

Abstract
Peroxynitrite (PN), generated from the reaction of nitric oxide (NO) and superoxide, is implicated in the pathogenesis of ischemic and neurodegenerative brain injuries. Mitochondria produce NO from mitochondrial NO synthases and superoxide by the electron transport chain. Our objective was to detect the generation of PN of mitochondrial origin and characterize its effects on mitochondrial respiratory function. Freshly isolated brain nonsynaptosomal mitochondria from C57Bl/6 (wild type, WT) and endothelial NO synthase knockout (eNOS-KO) mice were treated with exogenous PN (0.1, 1, 5 µmol/L) or a PN donor (SIN-1; 50 µmol/L) or a PN scavenger (FeTMPyP; 2.5 µmol/L). Oxygen consumption rate (OCR) was measured using Agilent Seahorse XFe24 analyzer and mitochondrial respiratory parameters were calculated. Mitochondrial membrane potential, superoxide, and PN were determined from rhodamine 123, dihydroethidium, and DAX-J2 PON green fluorescence measurements, respectively. Mitochondrial protein nitrotyrosination was determined by Western blots. Both exogenous PN and SIN-1 decreased respiratory function in WT isolated brain mitochondria. FeTMPyP enhanced state III and state IVo mitochondrial respiration in both WT and eNOS-KO mitochondria. FeTMPyP also elevated state IIIu respiration in eNOS-KO mitochondria. Unlike PN, neither SIN-1 nor FeTMPyP depolarized the mitochondria. Although mitochondrial protein nitrotyrosination was unaffected by SIN-1 or FeTMPyP, FeTMPyP reduced mitochondrial PN levels. Mitochondrial superoxide levels were increased by FeTMPyP but were unaffected by PN or SIN-1. Thus, we present the evidence of functionally significant PN generation in isolated brain mitochondria. Mitochondrial PN activity was physiologically relevant in WT mice and pathologically significant under conditions with eNOS deficiency.
NEW & NOTEWORTHY Mitochondria generate superoxide and nitric oxide that could potentially react with each other to produce PN. We observed eNOS and nNOS immunoreactivity in isolated brain and heart mitochondria with pharmacological inhibition of nNOS found to modulate the mitochondrial respiratory function. This study provides evidence of generation of functionally significant PN in isolated brain mitochondria that affects respiratory function under physiological conditions. Importantly, the mitochondrial PN levels and activity were exaggerated in the eNOS-deficient mice, suggesting its pathological significance.
Keywords: eNOS, mitochondrial nitric oxide synthase, nitrotyrosination, nNOS, oxygen consumption rate
INTRODUCTION
Mitochondria are key cellular organelles that play crucial roles in the energy production and regulation of cellular metabolism. The human brain accounts for 20% of glucose and oxygen consumption (1), and alterations in its intracellular energy production are associated with many neurodegenerative diseases (2, 3) and aging (4–7). Both oxidative and nitrative stress, mediated by reactive oxygen species (ROS) and reactive nitrogen species (RNS), respectively, play a critical role in the mitochondrial respiratory dysfunction (8–14). Peroxynitrite (PN) is a strong oxidative and nitrating molecule that mediates nitrative stress. PN is generated from the reaction between superoxide and nitric oxide (NO). The pathological effects of PN are induced by lipid peroxidation (15, 16), DNA damage, and protein modification including oxidation and nitration of protein moieties (17, 18). PN has been implicated in the pathogenesis of aging-induced neuronal death and associated disorders (19). Notably, PN plays a critical role in Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, amyotrophic lateral sclerosis (20), and ischemic injury (21, 22). Thus, mitochondrial dysfunction is likely to mediate the PN-induced detrimental actions in neurodegenerative diseases. Unlike NO, PN has been shown to irreversibly inhibit mitochondrial respiration by decreasing the activity of respiratory complexes (I, II, and III) and the enzymes involved in the tricarboxylic acid cycle (23, 24). Previously, treatment of isolated mitochondria with PN revealed nitration of various mitochondrial respiratory and nonrespiratory proteins (25). Likewise, PN treatment of isolated mouse brain mitochondria increased state II respiration with decreased respiratory control ratio and state III respiration with elevated nitrotyrosine levels (26, 27). Notably, isolated mitochondria from heart and kidney have been shown to generate PN after hypoxia and regeneration (28, 29). However, little is known about the effects of endogenous PN on brain mitochondria.
In the present study, we sought to investigate the actions of exogenous PN by direct exposure to PN solution and a PN donor (SIN-1) on respiration in isolated mitochondria from C57BL/6 wild-type (WT) mice by measuring oxygen consumption rates (OCRs) utilizing the Agilent Seahorse XFe24 analyze. In addition, to demonstrate the intrinsic ability of mitochondria to produce PN endogenously, we studied the effects of a PN scavenger, Fe(III)tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (FeTMPyP), on the respiratory parameters and PN levels in isolated brain mitochondria. FeTMPyP is a widely used PN decomposition catalyst that catalytically shunts PN to the innocuous nitrate form (30, 31). Lastly, to determine the role of endothelial nitric oxide synthase (eNOS) isoform in the regulation of PN activity, we studied the effects of SIN-1 and PN scavenger on mitochondrial respiration in eNOS knockout (eNOS-KO, OB6.129P2-NOS3tm1Unc/J) mice.
METHODS
Reagents
Sucrose (S33-500) and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, BP310-500) were purchased from Fisher Scientific (Waltham, MA). Mannitol (M4125), ethylene glycol-bis (2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA, 4370), fatty acid-free bovine serum albumin (BSA, A7030), Percoll (17-0891-01), sodium pyruvate (P2256), malate (M1000, Millipore Sigma), adenosine 5′-diphosphate sodium salt (A2754), antimycin A (A8674), rotenone (R8875, Millipore Sigma), magnesium chloride (M9272), and potassium dihydrogen phosphate (P5655) were purchased from Sigma-Aldrich (Missouri). Rhodamine 123 chloride (16672), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP, 15218), oligomycin (11341), PN (81565), SIN-1 chloride (82220), and FeTMPyP (75854) were purchased from Cayman Chemicals (Michigan). SDS-PAGE gels (4–20% gradient), transfer buffer, blocking buffer, and Immun-Blot PVDF Membrane were purchased from Bio-Rad Laboratories (Hercules, CA). Anti-nitrotyrosine primary antibody (No. 9691, dilution 1:1,000) and anti-HRP-linked anti-rabbit IgG secondary antibody (No. 7074S, dilution 1:10,000) were from Cell Signaling Technology (Danvers). Anti-VDAC1 antibody (ABCAM, ab14734, dilution 1:5,000) was purchased form Abcam (Cambridge). Previous literature has validated the antibody specificity and the dilutions to be used.
Isolation of Mouse Brain Nonsynaptosomal Mitochondria
All the animal protocols were approved by the Institutional Animal Care and Use Committee of Tulane University. WT (Stock No. 000664) and eNOS-KO (Stock No. 002684) mice were purchased from Jackson Laboratories (Maine). Brain nonsynaptosomal mitochondria were isolated and purified as previously described (32, 33). Briefly, after the dissection of tissues, the cerebral cortices were homogenized in ice-cold mitochondrial isolation buffer (MIB) consisting of (in mmol/L) 225 sucrose, 75 mannitol, 5 HEPES, 1 EGTA, and 0.5% BSA (pH 7.4). After a series of differential centrifugation steps, the crude mitochondrial pellet was resuspended in 15% Percoll and layered on top of 24% and 40% Percoll layers, respectively, so that a visible interface was present between layers. The reconstituted pellet was then centrifuged at 30,000 g for 8 min to collect the mitochondrial layer between the 40% and 24% Percoll layers. The layer containing mitochondria was then resuspended in MIB and centrifuged at 16,000 g for 10 min. The resulting pellet was resuspended in 1% BSA (in MIB) and centrifuged at 7,000 g for 10 min. The concentration of the obtained mitochondrial protein was determined using a BCA kit (Thermo Fisher Scientific, Waltham, MA). Quantification was conducted through absorbance measurements at 595 nm using BMG FLUOStar Optima (BMG Labtech, Ortenberg, Germany).
