

Abstract
Systemic lupus erythematosus (SLE) is the archetype of a systemic autoimmune disease, but the multifaceted pathogenic mechanisms leading to inflammation and organ damage are not fully understood. Homozygous deficiency of complement C1q, the first component of the classical pathway of complement, is strongly associated with the development of SLE, thus pointing at a primarily protective role of C1q. However, while most SLE patients do not have hereditary C1q deficiency, there is indirect evidence for the importance of C1q in the inflammatory processes of the disease, including hypocomplementemia as a result of activation via the classical pathway, deposition of C1q in affected tissues and the occurrence of autoantibodies against C1q (anti‐C1q). The growing body of knowledge on anti‐C1q led to the establishment of a biomarker that is used in the routine clinical care of SLE patients. Exploring the binding characteristics of anti‐C1q allows to understand the mechanisms, that lead to the expression of relevant autoantigenic structures and the role of genetic as well as environmental factors. Lastly, the analysis of the pathophysiological consequences of anti‐C1q is of importance because C1q, the target of anti‐C1q, is a highly functional molecule whose downstream effects are altered by the binding of the autoantibody. This review summarises current study data on anti‐C1q and their implications for the understanding of SLE.
Keywords: anti‐C1q antibodies, C1q, complement, SLE, systemic lupus erythematosus
Autoantibodies against complement C1q (anti‐C1q) are an exciting biomarker of disease activity and for the occurrence of active proliferative nephritis in patients with systemic lupus erythematosus. The exploration of binding characteristics of anti‐C1q and its functional consequences provides important insights into pathogenic mechanisms of the disease involving complement deposition and activation, the clearance mechanisms of apoptotic cell debris, phagocyte function and the role of genetic as well as environmental factors.
Introduction
Systemic lupus erythematosus (SLE) is the archetype of a systemic autoimmune disease, but the multifaceted pathogenic mechanisms leading to inflammation and organ damage are not fully understood. SLE is considered to be the consequence of intrinsic (genetic) as well as extrinsic (environmental) factors. 1 Variations in different genes, that are mostly involved in the regulation and function of the immune system, can affect the risk of developing SLE, and often multiple genetic factors are thought to play a role. Homozygous deficiency of complement C1q was found to be the most potent genetic disease susceptibility factor for human SLE, thus pointing at a critical role for complement C1q in the pathogenesis of SLE. 2 , 3 C1q is the recognition and starter molecule of the classical pathway of the complement system. It is a 460 kDa glycoprotein consisting of 18 polypeptide chains that have an N‐terminal collagen‐like domain. These chains form six triple helices assembling to a structure that resembles a bouquet of tulips with the stalks being formed by the collagen‐like regions while the C‐terminal parts form the flower‐like globular head regions of the molecule, which primarily mediate the binding of C1q. A comprehensive (but not exclusive) explanation for the role of C1q in SLE was provided by the so‐called ‘waste disposal hypothesis’. 2 , 4 This hypothesis assumes that systemic autoimmunity in SLE is driven by the defective clearance of apoptotic cells, that could become antigenic and, as a consequence, induce an autoimmune response. Typical autoantigens (e.g. nuclear antigens and phospholipid‐associated antigens), that are targeted in SLE, are found on the surface of apoptotic cells. 5 The non‐inflammatory phagocytosis of apoptotic cells is decreased in SLE patients, 6 , 7 and C1q has been shown to bind to apoptotic cells and to accelerate the clearance of self‐antigens generated during apoptosis. 8 , 9 , 10 , 11 As suggested by data from Ogden et al., 12 C1q binds to apoptotic cells via the globular heads and directly initiates their uptake by interacting with phagocyte receptors for its collagenous tail region. In addition to this function as a bridging molecule for phagocytes, the phagocytosing capacity of professional phagocytes is further increased by a priming effect of C1q, leading to an indirect enhancement of phagocytosis after exposure of phagocytes to bound C1q. 13 , 14 Moreover, macrophages from C1q‐deficient mice and C1q‐deficient patients show a delayed clearance of apoptotic cells in vitro. 15 Thus, C1q may be involved in preventing autoimmunity through a role in the disposal of dying and dead cells. Although C1q is not essential for the clearance of apoptotic cells, 16 but has an accelerating effect on phagocytosis, one needs to keep in mind that even small delays in clearance might have detrimental effects over an extended period of time, considering the estimation that approximately one million cells die per second in the course of physiological human tissue turnover. 17 In addition, the way how apoptotic cells are recognised and eliminated by the presence/absence of C1q is important, as C1q was shown to modulate the phenotype of professional phagocytes. 13 , 14 , 18
In the context of the ongoing autoimmune process, inflammation in SLE patients involves many components of the immune system, of which again complement C1q seems to be of importance. While most SLE patients do not have hereditary C1q deficiency, there is indirect evidence for the importance of C1q in the inflammatory processes of the disease:
Low levels of the components of the classical pathway of complement, including C1q, are frequently observed in SLE patients and often can be attributed to complement activation, particularly during flares. 19 , 20 Interestingly, low complement levels were found to be associated with a decreased uptake of dying cells in SLE patients. 21
C1q deposition is a specific histological finding in severe lupus nephritis where it is detected in electron‐dense deposits of the renal subendothelial space and/or the glomerular basement membrane. 22 , 23
In about one third of unselected SLE patients and in more than 90% of patients with proliferative lupus nephritis, autoantibodies targeting C1q (anti‐C1q) can be detected, and these anti‐C1q strongly correlate with disease activity and hypocomplementemia. 24
The growing body of knowledge on anti‐C1q autoantibodies (anti‐C1q) has led to the establishment of a biomarker that is used in the routine clinical care of patients with SLE. Additionally, exploring the binding characteristics of the antibody and its functional consequences also provides insights into the complex pathogenic mechanisms of the disease. This view is based on the observations that (1) the target of anti‐C1q is a highly functional molecule, 25 whose functions are likely to be altered by the binding of an autoantibody, (2) C1q itself has been shown to have a strong association with SLE as outlined before and (3) anti‐C1q show binding characteristics allowing insights into basic mechanisms of the disease. This review summarises the implications of current study data on anti‐C1q for the understanding of SLE.
