

Abstract
Current methods for Suzuki-Miyaura couplings of non-triflate phenol derivatives are limited by their intolerance of halides including aryl chlorides. This is because Ni(0) and Pd(0) often undergo oxidative addition of organohalides at a similar or faster rate than most Ar—O bonds. DFT and stoichiometric oxidative addition studies demonstrate that small phosphines, in particular PMe3, are unique in promoting preferential reaction of Ni(0) with aryl tosylates and other C—O bonds in the presence of aryl chlorides. This selectivity was exploited in the first Ni-catalyzed C—O-selective Suzuki-Miyaura coupling of chlorinated phenol derivatives where the oxygen-containing leaving group is not a fluorinated sulfonate such as triflate. Computational studies suggest that the origin of divergent selectivity between PMe3 and other phosphines differs from prior examples of ligand-controlled chemodivergent cross-couplings. PMe3 effects selective reaction at tosylate due to both electronic and steric factors. A close interaction between nickel and a sulfonyl oxygen of tosylate during oxidative addition is critical to the observed selectivity.
Keywords: Chemoselectivity, Cross-coupling, Density functional calculations, Ligand effects, Nickel
Graphical Abstract
INTRODUCTION
The Suzuki-Miyaura cross-coupling reaction is among the most widely used strategies for C—C bond formation in organic synthesis. This transformation is traditionally catalyzed by palladium and employs aryl halides or arenes substituted by highly labile fluorinated sulfonates (especially triflates) as electrophilic coupling partners.1 However, recent advances in nickel-catalyzed cross-couplings have facilitated the use of more robust phenol derivatives such as aryl sulfamates, tosylates, mesylates, carbamates, and esters.2 The ability to exploit less labile phenol derivatives as alternatives to aryl halides has several advantages. First, phenols can be cheaper or more accessible than organohalides. Second, many phenol derivatives can be carried through multiple synthetic steps, including those involving Pd-catalysis (e.g., Scheme 1A).3 Third, some phenol derivatives can direct other synthetic steps prior to their use in cross-coupling, such as ortho metalation, catalytic C—H functionalization, and electrophilic aromatic substitution.4,5
Scheme 1.
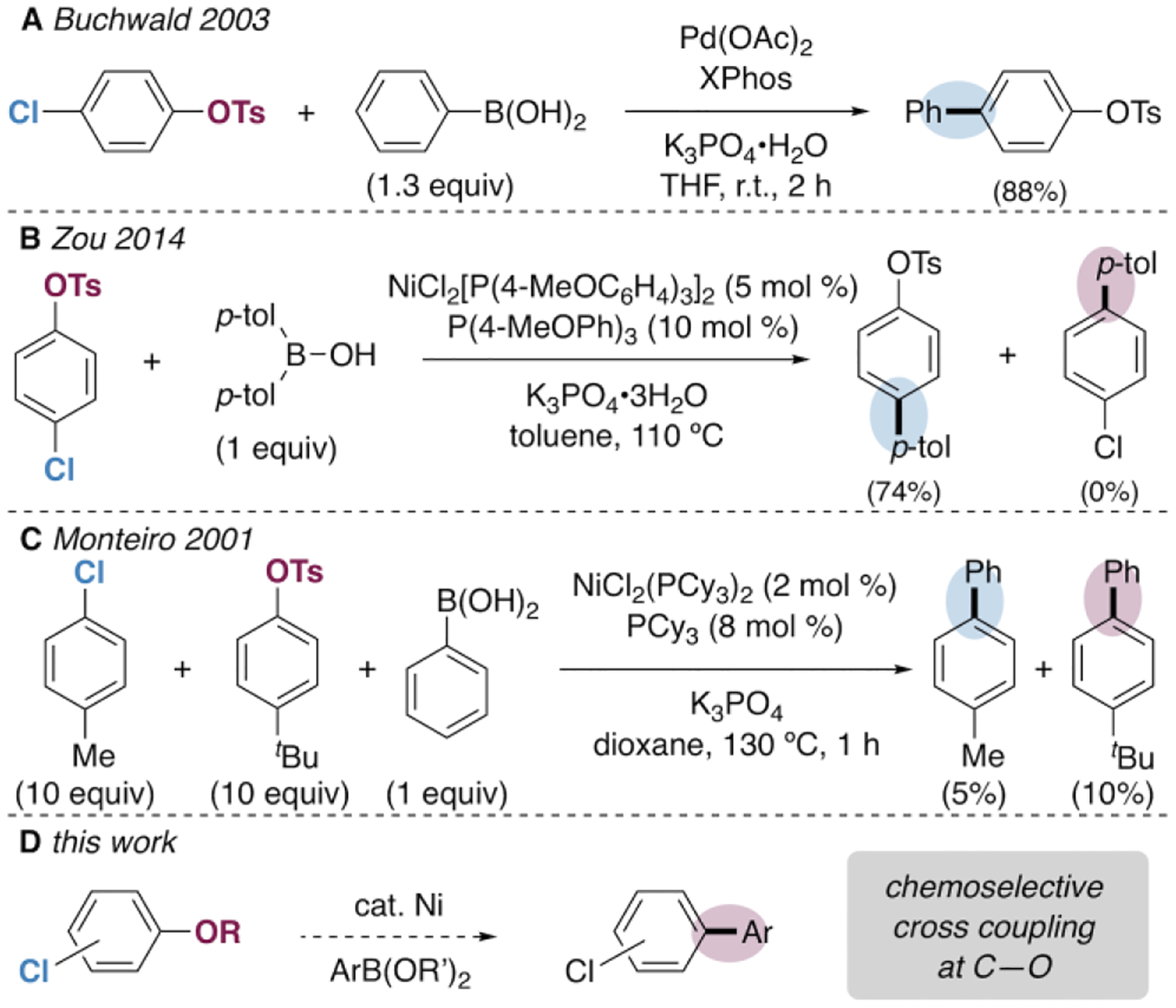
Ni- and Pd-Catalyzed Suzuki-Miyaura Couplings of Non-Triflate Chlorophenol Derivatives.
Despite numerous reports of Ni-catalyzed cross-coupling of robust phenol derivatives, a critical limitation of current methods is that aryl halides such as chloride are not tolerated. This is because organohalides tend to react with Ni(0) at a similar or faster rate than Ar—O bonds.6,7 Efforts to achieve Ni-catalyzed Suzuki-Miyaura coupling of chlorophenol derivatives have rarely been reported, but the few existing examples demonstrate preferential coupling of the chloride (e.g., Scheme 1B), poor yields, and/or low selectivity to form a mixture of products (e.g., Scheme 1C).8,9,10 This functional group incompatibility severely limits the utility of phenol-derived electrophiles, as myriad biologically-relevant synthetic targets contain halogens. For example, about 40% of drugs currently on the market or in clinical trials and about 50% of molecules in high-throughput screening are halogenated.11 Among these, a large percentage (~38%) are chlorinated.12 As such, development of methods for selective cross-coupling of chlorinated phenol derivatives via C—O cleavage (Scheme 1D) could streamline access to pharmacologically relevant targets.
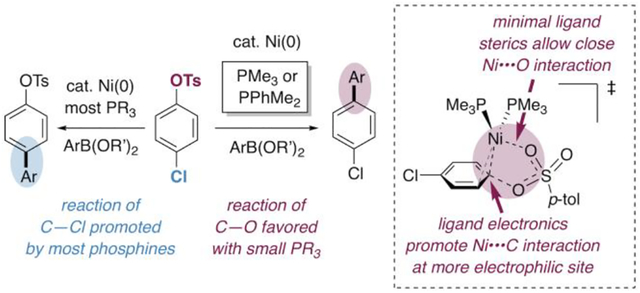
Herein we demonstrate that small phosphine ligands for Ni(0), especially PMe3, facilitate selective oxidative addition of Ar—OTs bonds in the presence of Ar—Cl bonds. This selectivity is accurately predicted by DFT calculations, and a series of stoichiometric studies confirm that methylphosphines are unique in enabling this C—O-selective oxidative addition. By using a Ni/PMe3 catalytic system, we provide proof-of-principle for a highly selective Suzuki-Miyaura cross-coupling of chloroaryl tosylates through C—O cleavage.10,13,14 Our DFT studies suggest that trimethylphosphine’s unique effect on selectivity can be attributed to a combination of electronic and steric factors. In particular, trimethylphosphine’s small size enables a close stabilizing interaction between a sulfonyl S=O and Ni during oxidative addition at C—OTs.
RESULTS AND DISCUSSION
DFT Calculations With PMe3.
We conducted density functional theory (DFT) calculations on oxidative addition at Ni(0) to gather insight into the effect of ligands on this step.15 Our initial DFT studies examined the reaction of Ni(PMe3)2 with 4-chlorophenyl tosylate (1). We chose to focus on aryl tosylates because they are generally reactive toward Ni(0), but are often inert toward other reaction conditions including Pd-catalysis (e.g., Scheme 1A). PMe3 was chosen as a model phosphine ligand due to its computational simplicity. Previous DFT studies suggested that bis-phosphine ligated nickel is likely favored over mono-ligated analogues during oxidative addition when dispersion is considered,16 so two PMe3 ligands were included in these calculations. Geometry optimizations were conducted using the functional MN15L,17 and energies were further refined with MN15L using a larger basis set and 1,4-dioxane as an implicit solvent (see SI for details).
Surprisingly, our initial calculations predict that oxidative addition of C—OTs at Ni(PMe3)2 should be significantly faster than oxidative addition of C—Cl (Figure 1). Transition structure 4a-TS is ~3.0 kcal mol−1 lower in energy than 3-TS, suggesting that selective cross-coupling at tosylate might occur with Ni/PMe3. We initially assumed that this prediction reflected error in the DFT energies, since it contradicts literature results with PCy3,8a 1,2-bis(diphenylphosphino)ethane,8h and triarylphosphines8f—ligands that are more conventional for catalysis. To our knowledge, PMe3 had not been experimentally evaluated in Ni-catalyzed Suzuki-Miyaura cross-couplings.18–20
Figure 1.
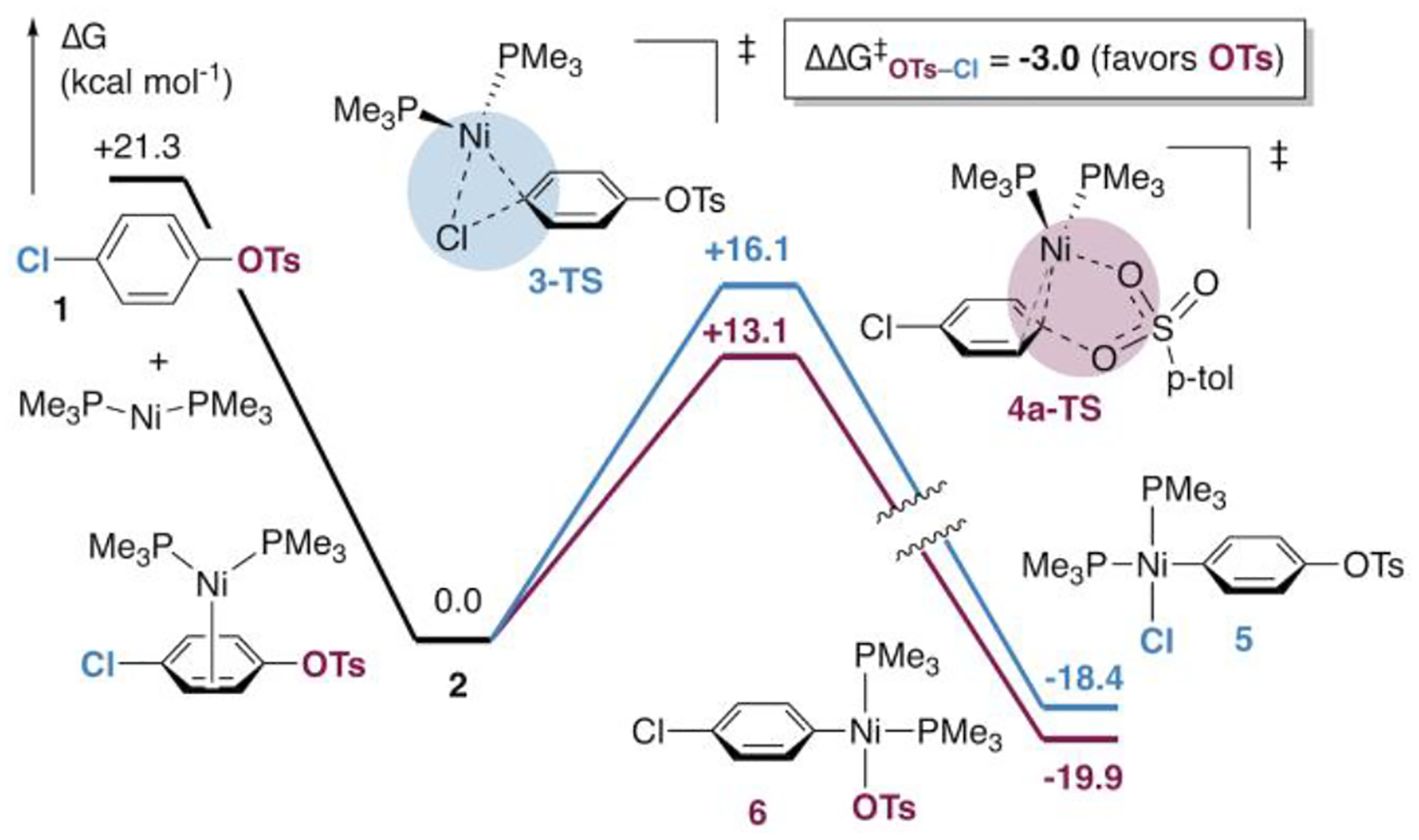
Free energy profile for oxidative addition of the C—O and C—Cl bonds of 1 at Ni(PMe3)2.
