

Abstract
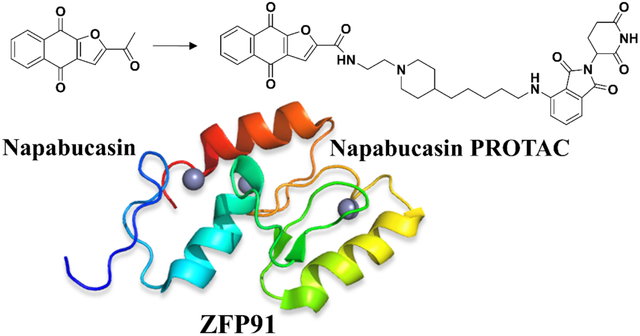
Napabucasin, undergoing multiple clinical trials, was reported to inhibit the signal transducer and transcription factor 3 (STAT3). To better elucidate its mechanism of action, we designed a napabucasin-based proteolysis targeting chimera (PROTAC), XD2–149 that resulted in inhibition of STAT3 signaling in pancreatic cancer cell lines without inducing proteasome-dependent degradation of STAT3. Proteomics analysis of XD2–149 revealed the downregulation of the E3 ubiquitin-protein ligase ZFP91. XD2–149 degrades ZFP91 with DC50 values in the nanomolar range. The cytotoxicity of XD2–149 was significantly, but not fully, reduced with ZFP91 knockdown providing evidence for its multi-targeted mechanism of action. The NQO1 inhibitor, dicoumarol rescued the cytotoxicity of XD2–149 but not ZFP91 degradation suggesting that the NQO1-induced cell death is independent of ZFP91. ZFP91 plays a role in tumorigenesis and is involved in multiple oncogenic pathways including NF-кB and HIF-1α.
Keywords: Napabucasin, STAT3, PROTAC, ZFP91
Graphical Abstract
INTRODUCTION
Naphthoquinone 2-acetylbenzo[f][1]benzofuran-4,9-dione (NPQ) was first discovered in 1982 as a plant-derived compound with anticancer activity.1 In 1998, NPQ was reported to have antipsoriatic, antibacterial and antifungal activities.2 Despite the promising preliminary data on NPQ, there was no interest in its development for almost a decade until 2009 when it was re-discovered by Boston Biomedical, Inc. and given the name napabucasin, also known as BBI608. Napabucasin was dubbed as the first-in-class cancer stemness inhibitor through inhibition of the signal transducer and transcription factor 3 (STAT3) signaling pathway.3, 4 Significant efforts were directed towards the development of napabucasin that resulted in clinical trials in several cancers as a single agent and in combination with chemotherapy (Table S1). Napabucasin was also formulated in polymeric vesicles that resulted in a decrease in the pancreatic cancer cell viability by 70%.5 Promising data from clinical trials accelerated the designation of napabucasin to orphan drug status for gastric/gastroesohphageal and pancreatic cancer in 2016 (Figure 1A).6
Figure 1.
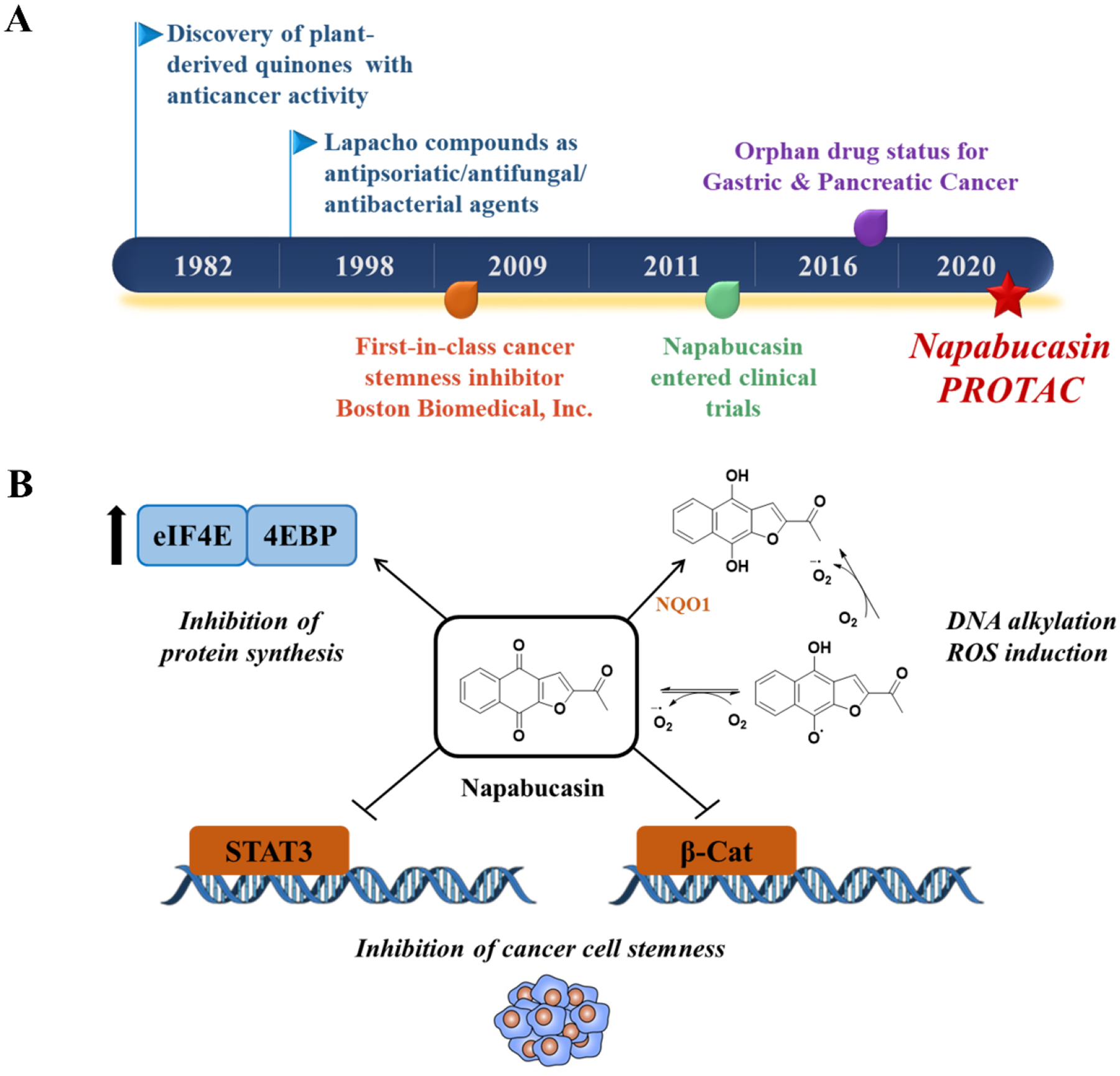
(A) Discovery timeline of napabucasin highlighting select milestones. (B) Reported mechanisms of action for napabucasin.
Pancreatic cancer is a devastating disease and is one of the leading causes of cancer-related deaths worldwide, therefore novel therapies are highly sought after.7 In vivo studies have shown that napabucasin is capable of blocking cancer metastasis and its relapse in pancreatic and colon cancers through inhibition of cancer stemness.3 Combination with nab-paclitaxel and gemcitabine in a Phase I/II clinical trial showed that napabucasin is well tolerated and displays activity against metastatic pancreatic adenocarcinoma (mPDAC).8 However, a Phase III study investigating the efficacy of napabucasin when given with the standard-of-care chemotherapy in patients with mPDAC was discontinued due to futility.9, 10
Several analogs of napabucasin were synthesized to improve its efficacy and/or physicochemical properties. Introducing a (piperidinyl)ethylamino-substitution enhanced the activity 10-fold and resulted in improved solubility.11 Another compound (LD-19) that possesses a methyl piperazine group displayed enhanced in vitro cytotoxicity in liver cancer cells and improved water solubility.12 However, none of the optimized analogs have been advanced to in-depth pre-clinical studies. While napabucasin is facing a lack of success in the clinic, there is still considerable interests in understanding its mechanism of action since it is reported as a STAT3 inhibitor.
STAT3 is a transcription factor that is implicated in the majority of human cancers and is associated with poor prognosis.13, 14 The constitutive activation of STAT3 in cancer cells is highly advantageous for proliferative and metastatic phenotypes. This master regulatory role allows for selective cancer cell killing, making it an attractive target for cancer treatment.15 Over the past 30 years, significant efforts were directed towards understanding the biological function of STAT3 and developing an effective inhibitor capable of blocking STAT3 activity. However, very few compounds including napabucasin made it to clinical trials. Most of these studies displayed insufficient efficacy and/or intolerable side effects.
Although napabucasin has been reported as a STAT3 inhibitor, its exact mechanism of action is not clearly understood. Napabucasin has been claimed to directly bind STAT3 by preventing its proteolytic degradation.16 Furthermore, a series of napabucasin analogs were reported to bind to STAT3 using surface plasmon resonance (SPR) analysis with a KD of 110 nM for the most potent analog.11 It is claimed that napabucasin inhibits STAT3 by binding to its SH2 domain and preventing its dimerization and subsequent activation but there is no crystallographic evidence to support that claim.11, 12, 16 Napabucasin is also reported to act on multiple other oncogenic pathways including WNT/ß-catenin pathway that is constitutively activated in tumor tissues.17 Napabucasin is a substrate for NAD(P)H: quinone oxidoreductase 1 (NQO1), and its activity is dependent on NQO1 levels.18 This enzyme catalyzes the reduction of a quinone to hydroquinone resulting in the generation of reactive oxygen species (ROS), DNA alkylation and subsequent cancer cell death.19 This suggests that part of napabucasin’s mechanism of action could be a result of ROS induction and/or DNA alkylation.20 Recently, we showed that napabucasin induces ROS, has similar transcriptomic signature as H2O2, and induces cancer cell death via necroptosis.21 Napabucasin was also reported to act through inhibition of protein synthesis via regulating the eukaryotic initiation factor 4E (Figure 1B).22
In an attempt to better understand napabucasin’s mechanism of action and its connection to STAT3, we designed a series of novel napabucasin-derived proteolysis targeting chimeras (PROTACs) and assessed their cytotoxicity in pancreatic cancer cell lines. PROTACs are heterobifunctional molecules that simultaneously bind an E3 ligase and a target protein, driving target protein ubiquitination by the E3 ligase complex leading to recognition and degradation by the 26S proteasome.23–25 The first-in-class STAT3 degrader, SD-36, was reported earlier this year by applying a cereblon (CRBN)-based PROTAC targeting the SH2 domain of STAT3.26, 27 SD-36 degraded STAT3 with nanomolar DC50 values in leukemia and lymphoma cell lines and elicited complete tumor regression within two weeks.27 This study validates the use of PROTACs in effective and selective targeting of STAT3. As napabucasin is also reported to target the SH2 domain of STAT3, we hypothesized that napabucasin-based PROTACs would be a novel and effective strategy to degrade STAT3. Herein, we describe the synthesis and evaluation of a series of napabucasin-PROTACs with cellular potency in pancreatic cancer cell lines. Proteomics and mechanistic studies did not reveal proteasome-dependent degradation of STAT3. Instead, we observed a significant degradation of ZFP91 with the napabucasin-PROTAC, 3–6d (XD2–149) (Figure 2). Our study provides further insight into the mechanism of action of napabucasin and for the first time, linking napabucasin to the oncogenic protein ZFP91.
Figure 2.
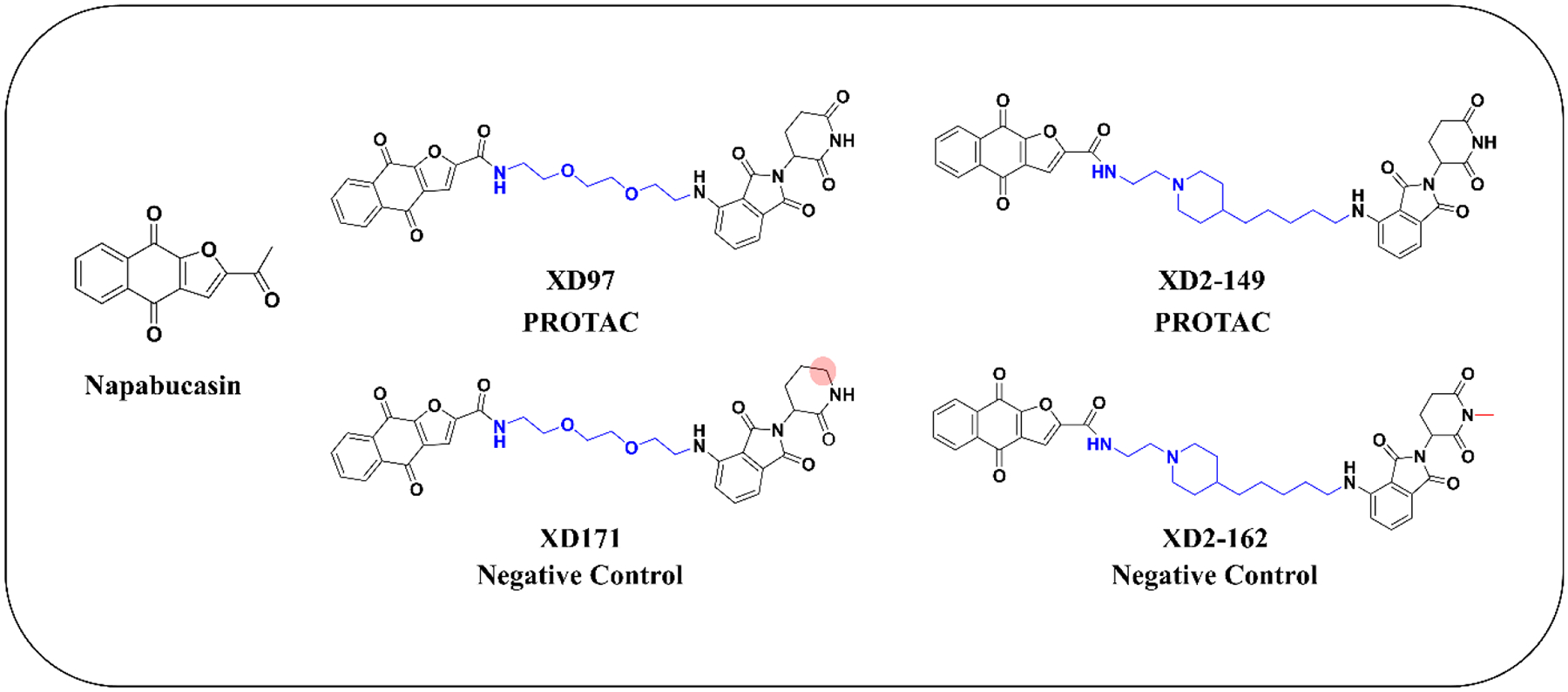
Chemical structures of napabucasin, the designed PROTACs, and their respective negative controls.
RESULTS AND DISCUSSION
Design of PROTACs Using Napabucasin as the Targeting Warhead.
In order to develop an effective PROTAC that can bind to and degrade the potential target of napabucasin, we designed a series of compounds applying three major optimization approaches. The design followed a systematic strategy that included the optimization of the linker length/composition, the position of the linker attachment to napabucasin and the choice of the E3 ligase ligand. It is critical to adjust for the distance between the two partner proteins to allow for efficient binding and degradation. CRBN and Von Hippel Lindau (VHL) E3 ligases have been successfully employed in the design of the PROTACs for degrading different proteins.27–31 CRBN E3 ligase ligands are composed of the low molecular weight immunomodulatory drugs (IMiDs) including pomalidomide, thalidomide and lenalidomide that makes them ideal for the design of PROTACs with good physicochemical properties.32 Therefore, we focused our studies on the design of CRBN-based PROTACs with exploration of the effect of VHL E3 ligase ligand. Pomalidomide has a high cellular stability compared to lenalidomide and thalidomide33, 34 so, we initially designed pomalidomide-based compounds for studying the choice of the optimal linker. The synthesized compounds were evaluated for their cytotoxicity and protein degradation in the two pancreatic cancer cell lines, BxPC-3 and MIA PaCa-2. We used BxPC-3 to guide our structure-activity relationship studies (SAR) due to the improved activity of the compounds in immunoblotting experiments, in addition to, its higher expression and activation of STAT3 (Figure 3).35
Figure 3.
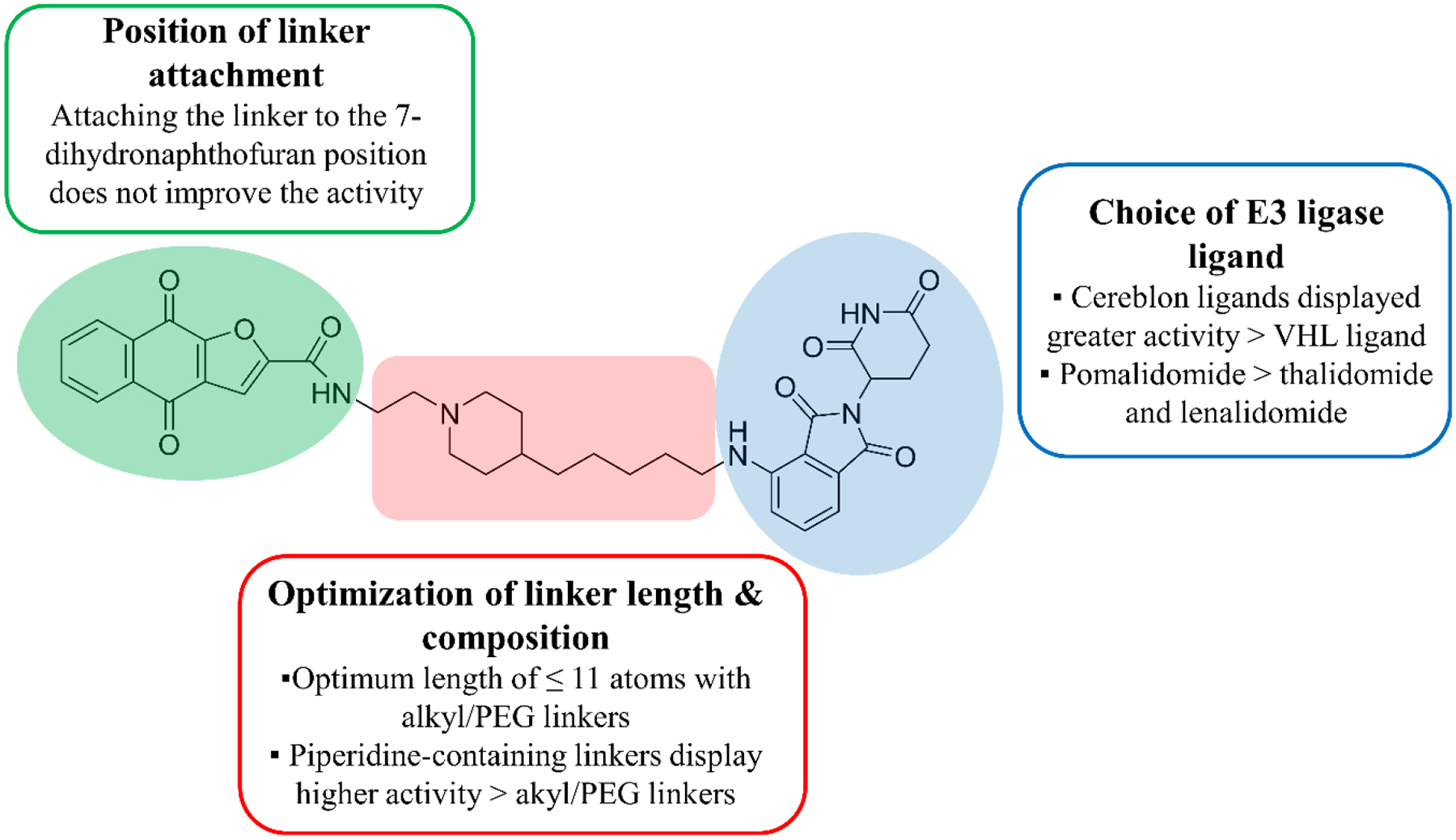
Summary for PROTAC optimization including linker length and composition, point of linker attachment to napabucasin, and the choice of the E3 ligase ligand.
In order to obtain the optimum PROTAC, it is important to consider the stability of the ternary compounds in which the linker plays an important role. With pomalidomide as the CRBN handle and napabucasin as the targeting warhead, we designed a series of PROTACs applying various linker lengths and compositions (Schemes 1 and 2). We synthesized compounds with alkyl and PEG linkers of different lengths (2 – 20 atoms) and evaluated their cytotoxicity (Table 1). Compound 2–5a with the 2-atom spacer displayed low micromolar cytotoxicity while compounds with linkers ranging between 3 and 11 atoms were 2-fold less potent than 2–5a. Increasing the linker length > 11 atoms in 2–5c and 2–5j resulted in decreased cytotoxicity. Our results show that the optimal linker length for the cytotoxicity of compounds is ≤ 11 atoms and that there is no significant difference between the hydrophobic and hydrophilic linkers.
Scheme 1. Synthesis of Compounds 5a-ja.
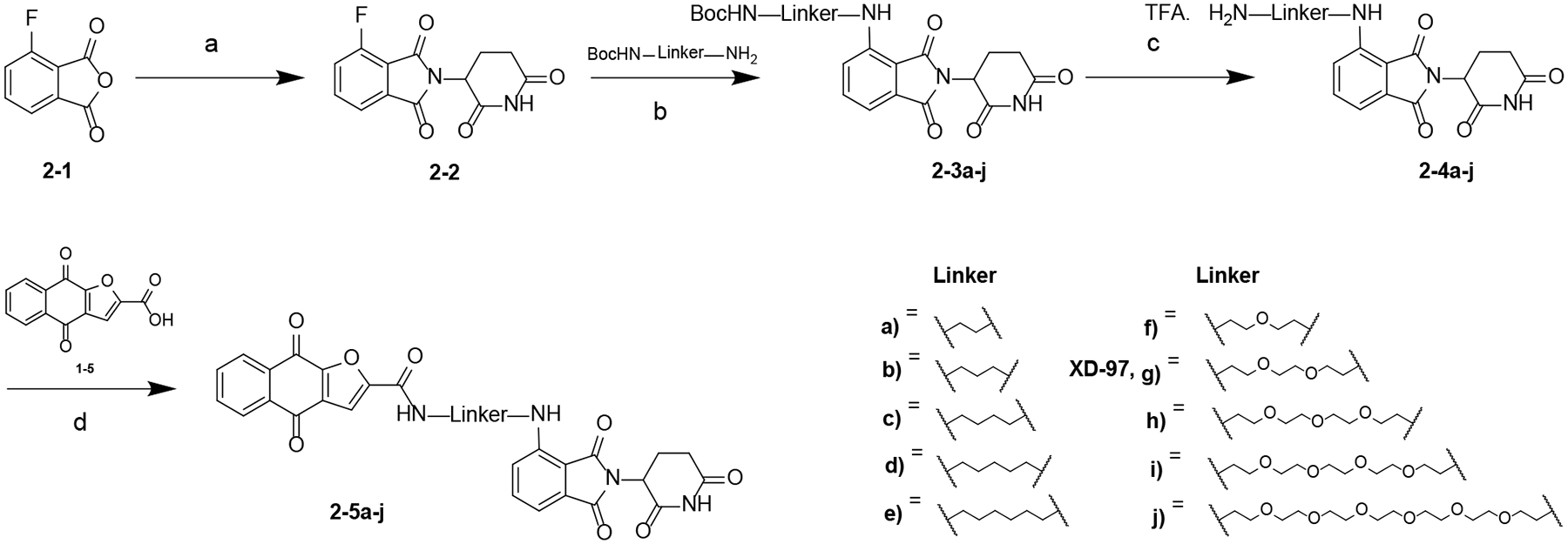
aReagents and conditions: (a) NaOAc, HOAc, 100 °C; (b) DIPEA, DMF, 80 °C; (c) TFA, DCM, rt; (d) HATU, DIPEA, DMF, rt.
