

Abstract
Environmental disruption of the circadian rhythm is linked with increased pain due to osteoarthritis (OA). We aimed to characterize the role of the clock gene in OA-induced pain more systemically using both genetic and pharmacological approaches. Genetically modified mice, (bmal1f/fNav1.8CreERT mice), generated by deleting the critical clock gene, bmal1, from Nav1.8 sensory neurons, were resistant to the development of mechanical hyperalgesia associated with OA induced by partial medial meniscectomy (PMM) of the knee. In wild-type mice, induction of OA by PMM surgery led to a substantial increase in BMAL1 expression in DRG neurons. Interestingly, pharmacological activation of the REV-ERB (a negative regulator of bmal1 transcription) with SR9009 resulted in reduction of BMAL1 expression, and a significant decrease in mechanical hyperalgesia associated with OA. Cartilage degeneration was also significantly reduced in mice treated with the REV-ERB agonist SR9009. Based on these data, we also assessed the effect of pharmacological activation of REV-ERB using a model of environmental circadian disruption with its associated mechanical hyperalgesia, and noted that SR9009 was an effective analgesic in this model as well. Our data clearly demonstrate that genetic disruption of the molecular clock, via deletion of bmal1 in the sensory neurons of the DRG, decreases pain in a model of OA. Furthermore, pharmacological activation of REV-ERB leading to suppression of BMAL1 expression may be an effective method for treating OA-related pain, as well as to reduce joint damage associated with this disease.
Keywords: Osteoarthritis, Pain, Clock Gene, SR9009, BMAL1, REV-ERB, OA, Circadian Disruption
Introduction:
The molecular clock is responsible for driving circadian rhythms (universal 24-h oscillation patterns) in metabolic, physiological and behavioral functions of almost all species (Burris et al., 2012; Butler and Burris, 2015; Crumbley and Burris, 2011; Kojetin and Burris, 2014). The master clock in the suprachiasmatic nucleus plays a key role in coordination of clocks in other tissues that subsequently play key roles in the transcriptional regulation of clock-controlled genes that regulate myriad cellular functions including cellular proliferation and differentiation and metabolism (Alibhai et al., 2017; Burris et al., 2012; Sitaula et al., 2017). Thus, disruption of the circadian rhythm/molecular clock has been associated with a range of pathologies including cancer, metabolic diseases, and behavioral disorders (Amador et al., 2016a; Amador et al., 2016b; Banerjee et al., 2014; Burris et al., 2012; Solt et al., 2012; Vieira et al., 2014; Wang et al., 2015).
However, circadian rhythm in cells is regulated by utilizing the period [Per1, Per2 (only having ability to bind with both BMAL1 and CLOCK) and Per3] and cryptochrome (Cry1 and -2; both only able to interact with BMAL1) as master clock genes, and circadian locomotor output cycles kaput (CLOCK), and brain and muscle aryl hydrocarbon receptor nuclear translocator-like 1 (BMAL1) as clock-controlled genes (Savvidis and Koutsilieris, 2012). Where, (i) CLOCK:BMAL1 together form a heterodimer transcriptional activator consisting of two Per-Arnt/AhR-Sim basic helix-loop-helix (bHLH-PAS) domain protein subunits for rapid induction of Per1 during phase resetting of the clock and (ii) CLOCK:BMAL2 form another heterodimer for increasing the expression of Dec1 and Dec2 genes, so, Dec1 and Dec2, basic helix-loop-helix transcription factors, are involved in the regulation of the mammalian circadian clock (Savvidis and Koutsilieris, 2012). Therefore, a delay between production and action of inhibitory clock gene products is regulated by nuclear export of the PER protein, resulting in production of stable oscillations of gene expression with a period of 24-hours (Burris et al., 2012; Butler and Burris, 2015; Savvidis and Koutsilieris, 2012; Solt et al., 2012).
We are particularly interested in how the circadian rhythm impacts the pathology of osteoarthritis (OA). OA, the most common form of arthritis (affecting articular cartilage of the joints), affects 27 million in the US alone (Neogi, 2013), imposes an annual economic burden exceeding $60 billion, and is a leading cause of disability (Carbo, 2013). There is no cure for OA or treatment to inhibit its progression. Furthermore, there is no OA-specific pain medication. How OA develops and progresses to painful and debilitating disease is unclear. However, recent work suggests that disruption of circadian rhythm is a risk factor for the development and progression of OA-like pathological changes in the mouse knee joint (Kc et al., 2015). Moreover, other investigators revealed that chondrocytes have autonomous clocks which regulate key pathways of cartilage development and these rhythms dampen with age in an experimental mouse OA model (Gossan et al., 2015). Additionally, circadian desynchronization is implicated in OA-like pathological changes in articular cartilage (Gossan et al., 2015; Kc et al., 2015).
Disruptions in the circadian rhythm are associated with pain hypersensitivity (Burris et al., 2012; Das, 2015; Kc et al., 2015). Here, we sought to determine if circadian clock dysfunction is involved in modulation of peripheral nerve trauma and pain in a model of OA. Furthermore, given recent advances in modulators of the molecular clock, we examined whether the REV-ERB agonist, SR9009, could modulate the pathology associated with OA in a mouse model. Therefore, we also examined the role that the circadian clock plays in OA by utilizing both genetic and pharmacological models. We used genetically modified transgenic mice (bmal1-f/f-Nav1.8CreERT mice) that have been generated by conditionally deleting the bmal1 gene from Nav1.8 sensory neurons to assess the role of bmal1 in OA, and we examined the role of pharmacological targeting of REV-ERB (an inhibitor of Bmal1 expression) in OA.
Methods:
Experimental animals:
Female C57BL6/J (n=59) and bmal 1f/fNav1.8 CreERT mice (25–30 g) [n=10; Cre+ (n=5) & Cre− (n=5)] were randomly assigned for studies. There were three types of experiments: (i) normal mice for circadian disruption (establishment of environmental disruption of circadian rhythm model), (ii) clock gene conditional knock out mice (genetically modified transgenic mice (bmal1f/fNav1.8CreERT) with OA and (iii) SR9009 analgesic efficacy in OA mice (reducing pain). Mice were housed under standard laboratory conditions (in a temperature-controlled (21±1°C) room with a normal 12-h light/dark cycle). The study protocols involving animal procedures were in agreement with the guidelines of the Rush Institutional Animal Care and Use Committee (IACUC).
Environmental disruption of circadian rhythm model in normal mice:
Young adult C57BL6/J (n=31) mice were randomly divided into two experiments. Experiment-1A (n=11), divided into two different groups: shift (Shifted 14 weeks; n=5) and preshift-nonshift (Shifted 6 weeks-nonshifted 8 weeks; n=6). In shift group, mice were exposed to 12h:12h light-dark (LD) cycle alternating weekly with a 12h:12h dark:light (DL) cycle in the circadian chamber, for environmental disruption of circadian rhythm in the “shift” group of mice (Kc, Li et al. 2015).The “preshift-nonshift,” mice were shifted in the chamber for 6 weeks (week1:LD, week2:DL, week3:LD, week4:DL, week5:LD and week6:DL); thereafter, these shifted mice were returned to a normal 12-h LD cycle (non-shift) for 8 weeks, to allow circadian rhythm dysfunction to reverse back into a constant circadian condition. In experiment-1B (n=20), mice were shifted in the circadian chamber for environmental disruption of circadian rhythm (Kc, Li et al. 2015). After 5 weeks, mice were randomized into two different groups: (i) SR9009 (100mg/kg; IP-injection (at 1:00 PM of the day); each day over 7 weeks; n=10) and (ii) vehicle treated group (n=10), and all mice followed for another 7 weeks, for a total of 12 weeks.
Loss of the clock gene Bmal1 in Nav1.8+ sensory neurons leads to reduced osteoarthritis pain:
In experiment-2, to assess the influence of a clock gene in OA pain we utilized a genetic knock-out mouse model where the key clock gene bmal1 could be conditionally deleted in Nav1.8+ sensory neurons (bmal1f/fNav1.8CreERT mice). Bmal1f/fNav1.8CreERT mice (Cre+ & Cre−) were created by crossing bmal1 flox-flox mice (Stock No. 007668; Jackson Laboratory, USA) with Nav1.8CreERT mice (Stock code: IVF/4204; The Mary Lyon Centre, Medical Research Council, OX11ORD, UK) allowing for deletion of the bmal1 gene from Nav1.8+ sensory neurons by administering tamoxifen [i.p; 100mg/kg (dissolved in corn oil); 7 days].
Thereafter, bmal1f/fNav1.8CreERT mice underwent PMM surgery at week 12 and validation of bmal1 specific deletion in DRG neuron were performed by implementing immunohistochemically (IHC) method that described in immunofluorescence in DRG section for BMAL1 vs. NeuN at 8 weeks after surgery. Nevertheless, behavioral pain assessments (PWT & PTT) were performed every two weeks concluding 8 weeks after surgery. At the end of the experiment 2 mice after 8 weeks, the animals were sacrificed and L3–6 DRGs were harvested at midday (12:00–2:00 PM) for immunohistochemically investigation (REV-ERBα, TRPV1, substance P, NGF, CGRP, TrkA and Nav1.7) that described in immunofluorescence in DRG section. In all cases CRE (+) bmal1 deletion mice were compared to CRE (−) control mice without the bmal1 deletion.
Surgical procedure for OA induction:
All the surgical operations were performed under a microscope in an aseptic setting. Mice were placed in a supine position and anesthetized with 1.5 % isoflurane (Abbott Laboratories, North Chicago, IL, USA) in oxygen via a facemask at a rate of 1 L/minute. The left hind leg hair was shaved, thoroughly scrubbed with a topical antiseptic solution (chlorhexidine gluconate), and draped in sterile fashion. After confirming adequate anesthesia, a 1 cm left knee incision was made with a #15 scalpel blade. The knee joint was identified from the tibia and femur; and the medial meniscotibial ligament was identified using anatomic landmarks. To induce partial medial meniscectomy (PMM), which destabilizes the ligaments, a microscalpel at a depth of 0.5–0.7 mm was used to remove 1 mm of cartilage at midline (Glasson et al., 2007; Thysen et al., 2015). The skin incision was then closed with 4–0 vicryl suture.
SR9009 localized drug treatment for OA-induced experiment −3 mice:
OA-induced experiment-3 mice had two parts, intrathecal (IT) or intraarticular (IA) injection. For each part, mice were randomized were into two groups: vehicle treated and SR9009 treated. Intrathecal or intraarticular injection was administered by using 30 μg drug in 5 μl volume. Intrathecal (IT) injection was carried out at 13 weeks post PMM at 9:00 AM. However, intraarticular (IA) injection was carried out twice a week from week 2 to 12 at 1:00 PM each day.
Animal pain behavioral tests:
Behavioral testing for pain in the circadian disruption (experiment 1) and OA (experiment 2 and 3) were performed weekly until the designated sacrifice time for each mouse. For IA injection, each behavioral test was performed at 24 hours after drug injection.