DFT Calculations Using PCy3 and PPh3.
We next calculated oxidative addition transition structures with the more experimentally relevant phosphine ligands PCy3 and PPh3 (Figure 2). In contrast to the calculations with PMe3, oxidative addition at Ni(PPh3)2 is predicted to be faster at chloride than tosylate (ΔΔG‡ = 2.0 kcal mol−1). Ni(PCy3)2 favors reaction at tosylate, but only by 1.2 kcal mol−1 (compared to 3.0 kcal mol−1 with PMe3). These predictions are consistent with experimental reports using PCy38a or triarylphosphines in catalysis,8f and contrast with our DFT results using PMe3.
Figure 2.
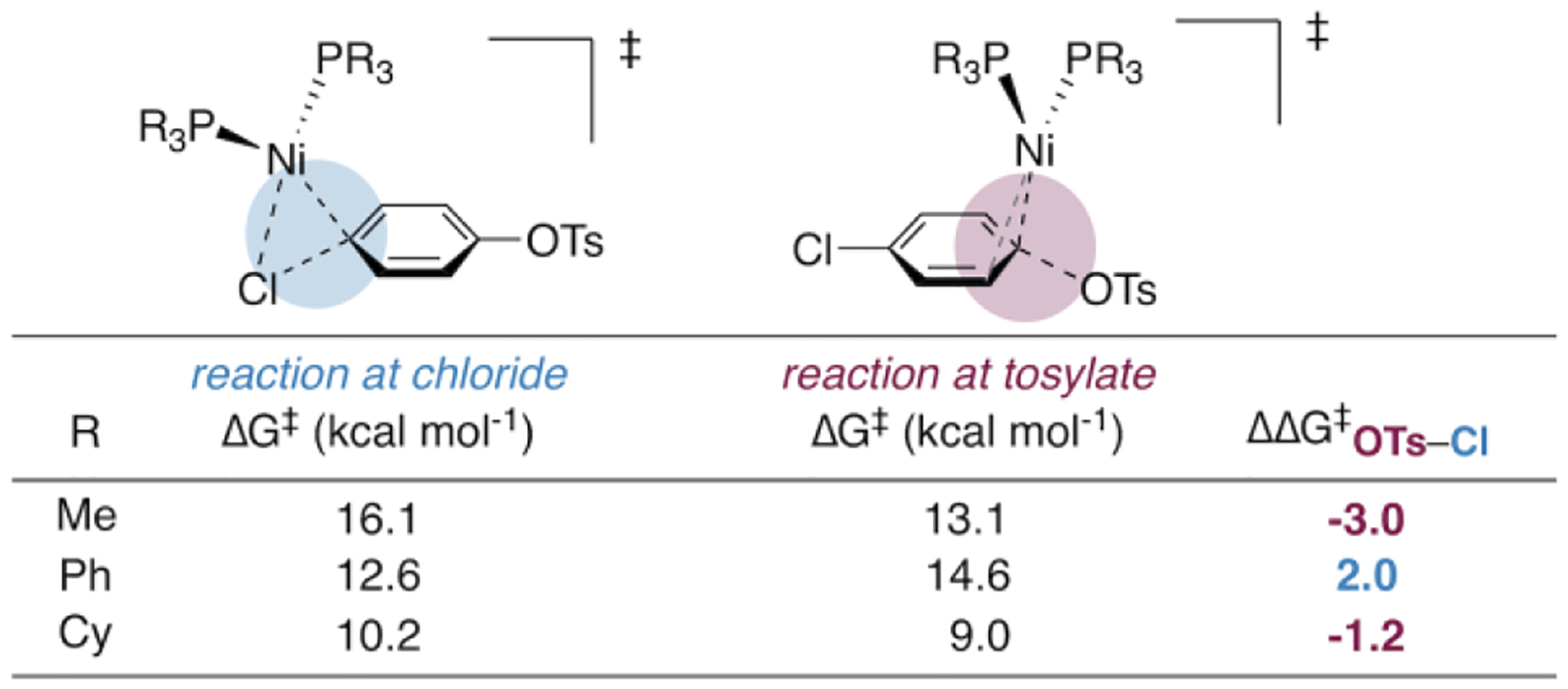
Calculated free energies of activation (in kcal mol−1) for oxidative addition with PMe3, PPh3, and PCy3, measured from the preceding π-complex.
Several additional DFT methods were evaluated with each of the ligands PMe3, PCy3, and PPh3. Most methods predict the same trend in preference for oxidative addition of tosylate (PMe3 > PCy3 > PPh3; see SI for details). With the majority of methods tested, Ni(PMe3)2 is predicted to favor reaction at tosylate, while Ni(PPh3)2 is usually predicted to react at chloride. As such, DFT calculations suggest that the small phosphine PMe3 is more likely to facilitate the desired chemoselectivity than traditional ligands like PCy3 and PPh3.
Stoichiometric Oxidative Addition Studies.
Stoichiometric oxidative addition studies with PCy3, PPh3, and PMe3 were undertaken and the results were compared to the DFT predictions. When Ni(cod)2 and PCy3 (1:2) are combined in 1,4-dioxane, a new signal appears in the 31P NMR spectrum assigned to a Ni(0)/PCy3 adduct (46.0 ppm, Figure 3A). Free PCy3 (9.9 ppm) is also still detectable after 2 h. Upon addition of 1-chloronaphthalene (7) a new signal grows in within 2 h (11.9 ppm) corresponding to the oxidative addition adduct 9 (Figure 3B). A smaller signal at 12.9 ppm was identified as the Ni(II)-2-naphthyl adduct resulting from oxidative addition of 2-chloronaphthalene, which is a contaminant in commercial 7.21 Alternatively, addition of aryl tosylate 8 leads to a new signal at 10.9 ppm corresponding to 10 (Figure 3C). Finally, when a Ni(cod)2/PCy3 solution is combined with a mixture of 7 and 8 (1.0 equiv each), signals for both 9 and 10 appear in a 1.5 : 1 ratio (Figure 3D).22 These results show that Ni(0)/PCy3 has a slight preference for oxidative addition of Ar—Cl over Ar—OTs in an intermolecular competition.
Figure 3.
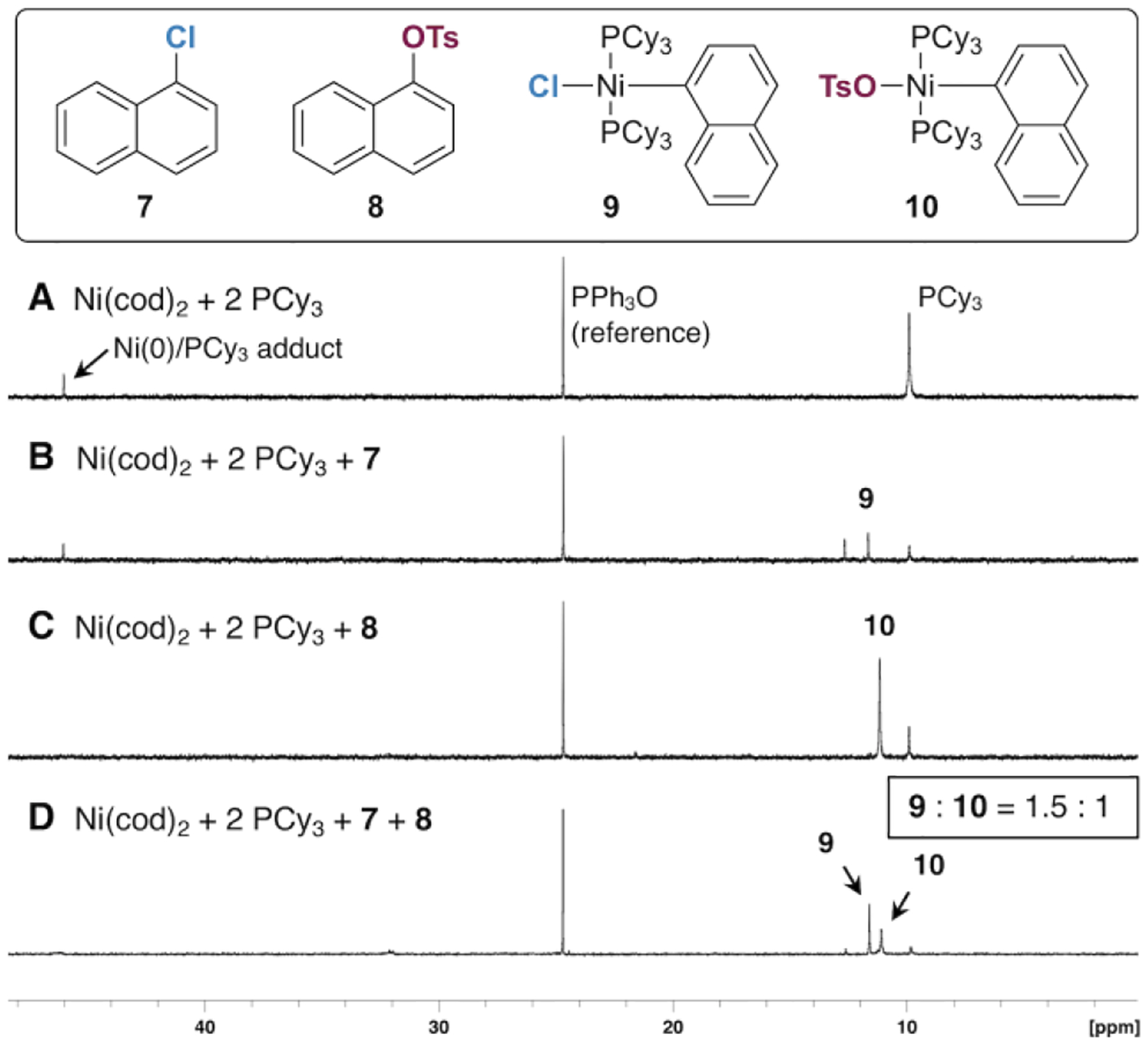
31P{1H} NMR studies on oxidative addition at Ni/PCy3 (reaction conditions = 2 h at room temperature in 1,4-dioxane).