Scheme 2. Synthesis of Compounds 3–6a-da.
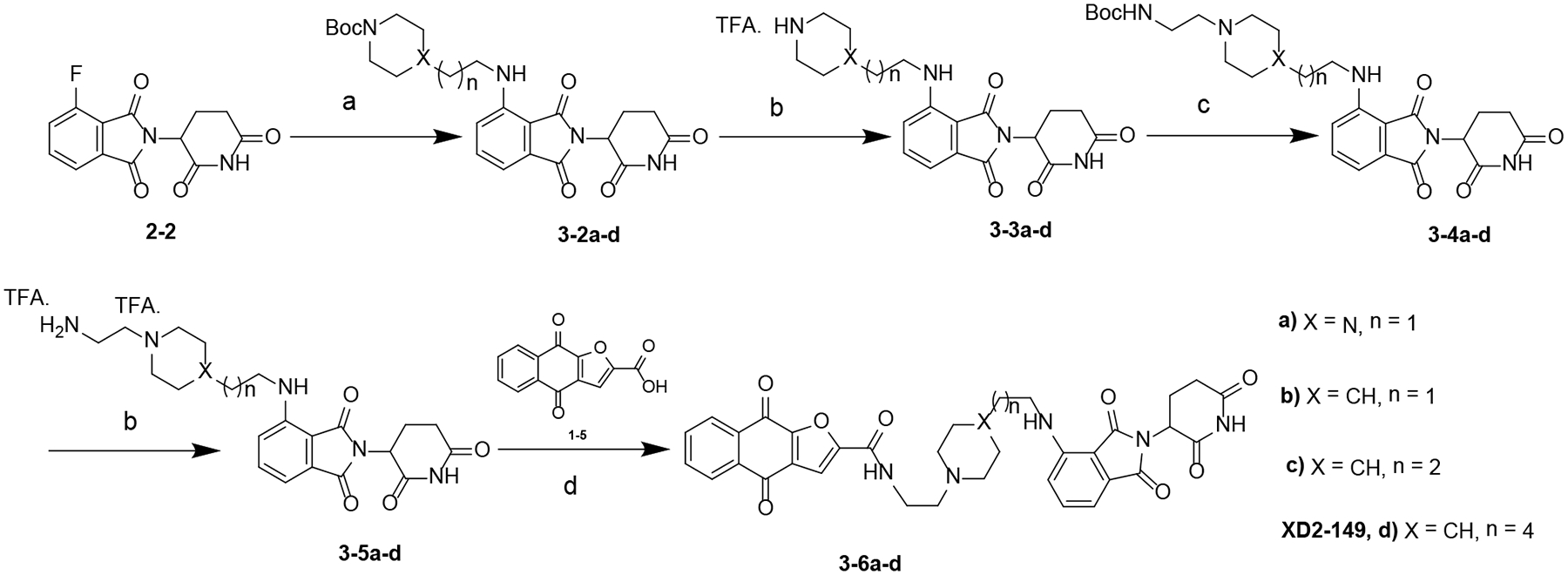
aReagents and conditions: (a) DIPEA, DMF, 80 °C; (b) TFA, DCM, rt; (c) NaBH(OAc)3, DCE, rt; (d) HATU, DIPEA, DMF, rt.
Table 1.
Cytotoxicity of CRBN-based PROTACs in Pancreatic Cancer Cell Lines
 | |||||
|---|---|---|---|---|---|
| Pomalidomide-based PROTACs | |||||
| ID | Linker | MW | cLogP | IC50 (μM) | |
| MIA PaCa-2 | BxPC-3 | ||||
| NPa | 240.21 | 2.19 | 1.2 ± 0.8 | 1.4 ± 0.3 | |
| 2–5a |  |
540.49 | 2.29 | 4.0 ± 0.9 | 1.7 ± 0.7 |
| 2–5b | 554.52 | 2.65 | 2.7 ± 0.4 | 3.5 ± 0.6 | |
| 2–5c | 567.54 | 2.61 | 1.60 ± 0.3 | 4.1 ± 1.1 | |
| 2–5d | 582.57 | 3.14 | 1.8 ± 0.7 | 3.2 ± 1.3 | |
| 2–5e | 596.60 | 3.67 | 3.0 ± 0.4 | 4.2 ± 1.4 | |
| 2–5f | 584.54 | 2.36 | 2.6 ± 0.9 | 4.6 ± 1.2 | |
| 2–5g, XD97 | 628.59 | 2.18 | 1.3 ± 0.5 | 4.3 ± 1.1 | |
| 2–5h | 672.65 | 2.01 | 2.1 ± 0.8 | 4.5 ± 0.9 | |
| 2–5i | 716.70 | 1.83 | 4.4 ± 0.7 | 10.1 ± 3.5 | |
| 2–5j | 804.29 | 1.48 | 5.0 ± 1.0 | 7.5 ± 0.4 | |
| 3–6a | 652.66 | 1.80 | 18.1 ± 11.4 | 25.6 ± 2.0 | |
| 3–6b | 651.68 | 3.55 | 1.1 ± 0.3 | 1.1 ± 0.5 | |
| 3–6c | 665.70 | 4.08 | 0.6 ± 0.3 | 0.6 ± 0.1 | |
| 3–6d, XD2–149 | 693.76 | 5.14 | 1.0 ± 0.1 | 0.8 ± 0.2 | |
| Thalidomide-based PROTACs | |||||
|---|---|---|---|---|---|
| ID | Linker | MW | cLogP | IC50 (μM) | |
| MIA PaCa-2 | BxPC-3 | ||||
| 4–5 | 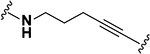 |
563.52 | 2.83 | 2.1 ± 1.0 | 5.3 ± 0.8 |
| 5–5a | 569.53 | 2.78 | 2.0 ± 0.3 | 4.2 ± 0.7 | |
| 5–5b | 629.58 | 2.24 | 3.8 ± 1.0 | 8.2 ± 4.1 | |
| 6–6a | 686.63 | 1.55 | 16.7 ± 3.8 | 20.4 ± 7.4 | |
| 6–6b | 730.68 | 1.38 | 3.7 ± 1.1 | 6.0 ± 5.5 | |
| 6–6c | 626.58 | 1.55 | 10.5 ± 3.0 | 23.8 ± 3.9 | |
| Lenalidomide-based PROTACs | |||||
|---|---|---|---|---|---|
| ID | Linker | MW | cLogP | IC50 (μM) | |
| MIA PaCa-2 | BxPC-3 | ||||
| 7–4 | 614.62 | 1.96 | 13.7 ± 1.0 | 10.5 ± 4.5 | |
| 8–1 | 553.57 | 3.37 | 6.4 ± 3.4 | 13.8 ± 7.7 | |
Napabucasin
2–5g (XD97) Reduces STAT3 Expression in a Proteasome-independent Manner.
We tested a selection of the compounds in a panel of cell lines outside of pancreatic cancer to determine if there is a specific cell line in which the compounds are selective (Table S2– S3). XD97 with an IC50 of ~ 0.3 μM in OVCAR-3 cells was the most cytotoxic. To determine whether the cytotoxicity of these compounds is STAT3-dependent, we used a pair of OVCAR-3 wildtype (WT) and STAT3 knockout (KO) cell lines (Table S3).36 Overall, the compounds were more potent in the WT cell line, among which XD97 displayed significant selectivity over the STAT3 KO cell line (> 6-fold). These data suggest that STAT3 is involved in the mechanism of cell death. Since the cytotoxicity was STAT3-dependent, we tested whether the napabucasin PROTAC XD97 degraded STAT3. XD97 at different concentrations and times caused near complete depletion of STAT3 after 12 h while decrease in p-STAT3 levels was observed after 6 h (Figure S1B).
To investigate if the degradation of STAT3 was proteasome-dependent, we employed three approaches: treatment in the presence of a proteasome inhibitor, competition with the E3 ligase ligand, and synthesis of a negative control incapable of binding CRBN. Upon treatment with the proteasome inhibitor MG132, we observed no blockade of XD97-induced decrease in STAT3 expression concluding this decrease is not dependent on the proteasome (Figure S1C). In fact, MG132 enhanced the activity of XD97 resulting in a more prominent reduction in STAT3 protein levels. It is worth noting that proteasome inhibitors like bortezomib have been reported to synergize with STAT3 inhibitors to promote cell death.37 However, we did not observe synergy between napabucasin and proteasome inhibitors in cell proliferation assays (data not shown). Similarly, competition experiments in the presence of pomalidomide did not antagonize the effect of XD97 indicating it is not a substrate of CRBN (Figure S1D). We synthesized 10–5 (XD171) as a negative control by replacing the glutarimide ring in pomalidomide by a δ-lactam ring that abrogates its binding to CRBN (Scheme 9) (Figure S1A).38 XD171 displayed comparable activity to XD97 (Figure S1E). Among the STAT family members, STAT1 has a high structural similarity to STAT3.39 Surprisingly, we observed a decrease in STAT1 levels that is even more prominent than that observed with STAT3 at the same concentrations of XD97 (Figure S1B). Taken together, the mechanism of action of XD97 is independent of the proteasome and the observed effects are possibly due to inhibitory activity or changes on the transcriptional level. We also conclude that the activity of XD97 is not specific to STAT3.
Scheme 9. Synthesis of Compound 10–5 (XD171)a.
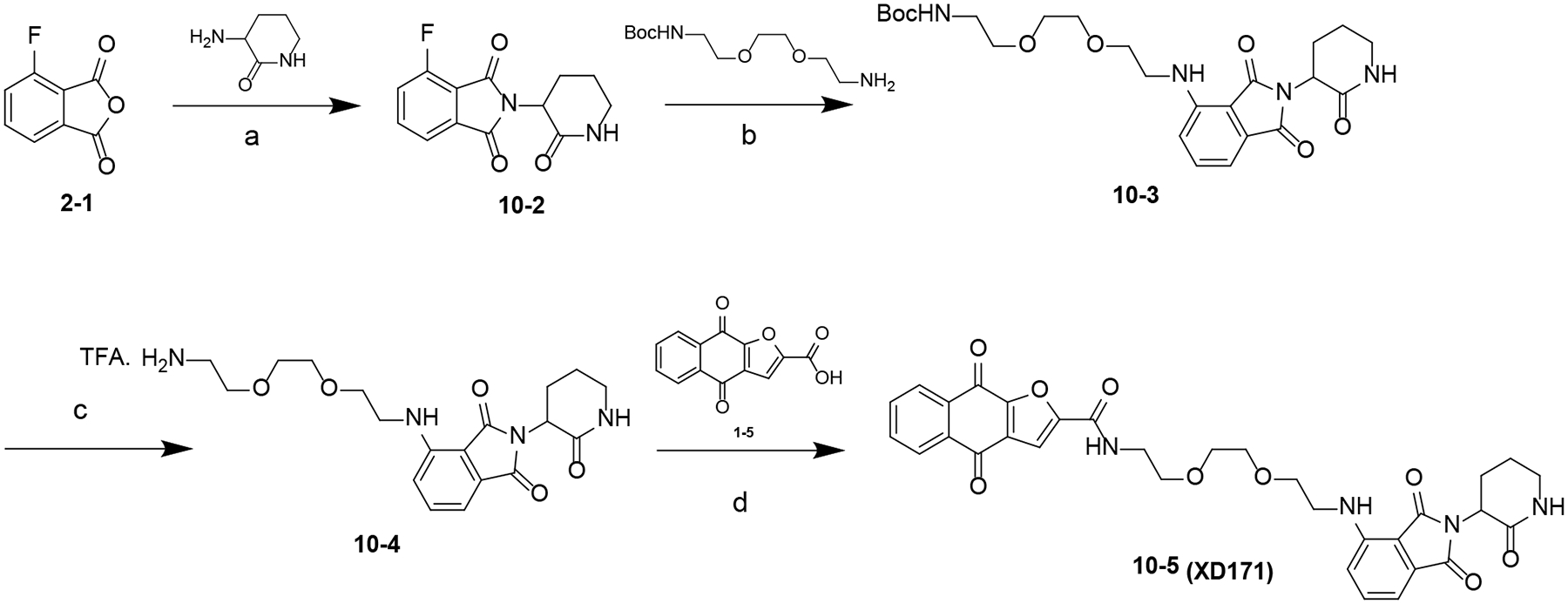
aReagents and conditions: (a) NaOAc, HOAc, 100 °C; (b) DIPEA, DMF, 80 °C; (c) TFA, DCM, rt; (d) HATU, DIPEA, DMF, rt.
We further tested XD97 for its effect on STAT3 expression in the pancreatic cancer cell line BxPC-3 to determine if it possessed a different mechanism of action and possibly act as a degrader. Similar to OVCAR-3, XD97 reduced the STAT3 level after 12 h but the effect in BxPC-3 was observed at a lower concentration (DC50 = 7.8 μM) despite of the greater cytotoxicity of XD97 in OVCAR-3 (IC50 of ~ 0.2 and 4.3 μM in OVCAR-3 and BxPC-3, respectively) (Figure S2A–B). This encouraged us to test XD97 in combination with the proteasome inhibitor bortezomib. Unfortunately, bortezomib did not block the activity of XD97 thereby proving its STAT3 inhibitory activity is independent of the proteasome-mediated degradation (Figure S2C). Interestingly, we observed reduced STAT3 expression with napabucasin (DC50 = 2.1 μM) that was not observed in OVCAR-3 (Figure S2B). Moreover, napabucasin and XD97 both reduced NQO1 levels (Figure S2D). It is possible that napabucasin has multiple targets or acts through interfering with the transcription/translation of target proteins.
Although PROTACs have the advantage of selectively degrading the target protein, recruitment of other neo-substrates can be a challenge. The surface of CRBN can change upon binding of IMiDs like pomalidomide and allow recruitment of other proteins such as the translation termination factor GSPT1 which has been previously reported as an off target substrate for CRBN-targeted PROTACs.40, 41 This change in the surface structure leads to creation of hotspots that cause the PROTAC molecule to act as a ‘molecular glue’ and bind non-specific proteins.42 The protein level of GSPT1 was not affected by XD97 or napabucasin treatment (Figure S2D). This provides evidence that napabucasin does not non-specifically cause degradation of proteins and that it is mechanism-based.
Optimization of Napabucasin-based PROTACs.
Moving forward with the linker optimization, we synthesized compounds with cyclized linkers using piperidine. The piperidine linkers displayed improved activity over the alkly/PEG linkers (Table 1). Piperidine groups have been previously incorporated into PROTACs to improve water solubility.43 3–6c and 3–6d (XD2–149) showed comparable potency with IC50 < 1 μM in BxPC-3. Compound 3–6a with a piperazine ring resulted in significant loss of activity (Table 1). Our data shows that piperidine-containing linkers display the best cytotoxicity and are superior to napabucasin suggesting these compounds might act as effective degraders. Therefore, XD2–149 was selected as a representative PROTAC for further mechanistic studies.
The small molecule 1–6 is a previously reported analog of napabucasin that displayed greater cytotoxicity (>10-fold) and was used to investigate the effect of the position of linker attachment (Scheme S1) (Table S4).11 Attaching the linker to the 7-dihydronaptho[2,3-b]furan position resulted in 13–11 did not result in improved cytotoxicity (Scheme S2) (Table S5). Changing the position of the linker attachment to the E3 ligase ligand from position 4 of the isoindoline-1,3-dione ring to position 5 (12–5) reduced the cytotoxicity as compared to XD97 (Scheme S3) (Table S5).
To examine the CRBN ligand portion, another set of compounds was designed using thalidomide as the CRBN E3 ligase ligand employing various linkers (Schemes 3–5) (Table 1). Amide-containing linkers displayed a significant decrease in activity with IC50 > 10 μM except for 6–6b with a linker that extends to > 13 atoms contributing to its enhanced activity. Compound 5–5b with a 9-atom PEG linker had an IC50 ~ 8 μM while 5–5a with a 4-atom alkyl linker was almost 2-fold more potent. Two other compounds with alkyl and PEG linkers conjugated to lenalidomide showed moderate cytotoxicity (IC50 range 6–14 μM) (Schemes 6 – 7) (Table 1).
Scheme 3. Synthesis of Compound 4–5a.
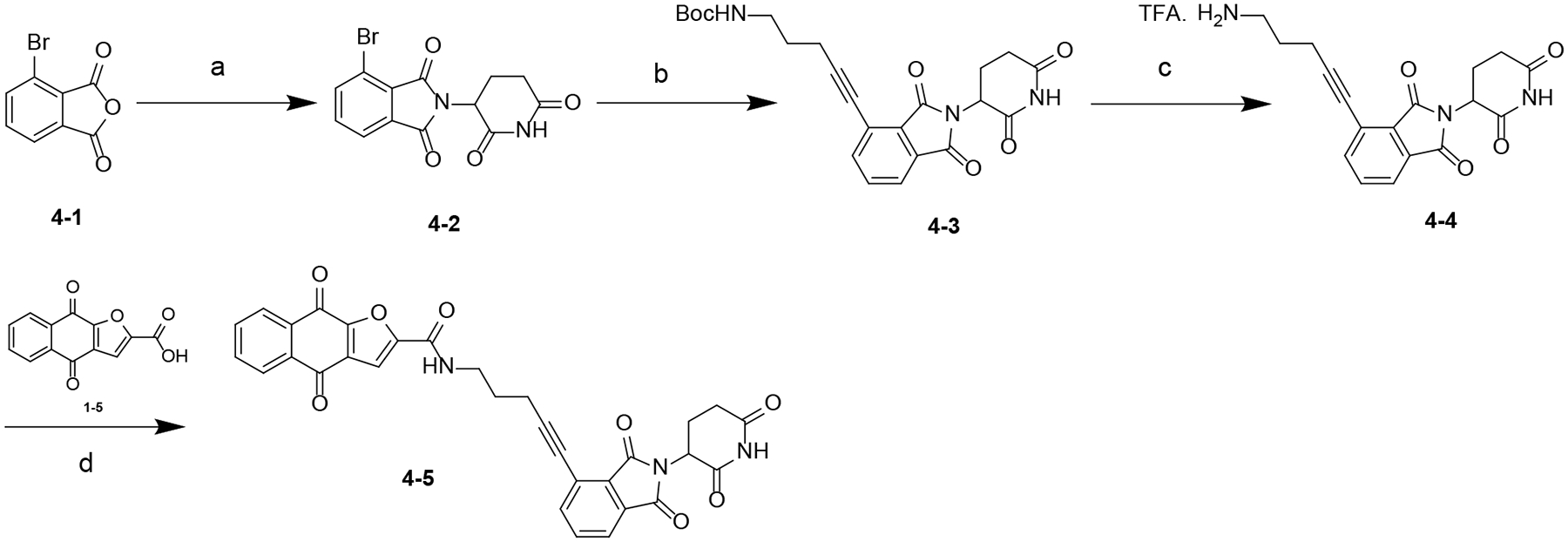
aReagents and conditions: (a) NaOAc, HOAc, 120 °C; (b) Pd(PPh3)2Cl2, CuI, TEA, DMF, 80 °C; (c) TFA, DCM, rt; (d) HATU, DIPEA, DMF, rt.
Scheme 5. Synthesis of Compounds 6–6a-ca.
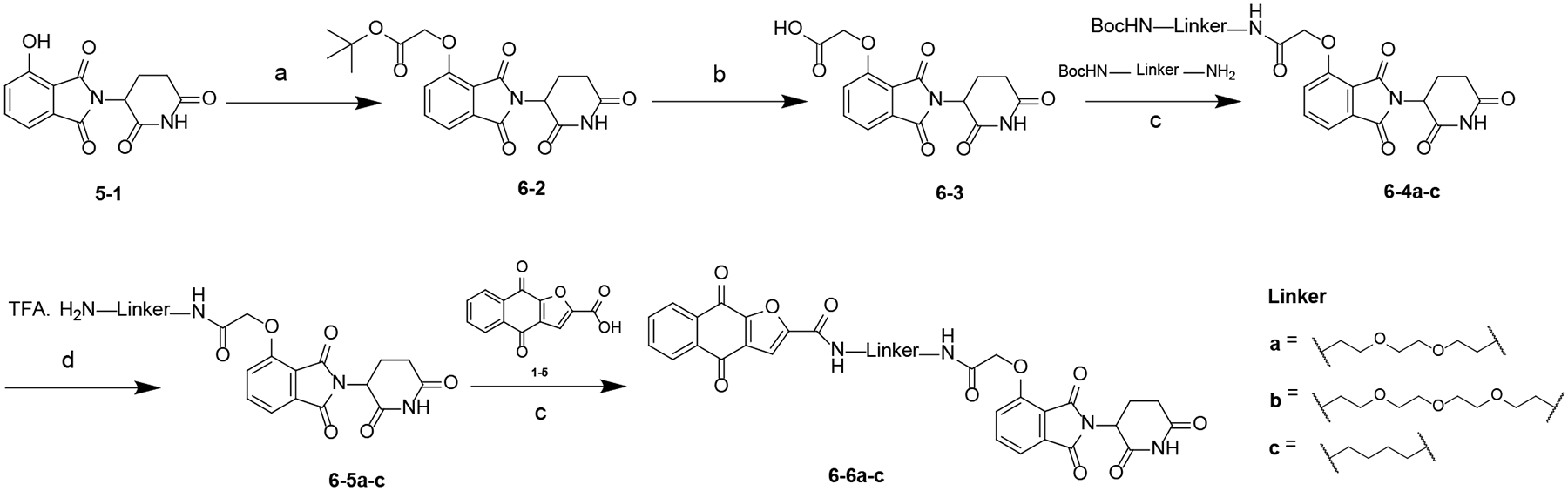
aReagents and conditions: (a) K2CO3, DMF, rt; (b) TFA, rt; (c) HATU, DIPEA, DMF, rt; (d) TFA, 50 °C.
Scheme 6. Synthesis of Compound 7–4a.
