

Abstract
Cardiopulmonary arrest (CA) is the leading cause of death and disability in the United States. CA-induced brain injury is influenced by multifactorial processes, including reduced cerebral blood flow (hypoperfusion) and neuroinflammation, which can lead to neuronal cell death and functional deficits. We have identified serum and glucocorticoid-regulated kinase-1 (SGK1) as a new target in brain ischemia previously described in the heart, liver, and kidneys (i.e., diabetes and hypertension). Our data suggest brain SGK1 mRNA and protein expression (i.e., hippocampus), presented with hypoperfusion (low cerebral blood flow) and neuroinflammation, leading to further studies of the potential role of SGK1 in CA-induced brain injury. We used a 6-min asphyxia cardiac arrest (ACA) rat model to induce global cerebral ischemia. Modulation of SGK1 was implemented via GSK650394, a SGK1-specific inhibitor (1.2 μg/kg icv). Accordingly, treatment with GSK650394 attenuated cortical hypoperfusion and neuroinflammation (via Iba1 expression) after ACA, whereas neuronal survival was enhanced in the CA1 region of the hippocampus. Learning/memory deficits were observed 3 days after ACA but ameliorated with GSK650394. In conclusion, SGK1 is a major contributor to ACA-induced brain injury and neurological deficits, while inhibition of SGK1 with GSK650394 provided neuroprotection against CA-induced hypoperfusion, neuroinflammation, neuronal cell death, and learning/memory deficits. Our studies could lead to a novel, therapeutic target for alleviating brain injury following cerebral ischemia.
NEW & NOTEWORTHY Upregulation of SGK1 exacerbates brain injury during cerebral ischemia. Inhibition of SGK1 affords neuroprotection against cardiac arrest-induced hypoperfusion, neuroinflammation, neuronal cell death, and neurological deficits.
Keywords: cerebral blood flow, cerebral ischemia, neuroinflammation, neuronal cell death, serum and glucocorticoid-regulated kinase
INTRODUCTION
Cardiopulmonary arrest (CA) is devastating to major organs in the body because of wide-spread ischemia, with only a 40% survival rate. Those who survive succumb to major disabilities, impacting the quality of life (20). The heart and brain are the most sensitive to ischemic injury since both organs rely heavily upon electrical ionic fluctuations that uses copious amounts of energy. Therefore, most of these disabilities are products of immense neuronal death, particularly in brain regions responsible for learning/memory formation, i.e., CA1 region of the hippocampus (17). Numerous clinical trials have been conducted to treat cerebral ischemia, but beneficial outcomes are still challenging (except for hypothermia) (7), further highlighting the great need to develop novel pharmacological interventions.
Serum/glucocorticoid-regulated kinases (SGKs) are members of serine/threonine protein kinase family expressed in various organs including the heart, liver, pancreas, lung, kidney, intestine, and brain (14). SGKs are encoded by three genes (SGK1–3) initially discovered via the activation by serum and glucocorticoids to regulate numerous patho-/physiological processes of homeostasis, inflammation, and apoptosis (14). However, SGKs also respond to stimuli such as oxidative stress, hormones, and heat shock (14) with affected organs found in the heart, liver, and kidney (14). SGK-mediated end-organ damage has been modestly investigated with conflicting results (3, 6, 10, 15, 18, 27) with little-to-no knowledge regarding ischemia-initiated end-organ brain damage. This led us to determine the role of SGKs relevant to brain metabolism under the auspice of cerebral ischemia.
We found that rat hippocampal SGK1 mRNA and protein levels were dramatically elevated following CA. We sought to inhibit SGK1 via GSK650394 (specific SGK1 inhibitor) (2) in an asphyxial cardiac arrest model (ACA, a rat model of cardiac arrest) to determine the role of SGK1 in CA-induced hypoperfusion, neuroinflammation, and resulting brain injuries. Our results suggest that 1) elevated SGK1 levels result in cortical hypoperfusion, neuroinflammation, and neuronal cell death in the CA1 region of the hippocampus and ultimately lead to learning/memory deficits and 2) inhibition of SGK1 via GSK650394 alleviated ACA-induced hypoperfusion, neuroinflammation, and neuronal cell death to preserve neurological function in the rats. Based on our findings, brain SGK1 was detrimental during ACA-induced cerebral ischemia.
MATERIALS AND METHODS
Chemicals
GSK650394 (Tocris Bioscience, Bristol, UK), a specific SGK1 inhibitor, was dissolved in a 0.02% dimethyl sulfoxide solution and administered (1.2 μg/kg) to rats. The specificity of GSK650394 for SGK1 over other related kinases is higher than 30-fold (22). Animals received a single intracerebroventricular injection of GSK650394 to the third ventricle (2.8 mm posterior to bregma, 0 mm lateral to midline, and 8.3 mm deep) 5 min before ACA/sham surgery. This is because diffusion of the drug can be detected in the hippocampus (Fig. 2B). Dosage of GSK650394 is derived from previous study (2) and results from laser speckle contrast imaging and capillary-based immunoassay studies suggesting that GSK650394 at 1.2 µg/kg is the optimal concentration that can inhibit hypoperfusion and neuroinflammation (Fig. 3) in vivo.
Fig. 2.
