lineage tracing in control and DXR-treated mice as indicated. At least 200 jejunal crypts were counted per mouse. Quantified control lineage trace events were obtained from Lgr5eGFP-CreERT2 reporter mice treated with TAM 5 or 6 days prior to sacrifice. Data are presented as mean ± SD. ∗∗∗∗P < .0001; 1-way analysis of variance followed by Tukey’s post hoc test.
Epithelial Regeneration After DXR Is Not Dependent on Crypt Populations Identified by Hopx, Bmi1, Dll1, or Defa6 Expression
Various genetic markers have been utilized to identify crypt cell populations that can regenerate the epithelium after loss of aISCs, including Hopx, Bmi1, mTert, Lrig1, and Sox9hi.8, 9, 10,35, 36, 37, 38 Further investigation of Bmi1-expressing cells has suggested that these cells primarily represent an enteroendocrine lineage and highly overlap with Prox1 expression.11 Additionally, recent publications have identified dedifferentiation of secretory progenitor cells (expressing Dll1), Paneth cells, and other dedicated secretory cells after DXR17,19,23 and IR-induced damage.12,18,19,39 This cellular remodeling process was driven by Notch activation.17, 18, 19
In order to examine the relative regenerative contributions of each of these populations, we crossed HopxCreERT2, Bmi1CreERT, Dll1eGFP-CreERT2, and Defa6iCre mice with Rosa26LSL-tdTomato mice.40 TAM was injected 1 day prior to DXR in order to permanently label each cell population and their respective progenies (Figure 5A). Dll1eGFP-CreERT2 labels secretory progenitor cells and mature secretory cells within the epithelium,12 and Defa6 expression is restricted to Paneth cells.41 Although the Defa6-driven Cre expression is constitutive, we maintained the same experimental design including TAM administration to ensure consistency with the other lineage trace experiments.
Figure 5.

Hopx+, Bmi1+, Dll1+, and Defa6+-expressing epithelial populations do not contribute to regeneration after DXR. (A) Experimental design to identify lineage tracing from Hopx+, Bmi1+, Dll1+, or Defa6+-expressing cells after DXR. Control mice were administered 1 TAM injection, and received no additional treatments. Experimental mice were injected with TAM, to initiate lineage tracing from the indicated cell population, then injected with DXR 1 day after TAM. All tissues were collected 6 days after TAM. (B–E) Representative images and quantification of lineage tracing from the indicated cell populations in control and DXR-treated reporter mice as indicated. Data are presented as mean ± SD. Student’s t test. Scale bar = 50 μm. ns, no significance.
Consistent with previous literature showing that Hopx- and Bmi1-expressing cells can interconvert to aISCs in homeostasis, tissues examined 6 days after TAM demonstrated low levels of lineage tracing (Figure 5B and 5C).9,40 By 6 days, most tdTomato+ cells were present near the villus tips in short contiguous runs of 3–5 positive cells, consistent with labeling of a TA cell at the time of TAM injection. Cells with a filamentous phenotype in the lamina propria were frequently labeled in the Bmi1+ transgenic mice. This is consistent with the overlap with Prox1+, which also identifies lymphatic endothelial cells.11 DXR damage did not alter the extent of lineage tracing from Hopx+ (P = .18) or Bmi1+ (P = .41) reporter mice (Figure 5B and 5C). Short runs of 3–5 tdTomato+ cells were observed near the villus tips, and occasional single Paneth cells were labeled.
Labeled Dll1+ cells in control mice that were phenotypically consistent with secretory cell types were spaced intermittently throughout the epithelial lining (Figure 5D). No lineage trace events were identified in homeostasis or after DXR (Figure 5D). Defa6+ reporter mice exhibited labeled crypt base cells with apically oriented granular contents in all jejunal crypts examined, consistent with the presence of Defensin-6–expressing Paneth cells (Figure 5E). After DXR, minimal (1.2 ± 1.7%) lineage trace events were observed from Defa6+ cells, which was not significantly different from control mice (P = .31) (Figure 5E). Taken together, these data demonstrate that cells expressing Hopx, Bmi1, Dll1, and Defa6 are not significant contributors to epithelial regeneration after aISC depletion with DXR.
Early Progeny of aISCs Provide the Majority of Epithelial Regeneration After DXR-Induced Damage in the Jejunum
The Lgr5+ aISC population has demonstrable heterogeneity in cell cycle frequency, gene expression, and sensitivity to damage. However, the significant reduction in lineage tracing from Lgr5+ aISCs after DXR and IR (Figure 3) indicated that the majority of regeneration was likely driven by a cell outside of the aISC compartment.26,42, 43, 44, 45 Committed Alpi1+ enterocyte precursors in the upper TA zone (crypt position +8 and above) can contribute to epithelial repair when aISCs are depleted using the Lgr5-iDTR model.16 We hypothesized that early progeny cells, which are TA daughter cells that have recently exited the crypt base, no longer express Lgr5eGFP+, and exist at the +4–7 cell position, would possess enhanced capacity to regenerate the intestinal epithelium after DXR. Under homeostatic conditions, aISCs cycle slightly more often than once per day, continually producing daughter cells that will enter the TA zone, downregulate stem cell markers, and migrate up the crypt-villus axis.31
One day after induction of lineage tracing in Lgr5eGFP-CreERT2 reporter mice, early progeny cells were observed as non-GFP tdTomato+ cells in the +4–7 cell position (Figure 6A, arrow). The appearance of early progeny cells within this time frame was consistent across all Lgr5eGFP-CreERT2 crypts and mice examined, with crypts containing an average of 1.2 ± 0.5 tdTomato+ early progeny cells/hemicrypt on jejunal cross-section (Figure 7A). Additionally, all GFP-expressing crypts had at least 1 double positive (GFP+/tdTomato+) cell at the crypt base after induction of lineage tracing (Figure 6A, arrowheads). GFP-only cells that did not express tdTomato after TAM were also present, which may be related to chromatin accessibility around the Loxp sites (Figure 6A, asterisks).46 However, these GFP-only cells appear to compose a limited subset of Lgr5eGFP+ cells that are actively contributing to the epithelial lining, given that lineage tracing was present in all Lgr5eGFP+ crypts in non–DXR-treated mice (Figure 7B).
Figure 6.

Early progeny of aISCs provide the majority of epithelial regeneration after DXR but not after IR. (A) Representative image of a jejunal crypt with a labeled early progeny cell (arrow), several double+ aISCs (arrowheads), and 2 GFP-only aISCs (asterisks) 1 day after TAM. Scale bar = 5 μm. (B) Experimental design to identify lineage tracing from early progeny of aISCs after DXR or IR. Control mice were administered 1 TAM injection, and received no additional treatments. Control mice for IR damage were the same as Figure 3D. Experimental mice were injected with TAM to initiate lineage tracing from aISCs and allow generation of labeled early progeny cells, then administered DXR or IR 1 day after TAM. All tissues were collected 6 days after TAM. (C, D) Representative images and quantification of lineage tracing from early progeny cells and surviving aISCs comparing control and DXR-treated mice as indicated. (E) Immunofluorescence (pseudo-colored white) was performed to identify the indicated differentiated cells co-localizing with lineage trace events (tdTomato+) originating from early progeny cells and surviving aISCs in Lgr5eGFP-CreERT2 reporter mice 5 days after DXR. Scale bar = 5 μm. (F) Representative images and quantification of lineage tracing from early progeny and surviving aISCs comparing control and DXR-treated Lgr5eGFP-CreERT2 reporter mice. The same control mice (n = 2) were used as in Figure 3D (analyzed at 5 days post-TAM). Data are presented as mean ± SD. ∗P < .05; ∗∗∗P < .001; ns: no significance; 1-way analysis of variance followed by Tukey’s post hoc test (compared with Figure 3B–D groups). Scale bar = 50 μm.
Figure 7.

One dose of TAM is sufficient for lineage tracing and does not alter DXR’s impact on aISCs in Lgr5eGFP-CreERT2reporter mice. (A) Enumeration of GFP+ aISCs, Dbl+ aISCs, and tdTomato+ early progeny cells in jejunal crypts of Lgr5eGFP-CreERT2 reporter mice 24 hours after TAM injection. Cells in at least 10 hemicrypts (identified by having a crypt lumen that extended from the crypt base to the crypt-villus junction) were counted per mouse. Data are presented as mean ± SD. (B) Quantification of GFP+ and tdTomato+ crypts 5 or 6 days after injection of TAM in Lgr5eGFP-CreERT2 reporter mice. Student’s t test. (C) Representative control (non-DXR/IR) images of GFP and tdTomato fluorescence in jejunal tissues at the time points indicated in Figure 6B’s experimental design. Images were obtained from Lgr5eGFP-CreERT2 reporter mice treated with no TAM (day 0), 1 day after TAM (day 1), and 6 days after TAM (day 6). (D) Quantification of Lgr5eGFP+ fluorescence in control Lgr5eGFP-CreERT2 reporter mice compared with Lgr5eGFP-CreERT2 reporter mice used for lineage tracing of aISC and early progeny cells in Figures 3B and 5C 5 days after DXR. This demonstrates that both experimental groups responded to DXR in an equivalent manner and that Lgr5eGFP+ fluorescence was persistently decreased out to 5 days post-DXR. Data are presented as mean ± SD.
