

Abstract
BACKGROUND AND AIMS:
Fibroblast growth factor 1 (FGF1) demonstrated protection against nonalcoholic fatty liver disease (NAFLD) in type 2 diabetic and obese mice by an uncertain mechanism. This study investigated the therapeutic activity and mechanism of a non-mitogenic FGF1 variant (FGF1ΔHBS) against NAFLD.
APPROACH AND RESULTS:
FGF1ΔHBS administration was effective in 9-month old db/db mice with NAFLD; liver weight, lipid deposition and inflammation declined and liver injury decreased. FGF1ΔHBS reduced oxidative stress by stimulating nuclear translocation of nuclear factor erythroid 2-related factor 2 (Nrf2) and elevation of antioxidant protein expression. FGF1ΔHBS also inhibited activity and/or expression of lipogenic genes, coincident with phosphorylation of AMP-activated protein kinase (AMPK) and its substrates. Mechanistic studies on palmitate exposed hepatic cells demonstrated that NAFLD-like oxidative damage and lipid accumulation could be reversed by FGF1ΔHBS. In palmitate-treated hepatic cells, siRNA knockdown of Nrf2 abolished only FGF1ΔHBS anti-oxidative actions but not improvement of lipid metabolism. In contrast, AMPK inhibition by pharmacological agent or siRNA abolished FGF1ΔHBS benefits on both oxidative stress and lipid metabolism that were FGF receptor 4 (FGFR4) dependent. Further support of these in vitro findings is that liver-specific AMPK knockout abolished therapeutic effects of FGF1ΔHBS against high-fat/high-sucrose diet-induced hepatic steatosis. Moreover, FGF1ΔHBS improved high-fat/high-cholesterol diet-induced steatohepatitis and fibrosis in apolipoprotein E knockout mice.
CONCLUSIONS:
These findings indicate that FGF1ΔHBS is effective for preventing and reversing liver steatosis and steatohepatitis and acts by activation of AMPK via hepatocyte FGFR4.
Keywords: Fibroblast growth factor 1, nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, AMP-activated protein kinase, nuclear factor erythroid 2-related factor 2
Introduction
Liver steatosis and nonalcoholic fatty liver disease (NAFLD), characterized by hepatic fat accumulation, inflammation and oxidative stress, are strongly linked with obesity, insulin resistance and dyslipidemia (metabolic syndrome) and type 2 diabetes (T2D) (1). NAFLD is especially serious in T2D because diabetic hyperglycemia provides excess glucose to the liver, leading to more hepatic oxidative stress and greater imbalance of hepatic metabolism. As the prevalence of NAFLD among T2D patients is up to 75% (2, 3) and the consequences are especially serious in diabetic patients, NAFLD is recognized as a major health burden to the T2D population. Currently there are no effective therapies for T2D associated NAFLD. Therefore, there is an urgent need to develop new therapeutic approaches to reduce the metabolic pathology of T2D responsible for the high prevalence of NAFLD.
Fibroblast growth factor 1 (FGF1), a well characterized mitogen belonging to the 18 member FGF family, is widely studied for its therapeutic benefits in cardiovascular disorders, nerve injury and wound healing (4, 5). Recently, FGF1 was shown to exert an unexpected metabolic activity by regulating lipid metabolism and glucose homeostasis in obesity and diabetes (6, 7). Furthermore, our recent study indicated that FGF1 prevents diabetic nephropathy in both type 1 diabetes and T2D animal models (8). However, because native FGF1 induces hyperproliferation, its use can potentially increase tumorigenic risk (9), which limits its widespread application, especially in cancer-prone diseases, including diabetes.
If the mitogenic and metabolic functions of FGF1 can be dissociated, then it may provide a safe and potent agent for prevention and/or therapy of diabetes and diabetic complications while minimizing risk of cancer. Thus, we engineered an FGF1 partial agonist with 3 substitutions (i.e., FGF1ΔHBS) that dramatically diminished its ability to induce heparan sulfate-assisted FGF receptor (FGFR) dimerization and activation (10). Moreover, FGF1ΔHBS has reduced proliferative potential, yet maintains full metabolic activity of wild type FGF1 (FGF1WT) in vitro and in vivo (10). Notably, FGF1ΔHBS 28 days treatment protects the liver from fatty acid accumulation in db/db mice (10), which is consistent with FGF1WT action to inhibit hepatic steatosis in high fat diet (HFD)-induced obesity (6) and in leptin-deficient ob/ob mice (7, 11). These exciting findings support the idea that FGF1ΔHBS is a potential safe and effective therapeutic approach for treatment of NAFLD especially in the setting of T2D. Nonetheless, the mechanism(s) underlying the beneficial activity of either FGF1WT or FGF1ΔHBS in NAFLD remains to be determined. In addition, the clinically important capacity of FGF1 to reverse established NAFLD or to prevent nonalcoholic steatohepatitis (NASH) has not been tested.
In the present study, we demonstrate the potent beneficial effects of FGF1ΔHBS on established NAFLD in T2D and on NASH in an apolipoprotein E knockout (ApoE-KO) mouse model. Importantly, we identify a likely mechanism required for FGF1ΔHBS actions on hepatic oxidative stress and lipid metabolism. Our data demonstrate that FGF1ΔHBS reverses NAFLD in late-stage T2D by inhibition of hepatic inflammation, oxidative stress and lipid dysregulation. Significantly, our data identifies a novel mechanism of FGF1ΔHBS protection against NAFLD via activation of hepatic adenosine monophosphate-activated protein kinase (AMPK). Furthermore, our in vivo and in vitro studies demonstrate that FGF1ΔHBS activation of nuclear factor erythroid 2-related factor 2 (Nrf2) is essential to prevent oxidative stress, but Nrf2 activation alone is insufficient for FGF1 prevention of hepatic lipid metabolic dysregulation. Moreover, activation of AMPK is essential to the ability of FGF1ΔHBS both to protect from dysregulated hepatic lipid metabolism and to inhibit oxidative stress.
Materials and Methods
db/db (BKS.Cg-Dock7m +/+ Leprdb/J) and ApoE-KO (B6.129P2-Apoe tm1Unc/J) mice were purchased from Jackson Laboratory (Bar Harbor, ME). Nine-month-old male db/db mice were administered FGF1ΔHBS (0.5 mg/kg body weight) or vehicle (phosphate buffered solution, PBS) via intraperitoneal injection every other day for 3 months in the therapeutic study. Liver-specific AMPK knockout (AMPK-LKO) mice were generated by crossing albumin-Cre recombinase transgenic mice with floxed AMPKα1/α2 mice as previously reported (12). Wide type (WT) littermates (albumin-Cre recombinase-negative, AMPKα1/α2flox/flox) were used as the control. At the age of 2 months, male mice were fed a high-fat/high-sucrose (HFHS) diet (Catalog No: D12327, Research Diets) for 5 months, and then supplemented with FGF1ΔHBS (0.5mg/kg body weight) or PBS every other day for 1 month. Eight-week-old male ApoE-KO mice were fed either a normal chow or a high-fat/high-cholesterol (HFHC) diet (Catalog No: TD88137, Envigo) to induce the development of nonalcoholic steatohepatitis (NASH). At the same time, these mice were administered FGF1ΔHBS (0.5 mg/kg body weight) or PBS via intraperitoneal injection every other day for 3 months in the preventive study. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Louisville or the Shanghai Institute of Nutrition and Health of the Chinese Academy of Sciences. The human hepatocellular carcinoma (HepG2) cell line was obtained from the ATCC. Mouse primary hepatocytes were isolated as described previously (13). Mechanistic studies were performed under the optimized experimental conditions established in pilot studies (Supporting Fig. S1 and S2). Details of methods used in this study are described in the Supporting Materials and Methods.
