

Abstract
Peripherally or centrally administered TNF-α elicits a prolonged sympathetically mediated pressor response, but the underlying molecular mechanisms are unknown. Activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) in cardiovascular regions of the brain has recently been recognized as a key mediator of sympathetic excitation, and ERK1/2 signaling is induced by activation of epidermal growth factor receptor (EGFR) tyrosine kinase activity. The present study examined the role of EGFR and ERK1/2 signaling in the sympathetic response to TNF-α. In urethane-anesthetized rats, intracarotid artery injection of TNF-α increased phosphorylation of EGFR and ERK1/2 in the subfornical organ (SFO) and the hypothalamic paraventricular nucleus (PVN); upregulated the gene expression of excitatory mediators in SFO and PVN; and increased blood pressure (BP), heart rate (HR), and renal sympathetic nerve activity (RSNA). A continuous intracerebroventricular infusion of the selective EGFR tyrosine kinase inhibitor AG1478 or the ERK1/2 inhibitor PD98059 significantly attenuated these responses. Bilateral PVN microinjections of TNF-α also increased phosphorylated ERK1/2 and the gene expression of excitatory mediators in PVN, along with increases in BP, HR, and RSNA, and these responses were substantially reduced by prior bilateral PVN microinjections of AG1478. These results identify activation of EGFR in cardiovascular regulatory regions of the forebrain as an important molecular mediator of TNF-α-driven sympatho-excitatory responses and suggest that EGFR activation of the ERK1/2 signaling pathway plays an essential role. These mechanisms likely contribute to sympathetic excitation in pathophysiological states like heart failure and hypertension, in which circulating and brain TNF-α levels are increased.
NEW & NOTEWORTHY Proinflammatory cytokines contribute to the augmented sympathetic nerve activity in hypertension and heart failure, but the central mechanisms involved are largely unknown. The present study reveals that TNF-α transactivates EGFR in the subfornical organ and the hypothalamic paraventricular nucleus to initiate ERK1/2 signaling, upregulate the gene expression of excitatory mediators, and increase sympathetic nerve activity. These findings identify EGFR as a gateway to sympathetic excitation and a potential target for intervention in cardiovascular disease states.
Keywords: brain, hypothalamic paraventricular nucleus, proinflammatory cytokines, renin-angiotensin system, subfornical organ, sympathetic nervous system
INTRODUCTION
Inflammation and immune activation have been implicated in the pathophysiology of hypertension and heart failure (1–4). Recent animal studies have revealed that inflammation augments the sympathetic nervous system activity that contributes to the development of these disorders (5, 6). Systemically and centrally administered proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β act upon the brain to elicit dramatic sympathetic and hemodynamic responses (7). In a rat model of heart failure induced by myocardial infarction, TNF-α receptor 1, the dominant receptor mediating TNF-α-elicited adverse effects, is upregulated in cardiovascular/autonomic brain regions including the hypothalamic paraventricular nucleus (PVN) and the subfornical organ (SFO) (8). In that experimental setting, interventions that block the influence of TNF-α in these brain regions significantly attenuate the augmented sympathetic outflow (8, 9). However, the cellular mechanisms by which TNF-α induces sympathetic activation and pressor responses remain unknown.
We have previously demonstrated that TNF-α promotes the activation of p44/42 mitogen-activated protein kinase (MAPK), also known as extracellular signal-regulated kinases 1 and 2 (ERK1/2), in the PVN (10). ERK1/2 activation evokes a series of downstream signaling cascades to amplify the production of excitatory neurochemical mediators including renin-angiotensin system (RAS) components (11), inflammatory mediators (12), and reactive oxygen species (13, 14). Moreover, we found that ERK1/2 signaling contributes substantially to the augmented sympathetic drive in heart failure (12, 15). These findings suggest that MAPK signaling via the ERK1/2 pathway may be an important mechanism underlying TNF-α-induced sympathetic excitation in heart failure, but how TNF-α initiates this process is unknown.
The present study explored the potential involvement of the epidermal growth factor receptor (EGFR). EGFR is a receptor protein tyrosine kinase composed of an extracellular ligand-binding domain, a transmembrane lipophilic domain, and an intracellular tyrosine kinase domain (16). Activation of EGFR regulates cell growth, differentiation, proliferation, and migration (17–19). Notably, the Raf/Ras/ERK MAPK signaling pathway is among the downstream molecular events triggered by EGFR tyrosine kinase (20). EGFR is activated by endogenous ligands of the epidermal growth factor family such as epidermal growth factor (EGF) and transforming growth factor α (TGF-α) but may also be transactivated by ligands for G-protein-coupled receptors (21), including angiotensin II (Ang II), and by cytokines, including TNF-α (22–24).
We hypothesized that transactivation of EGFR might be an important mechanism by which TNF-α upregulates ERK1/2 expression in cardiovascular regions of the brain. If true, then inhibition of EGFR tyrosine kinase activity in these brain regions would be expected to reduce the TNF-α-induced increases in ERK1/2 activity and mitigate the sympathetic and hemodynamic responses. To test this hypothesis, we examined the effect of the selective EGFR tyrosine kinase inhibitor AG1478 (25) on the central molecular and pressor responses to blood-borne TNF-α and to direct microinjection of TNF-α into brain tissue.
METHODS
Animals
Experiments were conducted on 10–12-wk-old male Sprague-Dawley rats (300–350 g) purchased from Envigo/Harlan (Indianapolis, IN). Animals were maintained in temperature-controlled (23 ± 2°C) and light-controlled (12:12-h light-dark cycle) animal housing facility at the University of Iowa Health Sciences Campus. Water and rodent chow were provided ad libitum. All experimental procedures were approved by the University of Iowa Institutional Animal Care and Use Committee. The experimental protocols were conducted in accordance with the National Institutes of Health, Guide for the Care and Use of Laboratory Animals.
Experimental Protocols
Urethane-anesthetized rats (n = 108) were studied in three experimental protocols:
Intracarotid artery (ICA) bolus injection of TNF-α (200 ng in 8 µL over 10 s) 10 min after initiating a 4-h intracerebroventricular (ICV) infusion (20 µL/h) of the EGFR tyrosine kinase inhibitor AG1478 (15.75 ng/µL, n = 18) or vehicle (VEH, n = 18). ICA injection of VEH in ICV VEH-treated animals (n = 18) served as control. Six rats in each group underwent recordings of arterial blood pressure (BP), heart rate (HR), and renal sympathetic nerve activity (RSNA). At the conclusion of the experimental protocol, 4–5 h after TNF-α injection, the rats were euthanized to collect brain tissues for molecular or immunohistochemical studies.
ICA bolus injection of TNF-α (200 ng in 8 µL over 10 s) 10 min after initiating a 4-h ICV infusion (20 µL/hr) of the MEK inhibitor PD98059 (10 ng/µL, n = 6) or VEH (n = 6). ICA injection of VEH in ICV VEH-treated animals (n = 6) served as control. BP, HR, and RSNA were recorded. The rats were euthanized 4–5 h after the TNF-α injection to collect brain tissues for molecular studies.
Bilateral PVN microinjection of TNF-α (25 ng/side) 10 min after bilateral PVN microinjection of the EGFR inhibitor AG1478 (100 ng/side, n = 12) or VEH (n = 12). PVN microinjection of VEH served as control (n = 12). Six rats in each group underwent recordings of BP, HR, and RSNA. Four-five hours after TNF-α microinjection, Pontamine sky blue (50 nL/side) was microinjected into the PVN, and rats were euthanized to collect brain tissues for molecular studies.
Drug Administered
TNF-α was purchased from Millipore (Billerica, MA) and dissolved in PBS for ICA injection and in artificial cerebrospinal fluid (aCSF) for PVN microinjection. The EGFR inhibitor AG1478 (25) was purchased from Sigma (St. Louis, MO) and the MEK1/2 inhibitor of the ERK1/2 MAPK signaling pathway PD98059 (26) was purchased from Selleckchem (Houston, TX). These drugs were first dissolved in dimethyl sulfoxide (DMSO) and then diluted in aCSF to make a 5% final DMSO concentration. The ICA dose of TNF-α was previously shown to elicit a robust sympathetic response (7), and the dose of TNF-α microinjected into PVN had elicited a similar pressor response when microinjected into SFO (27). The ICV doses of AG1478 were previously shown to effectively inhibit the effects of systemically administered aldosterone (28). The ICV dose of PD98059 significantly reduced RSNA, BP, and HR in rats with ischemia-induced heart failure in our previous study (12).
Surgical Preparations
General.
Rats were anesthetized with urethane (1.5 g/kg i.p.). Supplemental doses of urethane (0.1–0.3 g/kg i.p. or i.v.) were given as needed. The depth of anesthesia was periodically assessed by testing nociceptive reflex responses. PE-50 catheters were inserted in the left femoral artery for blood pressure (BP) recording and in the left femoral vein for intravenous (IV) injection. Body temperature was maintained at 37 ± 1°C with a temperature controller (model K-100; Baxter Healthcare, Valencia, CA).
Renal nerve isolation.
The left kidney was exposed via a flank incision and a branch of renal nerve was dissected from surrounding tissues. The renal nerve was placed on bipolar silver wire recording electrodes to record renal sympathetic nerve activity (RSNA). The nerve and electrodes were covered with Kwik-Cast silicon sealant (WPI, Inc., Sarasota, FL) before the incisions were sutured.
Intracarotid artery (ICA) injection.
A previous study (29) demonstrated that centrally directed intracarotid artery injections in the rat predominantly target the forebrain, and therefore provide a means of delivering drugs to that region by the natural blood-borne route. In our former studies, we have used this administration approach to examine the central effect of blood-borne substances on forebrain neuronal firing and sympathetic nerve activity in the rats (7, 30). In brief, a 2-cm midline incision was made on the ventral surface of the neck. After the omohyoideus muscle was retracted, the left carotid artery was isolated. A rostrally directed PE-50 cannula was inserted into the left common carotid artery, with the tip placed in the bifurcation.
Intracerebroventricular (ICV) injection.
