

SUMMARY
We describe a cellular contractile mechanism employed by fibroblasts and mesenchymal cancer cells to migrate in 3D collagen gels. During 3D spreading, fibroblasts strongly deform the matrix. They protrude, polarize, and initiate migration in the direction of highest extracellular matrix (ECM) deformation (prestrain). This prestrain is maintained through anterior cellular contractions behind the leading edge prior to protrusion, coordinating a distinct 3D migration cycle that varies between cell types. Myosin IIA is required for strain polarization, generating anterior contractions, and maintaining prestrain for efficient directional cell migration. Local matrix severing disrupts the matrix prestrain, suppressing directional protrusion. We show that epithelial cancer and endothelial cells rarely demonstrate the sustained prestrain or anterior contractions. We propose that mesenchymal cells sense ECM stiffness in 3D and generate their own matrix prestrain. This requires myosin IIA to generate polarized periodic anterior contractions for maintaining a 3D migration cycle.
In brief
Doyle et al. show that, in 3D environments, highly migratory mesenchymal cells use anterior contractions, localized between the leading edge and nucleus, to generate a large extracellular matrix deformation (prestrain) and enhance protrusion. Myosin II contractility and extensive integrin ligation mediate this migration phenotype.
Graphical Abstract
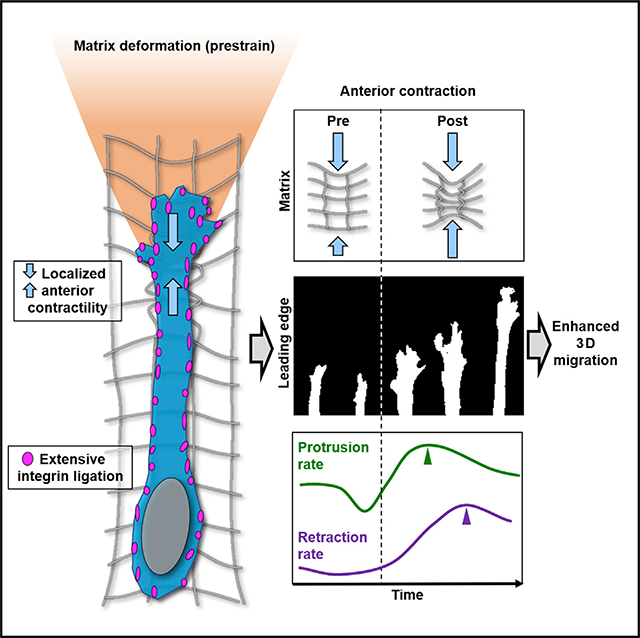
INTRODUCTION
Cell migration is critical for proper embryonic development, wound healing, and immunological responses. However, unchecked migration also contributes to cancer invasion and metastasis (Ridley et al., 2003). Migration requires a complex series of cellular processes. Three key general components have been identified that are considered to be crucial for cell translocation: protrusion of a leading edge, adhesion to the surrounding microenvironment, and retraction via a contractile mechanism; each process is a rate-limiting step (Lauffenburger and Horwitz, 1996). Most studies have been performed on rigid two-dimensional (2D) surfaces to identify specific temporal and spatial steps in motility. For mesenchymal fibroblasts, retraction precedes the leading-edge protrusion as part of the classic 2D cell migration cycle (Yamada and Sixt, 2019; Krause and Gautreau, 2014; Gupton and Waterman-Storer, 2006; Palecek et al., 1998; Chen, 1981, 1979).
Other internal and external factors can alter migration rates in this basic model of migration, including extracellular matrix (ECM) composition, ECM elasticity, ECM density, integrin expression, and cellular contractility (Gupton and Waterman-Storer, 2006; Palecek et al., 1997). For example, by increasing ECM density, the rate-limiting step switches from protrusion to retraction by affecting actin flow rate, adhesion turnover, and adhesion lifetime (Gupton and Waterman-Storer, 2006). Biophysical studies on 2D elastic surfaces have elucidated how cells respond to matrix stiffness. They can alter contractility to mimic their microenvironment as inferred from traction force measurements and migrate up a stiffness gradient in a process termed as durotaxis (DuChez et al., 2019; Lo et al., 2000). The process of transferring intracellular cytoskeletal contractility to the surrounding environment during mechanotransduction is intrinsically coupled to cell-ECM adhesion, as cells sample their physical surroundings to regulate cell migration (Plotnikov et al., 2012). While many of these intricate cellular processes have been thoroughly studied on a 2D matrix, there is a poor understanding of the spatiotemporal dynamics of mesenchymal cell migration in a 3D matrix.
In a 3D microenvironment, ECM-dependent factors such as matrix pore size, fibril alignment, ECM crosslinking, and whether an ECM is linear or nonlinear elastic (can undergo strain stiffening) can alter cell migration (Doyle et al., 2015; Charras and Sahai, 2014; Petrie et al., 2012). Matrix alignment enhances migration rate (Taufalele et al., 2019; Riching et al., 2014), while increasing matrix concentration associated with a concomitant decrease in matrix pore size reduces it (Taufalele et al., 2019; Wolf et al., 2013). The pore size limits translocation of the largest cellular organelle, the nucleus, as the main rate-limiting step for 3D migration in many cell types (Renkawitz et al., 2019; Yamada and Sixt, 2019; Wolf et al., 2013; Lämmermann et al., 2008). Analogous to research on 2D linear-elastic polyacrylamide gels, several studies have investigated the traction forces generated by cells in 3D microenvironments. A detailed 3D force reconstruction has shown contraction of epithelial breast cancer cells in collagen gels (Hall et al., 2016; Steinwachs et al., 2016). Although detailed spatial and temporal relationships during the 2D cell migration cycle have been established (Shafqat-Abbasi et al., 2016), little is known about a migration cycle in 3D environments.
Recent research has categorized 3D migration modes based on key cellular characteristics that include cell adhesiveness to the ECM mediated by integrins (Kechagia et al., 2019), contractility, type of protrusion at the leading edge, and involvement of ECM proteolysis (Doyle et al., 2015; Petrie et al., 2012; Sahai and Marshall, 2003). Highly adhesive fibroblasts are generally considered as classical mesenchymal migratory cells. Mesenchymal 3D migration is not exclusive to fibroblasts, as multiple embryonic and cancer cells can display a lamellipodial mesenchymal phenotype in 3D. Moreover, some cancer cells can switch their mode of 3D migration from mesenchymal to ameboid, where bleb-like protrusions dominate at the leading edge in association with low adhesion (Yamada and Sixt, 2019; Charras and Sahai, 2014). Other factors associated with the 3D mesenchymal migration phenotype include robust cell-matrix adhesion (Mekhdjian et al., 2017; Doyle et al., 2015; Cukierman et al., 2001) and low cell-cell adhesion for collective cell migration (Janiszewska et al., 2020), actin-myosin based contractility, spindle-shaped body, and a single main pseudopod directing migration. Several studies have focused on the dynamics of cell adhesions in 3D microenvironments, demonstrating how 3D ECM can alter adhesion turnover (Mekhdjian et al., 2017; Doyle et al., 2015; Kubow et al., 2013). However, there are substantial gaps in knowledge concerning the spatiotemporal dynamics of 3D mesenchymal migration, including whether a 3D cell migration cycle exists or differs from the canonical 2D cycle, as well as mechanistic insight into key components required for efficient 3D migration of mesenchymal and metastatic cancer cells.
Here, using live-cell 3D imaging and focusing on a primary human fibroblast model to characterize mesenchymal migration, we identify a curious interplay between fibroblasts and the 3D microenvironment. During cell polarization and directional migration, a major matrix deformation (a prestrain) develops strongly in the direction of cell protrusion that requires myosin IIA contractility and β1 integrin ligation. Local disruption of this matrix prestrain through ECM laser ablation results in cell repolarization, whereas inhibiting integrin ligation reduces the magnitude of the asymmetric prestrain. This matrix prestrain is established and maintained at nearly constant tension through a series of local anterior contractions (ACs) along the main pseudopod behind the leading edge. ACs are consistently followed by increased leading-edge protrusion as an important component of a distinctive polarized 3D cell migration cycle. Cells using this process have higher migration velocities. We also found that this dynamic mesenchymal migration depends on both the expression level of myosin II and the relative amount of integrin ligation, and it is shared to differing degrees by cancer cells.
RESULTS
3D cell spreading, polarization, and protrusion involve cell-generated anterior matrix deformation (oriented prestrain)
To fully characterize the spatiotemporal dynamics of individual cell interaction with the surrounding ECM during 3D cell migration, we performed live-cell 3D time-lapse spinning disk microscopy using primary human fibroblasts (human foreskin fibroblasts; HFFs) as the model mesenchymal cell. HFFs expressing TagGFP2 LifeAct and treated with SiR-DNA to visualize the cytoskeleton and nucleus, respectively, were polymerized within collagen hydrogels designed to have fibrillar architecture and a large pore size for permitting efficient 3D cell migration (see STAR methods and Doyle et al., 2015).
We first examined fibroblasts during the initial stages of cell spreading in a 3D matrix to characterize the origin of cell protrusions and polarity. In contrast to the pancake-like spreading of cells on 2D substrates, which break their circular isotropic state by retracting an edge, fibroblasts in 3D ECM primarily spread, polarize, and extend a protrusion along a single axis (Figures 1A and S1A). This tended to follow the orientation of several collagen fibrils to which the cell had initially attached (Figures S1B, S1F, and S1G). Immunostaining for activated β1 integrin (9EG7: indicative of ECM ligation), a key marker of cell-ECM interaction, during 3D spreading, showed integrin clustering at protrusion sites (Figure 1B) with an initially imbalanced pattern of ligation across the cell. However, as polarity and spreading continued, integrin clustering became widespread, with no significant differences between anterior, middle, and posterior regions of cells (Figure 1C). In fact, integrin clustering was observed at nearly every point of cell contact with the ECM.