Results
FGF1ΔHBS reverses NAFLD in late-stage T2D mice
To extend our previous observation (10) that either FGF1WT or FGF1ΔHBS treatment reduces early-stage NAFLD in 2-month old db/db mice, in the current study we tested if FGF1ΔHBS could reverse more severe liver injury in late-stage T2D mice. FGF1ΔHBS treatment was initiated at 9 months and continued through 12 months of age in db/db mice. Nine-month-old db/db mice had obvious T2D, as indicated by high blood glucose (Supporting Fig. S3A) and body weight (Supporting Fig. S3B). Prolonged diabetes coincided with development of liver pathological characteristic of NAFLD. Parameters of NAFLD found in db/db mice included dramatically elevated liver weight (Supporting Fig. S3C), abnormal morphology quantified as NAFLD activity score (NAS) (Supporting Figs. S3D and S3E), and hepatic lipid accumulation indicated both by greater Oil Red O staining (Supporting Fig. S3D) and higher hepatic triglyceride contents (Supporting Fig. S3F). In addition, 9-month-old db/db mice exhibited significantly higher plasma markers of liver injury: alanine aminotransferase (ALT, Supporting Fig. S3G) and aspartate aminotransferase (AST, Supporting Fig. S3H). However, there was no obvious hepatic fibrosis in this db/db NAFLD model (data not shown), which is consistent with previous reports that db/db mice rarely show any features of hepatic fibrosis when fed a normal chow diet (14, 15).
Three months of treatment with FGF1ΔHBS decreased diabetes as indicated by significantly lower blood glucose (Supporting Fig. S4A), improved glucose tolerance (Supporting Fig. S4B,C) and slightly reduced body weight (Supporting Fig. S4D). Most importantly FGF1ΔHBS reduced most characteristics of NAFLD: Liver size and weight were much lower (Fig. 1A,B). FGF1ΔHBS also improved hepatic histopathology (Fig. 1C, upper panel), evident as reduced NAS scores (Fig. 1D) and decreased lipid droplet deposition (Fig. 1C, lower panel). FGF1ΔHBS treatment reduced hepatic triglycerides (Fig. 1E) and non-esterified fatty acid (Fig. 1F) content. In addition, FGF1ΔHBS reduced the plasma markers of liver injury: ALT (Fig. 1G) and AST (Fig. 1H), decreased hepatic mRNA levels for inflammatory markers: tumor necrosis factor α, plasminogen activator inhibitor-1, monocyte chemoattractant protein-1 and intercellular adhesion molecule 1 (Fig. 1I–L); inhibited hepatic superoxide generation (Supporting Fig. S4I,J), and decreased hepatic malondialdehyde (MDA) content (Fig. 1M). There was a slight increase in plasma triglyceride concentration (Supporting Fig. S4E). Values that were not significantly affected by FGF1ΔHBS were hepatic cholesterol content (Supporting Fig. S4F), plasma total cholesterol (Supporting Fig. S4G) and plasma low-density lipoprotein concentration (Supporting Fig. S4H). As expected, FGF1ΔHBS did not increase hepatic cell proliferative marker proliferating cell nuclear antigen and Ki67 declined (Supporting Fig. S5).
Fig. 1. The therapeutic effects of FGF1ΔHBS on nonalcoholic fatty liver disease (NAFLD) in late-stage diabetic mice.

Nine-month-old db/db mice were treated with FGF1ΔHBS (0.5 mg/kg body weight) or phosphate buffered solution vehicle every other day for 3 months. (A) Liver size and (B) weight. (C) Representative images of H&E-stained and Oil Red O stained liver sections. (D) NAFLD activity score (NAS). (E) Triglyceride contents in liver. (F) Non-esterified fatty acid (NEFA) content in liver. (G and H) Plasma ALT and AST activity. (I-L) The mRNA levels of hepatic inflammatory factors (TNFα, PAI-1, MCP-1 and ICAM-1) in liver tissue. (M) MDA contents in liver tissue. Quantitative data are expressed as mean ± SEM, n=6–10. *P < 0.05, ** P < 0.01 vs. vehicle treated mice. ALT, alanine aminotransferase; AST, aspartate aminotransferase; TNFα, tumor necrosis factor α; PAI-1, plasminogen activator inhibitor-1; MCP-1, monocyte chemoattractant protein-1; ICAM-1, intercellular adhesion molecule 1.
FGF1ΔHBS treatment preserves antioxidant activity and normalizes lipid metabolism in db/db liver
The transcription factor Nrf2 plays a critical role in the antioxidant response by upregulating multiple antioxidant enzymes (16). Thus, we tested whether FGF1ΔHBS treatment of T2D mice activated hepatic Nrf2 and Nrf2-regulated enzymes. FGF1ΔHBS treatment significantly increased translocation of Nrf2 to the nucleus (Fig. 2A,B) and induced known targets of Nrf2 transcriptional upregulation, including NAD(P)H dehydrogenase (quinone 1) (Fig. 2A,C) and heme oxygenase-1 (Fig. 2A,D). These results suggest an important role of Nrf2-dependent upregulation of antioxidant activity as part of FGF1ΔHBS-mediated protection from NAFLD.
Fig. 2. FGF1ΔHBS prevents the hepatic oxidative stress and lipid disorder in late-stage diabetic mice.

Nine-month-old db/db mice were treated with FGF1ΔHBS (0.5 mg/kg body weight) or PBS vehicle every other day for 3 months. (A-D) The protein expressions of Nrf2 anti-oxidative signaling [nuclear (n)-Nrf2, NQO-1, and HO-1], (F-I) the lipogenic genes [mature/precursor (m/pre) SREBP-1, FAS and SCD-1], and (J-L) AMPK signaling [phosphorylated (p)-AMPK/AMPK and p-ACC/ACC] were detected by western blot and quantified by densitometry. (E) The mRNA expression of SREBP-1, FAS and SCD-1 genes was detected by RT-qPCR. GAPDH or Lamin B1 was used as loading controls for all western blot assays. Quantitative data are expressed as mean ± SEM, n=6–10. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle treated mice. Nrf2, nuclear factor erythroid 2-related factor 2; NQO-1, NAD(P)H dehydrogenase (quinone 1); HO-1, heme oxygenase-1; SREBP-1, sterol regulatory element-binding protein 1; FAS, fatty acid synthase; SCD-1, stearoyl-CoA desaturase-1; AMPK, AMP-activated protein kinase; ACC, acetyl-CoA carboxylase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
To identify the mechanism responsible for improved hepatic lipid metabolism by FGF1ΔHBS, several important components of hepatic fatty acid synthesis and oxidation were tested. Treatment with FGF1ΔHBS markedly reduced the mRNA level (Fig. 2E) and transcriptional activity of transcription factor SREBP-1 (as indicated by the ratio of mature SREBP-1 to precursor-SREBP-1 (13, 17), Fig. 2F,G). Changes in SREBP-1 were accompanied by significant reductions in its downstream targets, stearoyl-CoA desaturase 1 (SCD-1, Fig. 2F,H) and fatty acid synthase (FAS, Fig. 2F,I) at both mRNA and protein levels. In addition, FGF1ΔHBS treatment significantly upregulated three proteins in fatty acid β-oxidation: peroxisome proliferator-activated receptor α (Supporting Fig. S6A), peroxisome proliferator-activated receptor gamma coactivator 1α (Supporting Fig. S6B), and carnitine palmitoyltransferase 1α (Supporting Fig. S6C), but was without effect on fatty acid transporter CD36 (Supporting Fig. S6D) expression. These results indicate that both inhibition of lipogenesis and promotion of fatty acid oxidation contributed to FGF1ΔHBS suppression of hepatic lipid accumulation in T2D.