Mechanical allodynia (von Frey testing):
Allodynia was evaluated based on hindpaw withdrawal from mechanical stimuli (Chaplan et al., 1994). After allowing mice to accommodate for 30 min on a wire mesh grid, a calibrated set of von Frey filaments were applied from below to the plantar hind paw to determine the 50% force withdrawal threshold using an iterative method. The filament was applied to the skin with enough pressure to buckle. A brisk lifting of the foot was recorded as a positive response. If no response was observed, the filament with the next higher force was applied, while the filament with the next lower force was applied upon a positive response.
Heat hyperalgesia:
Response to noxious heat stimuli was determined using a hot plate at 55°C. Mice were placed on the hot plate inside an enclosure, and the timer started. Timing was terminated when any of the following behavioral events occurred: licking of hind paw, shaking of hind paw in air and jumping. A cut-off time of 30 seconds was used to avoid tissue damage. Two tests were carried out at 10 minutes intervals and then the mean value taken as the nociceptive threshold.
Activity Monitor:
Animals were tested in clean vivarium plastic cages (42× 25× 20 cm) enclosed in a cage rack Photobeam Activity System (San Diego Instruments, San Diego, CA) (Das et al., 2017). One set of photobeams were placed at foot level above the cage floor (with adjacent beams 5 cm apart) to automatically measure ambulation (counts of horizontal beam interruptions when an animal walks). Another set of photobeams were placed 5 cm above the cage floor (with adjacent beams 5 cm apart) to automatically measure rearing (counts of vertical beam interruptions when an animal stands). Activity was monitored in a dark room for 30 minutes at 2:00 PM of the day.
Histology:
At the end of 12 weeks (experiment 3), mice were sacrificed with CO2, the knee joint was dissected aseptically and fixed in 4% paraformaldehyde, decalcified (in EDTA, changed every 3 days for 4 weeks), washed under running water, dehydrated, paraffin-embedded and sectioned at 10 μm thickness. Thereafter, sections were deparaffinized (using xylene and a gradient of alcohol concentrations) and stained with Safranin-O/Fast green (SO/FG) to assess general morphology and for evaluation of degenerative changes according to OARSI (Osteoarthritis Research Society International) recommended semi-quantitative scoring system (Glasson et al, 2010).
Immunofluorescence in DRG:
At the end of the experiment (experiment 2 mice after 8 weeks and experiment 3 mice after 12 weeks), the animals were sacrificed with CO2 and L3–6 DRGs were harvested, fixed and embedded (Das et al., 2017) at midday (12:00–2:00 PM). All samples were cut at 4–5 μm thickness. After deparaffinization, the DRG sections were microwaved in a retrieval solution (10 mmol/L citrate, pH 6.0). The samples were then permeabilized with 0.5% Triton X-100 solution for 5 min. Non-specific binding was blocked by incubation in 5% normal goat or horse serum for 30 min at room temperature. After that, the DRG sections were incubated overnight with anti-rabbit TrkA antibody (1:200, Ab76291; Abcam), anti-rabbit NGF antibody (1:200; Sc-548; Santa Cruz Biotechnology), anti-rabbit Substance-P antibody (1:200; AB1566; Millipore Sigma), anti-rabbit CGRP antibody (1:500; Ab189786; Abcam), anti-rabbit Nav1.7 antibody (1:200; ASC-008; Alamone labs), anti-rabbit Nav1.8 antibody (1:200; ASC-016; Alamone labs), anti-guinea pig TRPV1 antibody (1:200; AB5566; Millipore), anti-mouse NeuN antibody (1:500; MAB377; Millipore), anti-rabbit Bmal1 antibody (1:100; NB1002288; Novus) and anti-mouse Rev-erb-α antibody (1:50,Sc-100910; Santa Cruz Biotechnology). The samples were then incubated with goat anti-rabbit Alexa 546 (1:250; A11035; Invitogen), goat anti-mouse Alexa 488 (1:250; A11029; Invitrogen) and goat anti-guinea pig Alexa488 (1:250; 106-545-003; Jackson Immuno Research Laboratories INC) fluorescent antibody conjugate. The immunoreactivity was examined using a fluorescence microscope. For each DRG section, the number of immunoreactive neurons was counted using Image J software (version1.8.0_60; Open Source).
RNA Isolation and Real-time Polymerase Chain Reaction (RT-PCR):
At 12 weeks’ time point, mice in each group were sacrificed with CO2. The L3–6 DRGs were harvested. Total RNA was isolated using TRIzol reagent (Invitrogen Corp., Carlsbad, CA) according to the manufacturer’s protocol. RT-PCR was performed to verify the differential expression of selected genes, which are related to circadian (rev-erb-α (Fw-CACCCAGTCTCAGTGATGATAG & Re-CGTTATAGAAGGAGGAGGAGGA), rev-erb-β (Fw-CCCAGGAACATGGAGCAATA & Re-GTAGTGAGAGGCAGCAACAT) and bmal (Fw-GCAGTGCCACTGACTACCAAG A & Re-TCC TGG ACA TTG CAT TGCAT)) clock gene and reference gene 18s (Fw-GTAACCCGTTGAACCCCATT & Re-CCATCCAATCGGTAGTAGCG) using a Roche Light Cycler system (Roche Diagnostics, GmbH Mannheim, Germany) and the SYBR Green method. Relative gene expression was determined by using the comparative CT method.
Statistical analysis:
Statistical analyses were performed using the GraphPadPrism (version 5.00, March7, 2007; Chicago, IL, USA) software. All data were presented as mean±SEM. Repeated measures two-way analysis of variance (ANOVA) followed by the Bonferroni test, was used to assess behavioral measures over time and between-group differences. The statistical significance criterion was P<0.05. For two-way ANOVA, F values are reported. The t test was used to assess PWT, PTT, protein levels, and locomotion between 2 groups at one time point (P<0.05). One-way ANOVA with post hoc Dunnett’s Multiple Comparison Test was used to assess OARSI Grade Score (histology for cartilage degeneration), protein levels, PTT, and relative gene expression between 3 or more groups.
Results:
Disruption of the circadian rhythm induces pain hypersensitivity.
We sought to establish a murine model of circadian disruption-induced pain hypersensitivity and utilized a paradigm where we induced a disruption by phase shifting the light:dark cycle in C57BL6/J female mice. Mice were maintained on a 12h:12h LD cycle (12:12 LD) and then exposed to alternate weekly 12h phase shifts for environmental disruption of circadian rhythm. Behavioral assessments of pain related measures were performed on both the shift (Shifted 14wks) and preshift-nonshift (Shifted 6wks-nonshifted 8wks) mice on multiple occasions until 14 weeks post shift. Results are shown in figures 1(A) for paw withdraw threshold (PWT) & 1(B) for paw thermal threshold (PTT). Mechanical hyperalgesia was measured using von Frey filaments. The PWT (figure 1(A); n=5–6) of the circadian disrupted mice progressively decreased lower than the initial baseline (hypersensitive), but the preshift-nonshift mice’s paw withdrawal thresholds then increased progressively (reversible hypersensitivity) after 8 weeks. Specifically, at 14 weeks PWT (mean, ±SEM) was different (P=0.0071; t- test) between the shift vs. preshift-nonshift mice.
Figure 1: Disruption of the circadian rhythm induces pain hypersensitivity.
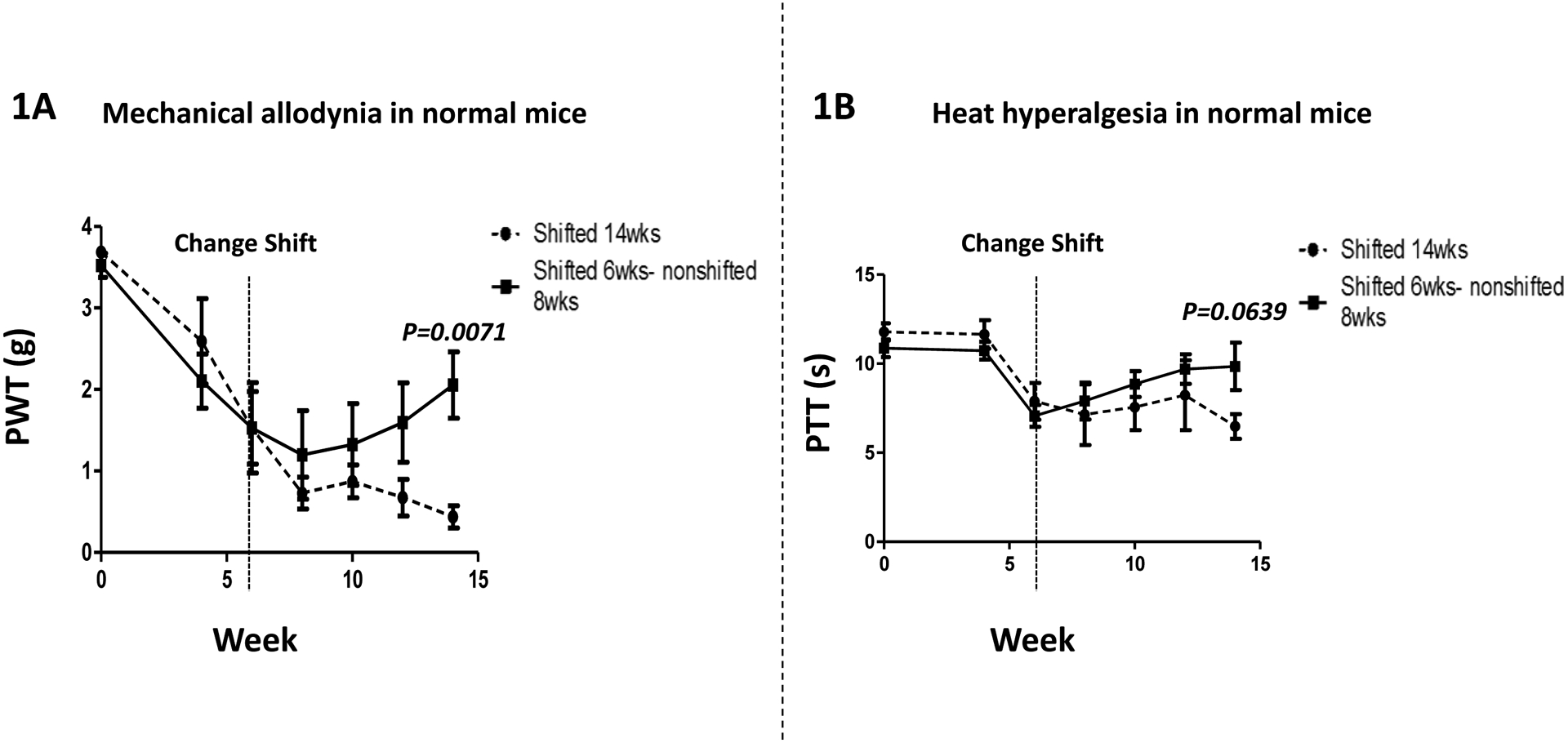
(Fig. 1A) Mechanical hyperalgesia was measured using von Frey filament test. The paw withdrawal threshold (PWT) of the Shift group (n = 5–6; Shifted 14wks) decreased throughout the 14 weeks, and was lower than the Preshift-Nonshift group (n = 5–6; Shifted 6wks-nonshifted 8wks) at week 14 (P=0.0071). (Fig. 1B) Heat hyperalgesia was measured using hot-plate test. The paw thermal threshold (PTT) in the Shift group (n = 5–6; Shifted 14wks) trended lower than the Preshift-Nonshift group (n = 5–6; Shifted 6wks-nonshifted 8wks) at week 14 (P=0.0639). Values are mean±SEM.