The animal was fixed in a stereotaxic frame and a 29-gauge stainless steel guide cannula was advanced to a final position 2 mm above the left lateral cerebral ventricle, using the following coordinates with reference to bregma: anteroposterior, −1.0 mm; dorsoventral, −2.5 mm; mediolateral, −1.5 mm (left). ICV injections were made via a 33-gauge injection cannula inserted into the guide cannula and extended 2 mm beyond its tip. The injection cannula was connected to a 10-µL microsyringe driven by a micropump (Harvard Apparatus, Holliston, MA). ICV injections were verified by 2% Pontamine sky blue (2 µL) that was injected in the same locations at the end of experiments.
Bilateral PVN microinjection.
As previously described (31), a 29-gauge guide cannula was placed 1.8 mm posterior to bregma, 0.4 mm left to midline for left PVN microinjection. Another 29-gauge guide cannula was placed 1.4 mm right to midline but angled 10° toward midline for right PVN microinjection. Both cannula tips were advanced to a final position 5.8 mm ventral to the cranial surface. Bilateral PVN microinjections were made via 35-gauge (128 μm outer diameter and 51.2 μm inner diameter) injection cannulas inserted into the guide cannulas and extended 2 mm beyond the tips of the guide cannulas. Bilateral PVN microinjections (0.2 µL over 10 s) were made simultaneously using 1 μL Hamilton microsyringes. The microinjection sites were verified by visualization of 2% Pontamine sky blue (100 nL) injected in the same locations at the end of experiments.
Hemodynamic and Sympathetic Nerve Recording
The recording of BP, HR, and RSNA has been described in detail previously (15). Briefly, a pressure transducer was connected to the femoral artery catheter to transfer the blood pressure signals to a Gould TA240S chart recorder (Gould Instruments, Valley View, OH).
The raw RSNA was amplified (model P511; Grass Instruments, Quincy, MA) and then was rectified and integrated by a Paynter filter (20-ms time constant; BAK Electronics, Germantown, MD). The noise level was determined at the end of experiment after ganglionic blockade with hexamethonium (30 mg/kg i.v.). The net value of RSNA was calculated by subtracting the background noise from the actual recorded value during the experiment.
The BP signal and the rectified and integrated RSNA voltage were fed into an online data acquisition system consisting of a Cambridge Electronics Design (CED, Cambridge, UK) 1401 Plus laboratory interface coupled with a personal computer. HR was derived from the arterial pressure waveform. Digitized data were stored for subsequent off-line analysis.
Molecular and Immunofluorescent Studies
Tissue preparations.
For molecular studies, rats were decapitated and the brains were immediately removed, frozen in liquid nitrogen, and stored at −80°C. To obtain tissues for real time PCR and Western blot measurements, the frozen brain was subsequently cut into 300-μm coronal sections and the target tissues including SFO, PVN, and cortex were obtained using a punch device (inner diameter 1.5 mm, Stoelting, Wood Dale, IL). The punched tissues were homogenized in cell lysis buffer (Cell Signaling Technology, Beverly, MA) to extract protein for Western Blot assay or in TRI reagent (Molecular Research Center, Cincinnati, OH) to extract total RNA for real-time PCR.
Real-time PCR.
The mRNA levels of angiotensin-converting enzyme (ACE), angiotensin II type 1a receptor (AT1aR), cyclooxygenase (COX)-2, nuclear factor-κB (NF-κB) p65 subunit, and the NADPH oxidase subunits p47phox and gp91phox were measured with real-time PCR following reverse transcription of total RNA. The sequences for primers and probes used are shown in Table 1. Real-time PCR was performed using the ABI prism 7700 Sequence Detection System (Applied Biosystems). GAPDH mRNA was quantified as an internal control for each sample. The value for each sample was normalized to housekeeping gene GAPDH (SYBR), and relative quantification was calculated using the cycle threshold [2−(ΔΔCt)] method (32). Quantitative mRNA data are expressed as a fold difference relative to the vehicle control.
Table 1.
Sequences for primers
| Gene | Sequences |
|---|---|
| ACE | |
| Forward primer:Reverse primer: | 5′-GTGTTGTGGAACGAATACGC-3′5′-CCTTCTTTATGATCCGCTTGA-3′ |
| AT1aR | |
| Forward primer:Reverse primer: | 5′-ACTCACAGCAACCCTCCAAG-3′5′-ATCACCACCAAGCTGTTTCC-3′ |
| COX-2 | |
| Forward primer:Reverse primer: | 5′-GGCACAAATATGATGTTCGCA-3′5′-CCTCGCTTCTGATCTGTCTTGA-3′ |
| NF-κB p65 | |
| Forward primer:Reverse primer: | 5′-GCATCCACAGCTTCCAGAAC-3′5′-GCACGGTTATCAAAAATCGG-3′ |
| p47phox | |
| Forward primer:Reverse primer: | 5′-AGGTTGGGTCCCTGCATCCTATTT-3′5′-TGGTTACATACGGTTCACCTGCGT-3′ |
| gp91phox | |
| Forward primer:Reverse primer: | 5′-CGGAATCTCCTCTCCTTCCT-3′5′-GCATTCACACACCACTCCAC-3′ |
| GAPDH | |
| Forward primer:Reverse primer: | 5′-AAGGTCATCCCAGAGCTGAA-3′5′-ATGTAGGCCATGAGGTCCAC-3′ |
ACE, angiotensin-converting enzyme; AT1aR, angiotensin II type 1a receptor; COX-2, cyclooxygenase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NF-κB p65, nuclear factor kappa B p65 subunit; p47phox, gp91phox, NADPH oxidase subunits.
Western blot.
The levels of phosphorylated(p-) p44/42 MAPK (p-ERK1/2) were measured using monoclonal antibodies (1:1,000; Cell Signaling Technology, Inc., Beverly, MA) to p-p44/42 MAPK (p-ERK1/2, Cat. No. 4370). Total ERK1/2 was detected using monoclonal antibodies (1:1,000; Cell Signaling Technology, Inc., Beverly, MA) to p44/42 MAPK (ERK1/2, Cat. No. 4695). c-Fos expression in the PVN was detected using rabbit mAb against c-Fos (Cat. No. 2250, 1:500, Cell Signaling Technology). The secondary antibodies were goat anti-rabbit IgG-HRP (SC-2004, 1:5,000, Santa Cruz). Immunoblots were visualized with an enhanced chemiluminescence reagent. Band intensities were quantified with Image Lab analysis software (Bio-Rad, Hercules, CA). The content of p-p44/42 (p-ERK1/2) were normalized to total p44/42 (ERK1/2). The protein levels of c-Fos were normalized by the total β-actin detected by rabbit monoclonal antibody (Cat. No. 4970, 1:1,000; Cell Signaling Technology).
Immunofluorescence.
Rats were transcardially perfused with 4% paraformaldehyde. Brains were removed and embedded in optimal cutting temperature compound and rapidly frozen in acetone chilled by dry ice. Coronal forebrain sections (16 µm) of target tissues were made using a cryostat and collected for immunofluorescent studies to determine the expression of phosphorylated (p-) EGFR in the SFO and PVN. Immunofluorescent staining of phosphorylated EGFR in the SFO and PVN was examined using primary antibodies, rabbit anti-EGFR (phospho Y1068) antibody (ab182618, 1:200, Abcam, MA) followed by secondary antibodies Alex Fluor 488 goat anti-rabbit IgG (A-11070, 1:200, Invitrogen). The labeling of neurons was performed using the mouse monoclonal antibody to neuronal marker NeuN (MAB377, 1:300, Millipore) followed by secondary antibodies Alex Fluor 568 goat anti-mouse IgG (A-11003, 1:200, Invitrogen). Immunofluorescent staining was visualized with a confocal laser-scanning microscope (Zeiss LSM 710, Carl Zeiss, Inc).
Statistical Analysis
The electrophysiological and hemodynamic data were analyzed with CED Spike2 software. Mean BP (MBP, mmHg), HR (bpm), and integrated RSNA (mV) were averaged over 5-min intervals after ICA injection or PVN microinjection of TNF-α and compared with baseline values averaged over 5-min intervals immediately before each intervention. Changes in integrated RSNA are reported as a percentage of baseline control.
All values are expressed as the means ± SEM. The significance of differences among groups was analyzed by one-way and two-way repeated-measure ANOVA followed by post hoc Fisher’s test. Student’s t test was used to determine statistical significance between paired data for a single comparison. P < 0.05 was considered to indicate statistical significance.
RESULTS
Effects of ICV EGFR and ERK1/2 Inhibitors on Responses to ICA TNF-α
ICA TNF-α activates EGFR and ERK1/2 in SFO and PVN.
ICA injection of TNF-α in rats receiving ICV VEH induced a substantial increase in immunoreactivity (fluorescent units) for p-EGFR in the SFO and PVN (Fig. 1A), indicating that EGFR is activated by blood-borne TNF-α in cardiovascular/autonomic regions of the brain. In SFO, the increased expression of p-EGFR was evenly distributed throughout the nucleus. In PVN, p-EGFR was expressed in all four commonly recognized subdivisions—the dorsal parvocellular (PVN-dp), the ventrolateral parvocellular (PVN-vlp), the medial parvocellular (PVN-mp), and the posterior magnocellular (PVN-pm), but with more intense staining in PVN-vlp, a region containing presympathetic neurons (Fig. 1B). The increased p-EGFR immunoreactivity was significantly attenuated in the SFO and PVN of the rats treated with ICV AG1478 (Fig. 1, A and B). The confocal immunofluorescent images revealed that the p-EGFR was expressed in neurons in the SFO and PVN (Fig. 1C).
Figure 1.