AMPK is a key energy sensor that plays a critical role in hepatic lipid metabolism in diabetic mice (18). Thus, we tested for a role of AMPK signaling in FGF1ΔHBS-mediated protection against hepatic steatosis. FGF1ΔHBS treatment significantly increased phosphorylation of hepatic AMPK (Fig. 2J,K) and upregulated phosphorylation of the AMPK target, acetyl-CoA carboxylase (ACC, Fig. 2J,L). These data indicate that activated AMPK signaling is likely important to FGF1ΔHBS regulation of hepatic lipid metabolism.
In hepatocytes Nrf2 is required for FGF1 protection from palmitate-induced oxidative stress but not for FGF1 protection from palmitate-induced lipid metabolic dysregulation
To explore the hepatocellular specific basis of FGF1 action, HepG2 cells were exposed to palmitate in vitro to mimic the diabetic milieu of oxidative damage and lipid dysregulation. Paralleling the hepatic response to diabetes in vivo (Figs. 1 and 2), palmitate exposure of HepG2 cells induced oxidative stress (Fig. 3A–C), inhibited Nrf2 nuclear translocation (Supporting Fig. S7A,B) and reduced expression of downstream antioxidant enzymes NAD(P)H dehydrogenase (quinone 1) (Supporting Fig. S7A,C) and heme oxygenase-1 (Supporting Fig. S7A,D). Palmitate exposure of HepG2 cells augmented lipid accumulation seen as increased lipid droplet formation (Fig. 3D,E) and increased triglyceride contents (Fig. 3F). Lipid accumulation coincided with activation of the lipogenic gene, SREBP-1 (Supporting Fig. S7E,F), and increased expression of the SREBP-1 target genes: FAS (Supporting Fig. S7E,G) and SCD-1 (Supporting Fig. S7E,H). Lipid accumulation also coincided with reduced phosphorylation of both AMPK (Supporting Fig. S7E,I) and ACC (Supporting Fig. S7E,J). Importantly, and as seen in diabetic livers, all of these changes induced by palmitate exposure of HepG2 cells were prevented by FGF1ΔHBS or FGF1WT treatment in an insulin-dependent manner (Fig. 3; Supporting Fig. S7).
Fig. 3. FGF1 protects against palmitate (Pal)-induced oxidative stress and lipid disorder in HepG2 cells.
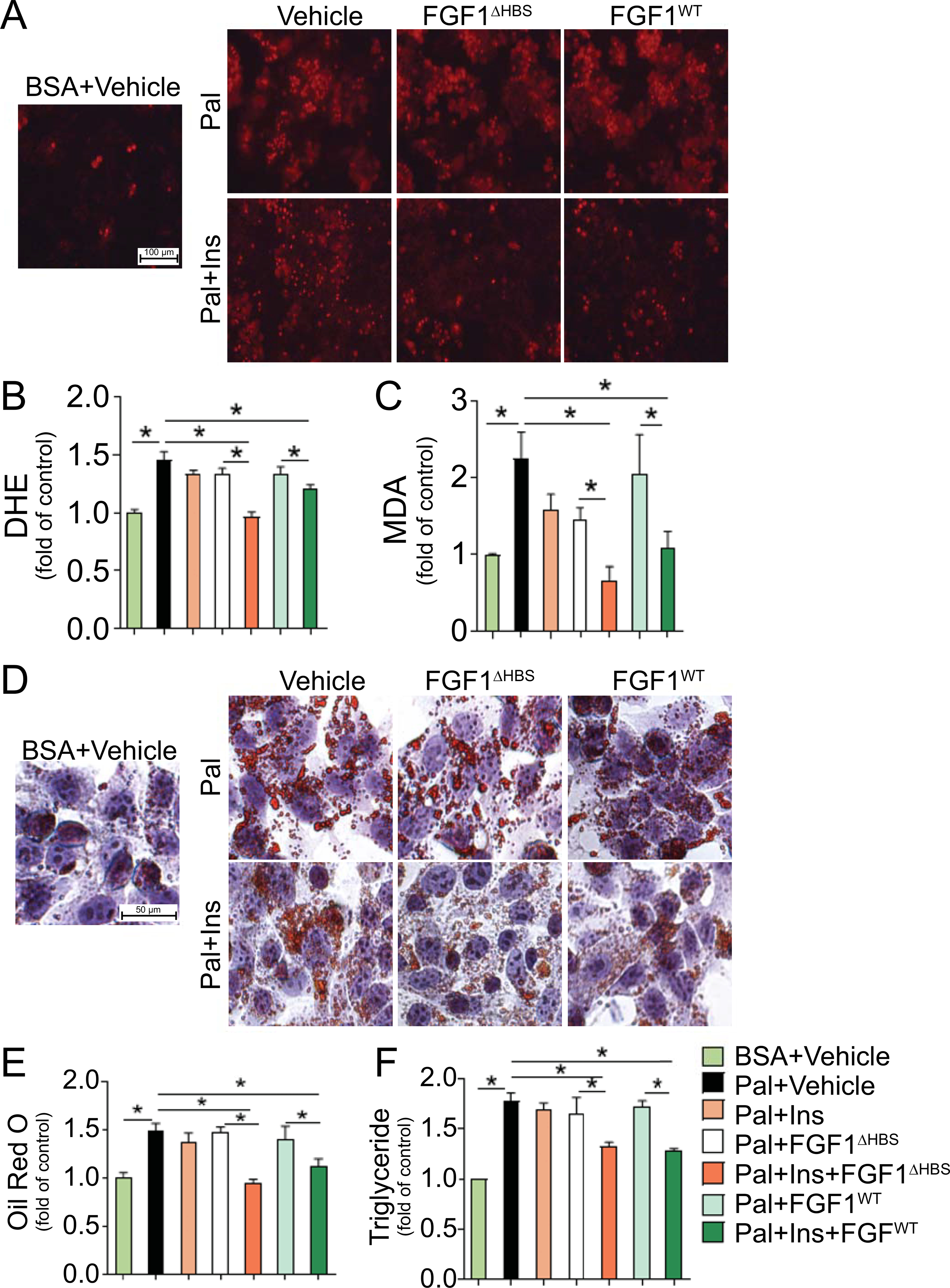
After serum starvation for 24 hours, HepG2 cells were treated with Pal-bovine serum albumin (BSA, 100 μmol/L) or control BSA at the presence or absence of insulin (Ins, 100 nmol/L) in fetal bovine serum (FBS) free medium for 12 hours, followed by incubation with FGF1WT and FGF1ΔHBS (1000 ng/ml) for additional 12 hours, and PBS was used as vehicle control. (A) Representative images of dihydroethidium (DHE) stained HepG2 cells. (B) Quantitative analysis of fluorescent intensity of DHE staining. (C) Malondialdehyde (MDA) contents in cell lysate. (D,E) Lipid deposition was determined by Oil Red O staining. (F) Triglyceride contents in cell lysate. Quantitative data are expressed as mean ± SEM, n=3–5. *P < 0.05.