The analogous experiments using PPh3 demonstrate much higher selectivity for oxidative addition of C—Cl. In a competition reaction between 7 and 8 with Ni(cod)2/PPh3 (1:2), the only oxidative addition adduct detected results from reaction at chloride (see SI for details). These results are consistent with the trends predicted by DFT, as well as Zou’s catalytic studies using a triarylphosphine (Scheme 1B).8f
In stark contrast to PCy3 and PPh3, the combination of a mixture of 7 and 8 with a 1:2 solution of Ni(cod)2/PMe3 gives preferential oxidative addition at C—OTs (Figure 4). The putative oxidative addition adducts 11 and 12 are formed in about a 1 : 6.3 ratio based on 31P NMR integrations. Taken together, the results of the intermolecular competition studies with the three ligands are consistent with the DFT-predicted trend in preference for oxidative addition at C—OTs (PMe3 > PCy3 > PPh3).
Figure 4.
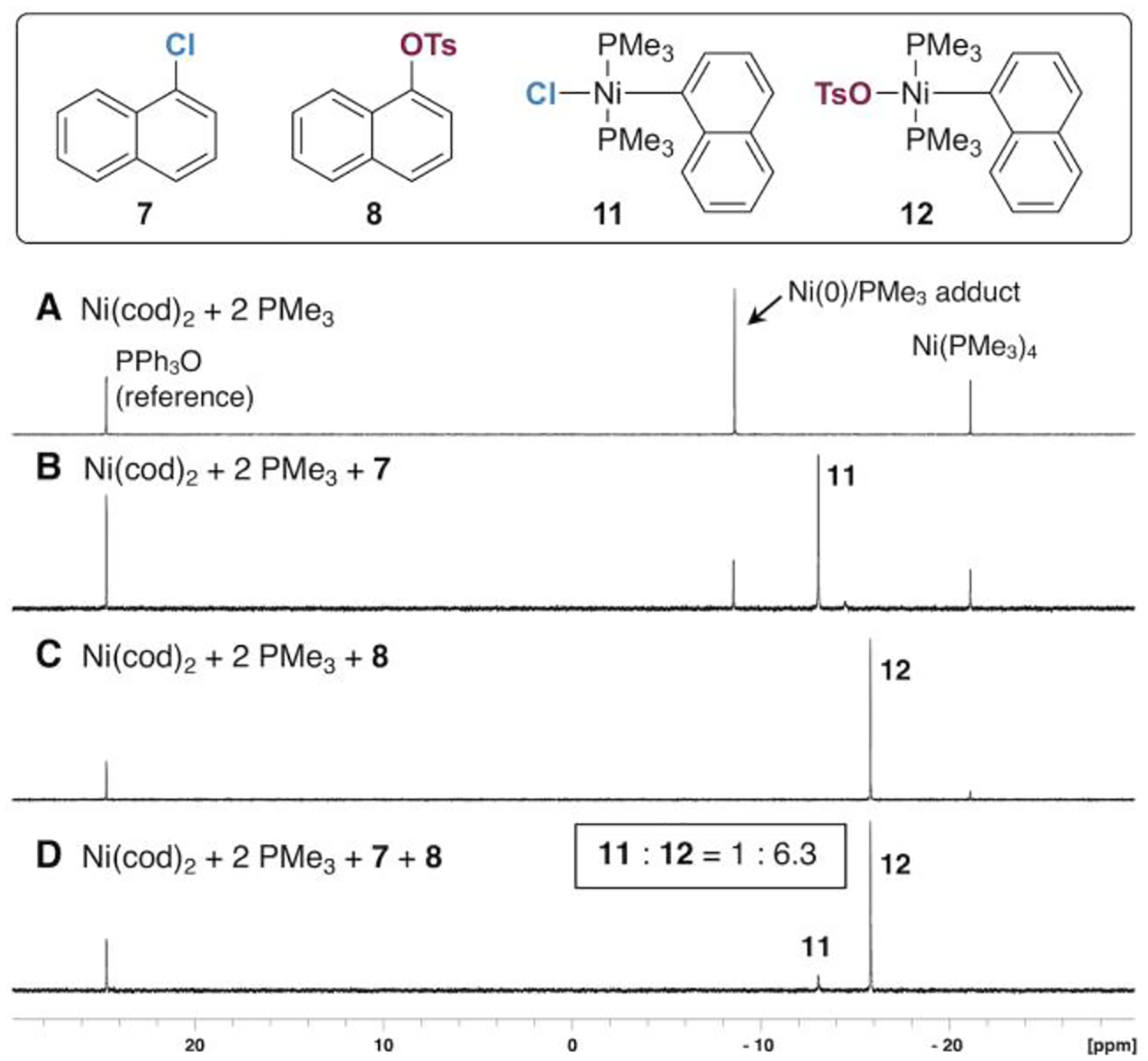
31P{1H} NMR studies on oxidative addition at Ni/PMe3 (reaction conditions = 2 h at room temperature in 1,4-dioxane).
A variety of additional phosphine ligands were evaluated for their effect on oxidative addition selectivity (Table 1). The results show a trend that triarylphosphines strongly favor reaction at chloride (entries 1–3), while most alkyl phosphines give a mixture of products (entries 4–12). The best selectivity for reaction at tosylate is obtained with PMe3 (entry 12), but PPhMe2 gives similar selectivity (entry 4). Interestingly, the analogous ethyl phosphines PPhEt2 and PEt3 slightly favor reaction at chloride instead of tosylate (entries 5 and 11), suggesting that the small size of a methyl group on phosphine is important.
Table 1.
Ligand Effect on Selectivity of Oxidative Addition.
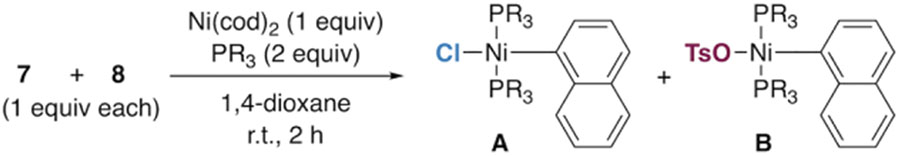 | |||||
|---|---|---|---|---|---|
| entry | ligand | A : B | entry | ligand | A : B |
| 1 | PPh3 | >99 : 1 | 7 | PCy3 | 1.5 : 1 |
| 2 |  |
>99 : 1 | 8 | P(n-Bu)3 | 1.1 : 1 |
| 9 | P(i-Bu)3 | 2.9 : 1 | |||
| 3 | 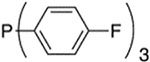 |
>99 : 1 | 10a | 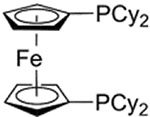 |
>99 : 1 |
| 4 | PPhMe2 | 1 : 5.7 | |||
| 5 | PPhEt2 | 2.2 : 1 | 11 | PEt3 | 1.8 : 1 |
| 6 | PPh2Me | 1 : 1 | 12 | PMe3 | 1 : 6.3 |
With 1 equiv of bisphosphine relative to nickel.
We next evaluated the selectivity when competing 1-chloronaphthalene 7 against a variety of other 1-naphthol derivatives for oxidative addition at Ni(0)/PMe3 (Table 2). Excitingly, reaction of an aryl triflate (entry 1), as well as the more robust phenol derivatives mesylate (entry 3) and sulfamate (entry 4) are preferred over reaction of naphthyl chloride when using PMe3. The use of PCy3 or PPh3 results in a slight preference for reaction of triflate over chloride, but oxidative addition of chloride is preferred over all other types of C—O bonds depicted in Table 2 with these ligands.
Table 2.
Selectivity of Ni/PR3 for Reaction of Various Phenol Derivatives in Competition with 1-Chloronaphthalene.
 | ||||
|---|---|---|---|---|
| entry | –OR | PMe3 A : B |
PCy3 A : B |
PPh3 A : B |
| 1 | –OTf | 1 : 88.6 | 1 : 2.3 | 1 : 2.1 |
| 2 | –OTs | 1 : 6.3 | 1.5 : 1 | >99 : 1 |
| 3 | –OMs | 1 : 10.0 | 2.7 : 1 | >99 : 1 |
| 4 | –OSO2NMe2 | 1 : 4.6 | 1.7 : 1 | >99 : 1 |
| 5 | –OCONEt2 | >99 : 1 | >99 : 1 | >99 : 1 |
| 6 | –OPiv | >99 : 1 | >99 : 1 | >99 : 1 |
The reported amounts of A include the Ni(Cl)(2-naphthyl) product resulting from the 2-Chloronaphthalene contaminant in 7, when detected.
The chemoselectivity of oxidative addition with PMe3 was further evaluated in an intramolecular competition. A 1:2 solution of Ni(cod)2/PMe3 was combined with chloronaphthyl tosylate 13 (Scheme 2). Tentative assignment of product signals was made by analogy to the reaction of Ni(cod)2/PMe3 with 7 or 8. The product resulting from reaction at tosylate is favored over the putative nickel chloride adduct by about 90:1, suggesting that tosylate selectivity can be even stronger in an intramolecular versus intermolecular competition.23 The major oxidative addition adduct 14 was isolated in 80% yield and its identity was confirmed by X-ray crystallography.
Scheme 2.
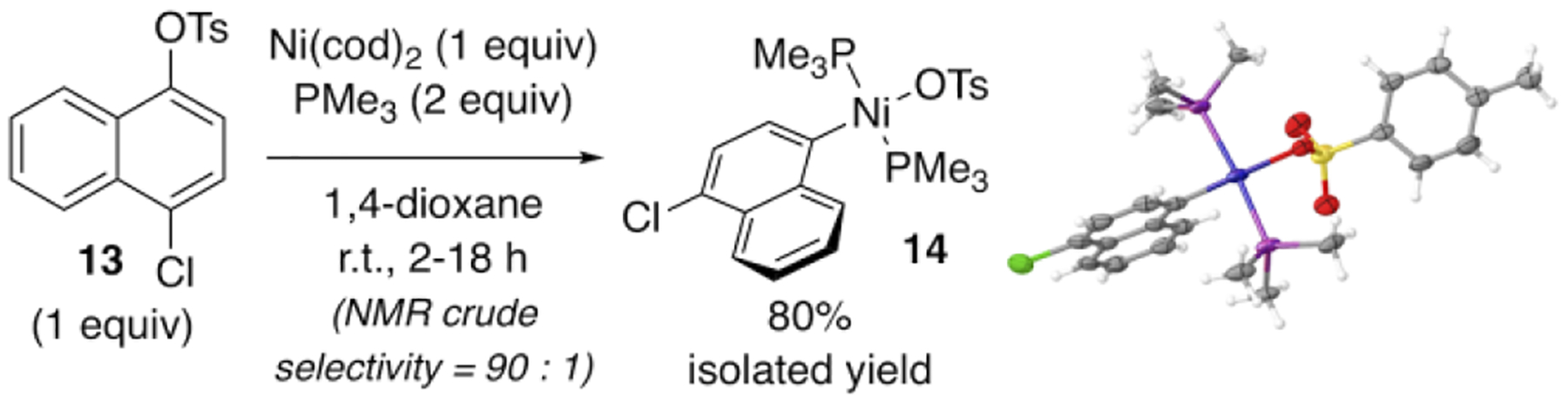
Chemoselective Oxidative Addition with PMe3.
Taken together, these stoichiometric studies indicate that PMe3 and PPhMe2 are unique in providing good selectivity for oxidative addition of aryl tosylates over aryl chlorides at Ni(0).