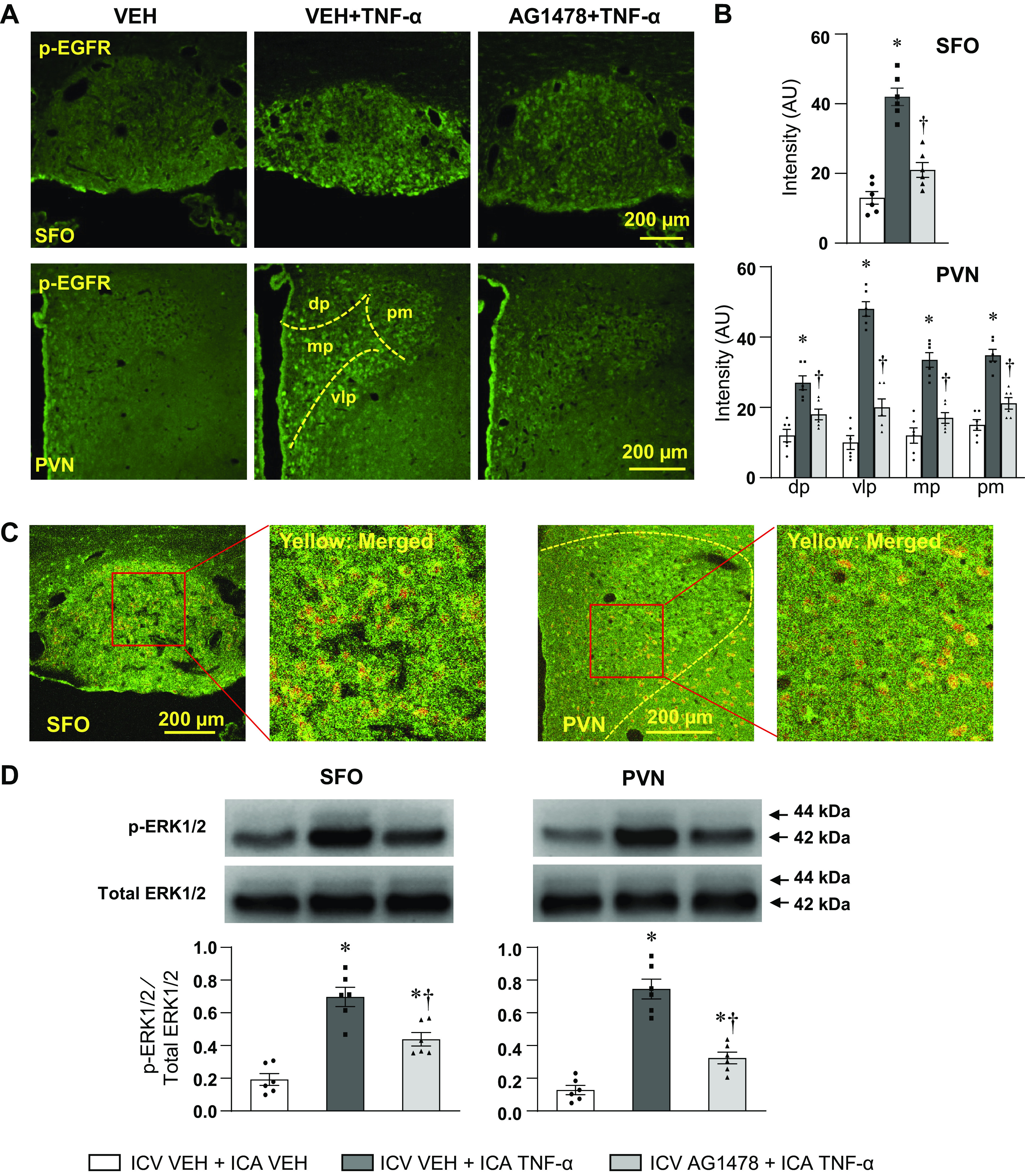
Laser confocal images (A) showing immunofluorescent staining (green) of phosphorylated-EGFR (p-EGFR) in the SFO (top) and PVN (bottom) in rats treated with ICA VEH or ICA TNF-α while receiving a continuous ICV infusion of VEH or AG1478. B: bar graphs showing the fluorescent intensity (in arbitrary units, AU) for p-EGFR in the SFO and in four subdivisions of the PVN: dp, dorsal parvocellular; mp, medial parvocellular; vlp, ventrolateral parvocellular; pm, posterior magnocellular. C: representative confocal images of the SFO (left) and PVN (right) showing the double staining of p-EGFR (green) and neuron marker NeuN (red). The merged images (yellow) show the expression of p-EGFR in neurons. D: Western blot analysis showing the protein expression of phosphorylated-ERK1/2 (p-ERK1/2) in PVN and SFO in rats treated with ICA VEH or ICA TNF-α while treated with ICV VEH or AG1478. Bar graphs show the group data and representative Western bands are shown above each bar. p-ERK1/2 is normalized to total ERK1/2. Values are expressed as mean ± SE (in B and D, n = 6 for each group). *P < 0.05, vs. ICV VEH + ICA VEH; †P < 0.05, ICV AG1478 + ICA TNF-α vs. ICV VEH + ICA TNF-α. EGFR, epidermal growth factor receptor; ICA, intracarotid artery; PVN, paraventricular nucleus; SFO, subfornical organ; VEH, vehicle.
The ratio of phosphorylated to total ERK1/2 protein in the PVN and SFO measured by Western blot was also markedly increased in the rats treated with ICA TNF-α, compared with the control rats treated with ICA VEH, and was significantly lower in the rats treated with ICV AG1478 (Fig. 1D). Total ERK1/2 was unaffected.
Inhibition of EGFR or ERK1/2 signaling blunts the TNF-α-induced pressor response.
In rats receiving an ICV infusion of VEH (aCSF with 5% DMSO), ICA injection of TNF-α (Fig. 2, A and C; Fig. 3, A and C) elicited significant increases in MBP, HR, and RSNA. These excitatory responses began within 20–30 min and persisted for the duration of the experiment, but the maximum increases in MBP (16.2 ± 1.9 mmHg), HR (51.1 ± 4.9 bpm), and RSNA (67.8 ± 7.2% changes) occurred 80–120 min after TNF-α injection. Treatment with a continuous ICV infusion of AG1478 (Fig. 2, B and C) significantly attenuated these TNF-α-induced increases in MBP (9.5 ± 1.1), HR (31.8 ± 4.3), and RSNA (38.8 ± 5.4% change). A continuous infusion of the MEK inhibitor PD98059 caused similar reductions in the maximal responses of MBP (19.3 ± 2.4 to 10.3 ± 2.3), HR (56.0 ± 8.6 to 31.2 ± 7.2), and RSNA (80.1 ± 9.1 to 43.5 ± 8.7) to TNF-α (Fig. 3, A–C).
Figure 2.
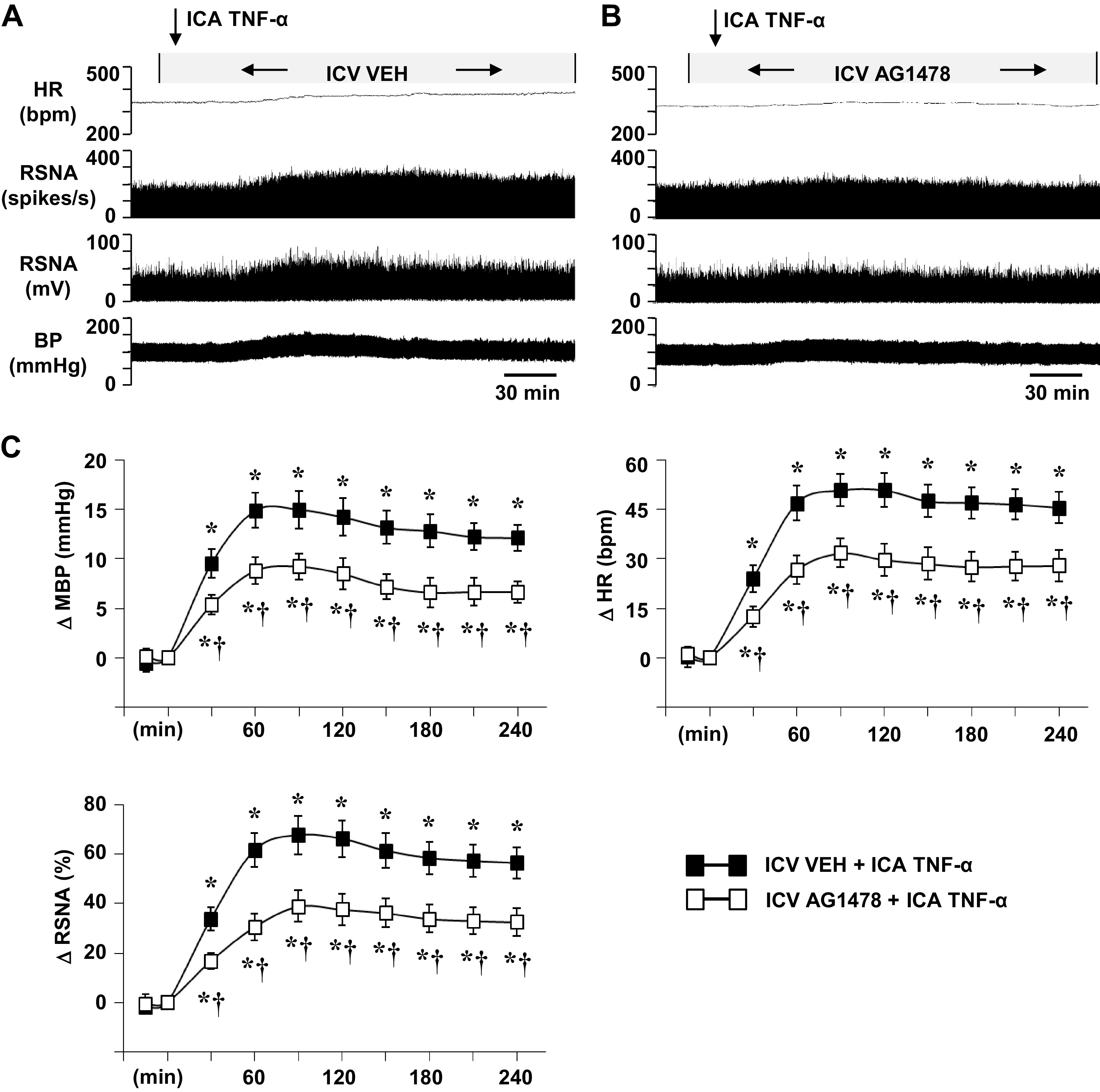
Representative tracings (A, B) and grouped data (C) showing the effects of intracarotid artery (ICA) injection of tumor necrosis factor (TNF)-α on blood pressure (BP, mmHg), heart rate (HR, bpm), and renal sympathetic nerve activity (RSNA), windowed (spikes/s) and integrated (mV), in rats receiving a continuous intracerebroventricular (ICV) infusion of vehicle (VEH) (A) or AG1478 (B). Arrows indicate the timing of the injections. Scale bar: 30 min. MBP, mean blood pressure; ΔRSNA (%), percent change from baseline in integrated RSNA. All values are expressed as the means ± SE (in C, n = 6 for each group). *P < 0.05 compared with baseline. †P < 0.05 compared with ICA TNF-α in ICV VEH-pretreated rats.