Paw thermal threshold (PTT) was similar to PWT, and the results are shown in Fig. 1B. At 14 weeks, there was a trend (P=0.0639;t test) toward higher PTT values in preshift-nonshift mice vs. shift mice.
Pharmacological Modulation of the circadian rhythm reverses pain hypersensitivity in normal mice.
As shown in Fig. 1A, circadian disruption results in pain hypersensitivity. Validation of this model to detect pain hypersensitivity in response to circadian disruption allowed us to assess the effects of the REV-ERB agonist, SR9009, a pharmacological modulator of the circadian clock. Using the identical model of circadian rhythm disruption, treatment of mice with SR9009 (100 mg/kg, i.p.) resulted in significant reduction in the pain hypersensitivity that is associated with circadian disruption. SR9009 significantly reduced the mechanical hyperalgesia (PWT; figure 2(A)). Specifically, mean (±SEM) PWT for pain hypersensitivity were different between groups after week 6; with group effect (F=28.63, DFn=1 & DFd=54; P<0.0001) and time effect (F=4.00, DFn=3 & DFd=54; P=0.0120) being significant. Importantly, at the 8 week (P<0.05), 10 week (P<0.001) and 12 week time points (P<0.001), PWT was higher in the SR9009 vs. vehicle treated mice.
Figure 2: Pharmacological Modulation of the circadian clock reverses pain hypersensitivity.
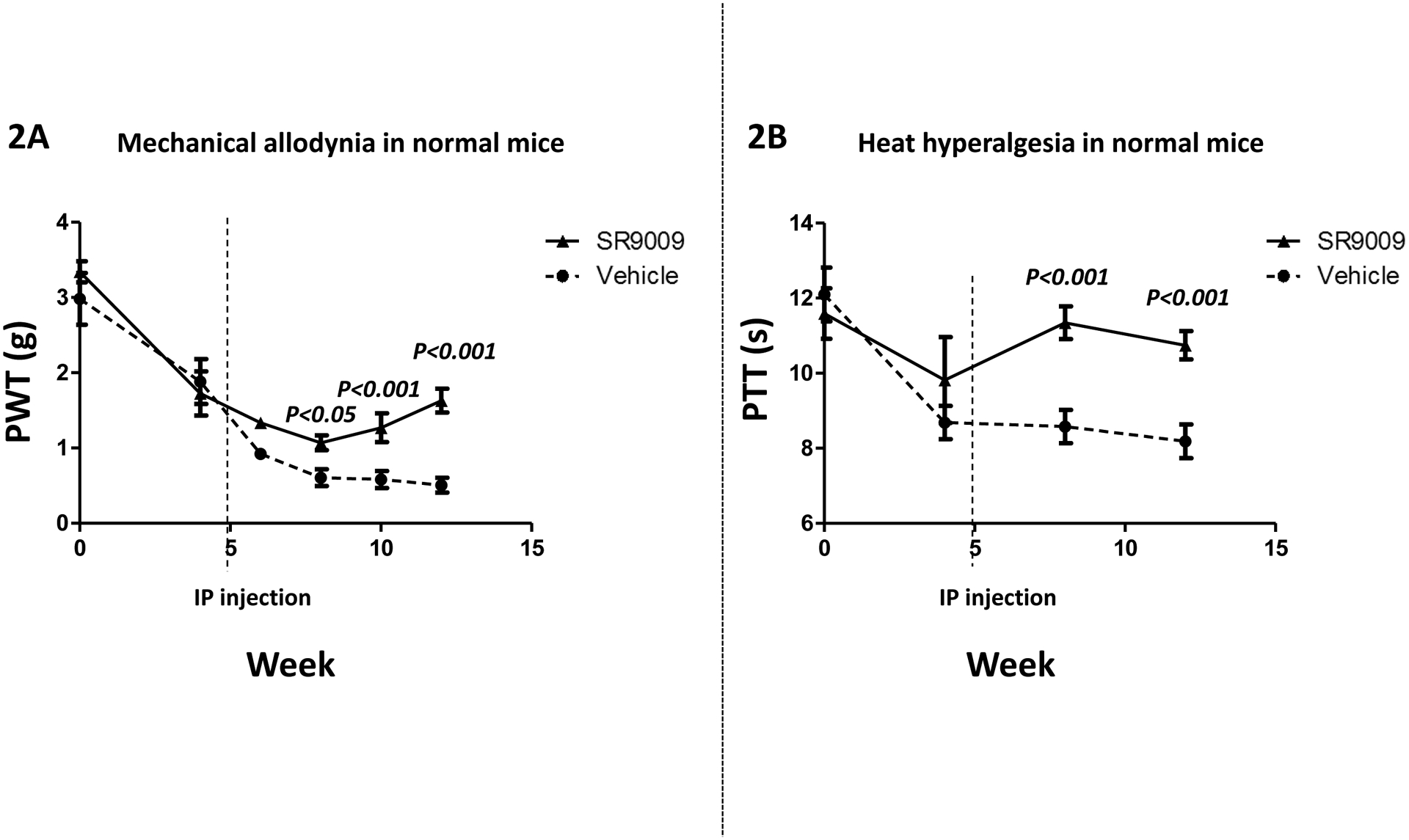
(Fig. 2A) Both groups had circadian disruption for 12 weeks. Mechanical hyperalgesia was measured using von Frey filament test. The paw withdrawal threshold (PWT) of the vehicle treated group (n = 10) progressively decreased throughout the 12 weeks, and were lower than SR9009 treated group (n = 10) at all time points after 6 weeks. (Fig. 2B) Heat hyperalgesia was measured using hot-plate test. The paw thermal threshold (PTT) in the vehicle treated group was reduced as compared with the SR9009 treated group after 6 weeks (n = 10 per group). Values are mean±SEM.
Moreover, SR9009 significantly reduce the paw thermal threshold (PTT; figure 2(B)) from week 6 to the end of the compared to vehicle-treated mice. Specifically, mean (±SEM) PTT for pain hyperalgesia were different between groups after week 6; with group effect (F=33.71, DFn=1 & DFd=18; P<0.0001) being significant, but, not time effect (F=1.58, DFn=1 & DFd=18; P=0.2245). Importantly, at the week 8 (P<0.001) and week 12 (P<0.001) time points, PTT was higher in the SR9009 vs. vehicle treated mice.
Loss of the clock gene bmal1 in Nav1.8+ sensory neurons leads to reduced osteoarthritis pain.
To assess the specificity deletion of a clock gene (bmal1) in sensory DRG neuron from genetically conditional knock-out mouse model, we implemented IHC methods for assessing the expression of BMAL1 protein level. The expression of BMAL1 protein (Fig.3G) in bmal1f/fNav1.8CreERT (+) mice was significantly very low in compare to bmal1f/fNav1.8CreERT (−) mice. Quantitative analyses, mean (±SEM) of BMAL1 [Fig.3 (A & C)] expression in DRGs clearly show a substantial reduction in bmal1f/fNav1.8CreERT (+) mice vs. bmal1f/fNav1.8CreERT (−) mice [Fig.3 (D & F)] undergoing the PMM surgery. Most importantly, the key clock gene bmal1 is conditionally deleted in sensory DRG neurons and that reduces the expression of BMAL1 protein (P=0.0005; unpaired t test) and shown in Fig.3G.
Figure 3: Loss of the clock gene bmal1 in Nav1.8+ sensory neurons leads to reduced BMAL1 protein in DRG.
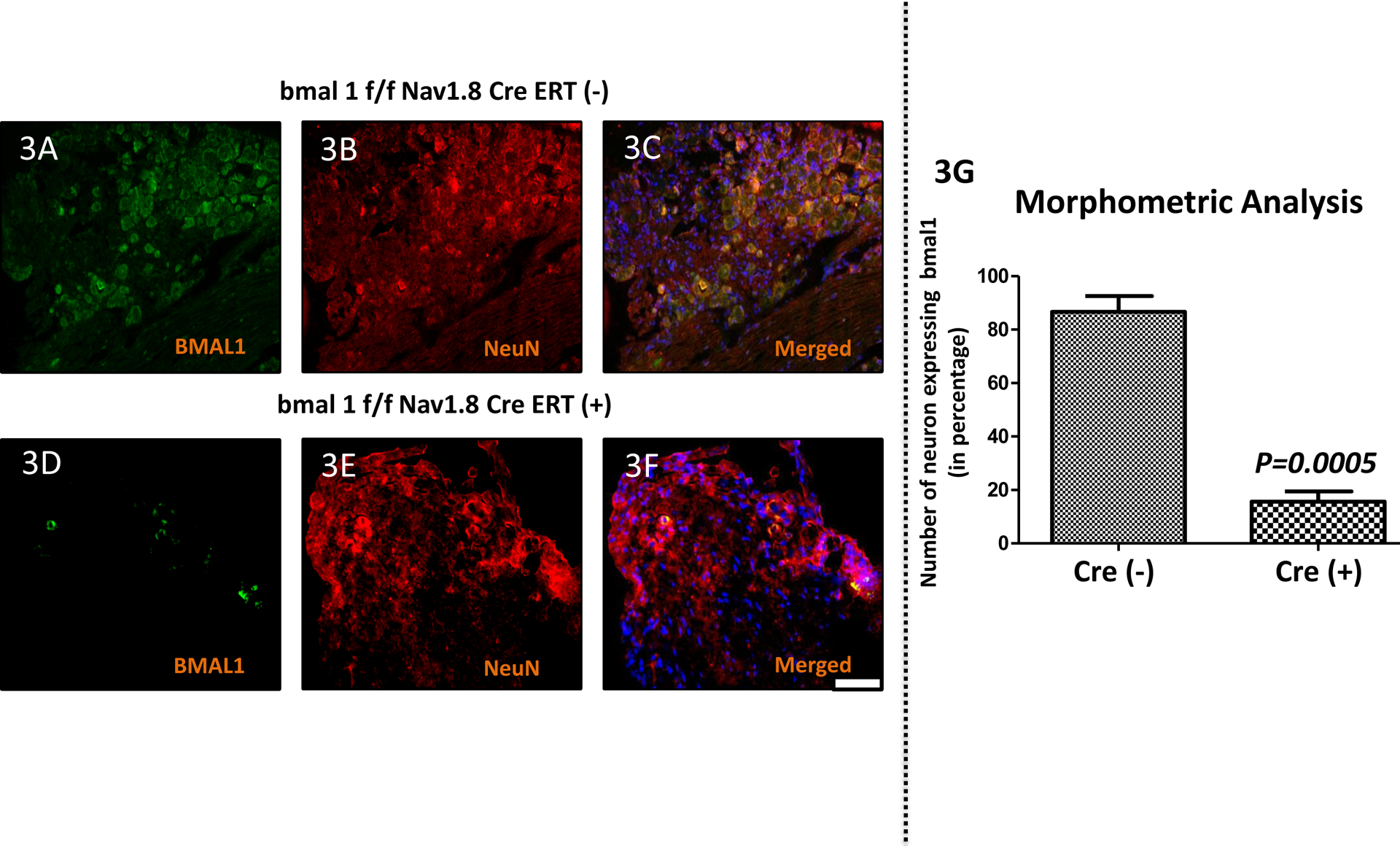
Expression of BMAL1 protein targets in the dorsal root ganglia (DRG) following OA surgery in bmal1f/f Nav1.8CreERT(−) & bmal1f/f Nav1.8CreERT(+) groups. Double immunofluorescence staining of BMAL1 (green; 3A & 3D) and NeuN (red; 3B & 3E) in DRG of bmal1f/f Nav1.8CreERT(−) & bmal1f/f Nav1.8CreERT(+) groups mice. Co-localization of the two stains appears yellow (Fig. 3C & 3F). A scale bar was 20μm and is shown in white in color for all images. Quantitative analyses of BMAL1 expression in DRGs shows lower expression in the bmal1f/f Nav1.8CreERT(+) group (Fig. 3G). Values are mean±SEM.