We then interrogated the early progeny cells’ contribution to epithelial regeneration after DXR or IR. Early -progeny cells were labeled 1 day prior to administration of DXR (20 mg/kg) in Lgr5eGFP-CreERT2 or Olfm4eGFP-CreERT2 -reporter mice, or IR (12 Gy total body irradiation) in Lgr5CreERT2 reporter mice (timeline: Figure 6B; representative images: Figure 7C). After DXR, labeled early progeny cells and surviving aISCs produced a similar percentage of lineage traced crypts as observed in control, non–DXR-treated tissues for Lgr5eGFP-CreERT2 (P < .05) (Figure 6C) and Olfm4eGFP-CreERT2 reporter mice (P = .2) (Figure 6D), with 25.4 ± 1.5% and 89.8 ± 2.2% of total crypts exhibiting lineage traces, respectively. The number of Lgr5eGFP+ crypts in Lgr5eGFP-CreERT2 reporter mice used for lineage-tracing experiments were not different after injury (P = .89), indicating that the increased lineage tracing was not due to higher retention and continued proliferation of Lgr5eGFP+ aISCs in the early progeny experimental group (Figure 7D). This suggests that the early progeny cells, which are not aISCs at the time of DXR-induced damage, substantially contribute to epithelial repair in this model.
The lineage trace events produced by early progeny cells and surviving aISCs included multiple differentiated cell types, including tdTomato-labeled enteroendocrine cells, goblet cells, and enterocytes (Figure 6E). Rare co-labeled Paneth cells were identified but the vast majority of Paneth cells were non-tdTomato+ in lineage-traced crypts (Figure 6E). This is likely a result of Paneth cells surviving through DXR-induced damage and thus not requiring replenishment, as demonstrated in previous literature and consistent with their role as long-lived postmitotic secretory cells.6,47 These data suggest that early progeny cells are able to produce differentiated cell populations, consistent with their regenerative potential after DXR.
We did not find a comparable contribution of early progeny cells to regeneration in IR-induced damage (Figure 6F), although the depletion of aISCs was similar to DXR (Figure 3D). Early progeny cells and surviving aISCs contributed to 38.9 ± 14.2% of all crypts after IR, whereas undamaged intestines exhibited labeling of 97.2 ± 1.2% of all crypts (Figure 6F) (P < .001). These data show that regenerative capacity of non–stem cell populations differs between damage models. IR has been a commonly utilized aISC injury model, with multiple populations contributing to epithelial regeneration.9, 10, 11, 12,15,17, 18, 19,23,36,42,48,49 It is unknown whether early progeny cells are more sensitive to IR-induced damage, and therefore limited in their regenerative capacity.
Early Progeny Cells Reacquire a Stem Cell–Like Transcriptional Profile After DXR
Early progeny cells seem to be poised to survive and regenerate the intestinal epithelium after DXR, and thus we pursued further investigation of these cells in homeostasis and within 24 hours after DXR damage. We hypothesized that early progeny cells would have increased stem cell gene expression in response to DXR, based on their enhanced regenerative capacity. Microfluidic quantitative reverse-transcription polymerase chain reaction (qRT-PCR) for a curated gene list was performed on aISCs and early progeny cells isolated from control crypt-enriched epithelium (24 hours post-TAM) (Figure 8A), as well as 6 hours post-DXR (30 hours post-TAM) and 24 hours post DXR (48 hours post-TAM) crypt-enriched epithelium (Figure 8C). As shown in Figure 6A, early progeny cells could be discriminated from aISCs in Lgr5eGFP-CreERT2 reporter mice by the presence of tdTomato fluorescence and lack of GFP fluorescence. Early progeny cells were isolated via fluorescent-activated cell sorting (FACS) using Lgr5eGFP-CreERT2 reporter mice within 1–2 days after induction of lineage tracing with or without DXR damage. Gating for live epithelial cells from dissociated jejunal crypts was performed, then cells were sorted based on GFP and tdTomato fluorescence: GFP only, tdTomato only, double positive, and double negative. The double negative population was collected as a reference population composed primarily of postmitotic epithelial cells, such as enterocytes, but also contained non-GFP+ crypt cells.
Figure 8.

Early progeny cells reacquire a stem cell–like transcriptional profile after DXR. (A) Experimental design to assess transcriptional differences in early progeny vs aISCs in control Lgr5eGFP-CreERT2 reporter mice. Control mice did not receive any additional treatment. Crypt-enriched epithelium was sorted for microfluidic PCR analysis by FACS on the basis of GFP and tdTomato fluorescence (GFP+, tdTomato+, GFP+/tdTomato+, negative). (B) Mean log2 fold change of aISC and reserve intestinal stem cell (rISC)–associated transcripts in the indicated populations isolated from Lgr5eGFP-CreERT2 reporter control mice 24 hours after TAM injection (n = 3, normalized to double negative epithelium). (C) Experimental design to assess early transcriptional responses to DXR treatment in early progeny vs aISCs. Mice were injected with DXR 1 day after TAM injection and FACS was performed on crypt-enriched epithelium at 6 or 24 hours post-DXR, similar to Figure 6A. (D) Mean log2 fold change of aISC or rISC-associated transcripts in the indicated populations 6 hours (n = 2) and 24 hours (n = 3) after DXR. Each cell population was normalized to its control population (Figure 6B) to demonstrate DXR-specific responses within the identified cell type. Statistically different gene expression (P values in text) was calculated by 2-way analysis of variance and the false discovery rate was controlled with the 2-stage step-up method of Benjamini, Krieger, and Yekutieli.
First, we were interested in comparing early progeny cells to their parent aISCs in homeostasis. Consistent with their role as aISCs, the GFP+ and the double positive (GFP+/tdTomato+) aISC populations expressed the highest levels of stem cell transcripts in homeostasis (Figure 8B). As expected, early progeny cells expressed lower stem cell transcripts than either aISC population, with significantly decreased genes including Lgr5 (P < .0001), Gkn3 (P < .0001), Olfm4 (P < .001), Agr3 (P < .01), and Ascl2 (P < .01) as compared with double+ aISCs (Figure 8B). This is consistent with their identity as early TA cells that have exited the crypt base.
Second, we examined aISCs and early progeny cells at 6 and 24 hours after DXR administration, in order to capture the immediate effects of DXR on each population (Figure 8C). The tdTomato+ early progeny cell population was unique in its response to DXR among the 4 populations studied (Figure 8D). Early progeny cells after DXR exhibited an upregulation of stem cell transcripts as compared with the expression in homeostatic early progeny cells, with significantly increased genes at 6 hours including Cd44 (P < .01) and Gkn3 (P < .05) (Figure 8D). At 24 hours after DXR, significantly increased genes included Cd44 (P < .001) and Ascl2 (P < .01) (Figure 8D). This upregulation of stem cell gene expression specifically in early progeny cells is consistent with the evidence for functional stemness presented in Figure 6, demonstrating their rapid reversion to a stem-like state as early as 24 hours after DXR. In contrast, DXR-damaged aISCs exhibited downregulation of stem cell transcripts (Figure 8D) as compared with homeostatic aISC populations. Significantly decreased gene expression in double+ aISCs were observed for Agr3 (P < .01) at 6 hours post-DXR, and Gkn3 (P < .0001), Olfm4 (P < .001), Agr3 (P < .0001), Prom1 (P < .05), and Bmi1 (P < .05) at 24 hours post-DXR.
Early Progeny Cells Are Not Quiescent in Homeostasis
Classically, chemoresistant populations have been associated with decreased cell cycle frequency or quiescence.50 Accordingly, we labeled early progeny cells and aISCs in S-phase with a 2-hour exposure to BrdU (5-bromo-2’-deoxyuridine), a thymidine analog, in Lgr5eGFP-CreERT2 reporter mice induced to lineage trace 24 hours prior to sacrifice (Figure 9A). Figure 9B demonstrates a BrdU+ early progeny cell outlined by the white dotted line. Early progeny cells had more BrdU uptake than GFP+ aISCs (P < .0001) and trended toward more BrdU uptake than double+ aISCs (P = .09), suggesting that quiescence is not the mechanism of chemoresistance in early progeny cells (Figure 9C). The lower BrdU incorporation in GFP+ aISCs suggests that GFP+ aISCs after TAM injection represent a distinct subset of aISCs, such as that of a mostly quiescent enteroendocrine lineage,51 or of noncycling aISCs at the crypt base.52
Figure 9.

