

Abstract
Exposure to pyrethroids, a popular insecticide class that targets voltage-gated Na+ (Nav) channels, has been correlated to an increase in diagnosis of neurodevelopmental disorders, such as attention deficit hyperactive disorder (ADHD), in children. Dysregulation of medium spiny neurons (MSNs) firing in the nucleus accumbens (NAc) is thought to play a critical role in the pathophysiology of ADHD and other neurodevelopmental disorders. The Nav1.6 channel is the primary molecular determinant of MSN firing and is sensitive to modification by pyrethroids. Building on previous studies demonstrating that deltamethrin (DM), a commonly used pyrethroid, leads to use-dependent enhancement of sodium currents, we characterized the effect of the toxin on long-term inactivation (LTI) of the Nav1.6 channel, a parameter known to affect neuronal firing, and characterized changes in MSN intrinsic excitability. We employed whole-cell patch-clamp electrophysiology to measure sodium currents in HEK-293 cells stably expressing Nav1.6 channels and intrinsic excitability of MSNs in the brain slice preparation. We found that in response to repetitive stimulation acute exposure to 10 μM DM potentiated a build-up of residual sodium currents and modified availability of Nav1.6 by inducing LTI. In the NAc, DM modified MSN intrinsic excitability increasing evoked action potential firing frequency and inducing aberrant action potentials with low amplitude and depolarized voltage threshold, phenotypes that could be explained by DM induced changes on the Nav1.6 channel. These results provide a potential initial mechanism of toxicity of DM that could lead to disruption of the NAc circuitry overtime, increasing the risk of ADHD and other neurodevelopmental disorders.
Keywords: deltamethrin, voltage-gated sodium channels, medium spiny neurons
Graphical Abstract
1. Introduction
Investigations into the underlying causes of neurodevelopmental disorders remain at the forefront of neuroscience research. In recent years, exposure to environmental toxicants, particularly insecticides, has emerged as a risk factor for neurodevelopmental delay and disorders such as attention deficit hyperactivity disorder (ADHD) and autism spectrum disorder (ASD) (Hisada et al., 2017; Lanphear, 2015; Quiros-Alcala et al., 2015; Richardson et al., 2015; Schettler, 2001; Shelton et al., 2014; Viel et al., 2017). This is especially relevant as pesticides remain within the top ten substances most frequently reported in both pediatric and adult exposures by the American Association of Poison Control Centers (Gummin et al., 2017). Pyrethroids are a class of insecticides derived from the natural pyrethrins found in chrysanthemum flowers (Busvine, 1960). This class of insecticides gained widespread use and popularity due to the Food Quality Protection Act of 1996 which led the U.S Environmental Protection Agency to phase out their perceived more toxic predecessor organophosphates (Power and Sudakin, 2007; Sudakin et al., 2007). Pyrethroids were intended as a safe alternative to organophosphates; however, recent studies have revealed adverse effects, especially in children (Hisada et al., 2017; Oulhote and Bouchard, 2013; Ray and Fry, 2006; Richardson et al., 2015; Viel et al., 2017). Early-life exposure to pyrethroids has been linked to neurodevelopment disorders like ADHD and ASD (Hisada et al., 2017; Richardson et al., 2015; Shelton et al., 2014; Viel et al., 2017). To begin investigating the possible causative links between pyrethroid exposure and the development of neurodevelopmental disorders, it is important to understand the initial injury to neurons induced by these pesticides.
Pyrethroids exert their toxicity in insects by modifying voltage-gated Na+ (Nav) channels (Davies et al., 2007; Du et al., 2015; James et al., 2017; Magby and Richardson, 2015; Motomura and Narahashi, 2001; Oliveira et al., 2013; Reilly et al., 2006; Soderlund et al., 2017; Tan et al., 2005; Zhorov and Dong, 2017). There are nine mammalian Nav channel isoforms (Nav1.1-Nav1.9) which are vital for the initiation and propagation of action potentials in excitable cells such as neurons and cardiomyocites (Goldin et al., 2000). Despite amino acid sequence differences, all Nav channels similarly respond to changes in membrane potential with conformational changes that correspond to three distinct functional states- open, closed, and inactivated (Catterall, 2000; Catterall et al., 2005; Yarov-yarovoy et al., 2001). Depolarization of the membrane potential triggers a conformational change of the Nav channel that leads to its transition from the closed to open state allowing the flow of sodium into the cell. In neurons, activation of Nav channels drives the rising phase of the action potential and is quickly followed by the transition to the inactivated state (open-state inactivation). Other forms of Nav channel inactivation have also been reported such as closed-state (steady-state) inactivation and long-term (slow) inactivation, the latter of which develops in response to repetitive stimulation over a prolonged period of time (Dover et al., 2010; Goldin, 2003; Hampl et al., 2016; Silva and Ruben, 2014; Vilin and Ruben, 2001). While both the closed and the inactivated states are non-conductive and stop the flow of sodium ions into the cell, they are mediated by distinct channel conformations with different ligand binding affinity for natural toxins (Catterall, 2000; Catterall et al., 2008, 2005; Goldin et al., 2000; Karoly et al., 2010; Wu et al., 2018; Yarov-yarovoy et al., 2001).
Pyrethroids directly bind to Nav channels which affects both activation and inactivation, resulting in a net increase in the flow of sodium ions into the cell, excitotoxicity, and eventual insect paralysis and death (Catterall, 2000; Du et al., 2015; James et al., 2017; Shafer et al., 2008). Although purportedly safe in humans, pyrethroids are able to modify human Nav channels due to the high evolutionary conservation between insect and mammalian Nav channels (Du et al., 2015; Kaneko, 2011; Oliveira et al., 2013; Vais et al., 2001; Zhorov and Dong, 2017).
Deltamethrin (DM), a popular and widely used pyrethroid, has been shown to alter the function of multiple Nav isoforms (Du et al., 2015; James et al., 2017; Oliveira et al., 2013; Tan et al., 2005). Disruption of these channels is associated with a variety of neuropsychiatric disorders (Y. Liu et al., 2019; Meisler and Kearney, 2005; Olson et al., 2015; Trudeau et al., 2006). For example, a mutation in Nav1.6 has been implicated in cognitive and behavioral deficits and ADHD (Trudeau et al., 2006). Nav1.6 is highly expressed in nucleus accumbens (NAc) medium spiny neurons (MSN), a neuronal population characterized by repetitive firing properties and vulnerability to excitotoxicity (Dichter et al., 2012; Gerfen and Surmeier, 2011; Lee et al., 2016; Rikani et al., 2014; Saxena and Caroni, 2011; Surmeier et al., 2007). In MSNs, Nav1.6 is concentrated in the soma and at the axonal initial segment, a specialized region of the axon which functions as the point of initiation for the action potential (Ali et al., 2018; Wang et al., 2017). In vivo gene silencing of the mRNA coding for Nav1.6 in the NAc leads to a significant decrease in persistent sodium current and intrinsic firing (Scala et al. 2018) in MSNs, indicating that Nav1.6 is the primary determinant of excitability in these cells. Additionally, disruption of repetitive firing in MSNs has been implicated in a variety of neuropsychiatric disorders including ADHD which have been linked to DM exposure (Francis and Lobo, 2017; Lanphear, 2015; Quiros-Alcala et al., 2015; Shelton et al., 2014; Venkataraman et al., 2017). Furthermore, DM exposure has been reported to induce changes in expression of MSN proteins important for NAc signaling (Magby and Richardson, 2017; Richardson et al., 2015), suggesting that these cells are especially sensitive to pyrethroid exposure.
Therefore, we chose to study Nav1.6 channel isoform and MSNs, as Nav1.6 is the molecular determinant of excitability in this highly relevant cell population. While electrophysiological studies have reported strong effects of DM on both persistent and tail current densities (James et al., 2017) as well as steady-state inactivation mediated by Nav1.6, findings that corroborate previous evidence (He and Soderlund, 2017, 2011; Soderlund et al., 2017; Tan and Soderlund, 2010), there remain unanswered questions on the role of DM in long-term inactivation (LTI) and the effect on MSN intrinsic excitability. Examining both LTI and changes to intrinsic excitability could potentially provide insights into the initial mechanism of injury in relevant neuronal populations like MSNs with repetitive firing properties following toxin exposure. Here, we utilized whole-cell patch-clamp electrophysiology to characterize effects of DM on Nav1.6 LTI and on intrinsic excitability of MSNs.