Figure 1. 3D cell polarization and protrusion are associated with an anterior matrix prestrain.

(A) Maximum intensity projection (MIP) of a fibroblast expressing TagGFP2-LifeAct (magenta) spreading in collagen (green).
(B and C) Activated β1 integrin (white) and F-actin (magenta) in fibroblasts at 30 min (B) and 6 h (C) of 3D spreading.
(D) Cell similar to (A) showing uniaxial polarization.
(E) PIV analysis of absolute (total) ECM deformations for the cell in (D).
(F) PIV analysis of relative (instantaneous) ECM deformations demonstrates an AC before reaching a steady state. Time: minutes.
(G) Schematic depicting how fibroblasts were observed to progress from an isotropic state to anisotropic polarization. Scale bars, (A) and (D), 20 μm; B, 10 μm.
We applied particle image velocimetry (PIV) to measure the ECM deformations generated by cells. For these experiments, the first time point was used as a relaxed matrix against which subsequent frames were calculated (see STAR methods). Fibroblasts gradually increase deformation of the surrounding microenvironment along a single axis as determined by quantification of strain (Figures 1D and 1E). Cell protrusion was associated with an increasingly polarized ECM strain over the first 3 h, and then, ECM deformation reached a steady state (Figure 1E). Interestingly, prior to establishment of polarization, ECM deformation, and subsequently the cell nucleus, demonstrated slow anterior-to-posterior oscillations similar to the force oscillations reported for 2D focal adhesions (Figure S1B) (Plotnikov et al., 2012).
High resolution imaging revealed that whereas collagen fibrils in front of the leading edge are pulled taut, regions between the leading edge and nucleus demonstrate fibril buckling consistent with a mechanical pinch between these two regions, which precedes protrusion (Figures S1D–S1G.). These equal-and-opposite contractile effects were only visible when tracking instantaneous relative deformations of the matrix, where adjacent time points were compared (Figures 1D–1F, magenta-outlined panel). Interestingly, we found that both overall long-term absolute and relative ECM deformations (occurring over 15 min of time Figure S1E) increase rapidly during fibril buckling and then level off after cell polarity is firmly established. While absolute total deformation at the cell anterior is maintained at a high anisotropic level, relative deformations are often transient and generally isotropic around the cell. These data suggest that during initial cell spreading in 3D, fibroblasts utilize anisotropic strain transmission to the ECM to polarize.
Mesenchymal fibroblasts maintain an anterior matrix prestrain during 3D migration using repetitive anterior contractions
Figure 1G illustrates the events we observed during 3D cell spreading and polarization: a local tension imbalance forms along a single axis, leading to an AC that initiates a matrix prestrain in the direction of protrusion. We investigated if fibroblasts deform the surrounding ECM similarly during 3D migration. Cells were lysed after each time-lapse series to acquire an unstrained, relaxed ECM image (see STAR methods). PIV analyses indicated that polarized absolute deformation of the surrounding matrix is maintained by migrating fibroblasts with high leading-edge, but a lower trailing-edge, deformation/pulling (Figures 2A, 2B, S2A, and S2B; Video S1). These leading/trailing-edge differences were also observed at different ECM concentrations and within ECMs having a smaller pore size (Figures S2C and S2D).
Figure 2. Fibroblasts sustain a matrix prestrain during cell migration.

(A) HFF expressing TagGFP2-LifeAct (magenta) embedded in 3D collagen (green) migrating directionally over time. “A” and “P” (ovals) represent the anterior (A) and posterior (P) deformations shown in (D and E).
(B and C) Absolute (B) and relative (C) deformation of the ECM of the same cell in (A). Time: minutes.
(D) Graphical plots of the data in (B) and (C). Top panel: sustained differences in anterior and posterior strain. Middle and bottom: absolute strain is sustained and high, whereas relative strain is variable and associated with contraction. Bottom: data normalized to the first time point.
(E) Mean (±SEM) deformations of matrix strain anterior and posterior to the cell (n = 17) and A/P ratio for all cells versus nonmotile cells (light gray: n = 17, N = 3).
(F) Tag-GFP2 LifeAct-expressing fibroblast (magenta) in collagen gel (green) prior to and after severing the matrix at anterior and posterior regions (red solid lines). Kymograph shows the local relaxation of the matrix. The yellow-dashed line indicates the time of severing. Red dashed lines show the movement of the matrix.
(G) Matrix displacements after anterior and posterior cuts (mean ± SEM; N = 7). **p ≤ 0.01. Scale bar, 20 μm.
Temporally, the deformations at the leading and trailing edges are maintained at a relatively constant level (Figure 2D) with approximately 2-fold differences during continuous migration (Figures 2B and 2E). These findings suggest that mechanical information is not directly transmitted equally between the ends of the cell. Fibroblasts consistently migrate into the region of highest ECM deformation in front of the cell toward the region of matrix prestrain (Figure 2E). To characterize ECM tension differences anterior and posterior to cells, we severed the ECM in a single Z plane with a two-photon laser and measured the ECM displacement after 40 s (Figures 2F and 2G). These data confirm anterior versus posterior differences in tension and strain.
Because these anterior ECM deformations are maintained during migration—analogous to isometric tension—we compared the relative, short-term deformations of 3D collagen. We found that such relative deformations vary greatly over time, often spiking 2-fold higher than at previous time points in the direction of migration (Figures 2C and 2D, bottom panels). Closer examination (Figure 3A) reveals an interesting contractile pattern. A small retrograde pull in front of the leading edge is followed by a larger AC approximately 20 μs behind the leading edge: the cell appears to pinch the matrix between these two points during migration (Figure 3A), similar to our findings during cell spreading (Figures 1D–1F). Tracking ECM movements compared with YFP-paxillin-containing adhesions showed that they were closely linked during these pinch-like contractions (Figures S2E–S2G). These unique matrix deformations could represent a contraction between adhesions at the leading edge of the cell body and the highly stable adhesions we had previously described (Doyle et al., 2015). To examine if this ECM deformation was due to cytoskeletal contraction, we transfected HFFs with mApple-paxillin and EGFP-α-actinin to highlight focal adhesions and organized cytoskeletal elements, respectively. Tracking of both components revealed dynamics in which the α-actinin and paxillin (to a lesser extent) moved from initial adhesions toward a central point between the leading edge and the main portion of the pseudopod corresponding to the low point of ECM deformation (Figure S3A). This finding suggests matrix pinching is associated with an anterior cytoskeletal contraction along a single axis; we will refer to this mechanism as an anterior contraction or an AC.
Figure 3. Anterior contraction enhances migration through increased protrusion.

(A) HFF expressing TagGFP2-LifeAct (LA, magenta) in 3D collagen (green) migrating directionally (white arrow). The white box (left) indicates the kymograph area to the right. Kymograph (center) shows a retrograde pull (red box) followed by an anterograde contraction (cyan box) that defines an AC, then protrusion.
(B and C) Effect of an AC on cell velocity (B) and mean-squared displacement (C; **p ≤ 0.01 t test, mean value ± SEM shown, N = 8, n = 40).
(D) A binary mask of the cell in (A) created to analyze protrusion (green box) and retraction (orange box). Time-lapse differential images show protruding (white) and rear-retracting (black) regions.
(E) Temporal analysis of the rate of protrusion (green), nuclear displacement (black), and retraction (orange) centered around AC initiation (time 0; *p ≤ 0.05, mean value ±SEM shown, N = 5, n = 18).
(F) Schematic illustration of how an anterior axial contraction drives matrix prestrain development and protrusion. Scale bar, 20 μm.
ACs are temporally associated with enhanced leading and trailing-edge dynamics to promote 3D migration
We found that a majority of HFFs underwent at least one AC over 3 h (~75%, n = 40). In addition, the presence of the AC phenotype correlates with enhanced migration rate but not cell persistence (Figures 3B and 3C; Video S2). Analyses indicated that protrusion rates decrease prior to an AC, while retraction rates remain relatively unchanged (Figure 3E). However, following an AC, protrusion rates increase ~2-fold, followed by a slow elevation of retraction rate at the trailing edge. Cross-correlation analysis around an AC demonstrated a ~12+ min lag for the trailing edge after leading-edge dynamics, suggesting a slow temporal coupling in which protrusion precedes retraction (Figure S3E). Including our 3D cell spreading data, we suggest that fibroblasts locally contract the ECM at the leading pseudopod through an AC (Figure 3F). Interestingly, this event occurs after a transient decrease in the leading-edge protrusion (Figure 3E).
Myosin IIA and integrin ligation are required for polarized cell tension gradients and AC development
To elucidate the roles of contraction and integrin engagement in ECM strain development (prestrain and AC) during 3D cell migration, we generated myosin IIA (MIIA) and myosin IIB (MIIB) CRISPR knockout fibroblasts and used blocking antibodies against β1 integrin. First, the loss of MIIA increased the rate of 3D cell spreading; cells spread even prior to our first time point and were unable to establish polarity (Figures 4A, S4A, S4C, and S4D). MIIB-null fibroblasts showed no significant deficits in spreading or cell polarization (Figures 4B and S4B–S4D). As cells spread, ECM deformations for MIIA-null cells plateaued quickly (Figure S4C), while MIIB nulls demonstrated a similar initial rate of ECM deformation that plateaued sooner than the wild type, which continued to rise (Figure S4C). Overall, MIIA-null cells strained the ECM ~6- and 5-fold less compared with wild-type and MIIB-null cells, respectively (Figure 4C). MIIA-null cells failed to strain the ECM much beyond the edge of the cell (Figures 4A and S4A), while MIIB nulls showed no major effects on ability to deform the ECM (Figures 4B and 4C). No ACs were observed after MIIA knockout, while MIIB nulls showed no difference in AC generation compared with scrambled controls (Figure 4D). Together, these data indicate that MIIA is necessary for creating ACs and the strain anisotropy required for cell polarization, while MIIB is required for further elevation of transmitted cellular contractility during spreading and sustaining the anisotropy.