Chronic oxidative stress plays an important role in the progression of fatty liver disease, possibly due to linkage between oxidative stress and dysregulated lipid metabolism (19). Nrf2, in addition to responding to oxidative stress, also has a critical role in lipid metabolism (16). To test if the antioxidant and/or anti-steatosis actions of Nrf2 were required for the response to FGF1 treatment, loss-of-function studies were performed using siRNA knockdown of Nrf2 in HepG2 cells. As intended, Nrf2 siRNA greatly reduced total Nrf2 (Fig. 4A) and nuclear Nrf2 (Fig. 4A,B). Knockdown of Nrf2 was accompanied by downregulation of Nrf2 target antioxidant genes (Fig. 4A,C,D) that resulted in a more severe oxidative stress response to palmitate exposure, as indicated by increased MDA production (Fig. 4E). Importantly, Nrf2 knockdown completely abolished the protective effects of treatment with FGF1ΔHBS or FGF1WT against palmitate-induced oxidative stress in HepG2 cells (Fig. 4E). However, knockdown of Nrf2 did not reduce the protective actions of either FGF1ΔHBS or FGF1WT treatment against lipid accumulation, as reflected by triglyceride contents (Fig. 4F) and lipogenic gene upregulation (Fig. 4G–J). These findings indicate that FGF1 protection from hepatic oxidative stress requires Nrf2; but in contrast, Nrf2 is unnecessary for FGF1-mediated protection from dysregulated lipid metabolism.
Fig. 4. The effects of siRNA Knockdown of Nrf2 on the beneficial effects of FGF1 on palmitate (Pal)-induced oxidative stress and lipid disorder in HepG2 cells.

After serum starvation for 24 hours, cells with or without siRNAs against Nrf2 or control (Ctrl) siRNA were treated with Pal-BSA (100 μmol/L) or control BSA at the presence or absence of insulin (Ins, 100 nmol/L) in fetal bovine serum (FBS) free medium for 12 hours, followed by incubation with FGF1WT and FGF1ΔHBS (1000 ng/ml) for additional 12 hours, and PBS was used as vehicle control. (A-D) The protein expressions of Nrf2 signaling [n-Nrf2, total (t)-Nrf2, NQO-1 and HO-1] were determined by western blot and quantified by densitometry. (E) MDA contents in cell lysate. (F) Triglyceride contents in cell lysate. (G-J) The protein expressions of lipogenic genes (m/pre SREBP-1, FAS, SCD-1) were determined by western blot and quantified by densitometry. GAPDH or LaminB1 was used as loading controls for all western blot assays. Quantitative data are expressed as mean ± SEM, n=3–5. *P < 0.05. NS, no significant difference.
In hepatocytes AMPK is required for FGF1 protection from both palmitate-induced lipid metabolic dysregulation and palmitate-induced oxidative stress
Previous studies in T2D indicated that hepatic metabolic dysregulation could be prevented by AMPK activation (20, 21). Based on our findings that FGF1 activates AMPK in vivo (Fig. 2) and in vitro (Fig. 4), we wondered whether AMPK was required for the protective effects of FGF1 against liver damage in T2D. To test this hypothesis, HepG2 cells were pretreated with Compound C, a selective AMPK inhibitor. Compared with dimethyl sulfoxide vehicle control, Compound C treatment significantly reduced AMPK expression (Supporting Fig. S8A,B) and phosphorylation (Supporting Fig. S8A,C). This was accompanied by reduced phosphorylation of the AMPK target, ACC (Supporting Fig. S8A,D) and greater maturation of SREBP-1 (Supporting Fig. S8A,E). Furthermore, inhibiting AMPK with Compound C significantly aggravated palmitate-induced lipid accumulation, as shown by increased triglyceride contents (Supporting Fig. S8F), and abolished the ability of either FGF1ΔHBS or FGF1WT treatment to inhibit lipid accumulation (Supporting Fig. S8F) and block upregulation of lipogenic genes (Supporting Fig. S8A,G,H). Surprisingly, Compound C inhibition of AMPK also exacerbated palmitate-induced oxidative stress in the absence or presence of FGF1 (Supporting Fig. S8I). This was evident by more MDA production (Supporting Fig. S8I), nearly complete attenuation of Nrf2 nuclear translocation (Supporting Fig. S8J,L) and reduced expression of Nrf2 downstream antioxidant genes (Supporting Fig. S8J,M,N). These changes occurred without significant effects on total Nrf2 expression (Supporting Fig. S8J,K). These results demonstrated that pharmacological inhibition of hepatic AMPK blocked FGF1 protection against both palmitate-induced lipid metabolic dysregulation and palmitate-induced oxidative stress.
To exclude potential non-specific actions of Compound C and to eliminate differences between HepG2 cells and primary hepatocytes, we next performed a loss-of-function study using AMPKα-siRNA in primary mouse hepatocytes. Compared with control-siRNA, AMPKα-siRNA knockdown of AMPKα almost completely eliminated AMPK expression (Fig. 5A) and phosphorylation (Fig. 5A,B) that was accompanied by reduced phosphorylation of AMPK downstream target gene, ACC (Fig. 5A,C). Furthermore, knockdown of AMPKα markedly elevated palmitate-induced lipid accumulation, reflected by increased triglyceride contents (Fig. 5H), and completely abolished the protective effects of both FGF1ΔHBS and FGF1WT against lipid accumulation (Fig. 5H) and lipogenic genes upregulation (Fig. 5D–G). Importantly, knockdown of AMPKα also aggravated palmitate-induced oxidative stress and completely abolished the protective effects of both FGF1ΔHBS and FGF1WT against oxidative stress induced by palmitate in primary hepatocytes, reflected by greater MDA production (Fig. 5M). These results coincided with a near complete attenuation of Nrf2 nuclear translocation (Fig. 5I,J) and less expression of Nrf2 downstream antioxidant genes (Fig. 5I,K,L) under basal and FGF1-stimulated conditions. These results support the notion that AMPK plays a central role in FGF1-mediated protection from palmitate-induced dysregulation of hepatic lipid metabolism and hepatic oxidative stress.
Fig. 5. The effects of siRNA knockdown of AMPKα on the protective effects of FGF1 against palmitate (Pal)-induced lipid disorder and oxidative stress in primary hepatocytes.

After serum starvation for 24 hours, cells with or without siRNAs against AMPKα1/2 or control (Ctrl) siRNA were treated with Pal-BSA (100 μmol/L) or control BSA at the presence or absence of insulin (Ins, 100 nmol/L) in FBS free medium for 12 hours, followed by incubation with FGF1WT and FGF1ΔHBS (1000 ng/ml) for additional 12 hours, and PBS was used as vehicle control. (A-C) Protein expressions of AMPK signaling pathway (p-AMPK/AMPK and p-ACC/ACC), the (D-G) lipogenic genes (m/pre SREBP-1, FAS, SCD-1), and (I-L) the Nrf2 signaling (n-Nrf2, NQO-1 and HO-1) were determined by western blot and quantified by densitometry. (H) Triglyceride contents in cell lysate. (M) MDA contents in cell lysate. β-actin or LaminB1 was used as loading controls for all western blot assays. Quantitative data are expressed as mean ± SEM, n=3–5. *P < 0.05. NS, no significant difference.