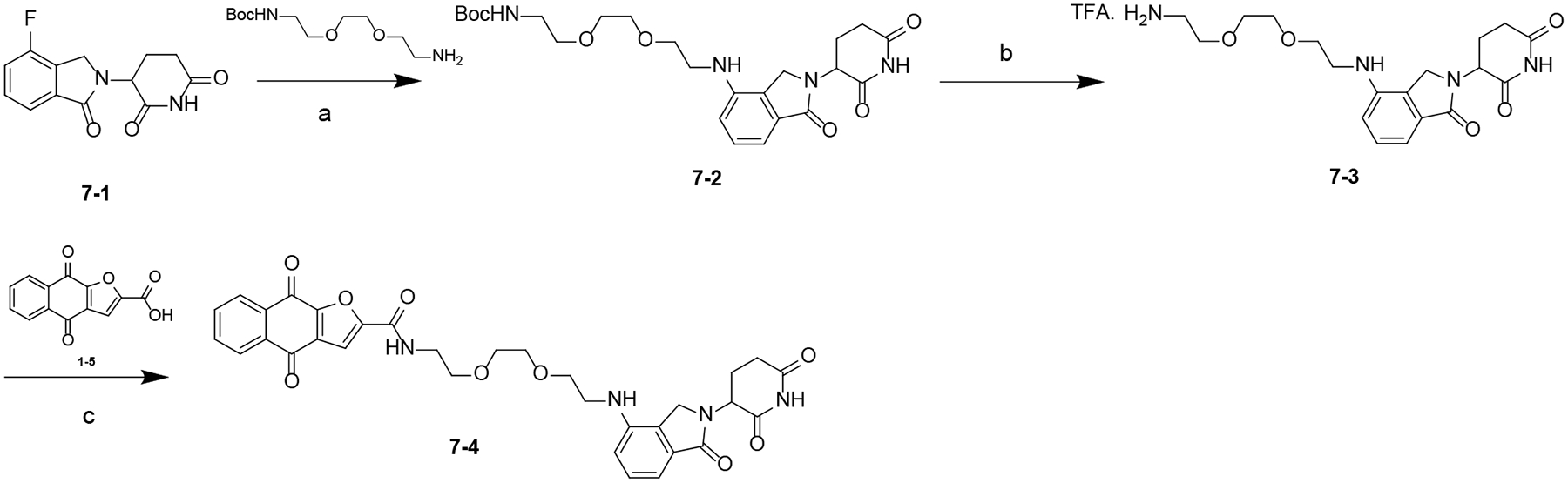
aReagents and conditions: (a) DIPEA, DMF, 80 °C; (b) TFA, DCM, rt; (c) HATU, DIPEA, DMF, rt.
Scheme 7. Synthesis of Compound 8–1a.
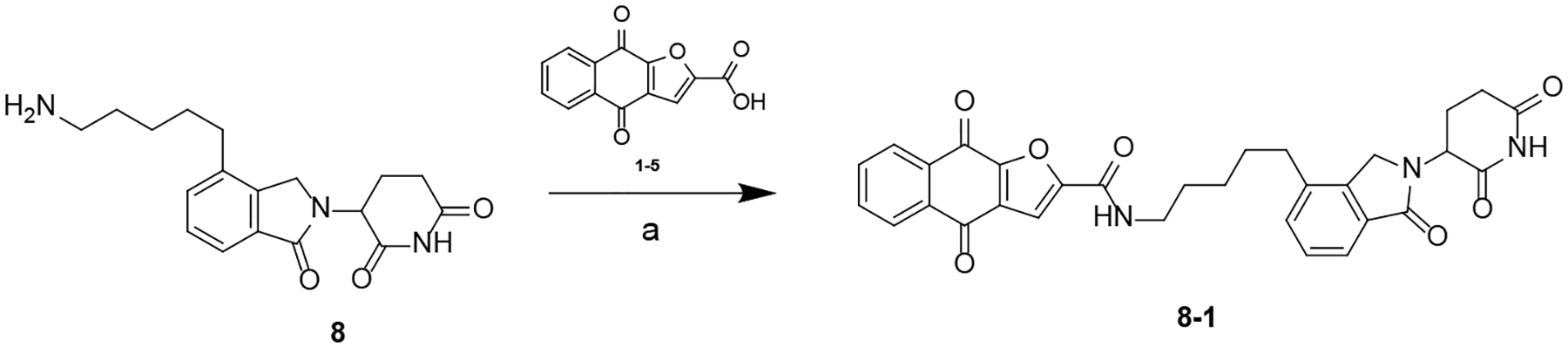
aReagents and conditions: (a) HATU, DIPEA, DMF, rt.
Our synthetic efforts extended to testing compounds with VHL as the E3 ligase ligand. Interestingly, the VHL-based compounds (Scheme 8) were inactive with IC50 > 30 μM suggesting VHL is not optimal for the design of napabucasin-based PROTACs (Table 2).
Scheme 8. Synthesis of Compounds 9–4a-ba.
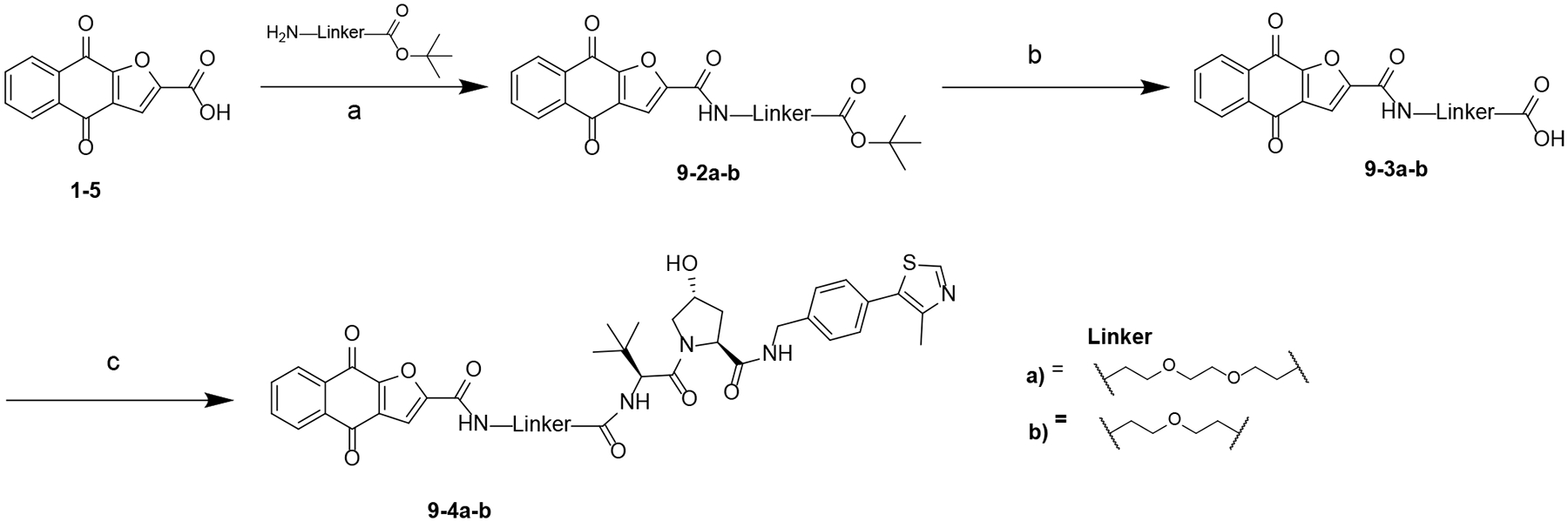
aReagents and conditions: (a) HATU, DIPEA, DMF, rt; (b) TFA, DCM, rt; (c) (S,R,S)-AHPC hydrochloride, HATU, DIPEA, DMF, rt.
Table 2.
Cytotoxicity of VHL-based PROTACs in Pancreatic Cancer Cell Lines
 | |||||
|---|---|---|---|---|---|
| ID | Linker | MW | cLogP | IC50 (μM) | |
| MIA PaCa-2 | BxPC-3 | ||||
| 9–4a | 799.90 | 3.82 | 14.5 ± 7.1 | >30 | |
| 9–4b |  |
755.84 | 3.99 | 15.8 ± 4.6 | >30 |
In conclusion, CRBN E3 ligases were more effective in the design of PROTACs compared to VHL. Thalidomide and lenalidomide-based compounds were less cytotoxic than pomalidomide-based conjugates suggesting that pomalidomide provides a more stable ternary complex with napabucasin and CRBN.
3–6d (XD2–149) is Cytotoxic in Pancreatic Cancer Cell Lines.
Previous studies on napabucasin have proven its activity against pancreatic cancer for which it was approved as an orphan drug. BxPC-3 cells has high protein levels of STAT3 and pSTAT3.35 We tested our compounds for their ability to inhibit cell growth in MIA PaCa-2 and BxPC-3 cell lines using MTT assay (Table 1–2, Table S5). Compound XD2–149 was among the most active compounds in both cell lines with an IC50 ~ 1 μM. We synthesized 11–7 (XD2–162) as a negative control by replacing the NH of the glutarimide ring in pomalidomide with an N-methyl group (Scheme 10) (Figure 4A). The N-methylated pomalidomide lacks the key H-bond donor involved in an interaction with His378 of CRBN, in addition to, the steric hindrance created by the methyl in the binding pocket.44, 45 XD2–149 is significantly more potent than XD2–162 in the MTT assay suggesting that the cell growth inhibition involves target degradation (Figure 4A–B). The colony formation assay (CFA), a cell survival assay that assesses the ability of the cells to survive and form colonies was also used to test the cytotoxicity of the compounds. Lead compound inhibits colony formation in different pancreatic cancer cells lines. XD2–149 displayed a greater inhibitory activity than napabucasin in BxPC-3 cell line (Figure 4C–D).
Scheme 10. Synthesis of Compound 11–7 (XD2–162)a.
aReagents and conditions: (a) NaOAc, HOAc, 100 °C; (b) DIPEA, DMF, 80 °C; (c) TFA, DCM, rt; (d) NaBH(OAc)3, DCE, rt; (e) HATU, DIPEA, DMF, rt.
Figure 4.
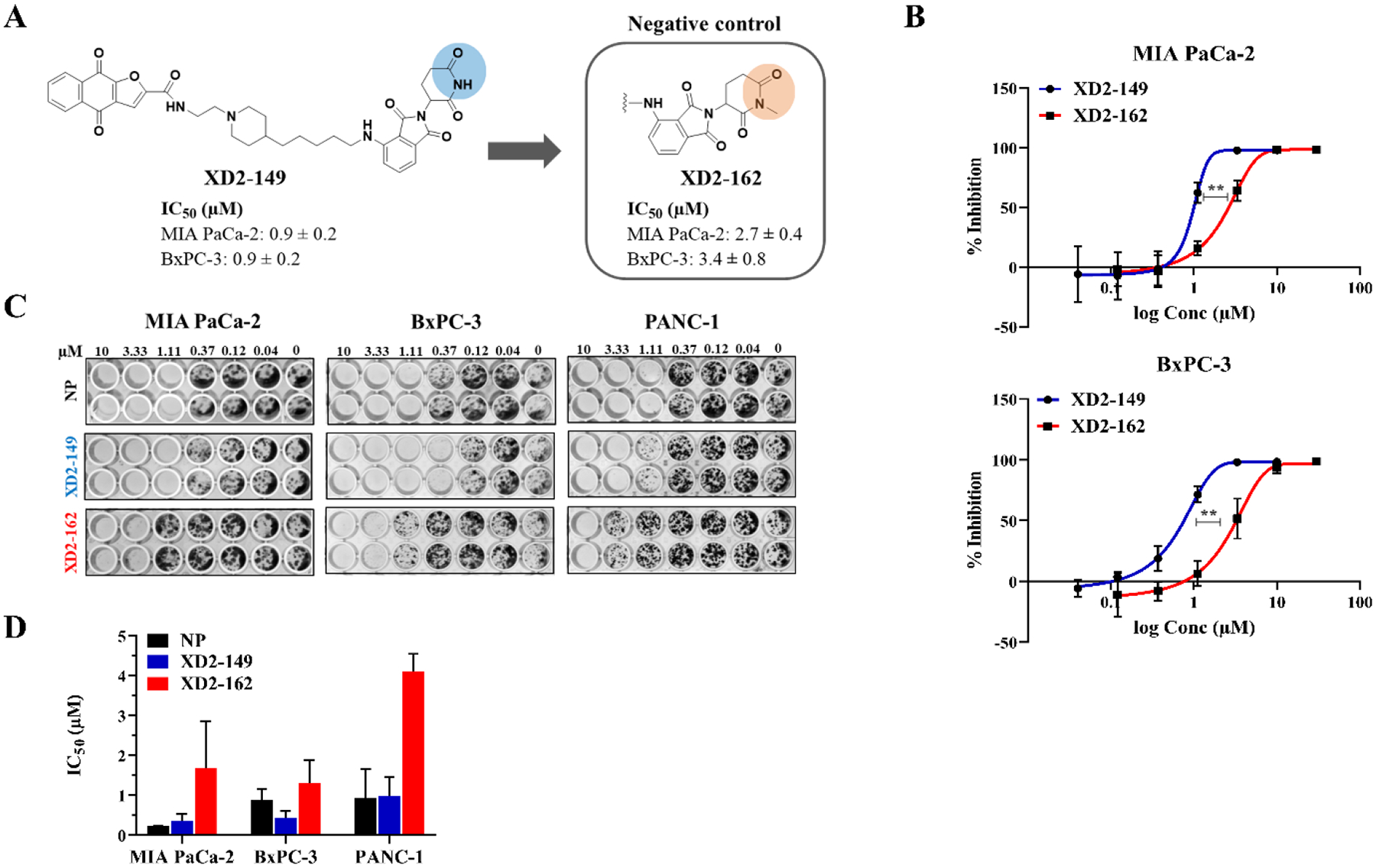
XD2–149 inhibits cell proliferation and colony formation in pancreatic cancer cell lines. (A) Design of XD2–162, the negative control for the PROTAC XD2–149. IC50 values correspond to the cytotoxicity of the compounds in the MTT assay. (B) Dose-response curves of the cytotoxicity of XD2–149 and XD2–162 in the MTT assay after 72 h. (C) Colony formation assay for napabucasin (NP), XD2–149 and XD2–162 in pancreatic cancer cell lines. Cells were treated for 7–10 days. (D) Quantification for the colony formation assay was done by dissolving the colonies using Sorensen’s buffer and reading the absorbance at 570 nm. All data presented was generated from three independent experiments and is presented as mean ± SD.
XD2–149 Inhibits IL6-dependent STAT3 Signaling.
To determine whether our compounds have an effect on the transcriptional activity of STAT3, we screened the compounds at 2 and 10 μM in the STAT3 luciferase assay using HEK-293 cell line. The luciferase reporter assay is commonly used in the discovery and evaluation of STAT3 inhibitors.26, 46 Following IL6-induced STAT3 activation, the cells were treated with the compounds for 24 h. The majority of the compound resulted in a reduced signal indicating inhibition of STAT3-driven transcription (Figure S3). All compounds were more potent in the luciferase assay than a 24-h MTT assay, indicating the signal loss was not due to cytotoxicity and cell death. Among the tested compounds, XD2–149 was the most efficient in the inhibition of STAT3-driven transcription with an IC50 ~ 1 μM which is almost 7-fold greater than its activity in the MTT assay (Figure 5).
Figure 5.
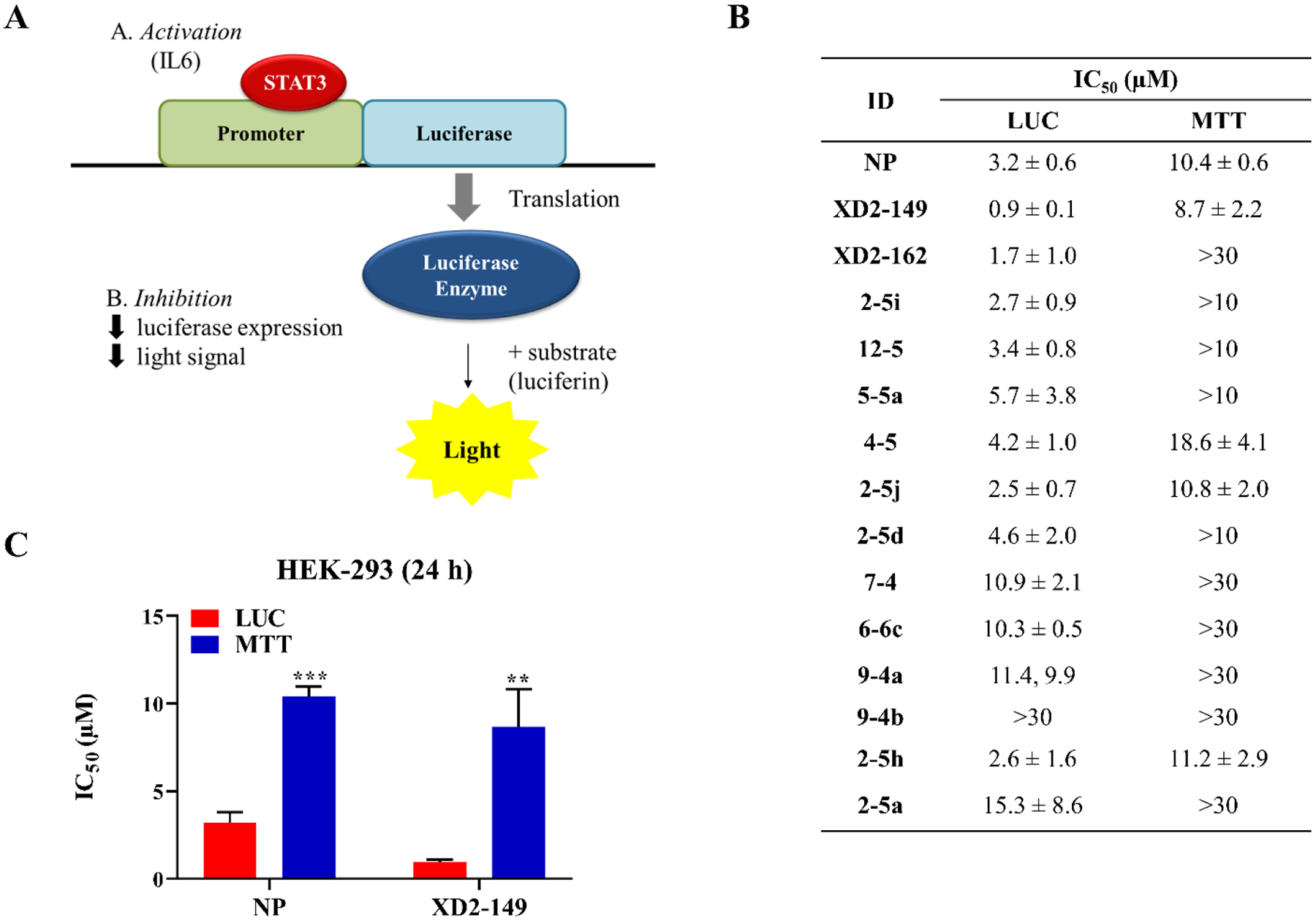
XD2–149 inhibits STAT3-driven gene transcription. (A) Diagram representing the principal of the STAT3 luciferase reporter assay. Transfected HEK-293 cells line that express renilla luciferase reporter under the transcriptional control of STAT3 were used. (B) IC50 values of the tested compounds in the luciferase assay after treatment for 24 h compared to their IC50 in the MTT assay at the same treatment time-point. For the luciferase assay, cells were pretreated with 100 ng/ml of IL6 for 24 h followed by treatment with the compounds for 24 h. The luciferin substrate was then added and the luminescence signal was measured. (C) IC50 values of napabucasin (NP) and XD2–149 in MTT and luciferase (LUC) assays after a 24 h-treatment. Data was generated from three independent experiments and is presented as mean ± SD. ** denotes p ≤ 0.01, *** denotes p ≤ 0.001.
XD2–149 Reduces STAT3 and Relevant Proteins’ Expression Independent of the Proteasome.
To evaluate the ability of XD2–149 to degrade STAT3, we treated MIA PaCa-2 and BxPC-3 cells with the compound at a range of concentrations up to 5 μM (~ 5X IC50). In BxPC-3, after a 16 h-treatment, we observed a decrease in the STAT3 protein expression at 5 μM while a significant decrease in the pSTAT3 level was achieved at 1 μM (Figure 6A). In contrast, XD2–162 did not have any effect on STAT3 expression but resulted in a decrease in pSTAT3 expression at 5 μM. This offset in concentration-dependent protein downregulation correlates well with the differences in MTT IC50 values. Interestingly, napabucasin alone was able to reduce the total STAT3 protein levels in addition to decreasing pSTAT3. The fact that the mechanism of action of the parent inhibitor involves reduction of STAT3 protein expression makes it challenging to study napabucasin PROTACs. The small-molecule analog of napabucasin 1–6 was also tested for its structural similarity to our compounds. Compound 1–6 did not have any effect on STAT3 expression at 0.5 μM (5X IC50) with a reduced pSTAT3 expression at the same concentration. As previously discussed, napabucasin is reported as a substrate of NQO1. Therefore, we tested the effect of XD2–149 on NQO1 as another possible target for degradation. We observed similar results to STAT3 which suggests that the activity of the compounds is not selective to STAT3 and the possibility of NQO1 degradation by the PROTAC given that napabucasin is a substrate of the enzyme. A reduction in STAT3 activation can result from increased ROS levels generated from NQO1-induced bioactivation of napabucasin.20
Figure 6.
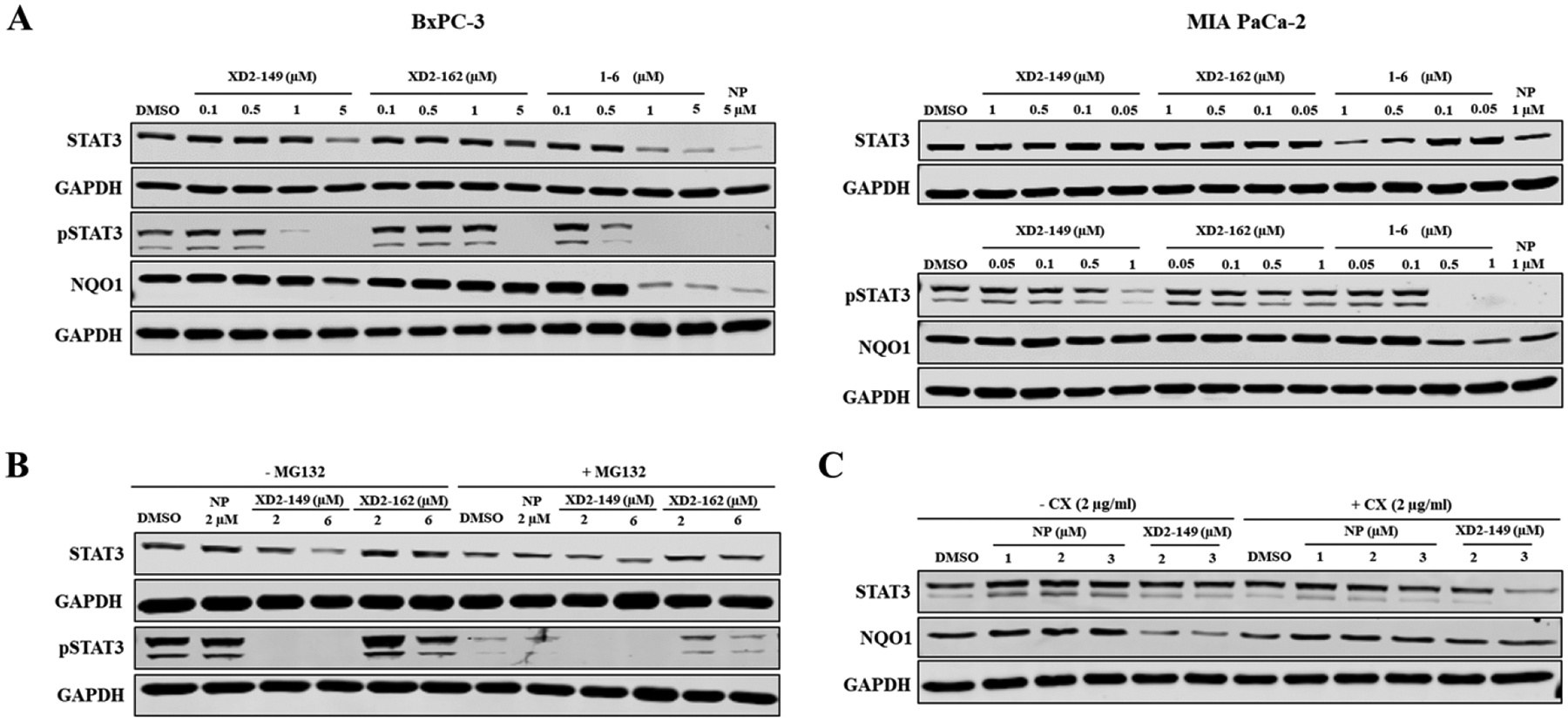
XD2–149 reduces the expression of STAT3, pSTAT3 and NQO1 in a proteasome-independent manner. (A) XD2–149 reduces the expression of STAT3, pSTAT3 and NQO1 proteins while XD2–162 showed effect only on pSTAT3. The small molecules 1–6 and napabucasin (NP) displayed similar effects to XD2–149. BxPC-3 and MIA PaCa-2 cells were treated with XD2–149, XD2–162, 1–6 or NP at the indicated concentrations for 16 h. (B) The effect of XD2–149 on STAT3/pSTAT3 expression is independent of the proteasome. BxPC-3 cells were pretreated with 10 μM MG132 for 2 h then with XD2–149, XD2–162 or NP for 12 h. (C) XD2–149 is possibly interfering with the protein synthesis of STAT3 and NQO1. BxPC-3 cells were pretreated with cycloheximide (CX) for 1 h followed by NP or XD2–149 for 16 h.