Peroxynitrite Treatment of Isolated Mitochondria
PN in 0.3 mol/L sodium hydroxide (Cayman Chemical, Ann Arbor, Michigan) was aliquoted and stored at −80°C. PN concentration in the aliquot was quantified spectrophotometrically before the PN assay. PN stock was diluted 40-fold using ice-cold 0.3 mol/L sodium hydroxide, and absorbance readings were taken at 302 nm using 0.3 mol/L NaOH as the blank. Concentrations were calculated from the extinction coefficient of the PN (1670 M−1 ·cm−1). After determining the stock concentration, further dilutions were made in the 0.3 mol/L NaOH right before PN was added to the mitochondria in the mitochondrial assay solution (MAS) buffer, consisting (in mmol/L) of 70 sucrose, 210 mannitol, 2 HEPES, 1 EGTA, 10 potassium phosphate, 5 magnesium chloride, and 0.2% BSA (pH 7.4) with 10 pyruvate, 2 malate, and 5 ADP, to ensure its stability (PN is stable in alkaline solutions). Following PN treatment, mitochondrial suspensions were mixed gently (except for the seahorse experiments) and incubated for the required amount of the time.
Mitochondrial Respiration Measurements
Mitochondrial respiration measurements utilizing the Agilent Seahorse XFe24 analyzer were executed as previously described (32–34). Mitochondrial suspension was prepared for measurement in MAS. Mitochondria (10 µg in 50 µL of MAS) were added to each well of the cell plate and centrifuged at 2,000 g for 20 min at 4°C. Later 50 µL of MAS was added followed by the 100 µL of MAS containing PN, SIN-1, or PN scavenger FeTMPyP. Concentrations of PN (0.1, 1.0, and 5.0 µmol/L), PN donor SIN-1 (50 µmol/L), and FeTMPyP (2.5 µmol/L) used in this study are based on the observations from previous studies and also from our preliminary experiments (35). The plate was incubated (0 min of PN, whereas 10 min for SIN-1 and FeTMPyP) and placed into the Agilent Seahorse XFe24 analyzer. The sensor cartridge was hydrated overnight in a non-CO2 incubator. Before determination of OCR, the cartridge was calibrated using purified water provided by Agilent. Later, the plate containing the mitochondria was placed in the analyzer, and OCR was measured sequentially by injecting 5 µmol/L oligomycin, 5 µmol/L FCCP, and antimycin A (10 µmol/L)-rotenone (2 µmol/L) to measure state II (basal), state III (in the presence of ADP), state IVo (oligomycin), and state IIIu (FCCP) respiration. The basal respiration was taken as 100 for each experiment, and other respiratory parameters were normalized to minimize the day-to-day variations between the seahorse experiments.
Mitochondrial Membrane Potential Measurements
Mitochondrial membrane potential (ψm) was determined by using the ψm-sensitive fluorescence dye rhodamine 123 (λex: 507 nm; λem: 529 nm). Mitochondrial suspensions were incubated with rhodamine 123 (1 µmol/L) in MAS for 20 min at 4°C. After centrifugation (10,000 g for 5 min), the mitochondrial pellet was resuspended in MAS and divided equally and treated with FCCP (5 µmol/L) in MAS, or treated with PN (0.1 µmol/L, 1 µM, 5 µmol/L), SIN-1 (50 µmol/L), or FeTMPyP (2.5 µmol/L) in MAS for 20–30 min in a non-CO2 incubator at 37°C. Later, the treated mitochondria were spun, the supernatant was discarded, and the mitochondrial pellet was resuspended in MAS. The mitochondria were loaded (10 µg per well with eight replicates) into a Falcon 96-well plate (353219; Life Sciences, Irving, TX). Fluorescence measurements were taken at 25°C and 527 nm with BMG FLUOStar Optima (BMG Labtech) spectrofluorometer.
Western Blot Analysis
Elevated nitrotyrosine levels in a given tissue indicate the local nitrative stress and PN generation. Following isolation, mitochondria were either left untreated or were treated with PN (0.1, 1.0, and 5.0 µmol/L), SIN-1 (50 µmol/L), or FeTMPyP (2.5 µmol/L). Mitochondrial samples were processed in NP-40 cell lysis buffer (Thermo Fisher Scientific) and subsequently boiled at 95°C and prepared for immunoblotting. Mitochondria homogenates (25 µg) were separated by SDS-PAGE under reducing conditions and transferred to a polyvinylidene difluoride sheet (Immun-Blot PVDF Membrane for protein blotting; Bio-Rad Laboratories). Membranes were treated with blocking buffer and then incubated with 1:1,000 dilution of anti-nitrotyrosine rabbit primary antibodies (Cell Signaling Technology) suspended in 2% BSA overnight at 4°C. Membranes were next incubated with a 1:10,000 dilution of anti-rabbit IgG, HRP-linked secondary antibody (Jackson ImmunoResearch, West Grove, PA). The membranes were visualized using enhanced chemiluminescence (SuperSignal West Pico; Pierce, Rockford, IL). Later, the membrane was then stripped, washed, and re-treated with VDAC1 (Abcam, ab14734, dilution 1:5,000) in 2% BSA overnight to be used as a loading control. 3-nitrotyrsoine levels were quantified using ImageJ software. Immunoband densities of all the lane were calculated (from 250 kDa to 20 kDa) and normalized to the respective voltage-dendent anion channel (VDAC) band density. In the present study, verification of the antibody validation and the antibody dilutions used was based on the published literature.
Mitochondrial Peroxynitrite Measurements
Mitochondrial PN measurements were performed using Cell Meter fluorometric intracellular peroxynitrite assay kit (AAT Bioquest, Sunnyvale, CA) (36). Briefly, mitochondria (25 μg/well, in triplicates) were incubated at room temperature for 1 h in MAS containing pyruvate (10 mmol/L) and malate (2 mmol/L) with or without the PN scavenger FeTMPyP (2.5 μmol/L). Fluorescence measurements (λex: 490 nm, λem: 530 nm) were taken at the 0- and 60-min time points. Readings from the wells with only scavenger and dye and without mitochondria were taken as the background control. 0-min readings were deducted from the 60-min readings to calculate the relative PN concentration.
Mitochondrial Reactive Oxygen Species Measurements
Mitochondrial ROS content was measured using dihydroethidium (DHE). Mitochondria were incubated (50 µg of mitochondria for treatment) with or without 2.5 µmol/L FeTMPyP (prepared in MAS with pyruvate/malate) for 30 min at 37°C. Mitochondria were pellet down by centrifugation at 10,000 g for 5 min, and the pellet was dissolved in 1 mL of 10 µmol/L DHE (prepared in MAS with pyruvate/malate), and 100 μL was loaded to eight wells of a microplate (eight replicate readings/treatment). Fluorescent kinetic readings were taken (λex: 510 nm; λem: 600 nm) for 2 min till 30 min; 10 µmol/L DHE solution was used as the blank. Rate of ROS production was measured from the slope. Blank-corrected fluorescence value from control mitochondria was taken as 100, and relative fluorescence values were calculated for scavenger-treated mitochondria.
