

Abstract
The total number of low‐molecular‐weight compounds in the plant kingdom, most of which are secondary metabolites, is hypothesized to be over one million, although only a limited number of plant compounds have been characterized. Untargeted analysis, especially using mass spectrometry (MS), has been useful for understanding the plant metabolome; however, due to the limited availability of authentic compounds for MS‐based identification, the identities of most of the ion peaks detected by MS remain unknown. Accurate mass values of peaks obtained by high accuracy mass measurement and, if available, MS/MS fragmentation patterns provide abundant annotation for each peak. Here, we carried out an untargeted analysis of compounds in the mature fruit of 25 tomato cultivars using liquid chromatography‐Orbitrap MS for accurate mass measurement, followed by manual curation to construct the metabolome database TOMATOMET (http://metabolites.in/tomato‐fruits/). The database contains 7,118 peaks with accurate mass values, in which 1,577 ion peaks are annotated as members of a chemical group. Remarkably, 71% of the mass values are not found in the accurate masses detected previously in Arabidopsis thaliana, Medicago truncatula or Jatropha curcas, indicating significant chemical diversity among plant species that remains to be solved. Interestingly, substantial chemical diversity exists also among tomato cultivars, indicating that chemical profiling from distinct cultivars contributes towards understanding the metabolome, even in a single organ of a species, and can prioritize some desirable metabolic targets for further applications such as breeding.
Keywords: bioinformatics, chemical diversity, metabolite annotation, metabolome, tomato cultivar, tomato fruit
1. INTRODUCTION
Untargeted analysis of metabolites using liquid chromatography‐mass spectrometry (LC‐MS) is a promising technology for understanding the metabolism of organisms of interest and for finding valuable metabolites for breeding and industrial uses such as medicines or new materials development (Tohge & Fernie, 2015; Wang, 2008; Wurtzel & Kutchan, 2016). Afendi et al. (2012) proposed that the total number of metabolites in the plant kingdom is over one million based on the species specificity of metabolites from a wide range of organisms appearing in KNApSAcK, a metabolite database that contains published chemical information of 51,179 metabolites. Although plant extracts from leaves, stems, roots, flowers and fruits of various plant species obtained from several growth stages or conditions have been analysed in an untargeted manner almost for two decades, we are still far away from understanding all features of plant metabolomes. As LC‐MS is applicable to the analysis of a wide range of metabolites with high sensitivity, except for volatiles, this technique has been used widely to detect metabolites in metabolomic studies. However, the major bottleneck of metabolome analyses is the lack of authentic chemical standards capable of identifying the most detected metabolites (Chaleckis et al., 2019; Viant et al., 2017; Wishart, 2009). Therefore, annotation of an ion peak detected by MS may not mean that the compound has been identified; rather, additional information about some chemical features must be linked to the detected ion. Various bioinformatics approaches have been developed for better annotation (Fukushima & Kusano, 2013; Hufsky & Böcker, 2017). Only 17%–25% of the compounds tested are identified correctly by in silico algorithms alone (Blaženović et al., 2017), although the accuracy and confidence of structural elucidation from tandem mass spectrometry (MS/MS) spectra and isotope ions have increased. Therefore, alternative ways of improving metabolite identification are needed.
Including specific information about the sample such as tissue specificity or the taxonomic relationship of the species with other plants improves metabolite identification significantly (Tsugawa, 2018). A large dataset of gas chromatography‐mass spectrometry (GC‐MS) profiles from 114,795 samples of various origins archived in the BinBase database was used successfully to discover some metabolites that accumulate specifically in cancer tissues (Lai et al., 2018). By combining information about taxonomy, known bioactivity, and the chemical relationship of compounds based on MS/MS spectra similarity (molecular network) with in‐house LC‐MS data from 292 plant species in the Euphorbiaceae analysed by the same LC‐MS procedures, seven bioactive natural compounds were discovered (Olivon et al., 2017). In contrast with GC‐MS, the difficulty in standardizing LC‐MS conditions hampers a direct comparison of LC‐MS data with those obtained by other analytical procedures. Nevertheless, accurate mass values with an error of a few ppm obtained by high‐resolution LC‐MS when the molecules are softly ionized to retain the intact form allow direct comparison of datasets with those of others, although a coincidental accurate mass value of a molecule with others is insufficient to consider them the same compound. Therefore, accumulation and publication of detected accurate mass values (hereafter we refer to them as “accurate mass records (AMRs)”) to compare samples of interest, is a meaningful concept in metabolomics.
Providing AMRs of an organism in public is a practical way for exploratory data comparison of untargeted metabolome data obtained by LC‐MS. It is known that a significant number of peaks detected by LC‐MS are those of derivative ions that originated from the same compounds, such as adduct ions, multimers and in‐source fragments, and the real number of unique compounds in a sample is much smaller than the observed peak numbers (Brown et al., 2011; Jankevics et al., 2012; Mahieu & Patti, 2017). Various bioinformatics tools to encapsulate the multiple derivative ions into the original compound have been reported, such as xMSannotator (Uppal et al., 2017), MS‐FLO (DeFelice et al., 2017) and CliqueMS (Senan et al., 2019). However, it is not practically feasible to perform such encapsulation in a similar quality for all data obtained by various researchers using various LC‐MS apparatus and conditions, because of the two reasons as follows: (a) as the encapsulation approaches are based on the co‐elution of peaks for peak annotation, the results depend on the MS sensitivity, sample concentration and peak detection parameters; (b) generation of the variation of adduct ions, multimers and fragments depend on the type of apparatus, setting of the apparatus, sample concentration, solvents and co‐eluting contaminants. In contrast with the difficulty or, in practice, the impossibility of applying equal quality of encapsulation for all peaks detected in the research community, a researcher can encapsulate and annotate the peak data for selected several peaks in their own equal quality when the raw data including AMRs and calculated information about adduct ion, multimer and fragmentation are provided as reference. The resource should be useful for future improvement of the encapsulation strategies and standardization of its quality.
However, well‐curated and reusable AMR data are limited, so far, to comparative studies between distinct species. Although untargeted high‐resolution MS analyses of strawberry (Fragaria x ananassa; Aharoni et al., 2002), Arabidopsis thaliana (Giavalisco et al., 2011), Medicago truncatula (Kera et al., 2018) and tomato (Iijima et al., 2008) have been reported, the MS data shown in the tables or the supplementary tables of these articles are not ready to direct comparison with other datasets. MetaboLights, a metabolomics data management system, contains high‐accuracy datasets from 2,851 analyses of 543 peaks from several plants (Steinbeck et al., 2012); however, a search function with mass values is not provided on the website. The metabolomics repository Metabolomics Workbench (Sud et al., 2016) is well‐designed to search metabolites by mass values; however, the distribution of matched peaks among species is not available from the search result page. The Medicinal Plant Metabolomics Resource (Wurtele et al., 2012) contains datasets of identified metabolites from 14 medicinal plant species and provides the capability to search for mass values with low resolution. The Global Natural Products Social Molecular Networking site (GNPS; Wang et al., 2016) is designed for the curation of mass spectrometry data but does not include a mass value search function. The metabolomics database KomicMarket (Sakurai et al., 2014) has assembled metabolome datasets for several plant species and has a mass value‐search function, but the search speed is slow for practical use. The Food Metabolome Repository (Sakurai & Shibata, 2017) covers a wide variety of food samples including 57 vegetables and 28 fruits, and provides search APIs; however, potential false‐positive peaks remain to be curated since sample preparation was not replicated. Therefore, efforts to generate curated and reusable AMRs for a wide range of organisms or foods are required for a better understanding of plant metabolism or the chemical world surrounding us.
We chose to use tomato fruits to construct an AMR dataset in this study. According to statistics provided by the Food and Agriculture Organization of United Nations (FAOSTAT, http://www.fao.org/faostat), the worldwide production of tomatoes has been the highest among 23 primary vegetables in the world since 1961. Tomato ingredients attract much attention due to their health benefits. Associations between tomato consumption and decreased risk of diseases such as cancer have been reported (Giovannucci, 1999; Martí et al., 2016; Raiola et al., 2014). Our previous study using mice also showed that certain oxo‐fatty acids in tomato act as potent agonists of a ligand‐activated transcription factor, peroxisome proliferator‐activated receptor α (PPARα), and possibly improve obesity‐induced dyslipidemia and hepatic steatosis (Kim et al., 2012). Only two resources provide tomato AMRs data for open access: (a) a dataset of 869 peaks of fruit metabolites from “Micro‐Tom”, a model tomato cultivar, detected by LC‐Fourier transform ion cyclotron resonance‐mass spectrometry (FT‐ICR‐MS) reported in a supplementary table of an article published by Iijima et al. (2008) and a dataset accessible at KomicMarket (http://webs2.kazusa.or.jp/komicmarket/). (b) Accurate mass data for 413 identified peaks from eight tomato studies are available at MetaboLights (https://www.ebi.ac.uk/metabolights/). Unfortunately, the accurate mass data for >2,000 peaks of tomato metabolites published by Perez‐Fons et al. (2014) are not accessible from public websites. A set of 2014 identified peaks from 10 tomato studies is deposited at the GNPS, although the data are not accessible by mass value searches. Therefore, the accumulation of curated and reusable tomato AMRs is required for understanding the fundamentals of metabolisms.