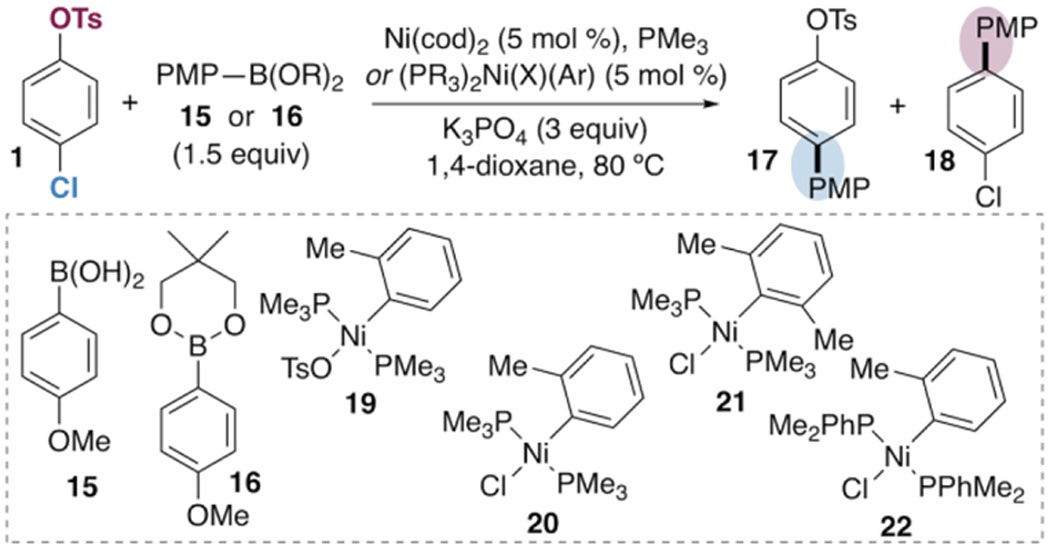
Catalytic Studies on Chemoselective Cross-coupling.
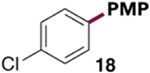
Trimethylphosphine was investigated as a ligand for a chemoselective Ni-catalyzed Suzuki-Miyaura coupling of chloroaryl tosylates. Initial conditions were modeled after previously reported cross-couplings of aryl tosylates.8a,24 Substrate 1, 4-methoxyphenylboronic acid (15), and K3PO4 were combined with Ni(cod)2 and PMe3 in 1,4-dioxane (Table 3). Excitingly, heating to 80 °C led to formation of products 17 and 18 in about a 1:13 ratio favoring C—OTs cleavage (entry 1). However, the yield of 18 is fair and mass balance is poor under these conditions. Moreover, PMe3 and Ni(cod)2 are both highly air-sensitive, and PMe3 is a volatile liquid that is difficult to measure accurately in small volumes. This latter feature of PMe3 proved to be particularly problematic for reproducibility, as the reaction outcome is sensitive to the PMe3/Ni(cod)2 ratio. The best ratio is PMe3:Ni ≈ 2:1 (9–10 mol % of PMe3, entries 1 and 3), but a higher or lower ratio gives worse yield of 18 and/or worse mass balance (entries 2, 4, and 5).
Table 3.
Optimization of a Chemoselective Suzuki-Miyaura Cross-Coupling of 4-Chlorophenyl Tosylate.a
 | ||||||
|---|---|---|---|---|---|---|
| entry | [B] | [Ni] | PMe3 (mol%) | 1 (%) | 17 (%) | 18 (%) |
| 1 | 15 | Ni(cod)2 | 10% | 9 | 4 | 54 |
| 2 | 15 | Ni(cod)2 | 6% | 27 | 7 | 39 |
| 3 | 15 | Ni(cod)2 | 9% | 12 | 6 | 62 |
| 4 | 15 | Ni(cod)2 | 12% | 3 | 4 | 55 |
| 5 | 15 | Ni(cod)2 | 15% | 14 | 10 | 38 |
| 6 | 16 | 19 | -- | 37 | 3 | 60 |
| 7b | 16 | 19 | -- | 5 | 2 | 88 (72)c |
| 8b | 16 | 20 | -- | 1 | 2 | 83 |
| 9b,d | 16 | 21 | -- | 21 | 3 | 61 (57)c |
| 10b,e | 16 | 22 | -- | 9 | 5 | 73 |
GC yields calibrated against undecane as the internal standard. Average of two runs. Small quantities of diarylation observed in all cases (~3–10%). PMP = para-methoxyphenyl.
With H2O (50 mol %).
Isolated yield in parentheses.
Set up on benchtop.
With 4 mol % of the mono-THF adduct of precatalyst 22.
For this reason, we synthesized the Ni(II) precatalyst 19. This precatalyst ensures a phosphine:nickel ratio of 2:1. Use of 19 together with boronic ester 16 led to much better mass balance and better reproducibility (entry 6). Finally, addition of a small quantity of water (0.5 equiv) enabled high yield of 18 with excellent mass balance (entry 7). Use of the chloride analogue of the precatalyst (20) gave similar results (entry 8).25 The reaction could be set up on the benchtop by using the semi-air-stable precatalyst 21 and sparging the reaction mixture briefly with N2 prior to heating, although the yield of 18 was somewhat eroded (entry 9).26 For comparison, a variety of other phosphine ligands were also evaluated in this catalytic system. Consistent with the stoichiometric studies, PPhMe2 gave comparable results to PMe3, albeit with slightly lower selectivity and yield (entry 10). All other phosphines evaluated in this catalytic reaction favored reaction at choride or gave poor selectivity and/or yield (see SI).
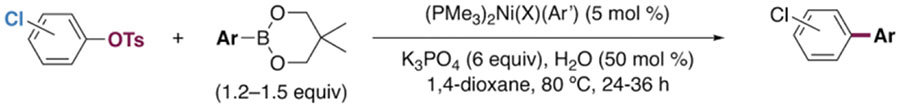
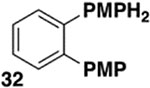
The generality of the Ni/PMe3 catalytic system was further explored with a modest scope of boronic ester coupling partners and chlorophenol derivatives (Table 4). Unsurprisingly, the high selectivity for reaction at tosylate was unaffected by the electronics of the arylboronic ester (entries 1–6), although in some cases larger amounts of diarylated products were detected (entries 1 and 6).27 High selectivity for reaction at tosylate was retained using chloroaryl tosylates with different substitution patterns (33 and 35). However, ortho-chlorophenyl tosylate 31 gives diarylation as the major product (32).28 4-Chlorophenyl dimethylsulfamate and triflate also provide 18 as the major product, resulting from oxidative addition of the C—O bond (entries 8–9). A considerable amount of unproductive substrate decomposition occurred when using the aryl triflate 30.29 An aryl tosylate was successfully cross-coupled with a chloro-substituted boronic ester (eq. 1). However, the attempted cross-coupling of heteroaryl substrates or boronic esters was unproductive and led to recovery of starting material or a complex mixture of products (see SI).30 We anticipate that the generality of this chemoselective cross-coupling can be improved in the future by better understanding the mechanisms of catalyst deactivation or conversion to less-selective catalytic species.31
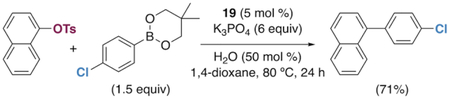 |
(1) |
Table 4.
Suzuki-Miyaura Cross-coupling of Chlorophenol Derivatives through Selective Cleavage of C—O Bonds.
 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | substrate | major product | % GC yield (selectivity)b | isolated yield (%) | entry | substrate | major product | % GC yield (selectivity)b | isolated yield (%) |
| 1c | 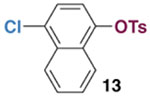 |
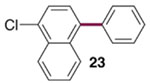 |
80 (>50:1) | 72 | 7d | 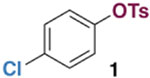 |
 |
88 (44:1) | 72 |
| 2 | 13 | 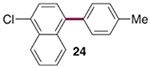 |
80 (>50:1) | 61 | 8d | 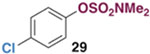 |
18 | 55 (8:1) | 39 |
| 3 | 13 | 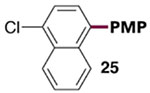 |
95 (>50:1) | 78 | 9d | 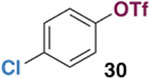 |
18 | 49 (>50:1) | 48 |
| 4 | 13 | 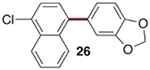 |
86 (>50:1) | 75 | 10 | 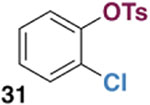 |
 |
48 | 35 |
| 5 | 13 | 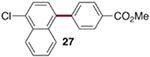 |
86 (>50:1) | 62 | 11 | 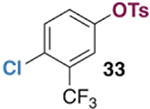 |
 |
73 (>50:1) | 35 |
| 6c | 13 |  |
87 (>50:1) | 64 | 12 |  |
 |
89 (30:1) | 67 |
Precatalyst for entries 1–6:19; entries 7–11:12; entry 12: 20. PMP = para-methoxyphenyl. Diarylated product observed in ≤10% estimated yield by GC unless noted.
Calibrated GC yield relative to undecane standard. Approximate selectivities of major product to minor monoarylated product based on calibrated GC yield of major product and uncalibrated GC yield of minor product, where the minor product results from cleavage of the C—Cl bond instead of C—OTs.
Diarylated product observed in ≤20% estimated yield by GC.
Selectivity based on calibrated GC yields of both the major and minor products.
Computational Analysis of Selectivity Origin.
Previous examples of ligand-controlled chemoselective cross-coupling have a mechanistic origin in the metal’s ligation state during oxidative addition. Perhaps the best-studied example is the case of Pd/phosphine-catalyzed Suzuki-Miyaura coupling of chloroaryl triflates.32 In the coupling of 30, electron-rich bisligated Pd(PCy3)2 prefers to react at C—OTf due to a stronger attractive interaction energy in the transition state (Figure 5A). In contrast, the less electron-rich monoligated Pd(PtBu3) reacts at C—Cl due to lower distortion energy.32b Ligation state is likely also the determining factor in other examples of Pd-catalyzed chemodivergent cross-couplings of chloro-33 or bromoaryl triflates.34 Very bulky monodentate ligands which promote a monoligated active catalyst can favor reaction at the halide, and bidentate or smaller monodentate ligands that promote a bisligated catalyst favor reaction at triflate. Similarly, bulky monodentate and bidentate phosphine ligands can effect divergent selectivity between activation of C(aryl)—O and C(acyl)—O bonds of aryl esters by Ni(0) (Figure 5B).35 DFT studies suggest that this selectivity relates to nickel’s coordination number by phosphine.35b,36
Figure 5.