Mechanical hyperalgesia was measured using von Frey filament (PWT). Paw withdrawal thresholds (figure 4(A); n=5) of Cre (+) mice were significantly increased relative to the thresholds of Cre (−) mice indicating that loss of bmal1 resulted in decreased OA pain. Specifically, mean (±SEM) PWT for pain hypersensitivity were different between groups over weeks 2–8; with group effect (F=41.07, DFn=1 & DFd=24; P=0.0002) being significant, but not time effect (F=2.06, DFn=3 & DFd=24; P=0.1317). Importantly, at each time point from week 2 to week 8, PWT was higher (P<0.05 to P<0.001) in the Cre (+) vs. Cre (−) mice.
Figure 4: Loss of the clock gene Bmal1 in Nav1.8+ sensory neurons leads to reduced OA (osteoarthritis) pain.
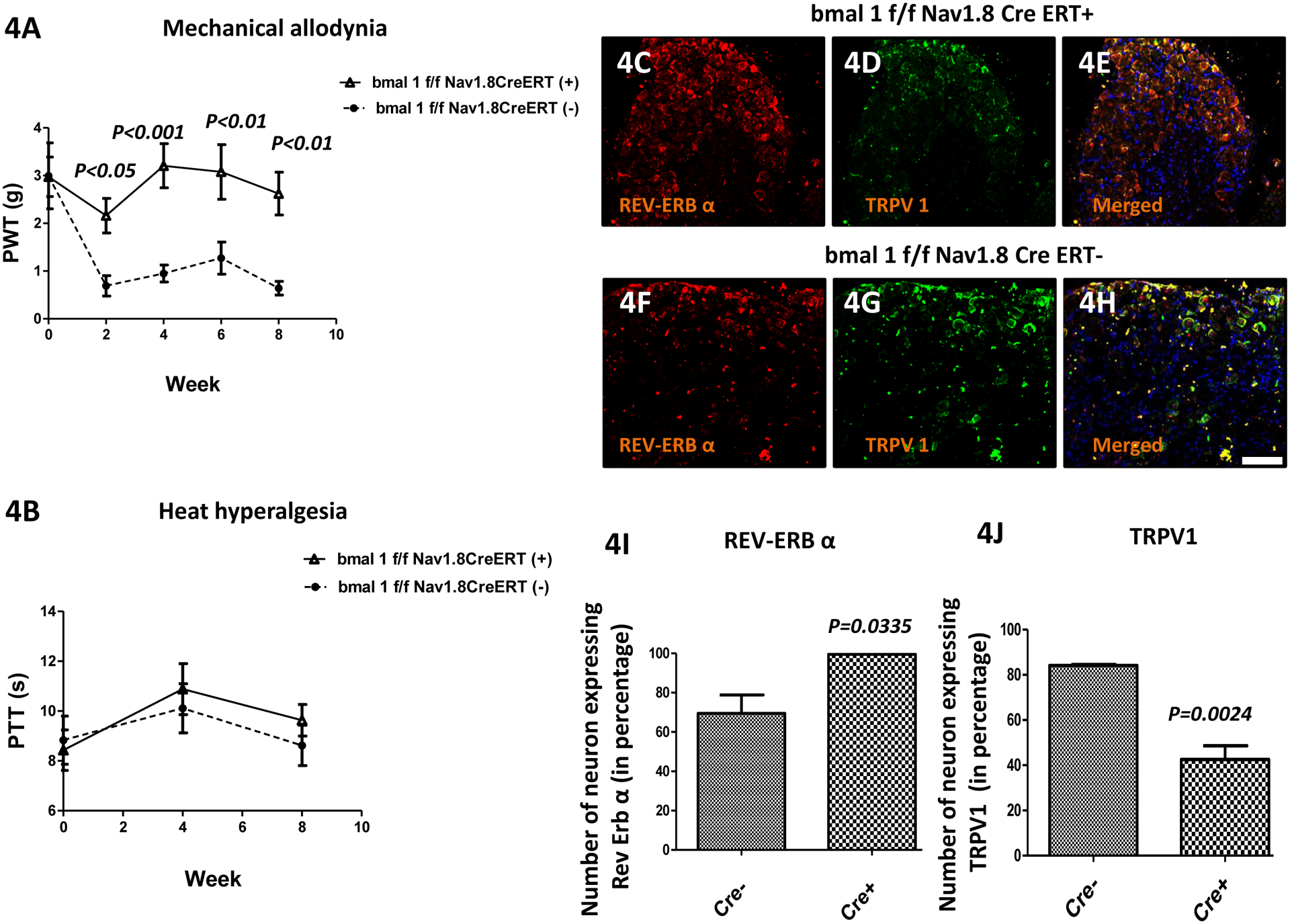
(Fig. 4A) Mechanical hyperalgesia was measured using von Frey filament test. The paw withdrawal threshold (PWT) of the bmal1f/f Nav1.8CreERT(−) group (n = 5) were low starting at 2 weeks after OA induction, and were lower than bmal1f/f Nav1.8CreERT(+) group (n = 5) at all time points. (Fig. 4B) Heat hyperalgesia was measured using hot-plate test. The paw thermal threshold (PTT) in the bmal1f/f Nav1.8CreERT(−) & bmal1f/f Nav1.8CreERT(+) group was not significantly changed. However, expression of Rev-Erb-α in the dorsal root ganglia (DRG) following OA surgery was higher and TRPV1 was lower in in bmal1f/f Nav1.8CreERT(+) vs. bmal1f/f Nav1.8CreERT(−) mice. Double immunofluorescence staining of Rev-Erb-α (red; Fig. 4C and Fig. 4F) and TRPV1 (green; Fig. 4D & Fig. 4G) in DRGs of bmal1f/f Nav1.8CreERT(−) & bmal1f/f Nav1.8CreERT(+) groups mice. Co-localization of the two stains appears yellow (Fig. Fig. 4E & Fig. 4H). A scale bar was 20μm and is shown in white in color for all images. Quantitative analyses of Rev-Erb-α and TRPV1 expression in DRG (Fig. 4I-4J). Values are mean±SEM.
Loss of bmal1 did not alter responsiveness in the hot plate test (Fig. 4B). Specifically, mean (±SEM) PTT for circadian disruption-induced pain hyperalgesia were not different between groups over weeks 2–8; with group effect (F=0.69, DFn=1 & DFd=8; P=0.4306) and time effect (F=4.86, DFn=1 & DFd=8; P=0.0586) not being significant.
Following the final behavioral test at week 8 post PMM surgery, we assessed the expression of both REV-ERBα and a nociception pain receptor (TRPV1) in peripheral sensory neurons (DRG) using double labeling immunohistochemistry (IHC). As illustrated in Figs 4C & 4F, REV-ERBα expression is elevated in the CRE (+) mice relative to the CRE (−) mice. Conversely, TRPV1 expression is reduced in the CRE (+) mice relative to the CRE (−) mice (Figs. 4D & 4G). Merged localization is illustrated in Figs. 4E & 4H and co-localization is indicated by yellow color. Quantitative analyses of REV-ERBα (Fig. 4(I) and TRPV1 (Fig. 4(J) expression in DRGs for Cre (+) and CRE (−) confirm that REV-ERBα expression is higher in the CRE (+) mice relative to the CRE (−) mice (P=0.0335), and that TRPV1 expression is lower in the CRE (+) mice relative to the CRE (−) mice (P=0.0024). Thus, loss of bmal1 results in increased REV-ERBα expression and decreased TRPV1 expression. These data are consistent with loss of bmal1 leading to decreased pain responsiveness (lower TRPV1 expression).
We further characterized the DRGs from these Cre (−) vs Cre (+) mice by double staining IHC for several pain-related proteins including substance P [red; Fig. 5(A) and 5(B)], NGF [red; Fig. 5(C) and 5(D)], CGRP [red; Fig. 5(E) and 5(F)], TrkA [red; Fig. 5(G) and 5(H)], Nav1.7 [red; Fig. 5(I) and 5(J)], NeuN [green; Fig. 5(A–J]. Co-localization appears yellow. The level of expression of substance-P (K; P=0.0004), NGF (L; P=0.0164), CGRP (M; P=0.0149), TrkA (N; P=0.0003) and Nav1.7 (O; P=0.0036) in DRGs in the Cre (+) mice were significantly lower than in the Cre (−) mice, which is again consistent with the loss of bmal1 being associated with decreased pain in a model of OA induced pain.
Figure 5: Loss of the clock gene Bmal1 in Nav1.8+ sensory neurons leads to reduced pain-related protein in DRG.
