

Abstract
We describe a free-radical footprinting approach for integral membrane protein (IMP) that extends significantly the “fast photochemical oxidation of proteins” (FPOP) platform. This new approach exploits highly hydrophobic perfluoroisopropyl iodide (PFIPI) together with tip sonication to ensure efficient transport into the micelle interior, allowing laser dissociation and footprinting of the transmembrane domains. In contrast to water soluble footprinters, PFIPI footprints both the hydrophobic intramembrane and the hydrophilic extramembrane domains of the IMP vitamin K epoxide reductase (VKOR). The footprinting is fast, giving high coverage for Tyr (100%) and Trp. The incorporation of the reagent with sonication does not significantly affect VKOR’s enzymatic function, and tyrosine iodination does not compromise protease digestion and the subsequent analysis. The locations for the modifications are largely consistent with the corresponding solvent accessibilities, recommending this approach for future membrane protein footprinting.
Keywords: Structural Proteomics, Protein Footprinting, Fluorinated Footprinter, Fast photochemical oxidation of proteins (FPOP), Membrane Protein, Transmembrane, Iodination, Iodine radical, Sonication, Micelle
Graphical Abstract
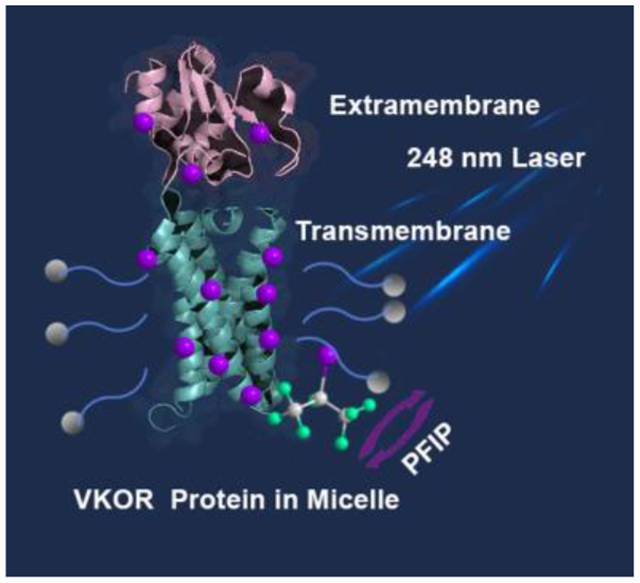
We describe a laser-mediated free-radical footprinting of an integral membrane protein (IMP). Compared to canonical footprinting, the approach exploits tip sonication to ensure efficient transport of a highly hydrophobic perfluoroalkyl iodide to footprint transmembrane domains. This approach, amenable to native IMP structures, yields 100% coverage for Tyr, and it is compatible for standard proteomic mass spectrometric analysis.
Integral membrane proteins (IMPs) play essential roles in cellular signaling, transport, and catalysis. Although they represent ~30% of the open reading frames of many genomes and an increasing number of important drug targets,[1] high-order structural information for IMPs is sparse and difficult to achieve. Less than 1% of all protein crystal structures are of IMPs, and structures of only ~200 human IMPs are known.[2] Furthermore, X-ray crystallography and Cryo-EM often fail to interrogate protein dynamics that is crucial for IMPs function.[3] Thus, a strong need exists for new methods.
Approaches based on mass spectrometry (MS) bridge the gap between low-resolution (e.g., circular dichroism, dynamic light scattering) and high-resolution methods (e.g., X-ray crystallography, Cryo-EM, NMR) and provide a broad readout of not only stoichiometry, conformation, dynamics but also the location of interactions.[4] Although MS-based footprinting is growing rapidly for soluble proteins, its application to IMPs has been slow and under-represented.[5] Its limitations are no longer instrumental[6] or determined by protein solubilization (e.g., in native vesicles, lipid nano- and picodiscs,[7] and MS-compatible surfactants), but rather chemical. A strategy is needed to introduce appropriate footprinters to the lipid phase where they can react with IMPs.[8]
“Footprinting” characterizes protein structure by using chemical reactions whose extent is a measure of solvent accessibility of the protein backbone or side chains and environment. Most current “footprinters” for soluble proteins are hydrophilic (e.g., D2O for HDX or •OH) and label protein surfaces according to solvent-accessibility[9]. IMPs, however, contain hydrophobic regions, buried in lipid bilayers.
In previous research, Konermann[10] and Gross[11] found that •OH (from photolysis of H2O2) selectively labels extramembrane regions of IMPs in vesicles and nanodiscs (Fig. 1). Radford and coworkers[12] identified that amphipol A8–35 reduces •OH labeling for OmpT and enhances the labeling efficiency for the boundary of the membrane-water interface. In a special case, Chance used X-ray mediated hydroxyl radical footprinting to define the outer membrane region of IMPs[13] by ionizing ordered, water molecules, leading to •OH footprinting of some transmembrane regions.[14] Implementing a carbene from an amphipathic source, Oldham and coworkers effectively labelled the outer surface of the bacterial membrane protein, OmpF.[15]
Figure 1.