Early progeny of aISCs are not quiescent in homeostasis. (A) Experimental design labeling early progeny cells and aISCs with BrdU in Lgr5eGFP-CreERT2 reporter mice not treated with DXR. Mice were injected with TAM 24 hours prior to collection to label aISCs and early progeny, and injected with BrdU 2 hours prior to collection to label cells in S-phase. (B) Representative single-color and composite images of a jejunal crypt after BrdU uptake. A BrdU+/tdTomato+ early progeny cell is outlined by the white dotted line. Scale bar = 5 μm. (C) Quantification of BrdU incorporation within the indicated populations. Data are presented as mean ± SD, n = 3 (10 crypts/mouse). ∗∗∗P < .001; ∗∗∗∗P < .0001; 1-way analysis of variance followed by Tukey’s post hoc test. ns, no significance.
Previous studies have demonstrated that aISCs have a high expression of homologous recombination transcripts.44,53,54 In aging aISCs, it has been suggested that a dampened DNA damage response is associated with decreased apoptosis.55 As quiescence does not explain early progeny cells’ chemoresistant phenotype, we questioned whether these cells would have different DNA damage response compared with aISCs prior to and after DXR damage. Therefore, we evaluated early progeny cells and aISCs for apoptosis and expression of genes involved in DNA damage response (DDR).
In contrast to the highly apoptotic aISC population, early progeny cells exhibited minimal apoptosis 6 and 24 hours after DXR (Figure 10A). At 6 hours, early progeny cells accounted for 2.5% of CASP3+ cells, whereas double+ aISCs made up 63.4% of the CASP3+ cells (P < .0001) (Figure 10B). A modest increase in apoptotic early progeny cells was observed at 24 hours after DXR (11.7%), although still significantly less than double+ aISCs (35%) (P < .01) (Figure 10B). This suggests that early progeny cells are resistant to DXR-mediated induction of apoptosis, unlike the highly sensitive aISCs.
To determine whether a dampened DDR was present in early progeny cells at the time of DXR injection, we evaluated these cells and aISCs in homeostasis. DDR transcripts were downregulated in the early progeny cells as compared with aISCs (Figure 10C). In particular, early progeny cells had significantly decreased expression of homologous recombination transcripts as compared with double+ aISCs, including Rad51 (P < .05), Brca1 (P < .001), and Chek2 (P < .01). Early progeny cells also expressed modestly reduced Top2a transcript (P = .06). Top2a encodes for the topoisomerase II alpha enzyme, which functions to relieve torsional stresses during transcription, and is poisoned by DXR.56 These data are supportive of distinct differences in homeostatic regulation of DDR between aISCs and early progeny cells despite close spatial location. We hypothesize that reduced DDR and reduced Top2a expression is associated with the early progeny cells’ intrinsic chemoresistance to DXR.
Cell population-specific alterations in DDR gene expression were also observed after DXR. Both aISC populations primarily upregulated transcripts associated with p53 activation by 6 hours after damage as compared with their control populations, including Puma (GFP+ and Dbl+, P < .001) and Mdm2 (GFP+, P < .01; Dbl+, P < .001) (Figure 10D). Mdm2 in aISCs was significantly increased over the upregulation observed in the double negative population at 6 hours (P < .05). Transcriptional upregulation of Puma classically occurs in response to p53 activation secondary to DNA damage and subsequently drives apoptosis by instigating the intrinsic apoptosis cascade.57 This is consistent with Figure 2A and B, in which aISCs were preferentially positive for CASP3 immunofluorescence after DXR. Upregulation of Cdkn1a was also present at 6 hours (GFP+ and Dbl+, P < .01) and 24 hours (GFP+, P < .01; Dbl+, P < .05) after DXR, which is indicative of cell cycle arrest or induction of senescence secondary to DNA damage.57
Early progeny cells had a modest upregulation of Puma and Mdm2, not significantly different from the double negative population (Puma, P = .95; Mdm2, P = .23) (Figure 10D). In addition, early progeny cells increased expression of homologous recombination transcripts by 6 and 24 hours after DXR as compared with control, including Brca1 (6 hours, P < .01; 24 hours, P < .05), Rad51 (6 hours, P < .05; 24 hours, P < .001), and Chek2 (24 hours, P < .001) at 24 hours after DXR. This upregulation in homologous recombination gene expression was not observed in the aISC populations after DXR. Early progeny cells, having survived the initial proapoptotic insult, respond to DXR by upregulating genes important for managing double-stranded DNA breaks. Presumably, this ability to sequentially avoid apoptosis and then upregulate DDR is important for early progeny cells’ capacity for regeneration after DXR.
Discussion
In this study, we demonstrate that a major source of epithelial regeneration after DXR-induced aISC depletion originates from early progeny cells that have recently exited the crypt base and no longer express stem cell–specific transcripts. These early progeny cells survived DXR-mediated apoptosis and began to express stem cell transcripts, as early as 6 and 24 hours after damage. DXR rapidly depleted aISCs via induction of p53-mediated cleavage of caspase-3, resulting in high levels of apoptosis in the aISCs. While we had previously suggested no reduction in the aISC population following DXR treatment,6 in retrospect, this was not quantitatively assessed on a per crypt basis, and likely did not capture the extent of aISC depletion that is progressive over the first few days after injury.
Owing to their role as long lived highly proliferative cells, aISCs have a high commitment to genomic integrity.58 When encountering high levels of DNA damage, these cells choose to undergo apoptosis, rather than perpetuate DNA mutations. DXR is a potent anthracycline chemotherapeutic with problematic off-target effects including cardiotoxicity, bone marrow suppression, and oral and intestinal mucositis. DXR undergoes rapid translocation to the nucleus, where it intercalates with nuclear and mitochondrial DNA, poisons topoisomerase II alpha, and produces free radicals.56,59 Consequently, we found that the majority of CASP3+ cells in the crypt at 6 hours after DXR injection are aISCs. This is similar to previous findings after IR injury, in which 40% of apoptotic cells were Lgr5eGFP+.60 We also observed expulsion of aISCs from enteroid buds in vitro after DXR. This is consistent with Andrade and Rosenblatt,61 in which apoptosis but not necrosis was the inciting event for cellular extrusion from an intestinal epithelial monolayer. We have demonstrated that DXR results in a rapid, persistent loss of Lgr5-expressing aISCs out to at least 5 days after insult. This was supported by the lack of lineage tracing when labeled was performed simultaneously with injury.
Although the regenerative response to IR has been extensively studied,9, 10, 11, 12,15,18,19,23,36,42,48 less is understood about regeneration after DXR.17,19,23 We were unable to identify contribution from Hopx+ and Bmi1+-expressing cells after DXR. Populations expressing these transcripts have exhibited an expanded capacity for regeneration after damage.8, 9, 10,37 These populations likely overlap with other crypt epithelial populations, as many of the proposed +4 markers are also expressed in active intestinal stem cells and Bmi1+ also labels enteroendocrine lineage cells.11,44,45 Additionally, Bmi1CreERT expression decreases from the duodenum to the ileum.37 However, there was sufficient jejunal labeling present to monitor for any expansion in regenerative contribution. In future experiments, it would be interesting to explore the regenerative hierarchy in other regions of the intestine.
As the early progeny cells in this study appear at position +4–7, it is likely we are labeling TA cells that are not yet differentiated enough to robustly express lineage specific programs, such as enterocyte-specific Alpi1.16 Given that the bulk of epithelial cells are enterocytes, and that secretory progenitors, labeled by Dll1+, did not contribute to epithelial regeneration in our model, we speculate that a majority of early progeny cells are destined for enterocyte identities under homeostatic conditions. Secretory progenitors require Notch activation to be able to reacquire stem-like properties after DXR damage in other studies.17, 18, 19 It is possible there is a dose-dependent response to DXR that alters Notch programming, as we did not observe recruitment of secretory cells or their progenitors in response to the dose of DXR used in this study.
The significant contribution to regeneration by early progeny cells after DXR, but not IR, was unexpected. A recent publication demonstrated that a subpopulation of aISCs in the Lgr5eGFP-CreERT2 mouse were retained after 10 Gy IR.27 While these cells downregulate GFP expression, they are able to produce significant percentages of lineage tracing postinjury. Other studies have used 12 Gy to induce crypt and aISC loss,10,42,48,54,62,63 and in this study we demonstrate reduced lineage tracing from aISCs at 12 Gy, suggestive of aISC depletion. Thus, the difference in regenerative contribution after IR between Sato et al27 and the data shown here may be due to severity of the injury. Ayyaz et al48 utilized single cell transcriptomics to identify a revival stem cell population which has regenerative potential in the face of IR. These cells arise infrequently from Lgr5+ cells, tend to be quiescent, and take up to a week after induction of labeling to appear.48 Similarly, a recent study by Murata et al49 demonstrated that aISC progeny generated within 4 days prior to IR contribute substantially to epithelial regeneration. The dedifferentiation potential of the progeny cells was dependent on the transcription factor Ascl2.49
The overlap of the previous studies with the early progeny cells identified here is unknown. Our study indicates that substantive repair after aISC depletion by DXR, but not IR, can be performed by cells generated within a day from Lgr5+ aISCs. The 1-day duration of labeling in our study is more restrictive than that of Murata et al’s49 experimental design. This suggests that early progeny cells are more sensitive to IR than progeny further away from the crypt base. However, we also found that early progeny cells expressed higher levels of Ascl2 after injury, suggesting a possible common regenerative pathway between damage models.49,64 Taken together, these studies and our current body of work suggest that the TA population exhibits a high degree of plasticity in response to crypt injury.