Here, we report a reusable dataset of 7,118 AMRs from tomato fruits of 25 tomato cultivars. The dataset was produced from peak information detected by accurate mass measurement and laborious manual curation. Remarkably, 71% of the mass values of the AMRs are not found in AMRs detected previously in A. thaliana, M. truncatula, or Jatropha curcas. A large diversity of compounds among the evaluated tomato cultivars is also revealed that should be considered in tomato breeding. To exemplify the suitability of the AMRs for prioritizing candidate metabolites, we annotated some tomato‐specific compounds as esculeoside‐ and tomatine‐derivatives. The AMR data are available at the TOMATOMET website (http://metabolites.in/tomato‐fruits) for open access.
2. EXPERIMENTAL PROCEDURES
2.1. Plant materials
The tomato cultivars used in this study are listed in Table S1. Cultivars No.1 to 23 were grown in the greenhouses of KAGOME CO., LTD. located in Tochigi and Fukushima Prefectures, Japan from summer 2013 to spring 2014. Cultivar No.24 (“Kyo‐temari”) was grown in the greenhouses of the Kyoto University experimental farm located in Takatsuki, Osaka Prefecture, Japan from winter 2013 to spring 2014. Cultivar No.25 (“Micro‐Tom”) was greenhouse‐grown at the Kazusa DNA Research Institute located at Kisarazu, Chiba Prefecture, Japan from spring 2014 to summer 2014. The fruits or seeds of cultivars No.1 to 23 were provided by KAGOME CO., LTD. Cultivars No. 24 and No. 25 were provided by the Experimental Farm of Kyoto University and the Tomato National BioResource Project (http://tomato.nbrp.jp/indexEn.html), respectively. Tomato fruits were harvested at the fully ripe stage by judging the fruit colour for each cultivar (red, orange, pink or black). The harvested fruits were immediately frozen in liquid nitrogen and stored at −80°C until use.
2.2. Liquid chromatography‐mass spectrometry analysis
Calyxes were removed from the frozen fruits. For each cultivar (Table S1), three frozen fruits (with both flesh and peel) were pooled, finely ground using a mortar and pestle under liquid nitrogen, and then lyophilized for 48 hr. The lyophilized powder (50 mg) was extracted with 1 ml of 80% v/v methanol containing 1.25 μM 7‐hydroxy‐5‐methylflavone (Sigma‐Aldrich) as an internal standard. After homogenizing the sample for 5 min using a bead crusher (Beads Crusher μT‐12, TAITEC) and a stainless‐steel bead (5.0 mm diameter, Bio Medical Science) in a 2 ml tube, the homogenates were centrifuged (20,400 g for 10 min at 4°C). The supernatant was filtered through a 0.2 μm polytetrafluoroethylene (PTFE) membrane (Millex‐LG, Merck Millipore) and used for LC‐MS analysis. The untargeted metabolome analysis was performed using an Agilent 1200 system (Agilent Technologies Ltd.) coupled to an LTQ Orbitrap XL (Thermo Fisher Scientific Co. Ltd.). The filtrate (5 μl) was applied to a TSK‐gel column ODS‐100V (3.0 × 50 mm, 5 μm, TOSOH Co. Ltd.). Water (HPLC grade; solvent A) and acetonitrile (HPLC grade; solvent B) were used as the mobile phase with 0.1% v/v formic acid added to both solvents. The gradient program was as follows: 3% B (0 min), 97% B (15 min), 97% B (20 min), 3% B (20.1 min) and 3% B (25 min). The flow rate was set to 0.4 ml/min, and the column oven temperature was set at 40°C. Compounds were detected in electrospray ionization (ESI)‐positive mode over the m/z range of 100–1,500, a mass resolution of 60,000 (at m/z 400) and a lock mass set at m/z 391.284286. For the four most intense ions of the precursor scan, MS/MS analyses were carried out using collision‐induced dissociation in a linear ion trap detector with a normalized collision energy of 35.0%. The frozen powder from a single cultivar was extracted and subjected to LC‐MS analysis as described above in triplicate. Mock samples were prepared as above without adding the frozen powdered material. Five series of LC‐MS runs for the triplicate 25 cultivars were carried out, in which three mock samples and five triplicate samples were analysed for each series. Data were acquired using Xcalibur software version 2.1 (Thermo Fisher Scientific). Further details of the procedures are available at the Metabolonote website (http://metabolonote.kazusa.or.jp/SE40:/; Ara et al., 2015).
2.3. Peak detection and peak alignment
Mass chromatogram data obtained by Xcalibur (.raw format) were converted to a text‐based format using MSGet software (http://www.kazusa.or.jp/komics/software/MSGet). Ion peaks were detected using the PowerFT module of the PowerGet software, version 3.5.4beta (http://www.kazusa.or.jp/komics/software/PowerGet; Sakurai et al., 2014). Information such as accurate m/z values, type of adduct ions and the ratio of the intensity of 13C1 isotopic peak to that of the monoisotopic peak was estimated by PowerFT. Peaks were aligned between the samples using the PowerMatch module of PowerGet based on m/z values, retention times, and similarity of MS/MS spectra if available. A file set of the setting parameters of PowerGet used in this study is available at TOMATOMET website.
2.4. Manual curation of the alignment results and characterization of peaks
The alignment results were manually curated using the alignment editing function in the PowerMatch module by checking the raw mass chromatogram for the identity of estimated m/z values, retention times, MS/MS spectra if available and for any other closely detected peaks. An in‐house Perl program was used to check these identities. Inappropriate ion peaks were removed from the alignment. Inappropriately separated aligned peak groups that were derived from the same putative compound were merged into a single alignment. Although retention times of most compounds on chromatograms are stable between experiments, misalignments that sometimes occurred by unpredictable drifts in retention times were corrected manually. Peaks associated with the MS/MS spectral data and peaks of higher intensity were prioritized for this manual curation. Peaks that were also detected in the mock samples were removed. Peaks that were reproducibly detected in at least two of the three analytical replications were regarded as valid peaks. After curation of the alignment results, the ion valence was checked and curated based on the distances between the 13C isotopic peaks using the MassChroViewer software (Sakurai & Shibata, 2017) whose 2‐dimensional mass chromatogram presentation and mass ruler function are suitable for checking this parameter. Peaks of more than or equal to pentavalent ions were ignored and were not analysed further.