Statistics
Data are presented as means ± SE. Seahorse data from the PN experiments and ROS data were analyzed by one-way ANOVA. Seahorse data from SIN-1 and PN scavenger, membrane potential, and Western blot data were analyzed by repeated-measures two-way ANOVA along with the mitochondrial PN levels (log-transformed if needed). Trend between the two groups in these experiments was analyzed by Student’s t test. P < 0.05 were considered to be statistically significant.
RESULTS
Effect of PN on Respiratory Parameters and ψm in Isolated Brain Mitochondria
PN dose-dependently decreased the various respiratory parameters in isolated brain mitochondria from the WT mice, but the statistically significant effect was observed at the highest concentration, 5 µmol/L (Fig. 1). PN at 5 µmol/L significantly decreased the basal or state II respiration by 55.5% in the isolated brain mitochondria (Fig. 1A). Similarly, 5 µmol/L PN significantly diminished the state III respiration by 47.8%, and state IIIu respiration by 58% in the isolated brain mitochondria (Fig. 1, B and D). State IVo respiration was also significantly decreased by 61% with PN (5 µmol/L) treatment in the isolated brain mitochondria from WT mice (Fig. 1C). Consistent with the observations with respect to respiratory parameters, PN (5 µmol/L) significantly reduced the ψm by 65.6% compared with untreated mitochondria (Fig. 1E).
Figure 1.

Peroxynitrite (PN; 5 µmol/L) impairs mitochondrial respiration and mitochondrial membrane potential. A: basal or state II respiration. B: state III respiration (ADP-induced). C: state IVo respiration (oligomycin-inhibited). D: state IIIu [carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP)-induced]. E: membrane potential. Mitochondrial respiratory parameters were measured using Agilent Seahorse XFe24 analyzer, and mitochondrial membrane potential (ψm) was measured fluorometrically using rhodamine 123 dye. Data were presented as means ± SE and analyzed by one-way ANOVA. *P < 0.05, statistically significant. n = 5–6 mice for seahorse experiments and n = 8 mice for the ψm studies.
Effect of SIN-1, PN Donor, on Respiratory Parameters
Compared with untreated mitochondria, SIN-1 treatment (50 µmol/L) did not alter the basal or state II respiration in WT isolated brain mitochondria, whereas it significantly decreased the same in the mitochondria from the eNOS-KO mice (16%, P = 0.04; Fig. 2B). However, SIN-1 treatment significantly decreased the state III respiration in both WT and eNOS-KO brain mitochondria (48.5%, P = 0.02; 33.6%, P = 0.009, respectively) compared with untreated mitochondria (Fig. 2, A and C). In contrast, State IVo respiration was unaltered by SIN-1 treatment in both WT and eNOS-KO brain mitochondria (Fig. 2D). Notably, mitochondrial uncoupler-induced or state IIIu respiration was unaltered by SIN-1 in the WT brain mitochondria compared with untreated mitochondria, whereas the mitochondria from eNOS-KO mice brain showed significant decrease (29.5%, P = 0.005; Fig. 2E).
Figure 2.

SIN-1-1 (50 µmol/L) decreases mitochondrial respiration in isolated brain mitochondria. A: oxygen consumption rate (OCR) versus time graph. B basal or state II respiration. C: state III respiration (ADP-induced). D: state IVo respiration (oligomycin-inhibited). E: state IIIu [carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP)-induced]. Mitochondria were isolated from wild-type (WT; n = 5 mice) and endothelial NO synthase knockout (eNOS-KO) mice (n = 6 mice) brains, treated with SIN-1 (50 µmol/L), and respiratory parameters were measured using Agilent Seahorse XFe24 analyzer. Data are presented as means ± SE and analyzed by repeated-measures two-way ANOVA (data were log-transformed for some parameters). *P < 0.05 and **P < 0.01, statistically significant.
Effect of FeTMPyP, PN Scavenger, on Respiratory Parameters in Isolated WT and eNOS-KO Brain Mitochondria
The PN scavenger, FeTMPyP, significantly elevated state II in the WT brain mitochondria (19.3%, P ≤ 0.05), whereas a trend toward increase was observed in the eNOS-KO brain mitochondria (119.5%, P = 0.10; Fig. 3, A and B). Compared with untreated mitochondria, both WT (29.2%, P = 0.03) and eNOS-KO brain mitochondria (52%, P ≤ 0.03) showed a significant increase in state III-respiration after treatment with the PN scavenger (Fig. 3C). Similarly, state IVo is significantly increased by FeTMPyP treatment in both WT (34.4%, P = 0.03) and eNOS-KO brain mitochondria (169%, P ≤ 0.05) versus untreated mitochondria (Fig. 3D). However, PN scavenger did not alter state IIIu respiration in the WT brain mitochondria, but it significantly increased it in the eNOS-KO brain mitochondria (91.3%, P = 0.01) compared with respective untreated mitochondria (Fig. 3E). These results indicate that scavenging PN produced by mitochondria endogenously modestly increases mitochondrial respiration in brain mitochondria from WT mice, whereas it increases respiration to a greater extent in brain mitochondria from eNOS-KO mice. Furthermore, these observations indicate that the generation of endogenous PN and its inhibitory effects on respiration are more pronounced in the absence of eNOS.
Figure 3.

FeTMPyP (2.5 µmol/L) increases respiration in isolated brain mitochondria. A: oxygen consumption rate (OCR) versus time graph. B: basal or state II respiration. C: state III respiration (ADP-induced). D: state IVo respiration (oligomycin-inhibited). E: state IIIu [carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP)-induced]. Mitochondria were isolated from wild-type (WT; n = 5 mice), and endothelial NO synthase knockout (eNOS-KO) mice (n = 5 mice) brains, treated with FeTMPyP (2.5 µmol/L), and respiratory parameters were measured using Agilent Seahorse XFe24 analyzer. Data are presented as means ± SE and analyzed by repeated-measures two-way ANOVA (data were log-transformed for some parameters). *P < 0.05, statistically significant.
Effect of SIN-1 and FeTMPyP on Inner Mitochondrial Membrane Potential
Agents that promote changes in mitochondrial respiration are likely to alter the ψm; thus, we determined the effects of SIN-1 and FeTMPyP on ψm of isolated brain mitochondria. Interestingly, SIN-1 treatment failed to alter the ψm in either the WT or eNOS-KO mitochondria, despite its effects on mitochondrial respiration (Fig. 4A). Similarly, FeTMPyP treatment failed to induce any significant changes in the ψm of either WT or eNOS-KO brain mitochondria, although it promoted changes in respiratory parameters (Fig. 4B).
Figure 4.

Neither the peroxynitrite (PN) donor (SIN-1) nor the PN scavenger (FeTMPyP) have a significant effect on mitochondrial membrane potential. A: SIN-1. B: FeTMPyP. Mitochondria were isolated from wild-type (WT) and endothelial NO synthase knockout (eNOS-KO) mice (n = 6 mice for SIN-1 and n = 5 mice for FeTMPyP) brains, treated with SIN-1 (50 µmol/L) or FeTMPyP (2.5 µmol/L), and mitochondrial membrane potential (ψm) was measured fluorometrically using rhodamine 123 dye. Data are presented as means ± SE and analyzed by repeated-measures two-way ANOVA. Values of P < 0.05 were considered to be statistically significant.
Effect of SIN-1 on Nitrotyrosination of Mitochondrial Proteins
As SIN-1 treatment altered the respiratory parameters in the isolated brain mitochondria, we went on to measure the nitrotyrosine content of the mitochondrial proteins, to check whether the decreased respiratory parameters associate with increased protein nitrotyrosination. Brain mitochondria from WT and eNOS-KO mice were not different with respect to the nitrotyrosinated protein content, and SIN-1 treatment had no significant effect on the nitrotyrosinated protein levels in both the phenotypes (Fig. 5).