Figure 3.
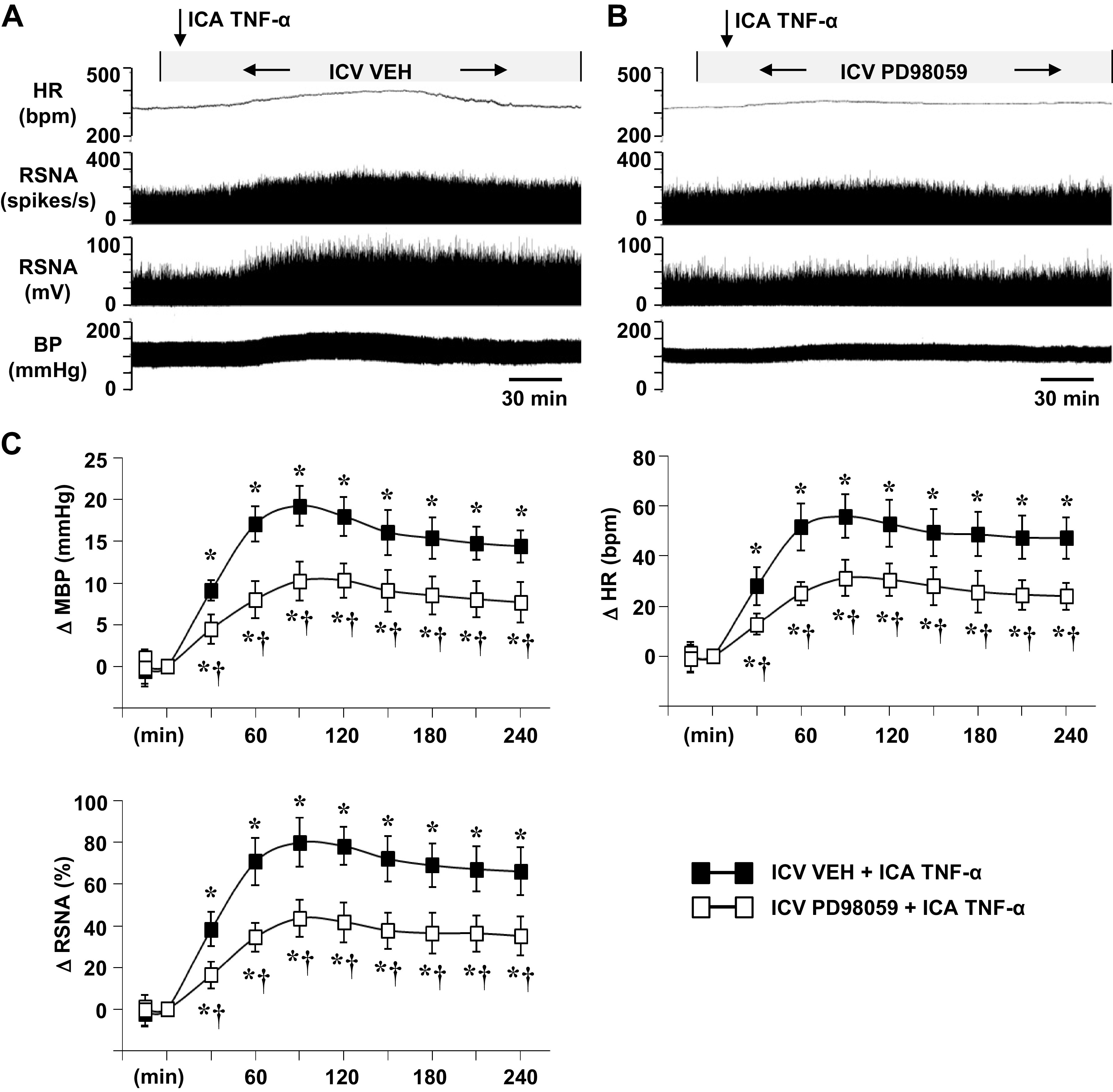
Representative tracings (A, B) and grouped data (C) showing the effects of intracarotid artery (ICA) injection of tumor necrosis factor (TNF)-α on blood pressure (BP, mmHg), heart rate (HR, bpm), and renal sympathetic nerve activity (RSNA), windowed (spikes/s) and integrated (mV), in rats receiving a continuous intracerebroventricular (ICV) infusion of vehicle (VEH) (A) or PD98059 (B). Arrows indicate the timing of the injections. Scale bar: 30 min. MBP, mean blood pressure; ΔRSNA (%), percent change from baseline in integrated RSNA. All values are expressed as the means ± SE (in C, n = 6 for each group). *P < 0.05 compared with baseline. †P < 0.05 compared with ICA TNF-α in ICV VEH-pretreated rats.
Neither inhibitor affected baseline MBP, HR, or RSNA. In rats treated with an ICV infusion of an equal volume of VEH (aCSF with 5% DMSO), ICA injections of VEH (PBS) had no obvious effects on baseline MBP (90.4 ± 2.4 mmHg), HR (328 ± 11 bpm), or integrated RSNA (12.8 ± 3.6 mV).
Inhibition of EGFR or ERK1/2 signaling reduces TNF-α-induced upregulation of excitatory mediators in SFO and PVN.
In rats treated with an ICV infusion of VEH (aCSF with 5% DMSO), ICA injection of TNF-α, versus ICA VEH (PBS), significantly increased the mRNA levels (fold changes) of the RAS components ACE and AT1aR, the inflammatory mediators COX-2 and NF-κB p65 subunit, and the NADPH oxidase subunits p47phox and gp91phox in SFO and PVN (Figs. 4 and 5). In rats treated with ICV infusion of either AG1478 (Fig. 4) or PD98059 (Fig. 5), compared with rats treated with an ICV infusion of VEH, these increases were significantly attenuated. ICA injection of TNF-α had no effect on the mRNA levels of any of these excitatory neurochemical mediators in brain cortex (Figs. 4 and 5).
Figure 4.

Quantitative analysis by real-time PCR showing the effects of intracarotid artery (ICA) injection of tumor necrosis factor (TNF)-α on the mRNA expression of (A) angiotensin-converting enzyme (ACE), (B) angiotensin II type-1a receptor (AT1a-R), (C) cyclooxygenase (COX)-2, (D) NF-κB p65 subunit, NADPH oxidase subunits (E) p47phox and (F) gp91phox in the cortex, subfornical organ (SFO), and paraventricular nucleus of hypothalamus (PVN) in rats receiving a continuous ICV infusion of vehicle (VEH) or AG1478. Values are mean ± SE (n = 6 for each group) and expressed as a fold change relative to VEH control. *P < 0.05, vs. ICV VEH + ICA VEH; †P < 0.05, ICV AG1478 + ICA TNF-α vs. ICV VEH + ICA TNF-α.
Figure 5.

Quantitative analysis by real-time PCR showing the effects of intracarotid artery (ICA) injection of tumor necrosis factor (TNF)-α on the mRNA expression of (A) angiotensin-converting enzyme (ACE), (B) angiotensin II type-1a receptor (AT1a-R), (C) cyclooxygenase (COX)-2, (D) NF-κB p65 subunit, NADPH oxidase subunits (E) p47phox and (F) gp91phox in the cortex, subfornical organ (SFO), and paraventricular nucleus of hypothalamus (PVN) in rats receiving a continuous ICV infusion of vehicle (VEH) or PD98059. Values are mean ± SE (n = 6 for each group) and expressed as a fold change relative to VEH control. *P < 0.05, vs. ICV VEH + ICA VEH; †P < 0.05, ICV PD98059 + ICA TNF-α vs. ICV VEH + ICA TNF-α.
Effects of PVN Microinjection of AG1478 on Responses to PVN Microinjection of TNF-α
Inhibition of EGFR signaling blunts the TNF-α-induced pressor response.
Bilateral PVN microinjection of TNF-α (25 ng/side), like ICA injection of TNF-α, induced substantial increases in MBP, HR, and RSNA (Fig. 6, A and C). This pressor response began within 15–20 min after microinjection of TNF-α and persisted for the duration of the recording session. Although the MBP response to TNF-α reached its highest point at 45–80 min (14.1 ± 1.8 mmHg), the HR (45.1 ± 4.6 bpm) and RSNA (56.8 ± 5.8% change) increases peaked 90–120 min after the TNF-α microinjection. In rats pretreated with bilateral PVN microinjections of AG1478, TNF-α-induced increases in MBP (7.1 ± 1.2 mmHg), HR (22.0 ± 4.4 bpm), and RSNA (29.7 ± 5.6% changes) were significantly attenuated (Fig. 6, B and C). In control rats treated with an equal volume of VEH (aCSF) in the PVN, there was no significant change from baseline values of MBP (88.6 ± 2.2 mmHg), HR (324 ± 12 bpm), or integrated RSNA (11.2 ± 3.1 mV).
Figure 6.