Compound database searches and predictions of elemental composition based on the average m/z values and the adduct ions of the alignment were performed using the MFSearcher tool (Sakurai et al., 2012). We search the candidates using the following three types of databases in this order and later ones were used when no candidate was found in the former: (a) The compound databases (KEGG, KNApSAcK, LIPID MAPS and HMDB, see below), (b) Pep1000 database in MFSearcher for prediction of linear peptides, and (c) EX‐HR2 database in MFSearcher for prediction of elemental compositions. The mass tolerances 1, 2 and 5 ppm were given for each search in this order and the larger tolerance was applied when no candidate was found with the smaller. Of the adduct ions predicted by PowerGet, ones of the same ion valence estimated using MassChroViewer were applied for calculation of the mass value of neutralized molecule for search. When no candidate was found, the adduct ions of the same ion valence out of the following adduct ions were used in this order and the later one was used when no candidate was found with the former: [M + H]+, [M + NH4]+, [M + K]+, [M + Na]+, [M + 2H]2+, [M + 2Na]2+, [M + Na + H]2+, [M + NH4 + H]2+, [M + K + H]2+, [M + 2NH4]2+, [M + 2K]2+, [M + 3H]3+, [M + 3Na]3+, [M + 3NH4]3+, [M + 3K]3+, [M + Na + 2H]3+, [M + 2Na + H]3+, [M + NH4 + 2H]3+, [M + 2NH4 + H]3+, [M + K + 2H]3+, [M + 2K + H]3+, [M + 4H]4+, [M + 4Na]4+, [M + 4NH4]4+, [M + 4K]4+, [M + Na + 3H]4+, [M + 2Na + 2H]4+, [M + 3Na + H]4+, [M + NH4 + 3H]4+, [M + 2NH4 + 2H]4+, [M + 3NH4 + H]4+. The existence of the candidates was judged after filtering the search results as below: In the case of compound database search, the candidates containing halogens and silicon were excluded; In the case of elemental composition prediction by EX‐HR2, the candidates with no hydrogen or oxygen/phosphorus ratio less than 2 assuming phosphate derived‐moieties were excluded. As exceptions, for the 14 peaks with the compound database results and the 52 peaks with the EX‐HR2 results, we manually selected appropriate candidates with considerations of accurate mass values and the predicted elemental compositions of MS2 product ions which were attributed by the observation of typical neutral losses of glycosides and so on. We used the following compound databases and the release date of the datasets: KEGG (Kanehisa et al., 2002), 5 December, 2018; KNApSAcK (Afendi et al., 2012), 28 June, 2017; LIPID MAPS (Fahy et al., 2007), 28 June, 2017; HMDB (Wishart et al., 2018), 26 November, 2018. The EX‐HR2 database contains possible elemental compositions as molecules that fulfil Senior‐ and Lewis‐ valent rules and the Seven Golden Rules (Kind & Fiehn, 2007) under the maximum number of the atoms: C,100; H, 200; O, 5; N, 10; P, 10 and S, 10 (Sakurai et al., 2012). The threshold values of the mass tolerances were determined based on the mass accuracy as approximately 1 ppm in our study in which a lock mass was applied for mass calibration and averaging of mass values in the alignment of the peaks from replicational analysis of multiple tomato cultivars. Using the results from 73 metabolites identified by authentic chemicals (see the Metabolite identification section) and their theoretical mass values ranging from 104 to 1,034, the mass accuracy was estimated as less than 0.83 and 0.41 ± 0.21 ppm on average. Therefore, we set 1 ppm for the first search. We set 2 ppm to capture the candidates excluded by the strict threshold value, and 5 ppm at the maximum by considerations of the cases of insufficient corrections by PowerGet for the mass shifts observed in the higher intensity ions and the mass fluctuations observed in the peaks with higher m/z values. We described the search and/or prediction results in Table S2 as follows. The compound IDs found in the compound database search, predicted peptides by Pep1000, and the number of predicted elemental compositions (0, 1 or multiple) by EX‐HR2 were described in the “Database hits” column. An elemental composition was described in the “Annotation” column when the candidates contain a single elemental composition.
2.5. Classification of chemical categories and metabolite annotation
In accordance with the study of Sano et al. (2012), the chemical structures of candidate compounds were manually checked, and if all of them shared a common structure, we classified the peak into one of the following chemical categories: alkaloids, aminocarboxylic acids, carotenoids, coumarins, fatty acid derivatives, flavonoids, glycolipids, iridoids, nucleotides, organic acids, phenolics, phospholipids, porphyrins, steroids, sugars and terpenoids. These categories have some overlapping relationships. For example, iridoids and flavonoids are subcategories of phenolics. In cases where all candidate structures were in a specific subcategory, we assigned the subcategory instead of the parent category. In cases where an MS/MS spectrum was obtained, spectrum similarity searches were conducted using MassBank (Horai et al., 2010) and MS‐MS Fragment Viewer (http://webs2.kazusa.or.jp/msmsfragmentviewer/; Sakurai et al., 2014). We assigned typical neutral losses, namely, NH3, H2O, CH2O2, C5H8O4 and C6H10O5 (Ma et al., 2014) from MS/MS spectra, and if the observed neutral loss fragments were not assumed for the candidate structures, the candidates were excluded from the above classification.
The number of glycosyl substituents was counted manually looking at the candidate chemical structures. If an MS/MS spectrum was available, the results were verified by checking the neutral loss fragments for ‐C6H10O5 and ‐C6H12O6. Only the number of hexoses was accounted for in this study because no candidates having only pentoses were found, despite careful checking. In case additional pentoses might be attached to hexoses, we described this possibility in Table S2.
Metabolite annotation and the annotation levels of MSI (Sumner et al., 2007) were assigned to peaks as follows: (a) if the peak was identified by authentic standards (MSI level 1, see next section), the compound name and chemical formula were assigned; (b) if the compound category and a single compound were candidates (MSI level 2), the category name, candidate compound name, presence and absence of phosphate and sulphate residues, and a chemical formula were assigned; (c) if the compound category was determined but a single candidate compound could not be predicted (MSI level 3), the category name was assigned; (d) if the compound category was not determined and a single elemental composition was predicted (MSI level 4), the elemental composition was assigned; and (e) if the compound category was not determined and multiple elemental compositions were predicted (MSI level 4), no term was assigned. In the MSI level 3 compounds, 16 peaks marked by superscript “a” in Table S2 were qualified by comparison with the authentic compounds but could not be identified due to detections of multiple isomers eluted closely at the retention times of the authentic compounds.
In cases in which the types and numbers of the substituents (such as glycosyl groups) were predicted by manual assignment of MS/MS spectral fragments, the information was added to the annotation (Table S2). If possible, the distinction of lipid subclass name (e.g. triacylglycerol [TG], phosphatidylethanolamine [PE]) was attached to peaks in the categories of fatty acid derivatives, glycolipids or phospholipids.
2.6. Metabolite identification
Metabolites were identified by checking the identities of m/z values, retention times and MS/MS spectra compared to those of the authentic standard compounds measured using the same LC‐MS conditions. The authentic standards were purchased from the suppliers as follows: 6‐hydroxycoumarin, adenine, adenosine, AMP, biotin, chlorogenic acid, citrate, CMP, cytidine, cytosine, GABA, gamma‐L‐glutamyl‐L‐cysteine, glutathione, GMP, guanine, guanosine, inosine, isocitrate, kaempferol 3‐O‐rutinoside, L‐arginine, L‐asparagine, L‐aspartate, L‐cysteine, L‐glutamate, L‐glutamine, L‐histidine, L‐lysine, L‐methionine, L‐phenylalanine, L‐proline, L‐serine, L‐threonine, L‐tryptophan, L‐tyrosine, NAD, nicotinamide, S‐adenosyl‐L‐methionine, trans‐feruloyltyramine, UMP, uracil and uridine were from Sigma‐Aldrich; 7‐hydroxycoumarin, anthranilic acid, caffeic acid, L‐kynurenine and tomatine were from Tokyo Chemical Industry Co., Ltd.; L‐norleucine, nicotinate, pantothenate, rutin, spermidine and spermine were from FUJIFILM Wako Pure Chemical Co.; cis‐aconitate and FMN were from Nakarai Tesque, Inc.; 13‐oxoODA and serotonin were from Cayman Chemical Co.; eriodictyol 7‐O‐glucoside, eriodictyol, fustin, hesperetin, kaempferol 3‐O‐glucoside, naringin and prunin were from Extrasynthese; p‐coumaric acid and succinate semialdehyde were from Santa Cruz Biotechnology, Inc.; N‐p‐trans‐coumaroyltyramine was from Wuhan ChemFaces Biochemical Co., Inc.; quinic acid was from Kanto Chemical Co., Inc.
2.7. Comparison of AMR data with those in previous reports
To compare AMRs with those previously reported, we surveyed the literature and databases for untargeted metabolome data obtained by positive ion mode ESI and with high‐resolution MS, namely FT‐ICR‐MS or Orbitrap‐MS (Thermo Fisher Scientific). Supplementary data in the following research papers were found and used in this study: Iijima et al. (2008) for Solanum lycopersicum; Krueger et al. (2011), Giavalisco et al. (2011), Gläser et al. (2014) and Cao et al. (2016) for A. thaliana; Kera et al. (2018) for M. truncatula and Sano et al. (2012) for J. curcas. The following studies were found in the MetaboLights database (Steinbeck et al., 2012) for Solanum lycopersicum: MTBLS36 (Beisken et al., 2014), MTBLS107 (Van Meulebroek et al., 2015) and MTBLS693 (Garbowicz et al., 2018). The files containing metabolite information were downloaded and used in this study. The mass values that were unique in each species and those shared among multiple species were calculated by grouping the mass values at a given 5‐ppm mass tolerance using an in‐house Perl program.
2.8. Statistical analysis
Principal component analysis (PCA) was performed using the prcomp function of the R program (version 3.1) based on the variance‐covariance matrix. The peak intensities were transformed to log‐based 10 and normalized by the average for each sample. Average values of the triplicate samples were used for PCA. The missing values were compensated with a small value that is 1/10 of the smallest intensities among all samples.
2.9. Database construction
TOMATOMET (http://metabolites.in/tomato‐fruits/) was constructed Java 8 (Oracle Corporation) and Spring Boot (Pivotal Software, Inc.).