Association of anti‐C1q with disease
Anti‐C1q autoantibodies (anti‐C1q) were first suspected by Agnello et al. in 1971 26 and eventually clearly described in 1988. 27 , 28 Anti‐C1q were mostly seen in patients with SLE and the Hypocomplementemic Urticaria Vasculitis Syndrome (HUVS), but they are not very specific for these diseases. Although to a lesser extent, anti‐C1q can also be found in patients with different autoimmune and/or renal diseases (e.g. mixed connective tissue disease, rheumatoid vasculitis, cryoglobulinemia, acute poststreptococcal glomerulonephritis, autoimmune thyroid disease and others), in HIV‐positive patients and even in healthy individuals. 29 , 30 , 31 , 32 , 33 In a normal population, positivity for anti‐C1q ranged from 4% in middle aged (40–49 years old) up to 18% in the elderly (70–79 years old). 34 Furthermore, positivity for anti‐C1q among normal blood donors strongly depends on the composition of the assay (usually, anti‐C1q are quantified by ELISA using a high molar salt buffer to avoid binding of immune complexes to plate‐bound C1q) and the definition of a positive test result. 35 Consequently, anti‐C1q cannot be regarded as a diagnostic marker of SLE, even though the highest titres of anti‐C1q were described in SLE patients and patients with the closely related HUVS. 36 , 37 However, anti‐C1q have been found to be a useful marker of disease activity in SLE. 38 In particular, anti‐C1q levels and the percentage of SLE patients being positive for anti‐C1q strongly dependent on the presence of active lupus nephritis at the time of sampling. 24 , 35 As a biomarker for the occurrence of proliferative lupus nephritis in patients with SLE, anti‐C1q seem to be superior to determining ‘classical’ parameters such as anti‐dsDNA or complement (C3, C4). 39 These observations also showed that anti‐C1q, in contrast to other autoantibodies in SLE, tend to disappear in patients with no or low disease activity (i.e. declining to titres being below the lower limit of detection). Most strikingly, in the absence of detectable anti‐C1q the development of severe lupus nephritis in the following months is very unlikely with a negative predictive value ranging up to 100%. 24 , 39 , 40 , 41 , 42 , 43 , 44 This is not only useful information for the clinician but also suggests a pathogenic role of anti‐C1q: Anti‐C1q seem to be an essential but not sufficient factor for the development of proliferative lupus nephritis. 44
Determining anti‐C1q may also be of clinical help in other diseases, for example acute poststreptococcal glomerulonephritis (APSGN), which shares several characteristics with lupus nephritis, and autoimmune thyroid disease (AITD). In both entities, anti‐C1q were found to correlate with disease severity. In children with APSGN anti‐C1q were associated with hypocomplementemia, proteinuria, elevated creatinine, occurrence of oliguria, hypertension and delayed resolution of the disease, and in patients with AITD with the thyroid function (hypo‐ and hyperthyroidism). 30 , 31 , 32 These findings suggest that anti‐C1q have pathogenic roles outside of SLE as well, but the role of anti‐C1q in those diseases is much less well studied, partially because of the relative rareness and/or the more difficult determination of disease activity, and therefore remains more speculative.
Characteristics of anti‐C1q
Anti‐C1q are mostly IgG with a predominance of the IgG1 and IgG2 subclasses. 45 , 46 , 47 , 48 In contrast to immune complexes, that bind to the globular heads of C1q, anti‐C1q mostly bind to the collagen‐like region of the molecule. 49 Their binding is mediated via the antigen‐binding fragments (Fab) and of high affinity. Interestingly, anti‐C1q do not or only weakly bind to unbound (soluble) C1q or to C1q within the C1 complex which consists of the three subcomponents C1q, C1r and C1s. 27 , 28 Thus, a possible pathogenic role might be limited to tissues or organs in which C1q is deposited and not associated with the serine proteases C1r and C1s. 50 In contrast to plasma C1q, which mainly assembles with C1r/s to form the C1 complex, free C1q is locally synthesised in tissues, mainly by dendritic cells and macrophages. 51 , 52 , 53 Such a situation would also be given in lupus nephritis by the presence of resident as well as infiltrating macrophages and dendritic cells, 54 , 55 and C1q might even be produced by resident kidney cells, for example glomerular mesangial and/or epithelial cells. 56 Of note, anti‐C1q were found to enhance the C1q production of macrophages in vitro. 57 In line with the hypothesis of a local effect, anti‐C1q could be isolated from glomerular basement fragments of patients with proliferative lupus nephritis, 58 and the deposition of anti‐C1q seems to occur via binding to deposited C1q. 50 , 59 Furthermore, C1q undergoes changes in conformation upon binding, 60 and Golan et al. 61 could show that binding of C1q to immune complexes or other C1q‐binding surfaces exposes new antigenic sites. These observations led to the conclusion that anti‐C1q bind to one or several cryptic epitopes that are exposed on the collagen‐like region of C1q as a result of changes of conformation of the molecule that occurs after binding to immune complexes or other specific surfaces. Exploring these conformational properties, anti‐C1q were found to specifically target C1q bound on early apoptotic cells, whereas anti‐C1q do not bind to C1q bound on immunoglobulins or artificial immune complexes. 62 Although C1q bound to immune complexes exposes neoepitopes as well, these neoepitopes were not recognised by patient‐derived anti‐C1q, suggesting that the conformational changes of C1q are complex and strongly dependent on the nature of the ligand. This observation does not only question the classical view on the role of immune complexes in SLE but also provides a direct link between SLE, apoptosis and C1q, thus supporting the hypothesis that SLE is driven by impaired clearance of apoptotic material.