Why do these early progeny cells survive DXR damage? Quiescence does not appear to be a major chemoresistant mechanism for these cells, as the percentage of early progeny cells incorporating BrdU was similar to double+ aISCs, and significantly more than GFP-only aISCs. It also seems surprising to us that cells that share a close spatial relationship, such as early progeny cells and aISCs, would have such disparate responses to DNA damage. Tao et al43 demonstrated that the Wnt signaling gradient is an important factor in sensitivity to DNA damage, in which cells that are further from the crypt base are less sensitive. Although that study was limited to Lgr5eGFP+ cells, it seems plausible that differences in the strength of Wnt signaling could drive the balance between chemosensitivity and resistance in our model.
We have identified differences between early progeny cells and aISCs with regard to expression of DDR transcripts. Atm, a mechanism by which cells sense and signal DNA breaks, is not expressed differently in our isolated populations. However, the early progeny cells do have reduced expression of transcripts encoding proteins that are involved in homologous recombination (HR). HR is critical for accurately repairing double-stranded DNA breaks. It seems counterintuitive that early progeny cells, which are able to survive DXR, would have reduced transcript levels of HR-associated genes. However, we speculate that the reduced expression of these DDR genes decreases predisposition to intrinsic apoptosis. In support of this hypothesis, Watanabe et al55 identified that reduced DDR is associated with reduced apoptosis, marked by CASP3, in aging ISC populations. Mutations in surviving and proliferating cells after DNA damaging insults could play a role in cancer initiation. We do not know at this time whether there is persistent DNA damage or mutations retained in the early progeny cells that make up the newly formed stem cell compartment after DXR-induced injury, and what impact this may have on potential development of preneoplastic or neoplastic lesions.
Finally, topoisomerase II alpha-DNA complexes, formed as TOP2A, introduces double-strand breaks after replication or to relieve stresses from coiled DNA, are stabilized by DXR.56 This stabilization leads to irreversible DNA damage, driving cells toward apoptosis. Early progeny cells express modestly lower levels of Top2a transcript as compared with aISCs, although whether this also relates to reduced TOP2A protein is unknown. If early progeny cells have decreased TOP2A, this could result in reduced poisoning of TOP2A-DNA complexes and therefore less irreversible DNA damage in early progeny cells than in aISCs. Thus, early progeny cells would have less sensitivity to DXR-induced damage. Further research is necessary to explore these hypotheses relating to chemoresistance in the intestinal epithelium.
Here, we have identified that early progeny cells of aISCs are a major contributing population to epithelial regeneration after chemotherapeutic insult to the small intestine. Rapid regeneration of the epithelium is critical to maintaining epithelial barrier function. Our study demonstrates that the highly flexible cellular identity of the crypt epithelium is critical to regeneration of the epithelium after chemotherapeutic injury. This finding could be harnessed to develop therapeutic strategies to minimize the severity of chemotherapy-associated mucositis and aISC damage.
Materials and Methods
Mice
Lgr5IRES-eGFP-CreERT2 (JAX stock # 008875; The Jackson Laboratory, Bar Harbor, ME)2 and Defa6iCre41 mice were bred in-house at North Carolina State University from established lines (all on C57BL/6 background). Defa6iCre mice were a gift from R. Blumberg at Brigham and Women’s Hospital (Boston, MA). HopxCreERT2 (JAX stock #017606; The Jackson Laboratory)9 and Bmi1CreERT (JAX stock #010531; The Jackson Laboratory)8 were obtained from JAX labs and maintained in-house. Rosa26LSL-tdTomato mice (JAX stock # 007914; The Jackson Laboratory) were crossed with the above Cre lines to generate mice for lineage-tracing experiments (reporter mice). Unstained cells and single-color control populations for flow cytometry were obtained from the jejunal epithelia of wild-type C57BL/6 mice. All animals were cared for under the North Carolina State University’s Institutional Animal Care and Use Committee guidelines. Experiments at North Carolina State University were performed by B. Sheahan or A. Freeman. Experiments were performed on 8- to 20-week-old mice, with male and female mice randomly allocated to experimental groups. Control mice were injected with TAM at the indicated time points.
Dll1eGFP-CreERT212 and Olfm4eGFP-CreERT23 mice were crossed with Rosa26LSL-tdTomato mice (all on a C57BL/6 background) at University of Michigan to generate mice for lineage-tracing experiments. All animals were cared for under the University of Michigan’s Institutional Animal Care and Use Committee guidelines. Experiments at University of Michigan were performed by T. Keeley. Experiments were performed on 8- to 16-week-old mice, with male and female mice randomly allocated to experimental groups. Control mice were injected with TAM at the indicated time points.
Lgr5IRES-CreERT232 mice were crossed with Rosa26LSL-tdTomato mice (all on a C57BL/6 background) at Duke University to generate mice for lineage-tracing experiments. All animals were cared for under Duke University’s Institutional Animal Care and Use Committee guidelines. Tissues were collected by B. Sheahan. Experiments were performed on 27-week-old mice, with male and female mice randomly allocated to experimental groups. The same control mice were analyzed 5 days after TAM injection to quantify lineage tracing to compare to the 2 experimental groups.
Mice were kept in grouped housing and maintained under a 12-hour light/dark cycle and fed regular free-choice chow through all experimental procedures. When possible, mice of similar age and sex (littermate control animals) were used for experimental and control groups. The n for each experiment is indicated either in the figure legend or graphically represented by symbols.
Mice were euthanized by cervical dislocation after anesthesia with isoflurane. Small intestine was immediately collected after euthanasia and flushed with ice-cold 1× phosphate-buffered saline (PBS) (Ca2+ and Mg2+ free). The small intestine proximal to the ligament of Treitz (signifying the duodenojejunal juncture) was discarded, and the proximal one-half (approximately 10–12 cm) of the remaining intestine was identified as jejunum.
Treatments
DXR Injection
Mice were injected once with 20 mg/kg DXR HCl (Actavis, Parsippany-Troy Hills, NJ) intraperitoneally. Mice were monitored for weight loss daily. If mice lost >20% of initial body weight, they were euthanized in accordance with Institutional Animal Care and Use Committee protocols. Control mice did not receive a vehicle injection in lieu of DXR.
Total Body Irradiation
Mice were exposed to 12 Gy of total body irradiation (TBI). TBI was performed 50 cm from the radiation source with a dose rate of 3.18 Gy/min with 320 kVp x-rays, using 12.5 mA and a filter consisting of 2.5-mm Al and 0.1-mm Cu (X-RAD 320 Biological Irradiator; Precision X-Ray, North Branford, CT). TBI was performed by S. Hasapis. The dose rate was measured with an ion chamber by members of the Radiation Safety Division at Duke University. Control mice were not irradiated.
Labeling of Cells in S-Phase
Mice were injected with 150 mg/kg BrdU dissolved in sterile water intraperitoneally 2 hours prior to collection of small intestinal tissues.
Induction of Lineage Tracing
Mice were injected with 50 mg/kg TAM (Sigma-Aldrich, St Louis, MO) in corn oil (Olfm4eGFP-CreERT2 and Dll1eGFP-CreERT2 reporter mice) or sunflower seed oil (all mice at North Carolina State University and Lgr5CreERT2 reporter mice) intraperitoneally at various time points prior to collection of small intestinal tissues. The TAM was reconstituted in 100% ethanol to 100 mg/mL, then diluted to 10 mg/mL with corn oil or sunflower seed oil prior to injection. We validated that all possible crypts were effectively labeled with this TAM dose using Lgr5eGFP-CreERT2 reporter mice. In these mice, the number of GFP+ crypts were not different from the number of tdTomato+ crypts (Figure 7B).