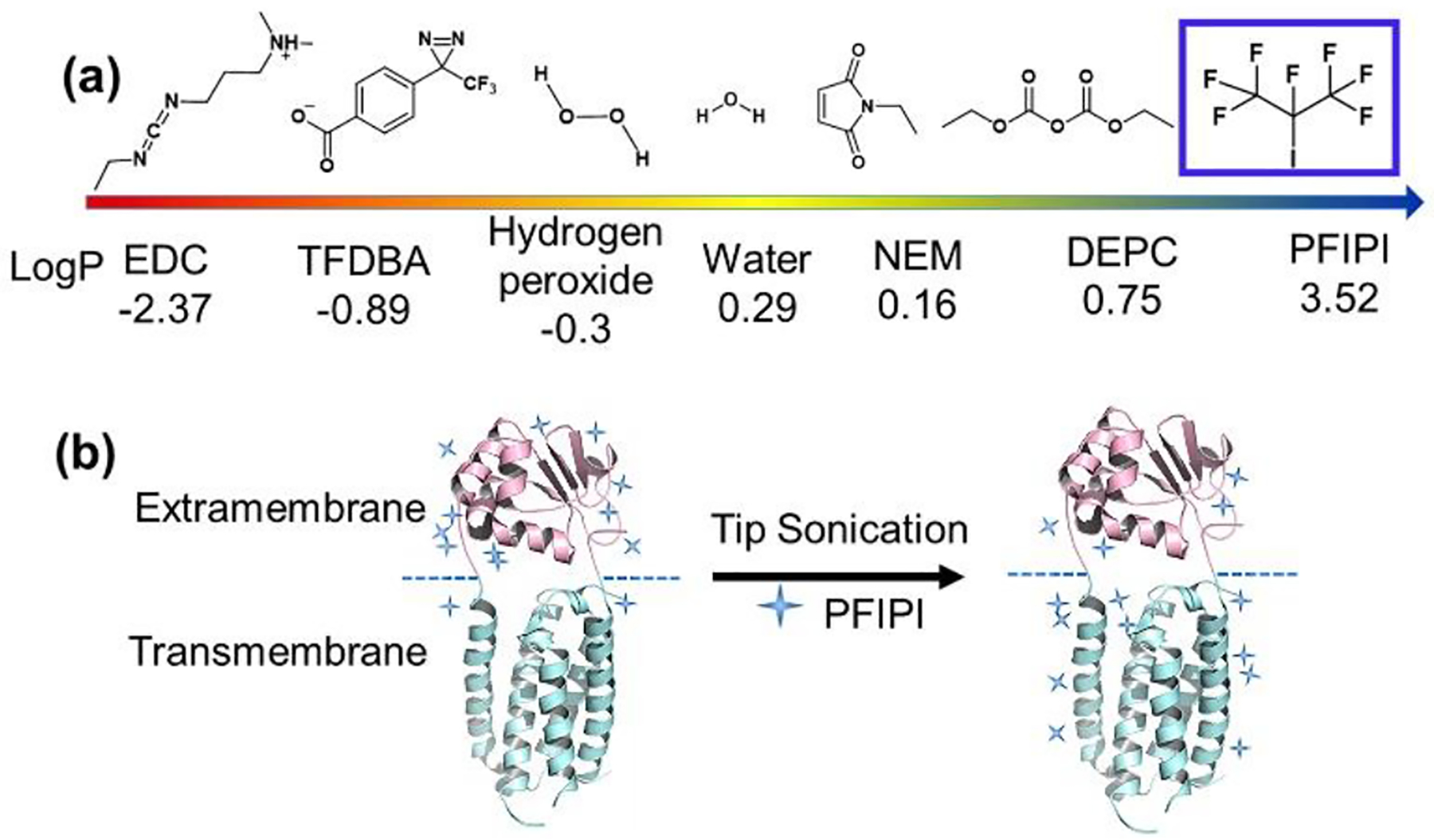
(a) Analysis of reagents and their logP values for utility in protein transmembrane footprinting. LogP values were simulated using the web-based interactive software from Molinspiration Cheminformatics. (b) Enhanced partition of PFIPI into a micelle with tip sonication showing improved yields of transmembrane footprinting (blue +). The region in magenta is the water-accessible extramembrane domain, whereas the region in cyan is hydrophobic transmembrane. The blue dashed line is the boundary dividing the two domains
Although residue-specific reagents (e.g., EDC,[16] NEM[17]) can also be applied to IMPs to footprint single, accessible residues (e.g., Cys, His), the reactions run the risk of perturbing protein high order structure.[18]
We reasoned that photolyzable reagents with high partition coefficients (P) into nonpolar solvents would be well-suited for transmembrane labeling. We chose PFIPI because it has a high positive logP (Fig. 1), small size, ready availability, and suitability to form radicals upon 248 nm laser photolysis on the fast photochemical oxidation of proteins (FPOP) platform.[19] FPOP, in its original conception, is a hydroxyl radical (·OH) protein footprinting approach that utilizes a pulsed KrF laser (248 nm) to homolyze hydrogen peroxide in solution to ·OH. The free radicals subsequently modify the side chains of protein in situ. FPOP uses a flow system and scavenger to limit the exposure of the reactive species to sub-milliseconds, thus enabling a fast “snapshot’ of protein events in solution. Because membrane regions are buried in a “sea” of C-H bonds from lipids or detergent tails, a reagent with C-F bonds, similarly sized as C-H bonds but with higher hydrophobicity,[20] should not only be accommodated in the hydrophobic phase but also introduce biorthogonal modifications.