3. RESULTS AND DISCUSSION
3.1. Construction of a dataset of tomato AMRs from fruits of 25 tomato cultivars
We analysed tomato extracts of mature fruits for accurate mass measurement by reversed phase‐LC and high‐resolution MS (LTQ‐Orbitrap, Thermo Fisher Scientific) in the electrospray ionization (ESI)‐positive mode. Twenty five cultivars suitable for fresh market, processing and ornamental uses were selected for the analysis (Table S1). The ion peaks of triplicate biological samples were detected and aligned using PowerGet software (Sakurai et al., 2014), resulting in a total of 505,662 alignments. As the automatically calculated results might have contained latent false positives (Mahieu & Patti, 2017), we corrected inappropriate peak detections, misalignments and misassignments of the adduct ions and removed noise peaks manually with the help of the editing function of PowerGet software and the mass chromatogram viewer software MassChroViewer (Sakurai & Shibata, 2017). In this study, we defined the peaks detected in more than two of the triplicate samples from each cultivar as unequivocally detected peaks. This designation was necessary because the quantity of some metabolites in biological samples may change significantly even if the sampling conditions were controlled as metabolite levels reach the detection limits of the instrument. Finally, the dataset that includes 7,118 peaks, in which 1,491 peaks (21%) containing MS/MS spectral data, was selected (Table S2). The mass values of the curated 7,118 peaks can be used as accurate mass records (AMRs) for comparison with those from other plant samples.
We annotated the peaks using the accurate mass values and if available, MS/MS spectral data. Using the accurate mass values, searches against compound databases were carried out with a defined 5‐ppm mass tolerance at the maximum, followed by manually evaluating the matching chemical structures for individual peaks (see EXPERIMENTAL PROCEDURES for the details). Of 7,118 peaks, 1,577 peaks (22.2%) were categorized into one of 16 metabolite categories (Sano et al., 2012; Table S3). The confidence in identifying these peaks corresponds to level 3, “putatively characterized compound classes”, as defined by the Metabolomics Standards Initiative (MSI; Sumner et al., 2007; Viant et al., 2017). Compound names were predicted for 142 peaks in the categorized chemical groups (identification level 2, “putatively annotated compounds”), and the metabolites for 73 peaks were further identified by comparison with authentic standard compounds (identification level 1, “identified compounds”). Information about the curated AMRs is available at the tomato database TOMATOMET (http://metabolites.in/tomato‐fruits/) that was constructed in this study (see below for Construction of a database for searching tomato AMRs). Our annotations could include false assignments due to unexpected adduct ions, although we considered 31 types of possible adduct ions and in‐source fragmentation products that are analytical artefacts produced during compound ionization.
In this study, we separated compounds by reversed‐phase LC under conditions that are suitable for separating a wide range of secondary metabolites that have medium to low polarity. Therefore, some metabolites with high polarity may be undetected. Analysis of high polarity compounds will be a subject of future studies. We chose the ESI positive mode for the MS analysis because in the well‐accessed public MS databases MassBank (Horai et al., 2010) and mzCloud (https://www.mzcloud.org/), ~70% of the datasets were obtained using the ESI positive mode. Future research should focus on obtaining datasets using the negative mode to complement present‐day datasets.
3.2. Comparison of the tomato AMRs with those from other plants
We compared the 7,118 AMRs directly with other plant AMRs to reveal any unique characteristics of the tomato datasets. From a thorough search of the literature and metabolome data repositories (see EXPERIMENTAL PROCEDURES), we collected the AMRs produced by FT‐ICR or Orbitrap‐MS of four plant species, namely, tomato (Beisken et al., 2014; Garbowicz et al., 2018; Iijima et al., 2008), A. thaliana (Cao et al., 2016; Giavalisco et al., 2011; Gläser et al., 2014; Krueger et al., 2011), M. truncatula (Kera et al., 2018) and J. curcas (Sano et al., 2012). For this comparison, we define "unique mass value(s)" as that(those) that has(have) the same mass value(s) when selected by a mass tolerance at 5‐ppm, although a unique mass value may correspond to more than one peak. The AMRs that did not match with any of the AMRs of other plants were considered to be specific to the plant. Although it is obvious that matching accurate mass values is not a sufficient criterion to consider two molecules to be the same chemical or to distinguish the difference between isomers and isobars; however, matches are useful for prioritizing some chemicals with the same mass for further study. The number of the unique mass values of this study was 4,417 of 7,118 AMRs (62.1%), whereas those of previously published studies of tomato, A. thaliana, M. truncatula and J. curcas were 598 of 727 (82.3%), 4,272 of 6,681 (63.9%), 401 of 511 (72.8%) and 4,340 of 6,778 (64.0%) respectively.
The tomato unique masses identified by our study overlapped with 334 unique masses (55.9%) identified by previous tomato studies. The low degree of matching between these studies suggests that cultivars, fruit maturity and growth conditions significantly affect tomato fruit metabolism. For example, the datasets of Iijima et al. (2008), which represent 70% of the AMRs used in this comparison, were obtained from fruits of maturing and fully mature stages of a single cultivar “Micro‐Tom”. The authors reported significant differences in metabolites during tomato maturation. Further metabolomic analyses of tomato samples grown under other conditions are needed to characterize unknown metabolic pathways.
A comparison of the 4,417 unique mass values revealed by this study with those of other studies resulted in no matches with 3,414 unique masses identified in A. thaliana (79.9%), 193 unique masses identified in M. truncatula (48.1%), and 3,066 unique masses identified in J. curcas (70.6%), respectively. The number of unique tomato mass values in our study that did not have any matches with any of the four plants was 3,113 unique mass values, corresponding to 4,239 AMRs. Of 3,113 unique mass values, only 328 unique mass values matched with known metabolites that are registered in the metabolism databases KEGG (Kanehisa et al., 2002), KNApSAcK (Afendi et al., 2012), LIPID MAPS (Fahy et al., 2007) and HMDB (Wishart et al., 2018).
We also compared the 4,417 unique mass values to accurate mass values archived in the Food Metabolome Repository (version 0.4.4), which has 969,352 peaks (149,310 unique mass values) from 222 foods (57 vegetables, 31 fishes, 28 fruits, 17 seasonings, 15 beverages, 11 cereals, 11 nuts and seeds, 11 beans, 10 milk products, 8 mushrooms, 8 meats, 5 potatoes, 5 sweets, 4 algae and 1 egg) detected in the ESI positive mode (Sakurai & Shibata, 2017). There were 3,813 unique mass values that matched in the repository. The 604 that did not match them corresponded to 951 tomato AMRs. This finding suggests that reporting AMRs from diverse samples should improve the prioritization of peaks for further investigation of plant metabolism based on sample specificity.
A comparison of the tomato AMRs with known metabolites was carried out using the metabolism databases TomatoCyc (version 4.0, https://plantcyc.org/databases/tomatocyc/4.0; 1,432 metabolites) and PlantCyc (version 12.0, https://www.plantcyc.org/databases/plantcyc/12.0; 3,208 metabolites), in which information about metabolites along with their published biosynthetic pathways has been collected (Schläpfer et al., 2017). The unique mass values of 189 and 323 AMRs were matched to those of TomatoCyc and PlantCyc, respectively. The intermediate metabolites and apolar compounds that are included in these databases were not matched as such compounds were not detected under the analytical conditions of our study.
As shown here, we compared the compound peaks based on the AMRs rather than the putative actual compounds by encapsulating the variety of adduct ions, multimers and fragments derived from the same compound, because the application and quality of the encapsulation in each data source are not uniform. The comparison based on the AMRs, we proposed here, is one of the ways to overcome this limitation and makes the untargeted metabolome data more useful to shed light on the unknown peaks for further metabolite annotation as exemplified below.
3.3. Examples of predicting unknown peaks
To demonstrate the use of the AMRs, especially for the tomato‐specific unknown peaks we annotated some peaks as candidate steroidal glycoalkaloids. From the 908 AMRs that had MS/MS fragmentation information but were not annotated with specific chemical names, 560 tomato‐specific peaks that did not match with the AMRs in the A. thaliana, M. truncatula or J. curcas datasets were selected. To analyse common metabolites of tomato, 62 peaks of the 560 tomato‐specific peaks that were found commonly in the 24 cultivars were selected (Figure 1). From the 62 selected peaks, we focused on KTP_024858 and KTP_019601 because their MS/MS fragmentation patterns were familiar to us as those of glycoalkaloids similar to tomatine, a steroidal alkaloid that is commonly found in maturing tomatoes. We found that the MS/MS fragmentation of tomatine that was obtained under the same LC‐MS conditions as those used in this study was similar to that of KTP_024858 in terms of neutral losses; the retention time of the peak when analysed by LC was also close to the retention time for tomatine. Combining this information with the predicted chemical formula C50H81NO21, we annotated this unknown peak as an isomer of dehydrotomatine. Only one isomer of dehydrotomatine was previously identified in tomato (Friedman et al., 1997; Ono et al., 1997), although some papers had predicted the presence of other possible isomers (Itkin et al., 2011; Mintz‐Oron et al., 2008; Schilmiller et al., 2010). Further studies on the isomer will determine its chemical structure.