Quantitative RT-PCR
Total RNA was isolated from jejunal tissues with the RNeasy Mini kit (Qiagen, Hilden, Germany) per the manufacturer’s protocol. Quality of messenger RNA was verified with a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). 500 ng cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA), with RNase A included in the reaction, following the manufacturer’s protocol. qRT-PCR analysis was performed with 25 ng complementary DNA/well using TaqMan Universal Master Mix II with UNG (Applied Biosystems), on a QuantStudio 6 PCR system (Thermo Fisher Scientific) for the following TaqMan probes: Actb (Mm02619580_g1), Lgr5 (Mm00438890_m1), Olfm4 (Mm01320260_m1), Hopx (Mm00558630_m1), and Bmi1 (Mm03053308_g1). All samples were run in triplicate. Signals were normalized to Actb for each sample, and relative fold changes were calculated via ΔΔCt analysis.
Crypt Culture for Time-Lapse Microscopy of GFP+ Cells
Chemical and mechanical dissociation was performed to obtain jejunal crypts as previously described with modifications.65 After filleting the length of the isolated jejunum, villi were removed by scraping the luminal side of the jejunum with a coverslip. The jejunal whole tissue was then incubated in 30 mM EDTA (pH 7.4) for 30 minutes on ice. Tissue was transferred to 1× PBS (Ca2+ and Mg2+ free), and mechanical dissociation (shaking) was performed to exfoliate crypts from the underlying lamina propria. Crypts were separated from intact villi by passage through a 70-μm cell strainer prior to counting. Approximately 100 isolated jejunal crypts were resuspended in 20 μL Matrigel (Corning, Corning, NY) and placed in 48-well tissue culture plates. After polymerization of the Matrigel, 250 μL of media was added per well. The media consisted of Advanced Dulbecco’s modified Eagle medium (DMEM)/F12 (Invitrogen, Carlsbad, CA) containing growth factors: 50 ng/mL recombinant mouse EGF (R&D Systems, Minneapolis, MN), 500 ng/mL R-spondin 1 (R&D Systems), 100 ng/mL recombinant mouse Noggin (PeproTech, Rocky Hill, NJ), 1× N2 supplement (Gibco, Gaithersburg, MD), 1× B27 (Gibco), 10 μM HEPES (Gibco), 1× Glutamax (Gibco), and 500 μg/mL penicillin-streptomycin (Gibco). Media were changed every other day. Enteroids were imaged daily for evidence of growth and budding. At 3 days postplating, 4 μL of DXR diluted in media was added to the media of treatment wells for a final concentration of 4 ng/μL. The same volume of fresh media was added to the media of control wells. GFP-expressing crypts were then monitored by time-lapse fluorescence microscopy (images obtained every 30 minutes for 12 hours after DXR addition) using a stage top incubator on the automated motorized stage of an inverted Olympus IX83 microscope (Olympus, Tokyo, Japan). Time-lapse images were processed with 2-dimensional deconvolution in CellSens (Olympus) prior to measurements. For each indicated time point, the distance (μm) of maximal GFP+ intensity (representing Lgr5eGFP+ cells) from the basolateral membrane of the enteroid bud was measured in the CellSens program (Olympus).
Transcriptional Analysis of Early Progeny Cells and aISCs
Prior to crypt-enriched epithelial cell isolation, 8- to 10-week-old Lgr5eGFP-CreERT2 reporter mice (n = 9) were injected with TAM. Control mice (n = 3) were analyzed 24 hours after TAM injection. DXR-treated mice were injected with DXR at 24 hours after TAM injection, then analyzed at either 6 hours (n = 3) or 24 hours (n = 3) after DXR. No mice were pooled for cell isolation.
Isolated jejunal crypts were centrifuged (1000 RPM × 5 minutes, 4°C) then resuspended in 1× Hank’s Balanced Salt Solution with 1 mg/mL dispase (Corning; 354235), and incubated at 37°C for 10 minutes with intermittent mechanical dissociation (shaking) to obtain a single cell suspension. After 10 minutes, 5 μg/mL of DNase I (Sigma-Aldrich), and 10% fetal bovine serum (FBS) (Gibco) were added to the suspension. The suspension was centrifuged (1000 RPM × 5 minutes, 4°C) and the pellet was resuspended in Advanced DMEM/F12 (Gibco) with 10% FBS and 5 μg/mL of DNase I. The cells were filtered through a 30-μm cell strainer prior to estimating the total number of cells per sample with a hemocytometer. The cells were centrifuged and resuspended at a concentration of 1 × 106 cells/100 μL in Advanced DMEM/F12 with 10% FBS and 5 μg/mL of DNase I. Cells were incubated with 2 μL 7-AAD (BioLegend, San Diego, CA; 420403) and 1 μL EPCAM-PE/Cy7 (BioLegend; 118215) per 1 × 106 cells for 20 minutes in the dark prior to sorting. Cells were kept on ice throughout antibody incubation and sorting.
FACS was performed with a Beckman Coulter MoFlow XDP cell sorter (Beckman Coulter, Brea, CA) in the Flow Cytometry and Cell Sorting Facility at North Carolina State University College of Veterinary Medicine. Events were plotted on forward scatter vs side scatter to exclude debris. Events were gated on side-scatter width vs side-scatter height to identify single-cell events. Single events were then gated for live epithelial cells, identified as 7-AAD negative and EPCAM-PE/Cy7 positive. Live epithelial cells were sorted on the basis of endogenous tdTomato and GFP fluorescence. Non–TAM-injected Lgr5eGFPCre-ERT2 reporter mice were examined to identify any background Cre activation. Background Cre activation in these reporter mice was minimal (0.03% tdTomato+ cells total in representative FACS plot). 500–30,000 cells per population of interest were sorted from each sample. Cells were sorted directly into Buffer RL (Norgen BioTek, Hamilton, Ontario, Canada; 17200) with 1% β-mercaptoethanol prior to freezing in liquid nitrogen and storage at –80°C. Total cells analyzed by the cell sorter ranged between 3.57–13.2 × 106/sample.
RNA was isolated from the sorted cell populations using Norgen Biotek RNA isolation kit (Norgen; 17200). 0.5 ng of RNA was transformed into complementary DNA using Fluidigm Reverse Transcription Master Mix (Fluidigm, South San Francisco, CA). One mouse (6 hours DXR group) was removed from analysis due to inadvertent TAM exposure from cagemate’s feces. At the Center for Gastroenterology Biology and Disease University of North Carolina Advanced Analytics Core facility, the complementary DNA samples underwent preamplification (Fluidigm) prior to loading on a 48.48 IFC plate for Ct analysis using the Fluidigm Biomark HD (Fluidigm) with TaqMan probes. Signals were normalized to Actb and Gapdh for each sample, and relative log2 fold changes were calculated, normalizing to the indicated Control transcript expressions for each panel in Figures 8 and 10.
Immunofluorescence
For immunofluorescence, jejunal tissues were fixed in 4% paraformaldehyde for 12–18 hours, dehydrated in 30% sucrose for 24 hours, embedded in optimal cutting temperature compound, and stored at –80°C until sectioning. The 7-μm sections were adhered to Superfrost Plus slides (Thermo Fisher Scientific; 4951PLUS4) and OCT was removed by immersion in 1× PBS after drying. Sections were mounted with Hard Set mounting medium with DAPI (Vector Laboratories, Burlingame, CA; H-1500) and imaged with an inverted Olympus IX83 microscope. Control sections were incubated with IgG of the same primary species or blocking solution (5% bovine serum albumin or 10% FBS in PBS) for all immunofluorescent experiments.
For lysozyme and RFP immunofluorescence, sections were antigen retrieved in sodium citrate buffer (2.94 g sodium citrate, 300 μL Tween 80, pH 6) using a pressure cooker. Sections were blocked in 10% FBS in PBS for 1 hour at room temperature, incubated overnight at 4°C with primary antibodies, washed in 1× PBS, and incubated for 1 hour at room temperature with secondary antibodies. Sections were immersed in 1× PBS and mounted as described previously. Antibodies included: goat anti-lysozyme (1:500, sc27958), rabbit anti-RFP (1:250, R10367), anti-goat DyLight 488 (1:500, SA5-10086), and anti-rabbit AF555 (1:500, A21428).
For cleaved caspase-3, OLFM4, sucrase-isomaltase, CHGA, MUC2, or EPCAM immunofluorescence, sections were lightly fixed and permeabilized with methanol/acetone (50%/50%) for 20 minutes at –20°C. After washing with 1× PBS, sections were blocked with 5% bovine serum albumin in PBS for 1 hour at room temperature, incubated overnight at 4°C with primary antibodies, washed in 1× PBS, and incubated for 1 hour at room temperature with secondary antibodies. Sections were immersed in 1× PBS and mounted as described previously. Primary antibodies included: rabbit anti-CASP3 (1:250, 9579S), rabbit anti-OLFM4 (1:500, 39141S), goat anti-sucrase-isomaltase (1:250, sc27603), rabbit anti-CHGA (1:250, ab15160), rabbit anti-MUC2 (1:250, sc15334), and rabbit anti-EPCAM (1:500, ab71916). Secondary antibodies included: anti-rabbit DyLight 649 (1:500, 406406), anti-rabbit AF488 (1:500, A21206), and anti-goat AF 647 (1:500, A21244). We performed EPCAM immunofluorescence to validate the quantification of total epithelial crypts in tissues to ensure correct blinded counting of crypts using DAPI fluorescence. The total number of crypts counted with DAPI fluorescence and EPCAM fluorescence was not different.