2. Materials and Methods
2.1. Chemicals
Deltamethrin (ab141019 >98% purity, ABCam, Cambridge, MA) aliquots of 100mM were stored at −20°C following suspension in DMSO (Sigma-Aldrich, St. Louis, MO). To achieve the final bath solution concentration of 0.01% DMSO or 10μM DM, aliquots were thawed and added to the solution on the experimental day. The concentration of DM used in this study was chosen on the basis of previous studies (James et al., 2017), and overt cytotoxicity at this concentration is not observed until 48hrs of exposure (Magby and Richardson, 2015), well after our exposure time of 1hr.
2.2. Animals
C57BL/6J male mice aged 30 days were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were housed in the University of Texas Medical Branch vivarium. The University of Texas Medical Branch operates in compliance with the United States Department of Agriculture Animal Welfare Act, the NIH Guide for the Care and Use of Laboratory Animals, the American Association for Laboratory Animal Science, and Institutional Animal Care and Use Committee approved protocols.
2.3. Electrophysiology
A PC-10 vertical Micropipette Puller (Narishige International Inc., East Meadow, NY) was used to make recording electrodes with resistances of 3–8 MΩ from borosilicate glass pipettes (GC150F-10, Harvard Apparatus, Holliston, MA). Recordings were obtained using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA), where both membrane capacitance and series resistance were estimated using the dial settings on the amplifier, and both capacitive transients and series resistances were compensated by 70–80%. Data acquisition and filtering occurred at 20 kHz and 5 kHz, respectively, before digitization and storage. Clampex 9 software (Molecular Devices) was used to set experimental parameters, and electrophysiological equipment was interfaced to this software using a Digidata 1200 analog–digital interface (Molecular Devices).
2.3.1. HEK-293 cell electrophysiology
HEK-293 cells stably expressing Nav1.6 (HEK-Nav1.6), which were chosen as an experimental model because they provide a direct test of compounds on human cells, were maintained as previously described (James et al., 2017) and dissociated and re-plated on glass coverslips at low-density. Details about HEK-Nav1.6 cell culturing can be found in Shavkunov et al., 2013. Cells were placed in a room temperature (20–22 °C) static extracellular bath solution (140 mM NaCl, 3 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, 10 mM glucose; Sigma-Aldrich, St. Louis, MO; pH 7.3) for 30 mins with a final concentration of either 10 µM DM or 0.01% DMSO. Recording electrodes were filled with intracellular recording solution (130 mM CH3O3SCs, 1 mM EGTA, 10 mM NaCl, 10 mM HEPES; Sigma-Aldrich, St. Louis, MO; pH 7.3). Recordings were then performed on each coverslip for up to one consecutive hour. To determine effects on LTI, a 4-sweep protocol with four 20 ms 0 mV pulses separated by 40 ms −90 mV interpulse recovery phases from a −90 mV holding potential was used (Barbosa et al., 2017; Dover et al., 2010; Z. Liu et al., 2019). Cells with a series resistance above 25 MΩ were excluded. Maximum current ratio and fraction of recovered channels were both determined through measuring the peak transient sodium current at each pulse divided by the peak transient sodium current at the first pulse. For maximum current ratio, peak sodium current was measured from i to ii as depicted in Fig. 1A. When measuring this parameter, a large residual current was identified which confounded attempts to draw conclusions about LTI. Residual current was measured from i to iii, after each depolarization cycle is complete at −90 mV, as depicted in Fig. 1A. To directly compare cells of varying size, current densities were calculated by dividing sodium current amplitude by individual membrane capacitances. To assess LTI induction, transient peak sodium current was isolated for fraction of recovered channels by measuring from i to ii for the first depolarization cycle and i to iv for subsequent depolarization cycles. The change in measurement between the first depolarization cycles (i to ii) and subsequent cycles (i to iv) is to account for the large residual current present in depolarization cycles 2, 3, and 4. All electrophysiology data were analyzed with Clampfit 9 software (Molecular Devices, San Jose, CA).
Figure 1).
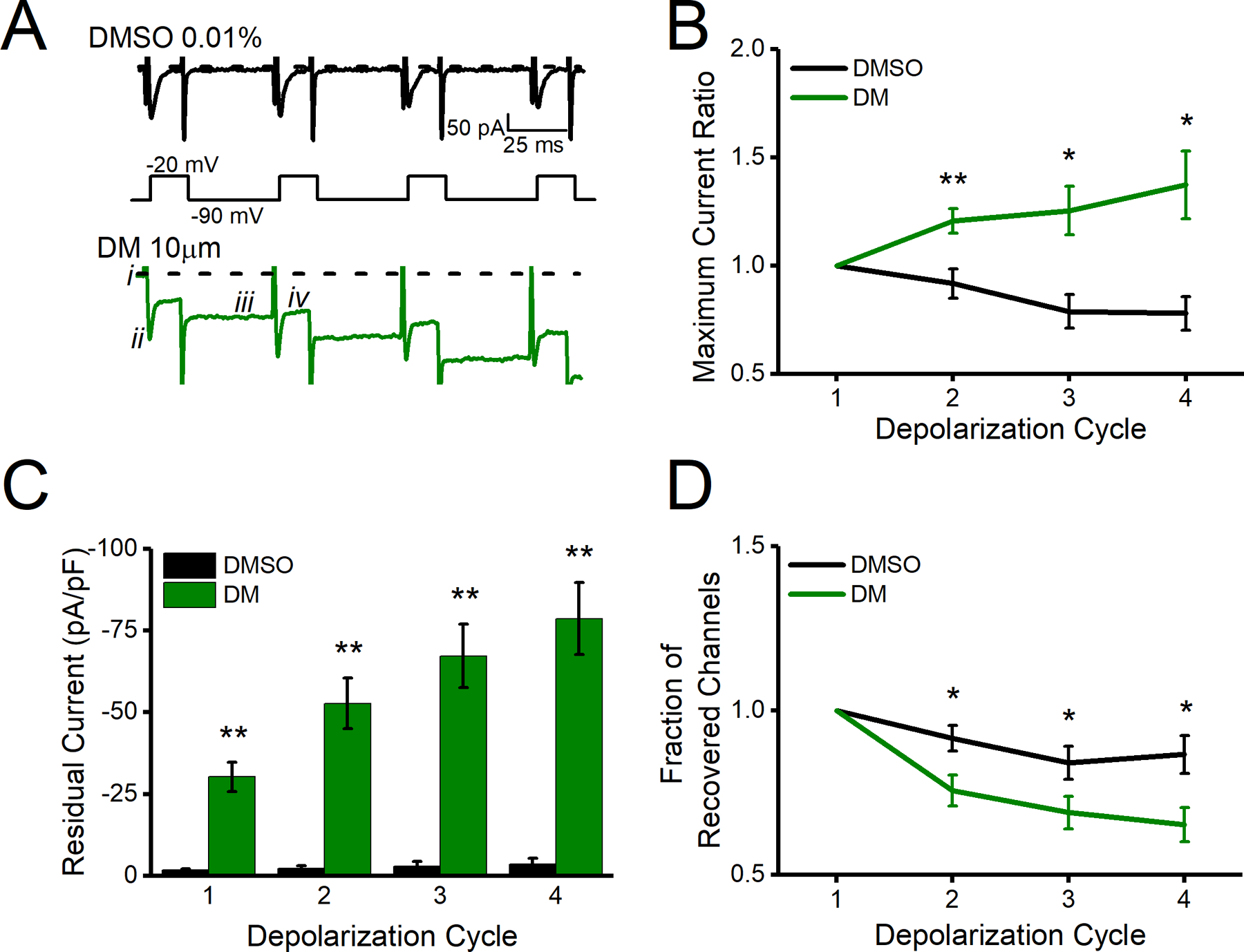
A. Representative traces of transient currents following repeated evoked changes in voltage (schematic under traces) with DMSO shown in black and DM shown in green. B. Maximum current ratio measured from i to ii for all depolarization cycles. C. Residual current measured as i to iii for all depolarization cycles. D. Fraction of recovered channels at measured from i to ii for the first depolarization cycle and ii to iv for subsequent cycles. All data are represented as mean ± SEM. *p<0.05, **p< 0.01.