We repeated the experiment in MIA PaCa-2 cells, however, we observed less prominent effects on the proteins’ expression than in BxPC-3 (Figure 6A). Notably, XD2–149 was less effective in inhibiting the phosphorylation of STAT3 in MIA PaCa-2 with no effect on the NQO1 level. It is worth noting that in MIA PaCa-2 cells, we were able to test napabucasin and XD2–149 only up to 1 μM due to their high cytotoxicity and narrow therapeutic window. Napabucasin did not have a significant effect on the STAT3 protein expression but completely depleted the pSTAT3 levels at 1 μM.
Among the tested cell lines, BxPC-3 displayed the best activity for XD2–149 in terms of cell growth inhibition and reduced STAT3 expression and activation. Therefore, further experiments were conducted in BxPC-3. To determine whether the reduction of STAT3 expression is due to proteasome-dependent degradation, we treated the cells with XD2–149 in the presence of the proteasome inhibitor MG132. Proteasome inhibition did not block the XD2–149-induced decrease in STAT3 protein (Figure 6B). In contrast, we observed an enhanced reduction in NQO1 expression with XD2–149 treatment in the presence of MG132.
To determine if these effects occur at the translational level, we tested the effect of XD2–149 on STAT3 and NQO1 in the presence of the protein synthesis inhibitor, cycloheximide. We observed an enhanced cycloheximide-mediated protein synthesis inhibition of STAT3 (Figure 6C). Similar results were previously reported on napabucasin in osteosarcoma cells where the study demonstrated the inhibitory effect of napabucasin on protein synthesis as a result of STAT3 inhibition.22 On the other hand, the reduced NQO1 expression caused by XD2–149 was blocked in the presence of cycloheximide suggesting that the compound-induced downregulation of NQO1 is dependent on the de novo protein synthesis. Moreover, we sought to determine whether napabucasin engages its downregulated proteins by performing cellular thermal shift assay (CETSA). We tested the effect of napabucasin on the thermal stability of STAT3, NQO1 and an irrelevant protein (PDI) and observed a slight shift in the melting curve with all tested proteins except for the control protein GAPDH (Figure S4).
Collectively, even though XD2–149 displayed pronounced cytotoxicity in various cell lines compared to the negative control, it proved to not degrade the STAT3 protein through the proteasome machinery. It interferes with the protein synthesis of NQO1 resulting in reduced NQO1 levels, which was restored in the presence of a protein synthesis inhibitor.
XD2–149 is a Bona Fide Degrader of ZFP91.
To further study the effect of XD2–149 at the protein level and determine proteins selectively altered upon treatment, we performed proteomics analysis in BxPC-3. We evaluated proteome changes after 16 h treatment with either XD2–149, napabucasin, or XD2–162. The negative control was included in the study for comparison and elimination of nonspecific proteins or proteins whose expression is affected as a result of the inhibitor only or other transcriptional changes. A total of 136 proteins were dysregulated (> 1.5 FC, p ≤ 0.05) by XD2–149 out of which 84 proteins were downregulated (Figure 7A). The top 25 upregulated and downregulated proteins for each treatment are presented in Tables S6–S11. Comparative analysis with napabucasin and XD2–162 showed 40 proteins that were exclusively downregulated by XD2–149 (Figure 7B, Tables 3–4). Interestingly, STAT3 was among the proteins exclusive for XD2–149 with a log2FC of −0.69. This downregulation was consistent with our Western blot results.
Figure 7.
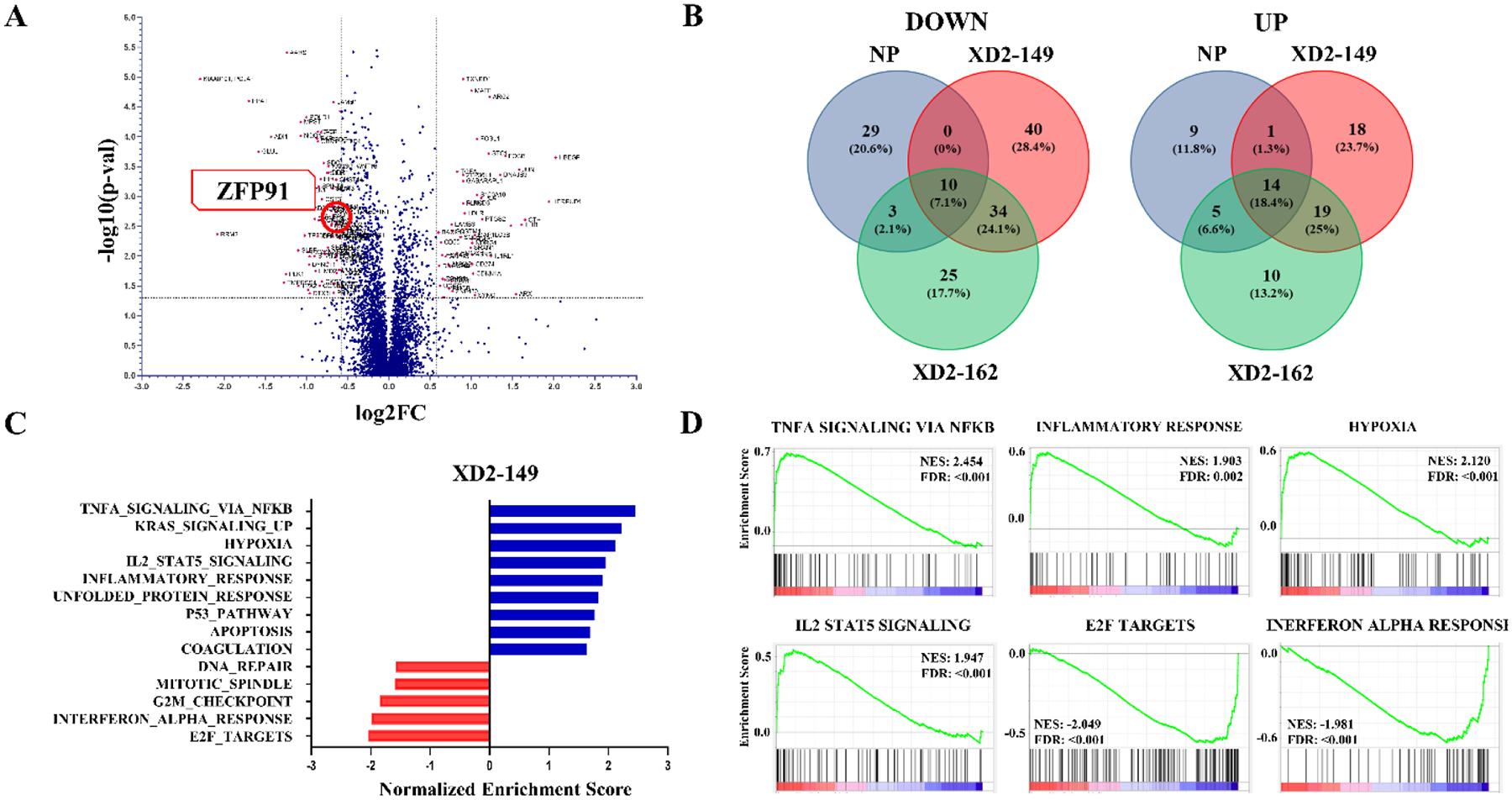
Transcriptomic profile of XD2–149 shows the exclusive downregulation of ZFP91 compared to napabucasin and the negative control in BxPC-3 cells with the enriched hallmark gene sets presented. BxPC-3 cells were treated with XD2–149 (2 μM), napabucain (2 μM) and XD2–162 (4 μM) for 16 h. Cell lysates were analyzed by multiplexed proteomics analysis (XD2–149: n = 3, napabucasin and XD2–162: n = 2). (A) Volcano plot for XD2–149 proteins. The significant upregulated and downregulated proteins are colored in pink. (fold-change (FC) > 1.5 and p-value ≤ 0.05). (B) Venn diagram showing the overlap of common proteins between the three treatments and the exclusive proteins for each. Significant proteins > 1.5 fold-change and p-value ≤ 0.05 were used to generate the diagram. (C) Upregulated and downregulated enriched hallmark gene sets from GSEA analysis of XD2–149. (D) Select enrichment plots for the top upregulated and downregulated enriched gene sets from GSEA analysis of XD2–149.
Table 3.
Downregulated Proteins Exclusive to XD2–149 Treatment in BxPC-3 Cells
| No | Gene Symbol | Name | Log2FCa | p-valueb |
|---|---|---|---|---|
| 1 | TMPRSS4 | Transmembrane protease serine 4 | −1.27 | 0.028 |
| 2 | PLK1 | Serine/threonine-protein kinase PLK1 | −1.25 | 0.020 |
| 3 | TP53BP2 | Apoptosis-stimulating of p53 protein 2 | −1.02 | 0.005 |
| 4 | DYNLT1 | Dynein light chain Tctex-type 1 | −0.97 | 0.014 |
| 5 | DTX3L | E3 ubiquitin-protein ligase DTX3L | −0.96 | 0.042 |
| 6 | MAGED1 | Melanoma-associated antigen D1 | −0.9 | 0.002 |
| 7 | STAT2 | Signal transducer and activator of transcription 2 | −0.9 | 0.010 |
| 8 | CBR1; SETD4 | Carbonyl reductase [NADPH] 1 | −0.86 | <0.001 |
| 9 | SPINT1 | Kunitz-type protease inhibitor 1 | −0.85 | 0.001 |
| 10 | CCNB1 | G2/mitotic-specific cyclin-B1 | −0.84 | 0.031 |
| 11 | EDF1 | Endothelial differentiation-related factor 1 | −0.82 | <0.001 |
| 12 | CST3 | Cystatin-C | −0.81 | 0.001 |
| 13 | NABP2 | SOSS complex subunit B1 | −0.78 | 0.008 |
| 14 | WDFY4 | WD repeat- and FYVE domain-containing protein 4 | −0.78 | 0.009 |
| 15 | CSDE1 | Cold shock domain-containing protein E1 | −0.75 | <0.001 |
| 16 | SHPRH | E3 ubiquitin-protein ligase SHPRH | −0.74 | 0.007 |
| 17 | FAM3C; WNT16 | Protein FAM3C | −0.73 | <0.001 |
| 18 | DDR1 | Epithelial discoidin domain-containing receptor 1 | −0.73 | <0.001 |
| 19 | DSP | Desmoplakin | −0.70 | 0.002 |
| 20 | GPR56; ADGRG1 | Adhesion G-protein coupled receptor G1 | −0.69 | 0.004 |
| 21 | STAT3 | Signal transducer and activator of transcription 3 | −0.69 | 0.001 |
| 22 | LTV1 | Protein LTV1 homolog | −0.69 | 0.003 |
| 23 | AGRN | Agrin | −0.68 | 0.001 |
| 24 | SDF4 | 45 kDa calcium-binding protein | −0.67 | 0.002 |
| 25 | LAMB1 | Laminin subunit beta-1 | −0.67 | <0.001 |
| 26 | UBE2T | Ubiquitin-conjugating enzyme E2 T | −0.66 | 0.029 |
| 27 | PHLDB1 | Pleckstrin homology-like domain family B member 1 | −0.64 | 0.009 |
| 28 | CHST14 | Carbohydrate sulfotransferase 14 | −0.64 | 0.001 |
| 29 | PLOD2 | Procollagen-lysine,2-oxoglutarate 5-dioxygenase 2 | −0.63 | 0.019 |
| 30 | PVRL1; NECTIN1 | Nectin-1 | −0.63 | 0.002 |
| 31 | PRRC2B | Protein PRRC2B | −0.63 | 0.003 |
| 32 | MGMT | Methylated-DNA--protein-cysteine methyltransferase | −0.63 | 0.004 |
| 33 | NSMCE2 | E3 SUMO-protein ligase NSE2 | −0.63 | 0.012 |
| 34 | ZFP91 | E3 ubiquitin-protein ligase ZFP91 | −0.62 | 0.002 |
| 35 | SERF2 | Small EDRK-rich factor 2 | −0.61 | 0.002 |
| 36 | POLD4 | DNA polymerase delta subunit 4 | −0.61 | 0.017 |
| 37 | PRDM16 | Histone-lysine N-methyltransferase PRDM16 | −0.6 | 0.033 |
| 38 | CIAPIN1 | Anamorsin | −0.59 | <0.001 |
| 39 | CCNDBP1 | Cyclin-D1-binding protein 1 | −0.59 | 0.005 |
| 40 | USP8 | Ubiquitin carboxyl-terminal hydrolase 8 | −0.59 | 0.008 |
FC > 1.5.
p-value ≤ 0.05.
Table 4.
Upregulated Proteins Exclusive to XD2–149 Treatment in BxPC-3 Cells
| No | Gene Symbol | Name | Log2FCa | p-valueb |
|---|---|---|---|---|
| 1 | JUN | Transcription factor AP-1 | 1.58 | <0.001 |
| 2 | ARX | Homeobox protein ARX | 1.54 | 0.043 |
| 3 | DNAJB9 | DnaJ homolog subfamily B member 9 | 1.35 | <0.001 |
| 4 | IL1RL1 | Interleukin-1 receptor-like 1 | 1.24 | 0.010 |
| 5 | SYNC | Syncoilin | 1.04 | 0.045 |
| 6 | RND3 | Rho-related GTP-binding protein RhoE | 0.98 | 0.001 |
| 7 | HMGCR | 3-hydroxy-3-methylglutaryl-coenzyme A reductase | 0.83 | 0.009 |
| 8 | ZNF430 | Zinc finger protein 430 | 0.77 | 0.039 |
| 9 | ECM1 | Extracellular matrix protein 1 | 0.76 | 0.026 |
| 10 | RHOB | Rho-related GTP-binding protein RhoB | 0.73 | 0.035 |
| 11 | GDF15 | Growth/differentiation factor 15 | 0.69 | 0.010 |
| 12 | ABCB6 | ATP-binding cassette sub-family B member 6, mitochondrial | 0.68 | 0.025 |
| 13 | AFF1 | AF4/FMR2 family member 1 | 0.67 | 0.049 |
| 14 | MPV17 | Protein Mpv17 | 0.66 | 0.049 |
| 15 | EPHB3 | Ephrin type-B receptor 3 | 0.65 | 0.024 |
| 16 | UGCG | Ceramide glucosyltransferase | 0.62 | 0.032 |
| 17 | AKR1C2 | Aldo-keto reductase family 1 member C2 | 0.6 | 0.004 |
| 18 | BACH1 | Transcription regulator protein BACH1 | 0.6 | 0.004 |
FC > 1.5.
p-value ≤ 0.05.
Among the downregulated proteins is the E3 ubiquitin-protein ligase ZFP91 that has been reported as a neo-substrate that can be recruited by CRBN-based PROTACs independent of the target protein.45 Previous studies show that the nonspecific recruitment of ZFP91 was observed with CRBN ligase but not VHL possibly due to differences in the protein surface that allows CRBN to create high-affinity binding interfaces.45 A pSILAC mass spectrometric study was performed for target identification of small molecules that can induce protein degradation.47 The study identified ZFP91 as a substrate of lenalidomide-induced CRBN exploring the possibility of IMiD-induced repurposing of CRBN to target new proteins for degradation. The structural basis and mechanism for lenalidomide-induced CRBN-mediated degradation of proteins were reported to prove that protein recruitment is dependent on the presence of the IMiD that remodels the CRBN surface for binding of these proteins.48 This intrigued us to test for the degradation of ZFP91 especially that XD2–149 did not result in proteasome-dependent degradation of STAT3; the intended target protein. We performed Western blot experiments treating BxPC-3 cells with XD2–149 and napabucasin for 16 h. Interestingly, ZFP91 protein was significantly downregulated by XD2–149 at 1 μM while we observed no effect with napabucasin up to 3 μM (Figure 8A). To determine whether this effect occurs at an earlier time point, we tested the effect of XD2–149 after 4, 16 and 24 h. A significant decrease in the expression of ZFP91 was observed at a comparable DC50 for the 16 and 24 h treatments while the earlier time point showed no effect (Figure 8B). Next, we repeated the experiment in the presence of the proteasome inhibitor carfilzomib to determine if the downregulation is a result of proteasome-dependent degradation. Gratifyingly, the results showed a restoration in the ZFP91 protein levels due to the blockade of the XD2–149-induced degradation (Figure 8C). We obtained similar results in MIA PaCa-2 cells (Figure S5). This confirms that our PROTAC results in proteasome-dependent degradation of ZFP91 and provides a possible explanation for the lack of degradation effect on the STAT3 protein. Combining napabucasin with XD2–149 did not affect the activity of the PROTAC suggesting it does not compete with napabucasin for its binding site (Figure 8D).
Figure 8.
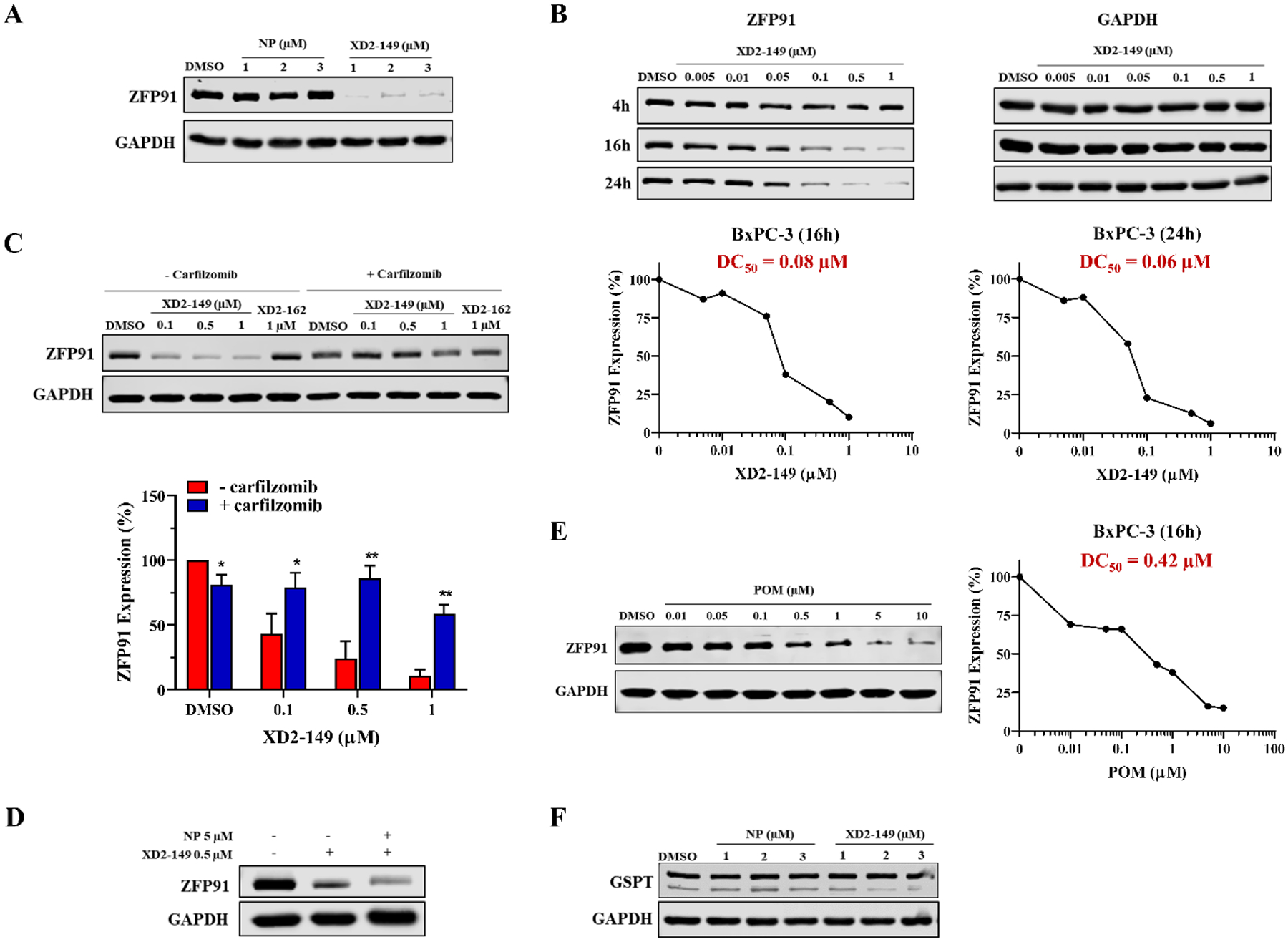
XD2–149 degrades ZFP91 protein in a proteasome-dependent manner. (A) XD2–149 reduces ZFP91 expression while napabucasin (NP) has no effect. BxPC-3 cells were treated with NP or XD2–149 at the indicated concentrations for 16 h. (B) XD2–149 degrades ZFP91 at 16 and 24 h with no effect at 4 h. Dose-response curves of ZFP91 degradation by XD2–149 are presented for the 16 and 24 h time-points. (C) The effect of XD2–149 on ZFP91 is proteasome-dependent. BxPC-3 cells were pretreated with 1 μM of the proteasome inhibitor carfilzomib for 1 h followed by XD2–149 or XD2–162 treatment for 16 h. (D) NP does not abolish the effect of XD2–149. BxPC-3 cells were treated with XD2–149 in presence or absence of NP for 16 h. (E) Pomalidomide (POM) induces ZFP91 degradation after 16 h. (F) XD2–149 have no effect on the expression of GSPT. BxPC-3 cells were treated with NP or XD2–149 for 16 h. Immunoblots were quantified using ImageJ and the DC50 values were calculated using GraphPad Prism 8. * denotes p ≤ 0.05, ** denotes p ≤ 0.01, *** denotes p ≤ 0.001.