As shown for most autoantibodies being detected in lupus, classical anti‐C1q were not yet found to cross‐react with other autoantigens. 43 , 63 More specifically, anti‐C1q do not cross‐react with collagen type II or the structurally related collectins (lung surfactant protein A, mannan‐binding lectin, bovine conglutinin). In addition, anti‐C1q usually do not bind to denatured C1q, suggesting that the antibody only recognises intact C1q molecules or fragments of the collagenous part expressing the same conformation‐dependent epitopes of bound C1q. 64 Up to now, knowledge on the precise epitope(s) of anti‐C1q is limited. In an early attempt to compare anti‐C1q derived from patients with different diseases, no striking differences in binding characteristics between anti‐C1q from SLE patients and patients with HUVS could be found. 65 In both diseases, anti‐C1q were found to bind to neoepitopes expressed on the collagen‐like region of C1q upon binding of C1q to a surface. However, Western blot analyses suggested differences in epitope specificity between the two entities. 64 Thus, assuming that more than one neoepitope is expressed on the collagen‐like region upon binding of C1q, it is possible that anti‐C1q may differ in their epitope specificity between different diseases. In a more recent attempt of an epitope mapping for anti‐C1q as occurring in SLE, Vanhecke et al. identified a major linear epitope of C1q targeted by anti‐C1q, the so‐called ‘A08’. 66 This epitope is cryptic and confirms the notion that anti‐C1q primarily bind to neoantigens that are only exposed after the binding of C1q to a target structure. 66 , 67 In addition, the identification of ‘A08’ as an epitope allowed establishing a peptide‐specific anti‐C1q ELISA that could be used as a diagnostic tool. 68 The ‘A08’ epitope is also of interest as it includes the so‐called arginine‐rich region of the collagen‐like region of the C1q A chain that had been shown to mediate the binding of non‐immunoglobulin molecules to C1q, such as lipopolysaccharide (LPS), C‐reactive protein (CRP), DNA, heparin, fibronectin, urate crystals, human serum amyloid P and von Willebrand factor. The binding of at least some of these molecules has even been shown to activate the classical pathway, 69 , 70 , 71 , 72 , 73 which could also be the case for the binding of anti‐C1q. Last, as a result of the position of the positively charged amino acid sequence, the peptide residues 14–26 of the C1q A chain (that are mostly identical with ‘A08’) could potentially be presented on HLA to be recognised by appropriate T‐cell receptors. 74
Pathogenic mechanisms leading to the generation of anti‐C1q
In the context of an impaired clearance of apoptotic material, it is plausible that C1q, bound to the surface of apoptotic bodies by its role as a bridging molecule, becomes antigenic in analogy to nuclear components that usually are not exposed to the immune system. Prolonged exposure of new epitopes to the immune system eventually could lead to an autoimmune response against C1q. In this respect, processes leading to the development of anti‐C1q would not fundamentally be different from those being believed to drive the generation of antinuclear and antiphospholipid antibodies. However, as a result of the specificity of anti‐C1q for apoptotic cell surface‐bound C1q, anti‐C1q provide specific support for the hypothesis that impaired clearance of apoptotic material indeed is a fundamental problem in patients with SLE. In addition, the analysis of human monoclonal anti‐C1q Fabs generated from a bone marrow‐derived phage display library of an SLE patient demonstrated that the development of anti‐C1q is the consequence of an antigen‐driven, affinity‐matured immune response. 75 Beyond this characteristic, the analysis of monoclonal anti‐C1q did not reveal antibody properties that would suggest the use of unusual germline sequences or abnormal maturation processes. Thus, as a starting point of the disease, it might not be the antibody response that is abnormal in SLE but the processes that allow the exposure of autoantigens such that they can be recognised by antibodies. 76 Understanding these processes will be of crucial importance for the understanding of SLE. It is of interest to note that anti‐C1q were more likely to be elevated in unaffected siblings of patients as well if the patient had elevated levels of that antibody. 77 Furthermore, anti‐C1q were also detectable in the healthy parents of the SLE probands, and there was a strong association between the presence of anti‐C1q in the (healthy) parents and their healthy, unaffected children. Therefore, anti‐C1q formation is at least partly genetically determined.
Apart from the expression of the relevant antigenic sites, additional triggers for the generation of anti‐C1q are necessary. In this regard, the striking sequence homology of ‘A08’ with an antigenic site of the Epstein–Barr virus (EBV) led to the hypothesis that anti‐C1q are the consequence of an immune response primarily targeting EBV. The observation was of interest since EBV infection seems to be of crucial importance for the development of SLE. This hypothesis is based on observational data demonstrating a 95–99% seropositivity for EBV in adult SLE patients, which exceeds the rate of seropositivity found in matched healthy controls (about 85 to 95%). 78 , 79 In addition, SLE patients were shown to have a more diverse antibody response against the EBV‐derived antigenic structure Epstein–Barr virus nuclear antigen‐1 (EBNA‐1) than controls, which includes a more pronounced immune response against the C‐terminal regions of EBNA‐1. 80 In fact, some C‐terminal antigenic sites of EBNA‐1 are able to trigger autoantibody production in vivo as observed in lupus patients (i.e. antibodies against double‐stranded DNA, Ro, the Smith antigen (Sm) and the U1 nuclear ribonucleoproteins (nRNP)). 81 , 82 , 83 In line with these findings, we could demonstrate, that anti‐C1q can be induced in vivo by the Epstein–Barr virus‐derived antigenic site ‘EBNA348’ (also being part of the C‐terminal EBNA‐1). In addition, while for example there was no sequence homology between Ro and the antigenic site of EBNA‐1, that was described to induce anti‐Ro, 81 ‘EBNA348’ has a short sequence homology with ‘A08’ of C1q that includes the amino acids being essential for the binding of anti‐C1q. 79 However, more study data are required to determine the role of EBV infection in the generation of anti‐C1q.
Taken together, besides the abnormal exposure of antigen, additional triggers for the generation of anti‐C1q are necessary. One such trigger could be a previous EBV infection with an aberrant antibody response against the virus leading to the development of the autoimmune response in SLE. This hypothesis is outlined in Figure 1b.
Figure 1.
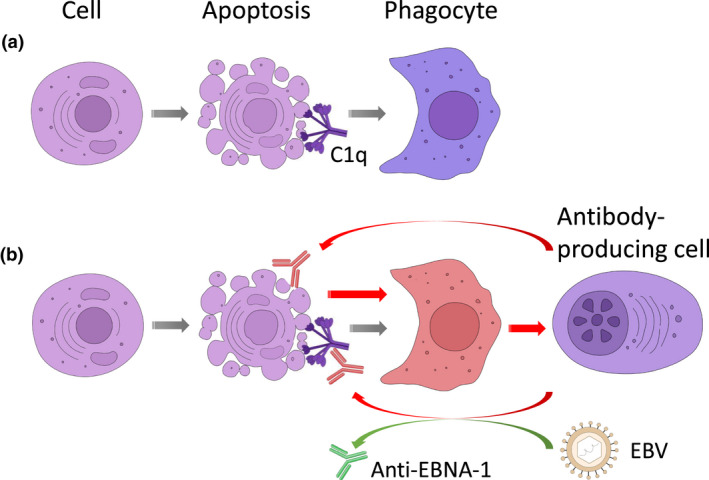
Conceptual model of the origin of anti‐C1q. (a) Physiologically, the clearance of apoptotic cells by professional phagocytes is an anti‐inflammatory process partially mediated by bound C1q. (b) In the context of an altered clearance of apoptotic cells by phagocytes, those phagocytes could induce an immune response, eventually leading to the production of autoantibodies targeting antigens that are exposed on the surface of apoptotic cells, such as many intracellular antigens as well as surface‐bound bridging molecules including C1q. The process of autoantibody generation against C1q seems to be facilitated by the previous infection with the Epstein–Barr virus (EBV) as a result of the presence of preformed antibodies cross‐reacting with EBNA‐1 of EBV and cryptic antigens being expressed on bound C1q.