Motivated by “like dissolves like”, we chose a goal is to achieve efficient incorporation of PFIPI into a micelle and minimize its perturbation on the micelle environment. PFIPI is stable in the “dark”, whereas the C-I bond in PFIPI can be dissociated on the FPOP platform in nanoseconds (likely limited by the width of the laser beam)[21] to generate simultaneously perfluoroisopropyl and iodine radicals for footprinting. Under these conditions, the PFIPI undergoes homolytic cleavage with a quantum efficiency of nearly 100% at 248 nm laser light, a quantum yield that is greater than that for photolysis of alkyl iodides.[21] Further, the laser serves as a “switch” to pulse on footprinting, a characteristic of FPOP.
Here we implement these ideas in a proof-of-principle transmembrane footprinting of an IMP, using the most hydrophobic IMP footprinter applied thus far (Fig. 1a). We also address whether the PFIPI perturbs the native protein structrure in the lipid environment by taking advantage of the enzymatic nature of our test protein. Furthermore, we evaluate the effectiveness of tip sonication to enhance PFIPI partition into the lipid bilayer.
We first tested the PFIPI reactivity with a derivatized and hydrophobic amino acid. We chose a protected tyrosine (PT, N-acetyl-L-tyrosine ethyl ester monohydrate) because it reacts with the iodine radical.[22] PT was mixed with different equivalents of PFIPI in 1:1 (v/v) acetonitrile/water and submitted to 248 nm laser irradiation. MS analysis showed mono- and di-iodinated PT but no perfluoropropylated PT (we are investigating the mechanism of this surprisingly highly specific reaction). Encouragingly, 50 equivalents PFIPI are enough to give 22% product yield with a single laser shot, suggesting that PFIPI is an appropriate choice for fast footprinting (Fig. S2 compares the fractional conversion of the PT to product with increased concentration of PFIPI). The footprinting reaction ultimately provides 70% coversion to mono-iodinated PT and 11% di-idodinated PT when using 103 equivalents of the PFIPI.
With vitamin K epoxide reductase (VKOR) as a model, we next determined whether PFIPI can diffuse into DDM (n-dodecyl-B-D-maltoside) detergent micelles and label transmembrane residues. In vertebrates, VKOR resides in the endoplasmic membrane and sustains blood coagulation. VKOR homologs in bacteria and archaea reside in the periplasmic membrane and promote oxidative protein folding. The bacterial homolog protein from Synechococcus sp. consists of a membrane-embedded domain carrying the VKOR activity, connected via a linker segment to the extracellular thioredoxin-like (Trx) domain. The VKOR domain contains five transmembrane helices, with four helix bundle that create a central pocket occupied by vitamin K or ubiquinone, and one helix passes through the membrane to connect to the fused periplasmic Trx-like domain.[23]
In this work, VKOR was solubilized in micelles consisting of DDM, a commonly used detergent for membrane proteins. We treated VKOR with several concentrations of PFIPI (see table S1) and determined the sites of modification by LC-MS/MS after digestion with chymotrypsin, selected because there is a dearth of tryptic sites. The sequence coverage achieved in the analysis, importantly, was ~97% for VKOR. The method is sensitive to residues containing an X-H bond in aromatic rings (i.e., Tyr, Trp, sometimes His). Based on MS analysis, we identified nine amino acids that were iodinated in the transmembrane region, including seven Tyr and two Trp (residues from 16–180 are assigned to be transmembrane based on X-ray crystallography). Notably, all Tyr’s in that region were labeled to different extents, giving 100% reactivity coverage of Tyr. With higher concentrations of the reagent, the modification ratio increased (Table S2). This behavior sharply contrasts with results of previous studies[10–11, 24] where both •OH and the water-soluble Langlois precursor to •CF3.footprinter did not label transmembrane residues, and a carbene produced only low modification extents[15].
Next, we explored whether sonication can further enhance the reagent partitioning into a membrane to improve the labeling efficiency. We used tip sonication pulsed to reduce heating the sample. For each cycle, we pulsed on sonication for 2 s, then off for 10 s (Fig. S1). The sonication led to increased footprinting of 9/16 residues (i.e., Y4, Y36, Y39, Y117, Y120, Y132, Y207, Y262 and Y277). Four of 16 residues (i.e., W99, Y163, Y178, and Y204) underwent comparable modification, whereas three of 16 residues (i.e., W64, Y228 and Y252) were slightly less modified (Fig. 2). Noteworthy, all transmembrane residues underwent relatively larger modification with tip sonication (except W64, which is not transmembrane but in a loop on top of the transmembrane domain). The results support tip sonication as an effective means of partitioning a suitable reagent to a lipid-like phase (of some relevance is a previous study[25] that shows pulsed tip sonication partially disrupts membrane vesicles, allowing an MS-compatible buffer into a vesicle lumen).
Figure 2.