FIGURE 1.
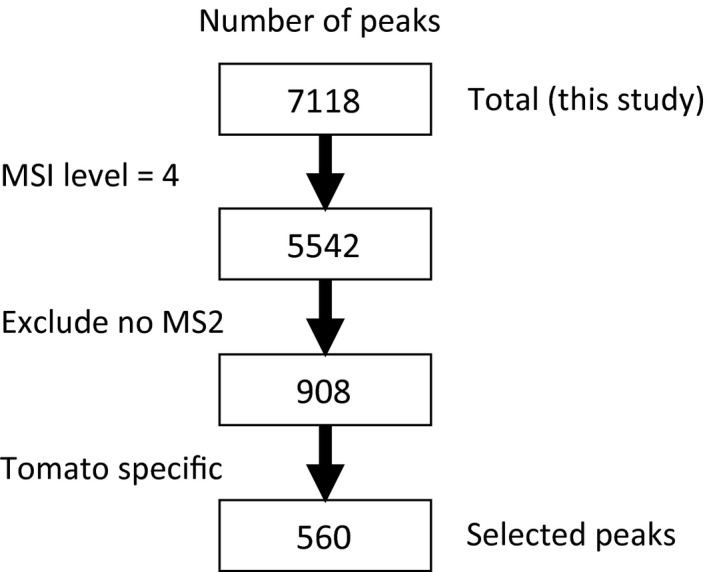
A workflow chart showing how peaks were selected for detailed manual annotation. The steps in selecting peaks for detailed manual annotation. For unknown peaks, MS/MS spectra and tomato‐specificity were used as criteria for selection
Expecting KTP_019601 to be a steroidal alkaloid from the MS/MS fragmentation, we found that KTP_019640, annotated as Ly‐coperoside F/G or Esculeoside A (C58H95NO29), had a similar MS/MS fragmentation pattern based on the neutral losses and retention time close to that of KTP_019601. Therefore, from the chemical formula C58H93NO29, KTP_019601 was annotated as an isomer of dehydro‐Lycoperoside F/G or dehydro‐Esculeoside A. As several peaks with similar m/z values to those of the glycoalkaloids exist near the retention time on the LC‐MS chromatogram, further studies should find additional isomers.
3.4. Secondary metabolites are diverse in tomato cultivars
To understand fundamentally why there was an increase in the number of new compounds using peak information, we further investigated the diversity of compounds in the tomato cultivars. Principal component analysis showed a large difference in metabolite profiles between cultivars (Figure 2a). The number of peaks also differed between cultivars, ranging from 1,322 peaks in cultivar #21 to 2,196 peaks in cultivar #12 (Figure 2b). As shown in Figure 2c, many peaks were commonly detected in small numbers of tomato cultivars, and more than half of the peaks were newly detected in comparison with those from previous reports (see the previous section). These results implied that sample‐specific metabolites exist within the tomato cultivars. We found that most of the primary metabolites resolved by LC‐MS that were identified using authentic compounds were detected in many cultivars (Table 1). The 67 metabolites identified after comparison to authentic standards were further classified into primary metabolites and secondary metabolites (Table S4). Among the 46 peaks classified as 42 primary metabolites, 31 (67.4%) were shared among 21–25 species, whereas, the 24 peaks classified as 23 secondary metabolites did not show such a tendency. This result implies that the secondary metabolites rather than the primary metabolites might contribute to the increase in metabolite diversity. Indeed, peak numbers that were annotated as putative flavonoids and steroids were very diverse among cultivars (Figure 3a,b). By manually checking the appropriateness of MS/MS fragments and the annotated chemical structure, we further assigned obvious O‐glycosides for the flavonoids and steroids and found that more than 90% of the flavonoids and steroids were putative glycosides (Figure 3).
FIGURE 2.
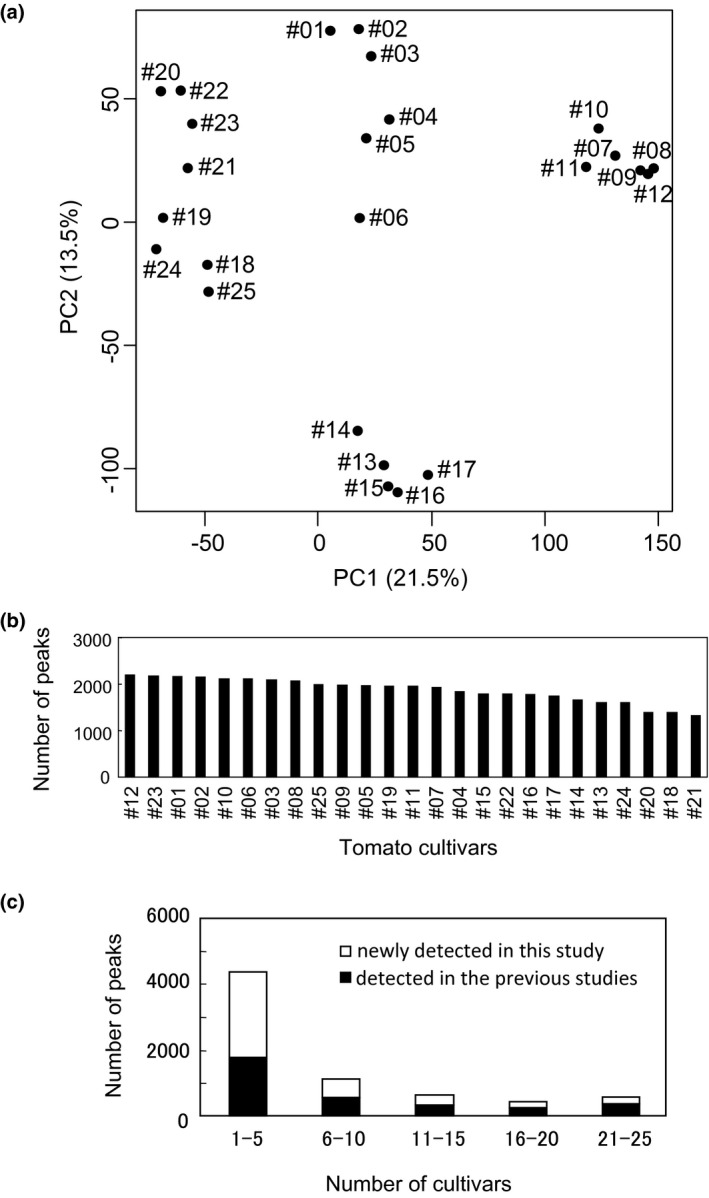
Comparison of accurate mass peaks detected among tomato cultivars. (a) PCA was used to compare peaks detected in all 25 cultivars. (b) The number of detected peaks in each cultivar. (c) Cultivar specificity of detected peaks. For the number of detected cultivars grouped into five classes (1–5, 6–10, 11–15, 16–20, 21–25), the number of peaks in common (black bars) or not in common (white bars) with a 5‐ppm margin to AMRs previously reported in other plant species (Arabidopsis thaliana, Medicago truncatula, tomato, Jatropha curcas)
TABLE 1.
Relationship between the number of peaks detected among cultivars and their annotation
| Annotated AMRs | Number of cultivars | ||||
|---|---|---|---|---|---|
| 1–5 | 6–10 | 11–15 | 16–20 | 21–25 | |
| Primary metabolites (MSI level 1 and 2) | 3 | 3 | 4 | 6 | 35 |
| Secondary metabolites (MSI level 1 and 2) | 10 | 10 | 8 | 14 | 21 |
| AMRs (MSI level 3) | 694 | 300 | 176 | 106 | 162 |
| Total | 707 | 313 | 188 | 126 | 218 |
FIGURE 3.