Figure 4. MIIA and integrin ligation are required for cell-matrix prestrain and AC development.

(A and B) Maximum intensity projection of TagGFP2 LifeAct (magenta)-expressing CRISPR knockout MIIA (A) or MIIB (B) fibroblasts in 3D collagen (green). Bottom panels show PIV imaging.
(C) 90th percentile ECM deformations in 3D collagen for scrambled control, MIIA, and MIIB knockouts (**p < 0.01 N = 3, n ≥ 13).
(D) Percent of the same cells undergoing anterior contraction during 3D migration (N = 3, n ≥ 17).
(E) Time-lapse and PIV images of HFF (LifeAct, magenta) migrating in collagen (green) responding to anti-β1 integrin antibody mAb13 addition (2 μg/ml shown).
(F) Graphical plots of mAb13 (10 μg/ml, red; 2 μg/ml, blue) addition on 90th percentile ECM deformations (N = 3, n ≥ 6).
(G) 90th percentile ECM deformations for the pre-, transition, and post-phases (N = 3, n ≥ 6). *p < 0.05 versus pre-phase. For all graphs, mean value ±SEM shown. Scale bars: 20 μm.
To assess the role of integrin ligation, we blocked integrin function during cell migration and observed transitions from integrin-ligated to integrin-inhibited migration (Figures 4E–4G; Video S3). 30 min after antibody mAb13 addition, cells slowly lost the ability to undergo an AC, and ECM prestrain was reduced, resulting in a virtually equal front-to-back ECM strain pattern (Figures 4E–4G). During this integrin-inhibited migration, cells demonstrated random and low ECM strains and a hypercontractile phenotype (mostly spherical) but were able to locomote slowly (Video S3). Together, these data indicate ECM strain pattern is dependent on both MIIA and integrin ligation.
Reduction of ECM prestrain initiates leading-edge retraction and stunts migration
We next tested whether the matrix prestrain is essential for directional migration. We used laser microablation (see STAR methods) to cut the collagen gel 20 μm in front of directionally migrating fibroblasts to disrupt their cell-generated prestrain (Figures 5A–5D; Video S4). ~94% of cells retracted their leading edge over 30 min (Figures 5C and 5D). A minority of the cells moved fully away from the area of reduced tension over 4 h. Those migrating away lost their leading edge and then actively probed the microenvironment and ultimately formed a new, distal leading edge (Figures 5D and 5E). In non-repolarizing cells, the changes at the leading edge were not propagated to the cell body or trailing edge, but the migration slowed, and cells never reached the severed ECM region (data not shown). Analyses of averaged directional deformations (summing all relative ECM Y-displacements) demonstrated that ECM displacements also undergo repolarization prior to establishment of a new migration direction (Figure 5F). In addition, analysis of leading edge (LE), nucleus (NUC), and trailing edge (TE) indicate that LE dynamics are temporally distinct—similar to our AC data (Figure 5G). Interestingly, NUC and TE dynamics coincide with the directional change in ECM deformation (Figure 5G), suggesting that for mesenchymal cells, propagation of cell contractility to the ECM as a prestrain is established prior to migration.
Figure 5. Reduction of ECM prestrain alters migration via cell mechanosensing.

(A) Pre- and post-ablation XY and ZY maximum intensity projections of an HFF (LifeAct, magenta) in 3D collagen (green). A 1003 80 μm (XZ) ECM laser “cut” in front of the cell.
(B) Relative ECM deformation maps for red dashed box region in (B) before and after ablation.
(C) Percent cells undergoing leading-edge retraction or migration away from the ECM cut. N = 7, n = 19.
(D) Time lapse of the cell shown in (A) (yellow-dashed line box).
(E) Kymograph (white-dashed box in D) of the ECM showing an AC (red arrows) before and after the change in ECM deformation from one end of the cell to the other.
(F) Averaged directional deformations (ADD) of the ECM. Arrows in (D) and (F) indicate ablation. Blue region corresponds to the blue box in (D).
(G) Forward migration index (FMI) for LE, NUC, and TE (N = 4).
(H) Cell severing at anterior or posterior regions shows significant differences in local ECM relaxation. Time lapse shows a cell cut anteriorly releasing ECM strain. White-dashed lines are fiduciary marks to visualize ECM displacements.
(I) Analysis of ECM movement for cells severed at the front and back. (N = 4, n = 11), *p ≤ 0.05. For (C), (G), and (I) mean value ±SEM shown. Scale bars, 10 μm.
To identify the source of cellular tension, we locally severed cell bodies at the front or the back and measured the ECM displacement. This cell severing reduces tension locally within seconds. Analyses show local disruption of contraction at the cell anterior leads to greater tension loss than at the back (Figures 5H and 5I). Together, these data indicate that fibroblasts require a self-generated matrix prestrain to migrate directionally and that the cellular tension within this prestrain is localized at the anterior of the cell.
Different cell types demonstrate different ECM strain characteristics in 3D microenvironments
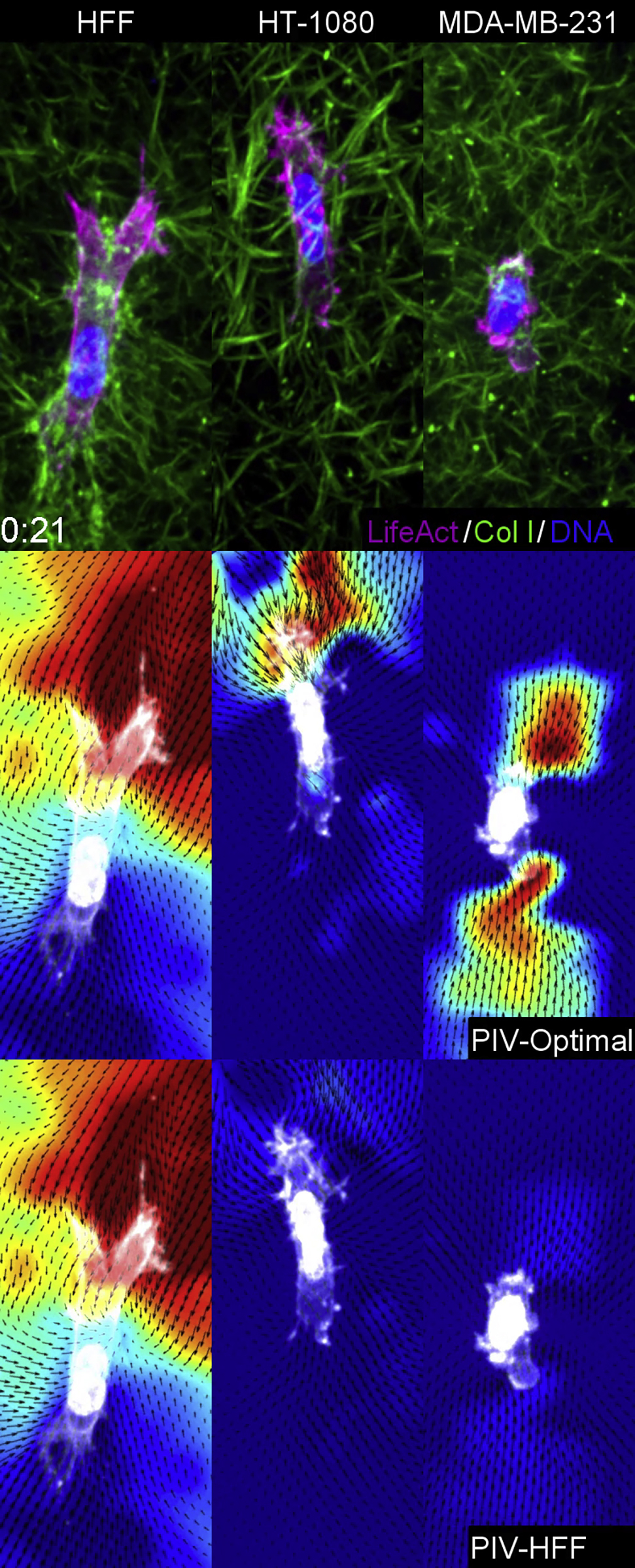
We next examined if anterior matrix prestrain and ACs were defining characteristics of mesenchymal 3D cell migration. We compared HT-1080 fibrosarcoma (malignant mesenchymal origin) and MDA-MB-231 breast cancer epithelial cells. We first characterized cell-ECM adhesion for activated β1 integrin. Both HT-1080s and HFFs demonstrate a similar level of activated, ligated β1 integrin in 3D collagen over the entirety of each cell. MDA cells instead showed integrin activation solely in extended lamellipodial regions and tail with little ligation visible in the mid-cell body (Figures 6A and 6B). Analyses of cell migration rates revealed fibroblasts and HT-1080 migrate at nearly a 3-fold faster rate than MDAs, but only fibroblasts show high directionality (Figures 6C and 6D). Interestingly, while all three cell types required contractility to migrate efficiently (inhibited by blebbistatin or Rho kinase inhibitor), both HFFs and HT-1080s demonstrated larger ~60% deficits compared with ~25% for MDAs (Figure 6C).