In hepatocytes FGF1 activation of AMPK and protection from palmitate-induced lipid accumulation is mediated by FGFR4
Among 4 major subtypes of FGFRs, FGFR4 was most abundant in isolated primary hepatocytes, followed by FGFR3 and FGFR1 (Fig. 6A). We next explored the role of FGFR4 in the hepatic actions of FGF1 on AMPK activation and lipid accumulation by knocking down FGFR4 expression using siRNA. Compared with control-siRNA, FGFR4-siRNA markedly reduced FGFR4 expression (Fig. 6B,C) that was accompanied by dramatically reduced phosphorylation of AMPK (Fig. 6B,D) and its downstream target gene, ACC (Fig. 6B,E) in the absence or presence of FGF1. Notably, knockdown of FGFR4 elevated palmitate-induced lipid accumulation, reflected by increased triglyceride contents (Fig. 6F), and completely abolished the protective effects of both FGF1ΔHBS and FGF1WT against triglyceride accumulation (Fig. 6F). Taken together, these findings suggest that the effects of FGF1 on hepatic AMPK activation and lipid accumulation are predominantly mediated by FGFR4.
Fig. 6. The effects of siRNA knockdown of FGFR4 on FGF1 activation of AMPK and protection from palmitate (Pal)-induced lipid accumulation in primary hepatocytes.

(A) The mRNA expressions of FGFRs (R1–4) in primary hepatocytes at 24 hours after isolation (n=3). (B-F) After serum starvation for 24 hours, cells with or without siRNAs against FGFR4 or control (Ctrl) siRNA were treated with Pal-BSA (100 μmol/L) or control BSA at the presence or absence of insulin (Ins, 100 nmol/L) in FBS free medium for 12 hours, followed by incubation with FGF1WT and FGF1ΔHBS for additional 12 hours, and PBS was used as vehicle control. (B) Protein expressions of FGFR4 and AMPK signaling pathway (p-AMPK, AMPK, p-ACC and ACC) were determined by western blot. (C-E) The quantitative analysis of FGFR4, p-AMPK/AMPK and p-ACC/ACC western blots by densitometry. (F) Triglyceride contents in cell lysate. Quantitative data are expressed as mean ± SEM, n=4–6. *P < 0.05. NS, no significant difference.
Liver-specific AMPK knockout abolishes FGF1ΔHBS reversal of NAFLD in HFHS fed mice
To ensure that the in vitro findings above also apply in NAFLD in vivo, studies with AMPK-LKO mice were performed. AMPK-LKO and WT mice were fed HFHS diet for 5 months followed by FGF1ΔHBS (0.5mg/kg body weight) or PBS treatment every other day for 1 month. The complete knockout of AMPK in the liver was verified by immunoblots (Fig. 7A). As expected, FGF1ΔHBS treatment significantly upregulated the phosphorylation of hepatic AMPK (Fig. 7A,B) and its downstream target ACC (Fig. 7A,C) in WT mice. But in AMPK-LKO mice, the expression and phosphorylation of AMPK (Fig. 7A,B) and its downstream target ACC (Fig. 7A,C) were almost completely abolished in the absence or presence of FGF1ΔHBS. Similar to FGF1ΔHBS protection observed in db/db mice (Fig. 1), FGF1ΔHBS treatment markedly reversed chronic HFHS diet feeding-induced liver steatosis in WT mice as evident in H&E- and Oil Red O-stained liver sections (Fig. 7D,E), liver triglyceride contents (Fig. 7F), reduced hepatic oxidative stress (Fig. 7G), and plasma markers of liver injury (Fig. 7H,I). Although hepatic AMPKα deficiency did not notably aggravate the HFHS diet-induced steatosis phenotype compared with HFHS-fed WT mice, AMPKα deficiency significantly reduced the protective effects of FGF1ΔHBS against hepatic lipid accumulation (Fig. 7E,F), MDA production (Fig. 7G) and plasma markers of liver injury (Fig. 7H,I). The ALT effect of FGF1ΔHBS (Fig. 7H) was almost normal perhaps because ALT can also come from non-hepatic tissues. Taken together, these findings strongly support an essential role of AMPK in mediating the protective effects of FGF1 that reduce both hepatic steatosis and liver injury.
Fig. 7. The therapeutic effects of FGF1ΔHBS on chronic NAFLD induced by high-fat/high-sucrose (HFHS) in mice.

At the age of 2 months, male wild type (WT) and liver specific AMPK knockout (AMPK-LKO) mice were fed on a HFHS diet (D12327, Research Diets) for 5 months, and then supplemented with FGF1ΔHBS (0.5mg/kg body weight) or PBS vehicle every other day for 1 month. (A) The protein expressions of AMPK signaling (p-AMPK/AMPK and p-ACC/ACC) were detected by western blot. (B,C) The quantitative analysis of western blots by densitometry. (D) Representative images of H&E- and (E) Oil Red O-stained liver sections. (F) Triglyceride contents in liver tissues. (G) MDA contents. (H,I) The plasma ALT and AST activity. β-actin was used as loading controls for all western blot assays. Quantitative data are expressed as mean ± SEM, n=6 for A-C, and n=7–11 for D-I. *P < 0.05 vs WT vehicle treated mice; #P < 0.05 vs WT FGF1ΔHBS treated mice.
FGF1ΔHBS attenuates the development of NASH in HFHC diet-fed ApoE-KO mice
To investigate the effects of FGF1ΔHBS on NASH, a HFHC diet-induced ApoE-KO mouse model was used (14, 22). Compared with normal chow-fed ApoE-KO mice, HFHC-fed mice had obvious morphological and pathological features of NASH, including increased liver size and weight (Fig. 8A,B), increased NAS score (Fig. 8C,D), elevated hepatic lipids accumulation (Fig. 8E,F; Supporting Fig. S9A,B), aggravated hepatic fibrosis indicated by collagen deposition (Fig. 8G,H) and fibrotic marker expression (Supporting Fig. S10A), and increased hepatic inflammation shown by macrophage infiltration (Fig. 8I) and hepatic inflammatory marker expression (Supporting Figs. S10B). These changes were accompanied by dramatic elevation of the plasma markers of liver injury: ALT and AST (Supporting Fig. S9F,G). Strikingly, FGF1ΔHBS treatment almost completely prevented the progression of HFHC-induced NASH in ApoE-KO mice (Fig. 8A–H; Supporting Fig. S9 and S10) that coincided with preservation of hepatic AMPK signaling (Fig. 8J–L). FGF1ΔHBS treatment also significantly decreased plasma concentrations of total cholesterol (Supporting Fig. S9C) and low-density lipoprotein fraction (Supporting Fig. S9D), yet without significant effect on plasma triglycerides (Supporting Fig. S9E). In line with the hepatic proliferation results in db/db mice, FGF1ΔHBS significantly reduced expression of cell proliferative markers proliferating cell nuclear antigen and Ki67 (Supporting Fig. S11) in livers of ApoE-KO mice. These results clearly demonstrate that FGF1ΔHBS can protect against NASH without stimulating hepatic proliferative potential in mice.