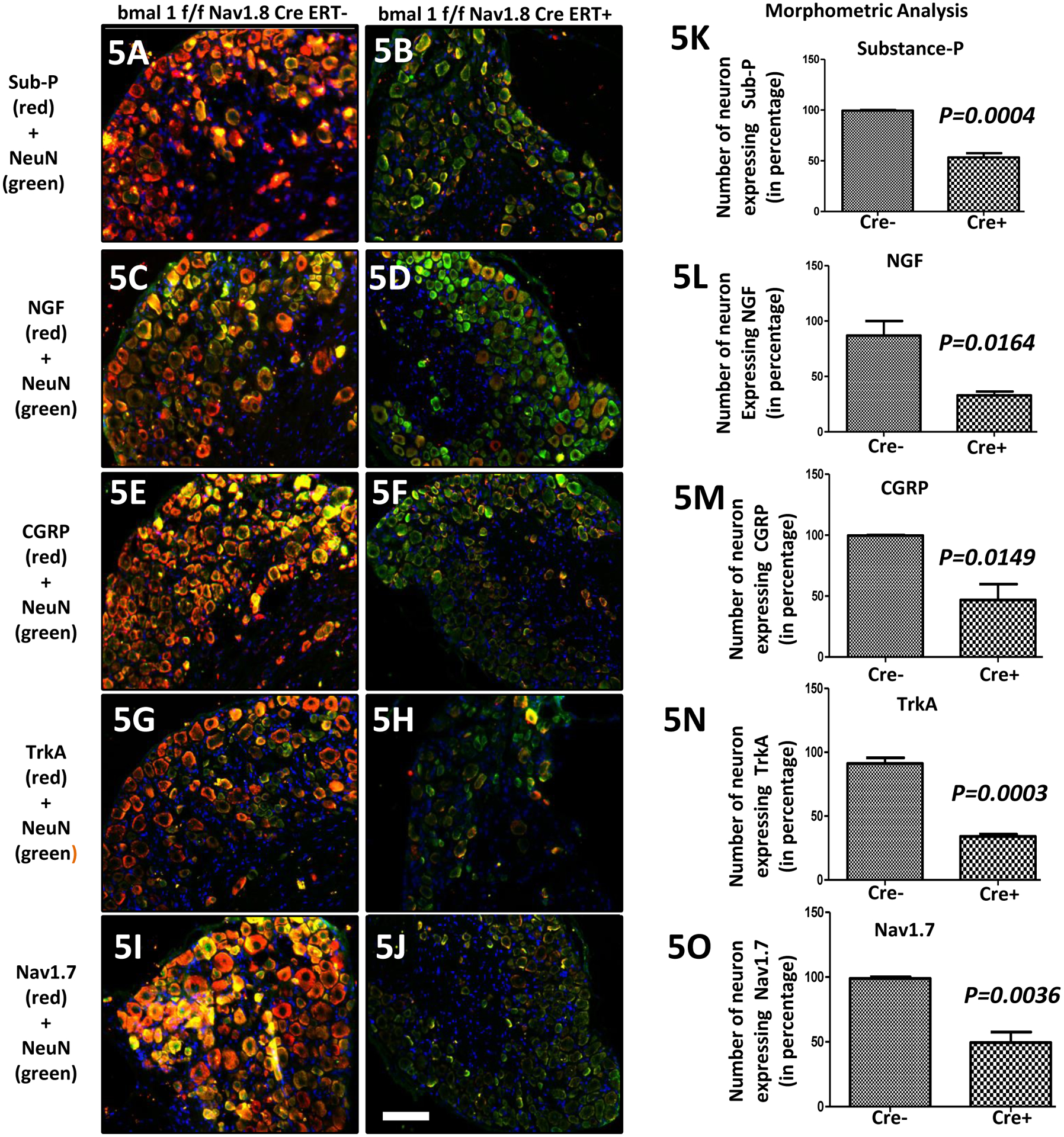
Expression of pain-related targets in the dorsal root ganglia (DRG) following OA surgery in bmal1f/f Nav1.8CreERT(−) & bmal1f/f Nav1.8CreERT(+) groups. Double immunofluorescence staining of substance P (red; 5A-5B), NGF (red; 5C-5D), CGRP (red; 5E-5F), TrkA (red; 5G-5H) and Nav1.7 (red; 5I-5J) and NeuN (green) in DRG of bmal1f/f Nav1.8CreERT(−) & bmal1f/f Nav1.8CreERT(+) groups mice. Co-localization of the two stains appear yellow. A scale bar was 20μm and is shown in white in color for all images. Quantitative analyses of substance P, NGF, CGRP, TrkA and Nav1.7 expression in DRGs shows lower expression in the bmal1f/f Nav1.8CreERT(+) group (Fig. 5K-4O). Values are mean±SEM.
REV-ERB agonist SR9009 reduces osteoarthritis induced pain.
REV-ERB is a critical regulator of the circadian rhythm and a key negative regulator of the expression of bmal1 (Amador et al., 2016a; Amador et al., 2016b; Burris et al., 2012; Butler and Burris, 2015; Crumbley and Burris, 2011; Kojetin and Burris, 2014; Sitaula et al., 2017; Wang et al., 2015). Given our data indicating that disruption of the circadian rhythm alters pain hyperalgesia and, more specifically, that loss of bmal1 is associated with reduced OA induced-pain, we hypothesized that a pharmacological activator of REV-ERB known to reduce the expression of bmal1 should have analgesic activity in the OA pain model. We administered SR9009 using two different routes: intrathecal (i.t.) and intraarticular (i.a) injection (both 30 μg/5 μl), in post-PMM surgical mice (C57BL6/J) to assess the effects of SR9009 on osteoarthritis-induced pain (Fig. 6). We first examined the effect of, single SR9009 i.t. administration (30 μg in 5 μl) on mice 13 weeks post PMM surgery (Fig. 6A). Highly significant mechanical hyperalgesia [P<0.001; paired t test] was noted in the OA mice compared to their pre-surgery baseline. Administration of SR9009 significantly reduced hyperalgesia (mean (±SEM) PWT) at 0.5 hour (P<0.01) and 1 hour (P<0.001) post SR9009 administration.
Figure 6: Analgesic activity of the REV-ERB agonist SR9009 in a model of osteoarthritis induced pain.
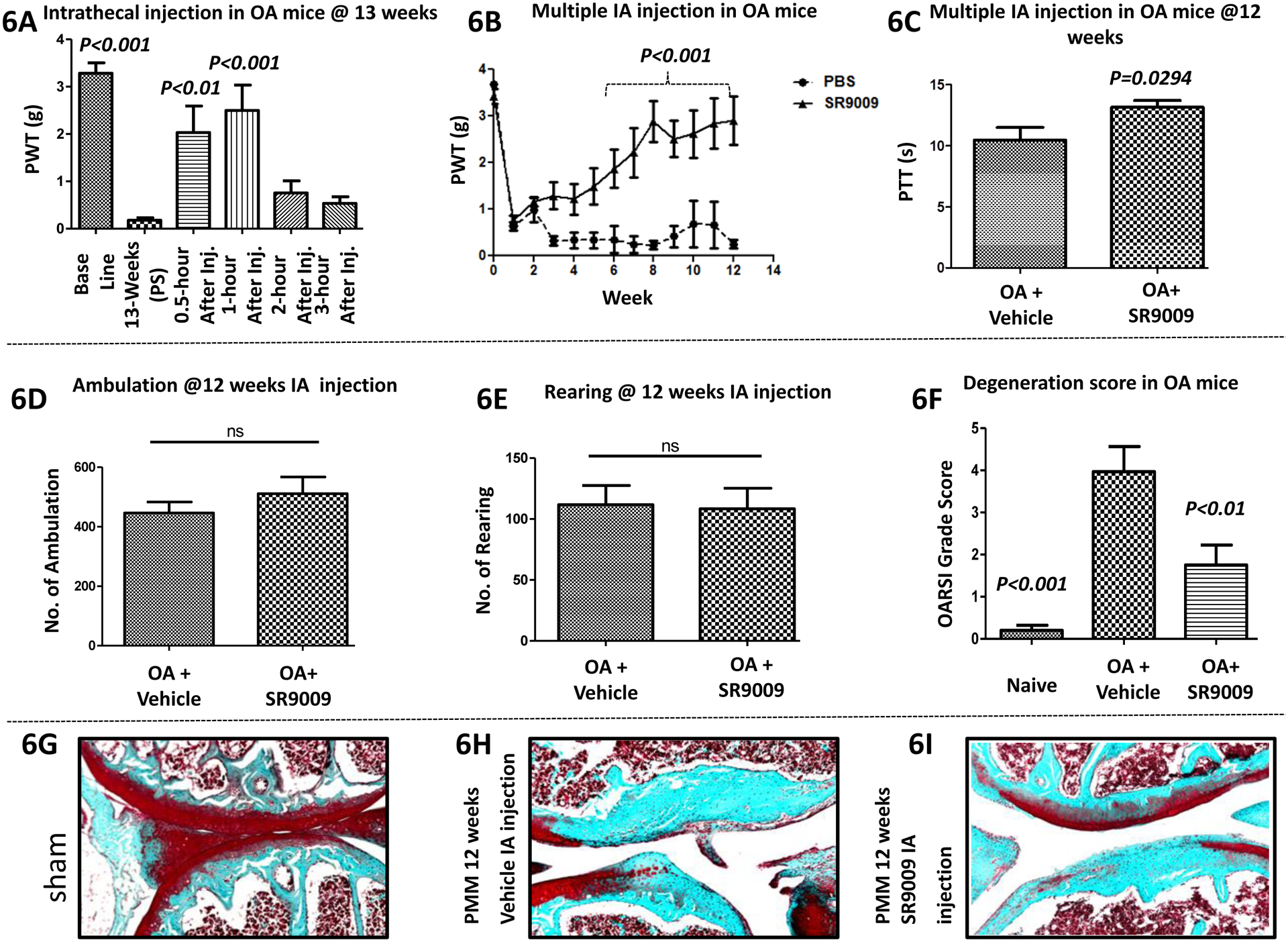
Fig.6A (single intrathecal injection; n=13) & Fig.6B (multiple intraarticular injections; n=7–8) where, mechanical hyperalgesia was measured using von Frey filament test. In OA mice, the paw withdrawal threshold (PWT) was elevated in the SR9009 treated group vs. the vehicle group. For Figs. C-I (intraarticular injection; n=7–8): (6C) Heat hyperalgesia was measured using hot-plate test. The paw thermal threshold (PTT) was elevated in the SR9009 treated group at 12 weeks (n = 7–8 per group). But, locomotor behavioral results (6D-6E) were not significant between groups. However, histological results (6F-6I) for cartilage protective and degenerative changes were shown in Fig. 6G (sham), Fig. 6H (PMM) and Fig. 6I (SR9009 treated group of mice), and histological scoring data (Fig. 6F) was better in the SR9009 treated group compared to vehicle treated OA mice. Values are mean±SEM.
We extended this line of experimentation by examining i.a. administration of SR9009 (30 μg in 5 μl at knee) in the identical OA model, followed by determination of PWT (von Frey) and PTT (hot plate), as well as additional behavioral assessments including burrowing and locomotion (ambulation and rearing). As shown in Fig. 6B, mechanical hyperalgesia (PWT) of the SR9009-treated group progressively resolved over time. Specifically, mean (±SEM) PWT for pain hypersensitivity were different over the 6–12 week period between SR9009 vs. PBS treated mice with both group effect (F=18.51, DFn=1 & DFd=108; P=0.0010)and time effect (F=4.92, DFn=9 & DFd=108; P<0.0001) being significant. Importantly, at each of the time points from week 6 to week 12 PWT was higher in the SR9009 group (P<0.05 to P<0.001). In addition, PTT (P=0.0294; t test) also was improved at 12 weeks post-surgery with SR9009 (Fig. 6C). SR9009 treatment did not alter spontaneous locomotion (Figs. 6D–E). Interestingly, SR9009 treatment was also associated with substantial reduction in cartilage degeneration as illustrated by histological examination of the joints (Figs. 6G–I) and quantitative analysis shown in Fig. 6F. Specifically, mean (±SEM) OARSI grading score for reduction in cartilage degeneration were different at the 12 week time point between the groups (F=14.46, P=0.0003), with both the naïve (P<0.001) and the OA + SR9009 (P<0.01) groups having better scores than the OA + vehicle group.