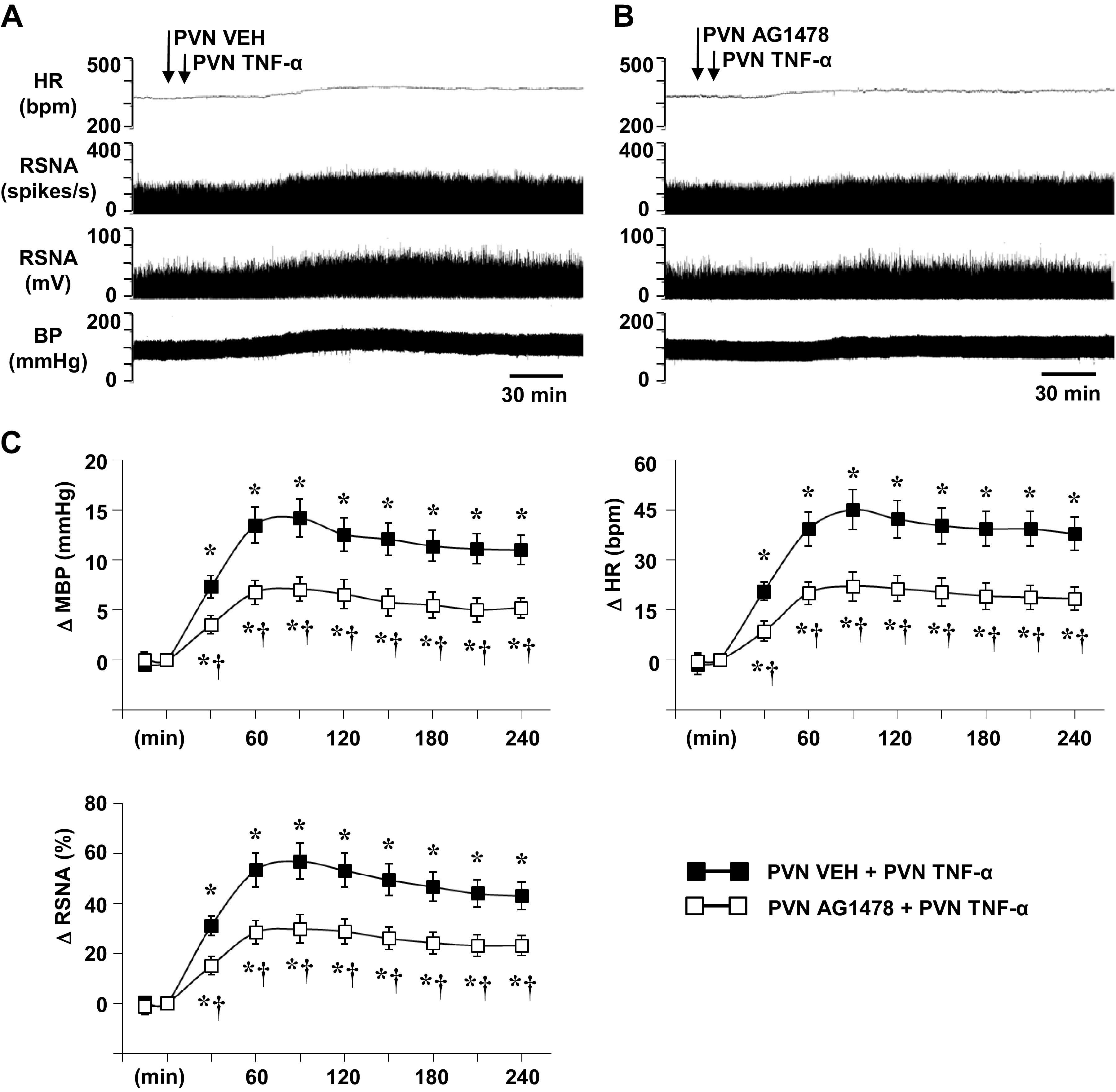
Representative tracings (A, B) and grouped data (C) showing the effects of hypothalamic paraventricular nucleus (PVN) microinjection of tumor necrosis factor (TNF)-α (25 ng/side) on blood pressure (BP, mmHg), heart rate (HR, bpm), and renal sympathetic nerve activity (RSNA), windowed (spikes/s) and integrated (mV), in rats pretreated with PVN microinjection of vehicle (VEH) (A) or AG1478 (B). Arrows indicate the timing of the injections. Scale bar: 30 min. MBP, mean blood pressure; ΔRSNA (%), percent change from baseline in integrated RSNA. All values are expressed as the means ± SE (in C, n = 6 for each group). *P < 0.05 compared with baseline. †P < 0.05 compared with PVN VEH + PVN TNF-α.
Inhibition of EGFR signaling reduces TNF-α-induced upregulation of p-ERK1/2 and c-Fos in the PVN.
Western blot analysis revealed that, compared with VEH, PVN microinjection of TNF-α markedly increased the level of p-ERK1/2 protein in the PVN (Fig. 7A). The level of total ERK1/2 was unchanged. The expression of c-Fos, a classical indicator of neuronal excitation, also increased in the PVN in response to microinjection of TNF-α (Fig. 7B). In rats pretreated with PVN microinjections of AG1478, the TNF-α-induced upregulation of p-ERK1/2 and c-Fos expression was significantly attenuated (Fig. 7, A and B).
Figure 7.
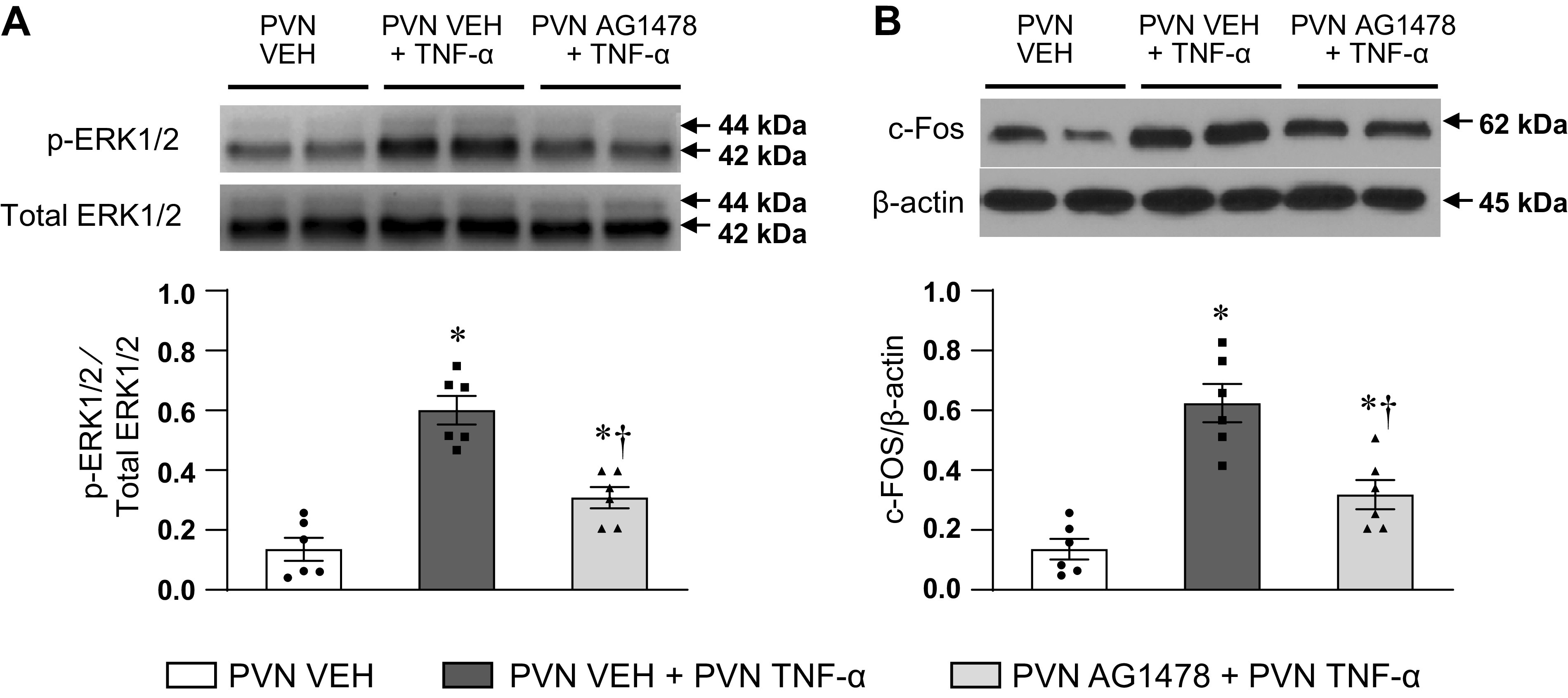
Western blot analysis showing the protein expression of phosphorylated (p)-ERK1/2 (A) and c-Fos (B) in hypothalamic paraventricular nucleus (PVN) in rats treated with bilateral PVN vehicle (VEH) and PVN TNF-α after pretreatment with PVN VEH or AG1478. Bar graphs show the group data, and representative Western bands are shown above each bar. Values are expressed as means ± SE. p-ERK1/2 is normalized to total ERK1/2, and c-Fos is normalized to β-actin (n = 6 for each group). *P < 0.05, vs. PVN VEH. †P < 0.05, PVN AG1478 + PVN TNF-α vs. PVN VEH + PVN TNF-α.
Inhibition of EGFR signaling reduces TNF-α-induced upregulation of excitatory mediators.
Bilateral PVN microinjection of TNF-α significantly increased the mRNA levels of the ACE, AT1aR, COX-2, NF-κB p65, p47phox, and gp91phox subunits, when compared with PVN microinjection of VEH (Fig. 8). Pretreatment with bilateral PVN microinjection of AG1478 significantly reduced these mRNA responses (fold change) to bilateral PVN microinjection of TNF-α (Fig. 8).
Figure 8.

Quantitative analysis by real-time PCR showing the effects of hypothalamic paraventricular nucleus (PVN) microinjection of tumor necrosis factor (TNF)-α (25 ng/side) on the mRNA expression of (A) angiotensin-converting enzyme (ACE), (B) angiotensin II type-1a receptor (AT1a-R), (C) cyclooxygenase (COX)-2, (D) NF-κB p65 subunit, and NADPH oxidase subunits (E) p47phox and (F) gp91phox in the cortex, subfornical organ (SFO), and PVN in rats pretreated with PVN vehicle (VEH) or AG1478. Values are mean ± SE (n = 6 for each group) and expressed as a fold change relative to VEH control. *P < 0.05 compared with the PVN VEH. †P < 0.05, PVN AG1478 + PVN TNF-α vs. PVN VEH + PVN TNF-α.