Fig. 8. The preventive effects of FGF1ΔHBS on nonalcoholic steatohepatitis (NASH) induced by high-fat/high-cholesterol (HFHC) in ApoE-KO mice.

Eight-week-old male ApoE-KO mice were fed on a HFHC diet and treated with FGF1ΔHBS (0.5 mg/kg body weight) or PBS vehicle every other day for 3 months. Age- and sex-matched ApoE-KO mice fed a normal chow and treated with PBS vehicle were used as controls. (A) Liver size and (B) weight. Representative images of (C) H&E, (E) Oil Red O, (G) Sirius Red, and (I) F4/80 staining of liver sections. (D) NAFLD activity score (NAS). (F) Triglyceride contents in liver tissues. (H) Quantitative analysis of Sirius Red positive-stained area of liver sections, normalized to “Chow+Vehicle” control group. (J-L) Protein expression for AMPK signaling (p-AMPK/AMPK and p-ACC/ACC) was detected by western blot and quantified by densitometry in liver tissues. α-tubulin was used as loading control. Quantitative data are expressed as mean ± SEM, n=6–9 for A-I, n=5 for J-L. (M) A mechanistic illustration of the basis of FGF1 preventection from metabolic disorder (hyperlipidemia and/or hyperglycemia)-induced hepatic oxidative stress and lipid disorder via FGFR4-mediated activation of AMPK signaling pathways. FGF1 enhances hepatic lipid metabolism and anti-oxidative signaling via FGFR4-mediated activation of AMPK to inhibit the lipid accumulation and activate Nrf2-mediated anti-oxidative signaling pathways, thus preventing metabolic syndrome-induced hepatic lipid disorder and oxidative stress, resulting in protection against NAFLD in T2D and obesity.
Discussion
T2D and NAFLD share common characteristics and risk factors, resulting in a high prevalence of NAFLD among individuals with T2D.(23) Because the severity of NAFLD is exacerbated by concurrent T2D, the lack of effective NAFLD treatments is most serious for patients with both NAFLD and T2D. The discovery in mouse models that FGF1 has positive effects on adipose remodeling (6), insulin resistance (7), hyperglycemia (7), and steatosis (11) suggests unique potential for FGF1 as a treatment for patients with NAFLD, T2D or both conditions. However, FGF1 has mitogenic properties that elevate cancer risk and present a major obstacle to FGF1’s therapeutic use in metabolic diseases (4). We previously reported that FGF1ΔHBS, a modified version of native FGF1, has markedly lower mitogenic activity, but retains full metabolic activity against diabetes with respect to lowering blood glucose, restoring insulin sensitivity and preventing hepatic lipid accumulation (10). Here we demonstrate that non-mitogenic FGF1ΔHBS is similarly effective as native FGF1 in vitro and in vivo for preventing hepatic oxidative stress, inhibiting lipogenesis, reducing lipid accumulation and decreasing inflammation. Furthermore, our mechanistic studies in hepatic cells and AMPK-LKO mice reveal that FGF1ΔHBS induction of antioxidant and metabolic responses requires hepatic AMPK activation via FGFR4. Moreover, non-mitogenic FGF1ΔHBS is able to ameliorate HFHC diet-induced NASH with hepatic fibrosis.
The most clinically relevant findings of this study are that FGF1ΔHBS treatment reverses NAFLD in late-stage of db/db T2D mice (Fig. 1) and prevents HFHC diet-induced NASH in ApoE-KO mice. Protection from liver steatosis by FGF1ΔHBS had previously been shown only in 3 months old db/db mice (10). This conclusion that FGF1ΔHBS can reverse NAFLD in late-stage of db/db T2D mice is based on the efficacy of FGF1ΔHBS to reduce multiple parameters of NAFLD in 9–12 months old db/db mice. The improved parameters include liver weight, size, lipid accumulation, inflammation and oxidative stress as well as plasma ALT and AST. Because NAFLD is usually diagnosed in patients long after NAFLD begins, the ability to reverse pre-existing disease is a likely prerequisite to clinical value of any new treatment in this large population of patients.
A previous study demonstrated that native FGF1 could prevent hepatic steatosis and inflammation in ob/ob NAFLD mice. However, in choline-deficient, L-amino acid-defined diet-induced NASH mice, FGF1 could only reduce hepatic inflammation but it could not prevent hepatic steatosis and fibrosis (11). The inability of FGF1 to prevent hepatic steatosis in the choline-deficient, L-amino acid-defined diet-induced model may have been due to the defect in hepatic lipid β-oxidation produced by choline deficiency (11). Herein we tested the anti-NASH effects of FGF1ΔHBS in a HFHC diet-induced ApoE-KO model. The results clearly indicate that FGF1ΔHBS can protect from NASH in this model as indicated by prevention of hepatocellular ballooning, steatosis, inflammation, and fibrosis (Fig. 8). The abilities of FGF1ΔHBS to prevent NASH and to reverse established NAFLD in mouse models provide a strong impetus to continue the evaluation of its clinical therapeutic potential.
As FGF1 effectively lowers blood glucose in obese, type 2 diabetic mice (6, 7, 10), an adequate mechanism to explain FGF1-protection against NAFLD may simply be that it normalizes elevated levels of blood glucose. However, recent studies demonstrate benefits of FGF1 on diabetic complications that do not depend on reduced hyperglycemia. Our group reported that FGF1 ameliorated diabetic nephropathy in type 1 diabetic mice despite no measurable effect on blood glucose (8), and previous studies have shown that FGF1 exhibited protective effects against diabetic cardiomyopathy in type 1 diabetic mice without lowering blood glucose (24). To assess whether FGF1 could protect liver cells by mechanisms independent of ambient glucose we utilized cultures of hepatic cells exposed to palmitate to mimic diabetic- or obesity-induced hyperlipidemia. The results demonstrate that treatment of cultured hepatocytes with FGF1WT or FGF1ΔHBS protects human HepG2 cells (Fig. 3; Supporting Fig. S7) and mouse primary hepatocytes (Fig. 5 and Fig. 6) from palmitate-induced oxidative stress and lipid disorders, independent of changes in exposure to glucose. Based on these direct actions on hepatocytes, it is evident that FGF1ΔHBS has actions independent of lowering blood glucose, which are integral to its ability to protect from NAFLD.
Oxidative stress is elevated in diabetic liver and the progression of NAFLD pathology is accelerated by oxidative stress (19). Therefore, reducing hepatic oxidative stress is an obvious therapeutic strategy to attempt to slow NAFLD onset in diabetes. In the present study, we observed that FGF1ΔHBS administration to db/db mice inhibited hepatic superoxide generation and MDA production (Fig. 1; Supporting Fig. S4) indicating a hepatic antioxidant response. Nrf2-mediated signaling plays a critical role activating the antioxidant response by upregulating multiple antioxidant enzymes (16). In livers of db/db mice, FGF1ΔHBS treatment activated Nrf2 signaling as indicated by increased nuclear translocation of Nrf2 and upregulation of the Nrf2 regulated antioxidant genes, heme oxygenase-1 and NAD(P)H dehydrogenase (quinone 1) (Fig. 2). These findings implicate hepatocyte Nrf2 as a key mediator of FGF1ΔHBS protection against NAFLD associated oxidative stress in T2D.