Consequences of the binding of anti‐C1q
With regard to potential secondary pathogenic mechanisms induced by the presence of anti‐C1q, the clinical findings mentioned above suggest that the presence of anti‐C1q is necessary but not sufficient for the development of severe lupus nephritis. This hypothesis is supported by observations made in more than 1000 individuals of families in which at least one member had SLE showing that anti‐C1q were associated with a history of lupus nephritis, but the mere presence of anti‐C1q, for example in healthy relatives, was not. 77 Thus, pathogenic effects of anti‐C1q appear to be limited to individuals that are susceptible to lupus, and in these individuals, anti‐C1q accelerate or exacerbate the disease.
There are no data showing that anti‐C1q directly activate the complement cascade in normal blood, but it is at least likely that binding of anti‐C1q to C1q leads to secondary amplification of complement activation, for example by increasing the amount of deposited IgG, which enhances complement activation and may eventually result in a self‐perpetuating mechanism. 84 This assumption is supported by observations made in an in vitro system in which bound anti‐C1q from SLE patients were found to secondarily activate the complement system via the classical and lectin pathways. 85 However, the effect of anti‐C1q on complement activation is controversial as another in vitro study found that affinity‐purified anti‐C1q, when compared to control IgG, inhibited the deposition of C3c on circulating immune complexes in a dose‐dependent manner. 86 Differences in observations can be explained by the different experimental settings that were used and that ‐ among other factors ‐ might lead to different exposure of binding sites of C1q as outlined before.
It is also possible that anti‐C1q interfere with the physiological roles of C1q that are not solely dependent on downstream complement activation, for example the uptake of immune complexes and/or apoptotic bodies. 50 As a result of binding characteristics of anti‐C1q, this interference is expected to inhibit or to alter the effects of bound C1q. In an in vivo model, Trouw et al. demonstrated that the injection of a monoclonal anti‐C1q antibody that is specific for the collagen‐like region of C1q resulted in glomerular deposition of the antibody together with C1q. This deposition was accompanied by mild neutrophil influx but could not induce severe renal damage. 87 However, the situation was different when additional glomerular immune complexes had been induced by a pre‐injection of subnephritogenic doses of a C1q‐fixing anti‐glomerular basement membrane (anti‐GBM) antibody. In this setting, the following injection of the anti‐C1q antibody could exacerbate the pre‐existing subclinical renal disease. Although the model does not precisely reflect the situation in SLE patients for several reasons, the authors could demonstrate that an anti‐C1q antibody can lead to disease exacerbation. In addition, concerning the molecular mechanisms, the study could demonstrate that deposition of C1q, complement activation involving C4 as well as C3, and Fcγ receptors mediate inflammation after binding of anti‐C1q, pointing to complex downstream effects. These findings were supported by more recent in vitro data in which anti‐C1q were found to significantly decrease the phagocytosis of early apoptotic cells being opsonised with C1q by macrophages. 86 In addition, Thanei et al. 14 could demonstrate that anti‐C1q induced a pro‐inflammatory phenotype in human monocyte‐derived macrophages, by reversing the anti‐inflammatory effects of C1q alone. This pro‐inflammatory effect was mediated by FcγRII, and macrophages that were exposed to C1q/anti‐C1q complexes had a significantly lower phagocytic activity of early and late apoptotic cells that was accompanied by a reduced Mer tyrosine kinase expression. Thus, anti‐C1q seem to not only exacerbate complement activation and to alter the function of C1q directly, but even enhance clearance defects of apoptotic material as being observed in SLE patients and, as a consequence, might induce a vicious circle. This concept is summarised in Figure 2.
Figure 2.
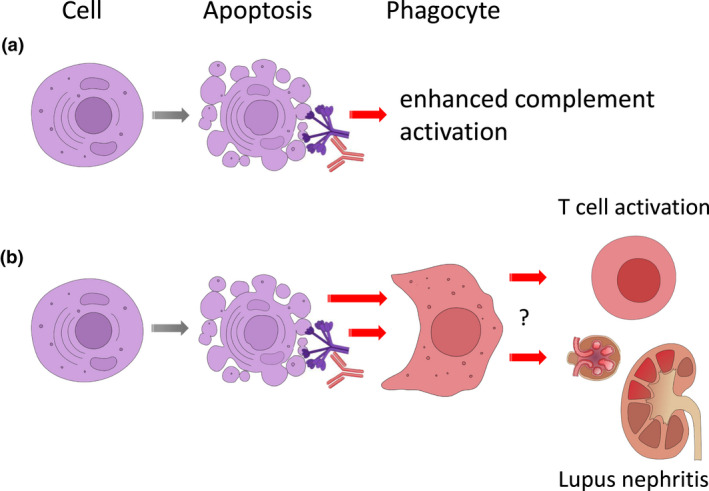
(a) Secondary effects of anti‐C1q. Once being present, anti‐C1q can enhance complement activation and (b) secondarily enhance the induction of pro‐inflammatory phagocytes with reduced phagocytic activity. These inflammatory cells together with complement activation may trigger further downstream inflammation including T‐cell activation, and lead to the development of proliferative lupus nephritis.
Open questions
Although our understanding of SLE and of anti‐C1q has clearly improved over the last decades, a number of unanswered questions remain. A more obvious one is the question of why anti‐C1q have a strong association with renal lupus, although many mechanisms outlined above would point to a rather systemic or at least not kidney‐specific effect. With regard to the expression of C1q epitopes, C1q deposited in glomeruli might express critical epitopes that are not exposed in other tissues. Expression of neoepitopes on bound C1q does not seem to have a simple yes/no character but to follow a more complex pattern with the appearance of certain neoepitopes being dependent on the binding partner for C1q. 62 Of note, accumulation of apoptotic cells in glomeruli of SLE patients or secondary deposition of apoptotic material in the glomerulus could provide such a critically important surface as a binding partner for C1q. 88 , 89 , 90 However, of course, additional factors might play an essential role as well.
Among the factors that have been described to be involved in the development of SLE, complement C1q is by far not the only one. Another one, whose important role is supported by increasing evidence, are type I interferons (IFN). The complex role of IFN type I in SLE has been reviewed recently. 91 Regarding C1q, an association between C1q deficiency and defective regulation of IFN‐α has been reported, 92 , 93 and C1q‐containing ICs were shown to markedly reduce the expression of the majority of IFN‐response genes. 94 However, how the presence of anti‐C1q influences the effects of C1q on the interferon signature remains to be examined.