PVN microinjections of TNF-α had no effect on mRNA levels of these excitatory and inflammatory mediators in the SFO and brain cortex.
DISCUSSION
Augmented sympathetic nerve activity has adverse effects on the heart, kidney, and vasculature that contribute to the pathophysiology and progression of cardiovascular and metabolic diseases (33–36). Over the past decade, the role of inflammation as driver of sympathetic overactivity and neurohumoral activation in the development of hypertension and heart failure has been increasingly appreciated (37, 38). However, the fundamental molecular mechanisms within the brain that account for the effect of inflammation on sympathetic excitation remain largely unknown.
The present study provides the first evidence that EGFR in cardiovascular regulatory regions of the brain plays an essential role in mediating inflammation-induced sympathetic and hemodynamic responses. The major findings are as follows: 1) ICA TNF-α (mimicking circulating TNF-α) activates EGFR and ERK1/2 signaling in SFO and PVN, increases gene expression of the RAS components ACE and AT1aR, the inflammatory mediators COX-2 and NF-κB, and the oxidative elements p47phox and gp91phox in SFO and PVN, and increases MAP, BP, and RSNA; 2) reducing EGFR activity with ICV administration of the selective EGFR tyrosine kinase inhibitor AG1478 attenuates the TNF-α-induced phosphorylation of ERK1/2, the TNF-α-induced upregulation of excitatory mediators in SFO and PVN, and the TNF-α-induced pressor response; 3) reducing ERK1/2 activity with ICV administration of the selective MEK inhibitor PD98059 also attenuates the TNF-α-induced upregulation of excitatory mediators in SFO and PVN and the TNF-α-induced pressor response; and 4) direct PVN microinjection of TNF-α increases p-ERK1/2 and mRNA for ACE, AT1aR, COX-2, NF-κB p65, p47phox, and gp91phox in the PVN, along with increases in MAP, HR, and RSNA, and these responses are ameliorated by PVN pretreatment with AG1478.
These findings demonstrate that the acute sympatho-excitatory response induced by TNF-α in cardiovascular regulatory regions of the forebrain is largely dependent upon EGFR and ERK1/2 signaling. They also have important implications for inflammation-induced sympathetic excitation in conditions like hypertension and heart failure, in which TNF-α levels are increased in the forebrain (39, 40). In those chronic settings, the ability of TNF-α to upregulate gene expression of RAS, inflammatory, and oxidative components via EGFR and ERK1/2 signaling, shown here, would be expected to perpetuate sympathetic activation and its adverse effects on the progression of disease. These downstream molecular influences of TNF-α to upregulate the expression of other excitatory mediators likely account for the failure of central administration of the TNF-α blocker etanercept to have an acute effect on Ang II-salt hypertension (41).
Since TNF-α is too large to readily cross the blood-brain barrier, the pressor response to ICA TNF-α observed in these studies is likely mediated primarily by its direct effects on circumventricular organs like the SFO, where the TNF receptor 1 (TNF-R1) is abundantly expressed (7, 42). This proposal is supported by prior studies showing that the sympathetic and hemodynamic responses to ICA injection of TNF-α are substantially reduced by lesions of the SFO (7) and that microinjection of TNF-α into SFO upregulates gene expression of RAS and inflammatory mediators in the SFO and downstream in the PVN while eliciting a pressor response (27), resembling the effects of ICA TNF-α in the present study. Moreover, in rats with ischemia-induced heart failure, knockdown in SFO of TNF-α receptor 1, which mediates most of the adverse effects of TNF-α (43), substantially reduces sympathetic nerve activity and the expression RAS and inflammatory mediators in SFO and PVN (8). The present study extends those observations by demonstrating that the excitatory influences of TNF-α in the SFO are largely dependent on EGFR and ERK1/2 signaling.
ICV administration of the EGFR and ERK1/2 inhibitors had similar effects on ICA TNF-α-induced molecular events in the PVN, but the mechanisms by which TNF-α activated EGFR-ERK1/2 signaling in the PVN are likely more complex. The PVN is located inside the blood-brain barrier, not readily accessible to blood-borne TNF-α. The PVN receives direct neuronal projections from the SFO (44, 45), and previous studies have shown that the actions of TNF-α within the SFO can modify the neurochemical milieu of the PVN, including the gene expression of RAS and inflammatory mediators (7, 27). In addition, TNF-α-induced EGFR-ERK1/2 activity in the SFO may stimulate the release in PVN of other mediators that can transactivate EGFR, such as ANG II (46, 47). Thus, although our ICV treatment data demonstrate that EGFR and downstream ERK1/2 signaling contributed to the molecular responses induced in PVN by ICA TNF-α, activation of PVN EGFR and ERK1/2 activity cannot be attributed to direct effects of TNF-α.
To circumvent this concern and determine the potential role of EGFR as a mediator of TNF-α effects in the PVN, we microinjected TNF-α into the PVN, with and without prior microinjection of the EGFR tyrosine kinase inhibitor. The results were quite similar to the effects of the ICA TNF-α: microinjected TNF-α increased ERK1/2 activity and the expression of excitatory mediators and elicited a pressor response, and these changes were all mitigated by pretreatment with AG1478. Together, the findings of the ICA injection and the PVN microinjection studies demonstrate that the sympathoexcitatory effects of TNF-α in SFO and PVN are largely mediated by transactivation of EGFR and suggest that transactivation of EGFR with subsequent ERK1/2 signaling are the key mechanisms involved.
TNF-α may transactivate EGFR by activating metalloproteinases to cleave endogenous ligands of EGFR, including TGF-α and EGF, from their transmembrane forms (48–50). The role of a disintegrin and metalloprotease 17 (ADAM17), also known as TNF-α-converting enzyme (TACE), is well studied in this regard (51). TNF-α enhances ADAM17/TACE activity in several cell types (52). We recently reported that ICV ANG II and IL-1β can increase ADAM17/TACE expression in PVN and SFO (31). The present findings raise the possibility that TNF-α may also enhance ADAM17 activity in these cardiovascular regulatory regions of the brain, resulting in the local production of EGFR agonists.
ERK1/2 MAPK signaling is a well-established outcome of the transactivation of EGFR (53–55). Phosphorylation of EGFR tyrosine kinase initiates a series of intracellular events culminating in activation of the Ras/Raf/MEK/ERK1/2 signaling pathway. The present study demonstrated that TNF-α-induced phosphorylation of ERK1/2 in PVN and SFO is dependent at least in part on transactivation of EGFR. Taken together, these observations support the hypothesis that TNF-α induces sympathetic and hemodynamic responses by stimulating ADAM17-mediated shedding of TGF-α or EGF to activate EGFR in cardiovascular regions of the brain, with subsequent activation of ERK1/2 signaling.
The mechanism(s) by which ERK1/2 signaling drives sympathetic activity remain elusive. ERK1/2-mediated upregulation of gene expression of RAS components and inflammatory mediators in the PVN and SFO may certainly contribute to sustained sympathetic overactivity in chronic settings like hypertension or heart failure. We have previously found that activation of brain ERK1/2 signaling and its downstream molecular events, particularly in the PVN, contributes to sympathetic overactivity in a rat model of heart failure (12, 56). However, the acute onset of sympatho-excitation with minutes of exposure to TNF-α cannot be explained by that mechanism. Nor can the rapid reduction in sympathetic nerve activity in rats with established heart failure treated with an intracerebroventricular infusion of an ERK1/2 inhibitor (15), occurring far too early to be explained by a reduction in excitatory proteins. These early effects of ERK1/2 manipulations demand an alternative explanation. One potential explanation is a direct channel of ERK1/2 to phosphorylate and inactivate the potassium channel subunit Kv4.2 (57), reducing A-current and thereby increasing the excitability of presympathetic PVN neurons. This possibility remains to be tested.
Although the mechanism(s) subserving EGFR- and ERK1/2-mediated sympathoexcitation remain to be elucidated, the time course of the sympathetic and hemodynamic responses to ICA injection or PVN microinjection of TNF-α, beginning within 15–30 min and lasting for at least 2 h, is consistent with the time course of TNF-α-induced EGFR transactivation, initiated within 10 min with peaks at 30 min to 6 h after stimulation (58, 59), and with the rapid EGFR-mediated phosphorylation of ERK1/2 (60).
Limitations of the Study
While our findings strongly support an important role for EGFR in TNF-α-induced sympathetic excitation via the ERK1/2 signaling pathway, inhibition of EGFR tyrosine kinase did not completely block sympathetic activation by TNF-α. Thus, other TNF-α-driven molecular pathways are likely involved. The effects of TNF-α are mediated by multiple signaling cascades to regulate a variety of molecular events (61), including alternate MAPK signaling pathways (62) that may affect the sympathetic response. In addition, it is possible that TNF-α, like other agonists (63), may activate ERK1/2 independently of EGFR. p-ERK1/2 remained higher than control in SFO and PVN following ICV AG1478 and the pressor response to TNF-α was only partially blocked, suggesting either an incomplete inhibition of EGFR with residual ERK1/2 activation or an alternative pathway to ERK1/2 activation (64). Another potential explanation for the partial pressor response to AG1478 is that this inhibitor selectively targets EGFR tyrosine kinase (25). It is conceivable that TNF-α may also activate the serine/threonine domain of EGRF as seen in human microvascular endothelial cells (58). Further, having demonstrated that PVN microinjection of AG1478 reduced p-ERK1/2 in the PVN, we did not test the effect of blocking PVN ERK1/2 on the pressor response to PVN microinjection of TNF-α. Thus, we cannot conclude with certainty that those responses were mediated by ERK1/2, though we have previously demonstrated that knockdown of ERK1/2 in the PVN reduces PVN neuronal excitation (i.e., c-Fos activity) and plasma norepinephrine in rats with heart failure (56). Finally, the cell types mediating the pressor effects of TNF-α were not addressed in this study. Although EGFR is generally considered to be primarily expressed in neurons in normal adult rats (65, 66), and is unlikely to have been upregulated and phosphorylated in glial cells early enough to participate in this acute pressor response to TNF-α in normal rats, the potential involvement of inflammatory cells in the ERK1/2-mediated prolonged pressor response to TNF-α cannot be excluded. These are issues for further investigation.