The same liver samples that revealed FGF1ΔHBS-induced activation of Nrf2 signaling also demonstrated improved lipid metabolism: Compared with untreated db/db liver samples, FGF1ΔHBS treated samples had much less lipid accumulation (Fig. 1), inhibition of lipogenic transcription factor SREBP-1 and lower expression of SREBP-1 target genes: FAS and SCD-1 (Fig. 2). Transcription factor Nrf2 not only upregulates expression of antioxidant genes, but it can also regulate expression of lipid metabolism genes leading to reduced lipogenesis and lipid accumulation (16, 25). Because known transcriptional actions of Nrf2 can mediate major antioxidant and lipid metabolic actions in db/db liver, we hypothesized that all protective actions of FGF1ΔHBS against NAFLD in db/db mice may be mediated by Nrf2. To test this hypothesis, Nrf2-siRNA knockdown studies were performed in HepG2 cells. These results showed that knockdown of Nrf2 completely abolished the ability of FGF1WT and FGF1ΔHBS to ameliorate palmitate-induced oxidative stress and to prevent palmitate-induced inhibition of antioxidant gene expression (Fig. 4). However, knockdown of Nrf2 had no significant effect on the ability of FGF1WT or FGF1ΔHBS to ameliorate palmitate-induced lipid accumulation or to prevent palmitate-induced modulation of lipogenic gene expression (Fig. 4). These findings indicate that both FGF1WT and FGF1ΔHBS prevent hepatic oxidative stress by activating Nrf2 signaling, but their anti-steatosis effects are independent of Nrf2 activation.
The fact that Nrf2 signaling does not mediate anti-steatosis effects of FGF1 leads to our second innovative finding that AMPK is a key downstream target for FGF1 protection against NAFLD. AMPK is a central metabolic sensor and regulator with a major role in many metabolic diseases. Activation of AMPK stimulates hepatic fatty acid oxidation and ketogenesis, inhibits cholesterol synthesis, lipogenesis, and triglyceride synthesis (26, 27). In addition, AMPK also acts as a redox sensor and regulator of redox balance through activation of multiple target proteins (27, 28). Several recent studies demonstrate crosstalk between Nrf2 and AMPK under different pathophysiological conditions (29, 30). In the present study, FGF1ΔHBS administration upregulated the phosphorylation of AMPK and ACC, and inhibited the transcription activity of SREBP-1 and the expression of its downstream target genes, FAS and SCD-1, in the liver of late-stage db/db mice (Fig. 2), and HFHS-fed mice (Fig. 7). Based on these findings, we hypothesized that FGF1 might reverse NAFLD in T2D by upregulating AMPK-mediated lipid metabolic and anti-oxidative pathways. Consistent with this hypothesis, pharmacological inhibition or gene specific knockdown of AMPK in human HepG2 cells or mouse primary hepatocytes eliminated the ability of FGF1WT or FGF1ΔHBS to suppress palmitate-induced lipid deposition and oxidative stress (Fig. 5; Supporting Fig. S8). AMPK inhibition or knockdown also eliminated the ability of FGF1WT and FGF1ΔHBS to inhibit SREBP-1-mediated lipogenic signaling and to protect Nrf2-mediated antioxidative signaling (Fig. 5; Supporting Fig. S8). Most importantly, liver specific knockout of AMPK completely abolished FGF1ΔHBS-reversal of NAFLD and hepatic lipogenic signal derangement (Fig. 7). All these findings support an essential role of AMPK in the protective antioxidant and lipid metabolic responses of FGF1 treatment in NAFLD. Interestingly, a recent study showed that activation of AMPK by an aminopropyl carbazole compound induces FGF1 in an HFD-induced NAFLD mouse model (31), suggesting a possible feedback mechanism by which FGF1 is able to activate AMPK, and then AMPK also upregulates FGF1 under metabolic disorder conditions. This potential feedback mechanism and its relevance to the pathogenesis and clinical treatment of NAFLD is worthy of further investigation.
There are at least seven FGFR proteins generated from FGFR1, FGFR2, FGFR3 and FGFR4 genes by alternative splicing (32). FGF1 is considered a universal ligand because it can bind and activate all of the receptors (4, 7, 33). In the present study, we found FGFR1, FGFR2, FGFR3 and FGFR4 isoforms in primary hepatocytes with highest abundance for FGFR4 (Fig. 6). However, the exact roles of FGFR4 in whole animal and liver metabolism remains controversial (34, 35). An earlier study found that global FGFR4-deficient mice exhibited systemic features of metabolic syndrome even on a normal diet (34). Surprisingly the same global FGFR4 deficiency alleviated HFD-induced fatty liver, while restoration of FGFR4 in hepatocytes restored sensitivity to HFD-induced fatty liver. In the current study, siRNA knockdown of FGFR4 in mouse primary hepatocytes (Fig. 6) markedly abolished FGF1 activation of AMPK and its downstream signaling and blunted FGF1 protection against palmitate-induced lipid accumulation. These findings demonstrate, at least in the hepatocyte, that FGFR4 mediates the direct protective actions of FGF1 against lipotoxicity. However, based on the earlier findings (34, 35) in global FGFR4-deficient mice, FGFR4 in different organs may exert complex (indirect) effects on the liver.
There are several limitations in our study. First, our observations are solely based on rodent models. In light of the fact that there is a difference in hepatic lipid metabolism between rodents and humans, the pathophysiological relevance of our findings remains to be confirmed in large animals and in clinical studies. Second, our data demonstrate that the lipid-lowering action of FGF1 requires hepatocyte FGFR4 to activate AMPK, but the complete signaling pathway and the defects that lead to impaired lipid metabolism in NAFLD need further investigation. Third, although our data demonstrate FGF1 reversal of palmitate-induced triglyceride accumulation in an insulin-dependent manner in hepatic cells in vitro, how insulin sensitizes this effect of FGF1 in vivo remains to be investigated in future studies.
In summary, our findings indicate that the novel, non-mitogenic variant FGF1ΔHBS exhibits therapeutic effects against NAFLD in T2D and HFHS model and NASH in HFHC model via effectively improving hepatic lipid metabolism while inhibiting hepatic oxidative stress, inflammation, and fibrosis. Thus, FGF1ΔHBS has great potential for treatment of NAFLD and NASH with low risk of cancer. Importantly, our studies provide new insight into the mechanism(s) by which FGF1ΔHBS prevents hepatic oxidative stress and steatosis in T2D. AMPK activates Nrf2-mediated antioxidative pathways and inhibits lipogenic pathways, and thus, is essential for FGF1 protection against NAFLD in T2D and obesity (Fig. 8M). These results advance the application of FGF1ΔHBS towards clinical use and reveal the potential of AMPK activation for protection from NAFLD and NASH.
Supplementary Material
Acknowledgments
The authors acknowledge the technical support of Drs. Jianxiang Xu and Jing Chen, and the insightful suggestions from Drs. Wenke Feng and Leah J. Siskind.
Financial Support: This study was supported in part by a Junior Faculty Award (1–13-JF-53) and a Basic Research Award (1–18-IBS-082) from American Diabetes Association; National Key R&D Program of China (2017YFA0506000); and NIH grants: GM127607 (D.J.C.), and R01 DE13686 (M.M.).