Pomalidomide has been reported to induce the degradation of CRBN neo-substrates like ZFP91.47 We sought to test its effect on ZFP91 in BxPC-3 cells to better understand the activity of our PROTAC. Pomalidomide displayed a DC50 of 0.42 μM, which is 5-fold less potent than the PROTAC (Figure 8E). This result shows that although the pomalidomide moiety contributes to ZFP91-induced degradation, the PROTAC might be involved in a more stable ternary complex that results in more effective degradation. Moreover, pomalidomide does not show any cytotoxicity in the MTT assay up to 30 μM (Table S4). We also tested for other targets like GSPT1 that might be recruited for proteasome-dependent degradation. However, XD2–149 did not degrade GSPT, excluding the possibility that our PROTAC is non-selectively targeting CRBN neo-substrates (Figure 8F). Other proteins that have also been reported as CRBN neo-substrates including IKZF1 and IKZF349, 50, CK1α48, and ZBTB1651 were not found among the proteins exclusively downregulated with XD2–149 treatment.
ZFP91 is involved in various biological processes and among its oncogenic properties is the regulation of NF-кB signaling pathway and HIF-1α promoting tumorigenesis.52, 53 It is also reported to play a role in ubiquitination and destabilization of FOXA1 to promote cancer cell survival.54 In-depth understanding of the exact role of ZFP91 is lacking. However, the published data so far suggests that its inhibition or degradation could be beneficial as an anti-cancer strategy.
Consistent with the napabucasin reported mechanism of action, proteomics results for XD2–149 show differential expression of proteins involved in cancer stemness and STAT3 signaling including downregulation of WNT16, DDR1, DSP, CST3, PLK1, DTX3L and CCNB1 (Table 3) and upregulation of JUN, IL1RL1, RND3, ECM1, RHOB, EPHB3, UCUG and AKR1C2 (Table 4). As previously discussed, XD2–149 inhibits STAT3-driven transcription in the luciferase assay indicating that it acts through inhibition of the IL6-STAT3 pathway. Altered proteins in common between napabucasin, XD2–149 and XD2–162 include 10 downregulated proteins and 14 upregulated proteins presented in Table S12–S13.
We performed gene set enrichment analysis (GSEA) to gain insight into the signaling pathways involved in the mechanism of action of XD2–149 (Table S23–S32). Enriched gene sets for napabucsain and XD2–162 are presented in Tables S14–S22 and S33–42, respectively. GSEA analysis for XD2–149 displayed nine upregulated and five downregulated enriched Hallmark gene sets (Figure 7C–D). Comparison with napabucasin and XD2–162 revealed enrichment of eight common upregulated Hallmark gene sets including TNFA signaling via NF-кB, hypoxia, IL2/STAT5 signaling, KRAS signaling, unfolded protein response, inflammatory response, apoptosis and coagulation (Figure S6). These pathways are commonly altered with STAT3 inhibition suggesting napabucasin and its PROTAC work through inhibition of STAT3 signaling. The altered pathways are also possibly cumulative as a result of multiple actions. We mentioned earlier that ZFP91 is involved in regulation of NF-кB and HIF-1α pathways and our GSEA data shows upregulation of both pathways suggesting that these effects can be secondary to degradation of ZFP91. On the other hand, there are seven downregulated gene sets in common including E2F targets, DNA replication, mismatch repair, and telomere maintenance via semiconservative replication (Figure S6). STAT3 plays a role in regulation of cell cycle through activation of E2F targets including Cyclin D1, Cyclin B1 and Cdc2.15, 55 This explains the observed downregulation in E2F targets that can result from STAT3 downregulation caused by napabucasin and XD2–149 treatment. STAT3 is known to play an important role in cell proliferation through mechanisms that include suppression of ATR and modulation of DNA repair.56 XD2–149 was also found to downregulate the hallmark interferon-alpha response gene set (Figure 7D). IFN-α engages with its receptor and triggers the activation of STAT3. Studies have shown that STAT3 itself plays a role in regulating the IFN-mediated response.57 This data is in accord with the reported experiments that STAT3 signaling is a major target pathway in the mechanism of action of napabucasin.
XD2–149 Cytotoxicity is Dependent on ZFP91.
Until now, there are no reported ZFP91-targeted anticancer therapies. A previous study on the kinase inhibitor foretinib demonstrated that GSPT1 and ZFP91 proteins are neo-substrates for the CRBN-based PROTAC but did not investigate their contribution to its anticancer activity.45 ZFP91 has been validated as an oncogenic target that regulates different pathways involved in tumorigenesis and its targeting will present a novel class of anti-cancer agents.53, 54, 58, 59 ZFP91 is highly expressed in pancreatic cancer cells and is associated with lower overall survival (Figure 9A).60 Knockdown studies of ZFP91 in pancreatic cancer cells demonstrated reduced cell growth.59 To determine whether the cytotoxicity of XD2–149 is dependent on ZFP91, we knocked down the protein in BxPC-3 using siRNA (Figure 9B). This resulted in a significant reduction (p < 0.01) in the cytotoxicity of XD2–149 after a 48-h treatment in MTT providing evidence to the importance of ZFP91 for its anti-cancer activity (Figure 9C).
Figure 9.
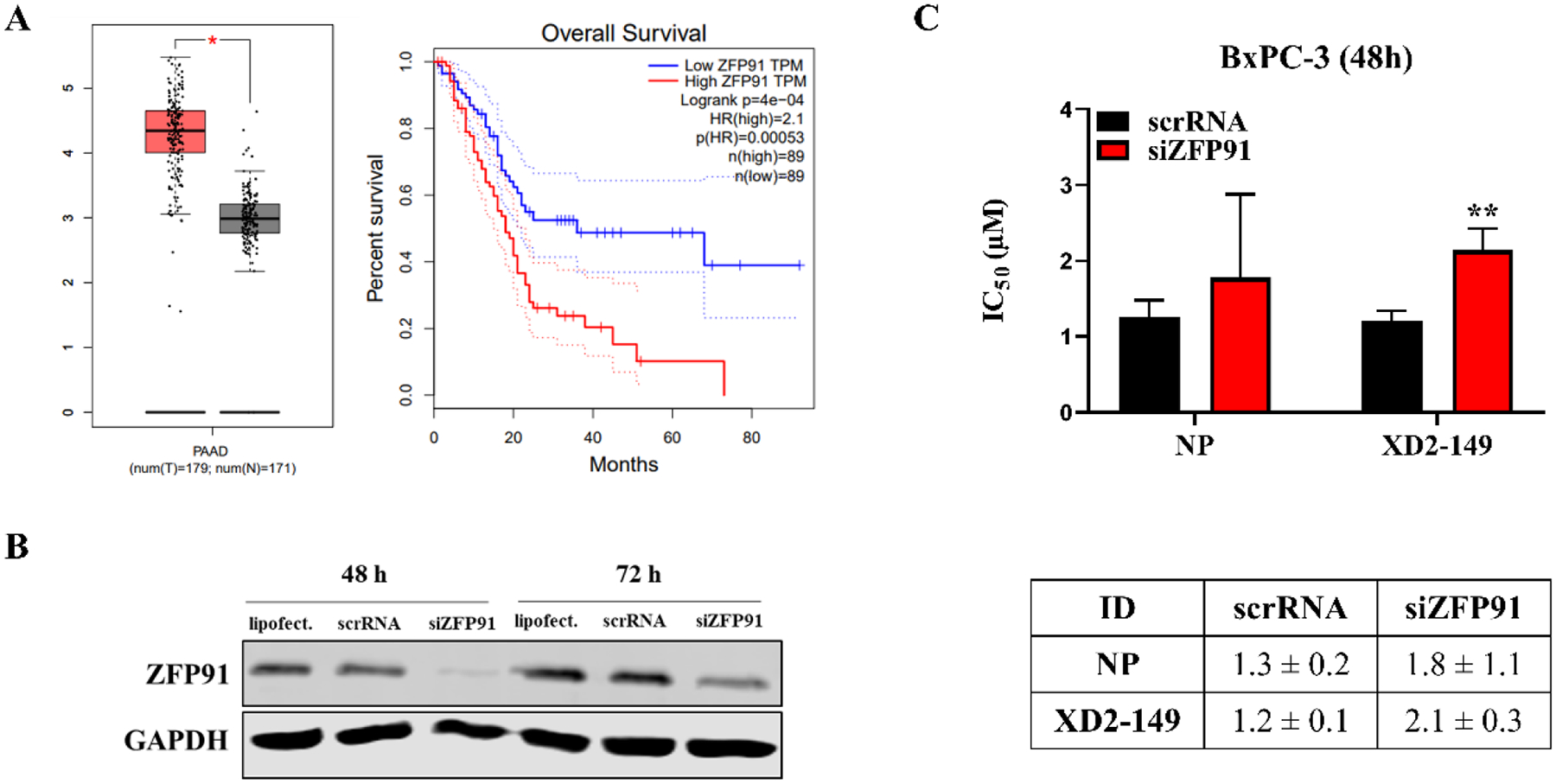
Cytotoxicity of XD2–149 is partially dependent on ZFP91. (A) Expression levels of ZFP91 in normal (grey) and pancreatic adenocarcinoma (PAAD) (red) tissues and the overall survival with low vs high ZFP91 expression in PAAD. The data was obtained from the GEPIA2 web server that is based on TCGA and GTEx databases. (B) siRNA knockdown of ZFP91 in BxPC-3 cells after 48 and 72 h. (C) IC50 values of XD2–149 show significantly reduction in its cytotoxicity with ZFP91 knockdown compared to the siRNA control cells (scrRNA). BxPC-3 cells were treated with siZFP91 and NP or XD2–149 for 48 h in the MTT assay. ** denotes p ≤ 0.01.
NQO1-mediated Cytotoxicity of XD2–149 is Independent of ZFP91.
To further study the cell death mechanisms that contribute to the cytotoxicity of XD2–149, we examined the mechanism induced by napabucasin to determine if they kill the cells similarly. As previously mentioned, NQO1 activates napabucasin to induce ROS and subsequently kill the cancer cells.20 In the presence of the NQO1 inhibitor, dicoumarol (DIC), napabucasin and XD2–149 displayed reduced colony formation in BxPC-3 and MIA PaCa-2 cells (Figure 10A). These results suggest that the cell death mechanism of XD2–149 is similar to napabucsain and is dependent on NQO1. To determine whether the NQO1-induced cytotoxicity is associated with ZFP91, we tested for the ZFP91 levels when treated with the PROTAC in the presence of DIC. The presence of DIC did not alter the ZFP91 levels suggesting a ZFP91-independent cell death mechanism (Figure 10B).
Figure 10.
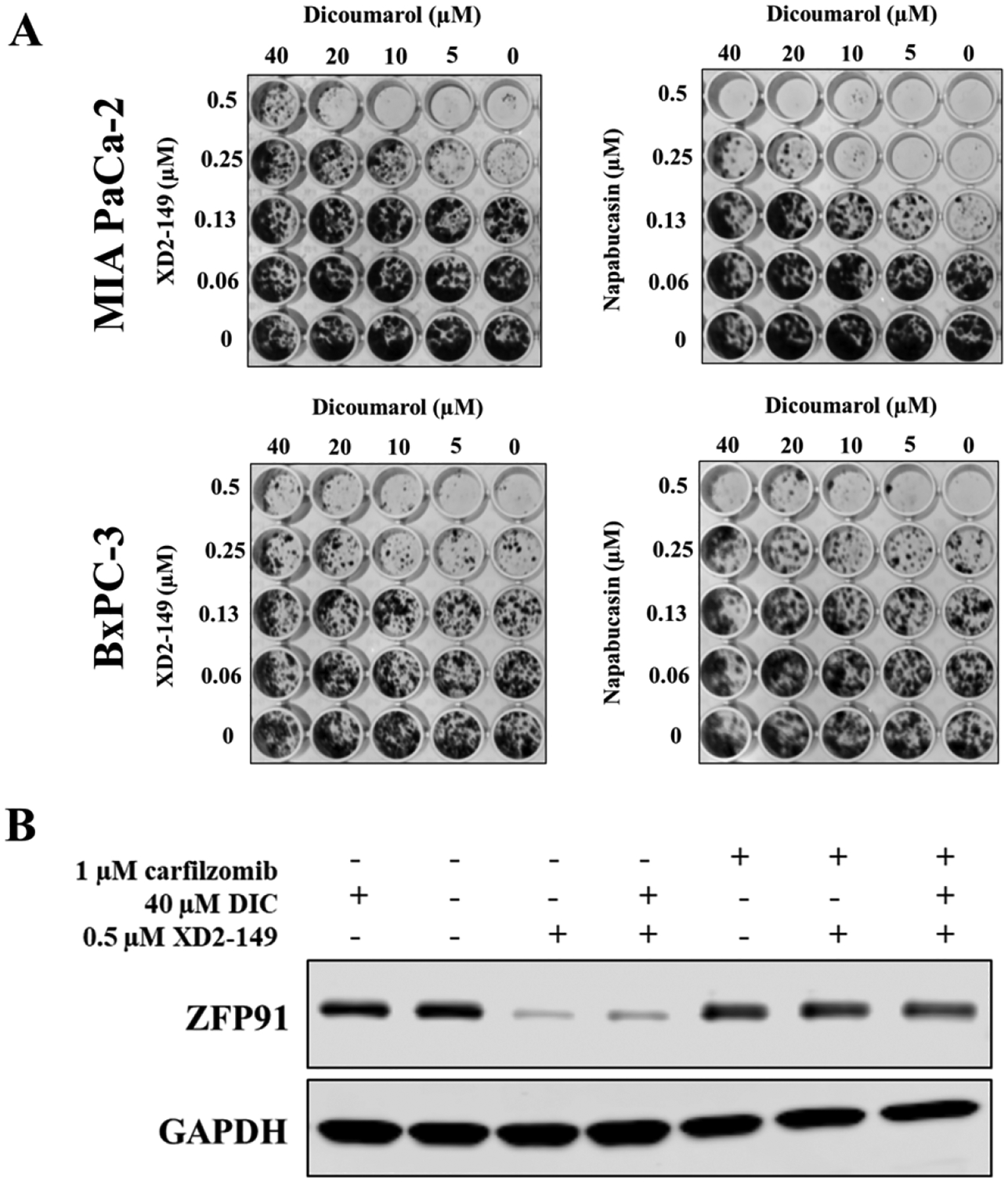
NQO1-induced cytotoxicity of XD2–149 is independent of ZFP91. (A) Dicoumarol partially rescues the cell death caused by XD2–149 in CFA. BxPC-3 and MIA PaCa-2 cells were treated with XD2–149 or napabucsain for 7–10 days. (B) Dicoumarol (DIC) effect is independent of ZFP91. BxPC-3 cells were treated with XD2–149 in absence or presence of DIC for 16 h. The proteasome inhibitor carfilzomib was added 1 h prior to the treatments.
CONCLUSIONS
We synthesized and tested a series of napabucasin-based PROTACs for targeting STAT3. We optimized the linker and the E3 ligase ligand to produce the lead compound XD2–149 that showed significant cytotoxicity in several cancer cell lines. Although XD2–149 resulted in inhibition of IL6/STAT3 pathway signaling, its effect was independent of the proteasome-mediated STAT3 degradation. GSEA of the proteomics data revealed several gene sets that indicated the STAT3 and other signaling pathways were targeted. Proteomics and Western blot analyses identified and validated a proteasome-dependent degradation of ZFP91 by XD2–149. The PROTAC was more effective in degrading ZFP91 than the IMiD pomalidomide. Furthermore, we confirmed that the cytotoxicity of XD2–149 is partially dependent on ZFP91. We also demonstrated that NQO1 contributes to the cytotoxicity of XD2–149 independent of ZFP91 which suggests that the PROTAC kills the cancer cells via multiple cell death mechanisms. ZFP91 is an oncogenic protein that has been studied for its potential as an anticancer target and further optimization may lead to the development of potent degraders of ZFP91 for anticancer therapy. Studies are in progress to validate other downregulated proteins as potential targets of our napabucasin-based PROTAC.
CHEMISTRY
Generation of pomalidomide-conjugated napabucasin PROTACS with varying linear linkers (5a-j) is summarized in Scheme 1. Intermediate 2–2 was obtained by condensation of 2–1 with 3-aminopiperidine-2,6-dione hydrochloride. A nucleophilic substitution reaction between 2–2 and corresponding amines in the presence of DIPEA yielded intermediate 2–3a-j. Boc-deprotection of 2–3a-j using TFA yielded 2–4a-j, which were converted to 2–5a-j by reacting with 1–5 (Scheme S1) in the presence of HATU and DIPEA.
Syntheses of pomalidomide-conjugated PROTACS with varying cyclized linkers (3–6a-d) are outlined in Scheme 2. A nucleophilic substitution reaction between 2–2 and corresponding amines in the presence of DIPEA yielded intermediates 3–2a-d, which were converted to 3–3a-d by deprotection of Boc group. Intermediates 3–4a-d were obtained through a reductive amination reaction using NaBH(OAc)3. Further removal of the Boc group yielded amine 3–5a-d. Compounds 3–6a-c and 3–6d (XD2–149) were obtained by reacting 5a-d with 1–5 in the presence of HATU and DIPEA.
The synthesis of compound 4–5 is outlined in Scheme 3. Condensation of 4–1 with 3-aminopiperidine-2,6-dione hydrochloride gave intermediate 4–2, which was converted to 4–3 through a Sonagashira reaction. Deprotection of Boc group of 4–3 yielded amine 4–4, which was converted to 4–5 by reacting with 1–5 in the presence of HATU and DIPEA.
The synthesis of compounds 5–5a and 5–5b are outlined in Scheme 4. Condensation of 5–1 with 3-aminopiperidine-2,6-dione hydrochloride gave intermediate 5–2, which was converted to 5–3a-b through a nucleophilic substitution. Deprotection of Boc groups of 5–3a-b yielded amine 5–4a-b, which were converted to 5–5a and 5–5b by reacting with 1–5 in the presence of HATU and DIPEA.
Scheme 4. Synthesis of Compounds 5–5a and 5–5ba.
aReagents and conditions: (a) NaOAc, HOAc, 110 °C; (b) KHCO3, KI, DMF, 70 °C; (c) TFA, DCM, rt; (d) HATU, DIPEA, DMF, rt.
The synthesis of compounds 6–6a-c are outlined in Scheme 5. A substitution reaction between 5–1 and t-butyl bromoacetate gave intermediate 6–2, which was converted to 6–3 using TFA. Condensation reaction between 6–3 and corresponding amines yielded 6–4a-c, which was converted to 6–5a-c by deprotection of Boc group. Compound 6–6a-cwere obtained by reacting 6–5a-c with 1–5 in the presence of HATU and DIPEA.
The synthesis of compound 7–4 is summarized in Scheme 6. A nucleophilic substitution reaction between 7–1 and corresponding amine in the presence of DIPEA yielded intermediate 7–2. Boc-deprotection of 7–2 using TFA yielded 7–3, which was converted to XD182 by reacting with 1–5 in the presence of HATU and DIPEA.
The synthesis of compound 8–1 is summarized in Scheme 7. 8–1 was obtained by reacting 8 with 1–5 in the presence of HATU and DIPEA.
The synthesis of compounds 9–4a-b are outlined in Scheme 8. A condensation reaction between 1–5 and corresponding amines gave intermediate 9–2a-b, which was converted to 9–3a-b using TFA. A condensation reaction between 9–3a-b and (S,R,S)-AHPC hydrochloride yielded 9–4a-b.
The synthesis of compound 10–5 (XD171) is summarized in Scheme 9. Intermediate 10–2 was obtained by condensation of 2–1 with 3-aminopiperidine-2,6-dione hydrochloride. A nucleophilic substitution reaction between 10–2 and corresponding amine in the presence of DIPEA yielded intermediate 10–3. Boc-deprotection of 10–3 using TFA yielded 10–4, which was converted to XD171 by reacting with 1–5 in the presence of HATU and DIPEA.
The synthesis of compound 11–7 (XD2–162) is outlined in Scheme 10. A nucleophilic substitution reaction between 11–2 and the corresponding amine in the presence of DIPEA yielded intermediate 11–3, which was converted to 11–4 by deprotection of the Boc group. Intermediate 11–5 was obtained through a reductive amination reaction using NaBH(OAc)3. Further removal of the Boc group yielded amine 11–6. Compounds XD2–162 was obtained by reacting 11–6 with 1–5 in the presence of HATU and DIPEA.
EXPERIMENTAL SECTION
General Methods.
Reagents and anhydrous solvents were purchased from commercial sources and used without further purification. Reaction progress was monitored by UV absorbance using thin-layer chromatography (TLC) on aluminum-backed precoated silica plates from Silicycle (SiliaPlate, 200 μm thickness, F254). Glassware for reactions were oven-dried in preparation, and reactions were performed using nitrogen or argon atmosphere using standard inert conditions. 1H NMR spectra were obtained using a Bruker (300 or 400 MHz) instrument. Spectral data are reported using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets, ddd = doublet of doublet of doublets, dt = doublet of triplets, ddt = doublet of doublet of triplets, dtd = doublet of triplet of doublets, and coupling constants are reported in Hz, followed by integration. Purifications using flash chromatography were performed using a Biotage Isolera chromatography system (25 μM spherical silica). Column chromatography was performed on silica gel (200–300 mesh), and preparative TLC was performed on UV 254 (0.5 mm thickness, Sigma-Aldrich). A Shimadzu LCMS 20–20 system was utilized for generating HPLC traces, obtaining mass spectrometry data, and evaluating purity. The system is equipped with a PDA UV detector and Kinetex 2.6 μm, XB-C18 100 Å, 75 mm × 4.6 mm column, which was used at room temperature. HPLC gradient method utilized a 1% to 90% MeCN in H2O with 0.01% formic acid over 15 min with a 0.50 mL/min flow rate. Purity of the final compounds is ≥95% and was assessed at 254 nm using the described column and method.
2-Hydroxy-3-(prop-1-en-1-yl)naphthalene-1,4-dione (1–2).
To a solution of reagent 1–1 (10.00 g, 54.41 mmol) in 170 mL of HOAc and 10 mL of concentrated HCl, was added propionaldehyde (20.81 mL, 287.09 mmol) at 80 °C, the reaction mixture was stirred at 80 °C for 1.5 h. After the reaction was completed, the reaction solution was poured into ice water and extracted with ethyl acetate (150 mL × 3). The organic layers were combined and extracted with saturated K2CO3 aqueous solution (150 mL × 6), the combined aqueous layer was acidized using concentrated HCl, the red solid formed was collected by filtration, which was used into next step without further purification (2.90 g). LCMS (ESI) 213 [M - H]−.