Perspectives
These data demonstrate that activation of EGFR receptor tyrosine kinase in the cardiovascular regulatory regions of the forebrain plays a major role in TNF-α-induced sympathetic excitation. EGFR-mediated activation of ERK1/2 signaling is likely a key mechanism in this process, since inhibiting EGFR reduces TNF-α-induced phosphorylation of ERK1/2 and inhibiting either EGFR or ERK1/2 activity reduces the sympathetic and hemodynamic responses to TNF-α to a similar degree. The effects of TNF-α-induced EGFR and ERK1/2 signaling to upregulate gene expression of brain RAS and inflammatory elements, while unlikely to mediate the acute pressor responses to TNF-α in normal rats, may play an important role in conditions like hypertension and heart failure, in which TNF-α-induced overexpression of excitatory mediators may contribute to sustained and detrimental increases in sympathetic nerve activity. Other factors that increase in the circulation and in the brain in hypertension and heart failure—e.g., ANG II (46)—may also transactivate EGFR to upregulate the sympathoexcitatory milieu of the SFO and PVN. Thus, targeting EGFR as a gateway to brain ERK1/2 signaling may be a promising therapeutic approach to treatment of the sympathetic overactivity that contributes to the progression of these diseases.
GRANTS
This study was supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, and by National Institutes of Health grants HL096671 and HL136149 (to R.B. Felder) and HL139521 (to S-G. Wei).
DISCLAIMER
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
R. B. Felder is party to a patent application for a microparticle preparation designed to reduce brain ERK1/2 signaling in heart failure. There are no other potential conflicts of interest.
AUTHOR CONTRIBUTIONS
R.B.F. and S.-G.W. conceived and designed research; S.-G.W. and Y.Y. performed experiments; S.-G.W. and Y.Y. analyzed data; S.-G.W. and R.B.F. interpreted results of experiments; S.-G.W. prepared figures; S.-G.W. and R.B.F. drafted manuscript; S.-G.W., Y.Y., and R.B.F. edited and revised manuscript; S.-G.W., Y.Y., and R.B.F. approved final version of manuscript.
REFERENCES
- 1.Glezeva N, Baugh JA. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart Fail Rev 19: 681–694, 2014. doi: 10.1007/s10741-013-9405-8. [DOI] [PubMed] [Google Scholar]
- 2.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension 57: 132–140, 2011. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montecucco F, Pende A, Quercioli A, Mach F. Inflammation in the pathophysiology of essential hypertension. J Nephrol 24: 23–34, 2011. doi: 10.5301/JN.2010.4729. [DOI] [PubMed] [Google Scholar]
- 4.Yndestad A, Damas JK, Oie E, Ueland T, Gullestad L, Aukrust P. Role of inflammation in the progression of heart failure. Curr Cardiol Rep 9: 236–241, 2007. doi: 10.1007/BF02938356. [DOI] [PubMed] [Google Scholar]
- 5.Levick SP, Murray DB, Janicki JS, Brower GL. Sympathetic nervous system modulation of inflammation and remodeling in the hypertensive heart. Hypertension 55: 270–276, 2010. doi: 10.1161/HYPERTENSIONAHA.109.142042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi P, Raizada MK, Sumner C. Brain cytokines as neuromodulators in cardiovascular control. Clin Exp Pharmacol Physiol 37: e52–e57, 2010. doi: 10.1111/j.1440-1681.2009.05234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei SG, Zhang ZH, Beltz TG, Yu Y, Johnson AK, Felder RB. Subfornical organ mediates sympathetic and hemodynamic responses to blood-borne proinflammatory cytokines. Hypertension 62: 118–125, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu Y, Wei SG, Weiss RM, Felder RB. TNF-alpha receptor 1 knockdown in the subfornical organ ameliorates sympathetic excitation and cardiac hemodynamics in heart failure rats. Am J Physiol Heart Circ Physiol 313: H744–H756, 2017. doi: 10.1152/ajpheart.00280.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang YM, Wang Y, Yang LM, Elks C, Cardinale J, Yu XJ, Zhao XF, Zhang J, Zhang LH, Yang ZM, Francis J. TNF-alpha in hypothalamic paraventricular nucleus contributes to sympathoexcitation in heart failure by modulating AT1 receptor and neurotransmitters. Tohoku J Exp Med 222: 251–263, 2010. doi: 10.1620/tjem.222.251. [DOI] [PubMed] [Google Scholar]
- 10.Wei SG, Zhang ZH, Yu Y, Felder RB. Central SDF-1/CXCL12 expression and its cardiovascular and sympathetic effects: the role of angiotensin II, TNF-alpha, and MAP kinase signaling. Am J Physiol Heart Circ Physiol 307: H1643–H1654, 2014. doi: 10.1152/ajpheart.00432.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei SG, Yu Y, Zhang ZH, Weiss RM, Felder RB. Mitogen-activated protein kinases mediate upregulation of hypothalamic angiotensin II type 1 receptors in heart failure rats. Hypertension 52: 679–686, 2008. doi: 10.1161/HYPERTENSIONAHA.108.113639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei SG, Yu Y, Weiss RM, Felder RB. Inhibition of brain mitogen-activated protein kinase signaling reduces central endoplasmic reticulum stress and inflammation and sympathetic nerve activity in heart failure rats. Hypertension 67: 229–236, 2016. [Erratum in Hypertension 72: e32, 2018]. doi: 10.1161/HYPERTENSIONAHA.115.06329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kilpatrick LE, Sun S, Li H, Vary TC, Korchak HM. Regulation of TNF-induced oxygen radical production in human neutrophils: role of delta-PKC. J Leukoc Biol 87: 153–164, 2010. doi: 10.1189/jlb.0408230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moon EJ, Sonveaux P, Porporato PE, Danhier P, Gallez B, Batinic-Haberle I, Nien YC, Schroeder T, Dewhirst MW. NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment. Proc Natl Acad Sci USA 107: 20477–20482, 2010. doi: 10.1073/pnas.1006646107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei SG, Yu Y, Zhang ZH, Weiss RM, Felder RB. Angiotensin II-triggered p44/42 mitogen-activated protein kinase mediates sympathetic excitation in heart failure rats. Hypertension 52: 342–350, 2008. doi: 10.1161/HYPERTENSIONAHA.108.110445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kovacs E, Zorn JA, Huang Y, Barros T, Kuriyan JA. Structural perspective on the regulation of the epidermal growth factor receptor. Annu Rev Biochem 84: 739–764, 2015. doi: 10.1146/annurev-biochem-060614-034402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andl CD, Mizushima T, Nakagawa H, Oyama K, Harada H, Chruma K, Herlyn M, Rustgi AK. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J Biol Chem 278: 1824–1830, 2003. doi: 10.1074/jbc.M209148200. [DOI] [PubMed] [Google Scholar]
- 18.Galvez-Contreras AY, Gonzalez-Castaneda RE, Campos-Ordonez T, Luquin S, Gonzalez-Perez O. Phenytoin enhances the phosphorylation of epidermal growth factor receptor and fibroblast growth factor receptor in the subventricular zone and promotes the proliferation of neural precursor cells and oligodendrocyte differentiation. Eur J Neurosci 43: 139–147, 2016. doi: 10.1111/ejn.13079. [DOI] [PubMed] [Google Scholar]
- 19.Yang X, Zhu MJ, Sreejayan N, Ren J, Du M. Angiotensin II promotes smooth muscle cell proliferation and migration through release of heparin-binding epidermal growth factor and activation of EGF-receptor pathway. Mol Cells 20: 263–270, 2005. doi: 10.1016/j.molcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 20.Katz M, Amit I, Yarden Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim Biophys Acta 1773: 1161–1176, 2007. doi: 10.1016/j.bbamcr.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liebmann C. EGF receptor activation by GPCRs: an universal pathway reveals different versions. Mol Cell Endocrinol 331: 222–231, 2011. doi: 10.1016/j.mce.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 22.Hobbs SS, Goettel JA, Liang D, Yan F, Edelblum KL, Frey MR, Mullane MT, Polk DB. TNF transactivation of EGFR stimulates cytoprotective COX-2 expression in gastrointestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 301: G220–G229, 2011. doi: 10.1152/ajpgi.00383.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin CC, Pan CS, Wang CY, Liu SW, Hsiao LD, Yang CM. Tumor necrosis factor-alpha induces VCAM-1-mediated inflammation via c-Src-dependent transactivation of EGF receptors in human cardiac fibroblasts. J Biomed Sci 22: 53, 2015. doi: 10.1186/s12929-015-0165-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pastore S, Mascia F, Mariani V, Girolomoni G. The epidermal growth factor receptor system in skin repair and inflammation. J Invest Dermatol 128: 1365–1374, 2008. doi: 10.1038/sj.jid.5701184. [DOI] [PubMed] [Google Scholar]
- 25.Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science 267: 1782–1788, 1995. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 26.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA 92: 7686–7689, 1995. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei SG, Yu Y, Zhang ZH, Felder RB. Proinflammatory cytokines upregulate sympathoexcitatory mechanisms in the subfornical organ of the rat. Hypertension 65: 1126–1133, 2015. doi: 10.1161/HYPERTENSIONAHA.114.05112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang ZH, Yu Y, Wei SG, Felder RB. Abstract 415: brain epidermal growth factor receptor and c-Src tyrosine kinase contribute to sympathetic excitation induced by systemically administered aldosterone in rats. Hypertension 60: A415, 2012. doi: 10.1161/hyp.60.suppl_1.a415. [DOI] [Google Scholar]