2.3.2. Brain slice electrophysiology
Coronal brain slices containing the NAc were prepared from C57BL/6J mice aged 33–50 days. The mouse slice preparation was chosen as an experimental model as it is a commonly used ex vivo model to investigate the effects of compounds on brain circuitry. Mice were anesthetized with isoflurane (Baxter, Deerfield, IL) and quickly decapitated before brains were dissected and 300 μm coronal slices containing the NAc were prepared with a vibratome (Leica Biosystems, Buffalo Grove, IL) in a continuously oxygenated (mixture of 95%/5% O2/CO2) chilled tris-based artificial cerebrospinal fluid (aCSF), consisting of the following: 72 mM Tris-HCl, 18 mM Tris-Base, 1.2 mM NaH2PO4, 2.5 mM KCl, 20 mM HEPES, 20 mM sucrose, 25 mM NaHCO3, 25 mM glucose, 10 mM MgSO4, 3 mM Na-pyruvate, 5 mM Na-ascorbate and 0.5 mM CaCl2 (Sigma-Aldrich, St. Louis, MO); 300–310 mOsm, pH 7.4. Slices were transferred to a 31°C recovery chamber with fresh tris-based aCSF for 15 minutes before being transferred to a 31°C chamber with continuously- oxygenated (mixture of 95%/5% O2/CO2) standard aCSF consisting of the following: 123.9 mM NaCl, 3.1 mM KCl, 10 mM glucose, 1 mM MgCl2, 2 mM CaCl2, 24 mM NaHCO3, and 1.16 mM NaH2PO4 (Sigma-Aldrich, St. Louis, MO); 300–310 mOsm, pH 7.4. MSN somatic recordings in standard aCSF were performed using recording electrodes filled with an internal solution containing 145 mM K-gluconate, 2 mM MgCl2, 0.1 mM EGTA, 2.5 mM Na2ATP, 0.25 mM Na2GTP, 5 mM phosphocreatine, and 10 mM HEPES (pH 7.2; 290 mOsm). After giga-seal formation and cell membrane rupture, MSNs were held in I=0 mode for approximately 1 minute to determine resting membrane potential before switching to current clamp mode to assess neuronal activity. Electrophysiological brain slice data analysis was performed as previously described with any minor changes and additions described below (Scala et al., 2018). Intrinsic neuronal excitability was assessed by measuring evoked action potentials with a range of current injections ranging from 10 pA to 220 pA with 800 msec 10 pA pulses. The action potential peak amplitude was defined as the highest mV elicited. The action potential current threshold (Ithr) was defined as the first current step at which at least one action potential was induced. The action potential voltage threshold (Vthr) was defined as the voltage at which the first-order derivative of the rising phase of the action potential exceeded 10 mV/ms. Aberrant action potentials were defined as action potentials with Vthr >−27mV or amplitude <45 mV (two standard deviations outside of the mean Vthr or amplitude of control neurons). For determination of percent aberrant action potentials per neuron, neurons that fired less than 3 action potentials at 120 pA current step were excluded as we were investigating changes in repetitive firing.
2.4. Statistical Analysis
Using pCLAMP9 software, electrophysiological parameters of DM-induced effects were quantified and compared to DMSO controls. Either a Student’s t-test or a Mann-Whitney U test was used depending on whether data met parametric assumptions for normality and homogeneity of variance. Statistical tests were performed with OriginPro 2017 software (OriginLab, Northampton, MA, USA).
3. Results
3.1. Effects of DM on long-term inactivation.
Whole-cell patch-clamp electrophysiology was utilized to explore the effects of DM on Nav1.6 channels stably expressed in HEK-293 cells (HEK-Nav1.6). Following a 30-min incubation with either DMSO (0.01%) or DM (10 μM), a standard step-wise protocol was used to assess properties of evoked transient sodium currents in HEK-Nav1.6 cells. The effects of DM on LTI, a process which dictates Nav channel availability over longer periods of time and therefore is vital in regulating repetitive firing (Dover et al., 2010; Hampl et al., 2016), has not yet been studied. Here we applied a series of repeated voltage pulses to measure any potential modulatory effect of DM on LTI. Representative traces of evoked Nav1.6-mediated sodium current in response to injected voltage steps (four 20 ms −20 mV depolarizations) are depicted in Fig. 1A. Following each stimulation, DM appeared to visually increase peak transient sodium current (Fig. 1B) due to a large build-up of residual current that prevented return to baseline (Fig. 1A, dotted line). This build-up of residual current was significantly increased at each depolarization step (Fig. 1C) and is in line with previously reported use-dependent effects of DM (James et al., 2017). However, when residual and transient currents were properly separated from each other by differentially adjusting the reference baseline (Fig. 1A, iii and iv, respectively) we were able to unmask true variations in peak transient sodium currents that were consistent with the channel entering LTI (Fig. 1D) as previously reported (Barbosa et al., 2017; Dover et al., 2010). We conclude that DM drives Nav1.6 to an irreversible, aberrant open state (residual current) while concomitantly inducing LTI, limiting the flux of sodium during transient opening of the channel.
3.2. Effects of DM on intrinsic excitability.
We previously established the ability of DM to modify Nav1.6 biophysical properties, such as persistent and tail currents and steady-state inactivation that together with the phenotypes described in Fig. 1 are known to influence action potential firing (James et al., 2017). To understand the effect of DM in the intact circuit, we investigated MSNs in the NAc because they have relatively high expression of Nav1.6, are highly vulnerable to excitotoxicity, are altered in neurodevelopmental disorders, and demonstrate protein expression changes following DM exposure (Y. Liu et al., 2019; Magby and Richardson, 2017; Olson et al., 2015; Rikani et al., 2014; Saxena and Caroni, 2011; Scala et al., 2018; Wang et al., 2017). Whole-cell patch-clamp electrophysiology in NAc slices of C57BL/6J mice was used to determine the effect of DM on intrinsic excitability of MSNs. Slices were incubated in a static bath of DMSO (0.01%) or DM (10 μM) for 1 hr prior to electrophysiological recordings. In Fig. 2A, representative traces of evoked action potentials from individual MSNs are depicted. MSNs exposed to DM appeared to have higher firing frequency than control cells across all injected current steps from 0 to 130 pA approaching statistical significance at 100 pA. However, average instantaneous firing frequency (IFF), which reflects how rapidly a neuron fires within a single injected current step, was unchanged (Fig. 2B). At 100 pA, DM significantly increased the number of action potentials fired 21 ± 3 (n=10) compared to control 12 ± 3 (n=10, p= 0.018, Student’s t-test) as seen in Fig. 2B. At the same current step, no changes were observed in average IFF for control condition (24.4 ± 5.2 Hz) vs DM (34.5 ± 3.9 Hz, n=10, p=0.397, Student’s t-test) as seen in Fig. 2C. In addition, no changes between the two groups were observed in the action potential current threshold (Ithr) (control 53 ± 10.11 pA vs DM 48 ± 9.8 pA, n=10, p=0.726, Student’s t-test) or voltage threshold (Vthr) (control −44.6 ± 4.4 mV vs DM −38.7 ± 2.4 mV p=0.255, Student’s t-test) as seen in Fig. 2D–F. However, inspection of action potential trains at current steps > 100 pA revealed that DM elicited a significant decrease in the amplitude of action potentials as depicted in Fig. 3A. Data from Fig. 3B summarizes the action potential amplitude average over injected current steps that were >100 pA. Despite action potential amplitude variability per cell the reduction was statistically significant at several current steps above 110 pA (Fig. 3C). For instance, at 180 pA, the peak amplitude of action potentials in DM exposed cells was 38.81 ± 7.8 mV (n=10) compared to controls 71.1 ± 6.4 mV (n=10, p=0.005, Student’s t-test). Our cursory analysis identified a possible group of uniquely aberrant action potentials. To better characterize these aberrant action potentials, we generated a histogram distribution of action potential amplitudes (Fig. 4A) and Vthr (Fig. 4B) at current step 120 pA. In addition to reduced amplitude, these action potentials exhibited a depolarizing shift in Vthr; the average Vthr for DM cells was more depolarized (−24.39 ± 3.02 mV) compared to controls (−33.69 ± 2.8 mV, n=10, p=0.03). We plotted the action potential Vthr vs amplitude and observed the distinct cluster of uniquely aberrant action potentials in the DM-exposed cells (Fig. 4C), with the DM-exposed neurons firing a significantly higher percentage of aberrant spikes that were > 2 standard deviations outside the control mean for both amplitude and Vthr, as shown in Fig. 4D (DM 55.7 ± 13.5 vs control 0.59 ± 0.6, n=8–10, p=0.004, Student’s t-test).