Figure 6. Different cell types demonstrate different integrin and ECM strain characteristics in 3D microenvironments.

(A and B) Immunostaining for activated β1 integrin (white) and actin (magenta) in collagen (green) for MDA-MB-231(A) and HT-1080(B).
(C and D) Migration rates (C) and mean-squared displacement (D) for HFF, HT1080 and MDA-MD-321 cells with control vehicle (DMSO) or 25 μM blebbistatin or 10 μM Y-27632. in (C), *p < 0.05 compared with the control (N ≥ 3, n ≥ 39). in (D), *p < 0.05 compared with HFF (N ≥ 3, n > 39).
(E) 90th % matrix deformation for HFF, HT-1080 (mesenchymal versus ameboid), HUVEC and MDA-MB-231 cells (N ≥ 3, n ≥ 8). **p < 0.0001 compared with all conditions. *p < 0.01.
(F and G) Time lapse of HT-1080 (F) or MDA cell (G) expressing TagGFP2 LifeAct (green) and accompanying absolute PIV images. Time in (D and E) is in minutes. For all graphs, mean value ±SEM shown. Scale bars, (A and B), 10 μm; (F and G), 20 μm.
PIV analyses of HT-1080 and MDAs during 3D migration demonstrated distinctly different dynamic ECM strain patterns (Figures 6F and 6G; Video S5). First, HT-1080s demonstrated 2-fold higher ECM deformations compared with MDAs (Figure 6E). Second, HT-1080s mimicked the high frontal matrix prestrain found in HFFs, while MDAs exhibited nearly equal displacements at the front and rear of the cell. Third, HT-1080s were able to maintain the matrix prestrain, while MDAs could do so for only several minutes, with ECM strain oscillating between the front and back (Video S5). During cell spreading, both MDAs and HT-1080s demonstrated ECM strains along cell protrusions, but unlike fibroblasts, they are not maintained (Figures S5A and S5B). MDAs showed random oscillations and high variability in shape, while HT-1080s became migratory very rapidly (within 3 h) and often transitioned between mesenchymal and ameboid phenotypes during spreading. Mesenchymal migration showed 43% greater ECM deformations compared with ameboid (axial ratio < 2 with numerous blebs; Figure 6E). Interestingly, cells transitioning to the ameboid migratory state switched from frontal matrix prestrain to equal and opposite, becoming similar to MDAs (Figures S5C and S5D).
Comparisons using a non-mesenchymal, non-malignant primary epithelial cell—human umbilical vein endothelial cells—revealed a pattern of single-cell motility that was entirely different. Cells with or without exogenous stimulation by VEGF and FGF2 displayed multiple transient protrusions but minimal ECM deformation, no evidence of local prestrain, and failure to migrate persistently (Figures 6E and S6A). These data suggest that mesenchymal HT-1080s share many, but not all, of the biophysical attributes of fibroblasts when migrating in 3D ECMs, unlike epithelial cancer and endothelial cells.
AC analyses revealed that HT-1080s demonstrate ACs in ~40% of the cells, while ACs occurred in only 10% of MDAs (Figures 7A–7C and 7F). Temporal analyses of protrusion and retraction rates indicated HT-1080s demonstrate enhanced protrusion following an AC, as for HFFs (Figure 6D). Comparisons of HFFs and HT-1080s showed that the presence of an AC in both cell types enhanced migration rates, though only HT-1080s demonstrated enhanced directionality (Figures 3B and 3C versus 7C). Together, these data indicate that both AC and matrix prestrain are part of a mesenchymal phenotype and that there are differing degrees of mesenchymal characteristics associated with different cell types.
Figure 7. Anterior contractions increase HT-1080 3D migration through control of protrusion-retraction cycles.

(A) HT-1080 cell expressing mApple α-actinin (magenta) migrating in 3D collagen (green). Time lapse (white-dashed region) illustrating a retrograde pull (yellow-dashed line) and anterograde contraction (red dashed line) of the ECM.
(B and C) Effect of an AC on cell velocity (B) and mean-squared displacement (C; N = 5, n = 41).
(D) Rate of protrusion (green), nuclear displacement (black), and rear retraction (orange) centered around HT-1080 AC initiation (N = 5, n = 10).
(E) Cross-correlation of the mean between leading-to-TE (LE-TE) and leading-edge-to-nuclear movements (LE-NUC) for HT-1080 cells.
(F) Percent cells demonstrating ACs under different conditions (N ≥ 3, n ≥ 10). Significantly different from HFF, **p ≤ 0.01; from parental (control) MDA cells. ##p ≤ 0.01.
(G) Pre- (left) and immediately post-ablation (middle) maximum intensity projections of an MDA-BR (LifeAct, magenta) cell migrating in 3D collagen (green). Kymograph shows the cell continues to migration toward cut without retraction, even though the collagen fibrils show buckling (lower insert).
(H) ablation data showing the leading edge and whole cell responses of HFF, HT-1080 and MDA-BR cells to ECM severing (N ≥ 3 n ≥ 9). **p ≤ 0.01. For (B–D), (F) and (H) mean value ±SEM shown. Scale bars, (A),10 μm; (G), 20 μm.
Strong myosin II contractility is a prerequisite for a true mesenchymal phenotype in 3D
We examined the relative amounts of MIIA and MIIB isoforms in each cell type, including myosin II expression in different MDA cell variants associated with metastatic tumors to brain or bone (Yoneda et al., 2001). As expected, HFFs expressed more MIIA and MIIB than either HT-1080s or MDA-WT (Figure S6A). Interestingly, MDAs from secondary brain tumors (MDA-BR) showed more MIIB expression while integrin patterns remained similar (data not shown). Comparisons of MDA-WT, MDA-BR, and MDA-BO 3D migration revealed no differences in overall migration velocity (Figure S6B), but MDA-BR showed a distinctly different migration pattern: cells displayed a lamellipodial pseudopod with frequent stress fibers and numerous blebs not seen in MDA-BO, which were maintained for hours (Figures S6C–S6E; Video S6). Significantly, MDA-BR cells were able to maintain an ECM prestrain at the front—though the magnitude did not change compared with controls—and a majority underwent an AC during 3D migration (~60%; Figures 7F and S6F).
To examine whether matrix prestress guided HT-1080 and MDA cell migration, we locally severed the matrix in front of migrating cells analogous to experiments performed on HFFs. MDA-BR cells were chosen due to their more consistent movement and ability to sustain ECM stress compared with the other MDA subtypes. Matrix ablation led to leading-edge retraction in the majority of HT-1080 (~77%), but not MDA-BR cells (~33%), while their migration away from the area was similar to HFFs (Figures 7G and 7H). Some MDA-BR cells (Figure 7G) showed an increased ability to displace the local matrix after ECM ablation, but that did not alter their protrusion or migration. These data suggest that a matrix prestrain is required for HT-1080 migration but is not necessary for MDA cells.
Because HT-1080 cells had integrin ligation patterns similar to HFFs, we overexpressed either eGFP-MIIA or eGFP-MIIB to test if that would enhance the contractile AC phenotype. Overexpression of MIIA or MIIB produced ~80% or 60% increases in ACs in HT0180s, respectively, and enhanced ECM prestrain (Figures 7F, 7G, and S7A–S7C; Video S7). These data suggest that MII expression is a key component of the mesenchymal phenotype, responsible for maintaining polarized ECM prestrain and AC formation during migration.
DISCUSSION
Our study provides a comprehensive characterization of the distinctive sequence of dynamic processes driving 3D cell migration by primary human fibroblasts. Based on this detailed blueprint for 3D fibroblast cell migration, we compared the 3D migration modes of two very different cancer cell types (mesenchymal versus epithelial). Their migration patterns differed from normal 3D mesenchymal migration and each other, depending on their ability to transmit sustained traction to the microenvironment. The two key elements were the relative amounts of activated integrin associated with cell adhesions to the matrix and the overall levels of expression of MIIA and MIIB isoforms. Experimentally increasing levels of MIIA (in HT-1080 cells) or MIIB (in HT-1080 and MDA-BR cells) enhanced the AC phenotype, whereas decreasing integrin ligation abolished it. An AC likely differs from the “hand-over-hand” movement of individual collagen fibrils shown by Meshel et al. (2005) in being highly dependent on MIIA, and not MIIB. Overall, our results identify a distinct mesenchymal migratory phenotype based on myosin II contractile machinery and integrins in cell adhesions, which are required for efficient cell migration in 3D microenvironments; this pattern is disrupted in malignant cells.
We emphasize that the deformations quantified by PIV represent a characterization of the local responses of the collagen matrix to cellular forces, and they are therefore only an indirect proxy for the magnitude and distribution of forces. Specifically, our data have identified evidence for focused actomyosin contractility at the anterior of the cell that results in a cone-shaped zone of matrix deformation extending ahead of the cell. We often observe much less matrix deformation visible at the rear of human fibroblasts, but because forces need to be equal and opposite, why are the latter deformations lower? The obvious additional mechanical factor is the presence of extensive cell-matrix attachments spread over the entire cell body mediated by large numbers of integrin-based cell-matrix adhesions. These many sites anchoring the cell body to the matrix will apply diffuse, general resistance or friction as the cell moves forward, representing diffuse opposing forces. Its effects are reflected in the slow, passive dragging forward of the nucleus and rear of the cell, and finally, in the relatively low collagen matrix deformation visible at the rear of the cell by PIV. We have observed a similar phenomenon in fibroblasts migrating along the upper surface of our 3D collagen gels, demonstrating a strong AC with reduced matrix deformation at the TE (data not shown).