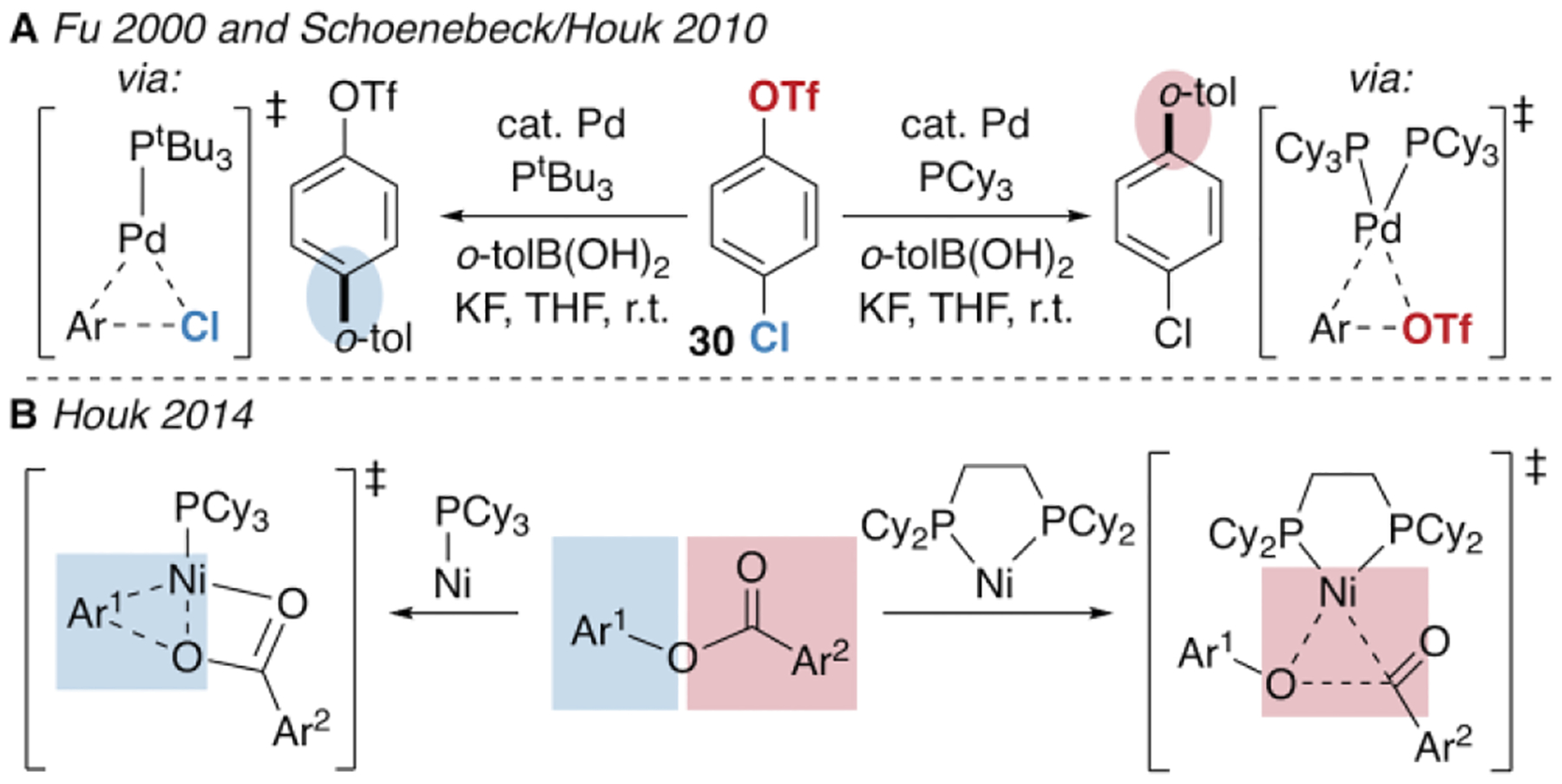
Chemodivergent oxidative addition with selectivity controlled by the metal’s ligation state.
In contrast, nickel’s ligation state is unlikely to be relevant to the selectivity observed in the current chemodivergent system.37,38 PMe3 and PPh3 give opposite preferences for reaction at tosylate and chloride, respectively, yet both are monodentate and neither ligand is particularly bulky. DFT calculations suggest that, for PMe3, PPh3, and PCy3, the lowest-energy transition state for oxidative addition at C—Cl or C—OTs involves bisphosphine-ligated nickel16 (moreover, a 2:1 ratio of phosphine to nickel was used in our experimental studies). As such, it appears that selectivity in this system has a different origin than prior examples of ligand-controlled chemodivergent cross-coupling. We undertook further DFT calculations to understand the unique selectivity preference of Ni/PMe3.
One hypothesis to explain the divergent behavior between PMe3 and PPh3 relates to the greater electron-donating character of PMe3.40 Nickel is calculated to be more negatively charged when ligated by PMe3 than by PPh3 (Figure 6A). Furthermore, Ni(PMe3)2 has a higher-energy HOMO than Ni(PPh3)2. Because C—O bonds are more polarized than C—Cl bonds, the carbon of C—OTs is more electrophilic (compare the charges at carbon in Figure 6B). A more electron-rich Ni(PMe3)2 might be expected to have a stronger attractive interaction at the more electrophilic site (C—OTs). A similar argument has been used to explain the divergent selectivity of mono- vs. bis-ligated palladium in the cross-coupling of chloroaryl triflates (Figure 5A).32b However, this electronic explanation is unsatisfying in the current system when considering PCy3, which is usually thought of as more electron-donating than either PPh3 or PMe3.40 Ni(PCy3)2 has a higher-energy HOMO than PMe3 and a more negatively-charged metal center, yet PCy3 effects only poor selectivity between tosylate and chloride. As such, ligand electronics are not sufficient to explain the unique selectivity observed with PMe3, especially when compared to other electron-rich alkyl phosphines like PCy3.
Figure 6.
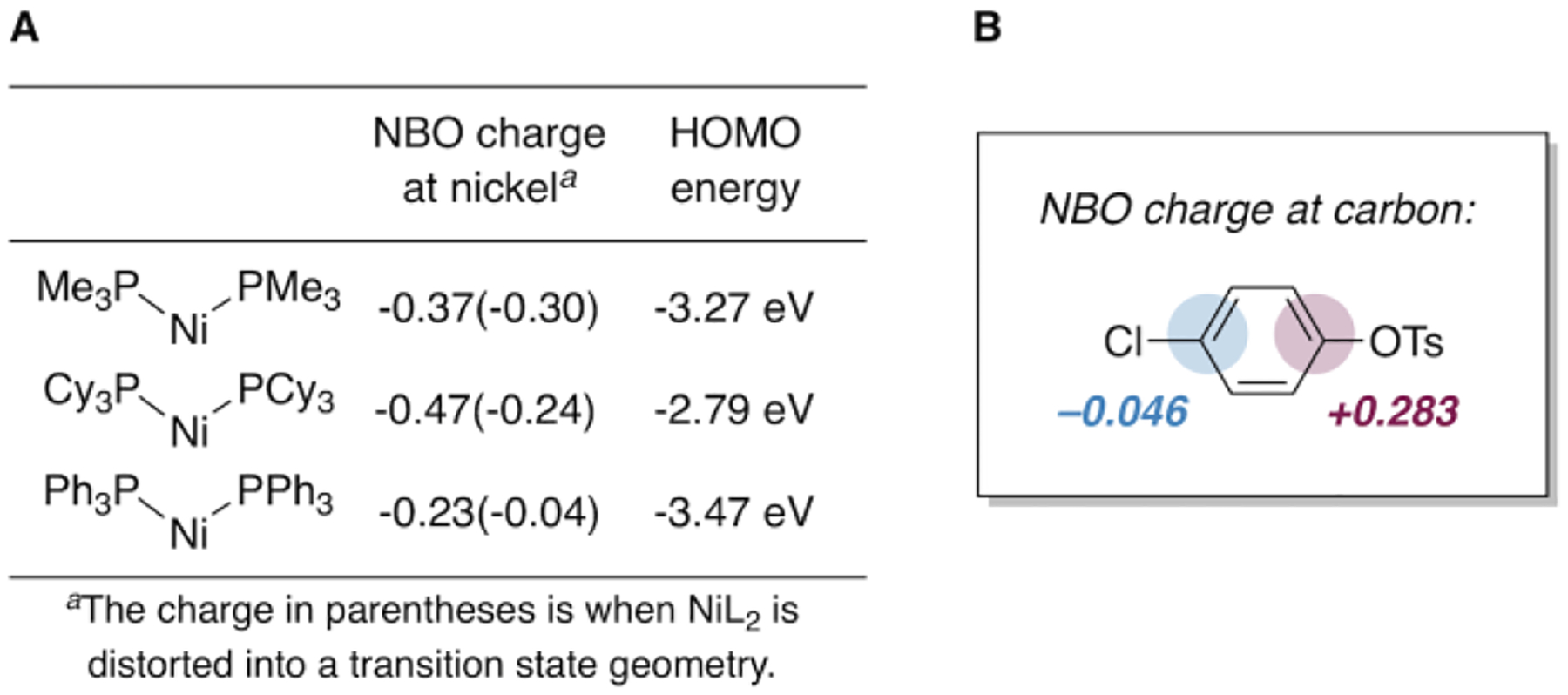
(A) Calculated NBO charges and HOMO energies for optimized Ni(PR3)2 structures, (B) NBO charges at carbon in chlorophenyl tosylate.39
To understand the difference between the behavior of PMe3 and PCy3, we more carefully examined the calculated oxidative addition transition structures using these ligands (Figure 7). With both of these ligands, insertion of Ni(0) into C—Cl proceeds through a concerted 3-centered transition structure in which nickel interacts with the departing chloride (3-TS and 38-TS). In contrast, 3-centered concerted transition structures for tosylate activation could not be located. Instead, C—O cleavage occurs through a nucleophilic displacement mechanism: nickel does not interact significantly with the departing oxygen. During reaction at tosylate, nickel has a closer association with the ortho carbon of the substrate: the Ni••••Cortho distances range from 2.10–2.31 Å in the transition structures for oxidative addition at C—OTs (4a-TS, 4b-TS, 37a-TS, and 37b-TS) compared to 2.33–2.37 Å for reaction at C—Cl (3-TS and 38-TS, Ni••••Cortho distances not labeled). Oxidative addition of tosylate can occur through a conformation in which the S=O bonds point away from nickel, referred to as a “dissociation” mechanism (4b-TS and 37b-TS), or through a conformation in which one of the sulfonyl oxygens interacts with nickel, referred to as a “5-centered” mechanism41 (4a-TS and 37a-TS). With PMe3, this Ni••••O=S interaction results in a lower energy structure compared to the dissociation mechanism. However, with PCy3, the two mechanisms for reaction at tosylate are energetically similar.
Figure 7.
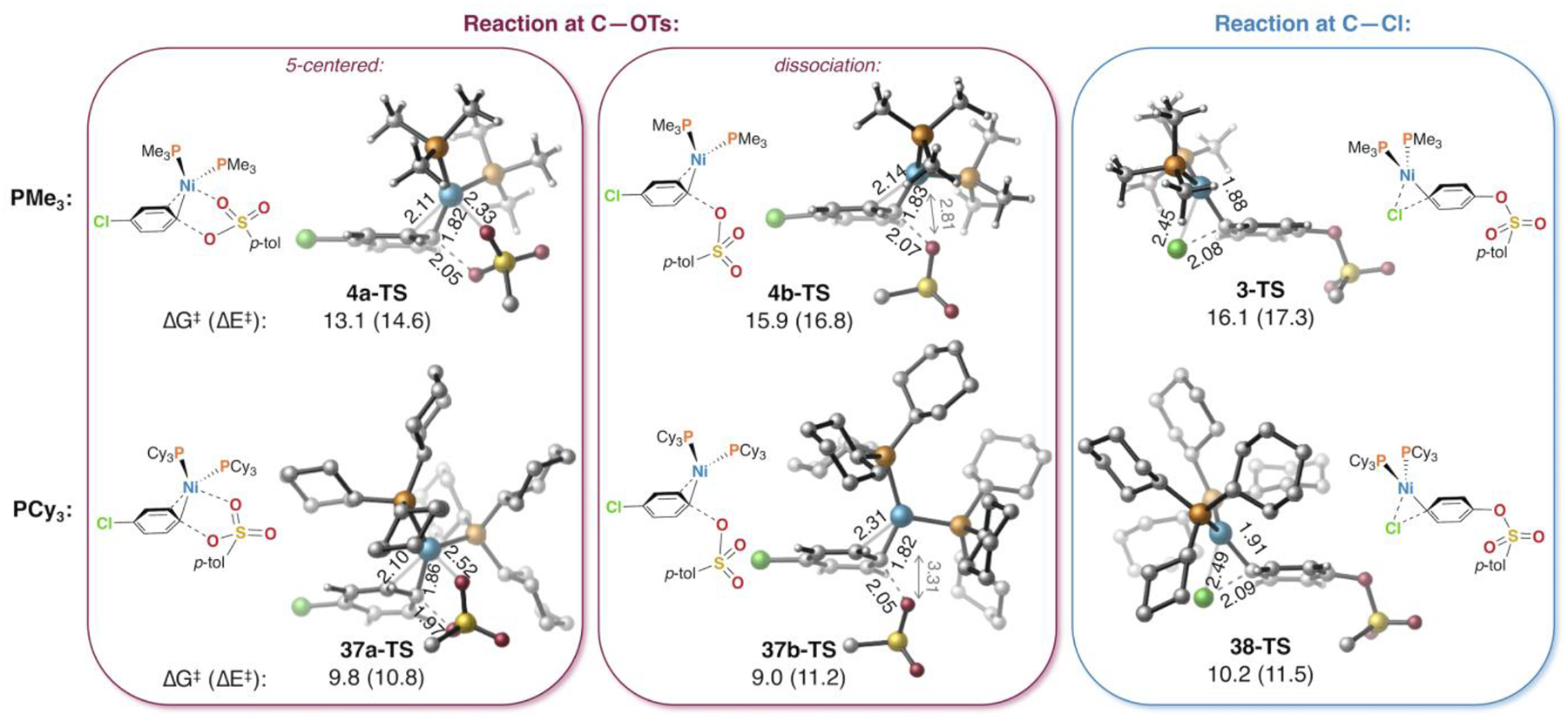
Oxidative addition transition structures using Ni(PMe)3 and Ni(PCy3)2. Most of the carbons and hydrogens of tosylate, as well as the hydrogens on PCy3, are hidden for clarity (see SI for complete structures).