Figure 2).
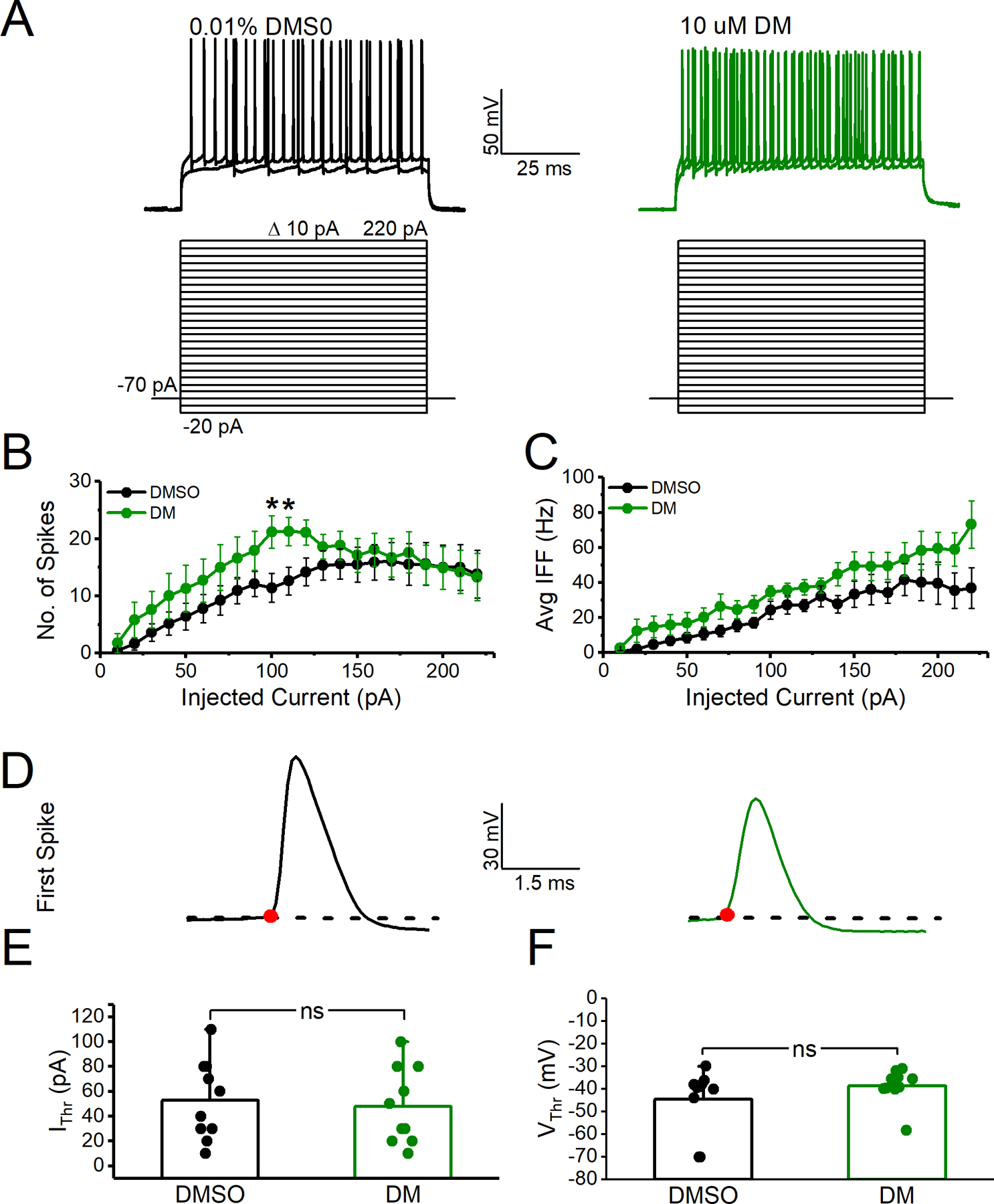
A. Representative traces of evoked action potential traces in response to current injection in MSNs of coronal mouse brain slices in control (DMSO 0.01%) or DM (10 μM). B. Number of action potentials versus injected current. C. Average instantaneous firing frequency (IFF) versus injected current. D. Representative traces of the first evoked action potential from DMSO (black) vs. DM exposed (green) MSN elicited at current threshold. E. Current threshold (Ithr) F. Voltage threshold (Vthr). All data are represented as mean ± SEM. *p<0.05.
Figure 3).
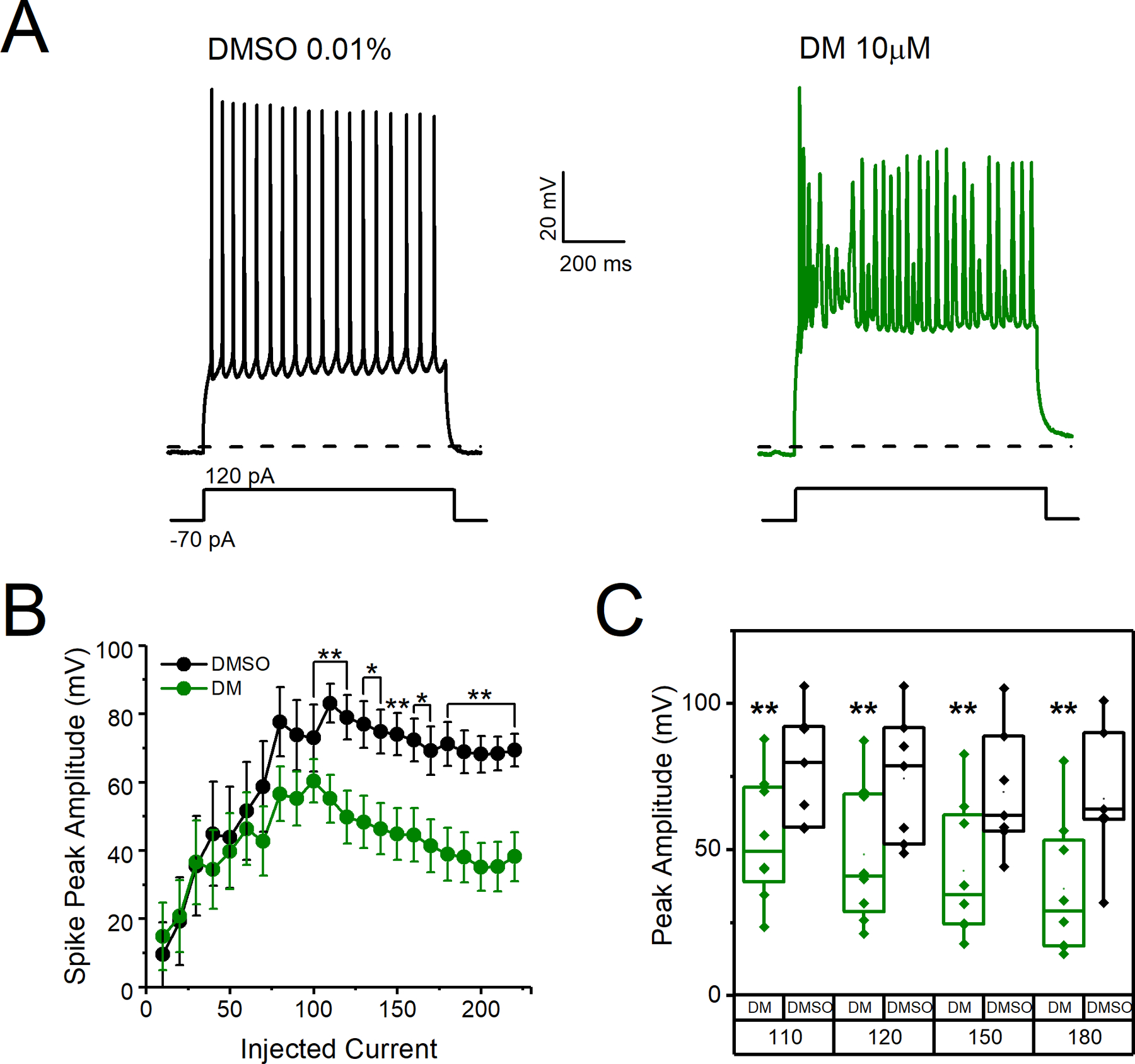
A. Representative traces of evoked action potential traces in response to current injection in MSNs of C57BL/6J mice following 30-minute incubation of coronal brain slices in DMSO (0.01%) or DM (10 μM). B. Peak amplitude of action potentials versus injected current steps. C. Peak amplitude of action potentials at 110, 120, 150, and 180 pA of injected current. Data here depict the amplitude of each cell at a particular current step and are extracted from Fig. 3B. All data are represented as mean ± SEM. *p<0.05, **p< 0.01.