Our findings and unified comprehensive model of 3D migration dynamics are generally consistent with previous studies focusing on descriptive subsets of cell contractile behavior in 3D environments. In fibrin gels, Owen et al. (2017) demonstrated similar contractions occurring in the main pseudopod of migrating fibroblasts, suggesting a similar AC can exist in other 3D matrices. Hetmanski et al. (2019) recently found that contraction and membrane tension at the cell rear are controlled via a caveolin/RhoA pathway with membrane tension being higher at the rear of the cell. The Friedl laboratory also demonstrated a strain stiffening of the matrix and a stiffness gradient in front of cancer cells and fibroblasts on top of, though not within, a 3Dcollagen gel (van Helvert and Friedl, 2016). They also described a pattern of 3D melanoma cell migration with collagen deformation at the LE, a delay between fiber translocation and cell movement, and intriguing oscillatory sequential movements of the cell rear (Starke et al., 2013) similar to the pattern we observed in malignant MDA-MB-231 cells and some HT-1080 cells, which contrasts markedly with the major anterior prestrain and prolonged, slow retraction of the cell rear characteristic of primary human fibroblasts. In addition, others have shown that a subset (~36%) of MDA-MB-231 cells can produce an asymmetric strain in the ECM at certain times during migration, though ECM strains mainly remained relatively symmetrical (Hall et al., 2016). Estimates of forces during MDA-MB-231 3D cell migration revealed varying asymmetries, though the images of strains appeared to indicate greater strain at the cell rear opposite to the end showing cell protrusions (Steinwachs et al., 2016). Although performed on a 2D substrate, experimental pulling and deformation of a substrate with a micro needle resulted in similar alterations in leading-edge dynamics–pulling the flexible substrate away from a cell promoted its migration toward the deformation and pushing it toward the cell resulted in withdrawal of the leading-edge protrusion within the same time frame as we observed (Lo et al., 2000). Overall, our current data demonstrate a spatiotemporal interplay of these previously described features with a specific chronology of polarized steps, plus key roles for β1 integrin ligation and myosin II during fibroblast 3D migration with alterations in cancer cells.
Is there a 3D cell migration cycle analogous to the cycle described previously for 2D migration? The cycle we describe for fibroblasts differs: within our matrix architecture, the protrusion rate of the leading-edge stalls coincident with retrograde pulling on the matrix. The next step is a local, anterior AC—as shown by the large forward movement of ECM between the leading edge and the nucleus that includes ECM fibril buckling or compression—with no involvement of the TE. After this compression, protrusion rate doubles within 12 min. TE retraction is gradual and occurs yet another 12 min later. This spatiotemporal sequence of steps with anterior contractile strain of the microenvironment followed by leading-edge protrusion and then more passive tail retraction defines a 3D mesenchymal cell migration cycle distinct from the canonical 2D cycle.
Underlying the morphological elements of the classical 2D cell migration cycle, Machacek et al. (2009) described spatiotemporal coordination between the Rho GTPases at the leading edge of migrating fibroblasts, where RhoA activation precedes Rac and Cdc42 for a temporal delay between contraction and protrusion-regulating events. Likewise, Ji et al. (2008) showed time a lag between force transduction that followed leading-edge protrusion of epithelial cells on 2D substrates. Our data reveal prolonged time lags associated with this type of coordination, which may be due to the dimensionality and nonlinear elasticity of the collagen matrix compared with rigid 2D surfaces.
The fact that we observe a localized AC in human fibroblasts indicates that they compartmentalize their contractility. There are delays between protrusion/retraction events at the front and rear, as well as temporal differences in front, nuclear, and rear cellular movements after disruption of prestrain through laser ablation. Contrary to the retraction-induced spreading model that can account for the majority of 2D cell migration (Chen, 1979), cells in 3D do not have a dorsal surface free of matrix interactions but are instead surrounded by numerous cell-matrix adhesions covering the entire cell surface. Hence, tail retractions do not lead directly to increases in protrusion. In fact, in contrast to the slingshot migration associated with 3D NIH 3T3 cell migration in a matrix of electrospun dextran methacrylate fibers (Wang et al., 2019), tail retractions of primary human fibroblasts in a 3D collagen environment did not launch cells forward or change prestrain tension in front of the LE. Our results demonstrate instead that the 3D migration pattern for fibroblasts centers around a polarized contractile event that enhances protrusion followed slowly by tail retraction, suggesting that 3D mesenchymal migration uses a “front wheel drive” with retraction that is relatively passive at the rear and notable for its slow anterior translocation, which is likely related to the extensive integrin-based cell adhesions. This front wheel drive migration was particularly evident for HT-1080 cells, where the cell body was often passively dragged behind a highly active leading edge.
Why are there large front-to-rear temporal delays in primary human fibroblast 3D cell migration? We propose it involves, at least in part, the pattern of integrin ligation. Our data here and elsewhere (Doyle et al., 2015) demonstrate that during migration, fibroblasts display active/bound (ligated) integrins at nearly every point of adhesive contact with adjacent collagen fibrils along the entire length of the cell. We speculate that many of these bound integrins, especially those distal to the leading or trailing edges, are low-strength adhesive binding sites, much like fibrillar adhesions beneath cells on 2D surfaces (Lu et al., 2020). Such adhesions would be expected to provide passive or frictional drag. This extensive binding to the ECM would not only account for delay between protrusion and retraction events but also the asymmetric strain distribution in both fibroblasts and HT-1080 cells. Reduction of integrin ligation reduced overall ECM strain and led to loss of the anterior prestrain. In contrast, in all MDA cell types we tested, integrin ligation was intrinsically exceptionally low and localized solely to lamellipodia and trailing edges. This phenotypic difference in relative integrin abundance and localization likely contributes to the overall low but relatively symmetrical ECM strains that we and others (Hall et al., 2016; Steinwachs et al., 2016) observe in this malignant cell type.
Our results suggest that prior to becoming fully motile, cells locally strain the ECM to initiate a tension gradient in a uniaxial fashion. We emphasize that our analyses for semi-quantifying strain patterns using PIV depended on our collagen polymerization conditions, which were designed with large pore size to promote migration and showed limited viscoelasticity/plasticity. When migrating HT-1080 cells could be observed to enter and leave a specific region, the collagen matrix fibrils generally reverted to their original null state (Figure S7D). Human fibroblasts, however, generated such enormous strain fields (>120 μm) that we could not establish pre-migration null states. Instead, we chemically disrupted all migrating cells to allow the matrix to return to its null state, thereby permitting measurements of local absolute matrix deformation.
Our laser ablation experiments demonstrate that once the prestrain, or tension, within the matrix is severed, cells retract their leading edge over a span of tens of minutes and subsequently fail to stabilize new protrusions in the same direction. Their leading edges slip backward as the cells undergo an AC, which also leads to further matrix buckling. These findings are consistent with the concept that tension within the matrix is required to stabilize cell adhesions, just as stiffness is needed to stabilize focal adhesions in 2D and 3D settings (Doyle et al., 2015; Schwarz and Gardel, 2012). Perhaps to initially become motile, certain cells may need to generate their own local durotactic tension gradient. Others, and we, previously demonstrated that substrate adhesion feeds forward to regulate leading-edge protrusion through the molecular clutch, which is mechanosensitive and plays a key role in establishing LE dynamics (Rothenberg et al., 2018; Doyle et al., 2012).
Human fibroblasts and HT-1080 cells create significantly larger strains leading to fibril buckling than all MDA variants. Consistent with this apparent difference in contractility, both fibroblasts and HT-1080 cells express several-fold higher MIIA levels than MDA cells, as well as an abundance of activated integrins, two key components of the 3D mechanotransduction machinery. Instead, in MDA cells the focal, point-like, weak, and fluctuating strain patterns mimic the localization of activated β1 integrins in small cell adhesions. In contrast, robust sustained matrix prestrain, ACs, relatively higher myosin II levels, and a polarized cycle of contraction-protrusion retraction that we observe are characteristic of normal mesenchymal 3D cell migration. These properties are partially reduced or lost in the two cancerous cell types we examined. Interestingly, in a fraction of the MDA-MB-231 brain-metastasizing cells shown here to express MIIB, some cells squeezed through ECM pores using an elongated pseudopod and displayed a “nuclear piston” type of migration (Petrie et al., 2014) distinct from the 3D mesenchymal migration we describe here.
Here we have established that in 3D microenvironments, fibroblasts have a migration cycle distinctly different from on a 2D surface. A “front wheel drive” mechanism directs 3D migration and is associated with a local, AC. The ability of fibroblasts to increase the local polarized tension within the microenvironment is crucial for migration and could be considered a biophysical attribute associated with a mesenchymal phenotype.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Doyle (Andrew.Doyle@nih.gov).
Materials availability
All plasmids are now available from Addgene. MYH9 and MYH10 lentivirus are available through Matthew Kutys(Matthew.Kutys@ucsf.edu). Collagen type I (in house preparation) is restricted due to low abundance.
Data and code availability
The datasets supporting the current study have not been deposited in a public repository because of overall size (>5 TB) but are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture conditions
Primary human foreskin dermal fibroblasts (HFF) were a kind gift from Susan Yamada (NIDCR/NIH) and were derived from human foreskin tissue samples provided by the Cooperative Human Tissue Network (funded by the National Cancer Institute). HT-1080 fibrosarcoma and MDA-MB-231 cells were purchased from ATCC. Brain and bone-tropic MDA-MB-231 cell lines were a kind gift from Kandice Tanner. All cells were cultured in phenol red-free DMEM (Hyclone) containing 10% fetal bovine serum (Hyclone), 100 U/ml penicillin/streptomycin (GIBCO), and 2 mM L-glutamine (GIBCO) at 37°C with 10% CO2.