Another question that needs to be studied is the long‐term effect of anti‐C1q on the morbidity of SLE patients. Thus far, studies on anti‐C1q in SLE focused on overall disease activity, particularly at the time of sampling. Little is known about how the presence of anti‐C1q might affect the physiological and pathogenic role of C1q in an extended period of time. For example, the chronic presence of anti‐C1q might affect the development of atherosclerosis in SLE patients by interfering not only with deposited C1q but also with components of the haemostatic system and macrophages. This topic is of relevance since SLE is associated with considerable cardiovascular morbidity. 95 As demonstrated in a mouse model of atherosclerosis, C1q has protective effects for early atherosclerosis, and human in vitro data show C1q deposition on cholesterol crystals that modulate the phenotype of phagocytosing macrophages. The binding of C1q to cholesterol crystals also leads to the exposure of new binding sites that might allow the binding of anti‐C1q with potential impact on disease progression. 96 , 97
Last, although this review focused on the role of C1q in apoptotic cell clearance, the role of C1q in SLE seems to be complex. Previous work described a direct effect of C1q on T‐cell proliferation. 98 , 99 In a more recent study using a mouse model of autoimmunity, C1q was found to control the response to self‐antigens by modifying the mitochondrial metabolism of CD8+ T cells, which can themselves propagate autoimmunity. 100 These data suggest not only a link between C1q and CD8+ T‐cell metabolism but provide an alternative (or additional) explanation of how C1q protects against lupus. In addition, the observation might have implications for the role of viral infections in the maintenance of autoimmunity. It will be interesting to study whether anti‐C1q play a role in this concept as well.
Conclusions
Anti‐C1q are an exciting biomarker of SLE disease activity and for the occurrence of active proliferative lupus nephritis in patients. In addition, the exploration of binding characteristics of anti‐C1q and its functional consequences provides important insights into pathogenic mechanisms of the disease involving complement deposition and activation, the clearance mechanisms of apoptotic cell debris, phagocyte function and the role of genetic as well as environmental factors such as EBV infection.
Conflict of interest
The author declares no conflict of interest.
Author Contribution
Marten Trendelenburg: Conceptualization; Formal analysis; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing.
Acknowledgments
Many thanks go to the lab members and Dr Sophie Sampson for critically revising the manuscript and, in particular, to Mrs Katharina Fick, who helped creating the figures in this article. The author is recipient of a research grant by the Swiss National Science Foundation (No 310030_172965). He has research collaborations with Roche, Novartis and Idorsia Pharmaceuticals (all Basel, Switzerland).
References
- 1. Tsokos GC. Autoimmunity and organ damage in systemic lupus erythematosus. Nat Immunol 2020; 21: 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol 2000; 76: 227–324. [DOI] [PubMed] [Google Scholar]
- 3. Stegert M, Bock M, Trendelenburg M. Clinical manifestations of human C1q deficiency: how much of a lupus ? Mol Immunol 2015; 67: 3–11. [DOI] [PubMed] [Google Scholar]
- 4. Botto M, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiol 2002; 205: 395–406. [DOI] [PubMed] [Google Scholar]
- 5. Casciola‐Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med 1994; 179: 1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte‐derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum 1998; 41: 1241–1250. [DOI] [PubMed] [Google Scholar]
- 7. Tas SW, Quartier P, Botto M, Fossati‐Jimack L. Macrophages from patients with SLE and rheumatoid arthritis have defective adhesion in vitro, while only SLE macrophages have impaired uptake of apoptotic cells. Ann Rheum Dis 2006; 65: 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol 1997; 158: 4525–4528. [PubMed] [Google Scholar]
- 9. Botto M, Dell'Agnola C, Bygrave AE et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet 1998; 19: 56–59. [DOI] [PubMed] [Google Scholar]
- 10. Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement‐dependent clearance of apoptotic cells by human macrophages. J Exp Med 1998; 188: 2313–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Navratil JS, Watkins SC, Wisnieski JJ, Ahearn JM. The globular heads of C1q specifically recognize surface blebs of apoptotic vascular endothelial cells. J Immunol 2001; 166: 3231–3239. [DOI] [PubMed] [Google Scholar]
- 12. Ogden CA, deCathelineau A, Hoffmann PR et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic bodies. J Exp Med 2001; 194: 781–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fraser DA, Laust AK, Nelson EL, Tenner AJ. C1q differentially modulates phagocytosis and cytokine responses during ingestion of apoptotic cells by human monocytes, macrophages, and dendritic cells. J Immunol 2009; 183: 6175–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thanei S, Trendelenburg M. Anti‐C1q Autoantibodies from systemic lupus erythematosus patients induce a proinflammatory phenotype in macrophages. J Immunol 2016; 196: 2063–2074. [DOI] [PubMed] [Google Scholar]
- 15. Taylor PR, Carugati A, Fadok VA et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic bodies in vivo . J Exp Med 2000; 192: 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pickering MC, Fischer S, Lewis MR, Walport MJ, Botto M, Cook HT. Ultraviolet‐radiation‐induced keratinocyte apoptosis in C1q‐deficient mice. J Invest Dermatol 2001; 117: 52–58. [DOI] [PubMed] [Google Scholar]
- 17. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009; 9: 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benoit ME, Clarke EV, Morgado P, Fraser DA, Tenner AJ. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol 2012; 188: 5682–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fremeaux‐Bacchi V, Weiss L, Demouchy C, Blouin J, Kazatchkine MD. Autoantibodies to the collagen‐like region of C1q are strongly associated with classical pathway‐mediated hypocomplementemia in systemic lupus erythematosus. Lupus 1996; 5: 216–220. [DOI] [PubMed] [Google Scholar]