Figure 4).
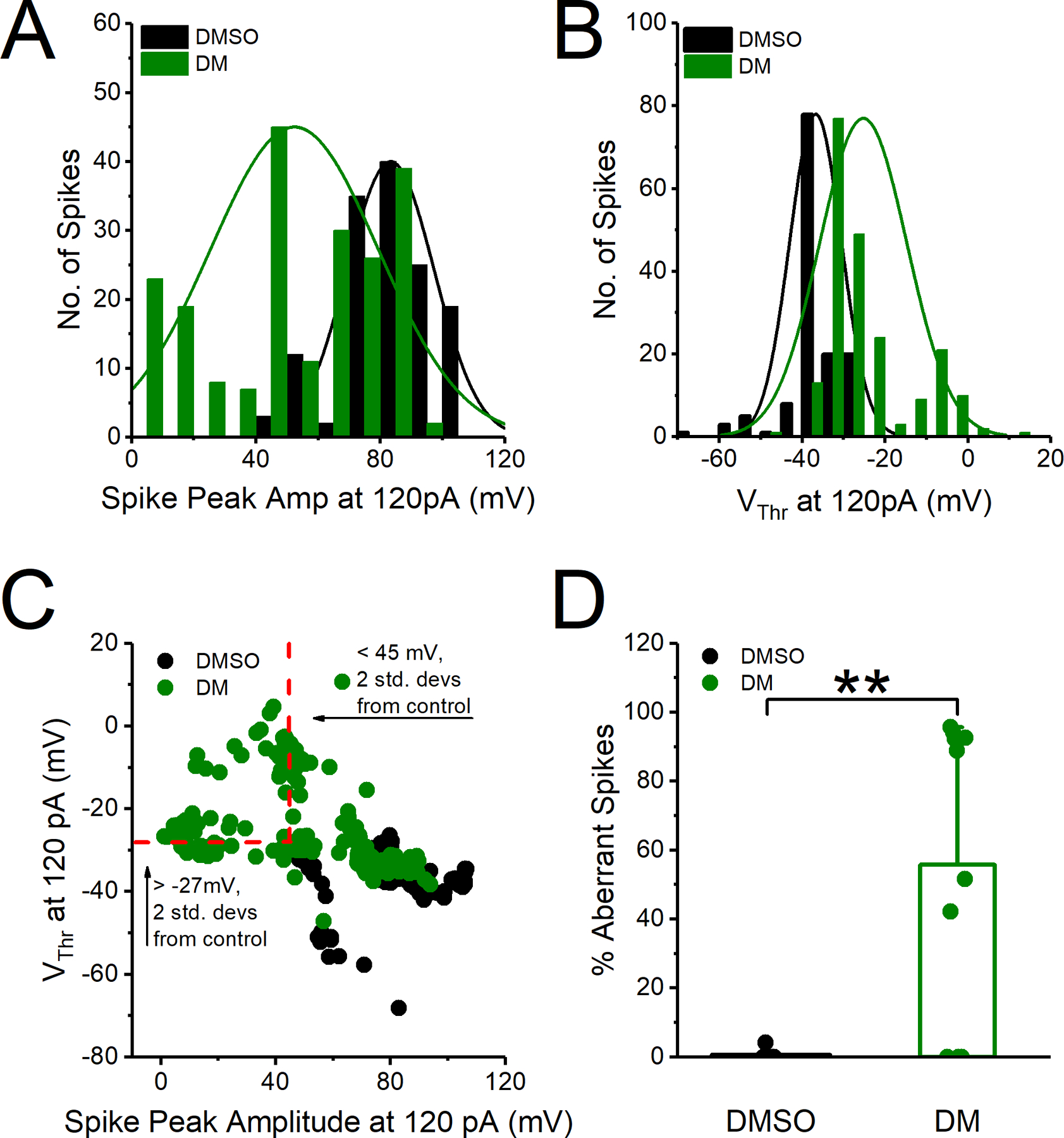
A. Histogram distribution of action potentials (spikes) peak amplitude at 120 pA injected current step. B. Histogram distribution of action potential voltage threshold (Vthr) at 120 pA of injected current step. C. Voltage threshold plotted against amplitude for action potentials at 120 pA injected current step. Red dotted lines represent cut off for aberrant spikes with voltage threshold (Vthr) >−27 mV or amplitude <45 mV (two standard deviations outside the mean of control neurons). Spikes in the upper left quadrant are therefore considered aberrant. D. Percent aberrant spikes at 120 pA injected current step. All data are represented as mean ± SEM. *p<0.05, **p< 0.01.
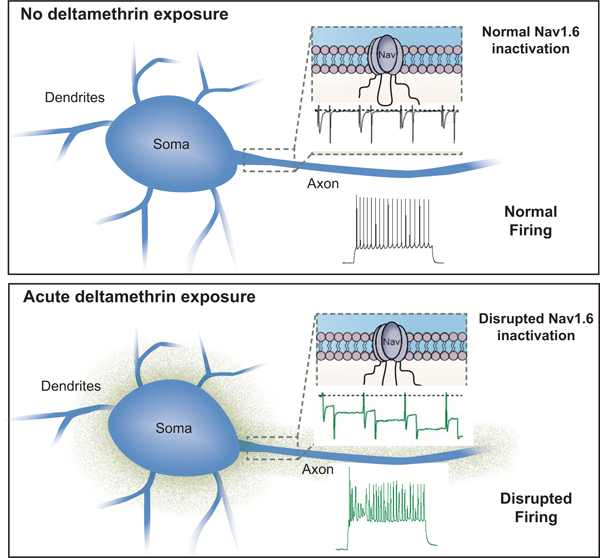
A schematic diagram that summarizes the results of this study is depicted in Fig. 5 showing a possible correlation between disrupted Nav1.6 function and aberrant firing pattern in MSNs following toxin exposure.
Figure 5).