2-Methylnaphtho[2,3-b]furan-4,9-dione (1–3).
A mixture of 1–2 (2.20 g, 10.28 mmol) and Hg(OAc)2 (6.45 g, 20.56 mmol) in 250 mL of HOAc was stirred at rt for 3 h, then filtered and the filtrate was concentrated under reduced pressure to obtain a red solid, which was dissolved in 40 mL of HOAc and 40 mL of concentrated HCl, the resulting solution was stirred at 80 °C for 1 h. After the reaction was completed, the reaction solution was diluted with water at 0 °C, the yellow solid formed was collected by filtration (2.30 g, 82%). 1H NMR (300 MHz, chloroform-d) δ 8.23–8.12 (m, 2H), 7.78–7.67 (m, 2H), 6.61 (q, J = 0.9 Hz, 1H), 2.52 (d, J = 0.9 Hz, 3H).
4,9-Dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carbaldehyde (1–4).
A mixture of intermediate 1–3 (1.80 g, 8.49 mmol), SeO2 (4.70 g, 67.92 mmol) and SiO2 (18.00 g) was dissolved in DCM (100 mL), then removed the solvent under reduced pressure, the residue obtained was stirred at 150 °C for 6 h. The crude product was purified with column chromatography (hexane/ethyl acetate = 4:1) to yield intermediate 1–4 as a yellow solid (1.10 g, 57%). 1H NMR (300 MHz, chloroform-d) δ 9.98 (s, 1H), 8.36–8.19 (m, 2H), 7.91–7.76 (m, 2H), 7.69 (s, 1H).
4,9-Dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxylic acid (1–5).
To a solution of intermediate 1–4 in HOAc (60 mL) was added H2O2 (22 mL) at 75 °C, the resulting mixture was stirred at 75 °C for 2 h. After cooling down, water (50 mL) was added, the solution was concentrated under reduced pressure, the solid formed was collected by filtration, washed with water (5 mL × 3). Intermediate 1–5 was obtained as a yellow solid, which was used into next step without further purification (470 mg, 73%). 1H NMR (300 MHz, DMSO-d6) δ 8.25–8.04 (m, 2H), 7.97–7.82 (m, 2H), 7.68 (s, 1H).
2-(2,6-Dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (2–2).
A mixture of 2–1 (2.20, 13.25 mmol), 3-aminopiperidine-2,6-dione hydrochloride (2.17, 13.25 mmol) and NaOAc (3.30, 40.24 mmol) was dissoved in HOAc (50 mL), the resulting mixture was stirred at 100 °C for 16 h. After the reaction was completed, the solution was concentrated under reduced pressure to obtain a residue, which was diluted with NaHCO3 aqueous solution and extracted with ethyl acetate (150 mL × 3), the combined organic phase was washed with NaHCO3 aqueous solution (150 mL) and brine (150 mL), dried over Na2SO4, filtered and concentrated to yield a yellow solid, which was used in the next step without further purification (2.30 g, 63%). 1H NMR (300 MHz, DMSO-d6) δ 11.16 (s, 1H), 7.94 (ddd, J = 8.4, 7.3, 4.6 Hz, 1H), 7.79 (d, J = 7.3 Hz, 1H), 7.73 (dd, J = 9.3, 8.4, 1H), 5.16 (dd, J = 12.9, 5.4 Hz, 1H), 2.89 (m, 1H), 2.67–2.50 (m, 2H), 2.14–2.00 (m, 1H).
General procedure for the preparation of 2–3a–j.
A mixture of intermediate 2–2 (1 equiv.), tert-butyl (2-aminoethyl)carbamate (1 equiv.), DIPEA (2 equiv.) and DMF (2 mL) was stirred at 80 °C. After 12 h, the reaction mixture was diluted with water (10 mL), extracted with ethyl acetate (2 × 15 mL), washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with preparative TLC (DCM/MeOH = 15:1) to obtain intermediate 2–3aj or was used in the next step without further purification.
tert-Butyl (2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)carbamate (2–3a).
Yellow solid (20 mg, 27%). 1H NMR (300 MHz, chloroform-d) δ 8.43 (s, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.10 (d, J = 7.1 Hz, 1H), 6.97 (d, J = 8.5 Hz, 1H), 6.39 (t, J = 5.5 Hz, 1H), 5.09–4.80 (m, 2H), 3.52–3.24 (m, 4H), 2.90–2.71 (m, 3H), 2.19–2.05 (m, 1H), 1.43 (s, 9H).
General procedure for the preparation of 2–4a–j.
Intermediate 2–3 was dissolved in DCM (2 mL), TFA (1 mL) was added to the reaction mixture, and the mixture was stirred at rt. After 2 h, the reaction mixture was concentrated under reduced pressure. The residue thus obtained was used in the next step without further purification.
N-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (2–5a).
To a stirred solution of intermediate 1–5 (12 mg, 0.048 mmol) in DMF (2 mL) was added HATU (27 mg, 0.071 mmol), DIPEA (16 mg, 0.15 mmol) and intermediate 2–4a (20 mg, 0.048 mmol), the reaction mixture was stirred at rt for 12 h. The reaction solution was diluted with water (10 mL), extracted with ethyl acetate (15 mL × 2), washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with preparative TLC (DCM/MeOH = 25:1) to yield compound 2–5a as a yellow solid (5 mg, 18%). 1H NMR (300 MHz, chloroform-d) δ 8.63 (s, 1H), 8.26–8.12 (m, 2H), 7.84–7.70 (m, 2H), 7.62 (s, H), 7.58 (s, 1H), 7.48 (t, J = 7.9 Hz, 1H), 7.08 (d, J = 7.0 Hz, 1H), 6.98 (d, J = 8.2 Hz, 1H), 6.49 (s, 1H), 5.17–4.92 (m, 1H), 3.79–3.63 (m, 2H), 3.63–3.47 (m, 2H), 2.97–2.73 (m, 3H), 2.21–2.06 (m, 1H). LCMS (ESI) 541 [M + H]+. HPLC purity at 254 nm.
N-(3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)propyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (2–5b).
Compound 2–5b was obtained as a yellow solid using a method similar to that described for compound 2–5a. 1H NMR (300 MHz, chloroform-d) δ 8.27–8.18 (m, 2H), 8.02 (s, 1H), 7.83–7.78 (m, 2H), 7.62 (s, 1H), 7.57–7.45 (m, 1H), 7.11 (d, J = 7.0 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 6.48 (s, 1H), 3.71–3.53 (m, 2H), 3.49–3.36 (m, 3H), 3.01–2.56 (m, 2H), 2.20–1.93 (m, 4H). LCMS (ESI) 555 [M + H]+. HPLC purity at 254 nm.
N-(4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)butyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (2–5c).
Compound 2–5c was obtained as a yellow solid using a method similar to that described for compound 2–5a. 1H NMR (300 MHz, chloroform-d) δ 8.29–8.16 (m, 2H), 8.02 (s, 1H), 7.85–7.75 (m, 2H), 7.60 (s, 1H), 7.50 (dd, J = 8.4, 7.3 Hz, 1H), 7.09 (d, J = 7.0 Hz, 1H), 6.92–6.81 (m, 2H), 6.26 (s, 1H), 5.04–4.78 (m, 1H), 3.65–3.47 (m, 2H), 3.42–3.24 (m, 2H), 2.97–2.67 (m, 3H), 2.27–2.06 (m, 1H), 1.83–1.74 (m, 4H). LCMS (ESI) 569 [M + H]+. HPLC purity at 254 nm.
N-(5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)pentyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (2–5d).
Compound 2–5d was obtained as a yellow solid using a method similar to that described for compound 2–5a. 1H NMR (300 MHz, chloroform-d) δ 8.32–8.14 (m, 2H), 8.05 (s, 1H), 7.89–7.71 (m, 2H), 7.60 (s, 1H), 7.50 (dd, J = 8.5, 7.2 Hz, 1H), 7.09 (d, J = 7.1 Hz, 1H), 6.89 (d, J = 8.5 Hz, 2H), 5.03–4.75 (m, 1H), 3.51 (q, J = 7.3 Hz, 2H), 3.38–3.15 (m, 2H), 3.00–2.65 (m, 3H), 2.22–2.01 (m, 1H), 1.81–1.67 (m, 4H), 1.08–0.66 (m, 2H). LCMS (ESI) 583 [M + H]+. HPLC purity at 254 nm.
N-(6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)hexyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (2–5e).
Compound 2–5e was obtained as a yellow solid using a method similar to that described for compound 2–5a. 1H NMR (300 MHz, chloroform-d) δ 8.29–8.13 (m, 2H), 8.09 (s, 1H), 7.88–7.71 (m, 2H), 7.59 (s, 1H), 7.55–7.41 (m, 1H), 7.08 (d, J = 7.1 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H), 6.87–6.75 (m, 2H), 6.24 (s, 1H), 5.00–4.79 (m, 1H), 3.47 (q, J = 6.3 Hz, 2H), 3.28 (q, J = 6.8 Hz, 2H), 2.98–2.65 (m, 3H), 2.22–2.07 (m, 1H), 1.68 (dd, J = 13.9, 6.5 Hz, 4H), 1.51–1.40 (m, 4H). LCMS (ESI) 597 [M + H]+. HPLC purity at 254 nm.
N-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (2–5f).
Compound 2–5f was obtained as a yellow solid using a method similar to that described for compound 2–5a. 1H NMR (300 MHz, chloroform-d) δ 8.29–8.08 (m, 3H), 7.85–7.74 (m, 2H), 7.61 (s, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.21 (s, 1H), 7.06 (d, J = 7.1 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 6.55 (s, 1H), 5.09–4.94 (m, 1H), 3.88–3.62 (m, 6H), 3.60–3.34 (m, 2H), 2.96–2.77 (m, 3H), 2.24–2.08 (m, 1H). LCMS (ESI) 585 [M + H]+. HPLC purity at 254 nm.
N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (2–5g, XD97).
Compound XD97 was obtained as a yellow solid using a method similar to that described for compound 2–5a. 1H NMR (300 MHz, chloroform-d) δ 8.47 (s, 1H), 8.27 – 8.12 (m, 2H), 7.85–7.68 (m, 2H), 7.52 (s, 1H), 7.33 (t, J = 7.7 Hz, 2H), 6.97 (d, J = 7.1 Hz, 1H), 6.83 (d, J = 8.5 Hz, 1H), 6.52 (s, 1H), 4.99–4.83 (m, 1H), 3.81 (t, J = 5.2 Hz, 2H), 3.76–3.59 (m, 8H), 3.48 (q, J = 5.4 Hz, 2H), 2.93–2.72 (m, 3H), 2.25 – 2.10 (m, 1H). LCMS (ESI) 629 [M + H]+. HPLC purity at 254 nm.
N-(2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (2–5h).
Compound 2–5h was obtained as a yellow solid using a method similar to that described for compound 2–5a. 1H NMR (300 MHz, chloroform-d) δ 8.27 (s, 1H), 8.23–8.14 (m, 2H), 7.83–7.75 (m, 2H), 7.56 (s, 1H), 7.49–7.38 (m, 2H), 7.05 (d, J = 7.2 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 6.46 (d, J = 5.1 Hz, 1H), 5.04–4.80 (m, 1H), 3.75–3.64 (m, 14H), 3.51–3.39 (m, 2H), 3.01–2.60 (m, 3H), 2.24–2.09 (m, 1H). LCMS (ESI) 673 [M + H]+. HPLC purity at 254 nm.
N-(14-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12-tetraoxatetradecyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (2–5i).
Compound 2–5i was obtained as a yellow solid using a method similar to that described for compound 2–5a. 1H NMR (300 MHz, chloroform-d) δ 8.46 (s, 1H), 8.28–8.16 (m, 2H), 7.83–7.72 (m, 2H), 7.58 (s, 2H), 7.46 (t, J = 8.0 Hz, 1H), 7.07 (d, J = 7.2 Hz, 1H), 6.87 (d, J = 8.6 Hz, 1H), 6.47 (s, 1H), 5.03–4.86 (m, 1H), 3.78–3.63 (m, 18H), 3.50–3.38 (m, 2H), 2.99–2.71 (m, 3H), 2.26–2.05 (m, 1H). LCMS (ESI) 717 [M + H]+. HPLC purity at 254 nm.
N-(20-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15,18-hexaoxaicosyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (2–5j).
Compound 2–5j was obtained as a yellow solid using a method similar to that described for compound 2–5a. 1H NMR (300 MHz, chloroform-d) δ 8.51 (s, 1H), 8.29–8.12 (m, 2H), 7.85–7.72 (m, 2H), 7.58 (s, 1H), 7.48 (q, J = 8.3, 7.3 Hz, 2H), 7.07 (d, J = 7.1 Hz, 1H), 6.89 (d, J = 8.6 Hz, 1H), 6.48 (t, J = 6.5 Hz, 1H), 5.01–4.86 (m, 1H), 3.70–3.62 (m, 22H), 3.50–3.38 (m, 2H), 2.96–2.66 (m, 3H), 2.19–2.04 (m, 1H). LCMS (ESI) 805 [M + H]+. HPLC purity at 254 nm.
tert-Butyl 4-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)piperazine-1-carboxylate (3–2a).
A mixture of 2–2 (200 mg, 0.72 mmol), tert-butyl 4-(2-aminoethyl)piperazine-1-carboxylate (174 mg, 0.76 mmol), and DIPEA (187 mg, 1.45 mmol) in DMF (5 mL) was stirred at 80 °C for 12 h. The reaction mixture was diluted with water (20 mL), extracted with ethyl acetate (25 mL × 3). The organic layers were combined and washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with column chromatography (DCM/MeOH = 15:1) to yield intermediate 3–2a as a yellow solid (80 mg, 23%). 1H NMR (300 MHz, chloroform-d) δ 9.11 (s, 1H), 7.45 (t, J = 7.9 Hz, 1H), 7.05 (d, J = 7.2 Hz, 1H), 6.83 (d, J = 8.4 Hz, 1H), 6.65 (s, 1H), 4.99–4.80 (m, 1H), 3.48–3.40 (m, 4H), 3.37–3.27 (m, 2H), 2.87–2.51 (m, 5H), 2.46–2.38 (m, 4H), 2.16–2.03 (m, 1H), 1.41 (s, 9H).
2-(2,6-Dioxopiperidin-3-yl)-4-((2-(piperazin-1-yl)ethyl)amino)isoindoline-1,3-dione trifluoroacetate (3–3a).
Intermediate 3–2a (60 mg, 0.12 mmol) was dissolved in DCM (2 mL), TFA (1 mL) was added, the mixture was stirred at rt for 2 h. The reaction solution was concentrated under reduced pressure to obtain 3–3a as a yellow solid, which was used into next step without further purification (60 mg, 100%).
tert-Butyl (2-(4-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)piperazin-1-yl)ethyl)carbamate (3–4a).
Intermediate 3–3a (60 mg, 0.12 mmol) and tert-butyl (2-oxoethyl)carbamate (40 mg, 0.24 mmol) were dissolved in DCE (10 mL), the mixture was stirred at rt for 0.5 h, then NaBH(OAc)3 (106 mg, 0.50 mmol) was added, the mixture was stirred at rt for 12 h. The reaction mixture was diluted with water (20 mL), extracted with DCM (15 mL × 3). The organic layers were combined and washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with column chromatography (DCM/MeOH = 10:1) to yield intermediate 3–4a as a yellow solid (60 mg, 91%). 1H NMR (300 MHz, methanol-d4) δ 7.55 (t, J = 8.0 Hz, 1H), 7.05 (d, J = 7.8 Hz, 2H), 5.06 (dd, J = 12.3, 5.2 Hz, 1H), 3.69–3.52 (m, 4H), 3.41 (dt, J = 14.7, 6.5 Hz, 4H), 3.16 (q, J = 6.9 Hz, 4H), 3.05–2.84 (m, 4H), 2.83–2.59 (m, 3H), 2.15–2.10 (m, 1H), 1.44 (s, 9H).
4-((2-(4-(2-Aminoethyl)piperazin-1-yl)ethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione trifluoroacetate (3–5a).
Intermediate 3–4a (40 mg, 0.076 mmol) was dissolved in DCM (2 mL), TFA (1 mL) was added, the mixture was stirred at rt for 2 h. The reaction solution was concentrated under reduced pressure to obtain 3–5a as a yellow solid, which was used in the next step without further purification (48 mg, 100%).
N-(2-(4-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)piperazin-1-yl)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (3–6a).
To a stirred solution of intermediate 1–5 (37 mg, 0.015 mmol) in DMF (2 mL) was added HATU (88 mg, 0.23 mmol), DIPEA (60 mg, 0.46 mmol) and intermediate 3–5a (48 mg, 0.077 mmol), the reaction mixture was stirred at rt for 12 h. The reaction solution was diluted with water (10 mL), extracted with ethyl acetate (15 mL × 2), washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with preparative TLC (DCM/MeOH = 15:1) to yield compound 3–6a as a yellow solid (12 mg, 24%). 1H NMR (400 MHz, DMSO-d6) δ 11.08 (s, 1H), 8.85 (t, J = 5.8 Hz, 1H), 8.18–8.07 (m, 2H), 7.94–7.87 (m, 2H), 7.62 (s, 1H), 7.60–7.54 (m, 1H), 7.09 (d, J = 8.5 Hz, 1H), 7.02 (d, J = 7.0 Hz, 1H), 6.75 (t, J = 5.1 Hz, 1H), 5.07 (dd, J = 12.9, 5.5 Hz, 1H), 3.39 (dt, J = 18.3, 6.1 Hz, 4H), 2.88 (ddd, J = 17.4, 14.0, 5.3 Hz, 1H), 2.64–2.52 (m, 4H), 2.50–2.35 (m, 10H), 2.07–1.99 (m, 1H). LCMS (ESI) 653 [M + H]+. HPLC purity at 254 nm.
N-(2-(4-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)piperidin-1-yl)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (3–6b).
Compound 3–6b was obtained as a yellow solid using a method similar to that described for compound 3–6a. 1H NMR (400 MHz, DMSO-d6) δ 11.08 (s, 1H), 8.84 (t, J = 5.7 Hz, 1H), 8.25–8.06 (m, 2H), 8.03–7.85 (m, 2H), 7.62 (s, 1H), 7.58 (dd, J = 8.6, 7.1 Hz, 1H), 7.09 (d, J = 8.6 Hz, 1H), 7.02 (d, J = 7.0 Hz, 1H), 6.49 (t, J = 5.8 Hz, 1H), 5.05 (dd, J = 12.9, 5.4 Hz, 1H), 3.40 (q, J = 6.4 Hz, 2H), 3.36–3.32 (m, 2H), 2.96–2.76 (m, 3H), 2.63–2.53 (m, 2H), 2.49–2.43 (m, 2H), 2.07–1.99 (m, 1H), 2.00–1.90 (m, 2H), 1.69 (d, J = 12.5 Hz, 2H), 1.57–1.45 (m, 2H), 1.41–1.27 (m, 1H), 1.21–1.16 (m, 2H). LCMS (ESI) 652 [M + H]+. HPLC purity at 254 nm.
N-(2-(4-(3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)propyl)piperidin-1-yl)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (3–6c).
Compound 3–6c was obtained as a yellow solid using a method similar to that described for compound 3–6a. 1H NMR (400 MHz, DMSO-d6) δ 11.08 (s, 1H), 8.83 (t, J = 5.7 Hz, 1H), 8.19–8.05 (m, 2H), 7.94–7.86 (m, 2H), 7.62 (s, 1H), 7.57 (dd, J = 8.6, 7.0 Hz, 1H), 7.08 (d, J = 8.6 Hz, 1H), 7.01 (d, J = 7.0 Hz, 1H), 6.52 (t, J = 5.9 Hz, 1H), 5.05 (dd, J = 12.9, 5.4 Hz, 1H), 3.39 (q, J = 6.4 Hz, 2H), 3.29–3.23 (m, 2H), 2.95–2.81 (m, 3H), 2.64–2.52 (m, 2H), 2.46 (t, J = 6.8 Hz, 2H), 2.07–2.00 (m, 1H), 1.97–1.86 (m, 2H), 1.67–1.52 (m, 4H), 1.31–1.20 (m, 3H), 1.16–1.05 (m, 2H). LCMS (ESI) 666 [M + H]+. HPLC purity at 254 nm.
N-(2-(4-(5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)pentyl)piperidin-1-yl)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (3–6d, XD2–149).
Compound XD2–149 was obtained as a yellow solid using a method similar to that described for compound 3–6a. 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 8.86 (t, J = 5.7 Hz, 1H), 8.18 – 8.06 (m, 2H), 7.90 (ddt, J = 6.8, 4.7, 3.4 Hz, 2H), 7.62 (s, 1H), 7.57 (dd, J = 8.5, 7.0 Hz, 1H), 7.09 (d, J = 8.6 Hz, 1H), 7.01 (d, J = 7.0 Hz, 1H), 6.53 (t, J = 6.1 Hz, 1H), 5.10 – 4.99 (m, 1H), 3.58 – 3.34 (m, 10H), 2.94 – 2.76 (m, 3H), 2.63 – 2.55 (m, 1H), 2.05 – 1.98 (m, 1H), 1.92 (d, J = 10.2 Hz, 1H), 1.58 (q, J = 9.5, 6.5 Hz, 4H), 1.38 – 1.24 (m, 6H). LCMS (ESI) 694 [M + H]+. HPLC purity at 254 nm.
4-Bromo-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (4–2).
A mixture of 4–1 (2.00 g, 8.81 mmol), 3-aminopiperidine-2,6-dione hydrochloride (1.44 g, 8.81 mmol) and NaOAc (2.17, 26.46 mmol) was dissoved in HOAc (50 mL), the resulting mixture was stirred at 120 °C for 24 h. After the reaction was completed, the solution was concentrated under reduced pressure to obtain a residue, which was diluted with water (100 mL), the grey solid formed was collected by filtration, which was used into next step without further purification (2.20 g, 74%). 1H NMR (300 MHz, DMSO-d6) δ 11.17 (s, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.93 (d, J = 7.3 Hz, 1H), 7.77 (t, J = 7.8 Hz, 1H), 5.17 (dd, J = 12.6, 4.8 Hz, 1H), 3.02–2.78 (m, 1H), 2.67–2.51 (m, 2H), 2.17–1.96 (m, 1H).
tert-Butyl (5-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)pent-4-yn-1-yl)carbamate (4–3).
A mixture of 4–2 (80 mg, 0.24 mmol), tert-butyl pent-4-yn-1-ylcarbamate (44 mg, 0.24 mmol), Pd(PPh3)2Cl2 (17 mg, 0.024 mmol), and CuI (9 mg, 0.048 mmol) in DMF/TEA (2 mL/1 mL) was purged with nitrogen several times, and then stirred at 80 °C for 12 h under nitrogen atmosphere. The reaction mixture was then filtered, and the filtrate was concentrated under reduced pressure to obtain a residue, which was diluted with water (10 mL) and extracted with ethyl acetate (15 mL × 3). The organic layers were combined and washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with column chromatography (DCM/MeOH = 25:1) to yield intermediate 4–3 as a yellow solid (14 mg, 13%). 1H NMR (300 MHz, chloroform-d) δ 8.25 (s, 1H), 7.86 (t, J = 7.7 Hz, 1H), 7.78 (d, J = 6.8 Hz, 1H), 7.64 (d, J = 8.3 Hz, 1H), 5.06–4.89 (m, 2H), 3.42–3.27 (m, 2H), 2.90–2.75 (m, 3H), 2.57 (t, J = 6.8 Hz, 2H), 2.22–2.07 (m, 1H), 1.95–1.78 (m, 2H), 1.41 (s, 9H).