Figure 5.

SIN-1 (50 µmol/L) has no significant effect on nitrotyrosination of mitochondrial proteins. A: representative Western blot for nitrotyrosine content and VDAC (loading control). B: representative bar diagram. Mitochondria were isolated from wild-type (WT) and endothelial NO synthase knockout (eNOS-KO) mice (n = 8–11 mice) brains, treated with SIN-1 (50 µmol/L). Mitochondrial pellets were solubilized with NP 40 buffer and subjected to SDS-PAGE (4%–20% gradient). Nitrotyrosine content in the proteins was detected on Immun-Blot using enhanced chemiluminescence. Total band intensity was measured using ImageJ. Data are presented as means ± SE and analyzed by repeated-measures two-way ANOVA.
Effect of FeTMPyP on Protein Nitrotyrosination and Peroxynitrite Levels in the Isolated Brain Mitochondria
As FeTMPyP elevated the mitochondrial respiration, we measured the protein nitrotyrosination and PN levels in the WT and eNOS-KO brain mitochondria. FeTMPyP treatment did not affect the nitrotyrosination of mitochondrial proteins (Fig. 6, A–B). Basal PN levels were significantly higher by an average of ∼26.8% in eNOS-KO mitochondria when compared with WT mice (Fig. 6C). FeTMPyP significantly decreased the PN levels in both WT and eNOS-KO brain mitochondria (Fig. 6C). eNOS-KO mitochondria have higher PN levels when compared with WT mitochondria, even after inhibition with FeTMPyP (Fig. 6C).
Figure 6.

FeTMPyP (2.5 µmol/L) has no significant effect on nitrotyrosination of mitochondrial proteins but decreases peroxynitrite (PN) production. A: representative Western blot for nitrotyrosine content and VDAC (loading control). B: representative bar diagram. C: mitochondrial PN production. Mitochondria were isolated from wild-type WT and endothelial NO synthase knockout (eNOS-KO) mice (n = 4 mice for Western, n = 4–8 mice for PN measurements) brains, treated with FeTMPyP (2.5 µmol/L) for 60 min. Mitochondrial pellets were solubilized with NP 40 buffer and subjected to SDS-PAGE (4%–20% gradient). Nitrotyrosine content in the proteins was detected on anti-nitrotyrosine Immun-Blot using enhanced chemiluminescence. Total band intensity was measured using ImageJ. PN measurements were taken using DAX-J2 PON green (excitation: 490 nm and emission: 530 nm) Data are presented as means ± SE and analyzed by repeated-measures two-way ANOVA. *P < 0.05, statistically significant. C, Control; S, FeTMPyP.
Effect of PN, SIN-1, and FeTMPyP on ROS Levels in the Isolated Brain Mitochondria
As exogenous and endogenous PN decreased the mitochondrial respiration, we measured the levels of ROS in the isolated brain mitochondria after treating with the exogenous PN (SIN-1 and PN) and the FeTMPyP. Both PN and SIN-1 failed to change the ROS levels, whereas the FeTMPyP significantly increased the ROS levels by an average of ∼60.5% (Fig. 7).
Figure 7.

FeTMPyP increases the reactive oxygen species (ROS) levels in the isolated brain mitochondria. Mitochondria were isolated from the wild-type (WT) mice brains and incubated with peroxynitrite (PN) (5 µmol/L), SIN-1 (50 µmol/L), and FeTMPyP (2.5 µmol/L) for 30 min (for ROS, n = 8–10 mice). ROS levels were measured using dihydroethidium (excitation: 510 nm and emission: 600 nm). Data are presented as means ± SE and analyzed by one-way ANOVA. *P ≤ 0.05, statistically significant.
DISCUSSION
The major findings of the present study investigating the effects of endogenous and exogenous PN on the respiration of isolated brain mitochondria are as follows: First, acute exposure to exogenous PN as well as prolonged PN exposure via SIN-1 was inhibitory to isolated mouse brain mitochondrial respiratory function. Second, scavenging the PN enhanced the mitochondrial respiratory function of isolated brain mitochondria, indicating the ability of mitochondria to produce PN independently. Third, SIN-1 and PN scavenger exerted a significantly greater impact on mitochondrial respiratory function in eNOS-KO than in WT control mice, suggesting greater PN activity and sensitivity in the absence of eNOS. Measurement of PN levels also showed increase in eNOS KO versus WT mice. Fourth, PN scavenger increased mitochondrial superoxide generation although PN and SIN-1 had no effect, suggesting that PN of mitochondrial origin inhibits mitochondrial superoxide levels. Lastly, relatively low concentration of SIN-1 or PN scavenger did not significantly increase the nitrotyrosine content of mitochondrial proteins despite the effect on mitochondrial respiratory function. The significance of these observations is that our study provides the first functional evidence of endogenous generation of PN in isolated brain mitochondria. Furthermore, we demonstrate the inhibitory regulation of eNOS on mitochondrial PN levels. Notably, our study identified both physiological and pathological endogenous mitochondrial PN signaling regulating mitochondrial respiratory function.
NO is synthesized from l-arginine by three isoforms of NO synthase (NOS), endothelial (eNOS), neuronal (nNOS), and inducible (iNOS) (14). In the extra mitochondrial locations, NOS isoforms and ROS-generating enzymes, such as NADPH oxidase (NOX) and xanthine oxidase, contribute to the formation of PN. Additionally, in intact cells, PN generation within the mitochondria is likely to occur from the reaction between ROS originating from electron transport chain (ETC). The mitochondrial ETC contains several redox complexes that contribute to superoxide formation; however, the major source remains the electron leakage at Complex I (10, 11) and NO, which may likely originate from three major sources (Fig. 8). First, NO-derived from eNOS and nNOS may enter mitochondria through diffusion through mitochondrial membranes. Second, NO may originate from mitochondrial NOS (mtNOS). However, the existence of mtNOS and the ability of the mitochondria to generate NO have been intensely debated (37–39). Nevertheless, studies from our laboratory and those of others have reported the presence of mtNOS identified by immunoreactivity of nNOS and eNOS in isolated heart and brain mitochondria (40). Third, denitrosylation of nitrosylated proteins are a likely source of mitochondrial NO owing to the activity of denitrosylases such as mitochondrial thioredoxin system (41, 42). S-nitrosylation of mitochondrial proteins may be induced by NO originating from endothelial cells (eNOS), neurons (nNOS), and possibly mitochondria (mtNOS) (Fig. 8). We previously reported the decrease in protein S-nitrosylation in isolated mouse mitochondria by inhibitors of both eNOS and nNOS, providing the evidence in support of the mitochondrial production of NO and nitrosylation of mitochondrial proteins (40).
Figure 8.