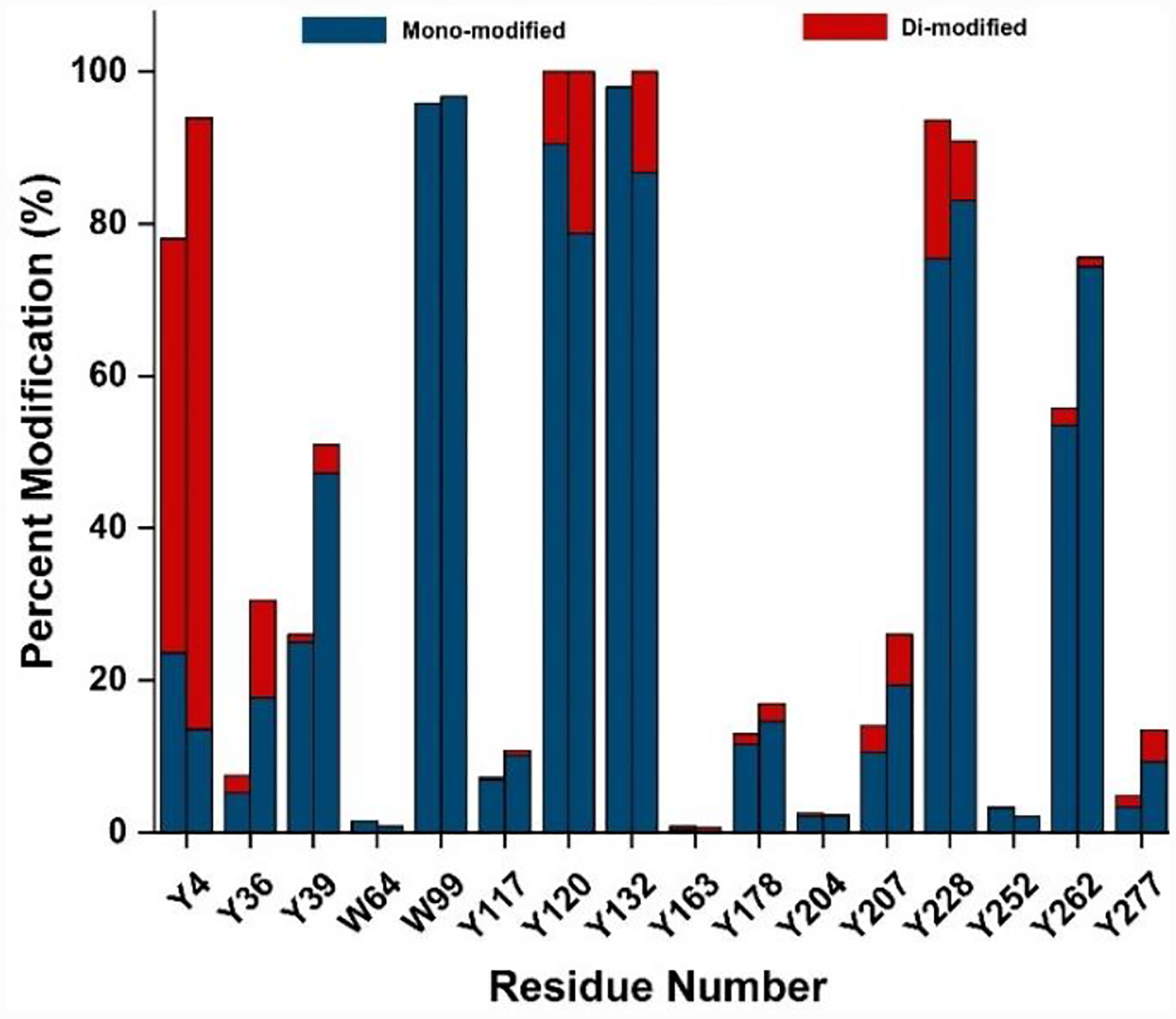
PFIPI membrane modification increased by tip sonication. The two bars in each cluster represent 500 equiv. of PFIPI without (left bar) and with (right bar) two cycles of tip sonication, respectively. Mono and di-modified residues are coded in blue and red, respectively.
Despite widespread use of sonication for protein extraction, there is always concern that it perturbs high order structure or induces protein aggregation.[26] To address these issues, we submitted VKOR to different rounds of sonication and used an enzyme assay to assess any critical change in activity. The assay, dithiothreitol (DTT)-dependent conversion of vitamin K to vitamin K hydroquinone (Fig. S4, a plot of vitamin K hydroquinone fluorescence intensity vs. reaction time) showed that sonication does not signifcantly affect the protein activity. The reaction rate for 1 cycle of sonication, as measured by the slope of reaction curve, is nearly identical with that of the control where no sonication was applied. With 2 cycles of sonication, the reaction rate decreased by ~10%, which is nearly identical to that produced by four cycles of sonication. Although no significant decrease of enzymatic activity occurs upon sonication, we don’t know if other structural properites of the protein are affected [25].
Given that footprinting of any given residue of soluble proteins depends in part on solvent accessibility, we examined the relationship between the modification extent and the accessibility of I• to the residues, measured as the accessible surface area (%ASA) (Table S3). In this evaluation, instead of using the radius of water (1.4 Å), we utilized the radius of the reactive iodine radical (1.98 Å) to probe the surface area of VKOR (see Table S3 for the ASA calculations).[27]
We classify residues in the transmembrane region according to their reactivities (Fig. 3 and Fig. S3) and locations in the transmembrane region. Category 1 residues are close to the aqueous interface in an “aromatic belt”.[28] They are at the end of transmembrane helices, adjoining loops or turns. Examples are Y120, Y132, Y178 and W99 (marked in red color). Their location imparts considerable reactivity that is reflected in their high modification extents, approaching nearly 100% for Y120, Y132, and W99. Y178 is also located near the interface, but its %ASA is nearly zero, yet its modification extent is ~ 20%. Like the other residues in this category, the reactivity is higher than may be expected because it is also dynamic.
Figure 3.

Footprinted residues as mapped on crystal structure (PDB file 3KP9). Labeled residues, indicated by circle in central structure, include 16 residues: 8 transmembrane residues, 1 residue (W64) on the water-micelle interface, and 6 extramembrane residues. Region containing Y4 is not marked as the N-terminus is not seen in crystal structure but can be footprinted, underscoring one of its advantages. The values in parentheses are %modification/%ASA (e.g., (100/29) for Y132 indicates (~100% modification/29%ASA)).