The geometries and energetics of these transition structures are consistent with a steric argument for the different selectivity seen with PMe3 compared to PCy3. If reaction at tosylate is desired, then PCy3 has the advantage of being more electron-donating than PMe3 (see discussion above). However, PMe3 is also electron-donating—albeit perhaps not as strongly as PCy3—and it has the added advantage of minimal sterics. The reason why small ligand size promotes reaction at tosylate is because of the possibility for a close stabilizing interaction between a sulfonyl S=O and Ni during the 5-centered transition state (4a-TS). This interaction is also possible with PCy3 (37a-TS), but it is less stabilizing due to a longer Ni••••O=S distance (2.52 vs. 2.33 Å). Apparently, PCy3 sterically shields nickel from having a close interaction with oxygen. As a result, the 5-centered mechanism with PCy3 is not more facile than the dissociation mechanism. In contrast, the 5-centered mechanism with PMe3 is about 3 kcal mol−1 lower in energy than both the corresponding dissociation mechanism and the mechanism for reaction at chloride.
We conducted a distortion-interaction analysis on the relevant PMe3- and PCy3-containing transition structures to gather further evidence for the role of ligand sterics on selectivity. This type of analysis, also called an activation-strain analysis, involves dissecting the activation energy (ΔE‡) into a distortion (ΔEdist) and an interaction term (ΔEint, Figure 8A).42 The distortion term is typically a positive value (unfavorable) and represents the energy penalty for distorting the reactants into the transition state geometry. The interaction term is usually attractive (a negative value), and represents the favorable interaction between substrate and catalyst due to factors like orbital mixing, electron correlation, and coulombic pairing of opposite charges. The relationship between distortion and interaction is defined as ΔE‡ = ΔEdist + ΔEint. Distortion and interaction energies of the transition structures were measured from the lowest-energy preceding π complexes.
Figure 8.
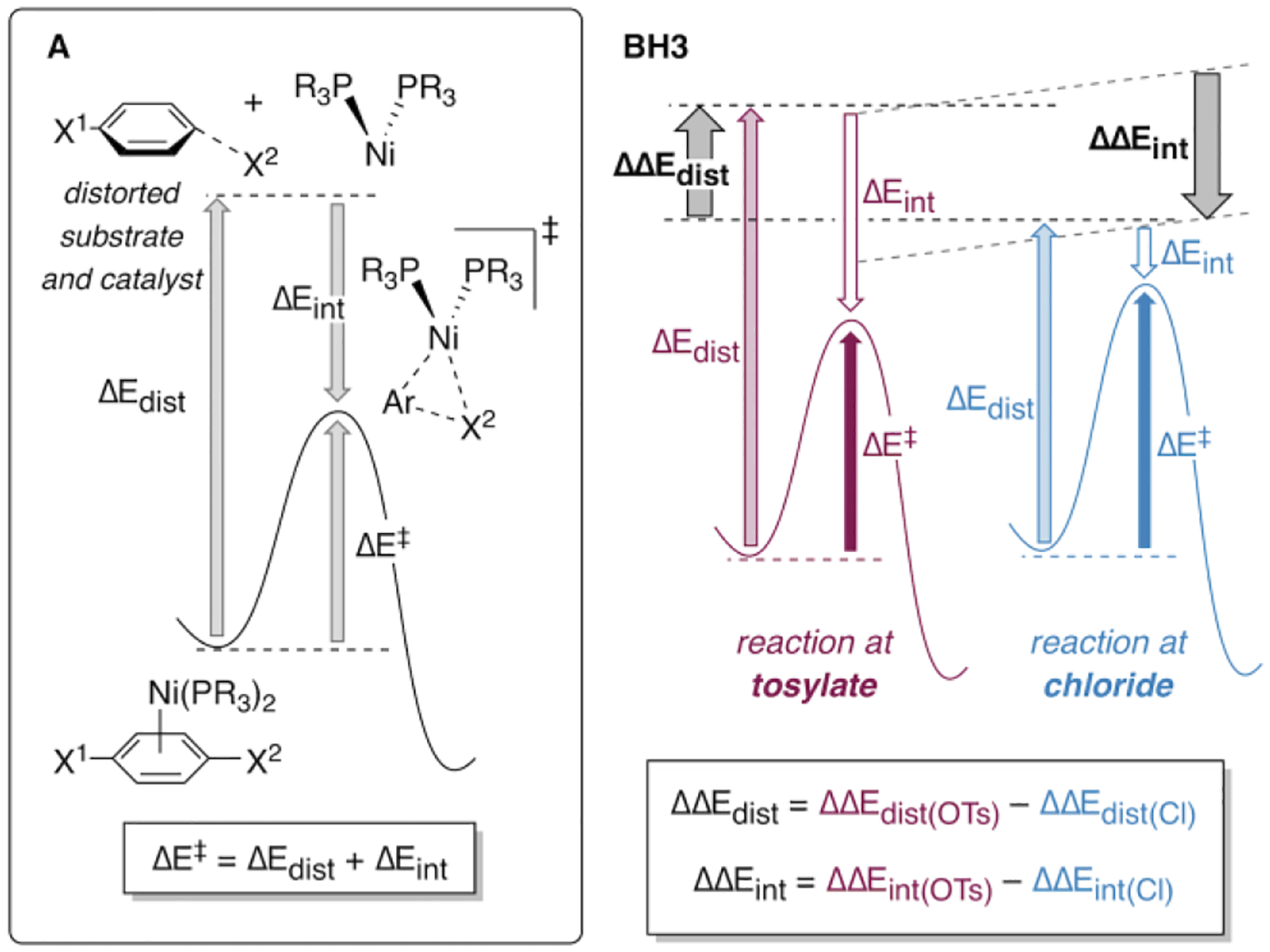
(A) Definition of distortion and interaction energy. (B) Selectivity is determined by the difference in distortion energy (ΔΔEdist) and interaction energy (ΔΔEint) when comparing oxidative addition at tosylate vs. chloride.
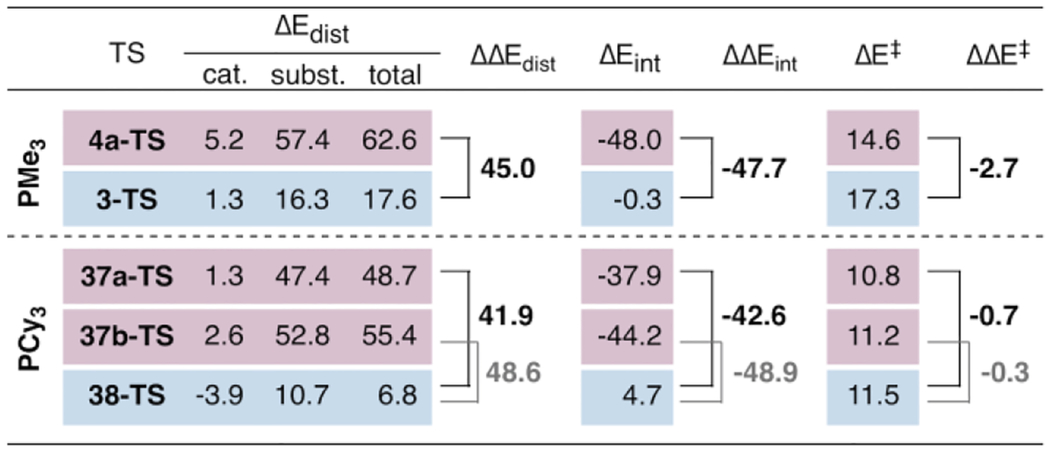
With both PCy3 and PMe3, reaction at tosylate involves greater distortion energy than reaction at chloride (compare total ΔEdist values in Table 5). This is due to greater distortion of both the substrate and the catalyst. The substrate is more distorted during reaction at tosylate because of nickel’s more extensive interaction with the arene π system (both the ipso and the ortho carbons) as well as distortion of several bond angles in the departing tosylate group. The larger catalyst distortion energies during reaction at tosylate reflect the more constrained geometry of NiL2. During reaction at chloride, the ligands have more room to spread out because Ni is further away from the substrate aryl ring. In fact, the catalyst distortion energy is actually slightly negative (favorable) during insertion of Ni(PCy3)2 into C—Cl (38-TS), reflecting a relaxation of the P–Ni–P bond angle in the TS compared to the preceding π complex (122° in 38-TS vs. 115° in the corresponding π-complex). Although distortion energy favors reaction at chloride for both ligands, interaction energy strongly favors (is more negative) reaction at tosylate. This can be rationalized by (1) the stronger coulombic attraction between Ni and the electron-deficient carbon of C—OTs and (2) the ability for another oxygen of S=O to interact with Ni during a 5-centered mechanism.
Table 5.
Distortion-Interaction Analysis of Relevant PMe3- and PCy3-Containing Transition Structures.
|
Selectivity of oxidative addition depends on the differences between the distortion and interaction energies for reaction at the two possible sites. These differences are defined as ΔΔEdist = ΔEdist(OTs) – ΔEdist(Cl) and ΔΔEint = ΔEint(OTs) – ΔEint(Cl) (Figure 8B). ΔΔEdist is always a positive value, and a larger ΔΔEdist signifies a more powerful influence of distortion energy in favor of reaction at chloride. Conversely, ΔΔEint is always negative. A more negative value of ΔΔEint indicates a more powerful influence of interaction energy in favor of reaction at tosylate.
If reaction at tosylate is desired, the dissociation mechanism with Ni(PCy3)2 (37b-TS) has a larger distortion disadvantage than either 5-centered mechanism 37a-TS or 4a-TS (compare the values of ΔΔEdist in Table 5). This may be due to the larger bending of tosylate out-of-plane of the arene in 37b-TS. However, this bending also leads to a large interaction energy advantage for the dissociation mechanism (ΔΔEint), likely due to better orbital overlap between nickel’s HOMO and the C—OTs σ* (the Ni–C–O bond angle is 117° in 37b-TS compared to 107° in 37a-TS and 101° in 4a-TS). The effects of ΔΔEdist and ΔΔEint nearly cancel each other out for 37b-TS, and this dissociation mechanism for reaction at tosylate using PCy3 is only slightly lower-energy than reaction at chloride. The 5-centered mechanism with PCy3 (37a-TS) has a much smaller distortion disadvantage than either 37b-TS or 4a-TS due to the earliness of the transition structure 37a-TS.43 However, 37a-TS also has less of an interaction energy advantage than the corresponding transition structure using PMe3 (4a-TS). This is because the stabilizing Ni••••O=S interaction in 37a-TS is less significant, as evidenced by the longer distance. In fact, when the intrinsic reaction coordinate for 37a-TS is followed, the Ni••••O distance remains longer than that of 4a-TS until well after the transition state (when the breaking C••••OTs bond is elongated to 2.55 Å). As a result, the effects of ΔΔEdist and ΔΔEint again nearly cancel out and 37a-TS has a similar energy barrier as that for reaction at chloride (38-TS). In contrast, the 5-centered mechanism with Ni(PMe3)2 has a stronger interaction between Ni and O=S, resulting in a value of ΔΔEint for 4a-TS that outweighs the influence of distortion energy (ΔΔEdist) and makes reaction at tosylate more facile than reaction at chloride.