- 20. Martin M, Smoląg KI, Björk A et al. Plasma C4d as marker for lupus nephritis in systemic lupus erythematosus. Arthritis Res Ther 2017; 19: 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bijl M, Reefman E, Horst G, Limburg PC, Kallenberg CG. Reduced uptake of apoptotic cells by macrophages in systemic lupus erythematosus: correlates with decreased serum levels of complement. Ann Rheum Dis 2006; 65: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jennette JC, Hipp CG. Immunohistopathologic evaluation of C1q in 800 renal biopsy specimens. Am J Clin Pathol 1985; 83: 415–420. [DOI] [PubMed] [Google Scholar]
- 23. Sjöwall C, Olin AI, Skogh T et al. C‐reactive protein, immunoglobulin G and complement co‐localize in renal immune deposits of proliferative lupus nephritis. Autoimmunity 2013; 46: 205–214. [DOI] [PubMed] [Google Scholar]
- 24. Siegert C, Daha M, Westedt ML, van der Voort E, Breedveld F. IgG autoantibodies against C1q are correlated with nephritis, hypocomplementemia, and dsDNA antibodies in systemic lupus erythematosus. J Rheumatol 1991; 18: 230–234. [PubMed] [Google Scholar]
- 25. Reid KBM. Complement component C1q: historical perspective of a functionally versatile, and structurally unusual, serum protein. Front Immunol 2018; 9: 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Agnello V, Koffler D, Eisenberg JW, Winchester RJ, Kunkel HG. C1q precipitins in the sera of patients with systemic lupus erythematosus and other hypocomplementemic states: characterization of high and low molecular weight types. J Exp Med 1971; 134(Suppl.): 228–241. [PMC free article] [PubMed] [Google Scholar]
- 27. Antes U, Heinz HP, Loos M. Evidence for the presence of autoantibodies to the collagen‐like portion of C1q in systemic lupus erythematosus. Arthritis Rheum 1988; 31: 457–464. [DOI] [PubMed] [Google Scholar]
- 28. Uwatoko S, Mannik M. Low‐molecular weight C1q‐binding immunoglobulin G in patients with systemic lupus erythematosus consists of autoantibodies to the collagen‐like region of C1q. J Clin Invest 1988; 82: 816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trendelenburg M. Antibodies against C1q in patients with systemic lupus erythematosus. Springer Semin Immunopathol 2005; 27: 276–285. [DOI] [PubMed] [Google Scholar]
- 30. Kozyro I, Perahud I, Sadallah S et al. Clinical value of autoantibodies against C1q in children with glomerulonephritis. Pediatrics 2006; 117: 1663–1668. [DOI] [PubMed] [Google Scholar]
- 31. Kozyro I, Korosteleva L, Chernoshej D, Danner D, Sukalo A, Trendelenburg M. Autoantibodies against complement C1q in acute post‐streptococcal glomerulonephritis. Clin Immunol 2008; 128: 409–414. [DOI] [PubMed] [Google Scholar]
- 32. Potlukova E, Jiskra J, Limanova Z et al. Autoantibodies against complement C1q correlate with the thyroid function in patients with autoimmune thyroid disease. Clin Exp Immunol 2008; 153: 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wisnieski JJ, Jones SM. IgG autoantibodies to the collagen‐like region of C1q in hypocomplementemic urticarial vasculitis syndrome, systemic lupus erythematosus and six other skeletal or rheumatic diseases. J Rheumatol 1992; 19: 884–888. [PubMed] [Google Scholar]
- 34. Siegert CEH, Daha MR, Swaak AJG, van der Voort EAM, Breedveld FC. The relationship between serum titres of autoantibodies against C1q and age in the general population and in patients with systemic lupus erythematosus. Clin Immunol Immunopathol 1993; 67: 204–209. [DOI] [PubMed] [Google Scholar]
- 35. Trendelenburg M, Lopez‐Trascasa M, Potlukova E et al. High prevalence of anti‐C1q antibodies in biopsy‐proven active lupus nephritis. Nephrol Dial Transplant 2006; 21: 3115–3121. [DOI] [PubMed] [Google Scholar]
- 36. Wisnieski JJ, Baer AN, Christensen J et al. Hypocomplementemic urticarial vasculitis syndrome. Clinical and serological findings in 18 patients. Medicine (Baltimore) 1995; 74: 24–41. [DOI] [PubMed] [Google Scholar]
- 37. Trendelenburg M, Courvoisier S, Spaeth PJ et al. Hypocomplementemic urticarial vasculitis or systemic lupus erythematosus? Am J Kidney Dis 1999; 34: 745–751. [DOI] [PubMed] [Google Scholar]
- 38. Bock M, Heijnen I, Trendelenburg M. Anti‐C1q antibodies as a follow‐up marker in SLE patients. PLoS One 2015; 10: e0123572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moroni G, Trendelenburg M, Del Papa N et al. Anti‐C1q antibodies may help in diagnosing renal flare in lupus nephritis. Am J Kidney Dis 2001; 37: 490–498. [DOI] [PubMed] [Google Scholar]
- 40. Gunnarson I, Ronnelid J, Huang YH et al. Association between ongoing anti‐C1q antibody production in peripheral blood and proliferative nephritis in patients with active systemic lupus erythematosus. Br J Rheumatol 1997; 36: 32–37. [DOI] [PubMed] [Google Scholar]
- 41. Horvath L, Czirjak L, Fekele B et al. High levels of antibodies against Clq are associated with disease activity and nephritis but not with other organ manifestations in SLE patients. Clin Exp Rheumatol 2001; 19: 667–672. [PubMed] [Google Scholar]
- 42. Siegert CE, Daha MR, Tseng CM, Coremans IE, van Es LA, Breedveld FC. Predictive value of IgG autoantibodies against C1q for nephritis in systemic lupus erythematosus. Ann Rheum Dis 1993; 52: 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sjoholm AG, Martensson U, Sturfelt G. Serial analysis of autoantibody responses to the collagen‐like region of C1q, collagen type II, and double‐stranded DNA in patients with systemic lupus erythematosus. J Rheumatol 1997; 24: 871–878. [PubMed] [Google Scholar]
- 44. Trendelenburg M, Marfurt J, Gerber I, Tyndall A, Schifferli JA. Lack of occurrence of severe lupus nephritis among anti‐C1q autoantibody negative patients. Arthritis Rheum 1999; 42: 187–188. [DOI] [PubMed] [Google Scholar]
- 45. Haseley LA, Wisnieski JJ, Denburg MR et al. Antibodies to C1q in systemic lupus erythematosus: chracteristics and relation to FcRIIA alleles. Kidney Int 1997; 52: 1375–1380. [DOI] [PubMed] [Google Scholar]