The rAV-CMV-TagGFP2 LifeAct Adenoviral Vector was purchased from ibidi. For infection, 1.5×105 (HFF) or 3×105 (HT1080, MDA-MB-231) were plated in a 35mm dish with virus at a MOI of 5–10 along with 8 μg/ml polybrene (Sigma). The following day, cells were rinsed with fresh media and replated into collagen gels as described below 24–48 h after infection. CRISPR/Cas9-mediated genetic modifications were generated as previously described (Chopra et al., 2018). Stable CRISPR knockout cell lines were generated using the lentiCRISPRv2 system (Addgene plasmid #52961). Specific gRNAs (SCR: GCACTACCAGAGCTAACTCA, MYH9: TCAAGGAGCGTTACTACTCA, MYH10: TGGATTCCATCAGAACGCCA) were cloned into the BsmBI site of plentiCRISPRv2. Individual gRNA-containing plentiCRISPRv2 plasmids were co-transfected with pVSVG, pRSV-REV, and pMDLg/pRRE packaging plasmids into HEK-293T cells using calcium phosphate transfection. After 48 h, viral supernatants were collected, concentrated using PEG-IT viral precipitator (SBI), and re-suspended in PBS. HFFs were transduced in culture overnight and media was replaced the following morning. 48 h post infection; cells were selected with 2 μg/ml puromycin for 3–4 days. eGFP-MIIA or eGFP-MIIB (from Robert Adelstein) was expressed in HFFs by lentiviral transduction. eGFP-MIIB was cloned between XbaI/SalI sites in pLenti CMV GFP Puro (Addgene #17448). eGFP-MIIA (from Robert Adelstein) was cloned into a modified pRRL vector between MluI/NheI sites. Generation of lentiviral particles and cell transduction was performed as described above. pmApple-paxillin was from Michael Davidson (Florida State University). EGFP-α-actinin was gift from Benjamin Geiger. pmApple α-actinin was generated by subcloning from pmApple-paxillin using Hindi II and Xba1 sites. Plasmids were transfected into fibroblasts by electroporation using a Bio-Rad Gene Pulsar TM at 170V, 960 μFd external capacitance and a time constant of 17–22 μs in 0.4 cm gap cuvettes.
METHOD DETAILS
Activation of glass imaging chambers
Glass-bottomed dishes (MatTek Corp., #1.5 thickness coverglass, 20 mm imaging area) were acid-washed with 68% nitric acid (Fisher Scientific) for 25 min, rinsed under a continuous flow of dH2O for 1 h, treated with 200 mM NaOH for 15 min, rinsed twice with dH2O, then dried under forced air and kept covered until needed. Triethoxysilylbutraldehyde (Gelest Inc.) was diluted to 2% in 100% ethanol and then added to glass surfaces and incubated for 5 min. This silane solution was aspirated then rinsed 2X with 100% ethanol and once with dH2O. Surfaces were then blown dry with forced air and cured at 65°C for 2 h, and finally stored desiccated at 4°C.
PVA blocking of glass coverslips
To locally deter collagen attachment to the center of the 20 mm imaging area, custom-made O-rings (inner diameter of 10 mm and outer diameter of 16 mm) were mechanically punched from Press-to-Seal silicone sheets (Invitrogen). An O-ring was centered within each 20 mm imaging area and firmly pressed in place, creating a watertight seal. A marker was used on the underside of the dish to outline the inner edge of the washer for reference while imaging. PVA (molecular weight 98,000; 98% hydrolyzed; Sigma-Aldrich) was diluted in H2O to a 6.5% stock solution. This mixture was solubilized at 90°C in a water bath and was immediately 0.2-μm filtered to remove impurities.1,124 μl 2N HCl was added to 8,876 μl of the PVA solution (~6% PVA). 200–400 μl of the PVA solution was added to the center of each washer and incubated in a covered humid container for 40 min. The solution was gently aspirated from the surface and washed 3X with dH2O. The aldehyde-hydroxyl bonds were reduced by treatment with 800 μg/ml NaBH4 in 200 mM ethanolamine buffer for 8 min. After rinsing 3X with dH2O, the silicone washers were removed, and the dishes were kept in an enclosed humid environment and used within 48 h. Cells were only imaged within the 10 mm unattached region to rule out boundary effect issues.
Collagen gel formation
Rat tail collagen was prepared in the lab using methods similar to Chandrakasan et al. (1976). Briefly, rat tail tendons were dissected out under a dissecting microscope, taking special care to remove fragments of bone, cartilage, blood vessels, and even the tendon sheaths, all of which can contribute “contaminating” collagens (II, III, IV, etc.) and other ECM components (fibronectin, proteoglycans), which can cause lot-to-lot variation. Tendon fibers were then suspended in 0.5 M acetic acid and stirred at 4°C for 48 h. The resulting solution of solubilized collagen was filtered through several layers of gauze and centrifuged at 14,000 × g for 1 h. The supernatant was collected and then dialyzed against three changes of acidic acid (0.02 M) at 4°C over a three-day period. The final viscous solution (~5 mg/ml) is transparent, can be stored at 4°C for at least 2 years without any noticeable changes when tested in various cell-based assays, and has been shown to support >95% cell viability when diluted with 10x culture medium and neutralized with NaOH. Collagen protein concentration was determined by comparing pre and post weights following lyophilization of 5–6 tubes containing 1 ml of solution and was confirmed with a Sircol collagen assay kit (Biocolor).
A collagen I stock solution was generated on ice by mixing rat tail collagen I (6.03 mg/ml) with 10X DMEM (Sigma) and 10X reconstitution buffer (200 mM HEPES, 262 mM NaHCO3) in a 10:1:1 ratio. The pH was then adjusted to 7.4 with 1N NaOH. PBS++ (PBS containing both calcium and magnesium) and cells (5.0×105 cells/ml collagen) were finally added to bring the final gel concentration to 3.0 mg/ml. 150 μl of the gel was added to a 35 mm MatTek dish (20 mm glass, #1.5 thickness). All collagen gels were polymerized on an Echotherm chilling/heating dry bath (Torrey Pines Scientific) 12°C, to create a fibrillar-bundled collagen matrix (Doyle et al., 2015). Gel polymerization times were approximately 45 min, after which all gels were allowed to reach room temperature (~22°C) for 10 min before medium at the same temperature was added.
Fluorescent labeling of collagen
Fluorescently labeled collagen gels were previously described (Doyle, 2018; Doyle, 2016). Briefly, 5ml of a 3 mg/ml collagen I solution was polymerized at room temperature. After gelation, the gel was incubated with 50mMborate buffer (pH 9.0) for 10 min. The borate solution was aspirated and replaced by 5 ml of either an Atto-488 or Atto-565 NHS-ester dye solution in the same buffer and incubated at room temperature in the dark for 1 hour. The concentration of dye (diluted in DMSO) was adjusted to a 3-molar excess as recommended by AttoTech. The dye solution was aspirated, and any remaining NHS-esters were quenched with 10 ml of 50 mM TRIS buffer (pH 7.5) for 10 min. The gel was then rinsed 6–10 times with PBS++ over several hours. Gels were then acidified in 200mMHCl and stirred until the gel was completely solubilized. The collagen solution was then dialyzed against 20mMglacial acetic acid (Fisher Scientific) for 4 h at a 1:1000 ratio. Collagen concentration was measured using a Sircol collagen assay kit. A fluorescent collagen stock solution was created and mixed in bulk (6 ml at a time) for consistency. 2–4% of the unlabeled collagen I solution was removed and replaced with the same amount of labeled collagen (calculated based on protein weight). The final protein concentration of this fluorescent collagen I stock solution was then calculated and used for all experiments.
Immunofluorescence staining
All fixation and permeabilization steps were performed at 37°C. Cells were permeabilized and fixed in 3% paraformaldehyde (Electron Microscopy Sciences), 0.5% Triton X-100 (Sigma), with 10 μg/ml nonfluorescent phalloidin (Invitrogen) in cytoskeletal buffer (CBS: 10 mM MES, 138 mM KCl, 2 mM EGTA, 3 mM MgCl2 plus 5% sucrose) for 90 seconds, post-fixed in 4% paraformaldehyde in CBS for 15 min. Cells were rinsed 3X in PBS++ and permeabilized with 0.5% Triton X-100 in CBS for 5 min. Cells were rinsed 5X over 40 min with PHEM+glycine (60 mM PIPES, 2 mM HEPES, 10 mM EGTA, 2 mM MgCl2, 100 mM glycine, pH 6.9). Non-specific sites were blocked with 20% donkey serum (Jackson ImmunoResearch Laboratories), together with M.O.M. reagent (Vector Laboratories), in PHEM+glycine buffer for 1 h. Cells were rinsed 3X with PHEM+glycine over 30 min. For MDA-MB-231 cells, the combined fixation and permeabilization first step was skipped. Primary and secondary antibodies were diluted in PHEM+glycine with 10% donkey serum and incubated for 45 and 25 min, respectively. Secondary antibodies were from Jackson Immuno Research Laboratories.