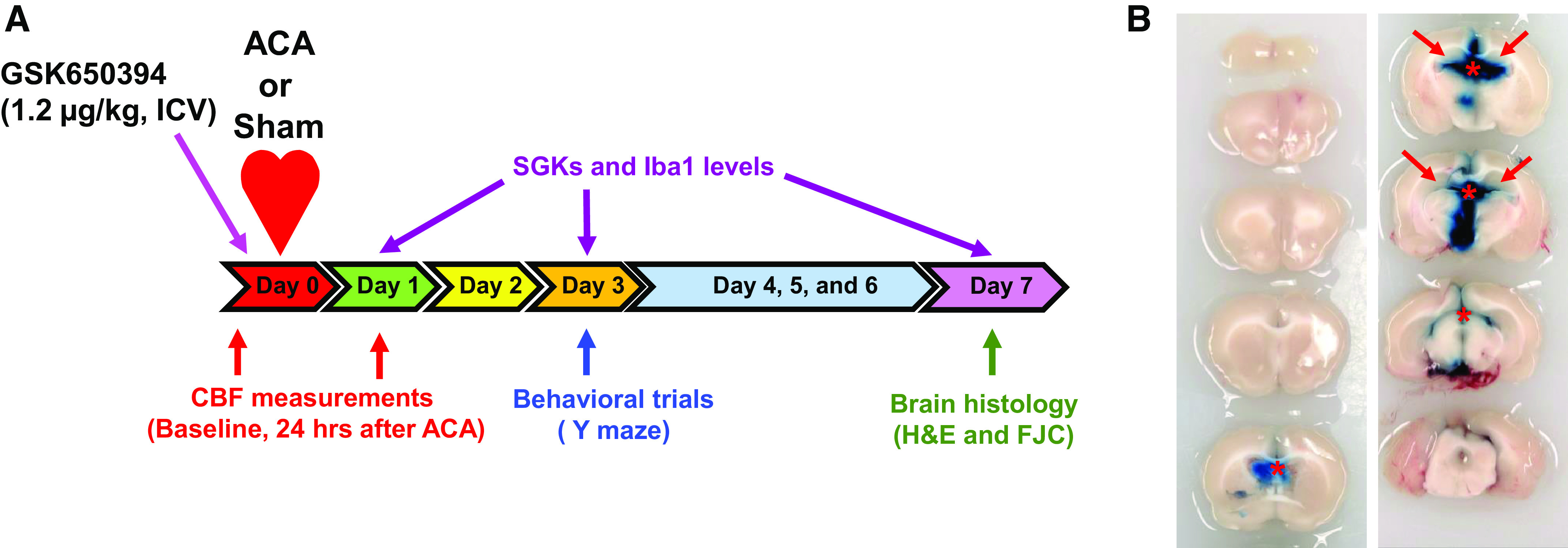
Schematic diagram of the experimental design. A: rats received an intracerebroventricular (icv) injection of GSK650394 (1.2 μg/kg) 5 min before asphyxia cardiac arrest (ACA). H&E, hematoxylin and eosin; FJC, Fluoro-Jade C. Cortical cerebral blood flow (CBF) was measured via laser speckle contrast imaging 30 min before and 24 h after ACA to examine the hypoperfusion event. Y maze was implemented 3 days after ACA/sham surgery. Upon completion of behavioral trials, rats were euthanized 7 days after ACA/sham surgery for brain histology. In a separate set of experiments, rats were euthanized 1, 3, and 7 days after ACA/sham for protein and mRNA analyses. SGK1, serum and glucocorticoid-regulated kinase-1. B: representative images of brain sections (2 mm thick) after intracerebroventricular injection of Evans blue. GSK650394 was injected the into third ventricle based on the fact that diffusion of Evans blue (tracer dye) through the ventricular system can be detected in the hippocampus (red arrows). *Evans blue was injected into the third ventricle. The rat was euthanized 5 h after injection.
Fig. 3.
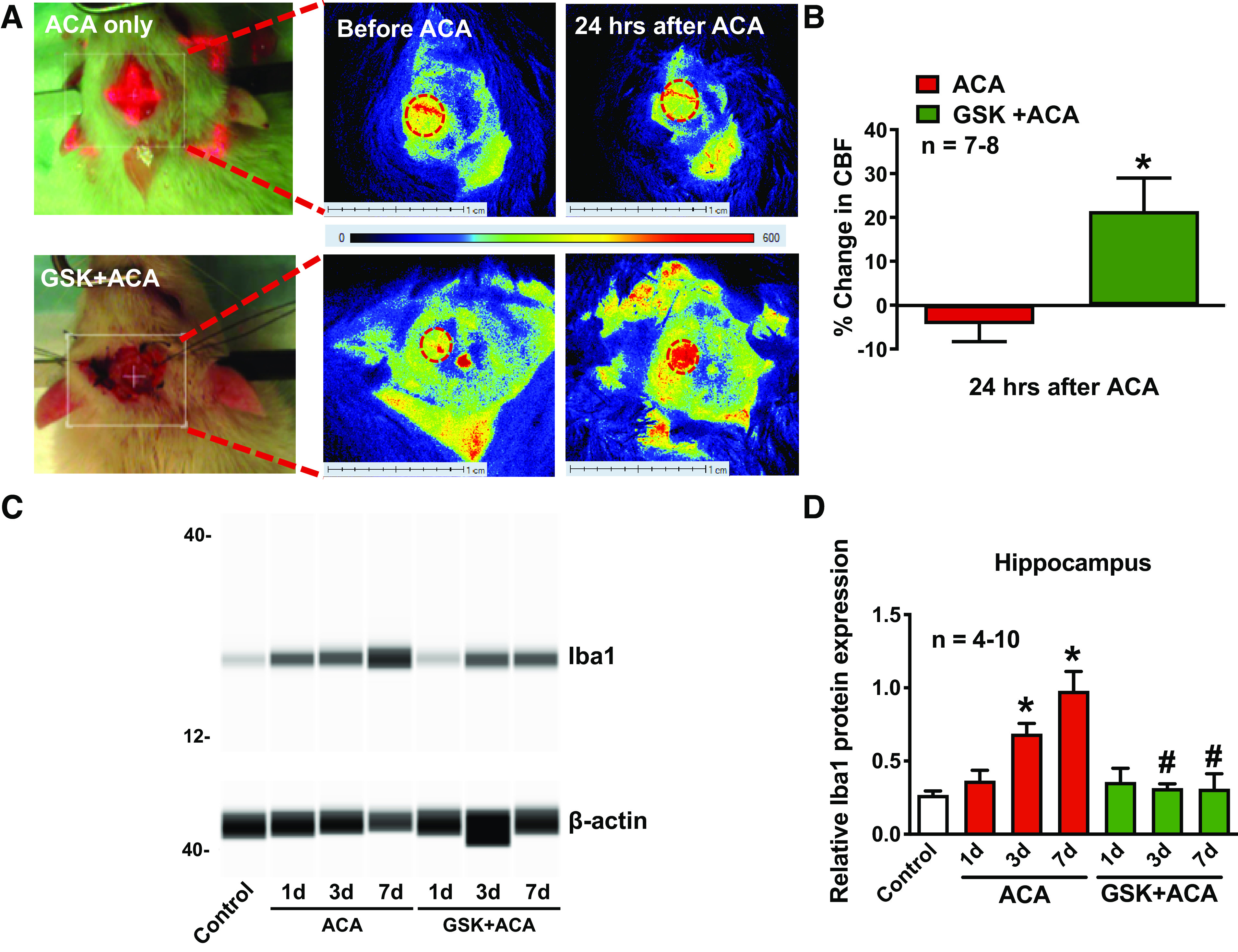
GSK650394 (GSK) alleviated asphyxia cardiac arrest (ACA)-induced hypoperfusion and neuroinflammation. Cerebral blood flow (CBF) was measured via laser speckle contrast imaging. A: representative flux images of cortical vasculature 30 min before and 24 h after ACA and GSK + ACA treatment. Dashed ovals represent the cranial window where CBF was measured. Changes in CBF were summarized in B and presented as percent change from baseline (CBF, 30 min before ACA). In a separate set of experiments, total hippocampal protein was extracted 1, 3, and 7 days (d) after ACA for Iba1 measurements. C: representative images of synthetic bands from capillary-based immunoassay. Iba1 bands were detected at 17 kDa, with β-actin present at 45 kDa. Iba1 protein levels before/after ACA were normalized to β-actin and summarized in D. Each lane corresponds to an individual capillary electrophoresis protein sample in which the conditions of adjacent lanes are fully independent. *P < 0.05, as compared with control; and #P < 0.05, as compared with respective days after ACA; n, number of experiments.
Animal Preparation