- 29.Haywood JR, Fink GD, Buggy J, Phillips MI, Brody MJ. The area postrema plays no role in the pressor action of angiotensin in the rat. Am J Physiol 239: H108–H113, 1980. doi: 10.1152/ajpheart.1980.239.1.H108. [DOI] [PubMed] [Google Scholar]
- 30.Zhang ZH, Francis J, Weiss RM, Felder RB. The renin-angiotensin-aldosterone system excites hypothalamic paraventricular nucleus neurons in heart failure. Am J Physiol Heart Circ Physiol 283: H423–H433, 2002. doi: 10.1152/ajpheart.00685.2001. [DOI] [PubMed] [Google Scholar]
- 31.Yu Y, Cao Y, Bell B, Chen X, Weiss RM, Felder RB, Wei SG. Brain TACE (tumor necrosis factor-alpha-converting enzyme) contributes to sympathetic excitation in heart failure rats. Hypertension 74: 63–72, 2019. doi: 10.1161/HYPERTENSIONAHA.119.12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Esler M, Kaye D. Increased sympathetic nervous system activity and its therapeutic reduction in arterial hypertension, portal hypertension and heart failure. J Auton Nerv Syst 72: 210–219, 1998. doi: 10.1016/S0165-1838(98)00107-6. [DOI] [PubMed] [Google Scholar]
- 34.Grassi G. Sympathetic overdrive and cardiovascular risk in the metabolic syndrome. Hypertens Res 29: 839–847, 2006. doi: 10.1291/hypres.29.839. [DOI] [PubMed] [Google Scholar]
- 35.Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev 90: 513–557, 2010. doi: 10.1152/physrev.00007.2009. [DOI] [PubMed] [Google Scholar]
- 36.Masuo K, Lambert GW, Esler MD, Rakugi H, Ogihara T, Schlaich MP. The role of sympathetic nervous activity in renal injury and end-stage renal disease. Hypertens Res 33: 521–528, 2010. doi: 10.1038/hr.2010.35. [DOI] [PubMed] [Google Scholar]
- 37.Diaz HS, Toledo C, Andrade DC, Marcus NJ, Rio DR. Neuroinflammation in heart failure: new insights for an old disease. J Physiol 598: 33–59, 2020. doi: 10.1113/JP278864. [DOI] [PubMed] [Google Scholar]
- 38.Haspula D, Clark MA. Neuroinflammation and sympathetic overactivity: mechanisms and implications in hypertension. Auton Neurosci 210: 10–17, 2018. doi: 10.1016/j.autneu.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 39.Francis J, Chu Y, Johnson AK, Weiss RM, Felder RB. Acute myocardial infarction induces hypothalamic cytokine synthesis. Am J Physiol Heart Circ Physiol 286: H2264–H2271, 2004. doi: 10.1152/ajpheart.01072.2003. [DOI] [PubMed] [Google Scholar]
- 40.Sriramula S, Cardinale JP, Francis J. Inhibition of TNF in the brain reverses alterations in RAS components and attenuates angiotensin II-induced hypertension. PLoS One 8: e63847, 2013. doi: 10.1371/journal.pone.0063847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bardgett ME, Holbein WW, Herrera-Rosales M, Toney GM. Ang II-salt hypertension depends on neuronal activity in the hypothalamic paraventricular nucleus but not on local actions of tumor necrosis factor-alpha. Hypertension 63: 527–534, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nadeau S, Rivest S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience 93: 1449–1464, 1999. doi: 10.1016/S0306-4522(99)00225-0. [DOI] [PubMed] [Google Scholar]
- 43.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation 5: 45, 2008. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson JW, Smith PM, Ferguson AV. Subfornical organ neurons projecting to paraventricular nucleus: whole-cell properties. Brain Res 921: 78–85, 2001. doi: 10.1016/S0006-8993(01)03093-1. [DOI] [PubMed] [Google Scholar]
- 45.Bains JS, Ferguson AV. Paraventricular nucleus neurons projecting to the spinal cord receive excitatory input from the subfornical organ. Am J Physiol 268: R625–R633, 1995. doi: 10.1152/ajpregu.1995.268.3.R625. [DOI] [PubMed] [Google Scholar]
- 46.Bali A, Jaggi AS. Angiotensin II-triggered kinase signaling cascade in the central nervous system. Rev Neurosci 27: 301–315, 2016. doi: 10.1515/revneuro-2015-0041. [DOI] [PubMed] [Google Scholar]
- 47.Shah BH, Catt KJ. TACE-dependent EGF receptor activation in angiotensin-II-induced kidney disease. Trends Pharmacol Sci 27: 235–237, 2006. doi: 10.1016/j.tips.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 48.Jijon HB, Buret A, Hirota CL, Hollenberg MD, Beck PL. The EGF receptor and HER2 participate in TNF-alpha-dependent MAPK activation and IL-8 secretion in intestinal epithelial cells. Mediators Inflamm 2012: 207398, 2012. doi: 10.1155/2012/207398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kakiashvili E, Dan Q, Vandermeer M, Zhang Y, Waheed F, Pham M, Szaszi K. The epidermal growth factor receptor mediates tumor necrosis factor-alpha-induced activation of the ERK/GEF-H1/RhoA pathway in tubular epithelium. J Biol Chem 286: 9268–9279, 2011. doi: 10.1074/jbc.M110.179903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoo J, Rodriguez Perez CE, Nie W, Edwards RA, Sinnett-Smith J, Rozengurt E. TNF-alpha induces upregulation of EGFR expression and signaling in human colonic myofibroblasts. Am J Physiol Gastrointest Liver Physiol 302: G805–G814, 2012. doi: 10.1152/ajpgi.00522.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sunnarborg SW, Hinkle CL, Stevenson M, Russell WE, Raska CS, Peschon JJ, Castner BJ, Gerhart MJ, Paxton RJ, Black RA, Lee DC. Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J Biol Chem 277: 12838–12845, 2002. doi: 10.1074/jbc.M112050200. [DOI] [PubMed] [Google Scholar]
- 52.Bzowska M, Jura N, Lassak A, Black RA, Bereta J. Tumour necrosis factor-alpha stimulates expression of TNF-alpha converting enzyme in endothelial cells. Eur J Biochem 271: 2808–2820, 2004. doi: 10.1111/j.1432-1033.2004.04215.x. [DOI] [PubMed] [Google Scholar]
- 53.Hsieh M, Conti M. G-protein-coupled receptor signaling and the EGF network in endocrine systems. Trends Endocrinol Metab 16: 320–326, 2005. doi: 10.1016/j.tem.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 54.Najafi M, Ahmadi A, Mortezaee K. Extracellular-signal-regulated kinase/mitogen-activated protein kinase signaling as a target for cancer therapy: an updated review. Cell Biol Int 43: 1206–1222, 2019. doi: 10.1002/cbin.11187. [DOI] [PubMed] [Google Scholar]
- 55.Wetzker R, Bohmer FD. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol 4: 651–657, 2003. doi: 10.1038/nrm1173. [DOI] [PubMed] [Google Scholar]
- 56.Yu Y, Wei SG, Zhang ZH, Weiss RM, Felder RB. ERK1/2 MAPK signaling in hypothalamic paraventricular nucleus contributes to sympathetic excitation in rats with heart failure after myocardial infarction. Am J Physiol Heart Circ Physiol 310: H732–H739, 2016. doi: 10.1152/ajpheart.00703.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schrader LA, Birnbaum SG, Nadin BM, Ren Y, Bui D, Anderson AE, Sweatt JD. ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am J Physiol Cell Physiol 290: C852–C861, 2006. doi: 10.1152/ajpcell.00358.2005. [DOI] [PubMed] [Google Scholar]
- 58.Izumi H, Ono M, Ushiro S, Kohno K, Kung HF, Kuwano M. Cross talk of tumor necrosis factor-alpha and epidermal growth factor in human microvascular endothelial cells. Exp Cell Res 214: 654–662, 1994. doi: 10.1006/excr.1994.1303. [DOI] [PubMed] [Google Scholar]
- 59.Murthy S, Mathur SN, Field FJ. Tumor necrosis factor-alpha and interleukin-1beta inhibit apolipoprotein B secretion in CaCo-2 cells via the epidermal growth factor receptor signaling pathway. J Biol Chem 275: 9222–9229, 2000. doi: 10.1074/jbc.275.13.9222. [DOI] [PubMed] [Google Scholar]
- 60.Brown MD, Sacks DB. Protein scaffolds in MAP kinase signalling. Cell Signal 21: 462–469, 2009. doi: 10.1016/j.cellsig.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol 12: 49–62, 2016. doi: 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi J-H, Sun S-C. Tumor necrosis factor receptor-associated factor regulation of nuclear factor kappaB and mitogen-activated protein kinase pathways. Front Immunol 9: 1849, 2018. doi: 10.3389/fimmu.2018.01849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bobe R, Yin X, Roussanne MC, Stepien O, Polidano E, Faverdin C, Marche P. Evidence for ERK1/2 activation by thrombin that is independent of EGFR transactivation. Am J Physiol Heart Circ Physiol 285: H745–H754, 2003. doi: 10.1152/ajpheart.01042.2002. [DOI] [PubMed] [Google Scholar]
- 64.Roskoski R, Jr. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 66: 105–143, 2012. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 65.Ferrer I, Alcantara S, Ballabriga J, Olive M, Blanco R, Rivera R, Carmona M, Berruezo M, Pitarch S, Planas AM. Transforming growth factor-alpha (TGF-alpha) and epidermal growth factor-receptor (EGF-R) immunoreactivity in normal and pathologic brain. Prog Neurobiol 49: 99–123, 1996. doi: 10.1016/0301-0082(96)00009-3. [DOI] [PubMed] [Google Scholar]
- 66.Tavassoly O, Sato T, Tavassoly I. Inhibition of brain epidermal growth factor receptor activation: a novel target in neurodegenerative diseases and brain injuries. Mol Pharmacol 98: 13–22, 2020. doi: 10.1124/mol.120.119909. [DOI] [PubMed] [Google Scholar]