Immunohistological correlates to pain reduction.
The analgesic efficacy of SR9009 (i.a. injection) in osteoarthritis pain was further supported by immunohistological parameters indicating that the expression of a range of pain-related targets (TrkA, NGF, Nav1.7, Nav1.8 and TRPV1) were suppressed in peripheral sensory neurons (DRG) of OA mice by treatment with SR9009 as illustrated in Fig. 7 A–O.
Figure 7: Analgesic activity of the i.a. REV-ERB agonist SR9009 in a model of OA (osteoarthritis) induced pain.
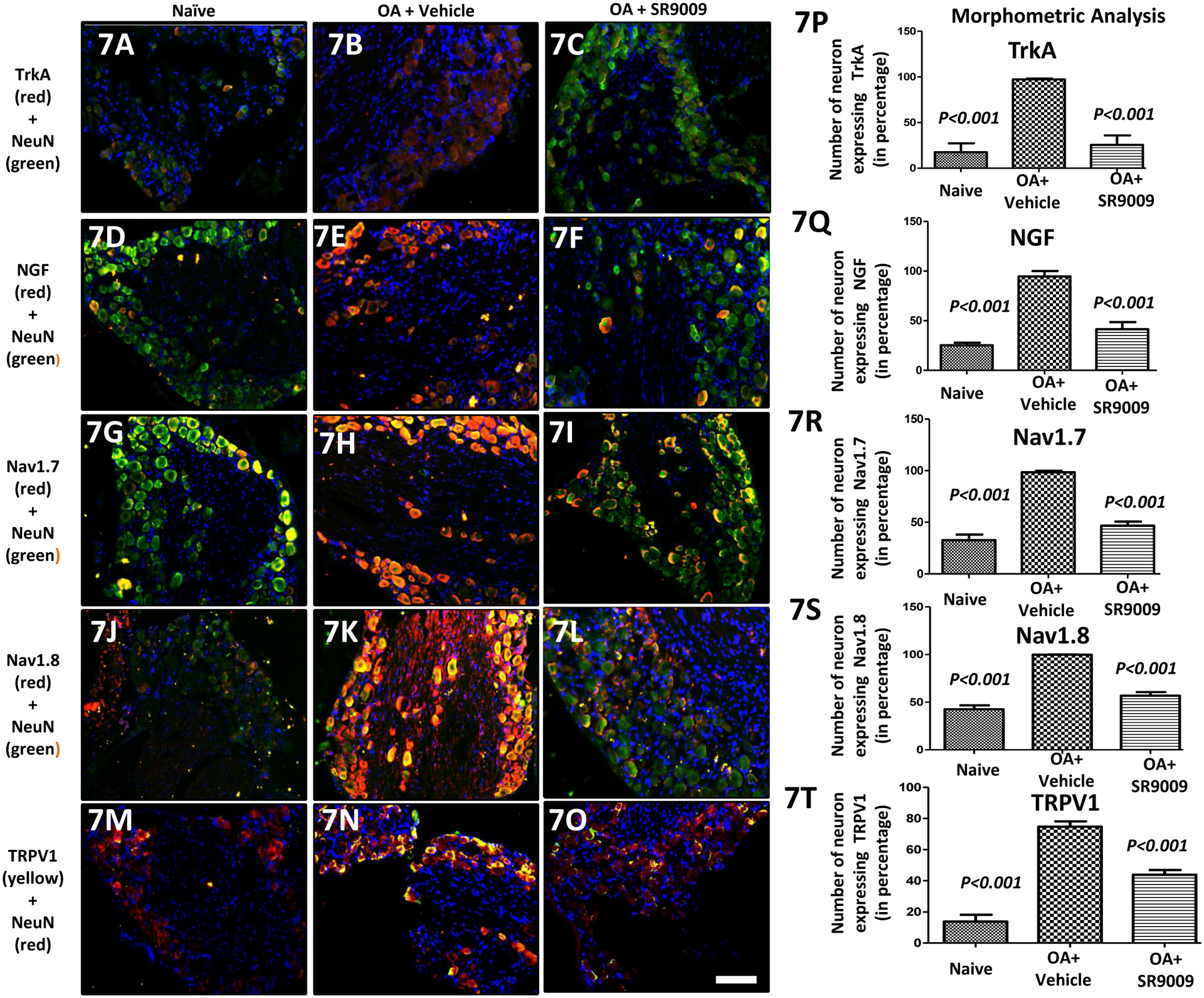
Expression of pain-related targets in the dorsal root ganglia (DRG) following groups of mice including naïve, PMM and SR9009 treated mice. Double immunofluorescence staining of TrkA (red; 7A-7C), NGF (red; 7D-7F), Nav1.7 (red; 7G-7I), Nav1.8 (red; 7J-7L) and TRPV1 (red; 7M-7O) and NeuN (green) in DRGs of Naïve, PMM and SR9009 treated groups mice. Co-localization of the two stains appears yellow. A scale bar was 20μm and is shown in white in color for all images. Quantitative analyses of TrkA, NGF, Nav1.7, Nav1.8 and TRPV1expression in DRGs (Fig. 7P-7T) showed that they were increased in OA mice, but that was reduced with i.a. SR9009. Values are mean±SEM.
Quantitative analyses, mean (±SEM) of protein (TrkA, NGF, Nav1.7, Nav1.8 & TRPV1 in Fig. 7(A–O)] expression in DRGs clearly show a substantial increase in the expression of these pain associated proteins in mice undergoing the PMM surgery vs. naïve mice (P<0.001), and most importantly, SR9009 treatment significantly reduces the expression of these proteins (P<0.001). Specifically, mean (±SEM) protein expression in sensory DRG neuron were different at the 12 week time point between the groups [TrkA (F=28.34, P=0.0001), NGF (F=40.47, P<0.0001), Nav1.7 (F=73.83, P<0.0001), Nav1.8 (F=95.43, P<0.0001), & TRPV1 (F=71.03, P<0.0001)], with both the naïve (P<0.001) and the OA + SR9009 (P<0.001) groups having better scores than the OA + vehicle group.
Our data presented above in Figs. 4 & 5 implicated Bmal1 in OA pain and given the role of REV-ERB in regulation of Bmal1 expression, we sought to characterize the relationship between Bmal1 expression of the analgesic activity of REV-ERB agonists in this model of OA-induced pain. As shown in Fig. 8, Bmal1 expression is elevated in response to the PMM surgery (Fig. 8D) where we detected significant mechanical hyperalgesia (Fig. 6B). Quantitative analyses, mean (±SEM) of BMAL1 [Fig. 8(J)] expression in DRGs clearly show a substantial increase in the expression of these pain associated proteins in mice undergoing the PMM surgery vs. naïve mice (P<0.001), and most importantly, SR9009 treatment significantly reduces the expression of these proteins (P<0.001). Similarly, mean (±SEM) of REV-ERBα [Fig. 8(K)] expression in DRGs clearly show a substantial decrease (P<0.001) in the expression of these pain associated proteins in mice undergoing the PMM surgery vs. naïve mice (P<0.001), and most importantly, SR9009 treatment significantly increase the expression of these proteins (P<0.001).
Figure 8: Analgesic activity of the i.a. REV-ERB agonist SR9009 in a model of OA (osteoarthritis) induced pain.
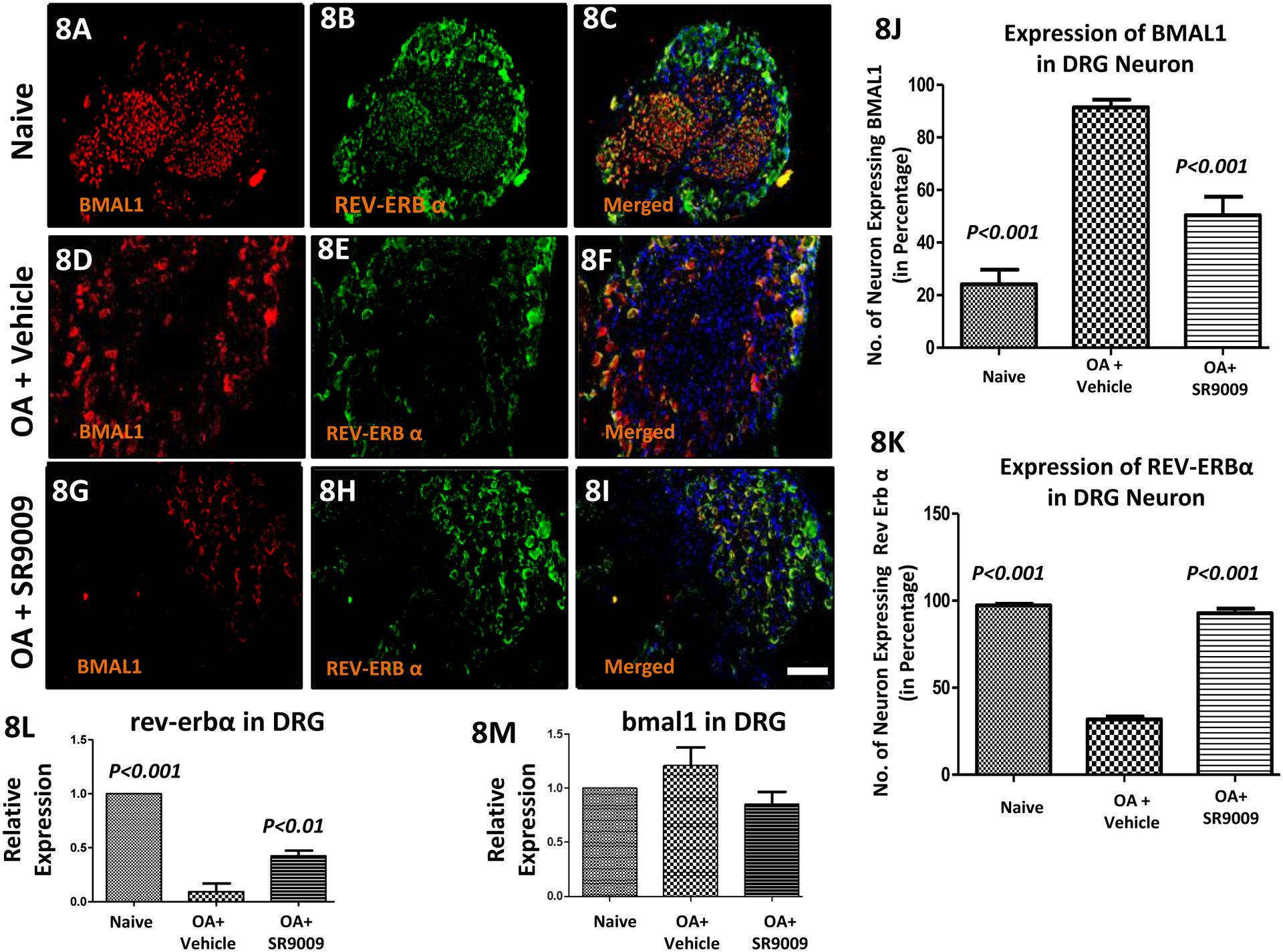
Expression of BMAL1 and REV-ERBα targets in the dorsal root ganglia (DRG) following groups of mice including naïve, OA and SR9009 treated OA mice. Double immunofluorescence staining of REV-ERBα (green; 8B, 8E & 8H) and BMAL1 (red; 8A, 8D & 8G) in DRGs of naïve, PMM and SR9009 treated groups of mice. Co-localization of the two stains appears yellow and red (Fig. 8C, 8F & 8I). A scale bar was 20μm and is shown in white in color for all images. Quantitative analyses of REV-ERBα and BMAL1 immunofluorescence expression in DRG (Fig. 8J & 8K). RT-PCR data in Fig. 8L (Rev-Erbα) and Fig. 8M (bmal1). Values are mean±SEM.