For co-immunofluorescence of BrdU, GFP, and tdTomato, sections were antigen retrieved in sodium citrate buffer as previous, then permeabilized with 0.3% Triton X-100 for 10 minutes, washed in 1× PBS, and blocked with 10% FBS in PBS for 1 hour at room temperature. Sections were incubated with rabbit anti-GFP (1:2000, ab183734) in Signal Stain Antibody Diluent (Cell Signaling Technology, Danvers, MA; #8112) for 1 hour at room temperature, followed by Signal Stain Boost (Cell Signaling Technology; #8114) for 30 minutes at room temperature. Fluorescein reagent (PerkinElmer, Waltham, MA; NEL741001KT) was applied to the sections for 10 minutes prior to the stripping procedure. Slides were boiled in citrate stripping solution (10 mM sodium citrate, pH 6) and held at a sub-boiling temperature (90°C) for 10 minutes then cooled to room temperature. Following stripping, rabbit anti-RFP (1:500, R10367) and rat anti-BrdU (1:250, NB-500-169) were applied to the sections overnight at 4°C. After washing in 1× PBS, the following secondary antibodies were applied for 1 hour at room temperature: anti-rabbit AF555 (1:500, A-21428) and anti-rat APC (1:500, A10540).
Lineage-Tracing Assessment
At least 200 crypts were counted per mouse for Lgr5eGFP-CreERT2, Lgr5CreERT2, Defa6iCre, HopxCreERT2 and Bmi1CreERT reporter mice. At least 50 crypts were counted per mouse for Dll1eGFP-CreERT2 and Olfm4eGFP-CreERT2 reporter mice. Total crypts were enumerated using DAPI fluorescence. tdTomato+ lineage-tracing events were considered positive if >5 contiguous tdTomato+ cells were emanating from a crypt base. Events limited to villi or unconnected to a crypt were not considered positive.
OLFM4 Scoring
OLFM4 is secreted apically into the lumen of the small intestine in mice, making it difficult to identify which cells are specifically positive for this protein.66 A blinded scoring system was adapted from Besson et al67 to capture 4 categories of OLFM4 immunopositivity. These were scored by the intensity of the fluorescence to approximate the number of cells positive for OLFM4. Only complete jejunal hemicrypts were scored (>10 crypts/mouse). The scoring scheme is as follows: 0 = negative; 1 = 1–2 positive cells/faint fluorescence; 2 = 3–5 positive cells/moderate fluorescence; 3 = 6+ positive cells/intense fluorescence.
In Situ Hybridization
For in situ hybridization, tissues were fixed in 10% zinc formalin for 12–18 hours and moved to 70% ethanol before embedding in paraffin. Then, 5 μm sections were adhered to Superfrost Plus slides (Thermo Fisher Scientific; 4951PLUS4). Sections underwent standard deparaffinization with Histo Clear II (Thermo Fisher Scientific; 50-899-90150). In situ hybridization for Olfm4 probe was performed using RNAscope chromogenic assay 2.5, with Ppib as positive control probe and Dapb as negative control probe according to manufacturer’s instructions (Advanced Cell Diagnostics, Newark, CA). Only epithelial crypt cells were considered to be cells of interest.
Statistical Analysis
All statistics and preparation of graphs were performed in GraphPad 8 (GraphPad Software, San Diego, CA). FIJI was utilized for image analysis and counting of cells (ImageJ version 1.52; National Institutes of Health, Bethesda, MD). No a priori calculations were performed for sample size analysis. Normality was assessed by Shapiro-Wilk and Q-Q plots prior to parametric testing by Student’s t test or 1- or 2-way analysis of variance followed by post hoc testing as appropriate for the number of groups. All microfluidic qPCR data was analyzed using 2-way analysis of variance. The false discovery rate for the microfluidic qPCR was controlled with the 2-stage step-up method of Benjamini, Krieger, and Yekutieli.
All authors had access to the study data and reviewed and approved the final manuscript prior to submission.
Acknowledgments
CRediT Authorship Contributions
Breanna Sheahan, DVM, PhD (Conceptualization: Equal; Formal analysis: Lead; Investigation: Lead; Methodology: Lead; Visualization: Equal; Writing – original draft: Lead; Writing – review & editing: Lead)
Ally N Freeman (Formal analysis: Supporting; Investigation: Supporting; Validation: Supporting; Writing – review & editing: Supporting)
Theresa M Keeley (Investigation: Supporting; Writing – review & editing: Supporting)
Linda C Samuelson, PhD (Funding acquisition: Supporting; Resources: Supporting; Supervision: Supporting; Writing – review & editing: Supporting)
Jatin Roper, MD (Funding acquisition: Supporting; Resources: Supporting; Supervision: Supporting; Writing – review & editing: Supporting)
Stephanie Hasapis (Investigation: Supporting; Writing – review & editing: Supporting)
Chang-Lung Lee, PhD (Funding acquisition: Supporting; Resources: Supporting; Supervision: Supporting; Writing – review & editing: Supporting)
Christopher Matthew Dekaney, PhD (Conceptualization: Lead; Funding acquisition: Lead; Resources: Lead; Supervision: Lead; Writing – review & editing: Lead).
Footnotes
Conflicts of Interest The authors disclose no conflicts.
Funding This work was supported by R01DK100508 from the National Institute of Diabetes and Digestive and Kidney Diseases (Christopher M. Dekaney), R01DK118023 from the National Institute of Diabetes and Digestive and Kidney Diseases (to Linda C. Samuelson), P30-DK34933 from the National Institute of Diabetes and Digestive and Kidney Diseases (to Linda C. Samuelson), and W81XWH-19-1-0170 from the Department of Defense (Chang-Lung Lee); the Whitehead Scholar Award from the Duke University School of Medicine (Jatin Roper); a pilot grant from the Opportunity Funds Management Core of the Centers for Medical Countermeasures against Radiation, National Institute of Allergy and Infectious Diseases (grant number U19AI067773 [to Chang-Lung Lee and Jatin Roper]); a Comparative Medicine and Translational Research Training Program fellowship (T32OD011130 [to Breanna J. Sheahan]); and an Undergraduate Summer Research Award through the North Carolina State UniversityCollege of Veterinary Medicine (to Ally N. Freeman).