Live-cell microscopy
For all live-cell fluorescence imaging experiments, FluoroBrite DMEM (GIBCO) with 10% FBS was used and supplemented with a 1:100 ratio of Oxyfluor (Oxyrase) with 10 mM DL-lactate (Sigma-Aldrich) as a substrate to reduce photobleaching and phototoxicity. Cells were imaged with a modified Yokogawa spinning-disk confocal scan head (CSU-21: modified by Spectral Applied Research, Inc.) on an automated Olympus IX-81 microscope using a 30X SAPO-Chromat silicone oil objective (N.A. 1.15) to reduce spherical aberration in 3D. A custom laser launch (built by A.D.D.) equipped with 445 nm (80 mW: Vortran Laser Technology), 488 nm (150 mW: Coherent), 514 nm (150 mW: Coherent), 568 nm (100 mW: Coherent), and 642 nm (110mW: Vortran Laser Technology) diode lasers supplied excitation wavelengths. A Gooch and Housego AOTF controlled shuttering and intensity for 488, 514, and 568 lines. 445 and 642 lines were shuttered and intensity controlled via TTL and direct voltage steps, respectively. The primary dichroics (442/568/647 and 405/488/568/647) were from Semrock (Rochester, NY). Images were captured using a backthinned EM CCD camera in 16-bit format using the 10 MHz digitization setting (Photometrics). EM gain was set between 400–600 (3X) with exposure times between 70–150 ms per image taken every 3 min for up to 6 h. Alternatively, a Prime 95B back-thinned CMOS camera (Photometrics) was used in 16-bit mode. A motorized Z-piezo stage was used to rapidly capture Z-stacks every 2 microns over a Z-distance of 60 microns (ASI, Eugene, OR). An environmental chamber surrounding the microscope maintained cells at a constant 37°C, with 10% CO2 and approximately 50% humidity (Precision Plastics, Beltsville MD). All components were controlled with MetaMorph imaging software (Molecular Devices, Downington, PA). Alternatively, in some cases a Zeiss LSM 880 with Fast AiryScan in super-resolution mode was used. A 40x (1.2 NA) water immersion objective was used with 488 nm, 561 nm and 633nm laser lines to illuminate TAGGFP2-lifeact, Atto565-labeled collagen I and SiR-DNA, respectively. The system was controlled with Zen Black software version 2.3.
Two-photon ablation
For two-photon ablation experiments, a Nikon A1R HD MP system was used (Nikon Instruments. Imaging used a 40X (1.15 N.A.) water immersion objective and 488 nm (0.5–1.5%) and 561 nm (1–2%) laser lines to illuminate TAGGFP2-LifeAct and Atto565-labeled collagen, respectively, using galvano mode and bidirectional scanning at 512X512. NIS-Elements (Nikon) controlled all equipment. Prior to imaging, a line ROI was created approximately 25–35 microns in front of the migrating cell. Using ND acquisition functions, three sequences were configured:1) a pre-ablation timelapse (0.5 micron Z spacing over 80 microns, 5-minute delay for 30 min), 2) an ablation sequence in multiple dimensions (single timepoint). and 3) a post-ablation sequence (0.5 micron Z spacing over 80 microns, 5-minute delay for 3 h). The Z-ablation sequence consisted of moving the Z position 40 microns below the cell’s Z-position, then using 70% power at 800 nm scanned 50 times for 10 seconds. This sequence was repeated every 5 microns in Z over an 80-micron range (16 times) before the timelapse was reinitiated. For the single Z-position cuts shown in Figures 2F and 2G, imaging was performed in resonant scanning mode at 7.5 frames/sec with a similar Z spacing but over only 20 microns. Images were collected every 3 seconds for 2–3 min using a similar routine in ND sequence. Kymographs were created and distances were measured at 40 seconds after ablation. For cell severing experiments, power was increased to 80% and a single point was chosen and ablated for 2 seconds. Imaging was performed in resonant mode. Using ND sequence Z stacks (0.5 micron spacing, 20 microns total) were taken every 10 seconds for 2 min, then a single Z-plane was imaged during ablation at 7.5 frames/sec for 2 min followed by Z stacks every 10 seconds for an additional 20 min. Kymographs were created and distances were measured at 2 and 15 seconds after the 2-second ablation.
Widefield time-lapse imaging
Widefield time-lapse phase contrast images were recorded on a microscope (Axiovert 135TV; Carl Zeiss, Inc.) fitted with a motorized XY- and Z-stage focus drive (Ludl Electronic Products Ltd.) using an enhanced contrast Plan-Neofluar 10 ×0.3 NA, a long working-distance Plan-Neofluar Korr 20 × 0.4 NA, or a long working-distance Plan-Neofluar Korr 40 × 0.6 NA phase objective (Carl Zeiss, Inc.). Images were acquired with a Flash 4 V2 CMOS camera set to 4X4 binning (Hamamatsu Photonics). MetaMorph imaging software was used to acquire images and control all hardware. A custom environmental chamber (Lucite) enclosed the microscope and maintained cells at 37°C with 10% CO2. A red filter (high-pass 600 nm) was used to block lower wavelengths of light during experiments using blebbistatin or Y-27632.
Particle image velocimetry
For particle image velocimetry imaging, images were collected in one of two ways: 1) For cell spreading, the first image acquired prior to spreading was used as what we define as the “Null” image, where the ECM was undisplaced or relaxed. 2) For cell migration assays, 3D 3-color data sets were first collected every 3–5 min (cell-type-dependent) over several hours (2–6 h), after which a 2X solution consisting of 10 mM EDTA and 1% Triton X-100 was added to disrupt cell-ECM adhesions and lyse the cell membranes. After 10 min at 37°C, the same XY position was used but a larger Z stack was acquired to account for Z-direction drift and was considered the “Null” image.
After image acquisition, each image stack was automatically aligned using the Fiji macro Correct 3D drift: briefly, the ECM channel was used for alignment and the pixel intensity was set to 7500. Following alignment, maximum intensity projections were created of the 4D ECM stacks (XYZ and time). Null frames were interlaced between timepoints, and stacks were converted to individual TIFF files. These files were then processed using the MATLAB (2018b) plugin PIVLAB v2.02 (ref) with the following settings; PIV algorithm: FFT window deformation; passes 1–4 were 128 (50% step), 64, 32, 16, respectively; window deformation interpolator was linear; subpixel estimator was Gauss 2X3-point.; 5X repeated correlation. PIVLAB text files were created and the 90th percentile deformations in microns were calculated. Some cells were not included in experiments based on our criteria: 1) aberrant matrix deformations not associated with cell migration and 2) observable local changes to matrix that were consistent with local matrix crosslinking by the cell.
QUANTIFICATION AND STATISTICAL ANALYSIS
Prism 4 by GraphPad software was used for all graphs and statistical analysis. One-way ANOVA, using a Tukey post-test for more than two data sets or Mann-Whitney t tests were used to establish statistically significant differences (P<0.05). All error bars indicate standard error of the mean. In figure legends, N = the number of independent experiments, and n = the number of data points.
Characterization of the anterior contraction
To accurately determine whether a cell demonstrated an anterior uniaxial contraction during 3D migration the following procedure was used: After live-cell imaging of TagGFP2 LifeAct expressing cell together with the fluorescent collagen matrix and the cell nucleus (using SIR-DNA), 2D maximum intensity projection (MIP) stacks were generated. Timepoints were then projected together to determine cell directionality and whether the cell was migratory or not. For HFFS and HT-1080, migration was considered if the cell moved one cell nucleus over 1 hr. For MDA-s it was half this distance. Line scans were drawn on the time projected image along the direction of migration, often multiple were used when migration was non directional. These regions of interest (ROI) were transferred to the 2D time stack and kymographs were generated of the all three imaging channels. From these kymographs it was determined by examining the ECM channel if an anterograde pull of the ECM near the leading edge followed a retrograde pull and was observed by the ECM being “pinched” together. The combination of an RP and a subsequent AP constituted the full anterior contraction and was counted. Figure S3 and Video S2 show examples of this.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| β1 integrin | Ken Yamada (NIDCR) | Clone mAb13 |
| β1 integrin | BD Biosciences | Clone 9EG7 |
| Alexa 647-Donkey anti Rat IgG | Jackson ImmunoResearch | 712-606-150 |
| Bacterial and virus strains | ||
| lentiCRISPRv2 system | Addgene | 52961 |
| rAV-CMV-TagGFP2 LifeAct Adenoviral | Ibidi | 60121 |
| Vector | ||
| EGFP-α-actinin plasmid | Geiger (Weizmann Institute) | N/A |
| pmApple-paxillin plasmid | Mike Davidson (Florida State Univ.) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Collagen I | A.Doyle and Greg Kitten (NIDCR) | N/A |
| Blebbistatin | Millipore | 203391 |
| Y27632 | Millipore | 509228 |
| DMSO | Sigma-Aldrich | D2650 |
| Oxyfluor | Fisher Scientific | 50-300-2016 |
| DL-Lactate | Sigma-Aldrich | L1375 |
| Experimental models: cell lines | ||
| Human foreskin fibroblasts (HFF) | Susan Yamada (NIDCR) | N/A |
| HT-1080 | ATCC | CCL-121 |
| MDA-MB-231 | ATCC | HTB-26 |
| MDA-MB-231 Parental | Kandice Tanner (NCI,NIH) | N/A |
| MDA-MB-231 Bone | Kandice Tanner (NCI,NIH) | N/A |
| MDA-MB-231 Brain | Kandice Tanner (NCI,NIH) | N/A |
| Software and algorithms | ||
| Fiji/ImageJ 2 | NIH | N/A |
| PIVLab | MatLab/William Thielicke | N/A |
| Prism 8 | GraphPad | N/A |
| MetaMorph | Molecular Devices | N/A |
| NIS-Elements | Nikon | N/A |
| Imaris | Oxford Instruments | N/A |
| Other | ||
| Atto-565 NHS ester dye | Sigma | 72464 |
| Atto-488 phalloidin | Sigma | 49409 |
| Phalloidin | Sigma | P2141 |
| SiR-DNA | Cytoskeleton | CY-SC007 |
Highlights.