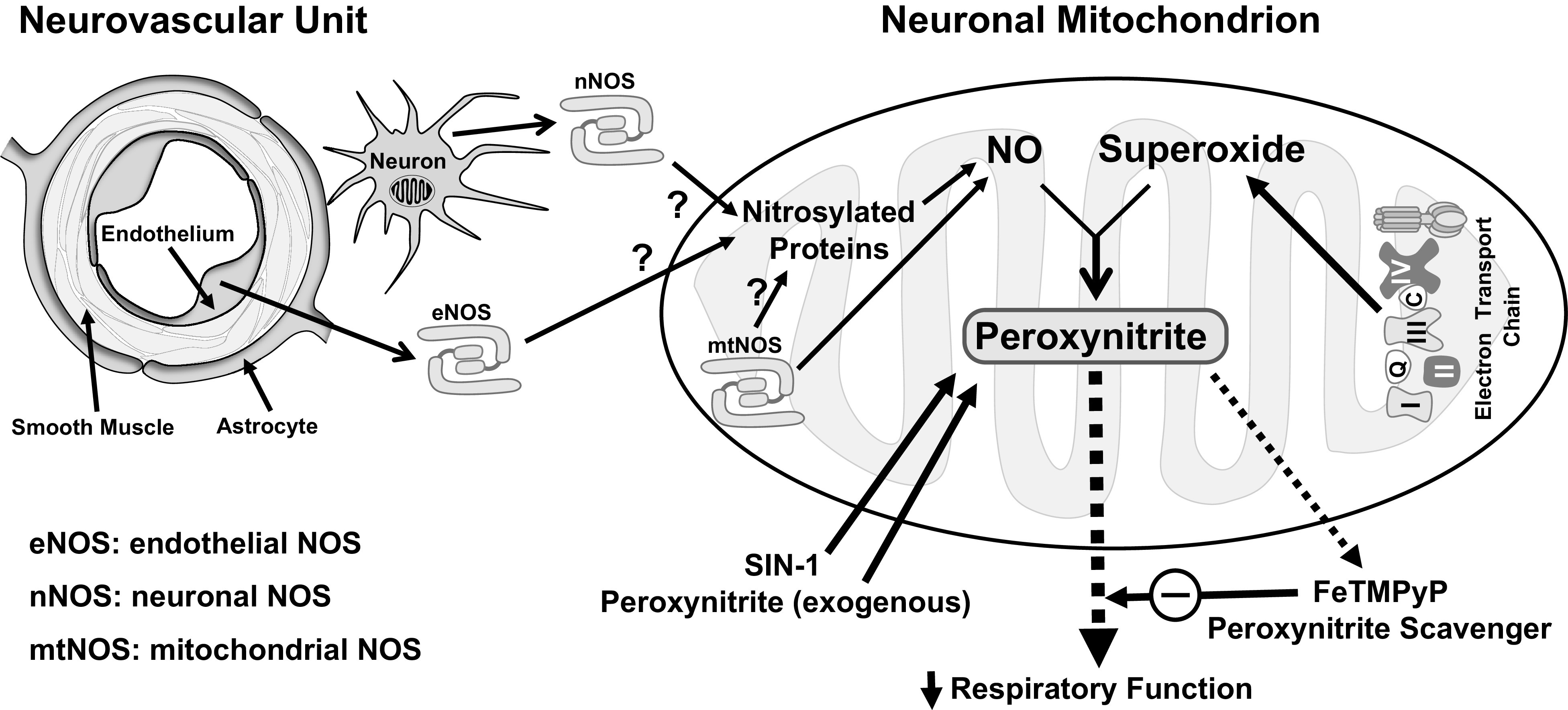
Schematic of peroxynitrite (PN) generation in isolated brain mitochondria and its activity on mitochondrial function. PN is generated when superoxide reacts with nitric oxide (NO). In isolated mitochondria, superoxide is derived from electron transport chain. There are two major mechanisms of NO generation in mitochondria. First, mitochondrial NOS (mtNOS) has been shown to produce NO (38, 39). Second, nitrosylated proteins in the mitochondria may release NO by the action of denitrosylases such as mitochondrial thioredoxin system (41, 42). Mitochondrial protein nitrosylation in turn could be induced by NO derived locally from mtNOS or NO derived extra-mitochondrially from eNOS (endothelial origin) and nNOS (neuronal origin) that diffuses through the mitochondrial membranes. SIN-1 and exogenous PN provide PN for studying the effects of PN on mitochondrial respiration. In contrast, FeTMPyP scavenges the PN and prevents its activity on mitochondrial function. Both exogenous and endogenous PN inhibit various aspects of mitochondrial respiratory function. Notably, increase in mitochondrial respiratory function in isolated mitochondria by FeTMPyP suggests intrinsic and physiological generation of PN in the mitochondria.
Mitochondrion is prone to nitrosative stress, and NO is known to play a regulatory role in mitochondrial functioning and oxidative phosphorylation. NO primarily inhibits Complex III and has also been shown to competitively inhibit Complex IV at the oxygen binding site (8, 12). Because PN (ONOO−) is produced when NO reacts nonenzymatically with ETC-derived superoxide (O2−), excessive generation of NO or ROS can result in irreversible mitochondrial damage mediated by PN (8, 43). SIN-1 has been shown to stimulate complex I-related state II respiration in response to the substrates pyruvate and malate together with inhibition of states III and V respiration (uncoupled respiration) (26). When studied in cultured hippocampal neurons, the exogenous NO from NO donors produced a rapid and reversible depolarization of ψm, as well as profound and irreversible energy depletion leading to neuronal death (44). In contrast, direct application of 200 µmol/L PN to isolated rat brain mitochondria irreversibly inhibited state IV respiration and decreased mitochondrial ψm resulting from the increase in proton leak (35). In the present study, direct application of PN inhibited all aspects of mitochondrial respiration and depolarized the mitochondrial ψm at a concentration (5 µmol/L) significantly lower than previous studies. We credit this to the improvements in the methods of mitochondrial preparation and the measurement of OCR that we recently reported (32, 33, 45). In contrast, SIN-1 significantly decreased state III and state IVo respiration of isolated brain mitochondria in mice. Interestingly, the same concentrations of SIN-1 had more profound effects in isolated brain mitochondria from eNOS-KO mice versus WT mice by decreasing the basal and the state III and state IIIu respiration. This confirms that SIN-1-derived PN exerts an inhibitory effect on the respiration of isolated mitochondria, and this is more pronounced in the absence of eNOS. Alternatively, enhanced PN activity in eNOS-KO mice, presumably due to increased PN generation and/or PN sensitivity, impacts a greater number of mitochondrial targets, for example, reduced IIIu, which represents the ability of the mitochondria to ramp up OCR in response to higher energy demand. Notably, we used low concentrations of PN and SIN-1 as the high concentrations are unlikely to simulate the actions of endogenous PN originating from extra-mitochondrial or mitochondrial sources. The actions of SIN-1 and exogenous PN showed some differences, which we believe may be attributed to the concentration and the kinetics of PN released from SIN-1. Similarly, it is difficult to predict the exact concentration of endogenous mitochondrial NO; thus, the actions of endogenous PN generated in mitochondria showed variation from SIN-1 and exogenous PN. Furthermore, PN depolarized mitochondria, but SIN-1 had no effect on the ψm of mice. Interestingly, SIN-1 inhibited respiration in eNOS-KO mitochondria like PN, indicating that eNOS-KO mitochondria have greater production/activity of PN. Consistent with this, PN measurements showed increased levels in the mitochondria from eNOS KO mice compared with those from WT mice.
Previous studies utilizing penicillamine, a scavenger of PN, showed the ability to partially protect respiratory function by improving Complex I-related state III and state V respiratory rates post-SIN-1 exposure of isolated brain mitochondria (26). This was accompanied by a decrease in nitrotyrosine content of SIN-1-exposed mitochondria. In the present study, PN scavenger increased mitochondrial respiratory function in WT and eNOS-KO mitochondria, suggesting that mitochondria ex vivo have the intrinsic ability to generate functionally active PN. Notably, PN scavenger induced significantly greater increase in both basal and state IVo respiration in mitochondria from eNOS-KO mice than in WT mice, suggesting that eNOS exerts inhibitory regulation on mitochondrial PN generation. Moreover, we have observed increased mitochondrial PN levels in eNOS KO mice compared with WT mice. These findings are consistent with previous reports showing increased production of basal PN in eNOS-KO mice compared with WT mice (46, 47). PN has also been proposed to act by oxidization of tetrahydrobiopterin (BH4), an essential cofactor for the eNOS. Loss of BH4 promotes eNOS uncoupling by diverting electrons flowing from eNOS to molecular oxygen rather than to l-arginine, leading to superoxide production instead of NO synthesis (46–48). RNS have been shown to reversibly inhibit mitochondrial respiration by competing with the oxygen to bind with cytochrome oxidase and inhibit respiratory complexes (8). It may be speculated that PN could induce nNOS uncoupling and promote similar oxidative stress.