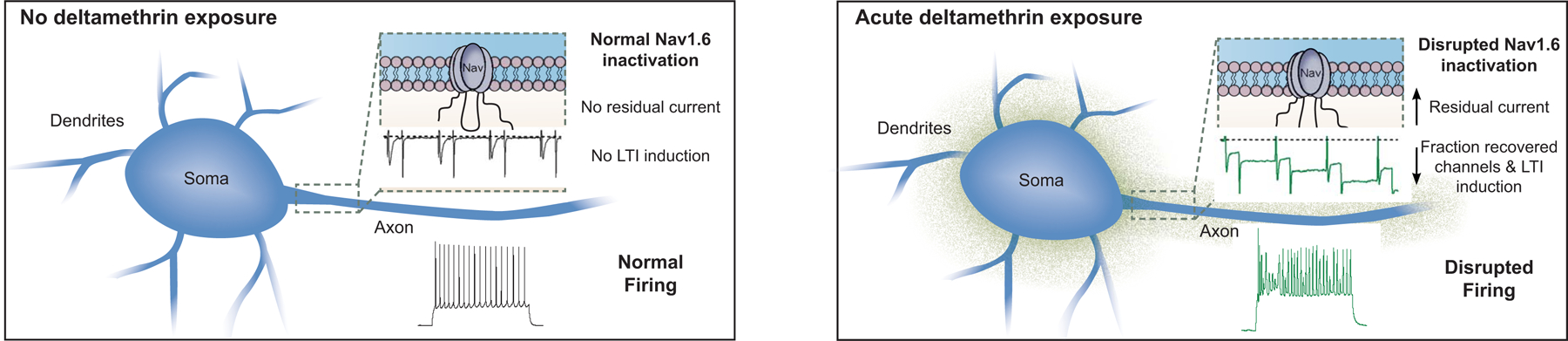
Schematic representation of acute DM induced changes. Nav1.6 inactivation is disrupted by increased build-up of residual current and a decreased fraction of recovered channels. The latter of which is indicative of LTI induction. This disruption is consistent with firing aberrations in MSNs of the NAc.
4. Discussion
The findings herein capture the description of DM modification of Nav1.6, such as increased build-up of residual current and induction of LTI elicited during repetitive stimulation, as well as its acute effect on intrinsic firing of MSNs in the NAc, a highly vulnerable cell type associated with reward-related disorders (Gerfen and Surmeier, 2011; Scala et al., 2018; Surmeier et al., 2007).
While complementing previous studies of DM effects on Nav1.6 our results demonstrated a significant use-dependent effect of DM on the channel leading to two opposite phenomena. First, in line with previous studies (James et al. 2017), we observed a progressive build-up of residual sodium currents (Soderlund, 2012; James et al., 2017; Soderlund et al., 2017; Tan and Soderlund, 2010) consistent with an overall potentiating effect of DM favoring an aberrant open channel conformation that persists even at negative membrane potentials (−90 mV). An opposite observed phenomenon was a use-dependent reduction of peak transient sodium currents (fraction of recovered channels) that resulted in ~40% reduction in peak current at the end of the conditioning train, which we interpreted as the ability of DM to induce Nav1.6 LTI. The magnitude of this reduction in peak transient current is consistent with previous studies on LTI (Barbosa et al., 2017; Dover et al., 2010). In neurons, the interplay of these two mechanisms could co-exist to shape action potential firing during repetitive stimulation. DM could increase firing at first, but then lead to action potential shape distortion or failure because of decreased channel availability induced in part by Nav1.6 LTI (Beyreuther et al., 2007).
Consistently, we found a pattern of aberrant firing in MSNs exposed to DM. Up to moderate current injections (100 pA), which led to an average firing frequency of <34 Hz (Fig. 2C) that is close to physiological firing rate of MSNs, DM increased the number of action potentials fired in these cells. However, at higher current injections (>100 pA, Fig. 3 and 4) DM caused a significant change in action potential amplitude and voltage threshold which resulted in overall aberrant firing. Because these changes in action potential amplitude and voltage threshold were prominent after the first 100–200 ms of neuron firing, it is plausible that they are induced by a use-dependent mechanism, possibly LTI, that progressively limits channel availability during repetitive firing. If reproduced in vivo, this acute effect of DM could potentially disrupt high-frequency cyclic firing of MSN that is part of cortical information processing in the basal ganglia network, a component of reward circuitry (Mahon et al., 2006).
This acute disruption of reward circuitry can manifest as durable long-term effects by means of the DM chemical structure and the window of exposure. With the presence of two phenyl rings that contribute to high lipophilicity and a cyclopropane moiety that lends to molecule stability, DM is prone to accumulation in lipid-rich organs like the brain (Chrustek et al., 2018). Thus, during development when the blood brain barrier is immature and thus far more susceptible to toxins, DM could bio-accumulate in the brain forming highly toxic reservoirs with age-dependent dosimetry in regions rich with highly vulnerable cells such as the NAc (Mortuza et al., 2017; Singh et al., 2016). Thus, changes induced by DM on Nav1.6 biophysical properties could potentially contribute to alterations of intrinsic excitability in these cells that are triggered acutely upon exposure to the toxin, but are expected to disrupt MSN firing with durable long-term effects on the reward circuit.
Acknowledgements
We acknowledge Dr. Bill T. Ameredes, Dr. Lance Hallberg, Dr. Thomas A. Green, and Dr. Paul Wadsworth for their substantial intellectual input and constructive discussions throughout the entirety of the project.
Funding Information
This work was supported by the Gulf Coast Center for Precision Environmental Health using funds from the National Institute of Environmental Health Sciences of the National Institute of Health under Award Number P30ES030285, National Institutes of Health Grants NIEHS Center Grant P30ES006676, National Institute of Environmental Health Sciences T32-ES007254 (Tapia) and the National Institute of Mental Health 1R01MH111107 (Laezza).