Male Sprague-Dawley rats (∼270–350 g) were purchased from Charles River Laboratories (Wilmington, MA). We chose to use male rats in these studies because continual variance of female hormones, i.e., 17β-estradiol, could have influence over the drug/treatment. It is well documented that females are less susceptible to postischemic brain damage in humans, as well as in experimental models of ischemia (1, 19). All animals were randomized to different treatment groups. Animals were housed for 1 wk within the animal facility at Louisiana State University Health Sciences Center-Shreveport. All experiments were approved by the Institutional Animal Care and Use Committee. Rats were fasted overnight before surgery. Animals were anesthetized using 4% isoflurane mixed with 30% O2-70% N2O. After endotracheal intubation, the maintenance dose of isoflurane was reduced to 1.5%. Respiration was sustained using a ventilator set to 60 breaths/min. The right femoral artery and vein were cannulated using single-lumen (PE-50) catheters to maintain readings for mean arterial pressure, analysis of arterial blood gas, blood glucose, and drug injections. Heating pads were used for the duration of the surgery to ensure head and body temperatures (36.5–37°C) were maintained. Detailed experimental design is provided in Fig. 2A.
ACA
Following catheterization, Nimbex (cisatracurium besylate, 0.27 mg/kg iv) was used to immobilize the rats. ACA was performed by removing the rats from the ventilator for 6 min. Cardiopulmonary resuscitation was executed by administrating epinephrine (0.005 mg/kg iv) and sodium bicarbonate (1 meq/kg iv), followed by mechanical ventilation with 100% O2 at 80 breaths/min. Manual chest compressions were given until the mean arterial pressure reached 60 mmHg. Animals in the control/sham-operated groups received similar surgical procedures without the induction of asphyxia. The survival rate after ACA is ∼80%. In this study, we did not observe any differences in survival rate between groups.
Laser Speckle Contrast Imaging
Laser speckle contrast imaging was used to measure cerebral blood flow (CBF). Rats were placed onto a stereotaxic frame following anesthesia. A 10-mm circular cranial window was created at the frontoparietal cortex, 1 mm lateral to bregma. Regional cortical blood flow was measured by illuminating the region of interest with a 785-nm laser via PeriCamPSIHR (Perimed, Järfälla, Sweden). The duration of laser exposure was 5 min, and blood perfusion was calculated based on pixel intensity via PIMSoft (Perimed, Järfälla, Sweden). CBF was measured 30 min before (baseline measurement) and 24 h post-ACA.