CONCLUSION
Prior Ni-catalyzed Suzuki-Miyaura cross-couplings of non-triflate phenol derivatives do not tolerate aryl chlorides, a feature that limits their synthetic utility. We have shown that DFT calculations accurately predict the following ligand trend for reaction of tosylate over chloride at Ni(0): PMe3 > PCy3 > PPh3. In stoichiometric studies evaluating a wider range of phosphine ligands, we demonstrate that methylphosphines are uniquely capable of promoting chemoselective oxidative addition of aryl tosylates in the presence of aryl chlorides. Good to excellent selectivity for reaction of aryl triflates, mesylates, and sulfamates over aryl chlorides was also observed. This selectivity was exploited to demonstrate proof-of-principle for a C—O-selective Suzuki-Miyaura coupling of chlorinated non-triflate phenol derivatives. We anticipate that a detailed understanding of catalyst decomposition mechanisms will lead to improvements to the scope of the catalytic process.
It appears that the unique behavior of methyl phosphines compared to other phosphines does not relate to the catalyst’s ligation state by ancillary ligands, and thus differs from prior examples of ligand-controlled chemodivergent cross-couplings. Computational analysis suggests that the unusual selectivity of Ni(PMe3)2 for insertion into C—OTs is due to both electronic and steric factors. In contrast to arylphosphines, the use of an alkyl phosphine results in an electron-rich active catalyst that can more effectively donate into the more electrophilic carbon of C—OTs. Furthermore, the small size of PMe3 allows for a close interaction between Ni and a sulfonyl oxygen of tosylate during a 5-centered oxidative addition mechanism. This interaction helps to stabilize the buildup of positive charge at nickel during its oxidation from Ni(0) to Ni(II), and is less significant with bulkier alkyl phosphines. Notably, the differences observed between PMe3 and PCy3 caution against relying on PMe3 as a computational model for more complex alkylphosphines.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by NSF CAREER (CHE-1848090) and Montana State University. Calculations were performed on Comet at SDSC through XSEDE (CHE-170089), which is supported by NSF (ACI-1548562). NMR spectra were recorded on an instrument funded by NSF (NSF-MRI:DBI-1532078) and the Murdock Charitable Trust Foundation (2015066:MNL). X-ray crystallographic data were collected at the University of Montana X-ray diffraction core facility (NIH, CoBRE NIGMS P20GM103546) using an instrument principally supported by NSF (MRI CHE-1337908). We thank Daniel A. Decato for his assistance in solving crystal structures. Troy Bearcomesout, Kayla Creelman, Rosalia Martinez, Mike Giroux, and Emily K. Reeves are gratefully acknowledged for early work. The authors thank Steven M. Rehbein for synthesizing PMe3 during early stages of the project.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental and computational details, NMR spectra, crystallographic details, and calculated energies (PDF). X-ray crystallographic data for 14 (CIF). Cartesian coordinates of minimum-energy calculated structures (XYZ).
The authors declare no competing financial interest.
REFERENCES
- 1.Selected reviews:; (a) Kotha S; Lahiri K; Kashinath D Recent Applications of the Suzuki-Miyaura Cross-Coupling Reaction in Organic Synthesis. Tetrahedron 2002, 58, 9633–9695; [Google Scholar]; (b) Suzuki A Cross-Coupling Reactions Of Organoboranes: An Easy Way To Construct C—C Bonds (Nobel Lecture). Angew. Chem. Int. Ed 2011, 50, 6722–6737. [DOI] [PubMed] [Google Scholar]
- 2.Selected reviews:; (a) Rosen BM; Quasdorf KW; Wilson DA; Zhang N; Resmerita A-M; Garg NK; Percec V Nickel-Catalyzed Cross-Couplings Involving Carbon-Oxygen Bonds. Chem. Rev 2011, 111, 1346–1416; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Han F-S Transition-metal-catalyzed Suzuki–Miyaura cross-coupling reactions: a remarkable advance from palladium to nickel catalysts. Chem. Soc. Rev 2013, 42, 5270–5298; [DOI] [PubMed] [Google Scholar]; (c) Tobisu M; Chatani N Nickel-Catalyzed Cross-Coupling Reactions of Unreactive Phenolic Electrophiles via C–O Bond Activation. Top. Curr. Chem. (Z) 2016, 374, 41; [DOI] [PubMed] [Google Scholar]; (d) Zarate C; van Gemmeren M; Somerville R; Martin R Phenol Derivatives: Modern Electrophiles in Cross-Coupling Reactions. In Advances in Organometallic Chemistry; Pérez PJ, Ed.; Elsevier, 2016, 66, 143–122; [Google Scholar]; (e) Zeng H; Qiu Z; Domínguez-Huerta A; Hearne Z; Chen Z; Li C-J An Adventure in Sustainable Cross-Coupling of Phenols and Derivatives via Carbon−Oxygen Bond Cleavage. ACS Catal 2017, 7, 510–519. [Google Scholar]
- 3.Nguyen HN; Huang X; Buchwald SL The First General Palladium Catalyst for the Suzuki-Miyaura and Carbonyl Enolate Coupling of Aryl Arenesulfonates. J. Am. Chem. Soc 2003, 125, 11818–11819. [DOI] [PubMed] [Google Scholar]
- 4.Snieckus V Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics. Chem. Rev 1990, 90, 879–933. [Google Scholar]
- 5.For examples, see; Chen Z; Wang B; Zhang J; Yu W; Liu Z; Zhang Y Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups. Org. Chem. Front 2015, 2, 1107–1295. [Google Scholar]
- 6.Everson DA; Jones BA; Weix DJ Replacing Conventional Carbon Nucleophiles with Electrophiles: Nickel-Catalyzed Reductive Alkylation of Aryl Bromides and Chlorides. J. Am. Chem. Soc 2012, 134, 6146–6159; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bajo S; Laidlaw G; Kennedy AR; Sproules S; Nelson DJ Oxidative Addition of Aryl Electrophiles to a Prototypical Nickel(0) Complex: Mechanism and Structure/Reactivity Relationships. Organometallics 2017, 36, 1662–1672. [Google Scholar]; (c) Huang L; Ackerman LKG; Kang K; Parsons AM; Weix DJ LiCl-Accelerated Multimetallic Cross-Coupling of Aryl Chlorides with Aryl Triflates. J. Am. Chem. Soc 2019, 141, 10978–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.In concurrent work, an exception was reported using a bipyridine ligand for nickel during a cross-electrophile coupling:; Kang K; Huang L; Weix DJ Sulfonate Versus Sulfonate: Nickel and Palladium Multimetallic Cross-Electrophile Coupling of Aryl Triflates with Aryl Tosylates. J. Am. Chem. Soc 2020, 142, 10634–10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zim D; Lando VR; Dupont J; Monteiro AL NiCl2(PCy3)2: A Simple and Efficient Catalyst Precursor for the Suzuki Cross-Coupling of Aryl Tosylates and Arylboronic Acids. Org. Lett 2001, 3, 3049–3051; [DOI] [PubMed] [Google Scholar]; (b) Guan B-T; Wang Y; Li B-J; Yu D-G; Shi Z-J Biaryl Construction via Ni-Catalyzed C—O Activation of Phenolic Carboxylates. J. Am. Chem. Soc 2008, 130, 14468–14470; [DOI] [PubMed] [Google Scholar]; (c) Baghbanzadeh M; Pilger C; Kappe CO Rapid Nickel-Catalyzed Suzuki-Miyaura Cross-Couplings of Aryl Carbamates and Sulfamates Utilizing Microwave Heating. J. Org. Chem 2011, 76, 1507–1510; [DOI] [PubMed] [Google Scholar]; (d) Li X-J; Zhang J-L; Geng Y; Jin Z Nickel-Catalyzed Suzuki−Miyaura Coupling of Heteroaryl Ethers with Arylboronic Acids. J. Org. Chem 2013, 78, 5078–5084; [DOI] [PubMed] [Google Scholar]; (e) Chen L; Lang H; Fang L; Yu J; Wang L Nickel-Catalyzed Desulfitative Suzuki–Miyaura Cross-Coupling of N,N-Disulfonylmethylamines and Arylboronic Acids. Eur. J. Org. Chem 2014, 6385–6389; [Google Scholar]; (f) Chen X; Ke H; Zou G Nickel-Catalyzed Cross-Coupling of Diarylborinic Acids with Aryl Chlorides. ACS Catal 2014, 4, 379–385; [DOI] [PubMed] [Google Scholar]; (g) LaBerge NA; Love JA Nickel-Catalyzed Decarbonylative Coupling of Aryl Esters and Arylboronic Acids. Eur. J. Org. Chem 2015, 5546–5553; [Google Scholar]; (h) Piontek A; Ochedzan-Siodlak W; Bisz E; Szostak M Nickel-Catalyzed C(sp2)—C(sp3) Kumada Cross-Coupling of Aryl Tosylates with Alkyl Grignard Reagents. Adv. Synth. Catal 2019, 361, 2329–2336. [Google Scholar]
- 9.An aryl chloride is tolerated in the Ni-catalyzed Suzuki coupling of a more reactive fluorosulfonate:; Hanley PS; Ober MS; Krasovskiy AL; Whiteker GT; Kruper WJ Nickel- and Palladium-Catalyzed Coupling of Aryl Fluorosulfonates with Aryl Boronic Acids Enabled by Sulfuryl Fluoride. ACS Catal 2015, 5, 5041–5046. [Google Scholar]
- 10. Palladium-catalyzed Suzuki-Miyaura couplings of chloroaryl tosylates also typically proceed through C—Cl cleavage. For examples see ref 3 and.; (a) Zhang L; Meng T; Wu J Palladium-Catalyzed Suzuki-Miyaura Cross-Couplings of Aryl Tosylates with Potassium Aryltrifluoroborates. J. Org. Chem 2007, 72, 9346–9349; [DOI] [PubMed] [Google Scholar]; (b) Ackermann L; Potukuchi HK; Althammer A; Born R; Mayer P Tetra-ortho-Substituted Biaryls through Palladium-Catalyzed Suzuki-Miyaura Couplings with a Diaminochlorophosphine Ligand. Org. Lett 2010, 12, 1004–1007; [DOI] [PubMed] [Google Scholar]; (c) Wang Z-Y; Chen G-Q; Shao L-X N‐Heterocyclic Carbene−Palladium(II)−1-Methylimidazole Complex-Catalyzed Suzuki−Miyaura Coupling of Aryl Sulfonates with Arylboronic Acids. J. Org. Chem 2012, 77, 6608–6614. [DOI] [PubMed] [Google Scholar]
- 11.Jiang S; Zhang L; Cui D; Yao Z; Gao B; Lin J; Wei D The Important Role of Halogen Bond in Substrate Selectivity of Enzymatic Catalysis. Scientific Reports 2016, 6, 34750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernandes MZ; Cavalcanti SMT; Moreira DRM; De Azevedo WF Jr.; Leite ACL Halogen Atoms in the Modern Medicinal Chemistry: Hints for the Drug Design. Curr. Drug Targets 2010, 11, 303–314. [DOI] [PubMed] [Google Scholar]
- 13. Selective C—O cleavage of chloroaryl tosylates has been observed in Pd-catalyzed Kumada couplings (see ref 14). However, in reported Pd-catalyzed Suzuki couplings, chloroaryl tosylates tend to react through C—Cl bond cleavage (Scheme 1a, ref 3,10).
- 14.Terao J; Naitoh Y; Kuniyasu H; Kambe N Pd-Catalyzed Cross-Coupling Reaction of Alkyl Tosylates and Bromides with Grignard Reagents in the Presence of 1,3-Butadiene. Chem. Lett 2003, 32, 890–891; [Google Scholar]; (b) Limmert ME; Roy AH; Hartwig JF Kumada Coupling of Aryl and Vinyl Tosylates under Mild Conditions. J. Org. Chem 2005, 70, 9364–9370; [DOI] [PubMed] [Google Scholar]; (c) Ackermann L; Althammer A Air-Stable PinP(O)H as Preligand for Palladium-Catalyzed Kumada Couplings of Unactivated Tosylates. Org. Lett 2006, 8, 3457–3460. [DOI] [PubMed] [Google Scholar]
- 15. Unless otherwise indicated in the Supporting Information, calculations were performed at the CPCM(1,4-dioxane)-MN15L/6–311++G(2d,p)/SDD(Ni)//MN15L/6–31G(d)/6–31+G(d)(Cl,O)/LANL2DZ(Ni) level of theory. See SI for details.