Category 2 residues are deeper in the “aromatic belt“ and more remote from the aqueous interface. Hence, they are less reactive. Examples are Y36, Y39, and Y117 (marked in green color), which are in stable helices, more remote from loops, comprising an interface with the polar moieties on lipids and micelles.The reactivities of these three residues correlate nearly linearly with the %ASA (Fig. S3). Indeed this region has properties intemediate to those near the bulk aqueous phase and those that make up the uniformly hydrophobic region of the protein interior. Furthermore, their aromatic side chains point to the exterior of VKOR protein, where they are poised to react with iodine radicals formed in the photolysis.
Category 3 residues are buried in the transmembrane region, and their key residues (-OH or -NH) whose H’s react with radicals point inward and may be stabilized by H-bonding. Only one residue so far is in the category (i.e., Y163 marked in blue color). Although it has reasonable exposure to an iodine radical (as reflected by its %ASA, its inward-pointing side chain points is H-bonded with Thr112 (Fig. 3), stabilizing the side chain and retarding removal of H• with respect to Category 1 and 2 residues.
Although the categorization of the transmembrane residues underpins an explanation for their reactivities, we cannot rule out specific interactions of the perfluoroisopropyl iodide precursor. Although it is risky to extrapolate from one study, these correlations may serve to locate residues in an unknown transmembrane region and provide insights on protein dynamics that cannot be seen in crystal and CryoEM structures.
Turning to footprinting of residues that are extramembrane (Fig. S3), we note that their reactivity is likely due to a high local concentration of the reagent at the interface of the protein and the micelle (and possibly to traces of PFIPI in the aqueous phase). PFIPI can attach to the protein surface by taking advantage of fluorine as a hydrogen bond acceptor.[29] For those residues, Y4 near the N-terminus is most heavily labeled (94% modification), likely reflecting a highly solvent-exposed and dynamic region, consistent with the need to remove the N terminus to enable VKOR crystallization. Residue Y277 near the C-terminus is much less modified (13%), consistent with tight folding of the soluble C-terminal domain, whereas Y228 and Y262 (Fig. S3) are highly reactive. The high reactivity of Y228 may reflect its location in a poorly structured, flexible loop that serves as a dynamic connecter for secondary structural elements, whereas Y262, appears to be less sterically hindered.
Because protein 3D structure is a network of residue interactions, the chemical reactivity of a footprinter is determined by not only intrinsic chemical reactivity and ASA but also by the protein microenvironment[30] (e.g, hydrogen bonding, reagent local concentration). Correlations of reactivity, structure, and ASA will require more detailed studies, and these should be productive and are planned in the future.