Reverse Transcriptase Polymerase Chain Reaction and Reverse Transcriptase Quantitative Polymerase Chain Reaction
RNA was isolated from the hippocampus using the RNeasy Mini Kit (Qiagen, Valencia, CA) and RNase-Free DNase Set. The isolated RNA was reverse transcribed using the SuperScript III First-Strand Synthesis System. PCR was performed using Platinum PCR SuperMix High Fidelity (Invitrogen, Carlsbad, CA). The thermal cycling (Bio-Rad, Hercules, CA) conditions were as follows: 2 min denaturation (94°C), 35 cycles of 94°C for 1 min, 60°C for 30 s, 72°C for 30 s, and 4°C at the end of reaction. In a separate set of experiments, qPCR reaction mixtures (20μl) were made based on iQ SYBR Green Supermix (Bio-Rad, Hercules, CA). The thermal cycling conditions were as follows: 3 min denaturation (95°C), 40 cycles of 95°C for 10 s, 60°C for 30 s, 2 cycles of 65°C for 5 s, and 95°C for 5 s. Triplicate reactions were quantified using the CFX96 Real-Time System (Bio-Rad, Hercules, CA). The cycle threshold (Ct) values were used for the calculation of fold changes in mRNA abundance according to the 2-ΔΔCt method. Primers for PCR are as follows; SGK1 forward 5′-TTGAGAGGGACTTGGAGGAGGT-3′, reverse 5′-CCAGAATGAGGGGAATGGTAGC-3′ (21); SGK2 forward 5′-AGTCAGGCGAGTGGGTGACAGG-3′, reverse 5′-TGGAAACCCCCAGGTTCCAGC-3′; SGK3 forward 5′-TCGCTATTTCTGATACCACCACAACC-3′ (8), reverse 5′-AGGCTCACTCCTGGTCTCAAGTTCA-3′ (8); and β-actin forward 5′-AGCCATGTACGTAGCCATCC-3′, reverse 5′-CTCACAGCTGTGGTGGTGAA-3′ (Invitrogen, Carlsbad, CA).
Capillary-Based Immunoassay
The hippocampus was collected from the brain tissue 1 and 3 days after ACA. Protein was extracted using T-PER (ThermoScientific, Waltham, MA), homogenized, centrifuged at 10,000 g for 5 min at 4°C. The supernatant was used to determine the protein levels of SGK1 and Iba1. Protein levels of SGK1 and Iba1 in hippocampal lysates were performed using a 12–230 kDa separation module on Wes system (Biotechne, Minneapolis, MN). The primary antibodies used included anti-SGK1 (1:50, ab32374, Abcam, Cambridge, UK), anti-Iba1 (1:50, GTX100042, GeneTex, Irvine, CA), and β-actin (1:200, 4970, CST, Danvers, MA). SGK1 hippocampal protein levels were normalized to the total protein of the sample using a Total Protein Detection Module (Biotechne, Minneapolis, MN), while experiments measuring Iba1 levels were normalized to β-actin.
Histology
Histological analysis of neuronal cell death in the CA1 region of the hippocampus was determined by hematoxylin and eosin (H&E) and Fluoro-Jade C (FJC) staining 7 days after ACA. Transcardial perfusion was performed with physiological saline (0.9%) for 2 min, followed by a mixture of 36% formaldehyde, glacial acetic acid, and methanol (1:1:8, FAM) for 15 min. The brain was extracted and immersed in FAM overnight at 4°C. Brains were embedded in paraffin and sectioned coronally (6 μm thickness) using a microtome (Leica, Wetzlar, Germany). Slides were stained with H&E and imaged at 40×. Separate brain sections were costained with 0.0001% FJC (AG325, MilliporeSigma, Burlington, MA) and 4′,6-diamidino-2-phenylindole (1:10,000, DAPI, D9542, MilliporeSigma, Burlington, MA) to determine CA1 hippocampal neuronal degeneration. FJC positive neurons were visualized at 20×. All investigators were blinded to the study. Dead or degenerative neurons as shown in Fig. 4, A and B, were qualitatively assessed (visual inspection), based on changes in morphological parameters, such as severe cellular shrinkage, cytoplasmic eosinophilia, pyknotic triangular-shaped nucleus, and bright green fluorescence.
Fig. 4.

Inhibition of serum and glucocorticoid-regulated kinase-1 alleviated asphyxia cardiac arrest (ACA)-induced neuronal deficits. Brain histopathology was performed 7 days after ACA. Representative images of coronal brain sections of the CA1 region of the hippocampus from control, ACA- and GSK650394 (GSK) +ACA-treated rats. Brain sections were stained with hematoxylin and eosin (H&E, purple and pink; A), FJC (green; B, left), and DAPI nuclear counterstain (blue; B, middle and right). A: healthy neurons were identified based on a lightly stained nucleus, a dark-stained nucleolus, and a red-stained cytoplasm. Dead/injured neurons contain a shrunken cytoplasm and a pyknotic nuclei. Red arrows indicate neuronal cell death, whereas green fluorescent-positive neurons in B represent degenerative neurons. Scale bars in H&E and FJC staining indicate 20 and 50 μm, respectively. C: rats were subjected to the Y-maze spontaneous alternation test 3 days after ACA to assess working/short-term memory. Control rats exhibited high alternation rates, which are indicative of intact learning/memory function. On the contrary, ACA-treated rats with learning/memory deficits displayed lower spontaneous alternation ratio (red bar). These ACA-induced learning/memory deficits can be ameliorated with GSK650394 (GSK; 1.2 μg/kg) treatment (green bar). *P < 0.05, as compared with control; #P < 0.05, as compared with respective days after ACA; n, number of experiments.
Y-Maze
Rats were subjected to the Y-maze spontaneous alternation test to assess short-term memory function 3 days after ACA (25). Rats were acclimated to the room for 10 min and placed in the center of the Y-maze to freely explore for 10 min. Trials were recorded using an overhead camera. Normal rats without cognitive deficits will explore different arms of the maze consecutively, called spontaneous alternation, because of their natural tendency to explore novelty. Spontaneous alternation ratio is calculated as the number of trials with consecutive entries into different arms of the maze divided by the total number of arm entries minus two (possible alternations).
Statistical Analysis
Results are expressed as means ± SE. Groups were tested for normal distribution using the Shapiro–Wilk test in SPSS (IBM, Armonk, NY). Statistical analysis was evaluated by one-way ANOVA (Tukey's post hoc test or Games–Howell post hoc test for neuroinflammation data sets) and independent t test as appropriate with SPSS. P < 0.05 was accepted as significance.