Our RT-PCR data for rev-erbα (Fig.8L) and bmal1 (Fig.8M) are consistent with the IHC results (Fig.8 (J–K). Again, OA reduces rev-erbα and treatment with the REV-ERB agonist SR9009 led to an increase in rev-erbα (Fig.8L) expression. Quantitative analyses shows that mean (±SEM) relative expression of rev-erbα in DRG clearly show a significantly substantial decrease in the expression due to progression of OA pain vs. naïve (P<0.001), and SR9009 treatment significantly increase the relative expression of that gene (P<0.01). Qualitatively, mean (±SEM) relative expression of bmal1 in DRG increases due to progression of OA pain in mice vs. naïve (Fig.8M), and SR9009 treatment reduces the expression of this gene, although these changes were not statistically significant. Therefore, we could hypotheses that OA increases bmal1 expression and treatment with the REV-ERB agonist SR9009 led to a decrease in bmal1 expression.
Discussion:
Osteoarthritis is a debilitating disease associated with cartilage degeneration and chronic pain, and it is very challenging to treat with currently approved medications. Given the link between the circadian rhythm and pain in a model of OA (Kc et al., 2015) and recent advances in the development of molecules that modulate the circadian rhythm, we sought to determine if there is a potential for development of novel therapeutics targeting components of the circadian clock for treatment of OA-associated pain.
Although environmental disruption of the circadian rhythm has been shown to be associated with increased pain in a model of OA, we utilized a genetic model to examine this effect in more detail. Tissue selective knock-out of the critical clock component Bmal1 in DRG sensory neurons led to mice that were significantly resistant to PMM-induced mechanical hyperalgesia. We noted that the expression of many pain associated proteins were also reduced in these knock-out mice. Thus, this led to the hypothesis that Bmal1 expression may be associated with increased pain in this model and that pharmacological agents that suppress Bmal1 may be useful in the treatment of OA-associated pain. Interestingly, recent developments in the field of circadian rhythms has led to ligands for several key components of the molecular clock and one of these, SR9009, is an agonist of the nuclear receptor REV-ERB which is a known inhibitor of Bmal1 expression (Butler and Burris, 2015; Crumbley and Burris, 2011; Kojetin and Burris, 2014; Sitaula et al., 2017; Wang et al., 2015).
However, from the SR9009 experiments, we hypothesized that this compound could be a novel analgesic for reducing osteoarthritis and circadian hypersensitivity (pain) because it showed a strong ability to modulate circadian clock genes (rev-erbα and bmal1) and reduce the pain related targets (TrkA, NGF, Nav1.7, Nav1.8 and TRPV1) significantly in DRG. However, from the Bmal1+ vs. Bmal1− comparison experiments, we found that the influence of clock gene (bmal1) in sensory neurons (Nav1.8) potentiate the osteoarthritis pain. Therefore, we could hypothesize that clock genes are strongly involved in osteoarthritis pain hypersensitivity for causing peripheral sensitization in sensory neurons.
Nevertheless, our established environmental disruption (12h:12h LD cycle & expose to 12h phase shifts) of circadian rhythm progressively developed significant mechanical hypersensitivity in shifted mice. Our results are consistent with recent findings where polysomnographic recording (electroencephalogram, EEG; electromyogram, EMG) showed that prolong forced activity and limited sleep opportunity (controlled environment) causes sleep loss in healthy mice (C57BL6/J) and increased sensitivity to noxious stimuli (Alexandre et al., 2017). Also, clinical data suggest that sleep and pain interact and/or are synergistic, data that comes from individuals with burn injuries, insomnia (at the time of discharge is a predictor for the development of chronic pain), fibromyalgia etc (Alexandre et al., 2017; Finan et al., 2013; Junker and Wirz, 2010; Lautenbacher et al., 2006). Tissue damage results in ectopic neural firing, neuroplasticity changes in peripheral and central sensitization in the nervous system via modulation of neuronal and non-neuronal mechanisms (Das, 2015; Pocock et al, 2007; Hutchinson et al, 2007; Thacker et al, 2007; Alford, 2007; Graham et al, 2006; Marchand et al, 2005; McMahon et al, 2005; Haydon, 2001; Raivich et al, 1996). Intensive research over the past two decades has revealed a vast array of ion channels, receptors, transporters and enzymes that are potential “druggable” targets for use in discovery programs aimed at developing the next generation of analgesic drugs (Das, 2015; Price et al., 2016; Smith et al., 2016; Smith et al., 2014; Smith et al., 2013; Viviani et al, 2007; Watkins et al, 2006; Wood et al, 2004; Watkins et al, 2003). Examples of ‘pain targets’ for potential modulation by novel analgesic agents include voltage-gated sodium (Nav) channels (Nav1.3, Nav1.7 and Nav1.8)(Wood et al., 2004), voltage-gated potassium (Kv) channels, voltage-gated calcium (Cav) channels (Cav2.2, CaV3.1, CaV3.2, CaV3.3)(Das, 2015; Gu and Lee, 2010), acid sensing ion channels (ASICs)(Gu and Lee, 2010), NMDA-type glutamate receptors(Brand et al., 2010), transient receptor potential type V1 (TRPV1) receptors(Park et al., 2008), neurokinin A, purinergic receptors, toll-like receptors, protease-activated receptors (PARs), opioid receptors, the norepinephrine transporter (NET), and cyclooxygenases (COX1/2).(Das, 2015).
But circadian rhythm dysfunction during osteoarthritis pain and other pain syndromes (fibromyalgia) is not yet validated to the level of hormones, receptors, and the consecutive signal transmission processes between and within cells, enzyme kinetics, and gene regulation. However, chronopathology finds strong links between diseases and disturbances in biological rhythm for regulation of cardiac and respiratory rhythm, the sleep/ wake pattern, the rhythm of the menstrual cycle, and disturbances in other rhythmic life processes (Junker and Wirz, 2010). Additionally, circadian rhythm dysfunction has been significant linked to many diseases in human pathophysiological events including cartilage degeneration in osteoarthritis, cancer, myocardial infarction, angina, strokes, asthma attacks, allergic reactions, rheumatic symptoms, and gastrointestinal ulcers among others, which do not occur with equal frequency over 24 h but follow a pronounced circadian pattern (Gossan et al., 2015; Junker and Wirz, 2010; Kc et al., 2015). Therefore, genetically modified, bmal1f/fNav1.8CreERT mice (Cre+ & Cre−) created by deleting bmal1 gene from Nav1.8 (sensory neuron) to validate the role of the circadian clock as a new insight into pathophysiological mechanisms underpinning osteoarthritis type of pain could have potential to identify novel targets for development of new analgesics to improve the clinical management of severe pain in patients with advanced osteoarthritis. One limitation of the current study is that we do not have activity measurements over multiple days within each L:D or D:L cycle. Another limitation of the current study is that, we do not have IHC or RT-PCR data corresponding to clock gene rhythmicity or results over multiple time points.
Similar to our results, recent literature reported that circadian rhythm dysfunction has a strong link to controlling pathological roles in articular cartilage homeostasis and degenerative joint disease including osteoarthritis (Gossan et al., 2015; Kc et al., 2015). Therefore, tissue injury and inflammation could cause vasoactive mediators (such as histamine, substance P (SP), serotonin (5-HT), nitric oxide (NO), prostaglandins and bradykinin) to be released which activate nociceptors resulting in nociception (Millan, 1999). This in turn induces release of pronociceptive neurotransmitters such as SP, calcitonin-gene-related peptide (CGRP), dynorphin (Dyn), neurokinin A (NKA), glutamate, adenosine triphosphate (ATP), nitric oxide (NO), prostaglandins (PGs), and neurotrophins such as nerve growth factor (NGF) and brain derived neurotropic factor (BDNF), from primary afferents at the first synapse in the dorsal horn of the spinal cord (Dubner, 2004; Millan, 1999; Millan, 2002; Yaksh, 2006). More recently, the important role of pro-inflammatory cytokines (e.g. tumor necrosis factor-alpha (TNF-α), interleukin-1β, interleukin-18 etc.) in peripheral sensitization mechanisms associated with persistent pain states, has begun to be appreciated (Griffis et al., 2006).
Conclusion:
We hypothesized that pharmacological targeting of clock related genes (rev-erb and bmal1) may lead to the development of novel therapeutics to treat chronic pain (osteoarthritis and fibromyalgia), sleep disorders, anxiety and stress in relation to pain. Importantly, our findings relating to circadian clock gene modulatory effects in pain (during osteoarthritis and environmental disruption of circadian rhythm) have been validated by introducing environmental disruption of circadian rhythm in mice, an osteoarthritis model in mice, and genetically modified (bmal1f/fNav1.8CReERT) mice. Our hypothesis is supported by our results that the pharmacological activator (SR9009) reduces pain significantly in mice through modulating the clock gene; rev-erbα.
Acknowledgement:
The authors thank VA BLD&R Merit Review Award (I01BC002647) to HJI; an NIH NIAMS R01 (AR062136) and R21 grant (AR067935) to H.J.I.; Arthritis Foundation to HJI and NIH R01MH093429 to T.P.B. for supporting the pharmacological targeting of clock related genes (rev-erb and bmal1) research to find the novel therapeutics (SR9009) to treat OA pain.
Abbreviations list
- OA
Osteoarthritis
- f/f
floxed-floxed; lox P; sandwiching of a DNA sequence
- PMM
Partial medial meniscectomy
- h
hour
- j
Jackson Laboratory
- LD
light-dark
- DL
dark-light
- PWT
paw withdrawal threshold
- PTT
paw thermal threshold
- DRG
dorsal root ganglion
- IT
intrathecal
- IA
intraarticular
- SO/FG
safranin-O/fast green
- OARSI
Osteoarthritis Research Society International
- RT-PCR
Real-time polymerase chain reaction
- P
probability
- SEM
standard error of the mean
- DFn
degree of freedom
- DFd
degree of distribution
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: None declared.