References
- Ali SR, Liu Z, Nenov MN, Folorunso O, Singh A, Scala F, Chen H, James TF, Alshammari M, Panova-Elektronova NI, White MA, Zhou J, Laezza F, 2018. Functional Modulation of Voltage-Gated Sodium Channels by a FGF14-Based Peptidomimetic. ACS Chem. Neurosci 9, 976–987. 10.1021/acschemneuro.7b00399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa C, Xiao Y, Johnson AJ, Xie W, Strong JA, Zhang J, Cummins TR, 2017. FHF2 isoforms differently regulate Nav1.6 mediated resurgent sodium currents in dorsal root ganglion neurons. Pflugers Arch Eur. J. Physiol 469, 195–212. 10.1007/s00424-016-1911-9.FHF2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyreuther BK, Freitag J, Heers C, Krebsfanger N, Scharfenecker U, Stohr T, 2007. Lacosamide: A Review of Preclinical Properties. CNS Drug Rev 13, 21–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busvine JR, 1960. The importance of synthetic pyrethroids. Bull. World Health Organ 22, 590–592. https://doi.org/00/00/0C/5E/ [pii] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, 2000. From Ionic Currents to Molecular Mechanisms: The Structure and Function of Voltage-Gated Sodium Channels. Cell Press 26, 13–25. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Dib-hajj S, Meisler MH, Pietrobon D, 2008. Inherited Neuronal Ion Channelopathies: New Windows on Complex Neurological Diseases. J. Neurosci 28, 11768–11777. 10.1523/JNEUROSCI.3901-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL, Waxman SG, 2005. International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol. Rev 57, 397–409. 10.1124/pr.57.4.4.and [DOI] [PubMed] [Google Scholar]
- Chrustek A, Holynska-Iwan I, Dziembowska I, Bogusiewicz J, Wroblewski M, Cwynar A, Olszewska-Slonina D, 2018. Current Research on the Safety of Pyrethroids Used as Insecticides. Medicina (B. Aires) 54. 10.3390/medicina54040061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies TGE, Field LM, Usherwood PNR, Williamson MS, 2007. Critical Review DDT, Pyrethrins, Pyrethroids and Insect Sodium Channels. IUBMB 59, 151–162. 10.1080/15216540701352042 [DOI] [PubMed] [Google Scholar]
- Dichter GS, Damiano CA, Allen JA, 2012. Reward circuitry dysfunction in psychiatric and neurodevelopmental disorders and genetic syndromes: animal models and clinical findings. J. Neurodev. Disord 4, 19. 10.1186/1866-1955-4-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dover K, Solinas S, Angelo ED, Goldfarb M, 2010. Long-term inactivation particle for voltage-gated sodium channels 19, 3695–3711. 10.1113/jphysiol.2010.192559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Nomura Y, Zhorov BS, Dong K, 2015. Rotational Symmetry of Two Pyrethroid Receptor Sites in the Mosquito Sodium Channel. Mol. Pharmacol 88, 273–280. 10.1124/mol.115.098707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis TC, Lobo MK, 2017. Emerging Role for Nucleus Accumbens Medium Spiny Neuron Subtypes in Depression. Biol. Psychiatry 81, 645–653. 10.1016/j.biopsych.2016.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ, 2011. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 441–466. 10.1146/annurev-neuro-061010-113641.Modulation [DOI] [PMC free article] [PubMed]
- Goldin AL, 2003. Mechanisms of sodium channel inactivation. Curr. Opin. Neurobiol 13, 284–290. 10.1016/S0959-4388(03)00065-5 [DOI] [PubMed] [Google Scholar]
- Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA, 2000. Nomenclature of Voltage-Gated Sodium Channels. Neuron 28, 365–368. 10.1016/S0896-6273(00)00116-1 [DOI] [PubMed] [Google Scholar]
- Gummin DD, Mowry JB, Spyker DA, Brooks DE, Fraser MO, Banner W, Gummin DD, Mowry JB, Spyker DA, Brooks DE, 2017. 2016 Annual Report of the American Association of Poison Control Centers ‘ National Poison Data System (NPDS): 34th Annual Report. Clin. Toxicol 55, 1072–1254. 10.1080/15563650.2017.1388087 [DOI] [PubMed] [Google Scholar]
- Hampl M, Eberhardt E, Reilly AOO, Lampert A, 2016. per. Sci. Rep 6. 10.1038/srep25974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Soderlund DM, 2017. Effects of the β1 Auxiliary Subunit on Modification of Rat Nav 1.6 Sodium Channels Expressed in HEK293 Cells by the Pyrethroid Insecticides Tefluthrin and Deltamethrin. Toxicol. Appl. Pharmacol 58–69. 10.1016/j.taap.2015.12.007.Effects [DOI] [PMC free article] [PubMed]
- He B, Soderlund DM, 2011. Differential State-Dependent Modification of Rat Nav1.6 Sodium Channels Expressed in Human Kidney (HEK293) Cells by the Pyrethroid Insecticides Tefluthrin and Deltamethrin. Toxicol. Appl. Pharmacol 257, 377–387. 10.1016/j.taap.2011.09.021.Differential [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisada A, Yoshinaga J, Zhang J, Katoh T, Shiraishi H, Shimodaira K, Okai T, Ariki N, Komine Y, Shirakawa M, Noda Y, Kato N, 2017. Maternal Exposure to Pyrethroid Insecticides during Pregnancy and Infant Development at 18 Months of Age. Int. J. Environ. Res. Public Health 14, 1–9. 10.3390/ijerph14010052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James TF, Nenov MN, Tapia CM, Lecchi M, Koshy S, Green TA, Laezza F, 2017. Consequences of acute Nav1.1 exposure to deltamethrin. Neurotoxicology 1–11. 10.1016/j.neuro.2016.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, 2011. Pyrethroids: Mammalian Metabolism and Toxicity. J. Agric. Food Chem 59, 2786–2791. [DOI] [PubMed] [Google Scholar]
- Karoly R, Lenkey N, Juhasz AO, Sylvester Vizi E, Mike A, 2010. Fast- or slow-inactivated state preference of Na+ channel inhibitors: A simulation and experimental study. PLoS Comput. Biol 6, 1–13. 10.1371/journal.pcbi.1000818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanphear BP, 2015. The Impact of Toxins on the Developing Brain. Annu. Rev. Public Heal 36, 211–30. 10.1146/annurev-publhealth-031912-114413 [DOI] [PubMed] [Google Scholar]
- Lee HJ, Weitz AJ, Bernal-Casas D, Duffy BA, Choy MK, Kravitz AV, Kreitzer AC, Lee JH, 2016. Activation of Direct and Indirect Pathway Medium Spiny Neurons Drives Distinct Brain-wide Responses. Neuron 91, 412–424. 10.1016/j.neuron.2016.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Schubert J, Sonnenberg L, Helbig KL, Hoei-Hansen CE, Koko M, Rannap M, Lauxmann S, Huq M, Schneider MC, Johannesen KM, Kurlemann G, Gardella E, Becker F, Weber YG, Benda J, Møller RS, Lerche H, 2019. Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain 142, 376–390. 10.1093/brain/awy326 [DOI] [PubMed] [Google Scholar]
- Liu Z, Wadsworth P, Singh AK, Chen H, Wang P, Folorunso O, Scaduto P, Ali SR, Laezza F, Zhou J, 2019. Identification of peptidomimetics as novel chemical probes modulating fibroblast growth factor 14 (FGF14) and voltage-gated sodium channel 1.6 (Nav1.6) protein-protein interactions. Bioorganic Med. Chem. Lett 29, 413–419. 10.1016/j.bmcl.2018.12.031.Identification [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magby J, Richardson J, 2015. Role of Calcium and Calpain in the Downregulation of Voltage-Gated Sodium Channel Expression by the Pyrethroid Pesticide Deltamethrin. J Biochem Mol Toxicol 29, 129–134. 10.1002/jbt.21676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magby JP, Richardson JR, 2017. Developmental pyrethroid exposure causes long-term decreases of neuronal sodium channel expression. Neurotoxicology 60, 274–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon S, Vautrelle N, Pezard L, Slaght SJ, Deniau JM, Chouvet G, Charpier S, 2006. Distinct patterns of striatal medium spiny neuron activity during the natural sleep-wake cycle. J. Neurosci 26, 12587–12595. 10.1523/JNEUROSCI.3987-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Kearney JA, 2005. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest 115, 2010–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortuza T, Chen C, White CA, Cummings BS, Muralidhara S, Gullick D, Bruckner JV, 2017. Toxicokinetics of Deltamethrin: Dosage Dependency, Vehicle Effects, and Low-Dose Age-Equivalent Dosimetry in Rats. Toxicol. Sci 1–10. 10.1093/toxsci/kfx260 [DOI] [PubMed]
- Motomura H, Narahashi T, 2001. Interaction of Tetramethrin and Deltamethrin at the Single Sodium Channel in Rat Hippocampal Neurons. Neurotoxicology 22, 329–339. [DOI] [PubMed] [Google Scholar]