RESULTS
SGK1 and SGK3 were highly expressed in the hippocampus, whereas SGK2 mRNA expression was not detected (Fig. 1A). Hippocampal SGK1 mRNA levels were significantly increased 1 and 3 days after ACA (4.19 ± 0.40, 6.41 ± 1.35; fold change as compared with control) (Fig. 1B). SGK1 protein levels in the hippocampus were also significantly elevated 3 days after ACA (1.26 ± 0.09; fold-change as compared with control) (Fig. 1, C and D). Contrarily, SGK3 mRNA levels were decreased 1 day after ACA (0.71 ± 0.15; fold change as compared with control) (Fig. 1B). Since hypoperfusion is most prevalent 24 h after ACA (16, 17), we measured cortical CBF at this time point (Fig. 2). CBF decreased 24 h after ACA as compared with baseline (−4.33 ± 3.96%), whereas treatment with GSK650394 (1.2 μg/kg) significantly enhanced CBF (21.44 ± 7.53%) (Fig. 3, A and B). Blood pressure was continuously monitored during the laser speckle contrast imaging procedure. We found no changes in mean arterial pressure when GSK650394 was administered via intracerebroventricular injection (97 ± 5 mmHg vs. 94 ± 4 mmHg, comparing ACA vs. GSK+ACA-treated rats, n = 7–8). Iba1 protein levels (a marker for neuroinflammation) significantly increased 3 and 7 days after ACA (0.69 ± 0.07, 0.98 ± 0.13) as compared with control (0.27 ± 0.03), whereas GSK650394 (1.2 μg/kg) reduced Iba1 protein levels (0.31 ± 0.03, 0.31 ± 0.10) (Fig. 3, C and D). FJC-positive neurons in the CA1 region of the hippocampus were prominent 7 days after ACA as compared with control, suggesting delayed neuronal cell death, whereas inhibition of SGK1 via GSK650394 protected CA1 neurons from ACA. (Fig. 4, A and B). We further investigated whether inhibition of SGK1 could lead to more favorable neurological outcomes using the Y-maze. Rats subjected to ACA exhibited a significant decrease in their spontaneous alternation ratio (0.44 ± 0.05), whereas GSK650394 enhanced the spontaneous alternation ratio 3 days after ACA (0.61 ± 0.04) (Fig. 4C).
Fig. 1.
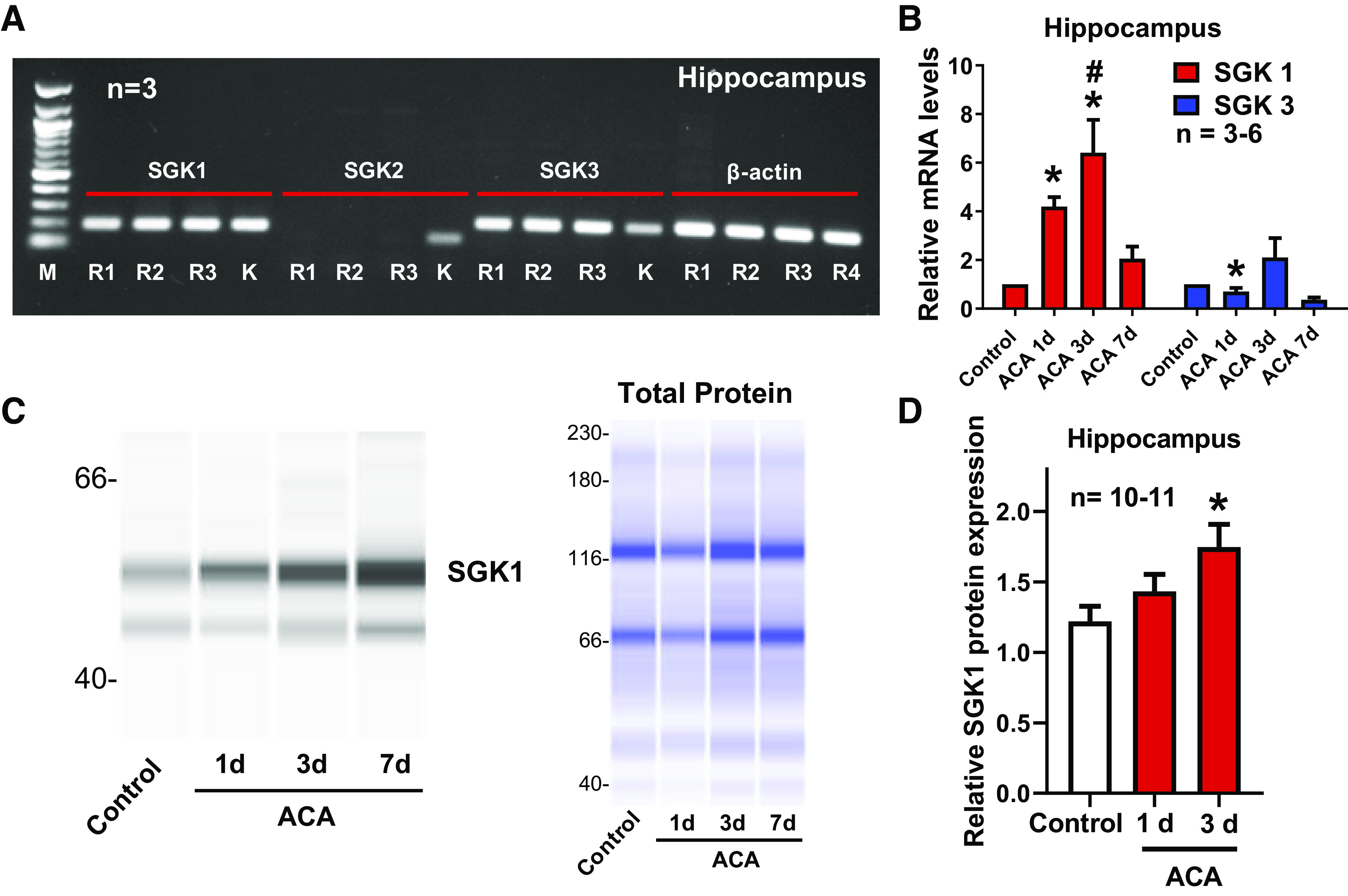
Hippocampal serum and glucocorticoid-regulated kinase-1 (SGK1) mRNA and proteins were enhanced 1 and 3 days after asphyxia cardiac arrest (ACA). A: representative images of gel electrophoresis from RT-PCR experiments. Total RNA was extracted from 3 rats denoted as R1, R2, and R3. SGK1 and SGK3 were detected in the hippocampus. SGK2 expression was not detected in the hippocampus. K indicates total RNA extracted from the rat kidney serving as a positive control, whereas β-actin served as an internal control. M indicates 100-base pair DNA ladder. B: total RNA was extracted 1, 3, and 7 days (d) after ACA. Relative hippocampal mRNA levels of SGK1 and SGK3 were measured by RT-qPCR. Results were normalized to the internal control (β-actin). C: representative images of the capillary-based immunoassay. Hippocampal total protein was extracted 1 and 3 days after ACA. SGK1 presented with bands at 49 and 55 kDa due to posttranslational modifications. Results from capillary-based immunoassay were summarized in D. SGK1 protein levels were normalized to total protein (C). *P < 0.05, as compared with control rats; and #P < 0.05, as compared with 7 days after ACA; n, number of experiments.
DISCUSSION
The major challenge in developing new therapies to manage brain injury after CA stems from the fact that CA-induced brain injuries involve multiple pathological processes, such as hypoperfusion, neuroinflammation, and cell death (23, 24, 26). These complicated mechanisms can ultimately result in severe neurological deficits. In this study, we examined the impact of SGK1 on these pathological processes in a rat model of cardiac arrest (ACA).