A matrix prestrain propels fibroblasts and cancer cells through 3D environments
Anterior contractions sustain the matrix prestrain through enhanced cell protrusion
Both require high myosin II contractility and substantial integrin ligation
The unbalanced front ECM prestrain is highly cell type dependent
ACKNOWLEDGMENTS
We thank Shayan Nazari and David Cruz Walma for critical reading of the manuscript, Kandice Tanner for helpful suggestions, and the NIDCR Imaging Core (ZIC-DE000750-01). This research was supported by the Intramural Research Program of the NIDCR, NIH.
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflict of interest.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.devcel.2021.02.017.
REFERENCES
- Chandrakasan G, Torchia DA, and Piez KA (1976). Preparation of intact monomeric collagen from rat tail tendon and skin and the structure of the nonhelical ends in solution. J. Biol. Chem 251, 6062–6067. [PubMed] [Google Scholar]
- Charras G, and Sahai E (2014). Physical influences of the extracellular environment on cell migration. Nat. Rev. Mol. Cell Biol 15, 813–824. [DOI] [PubMed] [Google Scholar]
- Chen WT (1979). Induction of spreading during fibroblast movement. J. Cell Biol 81, 684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WT (1981). Surface changes during retraction-induced spreading of fibroblasts. J. Cell Sci 49, 1–13. [DOI] [PubMed] [Google Scholar]
- Chopra A, Kutys ML, Zhang K, Polacheck WJ, Sheng CC, Luu RJ, Eyckmans J, Hinson JT, Seidman JG, Seidman CE, et al. (2018). Force generation via beta-cardiac myosin, titin, and alpha-actinin drives cardiac sarcomere assembly from cell-matrix adhesions. Dev. Cell 44, 87–96.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukierman E, Pankov R, Stevens DR, and Yamada KM (2001). Taking cell-matrix adhesions to the third dimension. Science 294, 1708–1712. [DOI] [PubMed] [Google Scholar]
- Doyle AD (2016). Generation of 3D collagen gels with controlled diverse architectures. Curr. Protoc. Cell Biol 72, 10.20.1–10.20.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle AD (2018). Fluorescent labeling of rat-tail collagen for 3D fluorescence imaging. Bio Protoc 8, e2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle AD, Carvajal N, Jin A, Matsumoto K, and Yamada KM (2015). Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat. Commun 6, 8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle AD, Kutys ML, Conti MA, Matsumoto K, Adelstein RS, and Yamada KM (2012). Micro-environmental control of cell migration–myosin IIA is required for efficient migration in fibrillar environments through control of cell adhesion dynamics. J. Cell Sci 125, 2244–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuChez BJ, Doyle AD, Dimitriadis EK, and Yamada KM (2019). Durotaxis by human cancer cells. Biophys. J 116, 670–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton SL, and Waterman-Storer CM (2006). Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell 125, 1361–1374. [DOI] [PubMed] [Google Scholar]
- Hall MS, Alisafaei F, Ban E, Feng X, Hui CY, Shenoy VB, and Wu M (2016). Fibrous nonlinear elasticity enables positive mechanical feedback between cells and ECMs. Proc. Natl. Acad. Sci. USA 113, 14043–14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetmanski JHR, de Belly H, Busnelli I, Waring T, Nair RV, Sokleva V, Dobre O, Cameron A, Gauthier N, Lamaze C, et al. (2019). Membrane tension orchestrates rear retraction in matrix-directed cell migration. Dev. Cell 51, 460–475.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewska M, Primi MC, and Izard T (2020). Cell adhesion in cancer: beyond the migration of single cells. J. Biol. Chem 295, 2495–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji L, Lim J, and Danuser G (2008). Fluctuations of intracellular forces during cell protrusion. Nat. Cell Biol 10, 1393–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kechagia JZ, Ivaska J, and Roca-Cusachs P (2019). Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol 20, 457–473. [DOI] [PubMed] [Google Scholar]
- Krause M, and Gautreau A (2014). Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol 15, 577–590. [DOI] [PubMed] [Google Scholar]
- Kubow KE, Conrad SK, and Horwitz AR (2013). Matrix microarchitecture and myosin II determine adhesion in 3D matrices. Curr. Biol 23, 1607–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, Keller M, Förster R, Critchley DR, Fässler R, and Sixt M (2008). Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453, 51–55. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, and Horwitz AF (1996). Cell migration: a physically integrated molecular process. Cell 84, 359–369. [DOI] [PubMed] [Google Scholar]
- Lo CM, Wang HB, Dembo M, and Wang YL (2000). Cell movement is guided by the rigidity of the substrate. Biophys. J 79, 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Doyle AD, Shinsato Y, Wang S, Bodendorfer MA, Zheng M, and Yamada KM (2020). Basement membrane regulates fibronectin organization using sliding focal adhesions driven by a contractile winch. Dev. Cell 52, 631–646.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, and Danuser G (2009). Coordination of Rho GTPase activities during cell protrusion. Nature 461, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekhdjian AH, Kai F, Rubashkin MG, Prahl LS, Przybyla LM, McGregor AL, Bell ES, Barnes JM, DuFort CC, Ou G, et al. (2017). Integrin-mediated traction force enhances paxillin molecular associations and adhesion dynamics that increase the invasiveness of tumor cells into a three-dimensional extracellular matrix. Mol. Biol. Cell 28, 1467–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshel AS, Wei Q, Adelstein RS, and Sheetz MP (2005). Basic mechanism of three-dimensional collagen fibre transport by fibroblasts. Nat. Cell Biol 7, 157–164. [DOI] [PubMed] [Google Scholar]
- Owen LM, Adhikari AS, Patel M, Grimmer P, Leijnse N, Kim MC, Notbohm J, Franck C, and Dunn AR (2017). A cytoskeletal clutch mediates cellular force transmission in a soft, three-dimensional extracellular matrix. Mol. Biol. Cell 28, 1959–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek SP, Huttenlocher A, Horwitz AF, and Lauffenburger DA (1998). Physical and biochemical regulation of integrin release during rear detachment of migrating cells. J. Cell Sci 111, 929–940. [DOI] [PubMed] [Google Scholar]
- Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, and Horwitz AF (1997). Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 385, 537–540. [DOI] [PubMed] [Google Scholar]
- Petrie RJ, Gavara N, Chadwick RS, and Yamada KM (2012). Nonpolarized signaling reveals two distinct modes of 3D cell migration. J. Cell Biol 197, 439–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Koo H, and Yamada KM (2014). Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science 345, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov SV, Pasapera AM, Sabass B, and Waterman CM (2012). Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 151, 1513–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkawitz J, Kopf A, Stopp J, de Vries I, Driscoll MK, Merrin J, Hauschild R, Welf ES, Danuser G, Fiolka R, and Sixt M (2019). Nuclear positioning facilitates amoeboid migration along the path of least resistance. Nature 568, 546–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching KM, Cox BL, Salick MR, Pehlke C, Riching AS, Ponik SM, Bass BR, Crone WC, Jiang Y, Weaver AM, et al. (2014). 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys. J 107, 2546–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, and Horwitz AR (2003). Cell migration: integrating signals from front to back. Science 302, 1704–1709. [DOI] [PubMed] [Google Scholar]
- Rothenberg KE, Scott DW, Christoforou N, and Hoffman BD (2018). Vinculin force-sensitive dynamics at focal adhesions enable effective directed cell migration. Biophys. J 114, 1680–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, and Marshall CJ (2003). Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol 5, 711–719. [DOI] [PubMed] [Google Scholar]
- Schwarz US, and Gardel ML (2012). United we stand: integrating the actin cytoskeleton and cell-matrix adhesions in cellular mechanotransduction. J. Cell Sci 125, 3051–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafqat-Abbasi H, Kowalewski JM, Kiss A, Gong X, Hernandez-Varas P, Berge U, Jafari-Mamaghani M, Lock JG, and Strömblad S (2016). An analysis toolbox to explore mesenchymal migration heterogeneity reveals adaptive switching between distinct modes. eLife 5, e11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke J, Maaser K, Wehrle-Haller B, and Friedl P (2013). Mechanotransduction of mesenchymal melanoma cell invasion into 3D collagen lattices: filopod-mediated extension-relaxation cycles and force anisotropy. Exp. Cell Res 319, 2424–2433. [DOI] [PubMed] [Google Scholar]
- Steinwachs J, Metzner C, Skodzek K, Lang N, Thievessen I, Mark C, Münster S, Aifantis KE, and Fabry B (2016). Three-dimensional force microscopy of cells in biopolymer networks. Nat. Methods 13, 171–176. [DOI] [PubMed] [Google Scholar]
- Taufalele PV, VanderBurgh JA, Muñoz A, Zanotelli MR, and Reinhart-King CA (2019). Fiber alignment drives changes in architectural and mechanical features in collagen matrices. PLoS One 14, e0216537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helvert S, and Friedl P (2016). Strain stiffening of fibrillar collagen during individual and collective cell migration identified by AFM nanoindentation. ACS Appl. Mater. Interfaces 8, 21946–21955. [DOI] [PubMed] [Google Scholar]
- Wang WY, Davidson CD, Lin D, and Baker BM (2019). Actomyosin contractility-dependent matrix stretch and recoil induces rapid cell migration. Nat. Commun 10, 1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, and Friedl P (2013). Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol 201, 1069–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, and Sixt M (2019). Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol 20, 738–752. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Williams PJ, Hiraga T, Niewolna M, and Nishimura R (2001). A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J. Bone Miner. Res 16, 1486–1495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study have not been deposited in a public repository because of overall size (>5 TB) but are available from the corresponding author on request.