Turning now to a practical consideration, we asked whether iodinated tyrosine compromise chymotryptic digestion, needed for these transmembrane regions that have sparse tryptic sites. Although we found no detectable negative effect of iodination on digestion, several PTM-based proteomics analyses reveal that modification of a cleavage site or a proximal position can reduce proteolytic efficiency. We analyzed the cleavage frequency using a nonspecific search setting, which enables us to calculate the number of unique peptides for all potential cleavage sites. Based on MS analysis, we find that chymotrypsin is not a highly specific enzyme as it cleaves at the C-terminal of 18 different amino acids. The prefered cleavage sites are Tyr, Leu, Phe, and Trp (Fig. 4, the cleavage frequency is greater than amino acid percent composition). This result is consistent with those previously reported by Heck[31] who also observed this nonspecificity. Iodination does not decrease the digestion efficency (compare Fig. 4a and 4b) and, if anything, enables a slightly higher cleavage frequency.
Figure 4.
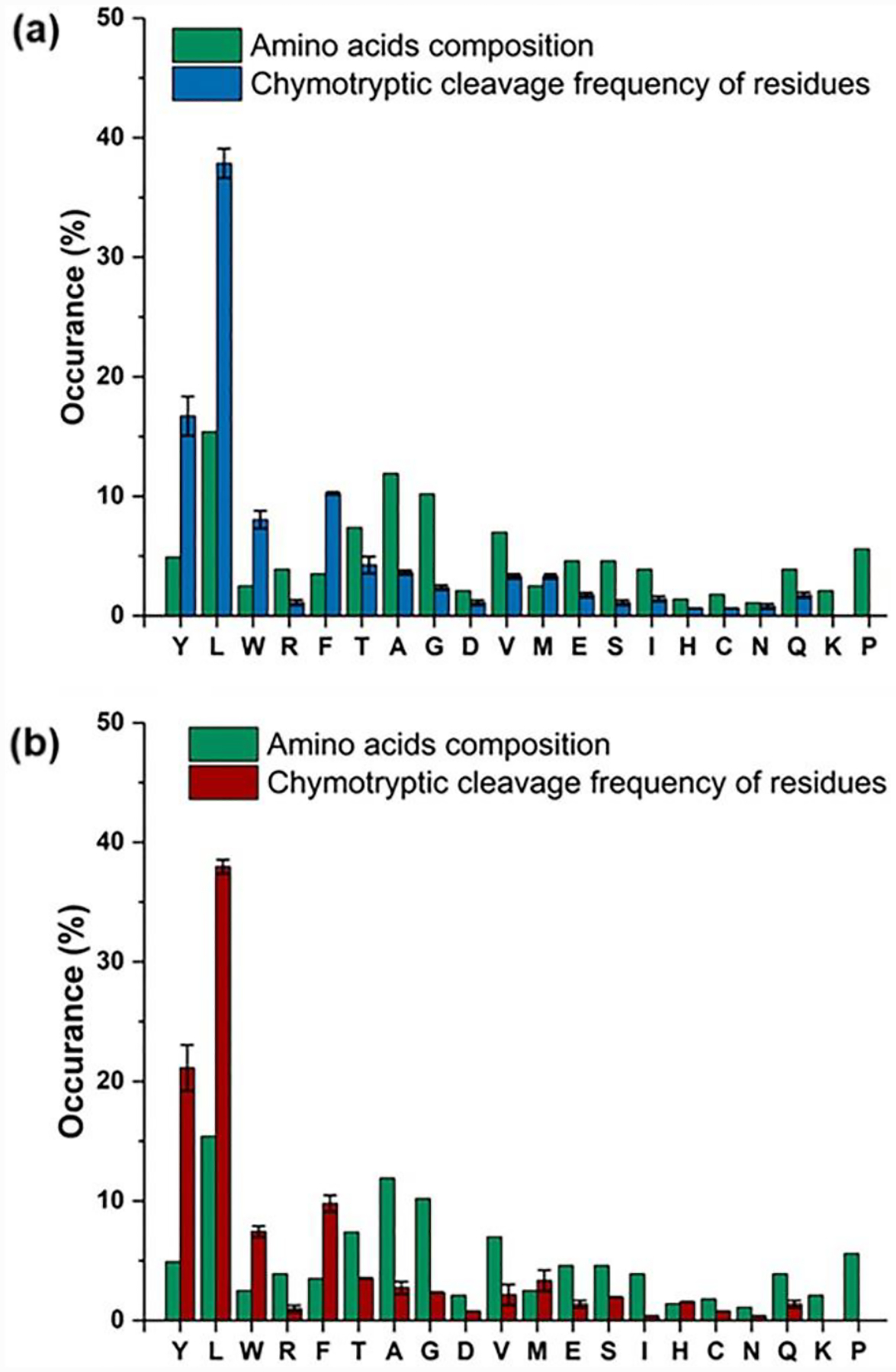
Comparison of VKOR digestion between non (top) and iodinated (bottom) forms of VKOR. The amino acids composition for VKOR are calculated using the Protparam tool, and the cleavage frequency is obtained with the workflow described in Fig. S5.
Another advantage of the approach is that with MS/MS, we can locate the modification sites of amino-acid residues. For example, LC-MS/MS analysis of peptide 197–204 (Fig. 5) convincingly reveals (1) that iodinated tyrosine does not interfere with chymotryptic proteolysis (Fig. 5b and c), and that mono- and even di-iodinated tryrosine can be enzymatically cleaved and measured by MS. (2) Iodination of tyrosine is stable and tolerant of CID given observation of both mono- and di-iodinated tyrosine as confirmed by the Yproduct Y ions and as immonium ions. These “signature“ ions further confirm the modification on tyrosine, enabling peptide identification. (3) The reversed-phase LC elutions shift to longer times (Fig. 5a) compared to those of unmodified peptides as the number of iodine substitutions increase (e.g., for Tyr204). This behavior is consistent with our study on CF3 footprinting[24] and contrasts with the effects of hydrophilic footprinters (e.g., •OH). Because iodine substitution increases retention time in reversed-phase LC and improves slightly digestion with chymotrypsin, which cleaves C-terminal to hydrophobic amino acids, iodination clearly increases hydrophobicity of the peptides from digestion.
Figure 5.
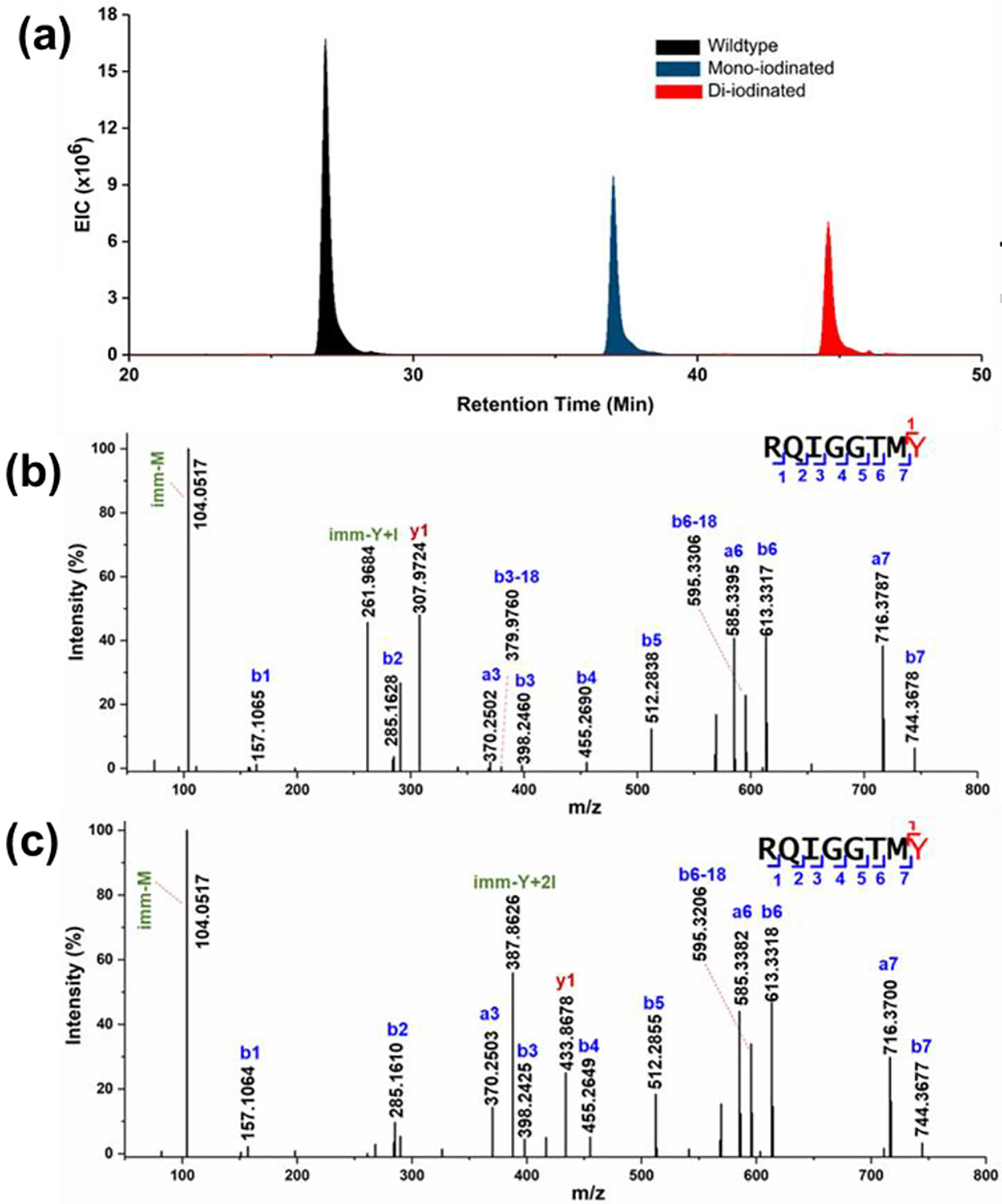
Representative LC-MS/MS spectra for peptide 197–204 (+2). (a) Extracted ion chromatogram for peptide. The incorporation of I increases the elution time in reversed phase HPLC. (b) Product-ion (MS/MS) spectrum of mono-iodinated 197–204, and (c) di-iodinated peptides. Chymotrypsin continues to cleave at the C-term side of Tyr bearing iodination. Iodinated Tyr was assigned by y and immonium ions (Tyr immonium ion is Yimm).