SGKs have been implicated in multiple cellular mechanisms, including inflammation and cell death and survival (14). Therefore, the expression of SGKs is enhanced under pathological conditions (i.e., myocardial ischemia, and neuronal excitotoxicity) (14). However, the role for SGK1 and other SGK isoforms in the context of brain ischemia have been under investigated. Interestingly, SGK1 has been identified as a potential target for diabetes (15, 18), a well-known risk factor associated with cardiovascular disease that could provide insight(s) converging to a common pathway.
In the brain, hippocampal SGK1 and SGK3 (not SGK2) were present under physiological conditions (Fig. 1A). Since SGK2 was not present in the brain, we measured the alterations of both hippocampal SGK1 and SGK3 mRNA and protein levels. Surprisingly, we found drastic changes in hippocampal SGK1 mRNA expression 1 and 3 days after ACA (Fig. 1B), whereas protein expression was enhanced 3 days after ACA (Fig. 1, C and D). Since phosphorylation of SGK1 at Thr256 and Ser422 is essential for SGK1 kinase activity (5), the discrepancy between mRNA and protein levels of SGK1 1 day after ACA may be due to the fact that SGK1 kinase activity was affected after ACA. With elevated SGK1 expression after ACA, rats presented with decreased CBF (hypoperfusion) (Fig. 3, A and B), elevated neuroinflammation and neuronal cell death (Figs. 3, C and D, and 4, A and B), and poor neurological outcomes (Fig. 4C), suggesting SGK1 plays a detrimental role in the progression of CA-induced brain injury. These results led us to further examine the impact of SGK1 on two major contributors (i.e., hypoperfusion and neuroinflammation) of brain ischemia, as well as incorporate potential therapeutic opportunities via SGK1 inhibition (GSK650394) against CA.
Although recent studies suggest SGKs can regulate vasoactive mediators (i.e., Ca2+/Na+), as well as nitric oxide production (4, 9, 14), the definitive role of SGK1 in CBF regulation during normal/pathological conditions is understudied. In fact, CBF is depressed concurrently with an enhanced SGK1 expression 24 h after ACA, whereas inhibition of SGK1 (GSK650394) prevented ACA-induced hypoperfusion (Fig. 3, A and B). To our knowledge, we are the first to identify that SGK1 can modulate brain circulation under pathological conditions; however, whether this modulation occurs through direct or indirect action on vascular tone remains unknown.
Neuroinflammatory responses are also a major contributor to ischemia-induced brain injury and neurological deficits (12, 13), with scarce research pertaining to SGK1 and neuroinflammation mainly describing peripheral organ targets. For example, upregulation of spleen SGK1 can cause T17 helper cells/regulatory T-cell imbalance, as a result of renal and cardiac inflammation and fibrosis (6). Additionally, knockout of SGK1 inhibits macrophage recruitment and polarization to attenuate cardiac inflammation and fibrosis (27), highlighting SGK1’s inflammatory role. On the contrary, pharmacological inhibition or CRISPR/Cas9-mediated knockout of SGK1 in microglial cell lines promotes microglia activation and inflammatory cytokine production in response to lipopolysaccharide exposure (3, 10). These results suggest a possible anti-inflammatory role of SGK1 in the microglia.