In WT mitochondria, PN scavenger promoted modest increase in basal and state IVo respiration, although it had no effect on ψm. This indicates that PN is required for physiological regulation of OCR related to ATP synthesis. Reduction in the ability to generate ATP independent of ψm changes suggests non-ETC-related mechanisms underlying the PN actions. In contrast, superoxide and NO have been known to depolarize the mitochondria by inhibiting ETC. Moreover, in eNOS-KO mitochondria, PN scavenger enhanced state IIIu respiration, suggesting additional mitochondrial sites of PN actions in the absence of eNOS. In this context, it has been shown that increases in either NO or superoxide alone results in equivalent fold-increase in PN, or elevation of both leads to exponential increase in PN (13). It is noted that inhibition of ETC complexes and slowing the electron transport is known to increase superoxide production (49, 50). We observed that exogenous PN depolarizes mitochondrial membrane potential only when applied at high concentration. Unlike PN, SIN-1-induced inhibition of mitochondrial respiration was not accompanied by depolarization of mitochondria, suggesting non-ETC inhibition-related mechanism underlying the mitochondrial effects of PN on respiration. Consistent with these findings, our measurements of ROS have observed that PN and SIN-1 had no effect on mitochondrial ROS generation. However, we observed increase in ROS levels in isolated mitochondria treated with PN scavenger. This suggests that mitochondrial PN can prevent ROS formation by an unknown mechanism. In addition, under our experimental conditions, potential PN or SIN-1 effects on ETC did not result in increased mitochondrial superoxide generation. Furthermore, at the concentrations used in the present study, PN did not appear to promote proton leak as PN reduced the basal respiration, which would have increased if PN promoted proton leak (uncouple respiration). Moreover, exogenous PN donor, SIN-1, did not change the mitochondrial membrane potential. This is inconsistent with proton leak, which would have depolarized the mitochondria. It may be speculated that PN may exert ETC-independent effect (Krebs cycle) on mitochondria to reduce the levels of electron donors for ETC (NADH and FADH2) that preempts ETC-derived superoxide generation.
nNOS-derived NO has been proposed to contribute to neuronal PN under conditions when succinate dehydrogenase or mitochondrial complex II is inhibited (51). Furthermore, the study also suggested the regulation of nNOS by eNOS that was found to be mediated by succinate dehydrogenase (SDH) (51). Nevertheless, it is unclear how exactly eNOS deficiency promotes increased PN activity/generation, and further studies are needed to identify the key underlying mechanisms. Interestingly, nitration of tyrosine residues is not a result of direct NO reactivity, but rather requires that the free radical reacts with superoxide anion to produce PN. Measurement of 3-nitrotyrosination of mitochondrial proteins in Western blot analysis of the eNOS-KO or WT was not sensitive enough to identify PN activity despite the functional evidence of PN actions. However, measurements of PN using commercially available kit was able to detect reduced PN levels in isolated mitochondria treated with PN scavenger, indicating that isolated mitochondria can generate PN ex vivo. Interestingly, recent studies examining the mitochondrial targets of PN identified numerous proteins and enzymes that were involved in Krebs cycle, ETC, and oxidative phosphorylation under normal (52) and ischemic (53) conditions. Consistent with these reports, our observations suggest the effects of PN involving multiple mechanistic targets in mitochondria. We plan to identify the specific protein targets of peroxynitrite in future studies in brain mitochondria. Taken together, mitochondrial PN has the significant ability to modulate mitochondrial function in physiological and pathological states. PN is a free radical with a short biological half-life estimated to be ∼10–20 ms (54); thus, the effects of PN scavenger in isolated mitochondria ex vivo could result from PN formed intrinsically in the isolated mitochondria. Nevertheless, the present study shows that isolated brain mitochondria exhibit the ex vivo ability to generate PN (Fig. 8).
Limitations
First, the isolated mitochondria used in this study were prepared from the homogenates of brain consistent with the practice of similar studies performed by us and others (33, 45). We isolated the mitochondria from the cerebral cortex containing numerous diverse cell types; however, we expect that predominant number of mitochondria are of neuronal origin. Second, the present study has made direct measurements of PN in the mitochondria using a commercially available DAX-J2 PON green fluorescence-based PN assay that has been widely used by others (36). Although the modest decrease in the green fluorescence following FeTMPyP treatment (Fig. 5) is disproportionately low considering the functional effect of FeTMPyP, the assay confirms the ability of isolated mitochondria to generate the PN. Currently, boronates have been proposed as fluorescent probes for PN (55) and are used in several commercial assays. However, a recent study by Rios et al. (2020) has shown that mitochondria-targeted phenyl boronate probe (oMitoPhB(OH)2) reacts with PN and other biologically relevant two-electron oxidants such as hydrogen peroxide, peroxymonocarbonate, and hypochlorite (56). Thus, boronate-based probes lack the specificity to identify PN in mitochondria or cultured cells and a reliably sensitive and specific assay to measure PN in mitochondria remains elusive. Third, it is likely that FeTMPyP could react with other oxidative radicals apart from PN (57); however, micromolar concentrations of FeTMPyP used in the present study predominantly catalyze the decomposition of PN at physiologically relevant pH and temperature (30, 58).
In conclusion, exogenous and endogenous PN is inhibitory to overall mitochondrial respiration. Importantly, isolated brain mitochondria generate endogenous PN under physiological and pathological conditions to exert significant modulation of mitochondrial respiration that appears to involve cross regulation of eNOS and nNOS.
GRANTS
This research project was supported by the National Institutes of Health: National Institute of Neurological Disorders and Stroke (NS094834 to P.V.G. Katakam and NS114286 to P.V.G. Katakam and R. Mostany) and National Institute of General Medical Sciences (NS094834 to P.V.G. Katakam). In addition, the study was supported by American Heart Association (National Center Scientist Development Grant, 14SDG20490359 to P.V.G Katakam; Greater Southeast Affiliate Predoctoral Fellowship Award, 16PRE27790122 to V.N.L.R. Sure; Predoctoral Fellowship Award, 20PRE35211153 to W.R. Evans. Lastly, this study was supported by Undergraduate Summer Research Fellowship by American Physiological Society (to A.L. Albuck).
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURE
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.L.A., S.S.S.V.P.S., J.A.S., W.R.E., L.K., and P.V.G.K. conceived and designed research; A.L.A., S.S.S.V.P.S., and W.R.E. performed experiments; A.L.A., S.S.S.V.P.S., W.R.E., and P.V.G.K. analyzed data; A.L.A., S.S.S.V.P.S., W.R.E., V.N.S. and P.V.G.K. interpreted results of experiments; A.L.A., S.S.S.V.P.S., W.R.E., and P.V.G.K. prepared figures; A.L.A., S.S.S.V.P.S., W.R.E., and P.V.G.K. drafted manuscript; A.L.A., S.S.S.V.P.S., J.A.S., W.R.E., L.K., V.N.S., R.M., and P.V.G.K. edited and revised manuscript; A.L.A., S.S.S.V.P.S., J.A.S., W.R.E., L.K., V.N.S., R.M., and P.V.G.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Ms. Sufen Zheng for technical help for the studies.