4-(5-Aminopent-1-yn-1-yl)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione trifluoroacetate (4–4).
Intermediate 4–4 was obtained as a yellow solid from 4–3 using a method similar as that described for intermediate 2–4a in quantitative yield.
N-(5-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)pent-4-yn-1-yl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (4–5).
To a stirred solution of intermediate 1–5 (8 mg, 0.033 mmol) in DMF (2 mL) was added HATU (18 mg, 0.047 mmol), DIPEA (16 mg, 0.12 mmol) and intermediate 4–4 (14 mg, 0.032 mmol), the reaction mixture was stirred at rt for 12 h. The reaction solution was diluted with water (10 mL), extracted with ethyl acetate (15 mL × 2), washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with preparative TLC (DCM/MeOH = 20:1) to yield compound 4–5 as a yellow solid (8 mg, 44%). 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 9.02 (t, J = 5.7 Hz, 1H), 8.16–8.08 (m, 2H), 7.94–7.87 (m, 2H), 7.86–7.75 (m, 3H), 7.64 (s, 1H), 5.14 (dd, J = 12.9, 5.4 Hz, 1H), 3.49 (q, J = 6.5 Hz, 2H), 2.89 (ddd, J = 17.1, 13.9, 5.3 Hz, 1H), 2.65–2.51 (m, 4H), 2.07 (ddd, J = 10.5, 5.4, 3.1 Hz, 1H), 1.89 (p, J = 6.9 Hz, 2H). LCMS (ESI) 564 [M + H]+. HPLC purity at 254 nm.
2-(2,6-Dioxopiperidin-3-yl)-4-hydroxyisoindoline-1,3-dione (5–2).
A mixture of 5–1 (3.00 g, 18.29 mmol), 3-aminopiperidine-2,6-dione hydrochloride (3.00 g, 18.29 mmol) and NaOAc (4.50, 54.88 mmol) was dissoved in HOAc (50 mL), the resulting mixture was stirred at 110 °C for 16 h. After the reaction was complete, the solution was concentrated under reduced pressure to obtain a residue, which was diluted with water (100 mL), the grey solid formed was collected by filtration, which was used in the next step without further purification (3.70 g, 74%). 1H NMR (300 MHz, DMSO-d6) δ 11.19 (s, 1H), 11.10 (s, 1H), 7.65 (dd, J = 8.4, 7.2 Hz, 1H), 7.32 (d, J = 7.1 Hz, 1H), 7.25 (dd, J = 8.4, 0.8 Hz, 1H), 5.07 (dd, J = 12.9, 5.4 Hz, 1H), 2.89 (ddd, J = 18.0, 14.0, 5.5 Hz, 1H), 2.67–2.49 (m, 2H), 2.10–1.92 (m, 1H).
tert-Butyl (4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)butyl)carbamate (5–3).
A suspension of 5–2 (100 mg, 0.36 mmol), tert-butyl (4-bromobutyl)carbamate (101 mg, 0.40 mmol), KHCO3 (110 mg, 1.10 mmol) and catalytic amount of KI in DMF (5 mL) was stirred at 70 °C for 12 h. The reaction mixture was diluted with water (20 mL), extracted with ethyl acetate (15 mL × 3). The organic layers were combined and washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with column chromatography (DCM/MeOH = 15:1) to yield intermediate 5–3 as a brown solid (60 mg, 37%). 1H NMR (300 MHz, chloroform-d) δ 8.68 (s, 1H), 7.64 (t, J = 7.9 Hz, 1H), 7.41 (d, J = 7.3 Hz, 1H), 7.19 (d, J = 8.5 Hz, 1H), 5.14–4.85 (m, 2H), 4.16 (t, J = 6.1 Hz, 2H), 3.30–3.08 (m, 2H), 2.92–2.66 (m, 3H), 2.18–2.02 (m, 1H), 1.89 (p, J = 6.5 Hz, 2H), 1.70 (p, J = 6.5 Hz, 2H), 1.40 (s, 9H).
4-(4-Aminobutoxy)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione trifluoroacetate (5–4).
Intermediate 5–4 was obtained as a yellow solid from 5–3 using a method similar as that described for intermediate 2–4a in quantitative yield.
N-(4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)butyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (5–5a).
To a stirred solution of intermediate 1–5 (20 mg, 0.083 mmol) in DMF (2 mL) was added HATU (39 mg, 0.10 mmol), DIPEA (27 mg, 0.21 mmol) and intermediate 5–4 (30 mg, 0.068 mmol), the reaction mixture was stirred at rt for 12 h. The reaction solution was diluted with water (10 mL), extracted with ethyl acetate (15 mL × 2), washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with preparative TLC (DCM/MeOH = 15:1) to yield compound 5–5a as a white solid (12 mg, 31%). 1H NMR (300 MHz, chloroform-d) δ 8.39 (s, 1H), 8.28–8.15 (m, 2H), 8.18–8.08 (m, 1H), 7.83–7.73 (m, 2H), 7.71 (t, J = 8.0 Hz, 1H), 7.60 (s, 1H), 7.48 (d, J = 7.3 Hz, 1H), 7.21 (d, J = 8.5 Hz, 1H), 5.45–5.20 (m, 1H), 4.24 (s, 2H), 3.78–3.44 (m, 2H), 22.88 (s, 2H), 2.38–2.19 (m, 1H), 2.12–1.86 (m, 4H). LCMS (ESI) 570 [M + H]+. HPLC purity at 254 nm.
N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)ethoxy)ethoxy)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (5–5b).
Compound 5–5b was obtained as a yellow solid using a method similar to that described for compound 5–5a. 1H NMR (300 MHz, chloroform-d) δ 8.43 (s, 1H), 8.19 (dd, J = 8.9, 3.8 Hz, 2H), 7.78 (t, J = 4.5 Hz, 2H), 7.58 (t, J = 7.9 Hz, 1H), 7.54 (s, 1H), 7.48–7.34 (m, 2H), 7.23 (d, J = 9.1 Hz, 1H), 5.05–4.86 (m, 1H), 4.39 (s, 2H), 4.01 (s, 2H), 3.82 (s, 2H), 3.75–3.60 (m, 6H), 2.95–2.71 (m, 3H), 2.21–2.07 (m, 1H). LCMS (ESI) 630 [M + H]+. HPLC purity at 254 nm.
tert-Butyl 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetate (6–2).
A mixture of 5–1 (1.50 g, 5.47 mmol), tert-butyl 2-bromoacetate (1.12 g, 5.74 mmol), and K2CO3 (1.13 g, 8.19 mmol) in DMF (20 mL) was stirred at rt for 12 h. Water was added slowly to the reaction solution, the precipitate formed was collected by filtration, washed with water (15 mL × 2). Intermediate 6–2 was obtained as a white solid (600 mg, 29%). 1H NMR (300 MHz, chloroform-d) δ 7.98 (s, 1H), 7.67 (t, J = 8.3 Hz, 1H), 7.51 (d, J = 7.4 Hz, 1H), 7.10 (d, J = 8.5 Hz, 1H), 5.03 – 4.88 (m, 1H), 4.79 (s, 2H), 2.99 – 2.63 (m, 3H), 2.22 – 2.03 (m, 1H), 1.48 (s, 9H).
2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetic acid (6–3).
Intermediate 6–2 (500 mg, 1.29 mmol) was dissolved in TFA (4 mL), the solution was stirred at rt for 3 h, then the reaction was diluted with DCM (10 mL), concentrated under reduced pressure to obtain a white solid, which was used into next step without further purification (500 mg, crude).
tert-Butyl (2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)ethoxy)ethyl)carbamate (6–4a).
To a stirred solution of 6–3 (86 mg, 0.27 mmol) in DMF (2 mL) was added HATU (152 mg, 0.40 mmol), DIPEA (0.80 mmol) and tert-butyl (2-(2-(2-aminoethoxy)ethoxy)ethyl)carbamate (69 mg, 0.28 mmol), the reaction was stirred at rt for 16 h. The reaction solution was diluted with water (10 mL), extracted with ethyl acetate (15 mL × 3). The organic layers were combined and washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with column chromatography (DCM/MeOH = 15:1) to yield intermediate 6–4a as a yellow solid (50 mg, 53%). 1H NMR (300 MHz, chloroform-d) δ 8.91 (s, 1H), 7.72 (t, J = 7.9 Hz, 1H), 7.62 (s, 1H), 7.53 (d, J = 7.3 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 5.16 (s, 1H), 5.05–4.90 (m, 1H), 4.65 (s, 2H), 3.66–3.46 (m, 10H), 3.28 (d, J = 5.6 Hz, 2H), 2.91–2.71 (m, 3H), 2.20–2.06 (m, 1H), 1.42 (s, 9H).
N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamide trifluoroacetate (6–5a).
Intermediate 6–4a (40 mg, 0.072 mmol) was dissolved in TFA (2 mL), the mixture was stirred at 50 °C for 1 h. The reaction solution was diluted with DCM (5 mL), concentrated under reduced pressure to obtain 6–5a as a yellow oil, which was used in the next step without further purification (40 mg, 100 %).
N-(2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)ethoxy)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (6–6a).
To a stirred solution of intermediate 1–5 (17 mg, 0.070 mmol) in DMF (2 mL) was added HATU (41 mg, 0.11 mmol), DIPEA (27 mg, 0.21 mmol) and intermediate 6–5a (40 mg, 0.072 mmol), the reaction mixture was stirred at rt for 12 h. The reaction solution was diluted with water (10 mL), extracted with ethyl acetate (15 mL × 2), washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with preparative TLC (DCM/MeOH = 15:1) to yield compound 6–6a as a white solid (14 mg, 29%). 1H NMR (300 MHz, chloroform-d) δ 8.83 (s, 1H), 8.29–8.10 (m, 2H), 7.78 (t, J = 4.5 Hz, 2H), 7.70 (q, J = 11.2, 9.6 Hz, 3H), 7.58–7.45 (m, 2H), 7.17 (d, J = 8.4 Hz, 1H), 5.15–4.95 (m, 1H), 4.64 (s, 2H), 3.81–3.49 (m, 12H), 3.02–2.64 (m, 3H), 2.26–2.13 (m, 1H). LCMS (ESI) 687 [M + H]+. HPLC purity at 254 nm.
N-(1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)-2-oxo-6,9,12-trioxa-3-azatetradecan-14-yl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (6–6b).
Compound 6–6b was obtained as a yellow solid using a method similar to that described for compound 6–6a. 1H NMR (300 MHz, chloroform-d) δ 8.21 (s, 2H), 7.79 (dd, J = 5.7, 3.4 Hz, 2H), 7.70 (s, 1H), 7.58 (s, 1H), 7.53 (d, J = 7.4 Hz, 1H), 7.20 (s, 1H), 4.64 (s, 2H), 3.71 (q, J = 6.3 Hz, 16H), 3.18 (d, J = 7.4 Hz, 4H). LCMS (ESI) 731 [M + H]+.HPLC purity at 254 nm.
N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)ethoxy)ethoxy)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (6–6c).
Compound 6–6c was obtained as a yellow solid using a method similar to that described for compound 6–6a. 1H NMR (300 MHz, DMSO-d6) δ 11.12 (s, 1H), 9.03 – 8.86 (m, 1H), 8.20 – 8.07 (m, 2H), 8.08 – 7.95 (m, 1H), 7.96 – 7.86 (m, 2H), 7.81 (t, J = 7.9 Hz, 1H), 7.63 (s, 1H), 7.48 (d, J = 7.4 Hz, 1H), 7.38 (d, J = 8.6 Hz, 1H), 5.11 (dd, J = 12.9, 5.4 Hz, 1H), 4.78 (s, 2H), 3.28 (d, J = 7.5 Hz, 4H), 3.18 (d, J = 6.4 Hz, 2H), 3.01 – 2.74 (m, 1H), 2.68 – 2.52 (m, 2H), 2.12 – 1.93 (m, 1H), 1.61 – 1.37 (m, 4H). LCMS (ESI) 649 [M + Na]+.HPLC purity at 254 nm.
N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (7–4).
Compound 7–4 was obtained as a yellow solid using a method similar to that described for compound 2–5a by using 3-(4-fluoro-1-oxoisoindolin-2-yl)piperidine-2,6-dione as starting material. 1H NMR (400 MHz, chloroform-d) δ 8.59 (s, 1H), 8.22 – 8.12 (m, 2H), 7.82 – 7.73 (m, 2H), 7.53 (s, 1H), 7.38 (t, J = 5.1 Hz, 1H), 7.23 (d, J = 7.7 Hz, 1H), 7.16 (dd, J = 7.6, 0.9 Hz, 1H), 6.73 (dd, J = 8.0, 0.9 Hz, 1H), 5.24 (dd, J = 13.2, 5.2 Hz, 1H), 4.32 (d, J = 15.6 Hz, 1H), 4.13 (d, J = 15.7 Hz, 1H), 3.77 (tt, J = 7.1, 3.7 Hz, 2H), 3.70 – 3.53 (m, 8H), 3.39 (q, J = 4.7 Hz, 2H), 2.94 – 2.77 (m, 2H), 2.34 (qd, J = 12.8, 5.7 Hz, 1H), 2.20 (dtd, J = 13.0, 5.1, 2.9 Hz, 1H). LCMS (ESI) 615 [M + H]+.HPLC purity at 254 nm.
N-(5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (8–1).
Compound 8–1 was obtained as a yellow solid using a method similar to that described for compound 4–5. 1H NMR (400 MHz, DMSO-d6) δ 10.99 (s, 1H), 8.93 (t, J = 5.7 Hz, 1H), 8.19 – 8.09 (m, 2H), 7.95 – 7.87 (m, 2H), 7.61 (s, 1H), 7.55 (dd, J = 6.4, 2.2 Hz, 1H), 7.49 – 7.41 (m, 2H), 5.12 (dd, J = 13.3, 5.1 Hz, 1H), 4.47 (d, J = 17.2 Hz, 1H), 4.31 (d, J = 17.1 Hz, 1H), 3.27 (d, J = 6.8 Hz, 2H), 2.99 – 2.84 (m, 1H), 2.64 (d, J = 8.0 Hz, 2H), 2.48 – 2.36 (m, 2H), 2.07 – 1.98 (m, 1H), 1.61 (dq, J = 21.6, 7.4 Hz, 4H), 1.37 (p, J = 7.8 Hz, 2H). LCMS (ESI) 554 [M + H]+.HPLC purity at 254 nm.
tert-Butyl 2-(2-(2-(4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamido)ethoxy)ethoxy)acetate (9–2a).
To a stirred solution of intermediate 1–5 (30 mg, 0.12 mmol) in DMF (2 mL) was added HATU (71 mg, 0.19 mmol), DIPEA (48 mg, 0.37 mmol) and tert-butyl 2-(2-(2-aminoethoxy)ethoxy)acetate (30 mg, 0.14 mmol), the reaction mixture was stirred at rt for 12 h. The reaction solution was diluted with water (10 mL), extracted with ethyl acetate (15 mL × 2), washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with column chromatography (DCM/MeOH = 15:1) to yield compound 9–2a as a yellow solid (28 mg, 51%). 1H NMR (400 MHz, chloroform-d) δ 8.23–8.16 (m, 2H), 7.81–7.73 (m, 2H), 7.56 (s, 1H), 7.40 (s, 1H), 4.09 (s, 2H), 3.76–3.63 (m, 8H), 1.44 (s, 9H).
2-(2-(2-(4,9-Dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamido)ethoxy)ethoxy)acetic acid (9–3a).
Intermediate 9–2a (28 mg, 0.063 mmol) was dissolved in DCM (2 mL), TFA (2 mL) was added, the mixture was stirred at rt for 2 h. The reaction solution was concentrated under reduced pressure to obtain 9–3a as a yellow oil, which was used into next step without further purification (30 mg, crude).
(2S,4R)-1-((S)-12-(tert-butyl)-1-(4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-yl)-1,10-dioxo-5,8-dioxa-2,11-diazatridecan-13-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (9–4a).
To a stirred solution of intermediate 9–3a (15 mg, 0.039 mmol) in DMF (2 mL) was added HATU (22 mg, 0.58 mmol), DIPEA (15 mg, 0.12mmol) and (S,R,S)-AHPC hydrochloride (17 mg, 0.039 mmol), the reaction mixture was stirred at rt for 12 h. The reaction solution was diluted with water (10 mL), extracted with ethyl acetate (15 mL × 2), washed with brine, dried over Na2SO4, filtered, and concentrated. The residue obtained was purified with preparative TLC (DCM/MeOH = 15:1) to yield compound 9–4a as a yellow solid (8 mg, 26%).1H NMR (400 MHz, DMSO-d6) δ 8.97 (t, J = 5.7 Hz, 1H), 8.94 (s, 1H), 8.55 (t, J = 6.0 Hz, 1H), 8.18 – 8.06 (m, 2H), 7.97 – 7.84 (m, 2H), 7.64 (s, 1H), 7.44 (d, J = 9.8 Hz, 1H), 7.38 (s, 4H), 5.14 (d, J = 3.5 Hz, 1H), 4.57 (d, J = 9.5 Hz, 1H), 4.48 – 4.31 (m, 3H), 4.24 (dd, J = 15.7, 5.7 Hz, 1H), 3.98 (s, 2H), 3.69 – 3.56 (m, 8H), 3.50 – 3.40 (m, 2H), 2.42 (s, 3H), 2.09 – 2.01 (m, 1H), 1.96 – 1.84 (m, 1H), 0.94 (s, 9H). LCMS (ESI) 800 [M + H]+. HPLC purity at 254 nm.
(4R)-1-((S)-2-(2-(2-(4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamido)ethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (9–4b).
Compound 9–4b was obtained as a yellow solid using a method similar to that described for compound 9–4a. 1H NMR (400 MHz, DMSO-d6) δ 9.12 (t, J = 5.8 Hz, 1H), 8.96 (s, 1H), 8.55 (t, J = 6.0 Hz, 1H), 8.19 – 8.06 (m, 2H), 7.94 – 7.86 (m, 2H), 7.66 (d, J = 1.3 Hz, 1H), 7.53 (d, J = 9.5 Hz, 1H), 7.39 (d, J = 1.3 Hz, 4H), 5.13 (d, J = 3.4 Hz, 1H), 4.57 (d, J = 9.4 Hz, 1H), 4.48 – 4.31 (m, 3H), 4.24 (dd, J = 15.7, 5.7 Hz, 1H), 4.02 (s, 2H), 3.71 – 3.58 (m, 4H), 3.55 – 3.46 (m, 2H), 2.43 (d, J = 1.4 Hz, 3H), 2.10 – 2.00 (m, 1H), 1.96 – 1.84 (m, 1H), 0.93 (s, 9H). LCMS (ESI) 756 [M + H]+.HPLC purity at 254 nm.
N-(2-(2-(2-((1,3-dioxo-2-(2-oxopiperidin-3-yl)isoindolin-4-yl)amino)ethoxy)ethoxy)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (10–5, XD171).
Compound XD171 was obtained as a yellow solid using a method similar to that described for compound 2–5a. 1H NMR (300 MHz, chloroform-d) δ 8.17 (t, J = 4.7 Hz, 2H), 7.82 – 7.63 (m, 3H), 7.51 (s, 1H), 7.32 (t, J = 7.8 Hz, 1H), 6.95 (d, J = 7.1 Hz, 1H), 6.81 (d, J = 8.5 Hz, 1H), 6.61 (s, 1H), 6.53 (s, 1H), 4.68 (dd, J = 12.1, 6.1 Hz, 1H), 3.77 (t, J = 5.4 Hz, 2H), 3.70 – 3.43 (m, 13H), 2.38 (q, J = 12.0 Hz, 1H), 2.09 (s, 1H), 1.94 (s, 1H). LCMS (ESI) 615 [M + H]+.HPLC purity at 254 nm.
N-(2-(4-(5-((2-(1-methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)pentyl)piperidin-1-yl)ethyl)-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-2-carboxamide (11–7, XD2–162).
Compound XD2–162 was obtained as a yellow solid using a method similar to that described for compound 3–6a. 1H NMR (400 MHz, DMSO-d6) δ 8.85 (t, J = 5.8 Hz, 1H), 8.18 – 8.08 (m, 2H), 7.96 – 7.85 (m, 2H), 7.62 (s, 1H), 7.58 (dd, J = 8.6, 7.0 Hz, 1H), 7.09 (d, J = 8.6 Hz, 1H), 7.01 (d, J = 7.1 Hz, 1H), 6.53 (t, J = 5.9 Hz, 1H), 5.19 – 5.01 (m, 1H), 3.42 – 3.34 (m, 10H), 3.01 (s, 3H), 2.94 – 2.70 (m, 3H), 2.44 (d, J = 6.7 Hz, 1H), 2.04 (dtd, J = 13.2, 5.5, 2.8 Hz, 1H), 1.91 (d, J = 2.1 Hz, 1H), 1.58 (t, J = 14.0 Hz, 4H), 1.21 – 1.01 (m, 6H). LCMS (ESI) 708 [M + H]+. HPLC purity at 254 nm.
Cell Culture.
Cell lines were maintained in RPMI-1640 or DMEM supplemented with 10% fetal bovine serum (FBS) (ThermoFisher Scientific). The cells were grown as monolayer cultures at 37 °C in a humidified atmosphere of 5% CO2 and tested for Mycoplasma contamination with the Mycoplasma detection kit, PlasmaTest (InvivoGen). BxPC-3 cell line was authenticated with STR DNS profiling (University of Michigan Sequencing Core) and matched to reference profiles from the ATCC database. The STAT3-knockout cell lines were generated in our laboratory using CRISPR Cas9.36
Cytotoxicity Assays.
For MTT assay, the experiments were carried out in 96-well plates where cells (3000–6000 cells/well) were seeded in the medium and incubated overnight. The compounds or the vehicle (DMSO) were added to the cells the following day and incubated for 3 days. MTT solution (3 mg/ml) was added to the wells and plates were incubated for 3h at 37 °C after which MTT was discarded and cells were dissolved in DMSO. The absorbance of the formazan crystals was read by a microplate reader (Molecular Devices) at 570 nm and the data were analyzed using GraphPad Prism 8 software. For colony formation assays, the cells (200–500 cells/well) were seeded in 96-well plates overnight and then treated with DMSO or the compound for 7–10 days until 80% confluency. After colony formation, the medium was removed and the cells were stained with 0.05% crystal violet, washed with water and imaged with iBright™ imaging system (Invitrogen). The colonies were dissolved in Sorensen buffer (0.1 M sodium citrate, pH 4.2) for colorimetric quantification.
Western Blot Analysis.