- Oliveira EE, Du Y, Nomura Y, Dong K, 2013. A residue in the transmembrane segment 6 of domain I in insect and mammalian sodium channels regulate differential sensitivities to pyrethroid insecticides. Neurotoxicology 42–50. 10.1016/j.neuro.2013.06.001.A [DOI] [PMC free article] [PubMed]
- Olson HE, Tambunan D, Lacoursiere C, Goldenberg M, Pinsky R, Martin E, Ho E, Khwaja O, Kaufmann WE, Poduri A, 2015. Mutations in epilepsy and intellectual disability genes in patients with features of Rett syndrome. Am. J. Med. Genet. Part A 167, 2017–2025. 10.1002/ajmg.a.37132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulhote Y, Bouchard MF, 2013. Urinary Metabolites of Organophosphate and Pyrethroid Pesticides and Behavioral Problems in Canadian Children. Environ. Health Perspect 121, 1378–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power LE, Sudakin DL, 2007. Pyrethrin and pyrethroid exposures in the United States: a longitudinal analysis of incidents reported to poison centers. J. Med. Toxicol 3, 94–99. 10.1080/15287390600755224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros-Alcala L, Mehta S, Eskenazi B, 2015. Pyrethroid pesticide exposure and parental report of learning disability and attention deficit/hyperactivity disorder in U.S. children: NHANES 1999–2002. Environ. Health Perspect 122, 1336–1342. 10.1289/ehp.1308031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray DE, Fry JR, 2006. A reassessment of the neurotoxicity of pyrethroid insecticides. Pharmacol. Ther 111, 174–193. 10.1016/j.pharmthera.2005.10.003 [DOI] [PubMed] [Google Scholar]
- Reilly AOO, Khambay BPS, Williamson MS, Field LM, Wallace BA, Davies TGE, 2006. Modelling insecticide-binding sites in the voltage-gated sodium channel. Biochem. J 369, 255–263. 10.1042/BJ20051925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JR, Taylor MM, Shalat SL, Guillot TS, Caudle WM, Hossain MM, Mathews TA, Jones SR, Cory-Slechta DA, Miller GW, 2015. Developmental pesticide exposure reproduces features of attention deficit hyperactivity disorder. FASEB J 29, 1960–1972. 10.1096/fj.14-260901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikani AA, Choudhry Z, Choudhry AM, Rizvi N, Ikram H, Mobassarah NJ, Tulli S, 2014. The mechanism of degeneration of striatal neuronal subtypes in Huntington disease. Ann. Neurosci 21, 112–114. 10.5214/ans.0972.7531.210308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Caroni P, 2011. Selective Neuronal Vulnerability in Neurodegenerative Diseases: From Stressor Thresholds to Degeneration. Neuron 71, 35–48. 10.1016/j.neuron.2011.06.031 [DOI] [PubMed] [Google Scholar]
- Scala F, Nenov MN, Crofton EJ, Singh KA, Folorunso O, Zhang Y, Chesson B, Wildburger N, Games T, Alshammari MA, Alshammari TK, Elfrink H, Grassi C, Kasper JM, Smith AE, Hommel JD, Lichti CF, Rudra JS, D’Ascenzo M, Green TA, Laezza F, 2018. Environmental enrichment and social isolation mediate neuroplasticity of medium spiny neurons through the GSK3 pathway. Cell Rep [DOI] [PMC free article] [PubMed]
- Schettler T, 2001. Toxic Threats to Neurologic Development of Children. Environ. Health Perspect 109, 813–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer TJ, Rijal SO, Gross GW, 2008. Complete inhibition of spontaneous activity in neuronal networks in vitro by deltamethrin and permethrin. Neurotoxicology 29, 203–212. 10.1016/j.neuro.2008.01.002 [DOI] [PubMed] [Google Scholar]
- Shavkunov AS, Wildburger NC, Nenov MN, James TF, Buzhdgyan TP, Panova-Elektonova NI, Green TA, Veselenak RL, Bourne N, Laezza F, 2013. The fibroblast growth factor 14 (FGF14)/voltage-gated sodium channel complex is a new target of glycogen synthase kinase 3 (GSK3). J. Biol. Chem 288, 19370–19385. 10.1074/jbc.M112.445924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton JF, Geraghty EM, Tancredi DJ, Delwiche LD, Schmidt RJ, Ritz B, Hansen RL, Hertz-Picciotto I, 2014. Neurodevelopmental disorders and prenatal residential proximity to agricultural pesticides: The charge study. Environ. Health Perspect 122, 1103–1109. 10.1289/ehp.1307044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, Ruben PC, 2014. Slow Inactivation of Na+ Channels, in: Voltage Gated Sodium Channels. Springer, Berlin. 10.1007/978-3-642-41588-3_3 [DOI] [Google Scholar]
- Singh A, Mudawal A, Maurya P, Jain R, Nair S, Shukla RK, Yadav S, Singh D, Khanna VK, Chaturvedi RK, Mudiam MKR, Sethumadhavan R, Siddiqi MI, Parmar D, 2016. Prenatal Exposure of Cypermethrin Induces Similar Alterations in Xenobiotic-Metabolizing Cytochrome P450s and Rate-Limiting Enzymes of Neurotransmitter Synthesis in Brain Regions of Rat Offsprings During Postnatal Development. Mol. Neurobiol 53, 3670–3689. 10.1007/s12035-015-9307-y [DOI] [PubMed] [Google Scholar]
- Soderlund DM, Tan J, He B, 2017. Functional reconstitution of rat Na v 1 . 6 sodium channels in vitro for studies of pyrethroid action. Neurotoxicology 60, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin DL, Power LE, Sudakin DL, Power LE, 2007. Organophosphate Exposures in the United States: A Longitudinal Analysis of Incidents Reported to Poison Centers Organophosphate Exposures in the United States: A Longitudinal Analysis of Incidents Reported to Poison Centers. J. Toxicol. Enviornmental Heal. Part A, 141–147. 10.1080/15287390600755224 [DOI] [PubMed]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W, 2007. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. TRENDS Neurosci 30, 228–235. 10.1016/j.tins.2007.03.008 [DOI] [PubMed] [Google Scholar]
- Tan J, Liu Z, Wang R, Huang ZY, Chen AC, Gurevitz M, Dong K, 2005. Identification of amino acid residues in the insect sodium channel critical for pyrethroid binding. Mol. Pharmacol 67, 513–522. 10.1124/mol.104.006205 [DOI] [PubMed] [Google Scholar]
- Tan J, Soderlund DM, 2010. Divergent Actions of the Pyrethroid Insecticides S-Bioallethrin, Tefluthrin and Deltamethrin on Rat Nav1.6 Sodium Channels. Toxicol. Appl. Pharmacol 247, 229–237. 10.1016/j.taap.2010.07.001.Divergent [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau MM, Dalton JC, Day JW, Ranum LPW, Meisler MH, 2006. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J. Med. Genet 527–530. 10.1136/jmg.2005.035667 [DOI] [PMC free article] [PubMed]
- Vais H, Williamson MS, Devonshire AL, Usherwood PNR, 2001. The molecular interactions of pyrethroid channels †. Pest Manag. Sci 57, 877–888. 10.1002/ps.392 [DOI] [PubMed] [Google Scholar]
- Venkataraman S, Claussen C, Dafny N, 2017. D1 and D2 specific dopamine antagonist modulate the caudate nucleus neuronal responses to chronic methylphenidate exposure. J. Neural Transm 124, 159–170. 10.1007/s00702-016-1647-x [DOI] [PubMed] [Google Scholar]
- Viel J-F, Rouget F, Warembourg C, Monfort C, Limon G, Cordier S, Chevrier C, 2017. Behavioural disorders in 6-year-old children and pyrethroid insecticide exposure: the PELAGIE mother–child cohort. Occup. Environ. Med. oemed-2016-104035 10.1136/oemed-2016-104035 [DOI] [PubMed]
- Vilin YY, Ruben PC, 2001. Slow inactivation in voltage-gated sodium channels: Molecular substrates and contributions to channelopathies. Cell Biochem. Biophys 35, 171–190. 10.1385/CBB:35:2:171 [DOI] [PubMed] [Google Scholar]
- Wang J, Ou S, Wang Y, 2017. Distribution and function of voltage-gated sodium channels in the nervous system. Channels 11, 534–554. 10.1080/19336950.2017.1380758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Ma H, Zhang F, Zhang C, Zou X, Cao Z, 2018. Selective Voltage-Gated Sodium Channel Peptide Toxins from Animal Venom: Pharmacological Probes and Analgesic Drug Development. ACS Chem. Neurosci 9, 187–197. 10.1021/acschemneuro.7b00406 [DOI] [PubMed] [Google Scholar]
- Yarov-yarovoy V, Brown J, Sharp EM, Clare JJ, Scheuer T, Catterall WA, 2001. Molecular Determinants of Voltage-dependent Gating and Binding of Pore-blocking Drugs in Transmembrane Segment IIIS6 of the Na+ Channel a Subunit *. J. Biol. Chem 276, 20–27. 10.1074/jbc.M006992200 [DOI] [PubMed] [Google Scholar]
- Zhorov BS, Dong K, 2017. Elucidation of pyrethroid and DDT receptor sites in the voltage-gated sodium channel. Neurotoxicology 60, 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]