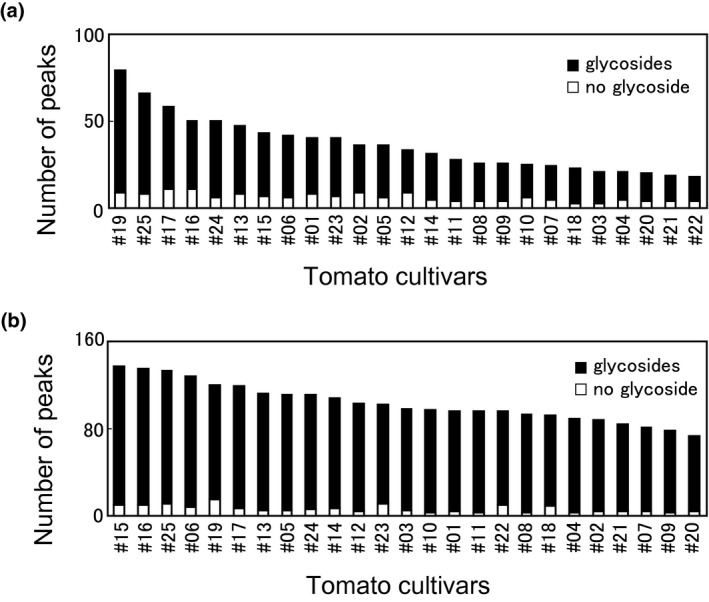
The number of peaks annotated as flavonoids or steroids among cultivars. The black bars indicate peaks annotated as glycosides and the white bars indicate annotated peaks without glycans. (a) flavonoids (b) steroids
These results suggested that the diversity of secondary metabolites largely contributes to the wide diversity of metabolites among tomato cultivars. Our results showed good agreement with a previous study by Slimestad and Verheul (2009), who reported some secondary metabolite diversity in tomato cultivars. Glycosylation has also been identified as a contributor to the increased diversity of secondary metabolites (Gachon et al., 2005; Tiwari et al., 2016). In this study, we also found other modifications such as methylation and acetylation. Because we could not determine if these substituents were directly attached to the core structure or indirectly attached via glycosyl groups, the diversity and magnitude of these other modifications are unclear. Similar to glycosylation, these modifications might contribute to the wide diversity of secondary metabolites among cultivars and species. To uncover the entire chemical space of unknown compounds in plants, similar protocols for metabolome analyses to enhance the number of AMRs should be performed using a wide range of species and cultivars.
3.5. Construction of a database for searching tomato AMRs
We constructed a searchable tomato database, “TOMATOMET”, containing the peaks detected in this study (http://metabolites.in/tomato‐fruits/). The peaks can be searched by their accurate mass values and information, such as compound annotation, category classification, distribution among cultivars cross‐species‐specificity, as well as retention times, MS/MS spectra and types of adduct ions. Peaks with types of variation in adduct ions that originate from a single putative metabolite were stored as individual records in TOMATOMET because these results will be useful for metabolite annotation based on cross‐species‐specificities and annotated adduct ion types. The search functions are available as application programming interfaces (APIs) in manners of representation state transfer (REST), and the results are available in a JavaScript object notation (JSON) format. These facilitate researchers to integrate the search function into other bioinformatics tools and to search a vast number of mass values. As the datasets are available in Table S2 in the Excel format, users can search them by user's parameters.
4. CONCLUSIONS
We constructed a set of peaks confidently detected in mature tomato fruits from 25 cultivars by manual curation and published the data via a searchable database, “TOMATOMET”. The dataset contributes a significant enhancement in the numbers of AMRs and unique mass values when compared to previous reports. A large portion of the newly detected AMRs is cultivar specific, and modification of secondary metabolites is undoubtedly involved to generate the wide diversity of metabolites found among cultivars. Using cross‐sample‐specificity, we annotated two previously unknown peaks as steroid glycoalkaloids. These results suggest that the enhancement of AMRs is useful for depicting the actual chemical space for mass‐based metabolite annotation and annotation based on cross‐sample‐specificity. Therefore, AMRs should be reported for diverse cultivars, tissue and organ types, and developmental stages even within a plant species.
ACCESSION NUMBERS
All raw MS data generated by this study were deposited in the metabolome data repository MassBase (http://webs2.kazusa.or.jp/massbase/) under accession numbers MDLC1_46828 to MDLC1_46929 and MDLC_146933 to MDLC1_46935.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
TA, ST and NW collected samples and performed experiments; TA and NS performed bioinformatics analyses and database development. DS, HS, KA, TK and YM designed the study. TA, NS and DS wrote the manuscript. All authors discussed the results and approved the final manuscript.
Supporting information
Table S1
Table S2
Table S3
Table S4
ACKNOWLEDGEMENTS
We thank Haruya Takahashi and Shinsuke Mohri (Kyoto University) for helping with sample preparation for mass spectrometry, Rihito Takisawa and Akira Kitajima (the Experimental Farm of Kyoto University) for providing tomato fruits of the “Kyo‐temari” cultivar, and Kunihiro Suda and Keishi Ozawa (Kazusa DNA Research Institute) for growing and harvesting “Micro‐Tom”. We thank Shigeo Tamura (KAGOME CO., LTD.) for supporting the research project. We thank the National BioResource Project (NBRP) of the Ministry of Education, Culture, Sports, Science and Technology (Japan) for providing “Micro‐Tom” seeds used in this research. This work is funded by KAGOME CO., LTD.
Ara T, Sakurai N, Takahashi S, et al. TOMATOMET: A metabolome database consists of 7118 accurate mass values detected in mature fruits of 25 tomato cultivars. Plant Direct. 2021;5:e00318. 10.1002/pld3.318
Takeshi Ara and Nozomu Sakurai contributed equally.
REFERENCES
- Afendi, F. M. , Okada, T. , Yamazaki, M. , Hirai‐Morita, A. , Nakamura, Y. , Nakamura, K. , Ikeda, S. , Takahashi, H. , Altaf‐Ul‐Amin, M. , Darusman, L. K. , Saito, K. , & Kanaya, S. (2012). KNApSAcK family databases: Integrated metabolite‐plant species databases for multifaceted plant research. Plant and Cell Physiology, 53, e1. 10.1093/pcp/pcr165 [DOI] [PubMed] [Google Scholar]
- Aharoni, A. , Ric de Vos, C. H. , Verhoeven, H. A. , Maliepaard, C. A. , Kruppa, G. , Bino, R. , & Goodenowe, D. B. (2002). Nontargeted metabolome analysis by use of Fourier transform ion cyclotron mass spectrometry. OMICS: A Journal of Integrative Biology, 6, 217–234. 10.1089/15362310260256882 [DOI] [PubMed] [Google Scholar]
- Ara, T. , Enomoto, M. , Arita, M. , Ikeda, C. , Kera, K. , Yamada, M. , Nishioka, T. , Ikeda, T. , Nihei, Y. , Shibata, D. , Kanaya, S. , & Sakurai, N. (2015). Metabolonote: A wiki‐based database for managing hierarchical metadata of metabolome analyses. Frontiers in Bioengineering and Biotechnology, 3, 38. 10.3389/fbioe.2015.00038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisken, S. , Earll, M. , Baxter, C. , Portwood, D. , Ament, Z. , Kende, A. , Hodgman, C. , Seymour, G. , Smith, R. , Fraser, P. , Seymour, M. , Salek, R. M. , & Steinbeck, C. (2014). Metabolic differences in ripening of Solanum lycopersicum 'Ailsa Craig' and three monogenic mutants. Scientific Data, 1, 140029. 10.1038/sdata.2014.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaženović, I. , Kind, T. , Torbašinović, H. , Obrenović, S. , Mehta, S. S. , Tsugawa, H. , Wermuth, T. , Schauer, N. , Jahn, M. , Biedendieck, R. , Jahn, D. , & Fiehn, O. (2017). Comprehensive comparison of in silico MS/MS fragmentation tools of the CASMI contest: Database boosting is needed to achieve 93% accuracy. Journal of Cheminformatics, 9, 32. 10.1186/s13321-017-0219-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M. , Wedge, D. C. , Goodacre, R. , Kell, D. B. , Baker, P. N. , Kenny, L. C. , Mamas, M. A. , Neyses, L. , & Dunn, W. B. (2011). Automated workflows for accurate mass‐based putative metabolite identification in LC/MS‐derived metabolomics datasets. Bioinformatics, 27, 1108–1112. 10.1093/bioinformatics/btr079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, J. , Li, M. , Chen, J. , Liu, P. , & Li, Z. (2016). Effects of MeJA on Arabidopsis metabolome under endogenous JA deficiency. Scientific Reports, 6, 37674. 10.1038/srep37674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaleckis, R. , Meister, I. , Zhang, P. , & Wheelock, C. E. (2019). Challenges, progress and promises of metabolite annotation for LC‐MS‐based metabolomics. Current Opinion in Biotechnology, 55, 44–50. 10.1016/j.copbio.2018.07.010 [DOI] [PubMed] [Google Scholar]