References:
- Alexandre C, Latremoliere A, Ferreira A, Miracca G, Yamamoto M, Scammell TE, and Woolf CJ. 2017. Decreased alertness due to sleep loss increases pain sensitivity in mice. Nature medicine. 23:768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alford L 2007. Findings of interest from immunology and psychoneuroimmunology. Manual Therapy. 12:176–180. [DOI] [PubMed] [Google Scholar]
- Alibhai FJ, LaMarre J, Reitz CJ, Tsimakouridze EV, Kroetsch JT, Bolz SS, Shulman A, Steinberg S, Burris TP, Oudit GY, and Martino TA. 2017. Disrupting the key circadian regulator CLOCK leads to age-dependent cardiovascular disease. Journal of molecular and cellular cardiology. 105:24–37. [DOI] [PubMed] [Google Scholar]
- Amador A, Huitron-Resendiz S, Roberts AJ, Kamenecka TM, Solt LA, and Burris TP. 2016a. Pharmacological Targeting the REV-ERBs in Sleep/Wake Regulation. PloS one. 11:e0162452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amador A, Wang Y, Banerjee S, Kameneka TM, Solt LA, and Burris TP. 2016b. Pharmacological and Genetic Modulation of REV-ERB Activity and Expression Affects Orexigenic Gene Expression. PloS one. 11:e0151014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Wang Y, Solt LA, Griffett K, Kazantzis M, Amador A, El-Gendy BM, Huitron-Resendiz S, Roberts AJ, Shin Y, Kamenecka TM, and Burris TP. 2014. Pharmacological targeting of the mammalian clock regulates sleep architecture and emotional behaviour. Nature communications. 5:5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A, Smith ES, Lewin GR, and Park TJ. 2010. Functional neurokinin and NMDA receptor activity in an animal naturally lacking substance P: the naked mole-rat. PloS one. 5:e15162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris TP, Busby SA, and Griffin PR. 2012. Targeting orphan nuclear receptors for treatment of metabolic diseases and autoimmunity. Chemistry & biology. 19:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AA, and Burris TP. 2015. Segregation of Clock and Non-Clock Regulatory Functions of REV-ERB. Cell metabolism. 22:197–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbo M 2013. Direct Anterior Approach Total Hip Arthroplasty: A Comparative Review of Surgical Approaches to the Hip. JBJS Journal of Orthopaedics for Physician Assistants. 1:17–22. [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, and Yaksh TL. 1994. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods. 53:55–63. [DOI] [PubMed] [Google Scholar]
- Crumbley C, and Burris TP. 2011. Direct regulation of CLOCK expression by REV-ERB. PloS one. 6:e17290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das V 2015. Chapter One - An Introduction to Pain Pathways and Pain “Targets”. In Progress in Molecular Biology and Translational Science. Vol. Volume 131. Theodore JP and Gregory D, editors. Academic Press. 1–30. [DOI] [PubMed] [Google Scholar]
- Das V, Kroin JS, Moric M, and Buvanendran A. 2017. Biochemical and Pharmacological Characterization of a Mice Model of Complex Regional Pain Syndrome. Regional anesthesia and pain medicine. [DOI] [PubMed] [Google Scholar]
- Dubner R 2004. The neurobiology of persistent pain and its clinical implications. Suppl Clin Neurophysiol. 57:3–7. [DOI] [PubMed] [Google Scholar]
- Finan PH, Goodin BR, and Smith MT. 2013. The association of sleep and pain: an update and a path forward. The journal of pain : official journal of the American Pain Society. 14:1539–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasson SS, Blanchet TJ, and Morris EA. 2007. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis and cartilage. 15:1061–1069. [DOI] [PubMed] [Google Scholar]
- Gossan N, Boot-Handford R, and Meng QJ. 2015. Ageing and osteoarthritis: a circadian rhythm connection. Biogerontology. 16:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JE, Robles TF, Kiecolt-Glaser JK, Malarkey WB, Bissell MG, and Glaser R. 2006. Hostility and pain are related to inflammation in older adults. Brain, behavior, and immunity. 20:389–400. [DOI] [PubMed] [Google Scholar]
- Griffis CA, Compton P, and Doering L. 2006. The effect of pain on leukocyte cellular adhesion molecules. Biol Res Nurs. 7:297–312. [DOI] [PubMed] [Google Scholar]
- Gu Q, and Lee L-Y. 2010. Acid-Sensing Ion Channels and Pain. Pharmaceuticals. 3:1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG 2001. GLIA: listening and talking to the synapse. Nature Reviews Neuroscience. 2:185–193. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, and Watkins LR. 2007. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. The Scientific World Journal. 7:98–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker U, and Wirz S. 2010. Review article: chronobiology: influence of circadian rhythms on the therapy of severe pain. Journal of oncology pharmacy practice : official publication of the International Society of Oncology Pharmacy Practitioners. 16:81–87. [DOI] [PubMed] [Google Scholar]
- Kc R, Li X, Voigt RM, Ellman MB, Summa KC, Vitaterna MH, Keshavarizian A, Turek FW, Meng QJ, Stein GS, van Wijnen AJ, Chen D, Forsyth CB, and Im HJ. 2015. Environmental disruption of circadian rhythm predisposes mice to osteoarthritis-like changes in knee joint. Journal of cellular physiology. 230:2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kojetin DJ, and Burris TP. 2014. REV-ERB and ROR nuclear receptors as drug targets. Nature reviews. Drug discovery 13:197–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenbacher S, Kundermann B, and Krieg JC. 2006. Sleep deprivation and pain perception. Sleep medicine reviews. 10:357–369. [DOI] [PubMed] [Google Scholar]
- Marchand F, Perretti M, and McMahon SB. 2005. Role of the immune system in chronic pain. Nat Rev Neurosci. 6:521–532. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Cafferty WB, and Marchand F. 2005. Immune and glial cell factors as pain mediators and modulators. Experimental neurology. 192:444–462. [DOI] [PubMed] [Google Scholar]
- Millan MJ 1999. The induction of pain: an integrative review. Progress in neurobiology. 57:1–164. [DOI] [PubMed] [Google Scholar]
- Millan MJ 2002. Descending control of pain. Prog Neurobiol. 66:355–474. [DOI] [PubMed] [Google Scholar]
- Milligan ED, and Watkins LR. 2009. Pathological and protective roles of glia in chronic pain. Nature Reviews Neuroscience. 10:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neogi T 2013. The epidemiology and impact of pain in osteoarthritis. Osteoarthritis and cartilage. 21:1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TJ, Lu Y, Juttner R, Smith ES, Hu J, Brand A, Wetzel C, Milenkovic N, Erdmann B, Heppenstall PA, Laurito CE, Wilson SP, and Lewin GR. 2008. Selective inflammatory pain insensitivity in the African naked mole-rat (Heterocephalus glaber). PLoS Biol. 6:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock JM, and Kettenmann H. 2007. Neurotransmitter receptors on microglia. Trends Neurosci. 30:527–535. [DOI] [PubMed] [Google Scholar]
- Price TJ, Das V, and Dussor G. 2016. Adenosine Monophosphate-activated Protein Kinase (AMPK) Activators For the Prevention, Treatment and Potential Reversal of Pathological Pain. Current drug targets. 17:908–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Bluethmann H, and Kreutzberg GW. 1996. Signaling molecules and neuroglial activation in the injured central nervous system. Keio J Med. 45:239–247. [DOI] [PubMed] [Google Scholar]
- Savvidis C, and Koutsilieris M. 2012. Circadian rhythm disruption in cancer biology. Molecular medicine. 18:1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitaula S, Zhang J, Ruiz F, and Burris TP. 2017. Rev-erb regulation of cholesterologenesis. Biochemical pharmacology. 131:68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MT, Anand P, and Rice AS. 2016. Selective small molecule angiotensin II type 2 receptor antagonists for neuropathic pain: preclinical and clinical studies. Pain. 157 Suppl 1:S33–41. [DOI] [PubMed] [Google Scholar]
- Smith MT, Lau T, Wallace VC, Wyse BD, and Rice AS. 2014. Analgesic efficacy of small-molecule angiotensin II type 2 receptor antagonists in a rat model of antiretroviral toxic polyneuropathy. Behavioural pharmacology. 25:137–146. [DOI] [PubMed] [Google Scholar]
- Smith MT, Wyse BD, and Edwards SR. 2013. Small molecule angiotensin II type 2 receptor (AT(2)R) antagonists as novel analgesics for neuropathic pain: comparative pharmacokinetics, radioligand binding, and efficacy in rats. Pain medicine. 14:692–705. [DOI] [PubMed] [Google Scholar]
- Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, Shin Y, Liu J, Cameron MD, Noel R, Yoo SH, Takahashi JS, Butler AA, Kamenecka TM, and Burris TP. 2012. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 485:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker MA, Clark AK, Marchand F, and McMahon SB. 2007. Pathophysiology of peripheral neuropathic pain: immune cells and molecules. Anesthesia & Analgesia. 105:838–847. [DOI] [PubMed] [Google Scholar]
- Thysen S, Luyten FP, and Lories RJ. 2015. Targets, models and challenges in osteoarthritis research. Disease models & mechanisms. 8:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira E, Burris TP, and Quesada I. 2014. Clock genes, pancreatic function, and diabetes. Trends in molecular medicine. 20:685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B, Gardoni F, and Marinovich M. 2007. Cytokines and neuronal ion channels in health and disease. International review of neurobiology. 82:247–263. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kojetin D, and Burris TP. 2015. Anti-proliferative actions of a synthetic REV-ERBalpha/beta agonist in breast cancer cells. Biochemical pharmacology. 96:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, and Maier SF. 2003. Glia: a novel drug discovery target for clinical pain. Nature Reviews Drug Discovery. 2:973–985. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wieseler-Frank J, Milligan ED, Johnston I, and Maier SF. 2006a. Chapter 22 Contribution of glia to pain processing in health and disease. Handb Clin Neurol. 81:309–323. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wieseler-Frank J, Milligan ED, Johnston I, and Maier SF. 2006b. Contribution of glia to pain processing in health and disease. Handbook of Clinical Neurology. 81:309–323. [DOI] [PubMed] [Google Scholar]
- Willis WD Jr, and Coggeshall RE. 2004. Sensory Mechanisms of the Spinal Cord: Volume 1 Primary Afferent Neurons and the Spinal Dorsal Horn. Springer. [Google Scholar]
- Wood JN, Boorman JP, Okuse K, and Baker MD. 2004. Voltage-gated sodium channels and pain pathways. Journal of Neurobiology. 61:55–71. [DOI] [PubMed] [Google Scholar]
- Yaksh TL 2006. Calcium Channels As Therapeutic Targets in Neuropathic Pain. The Journal of Pain. 7:S13–S30. [DOI] [PubMed] [Google Scholar]