- 46. Prada AE, Strife CF. IgG subclass restriction of autoantibody to solid‐phase C1q in membranoproliferative and lupus glomerulonephritis. Clin Immunol Immunopathol 1992; 63: 84–88. [DOI] [PubMed] [Google Scholar]
- 47. Siegert CE, Daha MR, van der Voort EAM, Breedveld FC. IgG and IgA antibodies to the collagen‐like region of C1q in rheumatoid vasculitis. Arthritis Rheum 1990; 33: 1464–1654. [DOI] [PubMed] [Google Scholar]
- 48. Siegert CE, Daha MR, Halma C, van der Voort EA, Breedveld FC. IgG and IgA autoantibodies to C1q in systemic and renal diseases. Clin Exp Rheumatol 1992; 10: 19–23. [PubMed] [Google Scholar]
- 49. Tsacheva I, Radanova M, Todorova N, Argirova T, Kishore U. Detection of autoantibodies against the globular domain of human C1q in the sera of systemic lupus erythematosus patients. Mol Immunol 2007; 44: 2147–2151. [DOI] [PubMed] [Google Scholar]
- 50. Uwatoko S, Gauthier VJ, Mannik M. Autoantibodies to the collagen‐like region of C1Q deposit in glomeruli via C1Q in immune deposits. Clin Immunol Immunopathol 1991; 61: 268–273. [DOI] [PubMed] [Google Scholar]
- 51. Rabs U, Martin H, Hitschold T, Golan MD, Heinz HP, Loos M. Isolation and characterization of macrophage‐derived C1q and its similarities to serum C1q. Eur J Immunol 1986; 16: 1183–1186. [DOI] [PubMed] [Google Scholar]
- 52. Castellano G, Woltman AM, Nauta AJ et al. Maturation of dendritic cells abrogates C1q production in vivo and in vitro . Blood 2004; 103: 3813–3820. [DOI] [PubMed] [Google Scholar]
- 53. Cao W, Bobryshev YV, Lord RS, Oakley RE, Lee SH, Lu J. Dendritic cells in the arterial wall express C1q: potential significance in atherogenesis. Cardiovasc Res 2003; 60: 175–186. [DOI] [PubMed] [Google Scholar]
- 54. Maria NI, Davidson A. Renal macrophages and dendritic cells in SLE nephritis. Curr Rheumatol Rep 2017; 19: 81. [DOI] [PubMed] [Google Scholar]
- 55. Castellano G, Trouw LA, Fiore N, Daha MR, Schena FP, van Kooten C. Infiltrating dendritic cells contribute to local synthesis of C1q in murine and human lupus nephritis. Mol Immunol 2010; 47: 2129–2137. [DOI] [PubMed] [Google Scholar]
- 56. Zhou W, Marsh JE, Sacks SH. Intrarenal synthesis of complement. Kidney Int 2001; 59: 1227–1235. [DOI] [PubMed] [Google Scholar]
- 57. Thanei S, Trendelenburg M. Anti‐C1q autoantibodies from patients with systemic lupus erythematosus induce C1q production by macrophages. J Leukoc Biol 2017; 101: 481–491. [DOI] [PubMed] [Google Scholar]
- 58. Mannik M, Wener M. Deposition of antibodies to the collagen‐like region of C1q in renal glomeruli of patients with proliferative lupus glomerulonephritis. Arthritis Rheum 1997; 40: 1504–1511. [DOI] [PubMed] [Google Scholar]
- 59. Trouw LA, Seelen MA, Duijs JMGJ, Benediktsson H, Van Kooten C, Daha MR. Glomerular deposition of C1q and anti‐C1q antibodies in mice following injection of anti‐mouse C1q antibodies. Clin Exp Immunol 2003; 132: 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gaboriaud C, Thielens NM, Gregory LA, Rossi V, Fontecilla‐Camps JC, Arlaud GJ. Structure and activation of the C1 complex of complement: Unraveling the puzzle. Trends Immunol 2004; 25: 368–373. [DOI] [PubMed] [Google Scholar]
- 61. Golan MD, Burger R, Loos M. Conformational changes in C1q after binding of immune complexes: detection of neoantigens with monoclonal antibodies. J Immunol 1982; 129: 445–447. [PubMed] [Google Scholar]
- 62. Bigler C, Schaller M, Perahud I, Trendelenburg M. Autoantibodies against complement C1q specifically target C1q bound on early apoptotic cells. J Immunol 2009; 183: 3512–3521. [DOI] [PubMed] [Google Scholar]
- 63. Martensson U, Thiel S, Jensenius JC, Sjoholm AG. Human autoantibodies against C1q: lack of cross‐reactivity with the collectins mannan‐binding protein, lung surfactant protein A and bovine conglutinin. Scand J Immunol 1996; 43: 314–320. [DOI] [PubMed] [Google Scholar]
- 64. Martensson U, Sjoholm AG, Sturfelt G, Truedsson L, Laurell AB. Western blot analysis of human IgG reactive with the collageneous portion of C1q: evidence of distinct binding specificities. Scand J Immunol 1992; 35: 735–744. [DOI] [PubMed] [Google Scholar]
- 65. Wisnieski JJ, Jones SM. Comparison of autoantibodies to the collagen‐like region of C1q in hypocomplementemic urticarial vasculitis syndrome and systemic lupus erythematosus. J Immunol 1992; 148: 1396–1403. [PubMed] [Google Scholar]
- 66. Vanhecke D, Roumenina LT, Wan H, Schaller M, Osthoff M, Trendelenburg M. Identification of a major linear C1q epitope allows the detection of Systemic Lupus Erythematosus associated anti‐C1q auto‐antibodies using a specific and sensitive peptide‐based assay. Arthritis Rheum 2012; 64: 3706–3714. [DOI] [PubMed] [Google Scholar]
- 67. Wu WJ, Tan Y, Liu XL, Yu F, Zhao MH. C1q A08 is a half‐cryptic epitope of anti‐C1q A08 antibodies in lupus nephritis and important for the activation of complement classical pathway. Front Immunol 2020; 11: 848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pang Y, Tan Y, Li Y et al. Serum A08 C1q antibodies are associated with disease activity and prognosis in Chinese patients with lupus nephritis. Kidney Int 2016; 90: 1357–1367. [DOI] [PubMed] [Google Scholar]
- 69. Ying SC, Gewurz AT, Jiang H, Gewurz H. Human serum amyloid P component oligomers bind and activate the classical complement pathway via residues 14–26 and 76–92 of the A chain collagen‐like region of C1q. J Immunol 1993; 150: 169–176. [PubMed] [Google Scholar]
- 70. Jiang H, Cooper B, Robey FA, Gewurz H. DNA binds and activates complement via residues 14–26 of the human C1q A chain. J Biol Chem 1992; 267: 25597–25601. [PubMed] [Google Scholar]
- 71. Jiang H, Robey FA, Gewurz H. Localization of sites through which C‐reactive protein binds and activates complement to residues 14–26 and 76–92 of the human C1q A chain. J Exp Med 1992; 175: 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kölm R, Schaller M, Roumenina LT et al. Von Willebrand factor interacts with surface‐bound C1q and induces platelet rolling. J Immunol 2016; 197: 3669–3679. [DOI] [PubMed] [Google Scholar]