- DeFelice, B. C. , Mehta, S. S. , Samra, S. , Čajka, T. , Wancewicz, B. , Fahrmann, J. F. , & Fiehn, O. (2017). Mass spectral feature list optimizer (MS‐FLO): A tool to minimize false positive peak reports in untargeted liquid chromatography‐mass spectroscopy (LC‐MS) data processing. Analytical Chemistry, 89, 3250–3255. 10.1021/acs.analchem.6b04372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy, E. , Sud, M. , Cotter, D. , & Subramaniam, S. (2007). LIPID MAPS online tools for lipid research. Nucleic Acids Research, 35, W606–W612. 10.1093/nar/gkm324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, M. , Kozukue, N. , & Harden, L. A. (1997). Structure of the tomato Glycoalkaloid Tomatidenol‐3‐beta‐lycotetraose (Dehydrotomatine). Journal of Agriculture and Food Chemistry, 45, 1541–1547. 10.1021/jf960875q [DOI] [Google Scholar]
- Fukushima, A. , & Kusano, M. (2013). Recent progress in the development of metabolome databases for plant systems biology. Frontiers in Plant Science, 4, 73. 10.3389/fpls.2013.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gachon, C. M. M. , Langlois‐Meurinne, M. , & Saindrenan, P. (2005). Plant secondary metabolism glycosyltransferases: The emerging functional analysis. Trends in Plant Science, 10, 542–549. 10.1016/j.tplants.2005.09.007 [DOI] [PubMed] [Google Scholar]
- Garbowicz, K. , Liu, Z. , Alseekh, S. , Tieman, D. , Taylor, M. , Kuhalskaya, A. , Ofner, I. , Zamir, D. , Klee, H. J. , Fernie, A. R. , & Brotman, Y. (2018). Quantitative trait loci analysis identifies a prominent gene involved in the production of fatty acid‐derived flavor volatiles in tomato. Molecular Plant, 11, 1147–1165. 10.1016/j.molp.2018.06.003 [DOI] [PubMed] [Google Scholar]
- Giavalisco, P. , Li, Y. , Matthes, A. , Eckhardt, A. , Hubberten, H. M. , Hesse, H. , Segu, S. , Hummel, J. , Köhl, K. , & Willmitzer, L. (2011). Elemental formula annotation of polar and lipophilic metabolites using 13C, 15N and 34S isotope labelling, in combination with high‐resolution mass spectrometry. The Plant Journal, 68, 364–376. 10.1111/j.1365-313X.2011.04682.x [DOI] [PubMed] [Google Scholar]
- Giovannucci, E. (1999). Tomatoes, tomato‐based products, lycopene, and cancer: Review of the epidemiologic literature. Journal of the National Cancer Institute, 91, 317–331. 10.1093/jnci/91.4.317 [DOI] [PubMed] [Google Scholar]
- Gläser, K. , Kanawati, B. , Kubo, T. , Schmitt‐Kopplin, P. , & Grill, E. (2014). Exploring the Arabidopsis sulfur metabolome. The Plant Journal, 77, 31–45. 10.1111/tpj.12359 [DOI] [PubMed] [Google Scholar]
- Horai, H. , Arita, M. , Kanaya, S. , Nihei, Y. , Ikeda, T. , Suwa, K. , Ojima, Y. , Tanaka, K. , Tanaka, S. , Aoshima, K. , Oda, Y. , Kakazu, Y. , Kusano, M. , Tohge, T. , Matsuda, F. , Sawada, Y. , Hirai, M. Y. , Nakanishi, H. , Ikeda, K. , … Nishioka, T. (2010). MassBank: A public repository for sharing mass spectral data for life sciences. Journal of Mass Spectrometry, 45, 703–714. 10.1002/jms.1777 [DOI] [PubMed] [Google Scholar]
- Hufsky, F. , & Böcker, S. (2017). Mining molecular structure databases: Identification of small molecules based on fragmentation mass spectrometry data. Mass Spectrometry Reviews, 36, 624–633. 10.1002/mas.21489 [DOI] [PubMed] [Google Scholar]
- Iijima, Y. , Nakamura, Y. , Ogata, Y. , Tanaka, K. , Sakurai, N. , Suda, K. , Suzuki, T. , Suzuki, H. , Okazaki, K. , Kitayama, M. , Kanaya, S. , Aoki, K. , & Shibata, D. (2008). Metabolite annotations based on the integration of mass spectral information. The Plant Journal, 54, 949–962. 10.1111/j.1365-313x.2008.03434.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itkin, M. , Rogachev, I. , Alkan, N. , Rosenberg, T. , Malitsky, S. , Masini, L. , Meir, S. , Iijima, Y. , Aoki, K. , de Vos, R. , Prusky, D. , Burdman, S. , Beekwilder, J. , & Aharoni, A. (2011). GLYCOALKALOID METABOLISM1 is required for steroidal alkaloid glycosylation and prevention of phytotoxicity in tomato. The Plant Cell, 23, 4507–4525. 10.1105/tpc.111.088732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankevics, A. , Merlo, M. E. , de Vries, M. , Vonk, R. J. , Takano, E. , & Breitling, R. (2012). Separating the what from the chaff: A prioritization pipeline for the analysis of metabolomics datasets. Metabolomics, 8, 29–36. 10.1007/s11306-011-0341-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M. , Goto, S. , Kawashima, S. , & Nakaya, A. (2002). The KEGG databases at GenomeNet. Nucleic Acids Research, 30, 42–46. 10.1093/nar/30.1.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kera, K. , Fine, D. D. , Wherritt, D. J. , Nagashima, Y. , Shimada, N. , Ara, T. , Ogata, Y. , Sumner, L. W. , & Suzuki, H. (2018). Pathway‐specific metabolome analysis with 18O2‐labeled Medicago truncatula via a mass spectrometry‐based approach. Metabolomics, 14, 71. 10.1007/s11306-018-1364-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. , Hirai, S. , Goto, T. , Ohyane, C. , Takahashi, H. , Tsugane, T. , Konishi, C. , Fujii, T. , Inai, S. , Iijima, Y. , Aoki, K. , Shibata, D. , Takahashi, N. , & Kawada, T. (2012). Potent PPARα activator derived from tomato juice, 13‐oxo‐9,11‐octadecadienoic acid, decreases plasma and hepatic triglyceride in obese diabetic mice. PLoS ONE, 7, e31317. 10.1371/journal.pone.0031317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind, T. , & Fiehn, O. (2007). Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics, 8, 105. 10.1186/1471-2105-8-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger, S. , Giavalisco, P. , Krall, L. , Steinhauser, M. C. , Büssis, D. , Usadel, B. , Flügge, U. I. , Fernie, A. R. , Willmitzer, L. , & Steinhauser, D. (2011). A topological map of the compartmentalized Arabidopsis thaliana leaf metabolome. PLoS One, 6, e17806. 10.1371/journal.pone.0017806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, Z. , Tsugawa, H. , Wohlgemuth, G. , Mehta, S. , Mueller, M. , Zheng, Y. , Ogiwara, A. , Meissen, J. , Showalter, M. , Takeuchi, K. , Kind, T. , Beal, P. , Arita, M. , & Fiehn, O. (2018). Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nature Methods, 15, 53–56. 10.1038/nmeth.4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. , Kind, T. , Yang, D. , Leon, C. , & Fiehn, O. (2014). MS2Analyzer: A software for small molecule substructure annotations from accurate tandem mass spectra. Analytical Chemistry, 86, 10724–10731. 10.1021/ac502818e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahieu, N. G. , & Patti, G. J. (2017). Systems‐level annotation of a metabolomics data set reduces 25,000 features to fewer than 1,000 unique metabolites. Analytical Chemistry, 89, 10397–10406. 10.1021/acs.analchem.7b02380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martí, R. , Roselló, S. , & Cebolla‐Cornejo, J. (2016). Tomato as a source of carotenoids and polyphenols targeted to cancer prevention. Cancers, 8, E58. 10.3390/cancers8060058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz‐Oron, S. , Mandel, T. , Rogachev, I. , Feldberg, L. , Lotan, O. , Yativ, M. , Wang, Z. , Jetter, R. , Venger, I. , Adato, A. , & Aharoni, A. (2008). Gene expression and metabolism in tomato fruit surface tissues. Plant Physiology, 147, 823–851. 10.1104/pp.108.116004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivon, F. , Allard, P. M. , Koval, A. , Righi, D. , Genta‐Jouve, G. , Neyts, J. , Apel, C. , Pannecouque, C. , Nothias, L. F. , Cachet, X. , Marcourt, L. , Roussi, F. , Katanaev, V. L. , Touboul, D. , Wolfender, J. L. , & Litaudon, M. (2017). Bioactive natural products prioritization using massive multi‐informational molecular networks. ACS Chemical Biology, 12, 2644–2651. 10.1021/acschembio.7b00413 [DOI] [PubMed] [Google Scholar]