References
- 1.van der Flier L.G., Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–260. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 2.Barker N., van Es J.H., Kuipers J., Kujala P., van den Born M., Cozijnsen M., Haegebarth A., Korving J., Begthel H., Peters P.J., Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 3.Schuijers J., van der Flier L.G., van Es J., Clevers H. Robust Cre-mediated recombination in small intestinal stem cells utilizing the Olfm4 locus. Stem Cell Rep. 2014;3:234–241. doi: 10.1016/j.stemcr.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.VanDussen K.L., Carulli A.J., Keeley T.M., Patel S.R., Puthoff B.J., Magness S.T., Tran I.T., Maillard I., Siebel C., Å Kolterud, Grosse A.S., Gumucio D.L., Ernst S.A., Tsai Y.-H., Dempsey P.J., Samuelson L.C. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development. 2012;139:488–497. doi: 10.1242/dev.070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Potten C.S. Extreme sensitivity of some intestinal crypt cells to X and γ irradiation. Nature. 1977;269:518–521. doi: 10.1038/269518a0. [DOI] [PubMed] [Google Scholar]
- 6.Dekaney C.M., Gulati A.S., Garrison A.P., Helmrath M.A., Henning S.J. Regeneration of intestinal stem/progenitor cells following doxorubicin treatment of mice. Am J Physiol Gastrointest Liver Physiol. 2009;297:G461–G470. doi: 10.1152/ajpgi.90446.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pritchard D.M., Watson A.J.M., Potten C.S., Jackman A.L., Hickman J.A. Inhibition by uridine but not thymidine of p53-dependent intestinal apoptosis initiated by 5-fluorouracil: evidence for the involvement of RNA perturbation. Proc Natl Acad Sci U S A. 1997;94:1795–1799. doi: 10.1073/pnas.94.5.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sangiorgi E., Capecchi M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915–920. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takeda N., Jain R., LeBoeuf M.R., Wang Q., Lu M.M., Epstein J.A. Interconversion between intestinal stem cell populations in distinct niches. Science. 2011;334:1420–1424. doi: 10.1126/science.1213214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan K.S., Chia L.A., Li X., Ootani A., Su J., Lee J.Y., Su N., Luo Y., Heilshorn S.C., Amieva M.R., Sangiorgi E., Capecchi M.R., Kuo C.J. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci U S A. 2012;109:466–471. doi: 10.1073/pnas.1118857109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan K.S., Gevaert O., Zheng G.X.Y., Anchang B., Probert C.S., Larkin K.A., Davies P.S., Cheng Z., Kaddis J.S., Han A., Roelf K., Calderon R.I., Cynn E., Hu X., Mandleywala K., Wilhelmy J., Grimes S.M., Corney D.C., Boutet S.C., Terry J.M., Belgrader P., Ziraldo S.B., Mikkelsen T.S., Wang F., Furstenberg RJ von, Smith N.R., Chandrakesan P., May R., Chrissy M.A.S., Jain R., Cartwright C.A., Niland J.C., Hong Y.-K., Carrington J., Breault D.T., Epstein J., Houchen C.W., Lynch J.P., Martin M.G., Plevritis S.K., Curtis C., Ji H.P., Li L., Henning S.J., Wong M.H., Kuo C.J. Intestinal enteroendocrine lineage cells possess homeostatic and injury-inducible stem cell activity. Cell Stem Cell. 2017;21:78–90.e6. doi: 10.1016/j.stem.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Es J.H., Sato T., van de Wetering M., Lyubimova A., Yee Nee A.N., Gregorieff A., Sasaki N., Zeinstra L., van den Born M., Korving J., Martens A.C.M., Barker N., van Oudenaarden A., Clevers H. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol. 2012;14:1099–1104. doi: 10.1038/ncb2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomic G., Morrissey E., Kozar S., Ben-Moshe S., Hoyle A., Azzarelli R., Kemp R., Chilamakuri C.S.R., Itzkovitz S., Philpott A., Winton D.J. Phospho-regulation of ATOH1 is required for plasticity of secretory progenitors and tissue regeneration. Cell Stem Cell. 2018;23:436–443.e7. doi: 10.1016/j.stem.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castillo-Azofeifa D., Fazio E.N., Nattiv R., Good H.J., Wald T., Pest M.A., de Sauvage F.J., Klein O.D., Asfaha S. Atoh1+ secretory progenitors possess renewal capacity independent of Lgr5+ cells during colonic regeneration. EMBO J. 2019;38 doi: 10.15252/embj.201899984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bohin N., Keeley T.M., Carulli A.J., Walker E.M., Carlson E.A., Gao J., Aifantis I., Siebel C.W., Rajala M.W., Myers M.G., Jones J.C., Brindley C.D., Dempsey P.J., Samuelson L.C. Rapid crypt cell remodeling regenerates the intestinal stem cell niche after notch inhibition. Stem Cell Rep. 2020;15:156–170. doi: 10.1016/j.stemcr.2020.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tetteh P.W., Basak O., Farin H.F., Wiebrands K., Kretzschmar K., Begthel H., van den Born M., Korving J., de Sauvage F., van Es J.H., van Oudenaarden A., Clevers H. Replacement of lost Lgr5-positive stem cells through plasticity of their enterocyte-lineage daughters. Cell Stem Cell. 2016;18:203–213. doi: 10.1016/j.stem.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Jones J.C., Brindley C.D., Elder N.H., Myers M.G., Rajala M.W., Dekaney C.M., McNamee E.N., Frey M.R., Shroyer N.F., Dempsey P.J. Cellular plasticity of Defa4Cre-expressing Paneth cells in response to notch activation and intestinal injury. Cell Mol Gastroenterol Hepatol. 2019;7:533–554. doi: 10.1016/j.jcmgh.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu S., Tong K., Zhao Y., Balasubramanian I., Yap G.S., Ferraris R.P., Bonder E.M., Verzi M.P., Gao N. Paneth cell multipotency induced by notch activation following injury. Cell Stem Cell. 2018;23:46–59.e5. doi: 10.1016/j.stem.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayakawa Y., Tsuboi M., Asfaha S., Kinoshita H., Niikura R., Konishi M., Hata M., Oya Y., Kim W., Middelhoff M., Hikiba Y., Higashijima N., Ihara S., Ushiku T., Fukayama M., Tailor Y., Hirata Y., Guha C., Yan K.S., Koike K., Wang T.C. BHLHA15-positive secretory precursor cells can give rise to tumors in intestine and colon in mice. Gastroenterology. 2019;156:1066–1081.e16. doi: 10.1053/j.gastro.2018.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ijiri K., Potten C.S. Response of intestinal cells of differing topographical and hierarchical status to ten cytotoxic drugs and five sources of radiation. Br J Cancer. 1983;47:175–185. doi: 10.1038/bjc.1983.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ijiri K., Potten C.S. Further studies on the response of intestinal crypt cells of different hierarchical status to eighteen different cytotoxic agents. Br J Cancer. 1987;55:113–123. doi: 10.1038/bjc.1987.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhan Y., Xu C., Liu Z., Yang Y., Tan S., Yang Y., Jiang J., Liu H., Chen J., Wu B. β -Arrestin1 inhibits chemotherapy-induced intestinal stem cell apoptosis and mucositis. Cell Death Dis. 2016;7:e2229. doi: 10.1038/cddis.2016.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buczacki S.J.A., Zecchini H.I., Nicholson A.M., Russell R., Vermeulen L., Kemp R., Winton D.J. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature. 2013;495:65–69. doi: 10.1038/nature11965. [DOI] [PubMed] [Google Scholar]
- 24.Smith N.R., Gallagher A.C., Wong M.H. Defining a stem cell hierarchy in the intestine: markers, caveats and controversies. J Physiol. 2016;594:4781–4790. doi: 10.1113/JP271651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gehart H., Clevers H. Tales from the crypt: new insights into intestinal stem cells. Nat Rev Gastroenterol Hepatol. 2019;16:19–34. doi: 10.1038/s41575-018-0081-y. [DOI] [PubMed] [Google Scholar]
- 26.Kim T.-H., Saadatpour A., Guo G., Saxena M., Cavazza A., Desai N., Jadhav U., Jiang L., Rivera M.N., Orkin S.H., Yuan G.-C., Shivdasani R.A. Single-cell transcript profiles reveal multilineage priming in early progenitors derived from Lgr5+ intestinal stem cells. Cell Rep. 2016;16:2053–2060. doi: 10.1016/j.celrep.2016.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sato T., Sase M., Ishikawa S., Kajita M., Asano J., Sato T., Mori Y., Ohteki T. Characterization of radioresistant epithelial stem cell heterogeneity in the damaged mouse intestine. Sci Rep. 2020;10:8308. doi: 10.1038/s41598-020-64987-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Becker C., Watson A.J., Neurath M.F. Complex roles of caspases in the pathogenesis of inflammatory bowel disease. Gastroenterology. 2013;144:283–293. doi: 10.1053/j.gastro.2012.11.035. [DOI] [PubMed] [Google Scholar]
- 29.Snippert H.J., van der Flier L.G., Sato T., van Es J.H., van den Born M., Kroon-Veenboer C., Barker N., Klein A.M., van Rheenen J., Simons B.D., Clevers H. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 30.Bach S.P., Renehan A.G., Potten C.S. Stem cells: the intestinal stem cell as a paradigm. Carcinogenesis. 2000;21:469–476. doi: 10.1093/carcin/21.3.469. [DOI] [PubMed] [Google Scholar]