When compared with the aforementioned studies, we investigated the impact of SGK1 on inflammation in vivo by measuring hippocampal Iba1 (a marker of microglia activation) protein levels after ACA. Iba1 protein levels were enhanced following ACA but were attenuated by GSK650394 (Fig. 3, C and D), suggesting SGK1 can induced neuroinflammation. These paradoxical results may be due to different methodologies used to induce inflammation, such as in vitro versus in vivo and peripheral versus central organs. SGK1 is actually detrimental and can activate microglial to potentiate neuroinflammatory processes.
CA-induced hypoperfusion and neuroinflammation can trigger delayed neuronal cell death 3–7 days after the initial ischemic injury (26) but can be alleviated with a specific SGK1 inhibitor, GSK650394 (Fig. 3, A and B), to suggest neuroprotection of SGK1 inhibition in promoting neuronal survival after CA. We assessed the potential neuroprotection of SGK1 inhibition on CA1 neurons using H&E and FJC staining 7 days after ACA. Inhibition of SGK1 in ACA-treated animals can alleviate neuronal cell death in the CA1 region of the hippocampus (Fig. 4, A and B).
Individuals who recover from CA typically experience learning and memory deficits as brain regions responsible for learning/memory formation (i.e., CA1 region of the hippocampus) is highly susceptible to ischemic injury (16, 17). Since treatment with GSK650394 alleviated neuronal cell death in the CA1 region of the hippocampus 7 days after ACA (Fig. 4, A and B), we used the Y-maze task to determine whether SGK1 inhibition protected against short-term learning and memory deficits after ACA. We evaluated short-term memory because patients suffer from short-term memory deficits as compared with healthy individuals (11). We observed alleviation of neurological deficits by inhibition of SGK1 via GSK650354 in ACA-treated rats (Fig. 4C), which suggests that SGK1 can decrease functional learning/memory in cerebral ischemia. In conclusion, SGK1 is detrimental in ACA, whereas inhibiting SGK1 can alleviate ACA-induced hypoperfusion and reduced neuroinflammation and promote neuronal survival, leading to better neurological outcomes. Inhibition of SGK1 could be a potential target for the treatment against CA.
GRANTS
This work was supported by American Heart Association Grants 19CDA34660032 (to R.H.C.), 17POST33660174 (to R.H.C.), 19TPA34850047 (to H.W.L.), 19POST34380784 (to C.Y.W.), 19PRE34380808 (to A.C.), and 17GRNT33660336 (to H.W.L.) and a Louisiana State University Research Council grant-in-aid (to R.H.C.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.H.L. conceived and designed research; R.H.L., M.S.G., C.Y.W., C.L., H.E.P., and J.T.N. performed experiments; R.H.L., M.S.G., C.Y.W., C.L., A.C., H.E.P., G.A.C., C.T.C., and J.T.N. analyzed data; R.H.L., D.P., D.L., D.D., H.L., M.S.G., A.C., G.A.C., and C.T.C. interpreted results of experiments; R.H.L., M.S.G., and C.Y.W. prepared figures; R.H.L. and M.S.G. drafted manuscript; R.H.L., D.D. and H.L. edited and revised manuscript; R.H.L., D.P., D.L., D.D., H.L., M.S.G., C.Y.W., C.L., A.C., H.E.P., G.A.C., C.T.C., and J.T.N. approved final version of manuscript.
REFERENCES
- 1.Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke 29: 159–165, 1998. doi: 10.1161/01.STR.29.1.159. [DOI] [PubMed] [Google Scholar]
- 2.Anacker C, Cattaneo A, Musaelyan K, Zunszain PA, Horowitz M, Molteni R, Luoni A, Calabrese F, Tansey K, Gennarelli M, Thuret S, Price J, Uher R, Riva MA, Pariante CM. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc Natl Acad Sci USA 110: 8708–8713, 2013. doi: 10.1073/pnas.1300886110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asai H, Inoue K, Sakuma E, Shinohara Y, Ueki T. Potential implication of SGK1-dependent activity change in BV-2 microglial cells. Int J Physiol Pathophysiol Pharmacol 10: 115–123, 2018. [PMC free article] [PubMed] [Google Scholar]
- 4.Böhmer C, Palmada M, Kenngott C, Lindner R, Klaus F, Laufer J, Lang F. Regulation of the epithelial calcium channel TRPV6 by the serum and glucocorticoid-inducible kinase isoforms SGK1 and SGK3. FEBS Lett 581: 5586–5590, 2007. doi: 10.1016/j.febslet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Chen W, Chen Y, Xu BE, Juang YC, Stippec S, Zhao Y, Cobb MH. Regulation of a third conserved phosphorylation site in SGK1. J Biol Chem 284: 3453–3460, 2009. doi: 10.1074/jbc.M807502200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Du YN, Tang XF, Xu L, Chen WD, Gao PJ, Han WQ. SGK1-FoxO1 Signaling Pathway Mediates Th17/Treg Imbalance and Target Organ Inflammation in Angiotensin II-Induced Hypertension. Front Physiol 9: 1581, 2018. doi: 10.3389/fphys.2018.01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ginsberg MD, Busto R. Combating hyperthermia in acute stroke: a significant clinical concern. Stroke 29: 529–534, 1998. doi: 10.1161/01.STR.29.2.529. [DOI] [PubMed] [Google Scholar]