Abbreviations:
- T2D
type 2 diabetes
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- FGF1
fibroblast growth factor 1
- FGFR
FGF receptor
- HFD
high fat diet
- ApoE-KO
apolipoprotein E knockout mice
- AMPK, AMP
activated protein kinase
- Nrf2
nuclear factor erythroid 2-related factor 2
- AMPK-LKO
liver-specific AMPK knockout mice
- HFHS
high-fat/high-sugar diet
- HFHC
high-fat/high-cholesterol diet
- HepG2
human hepatocellular carcinoma cell line
- PBS
phosphate buffered solution
- WT
wide type
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- H&E
hematoxylin and eosin
- MDA
malondialdehyde
- SREBP-1
sterol regulatory element-binding protein 1
- FAS
fatty acid synthase
- SCD-1
stearoyl-CoA desaturase 1
- siRNA
interfering RNA
- NAS
NAFLD activity score
Footnotes
Conflict of interest statement: No potential conflicts of interest relevant to this article were reported.
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record.
References and Notes:
- 1.Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003;37:917–923. [DOI] [PubMed] [Google Scholar]
- 2.Byrne CD, Olufadi R, Bruce KD, Cagampang FR, Ahmed MH. Metabolic disturbances in non-alcoholic fatty liver disease. Clin Sci (Lond) 2009;116:539–564. [DOI] [PubMed] [Google Scholar]
- 3.Cusi K Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology 2012;142:711–725 e716. [DOI] [PubMed] [Google Scholar]
- 4.Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov 2009;8:235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X The FGF metabolic axis. Front Med 2019;13:511–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jonker JW, Suh JM, Atkins AR, Ahmadian M, Li P, Whyte J, He M, et al. A PPARgamma-FGF1 axis is required for adaptive adipose remodelling and metabolic homeostasis. Nature 2012;485:391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suh JM, Jonker JW, Ahmadian M, Goetz R, Lackey D, Osborn O, Huang Z, et al. Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature 2014;513:436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang G, Song L, Chen Z, Qian Y, Xie J, Zhao L, Lin Q, et al. Fibroblast growth factor 1 ameliorates diabetic nephropathy by an anti-inflammatory mechanism. Kidney Int 2018;93:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korc M, Friesel RE. The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets 2009;9:639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Z, Tan Y, Gu J, Liu Y, Song L, Niu J, Zhao L, et al. Uncoupling the Mitogenic and Metabolic Functions of FGF1 by Tuning FGF1-FGF Receptor Dimer Stability. Cell Rep 2017;20:1717–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W, Struik D, Nies VJ, Jurdzinski A, Harkema L, de Bruin A, Verkade HJ, et al. Effective treatment of steatosis and steatohepatitis by fibroblast growth factor 1 in mouse models of nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2016;113:2288–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han Y, Hu Z, Cui A, Liu Z, Ma F, Xue Y, Liu Y, et al. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nat Commun 2019;10:623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Zhao C, Xiao J, Liu L, Zhang M, Wang C, Wu G, et al. Fibroblast growth factor 21 deficiency exacerbates chronic alcohol-induced hepatic steatosis and injury. Sci Rep 2016;6:31026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farrell G, Schattenberg JM, Leclercq I, Yeh MM, Goldin R, Teoh N, Schuppan D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019;69:2241–2257. [DOI] [PubMed] [Google Scholar]
- 15.Sahai A, Malladi P, Pan X, Paul R, Melin-Aldana H, Green RM, Whitington PF. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol 2004;287:G1035–1043. [DOI] [PubMed] [Google Scholar]
- 16.Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 2014;39:199–218. [DOI] [PubMed] [Google Scholar]
- 17.Horie T, Nishino T, Baba O, Kuwabara Y, Nakao T, Nishiga M, Usami S, et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat Commun 2013;4:2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 2011;13:376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spahis S, Delvin E, Borys JM, Levy E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid Redox Signal 2017;26:519–541. [DOI] [PubMed] [Google Scholar]
- 20.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 2006;3:403–416. [DOI] [PubMed] [Google Scholar]
- 21.Woods A, Williams JR, Muckett PJ, Mayer FV, Liljevald M, Bohlooly YM, Carling D. Liver-Specific Activation of AMPK Prevents Steatosis on a High-Fructose Diet. Cell Rep 2017;18:3043–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schierwagen R, Maybuchen L, Zimmer S, Hittatiya K, Back C, Klein S, Uschner FE, et al. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci Rep 2015;5:12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bril F, Cusi K. Management of Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes: A Call to Action. Diabetes Care 2017;40:419–430. [DOI] [PubMed] [Google Scholar]
- 24.Zhao YZ, Zhang M, Wong HL, Tian XQ, Zheng L, Yu XC, Tian FR, et al. Prevent diabetic cardiomyopathy in diabetic rats by combined therapy of aFGF-loaded nanoparticles and ultrasound-targeted microbubble destruction technique. J Control Release 2016;223:11–21. [DOI] [PubMed] [Google Scholar]
- 25.Slocum SL, Skoko JJ, Wakabayashi N, Aja S, Yamamoto M, Kensler TW, Chartoumpekis DV. Keap1/Nrf2 pathway activation leads to a repressed hepatic gluconeogenic and lipogenic program in mice on a high-fat diet. Arch Biochem Biophys 2016;591:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012;13:251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Day EA, Ford RJ, Steinberg GR. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol Metab 2017;28:545–560. [DOI] [PubMed] [Google Scholar]
- 28.Shirwany NA, Zou MH. AMPK: a cellular metabolic and redox sensor. A minireview. Front Biosci (Landmark Ed) 2014;19:447–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mo C, Wang L, Zhang J, Numazawa S, Tang H, Tang X, Han X, et al. The crosstalk between Nrf2 and AMPK signal pathways is important for the anti-inflammatory effect of berberine in LPS-stimulated macrophages and endotoxin-shocked mice. Antioxid Redox Signal 2014;20:574–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Wu D, Tian Y. Fibroblast growth factor 19 protects the heart from oxidative stress-induced diabetic cardiomyopathy via activation of AMPK/Nrf2/HO-1 pathway. Biochem Biophys Res Commun 2018;502:62–68. [DOI] [PubMed] [Google Scholar]
- 31.Hua X, Sun DY, Zhang WJ, Fu JT, Tong J, Sun SJ, Zeng FY, et al. P7C3-A20 alleviates fatty liver by shaping gut microbiota and inducing FGF21/FGF1, via the AMP-activated protein kinase/CREB regulated transcription coactivator 2 pathway. Br J Pharmacol 2020. DOI: 10.1111/bph.15008. [DOI] [PubMed] [Google Scholar]
- 32.Sarabipour S, Hristova K. Mechanism of FGF receptor dimerization and activation. Nat Commun 2016;7:10262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pellegrini L, Burke DF, von Delft F, Mulloy B, Blundell TL. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature 2000;407:1029–1034. [DOI] [PubMed] [Google Scholar]
- 34.Huang X, Yang C, Luo Y, Jin C, Wang F, McKeehan WL. FGFR4 prevents hyperlipidemia and insulin resistance but underlies high-fat diet induced fatty liver. Diabetes 2007;56:2501–2510. [DOI] [PubMed] [Google Scholar]
- 35.Ge H, Zhang J, Gong Y, Gupte J, Ye J, Weiszmann J, Samayoa K, et al. Fibroblast growth factor receptor 4 (FGFR4) deficiency improves insulin resistance and glucose metabolism under diet-induced obesity conditions. J Biol Chem 2014;289:30470–30480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.