- 16.Hooker LV; Neufeldt SR Ligation state of nickel during C—O bond activation with monodentate phosphines. Tetrahedron 2018, 74, 6717–6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu HS; He X; Truhlar DG MN15-L: A New Local Exchange-Correlation Functional for Kohn–Sham Density Functional Theory with Broad Accuracy for Atoms, Molecules, and Solids. J. Chem. Theory Comput 2016, 12, 1280–1293. [DOI] [PubMed] [Google Scholar]
- 18.Evaluation or use of PMe3 in other types of Ni-catalyzed couplings:; (a) Miller JA; Dankwardt JW; Penney JM Nickel Catalyzed Cross-Coupling and Amination Reactions of Aryl Nitriles. Synthesis 2003, 11, 1643–1648; [Google Scholar]; (b) Miller JA; Dankwardt JW Nickel catalyzed cross-coupling of modified alkyl and alkenyl Grignard reagents with aryl- and heteroaryl nitriles: activation of the C—CN bond. Tetrahedron Lett 2003, 44, 1907–1910; [Google Scholar]; (c) Dankwardt JW Nickel-Catalyzed Cross-Coupling of Aryl Grignard Reagents with Aromatic Alkyl Ethers: An Efficient Synthesis of Unsymmetrical Biaryls. Angew. Chem. Int. Ed 2004, 43, 2428–2432; [DOI] [PubMed] [Google Scholar]; (d) Yang X; Sun H; Zhang S; Li X Nickel-catalyzed C—F bond activation and alkylation of polyfluoroaryl imines. J. Organomet. Chem 2013, 723, 36–42; [Google Scholar]; (e) Tang J; Lv L; Dai X-J; Li C-C; Li L; Li C-J Nickel-catalyzed cross-coupling of aldehydes with aryl halides via hydrazone intermediates. Chem. Commun 2018, 54, 1750–1753; [DOI] [PubMed] [Google Scholar]; (f) Lv L; Zhu D; Tang J; Qiu Z; Li C-C; Gao J; Li C-J Cross-Coupling of Phenol Derivatives with Umpolung Aldehydes Catalyzed by Nickel. ACS Catal 2018, 8, 4622–4627; [Google Scholar]; (g) Lv L; Li J; Liu M; Li C-J N2H4 as traceless mediator for homo- and cross- aryl coupling. Nature Commun 2018, 9, 4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Use of PMe3 as a ligand for Pd-catalyzed cross-couplings:; (a) Widdowson DA; Wilhelm R Palladium catalysed cross-coupling of (fluoroarene)tricarbonylchromium(0) complexes. Chem. Commun 1999, 2211–2212; [Google Scholar]; (b) Wilhelm R; Widdowson DA; Palladium catalysed cross-coupling of (fluoroarene)tricarbonylchromium(0) complexes. J. Chem. Soc., Perkin Trans 1 2000, 3808–3813; [Google Scholar]; (c) Miller JA C—C Bond activation with selective functionalization: preparation of unsymmetrical biaryls from benzonitriles. Tetrahedron Lett 2001, 42, 6991–6993; [Google Scholar]; (d) Tang S; Li S-H; Yan W.-b. Palladium-catalyzed cross-coupling reaction of aryl(trialkyl)silanes with aryl nitriles. Tetrahedron Lett 2012, 53, 6743–6746; [Google Scholar]; (e) Balam-Villarreal JA; Sandoval-Chávez CI; Ortega-Jiménez F; Toscano RA; Carreón-Castro MP; López-Cortés JG; Ortega-Alfaro MC Infrared irradiation or microwave assisted cross-coupling reactions using sulfur-containing ferrocenyl-palladacycles. J. Organomet. Chem 2016, 818, 7–14. [Google Scholar]
- 20. Notably, only the product of C—O cleavage is reported in the cross-coupling of an umpolung aldehyde with 4-chlorophenyl tosylate in the presence of Ni/PMe3; see ref 18f.
- 21.1-Chloronaphthalene is commercially available from Acros Organics in 85% purity, with the remainder 2-chloronaphthalene.
- 22.When detected, the small amount of the Ni(II)(2-naphthyl) chloride adduct, resulting from oxidative addition of the contaminant 2-chloronaphthalene, was included in obtaining the ratio of reaction at C—Cl vs C—O in all stoichiometric studies we report.
- 23.Electronic differences between 13 and 7/8 may account for the enhanced selectivity in an intramolecular competition. Additionally, the stability of the Ni-arene π complexes that precede oxidative addition may contribute to differences in selectivity during an intra- vs. intermolecular competition.
- 24.Tang Z-Y; Hu Q-S Room-Temperature Ni(0)-Catalyzed Cross-Coupling Reactions of Aryl Arenesulfonates with Arylboronic Acids. J. Am. Chem. Soc 2004, 126, 3058–3059. [DOI] [PubMed] [Google Scholar]
- 25.Results with the chloride catalyst were noticed to worsen as the catalyst aged, despite storing it in a glovebox at −25 °C.
- 26.All reagents including the precatalyst 21 were weighed out open to air. However, yields and selectivities suffered with older batches of 21 that were stored under air.
- 27.Interestingly, diarylation tended to be more problematic when using older batches of Ni(II) precatalyst, even though the precatalysts were stored in a glovebox at −25 °C.
- 28.A time study indicated that the diarylation product is the major product even early on in the reaction.
- 29. The analogous reaction with 4-chlorophenyl mesylate led to about 40% GC yield of 18 with about 7:1 selectivity for reaction at mesylate over chloride (results not included in Table 4).
- 30. Reaction of 4-bromophenyl tosylate under Suzuki coupling conditions with Ni/PMe3 provides primarily the product of C—Br cleavage; see SI for details.
- 31.Beromi MM; Nova A; Balcells D; Brasacchio AM; Brudvig GW; Guard LM; Hazari N; Vinyard DJ Mechanistic Study of an Improved Ni Precatalyst for Suzuki−Miyaura Reactions of Aryl Sulfamates: Understanding the Role of Ni(I) Species. J. Am. Chem. Soc 2017, 139, 922–936; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beromi MM; Banerjee G; Brudvig GW; Charboneau DJ; Hazari N; Lant HMC; Mercado BQ Modifications to the Aryl Group of dppf-Ligated Ni σ-Aryl Precatalysts: Impact on Speciation and Catalytic Activity in Suzuki− Miyaura Coupling Reactions. Organometallics 2018, 37, 3943–3955; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Campeau L-C; Hazari N Cross-Coupling and Related Reactions: Connecting Past Success to the Development of New Reactions for the Future. Organometallics 2019, 38, 3–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Littke AF; Dai C; Fu GC Versatile Catalysts for the Suzuki Cross-Coupling of Arylboronic Acids with Aryl and Vinyl Halides and Triflates under Mild Conditions. J. Am. Chem. Soc 2000, 122, 4020–4028; [Google Scholar]; (b) Schoenebeck F; Houk KN Ligand-Controlled Regioselectivity in Palladium-Catalyzed Cross-coupling Reactions. J. Am. Chem. Soc 2010, 132, 2496–2497; [DOI] [PubMed] [Google Scholar]; (c) Niemeyer ZL; Milo A; Hickey DP; Sigman MS Parameterization of Phosphine Ligands Reveals Mechanistic Pathways and Predicts Reaction Outcomes. Nature Chem 2016, 8, 610–617. [DOI] [PubMed] [Google Scholar]
- 33.Representative examples:; (a) Proutiere F; Schoenebeck F Solvent Effect on Palladium-Catalyzed Cross-Coupling Reactions and Implications on the Active Catalytic Species. Angew. Chem. Int. Ed 2011, 50, 8192–8195; [DOI] [PubMed] [Google Scholar]; (b) Reeves EK; Humke JN; Neufeldt SR N-Heterocyclic Carbene Ligand-Controlled Chemodivergent Suzuki−Miyaura Cross-coupling. J. Org. Chem 2019, 84, 11799–11812; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Reeves EK; Bauman OR; Mitchem GB; Neufeldt SR Solvent Effects on the Selectivity of Palladium-Catalyzed Suzuki-Miyaura Couplings. Isr. J. Chem 2020, 60, 406–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Representative examples:; (a) Kamikawa T; Hayashi T Control of Reactive Site in Palladium-Catalyzed Grignard Cross-Coupling of Arenes Containing both Bromide and Triflate. Tetrahedron Lett 1997, 38, 7087–7090; [Google Scholar]; (b) Espino G; Kurbangalieva A; Brown JM Aryl bromide/triflate selectivities reveal mechanistic divergence in palladium-catalysed couplings; the Suzuki–Miyaura anomaly. Chem. Commun 2007, 1742–1744; [DOI] [PubMed] [Google Scholar]; (c) Ansari NN; Cummings MM; Söderberg BCG Chemoselectivity in the Kosugi-Migita-Stille coupling of bromophenyl triflates and bromo-nitrophenyl triflates with (ethenyl)tributyltin. Tetrahedron 2018, 74, 2547–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amaike K; Muto K; Yamaguchi J; Itami K Decarbonylative C−H Coupling of Azoles and Aryl Esters: Unprecedented Nickel Catalysis and Application to the Synthesis of Muscoride A. J. Am. Chem. Soc 2012, 134, 13573–13576; [DOI] [PubMed] [Google Scholar]; (b) Chatupheeraphat A; Liao H-H; Srimontree W; Guo L; Minenkov Y; Poater A; Cavallo L; Rueping M Ligand-Controlled Chemoselective C(acyl)−O Bond vs C(aryl)−C Bond Activation of Aromatic Esters in Nickel Catalyzed C(sp2)−C(sp3) Cross-Couplings. J. Am. Chem. Soc 2018, 140, 3724–3735. [DOI] [PubMed] [Google Scholar]
- 36.Hong Z; Liang Y; Houk KN Mechanisms and Origins of Switchable Chemoselectivity of Ni- Catalyzed C(aryl)−O and C(acyl)−O Activation of Aryl Esters with Phosphine Ligands. J. Am. Chem. Soc 2014, 136, 2017–2025. [DOI] [PubMed] [Google Scholar]
- 37. Cis-chelating bidentate ligands provide poor yields in our hands; see examples in Table S1 in the Supporting Information.
- 38.Another piece of evidence against ligation state being the determining factor in selectivity is that several smaller phosphines—albeit not as small as methylphosphines—that are unlikely to promote mono-ligated nickel also give poor selectivity, similar to PCy3 (e.g., PEt3, PPhEt2, and PBu3; see Table 1).
- 39. Calculated at the CPCM(dioxane)-MN15L/BS2//MN15L/BS1 level of theory (see SI). The NBO charge at Ni in parentheses is from NiL2 in the distorted geometry taken from the 5-centered C—OTs oxidative addition transition structure.
- 40.Tolman CA Steric Effects of Phosphorus Ligands in Organometallic Chemistry and Homogeneous Catalysis. Chem. Rev 1977, 77, 313–348. [Google Scholar]
- 41.Uthayopas C; Surawatanawong P Aryl C–O oxidative addition of phenol derivatives to nickel supported by an N-heterocyclic carbene via a Ni0 five-centered complex. Dalton Trans 2019, 48, 7817–7827. [DOI] [PubMed] [Google Scholar]
- 42.Bickelhaupt FM; Houk KN Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. When analyzing a later point on the IRC where the C---O distance is 2.06 Å, ΔΔEdist becomes very similar to that of the dissociation mech (47.2 kcal mol−1; see Table S21).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.