Cells were seeded (OVCAR-3: 500 × 103, MIA PaCa-2: 200 × 103, BxPC-3: 500 × 103, MCF-7: 300 × 103 cells/well) in 6-well plates. Following treatment, cells were lysed at 4 °C by sonication with lysis buffer (25 mM tris(hydroxymethyl)aminomethane, 150 mM NaCl, 17 mM Triton X-100, 3.5 mM SDS, pH 7.4) supplemented with protease inhibitor and phosphatase inhibitor. The cells were then spun at 12,000 rpm at 4 °C for 10 min and collected for determining the protein concentration with the BCA assay (ThermoFischer Scientific, Waltham, MO). Samples were then prepared and loaded onto 10 or 12 % acrylamide (BioRad, Hercules, CA) gels followed by the transfer of the proteins onto PVDF membranes (EMD Millipore, La Jolla, CA). The membranes were blocked for 1 h prior to incubation with the primary antibodies using Odyssey blocking buffer (LI-COR Biosciences). Membranes were then probed for STAT3/pSTAT3/STAT1/NQO1 (Cell Signaling, Danvers, MA, 1:1000), GAPDH (Cell Signaling, Danvers, MA, 1:4000), GSPT (Cell signaling, 1:1000), ZFP91 (Bethyl, 1:1000). Following overnight incubation with the primary antibodies at 4 °C, the membranes were incubated with the secondary antibodies (anti-rabbit, Cell Signaling, 1:7500 or anti-mouse, Cell Signaling, 1:5000) for 1 h and imaged with the Odyssey imaging system (LI-COR Biosciences).
STAT3 Luciferase Reporter Assay.
The GloResponse™ SIE-luc2P HEK-293 cells (10,000 cells/well) were seeded in a 96-well plate (white, opaque bottom) and incubated overnight. On day 2, IL6 (100 ng/ml) was added to the cells for 24 h to induce activation of the STAT3 pathway. The compounds were then added for 24 h after which the Bio-Glo™ luciferase assay reagent (Promega # G7940) was added to each well. The luminescence was measured after 5 min using a plate reader. An MTT assay in HEK-293 was performed in parallel treating the cells with the compounds for 24 h.
Protein Identification and Relative Quantitation by TMT Labeling and LC-Tandem MS.
BxPC-3 cells were treated with DMSO (control), napabucasin (2 μM), XD2–149 (2 μM) or XD2–162 (4 μM) for 16 h then collected and lysed in RIPA buffer (Pierce, #89900) at 4 °C. The control and XD2–149 treatments were carried out in triplicates while napabucasin and XD2–162 were carried out in duplicates. Protein samples were then collected and the concentrations were determined using the BCA assay (ThermoFisher Scientific). Samples were prepared in a concentration of 75 μg and submitted to the Proteomics Core in the Department of Pathology at University of Michigan. Tandem mass tag (TMT) labeling was performed using the TMT 10plex™ isobaric labeling kit (ThermoFisher Scientific, #90110) according to the manufacturer’s protocol with minor modifications. This involves reduction of the samples with DTT (1 h, 55 °C) followed by alkylation with 2-chloroacetamide (30 min, RT). Proteins were precipitated using cold acetone overnight. Next, the proteins were pelleted and resuspended in TEAB to which trypsin (Promega, V5113) was added for digestion (overnight, 37 °C). TMT reagents were reconstituted in anhydrous acetonitrile and the digested peptides were transferred to TMT reagent vials and incubated at RT for 1 h. The reaction was quenched with 5% hydroxylamine for 15 min then samples were combined and dried. Prior to MS analysis, two-dimensional separation of the sample was performed. For the first dimension, an offline fractionation of an aliquot of each sample into 10 fractions was performed following the manufacturer’s protocol (Pierce, #84868). Fractions were then dried and reconstituted in loading buffer (0.1% formic acid and 2% acetonitrile). For quantitation, the MultiNotch-MS3 method was employed61. MS was performed on the Orbitrap Fusion Tribrid with ETD (ThermoFisher Scientific) equipped with nano-LC system (Dionex RSLC-nano). Proteome discoverer (v2.1, ThermoFisher Scientific) was used for data analysis. MS2 spectra were searched against SwissProt human protein database. The abundance ratio data sets were transformed to log2FC values and the whole protein list was used for GSEA analysis (GSEA 4.0.3). For ranking, the proteins were filtered applying a fold-change (FC) cut-off of 1.5 and p-val ≤ 0.05. All proteins have experimental q-val of < 0.1.
siRNA Knockdown of ZFP91.
ZFP91 siRNA (hs.Ri.ZFP91.13.1, Integrated DNA Technologies) or scrambled negative control siRNA (#51-01-19-09, Integrated DNA Technologies) with Lipofectamine RNAiMAX transfection reagent (ThermoFisher Scientific) was added to BxPC-3 cells in Opti-MEM medium (ThermoFisher Scientific) for 48 and 72 h after which the cells were harvested for immunoblotting. The siRNA transfection reagent complex was prepared following the manufacturer’s instructions. Briefly, 30 pmol of siRNA in 150 μl of Opti-MEM medium were added to 9 μl of the lipofectamine reagent in 150 μl of Opti-MEM medium. After 5 min incubation at room temperature, the siRNA transfection complex was added to each well in a 6-well plate. For MTT assay, 5 pmol of siRNA in 25 μl of Opti-MEM medium were added to 1.5 μl of the lipofectamine reagent in 25 μl of Opti-MEM medium. After 5 min incubation at room temperature, 10 μl of the siRNA transfection complex was added to each well in a 96-well plate. XD2–149 or napabucasin was added to the cells simultaneously with the siRNA transfection reagent complex for 48 h to determine their cytotoxicity.
Statistical Analysis.
Significance levels for assays and immunoblots were calculated using unpaired Student’s t-test in GraphPad Prism. Results are shown as mean ± standard deviation from three independent experiments.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH grant R01 CA188252 and a grant from the University of Michigan Forbes Institute for Cancer Discovery. M.H. received a scholarship from the Egyptian government (Ministry of Higher Education and Scientific Research). We would also like to thank our colleagues Christine Cuthbertson, Daulat Khadka and Essam Osman for reading the manuscript and providing helpful suggestions.
ABBREVIATIONS USED
- CK1α
Casein Kinase 1α
- CRBN
cereblon
- CX
cycloheximide
- DIC
dicoumarol
- FC
fold-change
- FBS
fetal bovine serum
- GSEA
gene set enrichment analysis
- GSPT
G1 to S phase transition protein
- HIF-1α
Hypoxia inducible factor 1 subunit alpha
- IKZF1
IKAROS Family Zinc Finger 1
- IKZF3
IKAROS Family Zinc Finger 3
- IMiDs
immunomodulatory drugs
- mPDAC
metastatic pancreatic adenocarcinoma
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NF-кB
nuclear factor kappa B
- NP
napabucasin
- NQO1
NAD(P)H Quinone Dehydrogenase 1
- PROTAC
proteolysis targeting chimera
- SPR
surface plasmon resonance
- STAT3
signaling transducer and activator of transcription 3
- VHL
Von Hippel-Lindau
- ZBTB16
Zinc Finger And BTB Domain Containing 16
- ZFP91
zinc finger protein 91 homolog
Footnotes
Supporting Information
The Supporting Information is available free of charge at http://pubs.acs.org.
Additional figures described in the text; tables including clinical trials, activity of the compounds and proteomics results; additional synthetic schemes; additional methods; NMR and LC/MS charts.
Molecular formula strings and the associated biological data.
REFERENCES
- 1.Rao MM; Kingston DG, Plant anticancer agents. XII. Isolation and structure elucidation of new cytotoxic quinones from Tabebuia cassinoides. J. Nat. Prod 1982, 45, 600–604. [DOI] [PubMed] [Google Scholar]
- 2.Nagata K; Hirai KI; Koyama J; Wada Y; Tamura T, Antimicrobial activity of novel furanonaphthoquinone analogs. Antimicrob. Agents Chemother 1998, 42. 3, 700–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y; Rogoff HA; Keates S; Gao Y; Murikipudi S; Mikule K; Leggett D; Li W; Pardee AB; Li CJ, Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 1839–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang ZL, Chiang J,; Li W; Leggett D Novel STAT3 Pathway Inhibitors and Cancer Stem Cell Inhibitors WO 2009036059A3, July 2, 2009.
- 5.Karandish F; Mamnoon B; Feng L; Haldar MK; Xia L; Gange KN; You S; Choi Y; Sarkar K; Mallik S, Nucleus-targeted, echogenic polymersomes for delivering a cancer stemness inhibitor to pancreatic cancer cells. Biomacromolecules 2018, 19, 4122–4132. [DOI] [PubMed] [Google Scholar]
- 6.https://www.accessdata.fda.gov/scripts/opdlisting/oopd/listResult.cfm. (accessed October 10, 2020).
- 7.Neoptolemos JP; Kleeff J; Michl P; Costello E; Greenhalf W; Palmer DH, Therapeutic developments in pancreatic cancer: current and future perspectives. Nat. Rev. Gastroenterol. Hepatol 2018, 15, 332–347. [DOI] [PubMed] [Google Scholar]
- 8.Bekaii-Saab TS; Starodub A; El-Rayes BF; Shahda S; O’Neil BH; Noonan AM, Phase 1b/2 trial of cancer stemness inhibitor napabucasin (NAPA) plus nab-paclitaxel (nPTX) and gemcitabine (Gem) in metastatic pancreatic adenocarcinoma (mPDAC). J. Clin. Oncol 2018, 36, 4110–4110. [Google Scholar]
- 9.Sonbol MB; Ahn DH; Goldstein D; Okusaka T; Tabernero J; Macarulla T; Reni M; Li CP; O’Neil B; Van Cutsem E; Bekaii-Saab T, CanStem111P trial: a Phase III study of napabucasin plus nab-paclitaxel with gemcitabine. Future Oncol 2019, 15, 1295–1302. [DOI] [PubMed] [Google Scholar]
- 10.https://www.bostonbiomedical.com/news-and-media/20190701_boston-biomedical-inc-announces-update-canstem111p-study-following-interim-analysis/. (accessed August 1, 2019).
- 11.Li C; Chen C; An Q; Yang T; Sang Z; Yang Y; Ju Y; Tong A; Luo Y, A novel series of napabucasin derivatives as orally active inhibitors of signal transducer and activator of transcription 3 (STAT3). Eur. J. Med. Chem 2019, 162, 543–554. [DOI] [PubMed] [Google Scholar]
- 12.Zhou QF; Peng C; Du FY; Zhou LB; Shi YJ; Du Y; Liu DD; Sun WJ; Zhang MX; Chen GL, Design, synthesis and activity of BBI608 derivatives targeting on stem cells. Eur. J. Med. Chem 2018, 151, 39–50. [DOI] [PubMed] [Google Scholar]
- 13.Frank DA, STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett 2007, 251, 199–210. [DOI] [PubMed] [Google Scholar]
- 14.Turkson J; Jove R, STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613–6626. [DOI] [PubMed] [Google Scholar]
- 15.Bromberg JF; Wrzeszczynska MH; Devgan G; Zhao YX; Pestell RG; Albanese C; Darnell JE, Stat3 as an oncogene. Cell 1999, 98, 295–303. [DOI] [PubMed] [Google Scholar]
- 16.Locken H; Clamor C; Muller K, Napabucasin and related heterocycle-fused naphthoquinones as STAT3 inhibitors with antiproliferative activity against cancer cells. J. Nat. Prod 2018, 81, 1636–1644. [DOI] [PubMed] [Google Scholar]
- 17.Zhan T; Rindtorff N; Boutros M, Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang AY; Hsu E; Patel J; Li YQ; Zhang MJ; Iguchi H; Rogoff HA, Evaluation of tumor cell-tumor microenvironment component interactions as potential predictors of patient response to napabucasin. Mol. Cancer Res 2019, 17, 1429–1434. [DOI] [PubMed] [Google Scholar]
- 19.Parkinson EI; Hergenrother PJ, Deoxynyboquinones as NQO1-activated cancer therapeutics. Acc. Chem. Res 2015, 48, 2715–2723. [DOI] [PubMed] [Google Scholar]
- 20.Froeling FEM; Swamynathan MM; Deschenes A; Chio IIC; Brosnan E; Yao MA; Alagesan P; Lucito M; Li JY; Chang AY; Trotman LC; Belleau P; Park Y; Rogoff HA; Watson JD; Tuveson DA, Bioactivation of napabucasin triggers reactive oxygen species-mediated cancer cell death. Clin. Cancer Res 2019, 25, 7162–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu S; Sechi M; Singh PK; Dai L; McCann S; Sun D; Ljungman M; Neamati N, A novel redox modulator induces a GPX4-mediated cell death that is dependent on iron and reactive oxygen species. J. Med. Chem 2020, 63, 9838–9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuo DQ; Shogren KL; Zang J; Jewison DE; Waletzki BE; Miller AL; Okuno SH; Cai ZD; Yaszemski MJ; Maran A, Inhibition of STAT3 blocks protein synthesis and tumor metastasis in osteosarcoma cells. J. Exp. Clin. Cancer Res 2018, 37, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadd MS; Testa A; Lucas X; Chan KH; Chen W; Lamont DJ; Zengerle M; Ciulli A, Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol 2017, 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buckley DL; Crews CM, Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew. Chem. Int. Ed. Engl 2014, 53, 2312–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cromm PM; Crews CM, Targeted protein degradation: from chemical biology to drug discovery. Cell Chem. Biol 2017, 24, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai LC; Zhou HB; Xu RQ; Zhao YJ; Chinnaswamy K; McEachern D; Chen JY; Yang CY; Liu ZM; Wang M; Liu L; Jiang H; Wen B; Kumar P; Meagher JL; Sun DX; Stuckey JA; Wang SM, A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell 2019, 36, 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou HB; Bai LC; Xu RQ; Zhao YJ; Chen JY; McEachern D; Chinnaswamy K; Wen B; Dai LP; Kumar P; Yang CY; Liu ZM; Wang M; Liu L; Meagher JL; Yi H; Sun DX; Stuckey JA; Wang SM, Structure-based discovery of SD-36 as a potent, selective, and efficacious PROTAC degrader of STAT3 rotein. J. Med. Chem 2019, 62, 11280–11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salami J; Alabi S; Willard RR; Vitale NJ; Wang J; Dong HQ; Jin MZ; McDonnell DP; Crew AP; Neklesa TK; Crews CM, Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol 2018, 1, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li YB; Yang JL; Aguilar A; McEachern D; Przybranowski S; Liu L; Yang CY; Wang M; Han X; Wang SM, Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J. Med. Chem 2019, 62, 448–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang ZQ; He NZ; Guo ZW; Niu CL; Song T; Guo YF; Cao KK; Wang AH; Zhu JJ; Zhang XD; Zhang ZC, Proteolysis targeting chimeras for the selective degradation of Mcl-1/Bcl-2 derived from nonselective target binding ligands. J. Med. Chem 2019, 62, 8152–8163. [DOI] [PubMed] [Google Scholar]
- 31.Raina K; Lu J; Qian YM; Altieri M; Gordon D; Rossi AMK; Wang J; Chen X; Dong HQ; Siu K; Winkler JD; Crew AP; Crews CM; Coleman KG, PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chamberlain PP; Cathers BE, Cereblon modulators: Low molecular weight inducers of protein degradation. Drug Discov. Today Technol 2019, 31, 29–34. [DOI] [PubMed] [Google Scholar]
- 33.Girardini M; Maniaci C; Hughes SJ; Testa A; Ciulli A, Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem 2019, 27, 2466–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shahbazi S; Peer CJ; Polizzotto MN; Uldrick TS; Roth J; Wyvill KM; Aleman K; Zeldis JB; Yarchoan R; Figg WD, A sensitive and robust HPLC assay with fluorescence detection for the quantification of pomalidomide in human plasma for pharmacokinetic analyses. J. Pharm. Biomed. Anal 2014, 92, 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Debnath B; Xu SL; Neamati N, Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J. Med. Chem 2012, 55, 6645–6668. [DOI] [PubMed] [Google Scholar]
- 36.Lu T; Bankhead A; Ljungman M; Neamati N, Multi-omics profiling reveals key signaling pathways in ovarian cancer controlled by STAT3. Theranostics 2019, 9, 5478–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li C; Zang Y; Sen M; Leeman-Neill RJ; Man DS; Grandis JR; Johnson DE, Bortezomib up-regulates activated signal transducer and activator of transcription-3 and synergizes with inhibitors of signal transducer and activator of transcription-3 to promote head and neck squamous cell carcinoma cell death. Mol. Cancer Ther 2009, 8, 2211–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang HT; Dobrovolsky D; Paulk J; Yang G; Weisberg EL; Doctor ZM; Buckley DL; Cho JH; Ko E; Jang J; Shi K; Choi HG; Griffin JD; Li Y; Treon SP; Fischer ES; Bradner JE; Tan L; Gray NS, A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol 2018, 25, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin J; Buettner R; Yuan YC; Yip R; Horne D; Jove R; Vaidehi N, Molecular dynamics simulations of the conformational changes in signal transducers and activators of transcription, Stat1 and Stat3. J. Mol. Graph. Model 2009, 28, 347–356. [DOI] [PubMed] [Google Scholar]
- 40.Ishoey M; Chorn S; Singh N; Jaeger MG; Brand M; Paulk J; Bauer S; Erb MA; Parapatics K; Muller AC; Bennett KL; Ecker GF; Bradner JE; Winter GE, Translation termination factor GSPT1 Is a phenotypically relevant off-target of heterobifunctional phthalimide degraders. ACS Chem. Biol 2018, 13, 553–560. [DOI] [PubMed] [Google Scholar]
- 41.Matyskiela ME; Lu G; Ito T; Pagarigan B; Lu C-C; Miller K; Fang W; Wang N-Y; Nguyen D; Houston JJN, A novel cereblon modulator recruits GSPT1 to the CRL4 CRBN ubiquitin ligase. Nature 2016, 535, 252–257. [DOI] [PubMed] [Google Scholar]
- 42.Yang J; Li Y; Aguilar A; Liu Z; Yang CY; Wang S, Simple structural modifications converting a bona fide MDM2 PROTAC degrader into a molecular glue molecule: A cautionary tale in the design of PROTAC degraders. J. Med. Chem 2019, 62, 9471–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han X; Wang C; Qin C; Xiang W; Fernandez-Salas E; Yang CY; Wang M; Zhao L; Xu T; Chinnaswamy K; Delproposto J; Stuckey J; Wang S, Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem 2019, 62, 941–964. [DOI] [PubMed] [Google Scholar]
- 44.Akuffo AA; Alontaga AY; Metcalf R; Beatty MS; Becker A; McDaniel JM; Hesterberg RS; Goodheart WE; Gunawan S; Ayaz M; Yang Y; Karim MR; Orobello ME; Daniel K; Guida W; Yoder JA; Rajadhyaksha AM; Schonbrunn E; Lawrence HR; Lawrence NJ; Epling-Burnette PK, Ligand-mediated protein degradation reveals functional conservation among sequence variants of the CUL4-type E3 ligase substrate receptor cereblon. J. Biol. Chem 2018, 293, 6187–6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bondeson DP; Smith BE; Burslem GM; Buhimschi AD; Hines J; Jaime-Figueroa S; Wang J; Hamman BD; Ishchenko A; Crews CM, Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol 2018, 25, 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chau MN; Banerjee PP, Development of a STAT3 reporter prostate cancer cell line for high throughput screening of STAT3 activators and inhibitors. Biochem. Biophys. Res. Commun 2008, 377, 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.An J; Ponthier CM; Sack R; Seebacher J; Stadler MB; Donovan KA; Fischer ES, pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4(CRBN) ubiquitin ligase. Nat. Commun 2017, 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petzold G; Fischer ES; Thomä NH, Structural basis of lenalidomide-induced CK1α degradation by the CRL4(CRBN) ubiquitin ligase. Nature 2016, 532, 127–130. [DOI] [PubMed] [Google Scholar]
- 49.Kronke J; Udeshi ND; Narla A; Grauman P; Hurst SN; McConkey M; Svinkina T; Heckl D; Comer E; Li X; Ciarlo C; Hartman E; Munshi N; Schenone M; Schreiber SL; Carr SA; Ebert BL, Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gandhi AK; Kang J; Havens CG; Conklin T; Ning Y; Wu L; Ito T; Ando H; Waldman MF; Thakurta A; Klippel A; Handa H; Daniel TO; Schafer PH; Chopra R, Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br. J. Haematol 2014, 164, 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matyskiela ME; Zhu J; Baughman JM; Clayton T; Slade M; Wong HK; Danga K; Zheng X; Labow M; LeBrun L; Lu G; Chamberlain PP; Thompson JW, Cereblon Modulators Target ZBTB16 and Its Oncogenic Fusion Partners for Degradation via Distinct Structural Degrons. ACS Chem. Biol 2020. [DOI] [PubMed] [Google Scholar]
- 52.Jin X; Jin HR; Jung HS; Lee SJ; Lee JH; Lee JJ, An atypical E3 ligase zinc finger protein 91 stabilizes and activates NF-kappaB-inducing kinase via Lys63-linked ubiquitination. J. Biol. Chem 2010, 285, 30539–30547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma J; Mi CL; Wang KS; Lee JJ; Jin XJ, Zinc finger protein 91 (ZFP91) activates HIF-1 alpha via NF-kappa B/p65 to promote proliferation and tumorigenesis of colon cancer. Oncotarget 2016, 7, 36551–36562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang DE; Dai Y; Xu Y; Lin LW; Liu DZ; Hong XP; Ou ML; Jiang HW; Xu SH, The ubiquitinase ZFP91 promotes tumor cell survival and confers chemoresistance through FOXA1 destabilization. Carcinogenesis 2020, 41, 56–66. [DOI] [PubMed] [Google Scholar]
- 55.Sun JJ; Du YP; Song QL; Nan J; Guan PZ; Guo JH; Wang X; Yang JB; Zhao CY, E2F is required for STAT3-mediated upregulation of cyclin B1 and Cdc2 expressions and contributes to G2-M phase transition. Acta. Biochim. Biophys. Sin 2019, 51, 313–322. [DOI] [PubMed] [Google Scholar]
- 56.Koganti S; Hui-Yuen J; McAllister S; Gardner B; Grasser F; Palendira U; Tangye SG; Freeman AF; Bhaduri-McIntosh S, STAT3 interrupts ATR-Chk1 signaling to allow oncovirus-mediated cell proliferation. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 4946–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang WB; Deng HK; Levy DE; Lee CK, STAT3 negatively regulates antiviral responses through suppression of TLR and type I IFN-mediated responses. Cytokine 2009, 48, 126–126. [Google Scholar]
- 58.Xu SH; Zhu S; Wang Y; Huang JZ; Chen M; Wu QX; He YT; Chen; Yan GR, ECD promotes gastric cancer metastasis by blocking E3 ligase ZFP91-mediated hnRNP F ubiquitination and degradation. Cell Death Dis 2018, 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang W; Li N; Hu J; Wang L, Inhibitory effect of RNA-mediated knockdown of zinc finger protein 91 pseudogene on pancreatic cancer cell growth and invasion. Oncol. Lett 2016, 12, 1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang Z; Kang B; Li C; Chen T; Zhang Z, GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res 2019, 47, W556–W560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McAlister GC; Nusinow DP; Jedrychowski MP; Wuhr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP, MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 2014, 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.