In summary, this novel approach extends protein footprinting to transmembrane residues, as demonstrated for the IMP VKOR in DDM footprinted by the most hydrophobic reagent applied thus far. The gentle tip sonication enhances reagent partitioning into lipid-like media without significant protein high-order struture pertubation. The outcomes establish strategic merits of this approach: (1) it utilizes PFIPI, a small reagent that mimics the hydrophobic tails of lipids and detergents that constitute the transmembrane environment. The reagent can be introduced by gentle sonication that does not significantly perturb the protein as measured by a sensitive enzymatic assay. (2) This novel chemistry can be combined with •OH labeling, enabled by the same 248 nm laser on a standard FPOP platform, to footprint in both the transmembrane and extramembrane domains. (3) Modification by •I is both stable for canonical bottom-up MS/MS analysis and non-perturbing for proteolysis, a key advantage when exploring transmembrane domains that are often sparse in Arg and Lys for tryptic digestion. (4) The dissociation of the C-I bond is rapid (0.5–3 × 10−13 s),[21] and its quantum yield for homolysis bond is nearly 1.0 at 248 nm. [21] One piece of evidence supporting this latter feature is that iodination of transmembrane Tyr can occur with only 50 equivalents of PFIPI with respect to the protein and one pulse of the laser to enable chemistry that is complete presumably in submilliseconds.
An intriguing observation is that only iodination, not perfluoropropylation, occurs even though PFIPI presumably photolyzes to generate both •I and •C3F7. The •C3F7 does not label transmembrane residues, likely because it is consumed in the first step by generation of protein-centered radicals and C3F7H. Nevertheless, the incorporation of only •I reduces the complexity of the footprint and of the data interpretation but with a sacrifice of coverage. The mechanism is subject of ongoing research.
The outcome extends considerably a novel paradigm suggested in previous articles in this journal on footprinting membrane proteins[15] and on the use of fluorine chemistry in structrual proteomics[19, 24, 32]. Our design steps include (1) reagent selection in terms of LogP, (2) reagent transfer to the membrane domain (3) reagent photochemistry in the lipid-like medium to give free radicals (or other reactive species), and (4) tailored radical specifity/reactivity design to address the biochemical question at hand. The results suggest that this approach represents a productive means to achieve adequate-coverage footprinting for membrane proteins, to report dynamic regions and residue location, and to accommodate expansion in many ways to include new reactive species including free radicals from reagents modeled on the perfluoroalkyl system, carbenes[15], and possibly carbocations. Extension of the concept and evaluation of its generality are underway.
Supplementary Material
Acknowledgements
We thank Scot R. Weinberger for his suggestion of tip sonication, Dr. Haijun Liu for advice on tip sonication, Dr. George Mathai for insights on the iodination mechanism, and Shuang Li for advice on the VKOR bioactivity assay. We also thank PMI[33] for the data processing software. This work was supported by NIH Grants R01GM131008 to WL and MLG and by P41GM103422 and R24lGM136766 to MLG.