- 8.Heller BA, Ghidinelli M, Voelkl J, Einheber S, Smith R, Grund E, Morahan G, Chandler D, Kalaydjieva L, Giancotti F, King RH, Fejes-Toth AN, Fejes-Toth G, Feltri ML, Lang F, Salzer JL. Functionally distinct PI 3-kinase pathways regulate myelination in the peripheral nervous system. J Cell Biol 204: 1219–1236, 2014. doi: 10.1083/jcb.201307057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helms MN, Yu L, Malik B, Kleinhenz DJ, Hart CM, Eaton DC. Role of SGK1 in nitric oxide inhibition of ENaC in Na+-transporting epithelia. Am J Physiol Cell Physiol 289: C717–C726, 2005. doi: 10.1152/ajpcell.00006.2005. [DOI] [PubMed] [Google Scholar]
- 10.Inoue K, Sakuma E, Morimoto H, Asai H, Koide Y, Leng T, Wada I, Xiong ZG, Ueki T. Serum- and glucocorticoid-inducible kinases in microglia. Biochem Biophys Res Commun 478: 53–59, 2016. doi: 10.1016/j.bbrc.2016.07.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaszke-Psonka M, Piegza M, Ścisło P, Pudlo R, Piegza J, Badura-Brzoza K, Leksowska A, Hese RT, Gorczyca PW. Cognitive impairment after sudden cardiac arrest. Kardiochir Torakochirurgia Pol 13: 393–398, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jerome SN, Akimitsu T, Gute DC, Korthuis RJ. Ischemic preconditioning attenuates capillary no-reflow induced by prolonged ischemia and reperfusion. Am J Physiol 268: H2063–H2067, 1995. doi: 10.1152/ajpheart.1995.268.5.H2063. [DOI] [PubMed] [Google Scholar]
- 13.Kvietys PR, Granger DN. Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic Biol Med 52: 556–592, 2012. doi: 10.1016/j.freeradbiomed.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev 86: 1151–1178, 2006. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- 15.Lang F, Görlach A, Vallon V. Targeting SGK1 in diabetes. Expert Opin Ther Targets 13: 1303–1311, 2009. doi: 10.1517/14728220903260807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee RH, Couto E Silva A, Lerner FM, Wilkins CS, Valido SE, Klein DD, Wu CY, Neumann JT, Della-Morte D, Koslow SH, Minagar A, Lin HW. Interruption of perivascular sympathetic nerves of cerebral arteries offers neuroprotection against ischemia. Am J Physiol Heart Circ Physiol 312: H182–H188, 2017. doi: 10.1152/ajpheart.00482.2016. [DOI] [PubMed] [Google Scholar]
- 17.Lee RH, Couto E Silva A, Possoit HE, Lerner FM, Chen PY, Azizbayeva R, Citadin CT, Wu CY, Neumann JT, Lin HW. Palmitic acid methyl ester is a novel neuroprotective agent against cardiac arrest. Prostaglandins Leukot Essent Fatty Acids 147: 6–14, 2019. doi: 10.1016/j.plefa.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li P, Hao Y, Pan FH, Zhang M, Ma JQ, Zhu DL. SGK1 inhibitor reverses hyperglycemia partly through decreasing glucose absorption. J Mol Endocrinol 56: 301–309, 2016. doi: 10.1530/JME-15-0285. [DOI] [PubMed] [Google Scholar]
- 19.Murphy S, McCullough L, Littleton-Kearney M, Hurn P. Estrogen and selective estrogen receptor modulators: neuroprotection in the Women’s Health Initiative era. Endocrine 21: 17–26, 2003. doi: 10.1385/ENDO:21:1:17. [DOI] [PubMed] [Google Scholar]
- 20.Nichol G, Thomas E, Callaway CW, Hedges J, Powell JL, Aufderheide TP, Rea T, Lowe R, Brown T, Dreyer J, Davis D, Idris A, Stiell I; Resuscitation Outcomes Consortium Investigators . Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA 300: 1423–1431, 2008. doi: 10.1001/jama.300.12.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheng H, Sun T, Cong B, He P, Zhang Y, Yan J, Lu C, Ni X. Corticotropin-releasing hormone stimulates SGK-1 kinase expression in cultured hippocampal neurons via CRH-R1. Am J Physiol Endocrinol Metab 295: E938–E946, 2008. doi: 10.1152/ajpendo.90462.2008. [DOI] [PubMed] [Google Scholar]
- 22.Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W, Hammond M, Patterson JR, Thompson SK, Kazmin D, Norris JD, McDonnell DP. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res 68: 7475–7483, 2008. doi: 10.1158/0008-5472.CAN-08-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tahsili-Fahadan P, Farrokh S, Geocadin RG. Hypothermia and brain inflammation after cardiac arrest. Brain Circ 4: 1–13, 2018. doi: 10.4103/bc.BC_4_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolfson SK Jr, Safar P, Reich H, Clark JM, Gur D, Stezoski W, Cook EE, Krupper MA. Dynamic heterogeneity of cerebral hypoperfusion after prolonged cardiac arrest in dogs measured by the stable xenon/CT technique: a preliminary study. Resuscitation 23: 1–20, 1992. doi: 10.1016/0300-9572(92)90158-9. [DOI] [PubMed] [Google Scholar]
- 25.Wu CY, Lerner FM, Couto E Silva A, Possoit HE, Hsieh TH, Neumann JT, Minagar A, Lin HW, Lee RH. Utilizing the Modified T-Maze to Assess Functional Memory Outcomes After Cardiac Arrest. J Vis Exp (131), 56694, 2018. doi: 10.3791/56694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiang Y, Zhao H, Wang J, Zhang L, Liu A, Chen Y. Inflammatory mechanisms involved in brain injury following cardiac arrest and cardiopulmonary resuscitation. Biomed Rep 5: 11–17, 2016. doi: 10.3892/br.2016.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang M, Zheng J, Miao Y, Wang Y, Cui W, Guo J, Qiu S, Han Y, Jia L, Li H, Cheng J, Du J. Serum-glucocorticoid regulated kinase 1 regulates alternatively activated macrophage polarization contributing to angiotensin II-induced inflammation and cardiac fibrosis. Arterioscler Thromb Vasc Biol 32: 1675–1686, 2012. doi: 10.1161/ATVBAHA.112.248732. [DOI] [PubMed] [Google Scholar]