- 73. Ghebrehiwet B, Kandov E, Kishore U, Peerschke EIB. Is the A‐chain the engine that drives the diversity of C1q functions? Revisiting its unique structure. Front Immunol 2018; 9: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Trinder PK, Maeurer MJ, Kaul M, Petry E, Loos M. Functional domains of the human C1q A‐chain. Behring Inst Mitt 1993; 93: 180–188. [PubMed] [Google Scholar]
- 75. Schaller M, Bigler C, Danner D, Ditzel HJ, Trendelenburg M. Autoantibodies against C1q in systemic lupus erythematosus are antigen driven. J Immunol 2009; 183: 8225–8231. [DOI] [PubMed] [Google Scholar]
- 76. Schwartz RS. Some speculations on the origins of autoantibodies. Ann N Y Acad Sci 1987; 505: 8–11. [DOI] [PubMed] [Google Scholar]
- 77. Hunnangkul S, Nitsch D, Rhodes B et al. Familial clustering of non‐nuclear autoantibodies and C3 and C4 complement components in systemic lupus erythematosus. Arthritis Rheum 2008; 58: 1116–1124. [DOI] [PubMed] [Google Scholar]
- 78. Hanlon P, Avenell A, Aucott L, Vickers MA. Systematic review and meta‐analysis of the sero‐epidemiological association between Epstein‐Barr virus and systemic lupus erythematosus. Arthritis Res Ther 2014; 16: R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Csorba K, Schirmbeck LA, Tuncer E et al. Anti‐C1q antibodies as occurring in systemic lupus erythematosus could be induced by an Epstein‐Barr virus‐derived antigenic site. Front Immunol 2019; 10: 2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. McClain MT, Poole BD, Bruner BF, Kaufman KM, Harley JB, James JA. An altered immune response to Epstein‐Barr nuclear antigen 1 in pediatric systemic lupus erythematosus. Arthritis Rheum 2006; 54: 360–368. [DOI] [PubMed] [Google Scholar]
- 81. McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med 2005; 11: 85–89. [DOI] [PubMed] [Google Scholar]
- 82. Poole BD, Gross T, Maier S, Harley JB, James JA. Lupus‐like autoantibody development in rabbits and mice after immunization with EBNA‐1 fragments. J Autoimmun 2008; 31: 362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yadav P, Tran H, Ebegbe R et al. Antibodies elicited in response to EBNA‐1 may cross‐react with dsDNA. PLoS One 2011; 6: e14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Siegert CE, Daha MR, Lobatto S, van der Voort EA, Breedveld FC. IgG autoantibodies to C1q do not detectably influence complement activation in vivo and in vitro in systemic lupus erythematosus. Immunol Res 1992; 11: 91–97. [DOI] [PubMed] [Google Scholar]
- 85. Thanei S, Vanhecke D, Trendelenburg M. Anti‐C1q autoantibodies from SLE patients activate the complement system via both the classical and the lectin pathway. Clin Immunol 2015; 160: 180–187. [DOI] [PubMed] [Google Scholar]
- 86. Pang Y, Yang XW, Song Y, Yu F, Zhao MH. Anti‐C1q autoantibodies from active lupus nephritis patients could inhibit the clearance of apoptotic cells and complement classical pathway activation mediated by C1q in vitro . Immunobiology 2014; 219: 980–989. [DOI] [PubMed] [Google Scholar]
- 87. Trouw LA, Groeneveld TWL, Seelen MA et al. Anti‐C1q autoantibodies deposit in glomeruli but are only pathogenic in combination with glomerular C1q‐containing immune complexes. J Clin Invest 2004; 114: 679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Harrison DJ. Cell death in the diseased glomerulus. Histopathology 1988; 12: 679–683. [DOI] [PubMed] [Google Scholar]
- 89. Faurschou M, Penkowa M, Andersen CB, Starklint H, Jacobsen S. Renal cell apoptosis in human lupus nephritis: a histological study. Lupus 2009; 18: 994–999. [DOI] [PubMed] [Google Scholar]
- 90. Nielsen CT, Rasmussen NS, Heegaard NH, Jacobsen S. "Kill" the messenger: Targeting of cell‐derived microparticles in lupus nephritis. Autoimmun Rev 2016; 15: 719–725. [DOI] [PubMed] [Google Scholar]
- 91. Postal M, Vivaldo JF, Fernandez‐Ruiz R, Paredes JL, Appenzeller S, Niewold TB. Type I interferon in the pathogenesis of systemic lupus erythematosus. Curr Opin Immunol 2020; 67: 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lood C, Gullstrand B, Truedsson L et al. C1q inhibits immune complex‐induced interferon‐alpha production in plasmacytoid dendritic cells: a novel link between C1q deficiency and systemic lupus erythematosus pathogenesis. Arthritis Rheum 2009; 60: 3081–3090. [DOI] [PubMed] [Google Scholar]
- 93. Santer DM, Hall BE, George TC et al. C1q deficiency leads to the defective suppression of IFN‐alpha in response to nucleoprotein containing immune complexes. J Immunol 2010; 185: 4738–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Santer DM, Wiedeman AE, Teal TH, Ghosh P, Elkon KB. Plasmacytoid dendritic cells and C1q differentially regulate inflammatory gene induction by lupus immune complexes. J Immunol 2012; 188: 902–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Koenig KF, Ribi C, Radosavac M, Zulewski H, Trendelenburg M. Swiss SLE cohort study (SSCS). Prevalence of vascular disease in systemic lupus erythematosus compared with type‐1 diabetes mellitus: a cross‐sectional study of two cohorts. Lupus 2015; 24: 58–65. [DOI] [PubMed] [Google Scholar]
- 96. Bhatia VK, Yun S, Leung V et al. Complement C1q reduces early atherosclerosis in low‐density lipoprotein receptor‐deficient mice. Am J Pathol 2007; 170: 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Donat C, Thanei S, Trendelenburg M. Binding of von Willebrand Factor to complement C1q decreases the phagocytosis of cholesterol crystals and subsequent IL‐1 secretion in macrophages. Front Immunol 2019; 10: 2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chen A, Gaddipati S, Volkman DJ, Peerschke EIB, Ghebrehiwet B. Human T cells possess specific receptors for C1q: role in activation and proliferation. J Immunol 1994; 153: 1430–1440. [PubMed] [Google Scholar]
- 99. Zhao N, Wu J, Xiong S et al. Mannan‐binding lectin, a serum collectin, suppresses T‐cell proliferation via direct interaction with cell surface calreticulin and inhibition of proximal T‐cell receptor signaling. FASEB J 2017; 31: 2405–2417. [DOI] [PubMed] [Google Scholar]
- 100. Ling GS, Crawford G, Buang N et al. C1q restrains autoimmunity and viral infection by regulating CD8+ T cell metabolism. Science 2018; 360: 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]