- 31.Schepers A.G., Vries R., van den Born M., van de Wetering M., Clevers H. Lgr5 intestinal stem cells have high telomerase activity and randomly segregate their chromosomes: Lgr5 adult intestinal stem cell characteristics. EMBO J. 2011;30:1104–1109. doi: 10.1038/emboj.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huch M., Dorrell C., Boj S.F., van Es J.H., Li V.S.W., van de Wetering M., Sato T., Hamer K., Sasaki N., Finegold M.J., Haft A., Vries R.G., Grompe M., Clevers H. In vitro expansion of single Lgr5 + liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494:247–250. doi: 10.1038/nature11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bohin N., Carlson E.A., Samuelson L.C. Genome toxicity and impaired stem cell function after conditional activation of CreERT2 in the intestine. Stem Cell Rep. 2018;11:1337–1346. doi: 10.1016/j.stemcr.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jahn H.M., Kasakow C.V., Helfer A., Michely J., Verkhratsky A., Maurer H.H., Scheller A., Kirchhoff F. Refined protocols of tamoxifen injection for inducible DNA recombination in mouse astroglia. Sci Rep. 2018;8:5913. doi: 10.1038/s41598-018-24085-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montgomery R.K., Carlone D.L., Richmond C.A., Farilla L., Kranendonk M.E.G., Henderson D.E., Baffour-Awuah N.Y., Ambruzs D.M., Fogli L.K., Algra S., Breault D.T. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci U S A. 2011;108:179–184. doi: 10.1073/pnas.1013004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Powell A.E., Wang Y., Li Y., Poulin E.J., means A.L., Washington M.K., Higginbotham J.N., Juchheim A., Prasad N., Levy S.E., Guo Y., Shyr Y., Aronow B.J., Haigis K.M., Franklin J.L., Coffey R.J. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell. 2012;149:146–158. doi: 10.1016/j.cell.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian H., Biehs B., Warming S., Leong K.G., Rangell L., Klein O.D., de Sauvage F.J. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011;478:255–259. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Landeghem L., Santoro M.A., Krebs A.E., Mah A.T., Dehmer J.J., Gracz A.D., Scull B.P., McNaughton K., Magness S.T., Lund P.K. Activation of two distinct Sox9-EGFP-expressing intestinal stem cell populations during crypt regeneration after irradiation. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1111–G1132. doi: 10.1152/ajpgi.00519.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jadhav U., Saxena M., O’Neill N.K., Saadatpour A., Yuan G.-C., Herbert Z., Murata K., Shivdasani R.A. Dynamic reorganization of chromatin accessibility signatures during dedifferentiation of secretory precursors into Lgr5+ intestinal stem cells. Cell Stem Cell. 2017;21:65–77.e5. doi: 10.1016/j.stem.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li N., Yousefi M., Nakauka-Ddamba A., Jain R., Tobias J., A Epstein J., Jensen S., J Lengner C. Single-cell analysis of proxy reporter allele-marked epthelial cells establishes intestinal stem cell hierarchy. Stem Cell Rep. 2014;3:876–891. doi: 10.1016/j.stemcr.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adolph T.E., Tomczak M.F., Niederreiter L., Ko H.-J., Böck J., Martinez-Naves E., Glickman J.N., Tschurtschenthaler M., Hartwig J., Hosomi S., Flak M.B., Cusick J.L., Kohno K., Iwawaki T., Billmann-Born S., Raine T., Bharti R., Lucius R., Kweon M.-N., Marciniak S.J., Choi A., Hagen S.J., Schreiber S., Rosenstiel P., Kaser A., Blumberg R.S. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272–276. doi: 10.1038/nature12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barriga F.M., Montagni E., Mana M., Mendez-Lago M., Hernando-Momblona X., Sevillano M., Guillaumet-Adkins A., Rodriguez-Esteban G., Buczacki S.J.A., Gut M., Heyn H., Winton D.J., Yilmaz O.H., Attolini C.S.-O., Gut I., Batlle E. Mex3a marks a slowly dividing subpopulation of Lgr5+ intestinal stem cells. Cell Stem Cell. 2017;20:801–816.e7. doi: 10.1016/j.stem.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tao S., Tang D., Morita Y., Sperka T., Omrani O., Lechel A., Sakk V., Kraus J., Kestler H.A., Kühl M., Rudolph K.L. Wnt activity and basal niche position sensitize intestinal stem and progenitor cells to DNA damage. EMBO J. 2015;34:624–640. doi: 10.15252/embj.201490700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muñoz J., Stange D.E., Schepers A.G., van de Wetering M., Koo B.-K., Itzkovitz S., Volckmann R., Kung K.S., Koster J., Radulescu S., Myant K., Versteeg R., Sansom O.J., van Es J.H., Barker N., van Oudenaarden A., Mohammed S., Heck A.J.R., Clevers H. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012;31:3079–3091. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moossavi S. Heterogeneity of the level of activity of lgr5+ intestinal stem cells. Int J Mol Cell Med. 2014;3:216–224. [PMC free article] [PubMed] [Google Scholar]
- 46.Vooijs M., Jonkers J., Berns A. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep. 2001;2:292–297. doi: 10.1093/embo-reports/kve064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.King S.L., Mohiuddin J.J., Dekaney C.M. Paneth cells expand from newly created and preexisting cells during repair after doxorubicin-induced damage. Am J Physiol Gastrointest Liver Physiol. 2013;305:G151–G162. doi: 10.1152/ajpgi.00441.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ayyaz A., Kumar S., Sangiorgi B., Ghoshal B., Gosio J., Ouladan S., Fink M., Barutcu S., Trcka D., Shen J., Chan K., Wrana J.L., Gregorieff A. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature. 2019;569:121–125. doi: 10.1038/s41586-019-1154-y. [DOI] [PubMed] [Google Scholar]
- 49.Murata K., Jadhav U., Madha S., van Es J., Dean J., Cavazza A., Wucherpfennig K., Michor F., Clevers H., Shivdasani R.A. Ascl2-dependent cell dedifferentiation drives regeneration of ablated intestinal stem cells. Cell Stem Cell. 2020;26:377–390.e6. doi: 10.1016/j.stem.2019.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li L., Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327:542–545. doi: 10.1126/science.1180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sei Y., Lu X., Liou A., Zhao X., Wank S.A. A stem cell marker-expressing subset of enteroendocrine cells resides at the crypt base in the small intestine. Am J Physiol - Gastrointest Liver Physiol. 2011;300:G345–G356. doi: 10.1152/ajpgi.00278.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kozar S., Morrissey E., Nicholson A.M., van der Heijden M., Zecchini H.I., Kemp R., Tavaré S., Vermeulen L., Winton D.J. Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell. 2013;13:626–633. doi: 10.1016/j.stem.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 53.Merlos-Suárez A., Barriga F.M., Jung P., Iglesias M., Céspedes M.V., Rossell D., Sevillano M., Hernando-Momblona X., da Silva-Diz V., Muñoz P., Clevers H., Sancho E., Mangues R., Batlle E. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011;8:511–524. doi: 10.1016/j.stem.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 54.Hua G., Thin T.H., Feldman R., Haimovitz-Friedman A., Clevers H., Fuks Z., Kolesnick R. Crypt base columnar stem cells in small intestines of mice are radioresistant. Gastroenterology. 2012;143:1266–1276. doi: 10.1053/j.gastro.2012.07.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe K., Ikuno Y., Kakeya Y., Ikeno S., Taniura H., Kurono M., Minemori K., Katsuyama Y., Naka-Kaneda H. Age-related dysfunction of the DNA damage response in intestinal stem cells. Inflamm Regen. 2019;39:8. doi: 10.1186/s41232-019-0096-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burgess D.J., Doles J., Zender L., Xue W., Ma B., McCombie W.R., Hannon G.J., Lowe S.W., Hemann M.T. Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Natl Acad Sci U S A. 2008;105:9053–9058. doi: 10.1073/pnas.0803513105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vousden K.H., Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 58.Tsai R.Y.L. Balancing self-renewal against genome preservation in stem cells: how to have the cake and eat it too? Cell Mol Life Sci. 2016;73:1803–1823. doi: 10.1007/s00018-016-2152-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang F., Teves S.S., Kemp C.J., Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta. 2014;1845:84–89. doi: 10.1016/j.bbcan.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu Y., Huang Y.-F., Kek C., Bulavin D.V. Apoptosis differently affects lineage tracing of Lgr5 and Bmi1 intestinal stem cell populations. Cell Stem Cell. 2013;12:298–303. doi: 10.1016/j.stem.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 61.Andrade D., Rosenblatt J. Apoptotic regulation of epithelial cellular extrusion. Apoptosis Int J Program Cell Death. 2011;16:491–501. doi: 10.1007/s10495-011-0587-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gregorieff A., Liu Y., Inanlou M.R., Khomchuk Y., Wrana J.L. Yap-dependent reprogramming of Lgr5 + stem cells drives intestinal regeneration and cancer. Nature. 2015;526:715–718. doi: 10.1038/nature15382. [DOI] [PubMed] [Google Scholar]
- 63.Potten C.S. Protection of the small intestinal clonogenic stem cells from radiation-induced damage by pretreatment with interleukin 11 also increases murine survival time. Stem Cells. 1996;14:452–459. doi: 10.1002/stem.140452. [DOI] [PubMed] [Google Scholar]
- 64.Yan K.S., Kuo C.J. Ascl2 reinforces intestinal stem cell identity. Cell Stem Cell. 2015;16:105–106. doi: 10.1016/j.stem.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 65.Sato T., Vries R.G., Snippert H.J., van de Wetering M., Barker N., Stange D.E., van Es J.H., Abo A., Kujala P., Peters P.J., others Single Lgr5 stem cells build crypt villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 66.van der Flier LG., Haegebarth A., Stange D.E., van de Wetering M., Clevers H. OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology. 2009;137:15–17. doi: 10.1053/j.gastro.2009.05.035. [DOI] [PubMed] [Google Scholar]
- 67.Besson D., Pavageau A.-H., Valo I., Bourreau A., Bélanger A., Eymerit-Morin C., Moulière A., Chassevent A., Boisdron-Celle M., Morel A., Solassol J., Campone M., Gamelin E., Barré B., Coqueret O., Guette C. A quantitative proteomic approach of the different stages of colorectal cancer establishes OLFM4 as a new nonmetastatic tumor marker. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.009712. M111.009712. [DOI] [PMC free article] [PubMed] [Google Scholar]