REFERENCES
- 1.Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21: 1133–1145, 2001. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Giachin G, Bouverot R, Acajjaoui S, Pantalone S, Soler-Lopez M. Dynamics of human mitochondrial complex I assembly: implications for neurodegenerative diseases. Front Mol Biosci 3: 43, 2016. doi: 10.3389/fmolb.2016.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Onyango IK, Khan SM, Bennett JP, Jr.. Mitochondria in the pathophysiology of Alzheimer's and Parkinson's diseases. Front Biosci (Landmark Ed) 22: 854–872, 2017. doi: 10.2741/4521. [DOI] [PubMed] [Google Scholar]
- 4.Csiszar A, Yabluchanskiy A, Ungvari A, Ungvari Z, Tarantini S. Overexpression of catalase targeted to mitochondria improves neurovascular coupling responses in aged mice. Geroscience 41: 609–617, 2019. doi: 10.1007/s11357-019-00111-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong Y, Digman MA, Brewer GJ. Age- and AD-related redox state of NADH in subcellular compartments by fluorescence lifetime imaging microscopy. Geroscience 41: 51–67, 2019. doi: 10.1007/s11357-019-00052-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kiss T, Nyul-Toth A, Balasubramanian P, Tarantini S, Ahire C, Yabluchanskiy A, Csipo T, Farkas E, Wren JD, Garman L, Csiszar A, Ungvari Z. Nicotinamide mononucleotide (NMN) supplementation promotes neurovascular rejuvenation in aged mice: transcriptional footprint of SIRT1 activation, mitochondrial protection, anti-inflammatory, and anti-apoptotic effects. Geroscience 42: 527–546, 2020. doi: 10.1007/s11357-020-00165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicholatos JW, Robinette TM, Tata SV, Yordy JD, Francisco AB, Platov M, Yeh TK, Ilkayeva OR, Huynh FK, Dokukin M, Volkov D, Weinstein MA, Boyko AR, Miller RA, Sokolov I, Hirschey MD, Libert S. Cellular energetics and mitochondrial uncoupling in canine aging. Geroscience 41: 229–242, 2019. doi: 10.1007/s11357-019-00062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown GC, Borutaite V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc Res 75: 283–290, 2007. doi: 10.1016/j.cardiores.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 9.Durand MJ, Ait-Aissa K, Levchenko V, Staruschenko A, Gutterman DD, Beyer AM. Visualization and quantification of mitochondrial structure in the endothelium of intact arteries. Cardiovasc Res 115: 1546–1556, 2019. doi: 10.1093/cvr/cvy294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature 415: 96–99, 2002. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- 11.Barja G, Herrero A. Localization at complex I and mechanism of the higher free radical production of brain nonsynaptic mitochondria in the short-lived rat than in the longevous pigeon. J Bioenerg Biomembr 30: 235–243, 1998. doi: 10.1023/A:1020592719405. [DOI] [PubMed] [Google Scholar]
- 12.Ignarro LJ. Nitric Oxide: Biology and Pathobiology. San Diego: Academic Press; 2010. [Google Scholar]
- 13.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87: 315–424, 2007. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Nakayama T, Inoue N, Kato T. Quantitative analysis of nitric oxide synthase expressed in developing and differentiating rat cerebellum. Brain Res Dev Brain Res 111: 65–75, 1998. doi: 10.1016/S0165-3806(98)00123-0. [DOI] [PubMed] [Google Scholar]
- 15.Katz AI. Renal Na-K-ATPase: its role in tubular sodium and potassium transport. Am J Physiol 242: F207–F219, 1982. doi: 10.1152/ajprenal.1982.242.3.F207. [DOI] [PubMed] [Google Scholar]
- 16.King PA, Anderson VE, Edwards JO, Gustafson G, Plumb RC, Suggs JW. A stable solid that generates hydroxyl radical upon dissolution in solutions: reaction with aqueous proteins and nucleic acid. J Am Chem Soc 114: 5430–5432, 1992. doi: 10.1021/ja00039a068. [DOI] [Google Scholar]
- 17.Inoue S, Kawanishi S. Oxidative DNA damage induced by simultaneous generation of nitric oxide and superoxide. FEBS Lett 371: 86–88, 1995. doi: 10.1016/0014-5793(95)00873-8. [DOI] [PubMed] [Google Scholar]
- 18.Zhang M-Z, Yao B, McKanna JA, Harris RC. Cross talk between the intrarenal dopaminergic and cyclooxygenase-2 systems. Am J Physiol Renal Physiol 288: F840–F845, 2005. doi: 10.1152/ajprenal.00240.2004. [DOI] [PubMed] [Google Scholar]
- 19.Maruyama W, Kato Y, Yamamoto T, Oh-hashi K, Hashizume Y, Naoi M. Peroxynitrite induces neuronal cell death in aging and age-associated disorders: a review. J Am Aging Assoc 24: 11–18, 2001. doi: 10.1007/s11357-001-0002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torreilles F, Salman-Tabcheh S, Guérin M-C, Torreilles J. Neurodegenerative disorders: the role of peroxynitrite. Brain Res Rev 30: 153–163, 1999. doi: 10.1016/S0165-0173(99)00014-4. [DOI] [PubMed] [Google Scholar]
- 21.Forman LJ, Liu P, Nagele RG, Yin K, Wong PY-K. Augmentation of nitric oxide, superoxide, and peroxynitrite production during cerebral ischemia and reperfusion in the rat. Neurochem Res 23: 141–148, 1998. doi: 10.1023/A:1022468522564. [DOI] [PubMed] [Google Scholar]
- 22.GüRsoy-ÖZdemir Y, Bolay H, Saribaş O, Dalkara T. Role of endothelial nitric oxide generation and peroxynitrite formation in reperfusion injury after focal cerebral ischemia. Stroke 31: 1974–1980, 2000. doi: 10.1161/01.STR.31.8.1974. [DOI] [PubMed] [Google Scholar]
- 23.Alvarez B, Radi R. Peroxynitrite reactivity with amino acids and proteins. Amino Acids 25: 295–311, 2003. doi: 10.1007/s00726-003-0018-8. [DOI] [PubMed] [Google Scholar]
- 24.Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med 33: 1451–1464, 2002. doi: 10.1016/S0891-5849(02)01111-5. [DOI] [PubMed] [Google Scholar]
- 25.Kohutiar M, Eckhardt A, Miksik I, Santorova P, Wilhelm J. Proteomic analysis of peroxynitrite-induced protein nitration in isolated beef heart mitochondria. Physiol Res 67: 239–250, 2018. doi: 10.33549/physiolres.933608. [DOI] [PubMed] [Google Scholar]
- 26.Singh IN, Sullivan PG, Hall ED. Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. J Neurosci Res 85: 2216–2223, 2007. doi: 10.1002/jnr.21360. [DOI] [PubMed] [Google Scholar]
- 27.Xiong Y, Singh IN, Hall ED. Tempol protection of spinal cord mitochondria from peroxynitrite-induced oxidative damage. Free Radic Res 43: 604–612, 2009. doi: 10.1080/10715760902977432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viñas JL, Sola A, Hotter G. Mitochondrial NOS upregulation during renal I/R causes apoptosis in a peroxynitrite-dependent manner. Kidney Int 69: 1403–1409, 2006. doi: 10.1038/sj.ki.5000361. [DOI] [PubMed] [Google Scholar]
- 29.Zenebe WJ, Nazarewicz RR, Parihar MS, Ghafourifar P. Hypoxia/reoxygenation of isolated rat heart mitochondria causes cytochrome c release and oxidative stress; evidence for involvement of mitochondrial nitric oxide synthase. J Mol Cell Cardiol 43: 411–419, 2007. doi: 10.1016/j.yjmcc.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hunt JA, Lee J, Groves JT. Amphiphilic peroxynitrite decomposition catalysts in liposomal assemblies. Chem Biol 4: 845–858, 1997. doi: 10.1016/S1074-5521(97)90117-4. [DOI] [PubMed] [Google Scholar]