- Ono, H. , Kozuka, D. , Chiba, Y. , Horigane, A. , & Isshiki, K. (1997). Structure and cytotoxicity of dehydrotomatine, a minor component of tomato glycoalkaloids. Journal of Agriculture and Food Chemistry, 45, 3743–3746. 10.1021/jf970253k [DOI] [Google Scholar]
- Perez‐Fons, L. , Wells, T. , Corol, D. I. , Ward, J. L. , Gerrish, C. , Beale, M. H. , Seymour, G. B. , Bramley, P. M. , & Fraser, P. D. (2014). A genome‐wide metabolomic resource for tomato fruit from Solanum pennellii . Scientific Reports, 4, 3859. 10.1038/srep03859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiola, A. , Rigano, M. M. , Calafiore, R. , Frusciante, L. , & Barone, A. (2014). Enhancing the health‐promoting effects of tomato fruit for biofortified food. Mediators of Inflammation, 2014, 139873. 10.1155/2014/139873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai, N. , Ara, T. , Enomoto, M. , Motegi, T. , Morishita, Y. , Kurabayashi, A. , Iijima, Y. , Ogata, Y. , Nakajima, D. , Suzuki, H. , & Shibata, D. (2014). Tools and databases of the KOMICS web portal for preprocessing, mining, and dissemination of metabolomics data. BioMed Research International, 2014, 194812. 10.1155/2014/194812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai, N. , Ara, T. , Kanaya, S. , Nakamura, Y. , Iijima, Y. , Enomoto, M. , Motegi, T. , Aoki, K. , Suzuki, H. , & Shibata, D. (2012). An application of a relational database system for high‐throughput prediction of elemental compositions from accurate mass values. Bioinformatics, 29, 290–291. 10.1093/bioinformatics/bts660 [DOI] [PubMed] [Google Scholar]
- Sakurai, N. , & Shibata, D. (2017). Tools and databases for an integrated metabolite annotation environment for liquid chromatography‐mass spectrometry‐based untargeted metabolomics. Carotenoid Science, 22, 16–22. [Google Scholar]
- Sano, R. , Ara, T. , Akimoto, N. , Sakurai, N. , Suzuki, H. , Fukuzawa, Y. , Kawamitsu, Y. , Ueno, M. , & Shibata, D. (2012). Dynamic metabolic changes during fruit maturation in Jatropha curcas L. Plant Biotechnology, 29, 175–178. 10.5511/plantbiotechnology.12.0503a [DOI] [Google Scholar]
- Schilmiller, A. , Shi, F. , Kim, J. , Charbonneau, A. L. , Holmes, D. , Daniel Jones, A. , & Last, R. L. (2010). Mass spectrometry screening reveals widespread diversity in trichome specialized metabolites of tomato chromosomal substitution lines. The Plant Journal, 62, 391–403. 10.1111/j.1365-313X.2010.04154.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schläpfer, P. , Zhang, P. , Wang, C. , Kim, T. , Banf, M. , Chae, L. , Dreher, K. , Chavali, A. K. , Nilo‐Poyanco, R. , Bernard, T. , Kahn, D. , & Rhee, S. Y. (2017). Genome‐wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiology, 173, 2041–2059. 10.1104/pp.16.01942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senan, O. , Aguilar‐Mogas, A. , Navarro, M. , Capellades, J. , Noon, L. , Burks, D. , Yanes, O. , Guimerà, R. , & Sales‐Pardo, M. (2019). CliqueMS: A computational tool for annotating in‐source metabolite ions from LC‐MS untargeted metabolomics data based on a coelution similarity network. Bioinformatics, 35, 4089–4097. 10.1093/bioinformatics/btz207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slimestad, R. , & Verheul, M. (2009). Review of flavonoids and other phenolics from fruits of different tomato (Lycopersicon esculentum Mill.) cultivars. Journal of the Science of Food and Agriculture, 89, 1255–1270. 10.1002/jsfa.3605 [DOI] [Google Scholar]
- Steinbeck, C. , Conesa, P. , Haug, K. , Mahendraker, T. , Williams, M. , Maguire, E. , Rocca‐Serra, P. , Sansone, S. A. , Salek, R. M. , & Griffin, J. L. (2012). MetaboLights: Towards a new COSMOS of metabolomics data management. Metabolomics, 8, 757–760. 10.1007/s11306-012-0462-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sud, M. , Fahy, E. , Cotter, D. , Azam, K. , Vadivelu, I. , Burant, C. , Edison, A. , Fiehn, O. , Higashi, R. , Nair, K. S. , Sumner, S. , & Subramaniam, S. (2016). Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Research, 44, D463–470. 10.1093/nar/gkv1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner, L. W. , Amberg, A. , Barrett, D. , Beale, M. H. , Beger, R. , Daykin, C. A. , Fan, T. W. M. , Fiehn, O. , Goodacre, R. , Griffin, J. L. , Hankemeier, T. , Hardy, N. , Harnly, J. , Higashi, R. , Kopka, J. , Lane, A. N. , Lindon, J. C. , Marriott, P. , Nicholls, A. W. , … Viant, M. R. (2007). Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics, 3, 211–221. 10.1007/s11306-007-0082-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari, P. , Sangwan, R. S. , & Sangwan, N. S. (2016). Plant secondary metabolism linked glycosyltransferases: An update on expanding knowledge and scopes. Biotechnology Advances, 34, 714–739. 10.1016/j.biotechadv.2016.03.006 [DOI] [PubMed] [Google Scholar]
- Tohge, T. , & Fernie, A. R. (2015). Metabolomics‐inspired insight into developmental, environmental and genetic aspects of tomato fruit chemical composition and quality. Plant and Cell Physiology, 56, 1681–1696. 10.1093/pcp/pcv093 [DOI] [PubMed] [Google Scholar]
- Tsugawa, H. (2018). Advances in computational metabolomics and databases deepen the understanding of metabolisms. Current Opinion in Biotechnology, 54, 10–17. 10.1016/j.copbio.2018.01.008 [DOI] [PubMed] [Google Scholar]
- Uppal, K. , Walker, D. I. , & Jones, D. P. (2017). xMSannotator: An R package for network‐based annotation of high‐resolution metabolomics data. Analytical Chemistry, 89, 1063–1067. 10.1021/acs.analchem.6b01214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meulebroek, L. , Bussche, J. V. , De Clercq, N. , Steppe, K. , & Vanhaecke, L. (2015). A metabolomics approach to unravel the regulating role of phytohormones towards carotenoid metabolism in tomato fruit. Metabolomics, 11, 667–683. 10.1007/s11306-014-0728-9 [DOI] [Google Scholar]
- Viant, M. R. , Kurland, I. J. , Jones, M. R. , & Dunn, W. B. (2017). How close are we to complete annotation of metabolomes? Current Opinion in Chemical Biology, 36, 64–69. 10.1016/j.cbpa.2017.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. (2008). Needs for new plant‐derived pharmaceuticals in the post‐genome era: An industrial view in drug research and development. Phytochemistry Reviews, 7, 395–406. 10.1007/s11101-008-9092-6 [DOI] [Google Scholar]
- Wang, M. , Carver, J. J. , Phelan, V. V. , Sanchez, L. M. , Garg, N. , Peng, Y. , Nguyen, D. D. , Watrous, J. , Kapono, C. A. , Luzzatto‐Knaan, T. , Porto, C. , Bouslimani, A. , Melnik, A. V. , Meehan, M. J. , Liu, W. T. , Crüsemann, M. , Boudreau, P. D. , Esquenazi, E. , Sandoval‐Calderón, M. , … Bandeira, N. (2016). Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nature Biotechnology, 34, 828–837. 10.1038/nbt.3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart, D. S. (2009). Computational strategies for metabolite identification in metabolomics. Bioanalysis, 1, 1579–1596. 10.4155/bio.09.138 [DOI] [PubMed] [Google Scholar]
- Wishart, D. S. , Feunang, Y. D. , Marcu, A. , Guo, A. C. , Liang, K. , Vázquez‐Fresno, R. , Sajed, T. , Johnson, D. , Li, C. , Karu, N. , & Sayeeda, Z. (2018). HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Research, 46, D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtele, E. S. , Chappell, J. , Jones, A. D. , Celiz, M. D. , Ransom, N. , Hur, M. , Rizshsky, L. , Crispin, M. , Dixon, P. , Liu, J. , Widrlechner, M. P. , & Nikolau, B. J. (2012). Medicinal plants: A public resource for metabolomics and hypothesis development. Metabolites, 2, 1031–1059. 10.3390/metabo2041031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtzel, E. T. , & Kutchan, T. M. (2016). Plant metabolism, the diverse chemistry set of the future. Science, 353, 1232–1236. 10.1126/science.aad2062 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Table S4