- 31.Misko TP, Highkin MK, Veenhuizen AW, Manning PT, Stern MK, Currie MG, Salvemini D. Characterization of the cytoprotective action of peroxynitrite decomposition catalysts. J Biol Chem 273: 15646–15653, 1998. doi: 10.1074/jbc.273.25.15646. [DOI] [PubMed] [Google Scholar]
- 32.Sakamuri S, Sperling JA, Sure VN, Dholakia MH, Peterson NR, Rutkai I, Mahalingam PS, Satou R, Katakam PV. Measurement of respiratory function in isolated cardiac mitochondria using Seahorse XFe24 Analyzer: applications for aging research. Geroscience 40: 347–356, 2018. doi: 10.1007/s11357-018-0021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sperling JA, Sakamuri S, Albuck AL, Sure VN, Evans WR, Peterson NR, Rutkai I, Mostany R, Satou R, Katakam PV. Measuring respiration in isolated murine brain mitochondria: implications for mechanistic stroke studies. Neuromolecular Med 21: 493–504, 2019. doi: 10.1007/s12017-019-08552-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sure VN, Sakamuri SS, Sperling JA, Evans WR, Merdzo I, Mostany R, Murfee WL, Busija DW, Katakam PV. A novel high-throughput assay for respiration in isolated brain microvessels reveals impaired mitochondrial function in the aged mice. Geroscience 40: 365–375, 2018. doi: 10.1007/s11357-018-0037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brookes PS, Land JM, Clark JB, Heales SJ. Peroxynitrite and brain mitochondria: evidence for increased proton leak. J Neurochem 70: 2195–2202, 2002. doi: 10.1046/j.1471-4159.1998.70052195.x. [DOI] [PubMed] [Google Scholar]
- 36.Luo Z, Zhao Q, Liu J, Liao J, Peng R, Xi Y, Diwu Z. Fluorescent real-time quantitative measurements of intracellular peroxynitrite generation and inhibition. Anal Biochem 520: 44–48, 2017. doi: 10.1016/j.ab.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Lacza Z, Horn TF, Snipes JA, Zhang J, Roychowdhury S, Horvath EM, Figueroa JP, Kollai M, Szabo C, Busija DW. Lack of mitochondrial nitric oxide production in the mouse brain. J Neurochem 90: 942–951, 2004. doi: 10.1111/j.1471-4159.2004.02553.x. [DOI] [PubMed] [Google Scholar]
- 38.Navarro A, Boveris A. Brain mitochondrial dysfunction and oxidative damage in Parkinson's disease. J Bioenerg Biomembr 41: 517–521, 2009. doi: 10.1007/s10863-009-9250-6. [DOI] [PubMed] [Google Scholar]
- 39.Zaobornyj T, Ghafourifar P. Strategic localization of heart mitochondrial NOS: a review of the evidence. Am J Physiol Heart Circ Physiol 303: H1283–H1293, 2012. doi: 10.1152/ajpheart.00674.2011. [DOI] [PubMed] [Google Scholar]
- 40.Galea E, Regunathan S, Eliopoulos V, Feinstein DL, Reis DJ. Inhibition of mammalian nitric oxide synthases by agmatine, an endogenous polyamine formed by decarboxylation of arginine. Biochem J 316: 247–249, 1996. doi: 10.1042/bj3160247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 320: 1050–1054, 2008. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol 10: 721–732, 2009. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 43.Brown GG, Jacobus J, McKenna B. Structural imaging for addiction medicine: from neurostructure to neuroplasticity. Prog Brain Res 224: 105–127, 2016. doi: 10.1016/bs.pbr.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brorson JR, Schumacker PT, Zhang H. Nitric oxide acutely inhibits neuronal energy production. J Neurosci 19: 147–158, 1999. doi: 10.1523/JNEUROSCI.19-01-00147.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakamuri SS, Sperling JA, Evans WR, Dholakia MH, Albuck AL, Sure VN, Satou R, Mostany R, Katakam PV. Nitric oxide synthase inhibitors negatively regulate respiration in isolated rodent cardiac and brain mitochondria. Am J Physiol Heart Circ Physiol 318: H295–H300, 2020. doi: 10.1152/ajpheart.00720.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Förstermann U. Endothelial NO synthase as a source of NO and superoxide. Eur J Clin Pharmacol 62: 5–12, 2006. doi: 10.1007/s00228-005-0006-x. [DOI] [Google Scholar]
- 47.Zou MH, Cohen R, Ullrich V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium 11: 89–97, 2004. doi: 10.1080/10623320490482619. [DOI] [PubMed] [Google Scholar]
- 48.Cassuto J, Dou H, Czikora I, Szabo A, Patel VS, Kamath V, Belin de Chantemele E, Feher A, Romero MJ, Bagi Z. Peroxynitrite disrupts endothelial caveolae leading to eNOS uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients. Diabetes 63: 1381–1393, 2014. doi: 10.2337/db13-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kadlec AO, Beyer AM, Ait-Aissa K, Gutterman DD. Mitochondrial signaling in the vascular endothelium: beyond reactive oxygen species. Basic Res Cardiol 111: 26, 2016. doi: 10.1007/s00395-016-0546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katakam PV, Gordon AO, Sure VN, Rutkai I, Busija DW. Diversity of mitochondria-dependent dilator mechanisms in vascular smooth muscle of cerebral arteries from normal and insulin-resistant rats. Am J Physiol Heart Circ Physiol 307: H493–H503, 2014. doi: 10.1152/ajpheart.00091.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulz JB, Huang PL, Matthews RT, Passov D, Fishman MC, Beal MF. Striatal malonate lesions are attenuated in neuronal nitric oxide synthase knockout mice. J Neurochem 67: 430–433, 1996. doi: 10.1046/j.1471-4159.1996.67010430.x. [DOI] [PubMed] [Google Scholar]
- 52.Kohutiar M, Eckhardt A, Miksik I, Santorova P, Wilhelm J. Proteomic analysis of peroxynitrite-induced protein nitration in isolated beef heart mitochondria. Physiol Res 67: 239–250, 2018. doi: 10.33549/physiolres.933608. [DOI] [PubMed] [Google Scholar]
- 53.Liu B, Tewari AK, Zhang L, Green-Church KB, Zweier JL, Chen Y-R, He G. Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: mitochondria as the major target. Biochim Biophys Acta 1794: 476–485, 2009. doi: 10.1016/j.bbapap.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferrer-Sueta G, Radi R. Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem Biol 4: 161–177, 2009. doi: 10.1021/cb800279q. [DOI] [PubMed] [Google Scholar]
- 55.Zielonka J, Sikora A, Hardy M, Joseph J, Dranka BP, Kalyanaraman B. Boronate probes as diagnostic tools for real time monitoring of peroxynitrite and hydroperoxides. Chem Res Toxicol 25: 1793–1799, 2012. doi: 10.1021/tx300164j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rios N, Radi R, Kalyanaraman B, Zielonka J. Tracking isotopically labeled oxidants using boronate-based redox probes. J Biol Chem 295: 6665–6676, 2020. doi: 10.1074/jbc.RA120.013402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasternack RF, Skowronek WR, Jr.. Catalysis of the disproportionation of superoxide by metalloporphyrins. J Inorg Biochem 11: 261–267, 1979. doi: 10.1016/S0162-0134(00)80022-7. [DOI] [PubMed] [Google Scholar]
- 58.Stern MK, Jensen MP, Kramer K. Peroxynitrite decomposition catalysts. J Am Chem Soc 118: 8735–8736, 1996. doi: 10.1021/ja961279f. [DOI] [Google Scholar]