References
- [1].Overington JP, Al-Lazikani B, Hopkins AL, Nat. Rev. Drug Discov 2006, 5, 993. [DOI] [PubMed] [Google Scholar]
- [2].a) Cheng Y, Curr. Opin. Struct. Biol 2018, 52, 58; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Carpenter EP, Beis K, Cameron AD, Iwata S, Curr. Opin. Struct. Biol 2008, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Calabrese AN, Radford SE, Methods 2018, 147, 187. [DOI] [PubMed] [Google Scholar]
- [4].Lössl P, van de Waterbeemd M, Heck AJ, EMBO J 2016, 35, 2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kar UK, Simonian M, Whitelegge JP, Expert. Rev. Proteomic 2017, 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Harvey SR, Liu Y, Liu W, Wysocki VH, Laganowsky A, Chem. Commun 2017, 53, 3106; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhong Y, Han L, Ruotolo BT, Angew. Chem. Int. Ed. Engl 2014, 9209; [DOI] [PubMed] [Google Scholar]; c) Ambrose S, Housden NG, Gupta K, Fan J, White P, Yen HY, Marcoux J, Kleanthous C, Hopper JTS, Robinson CV, Angew. Chem. Int. Ed. Engl 2017, 56, 14463; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Leney AC, Heck AJR, J. Am. Soc. Mass Spectrom 2017, 28, 5. [DOI] [PubMed] [Google Scholar]
- [7].a) Reading E, Hall Z, Martens C, Haghighi T, Findlay H, Ahdash Z, Politis A, Booth PJ, Angew. Chem. Int. Ed. Engl 2017, 56, 15654; [DOI] [PubMed] [Google Scholar]; b) Reid DJ, Keener JE, Wheeler AP, Zambrano DE, Diesing JM, Reinhardt-Szyba M, Makarov, Marty MT, Anal. Chem 2017, 11189. [DOI] [PubMed] [Google Scholar]
- [8].Limpikirati P, Liu T, Vachet RW, Methods 2018, 144, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu XR, Zhang MM, Gross ML, Chemical Reviews 2020, 120, 4355–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pan Y, Ruan X, Valvano MA, Konermann L, J Am Soc Mass Spectrom 2012, 23, 889. [DOI] [PubMed] [Google Scholar]
- [11].Lu Y, Zhang H, Niedzwiedzki DM, Jiang J, Blankenship RE, Gross ML, Anal. Chem 2016, 88, 8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Watkinson TG, Calabrese AN, Ault JR, Radford SE, Ashcroft AE, J. Am. Soc. Mass. Spectrom 2017, 28, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Du Y, Duc NM, Rasmussen SGF, Hilger D, Kubiak X, Wang L, Bohon J, Kim HR, Wegrecki M, Asuru A, Jeong KM, Lee J, Chance MR, Lodowski DT, Kobilka BK, Chung KY, Cell 2019, 177, 1232–1242.e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Angel TE, Gupta S, Jastrzebska B, Palczewski K, Chance MR, Proc. Natl. Acad. Sci. U S A 2009, 106, 14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Manzi L, Barrow AS, Hopper JTS, Kaminska R, Kleanthous C, Robinson CV, Moses JE, Oldham NJ, Angew. Chem. Int. Ed. Engl 2017, 56, 14873. [DOI] [PubMed] [Google Scholar]
- [16].Wen J, Zhang H, Gross ML, Blankenship RE, Proc. Natl. Acad. Sci. U S A 2009, 106, 6134–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Calabrese AN, Jackson SM, Jones LN, Beckstein O, Heinkel F, Gsponer J, Sharples D, Sans M, Kokkinidou M, Pearson AR, Radford SE, Ashcroft AE, Henderson PJF, Anal.Chem 2017, 89, 8844; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kahsai AW, Xiao K, Rajagopal S, Ahn, Shukla AK, Sun J, Oas TG, Lefkowitz RJ, Nat. Chem. Biol 2011, 7, 692; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kahsai AW, Rajagopal S, Sun J, Xiao K, Nat. Protoc 2014, 9, 1301; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jones LN, Baldwin SA, Henderson PJF, Ashcroft AE, Rapid. Commun. Mass. Spectrom 2010, 24, 276–284; [DOI] [PubMed] [Google Scholar]; e) Weinglass AB, Whitelegge JP, Hu Y, Verner GE, Faull KF, Kaback HR, EMBO J 2003, 22, 1467; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Shen G, Cui W, Zhang H, Zhou F, Huang W, Liu Q, Yang Y, Li S, Bowman GR, Sadler JE, Gross ML, Li W, Nat. Struct. Mol. Biol 2017, 24, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pan X, Vachet RW, Mass Spectrom. Rev 10.1002/mas.21667 [DOI] [PMC free article] [PubMed]
- [19].Cheng M, Guo C, Gross ML, Angew. Chem. Int. Ed 2020, 59, 5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].O’Hagan D, Chem. Soc. Rev 2008, 37, 308.18197347 [Google Scholar]
- [21].Godwin FG, Gorry PA, Hughes PM, Raybone D, Watkinson TM, Whitehead JC, Chem. Phys. Lett 1987, 135, 163. [Google Scholar]
- [22].a) Dolbier WR, Chem. Rev 1996, 96, 1557; [DOI] [PubMed] [Google Scholar]; b) Chen J, Cui W, Giblin D, Gross ML, J. Am. Soc. Mass Spectrom 2012, 23, 1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA, Nature 2010, 463, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cheng M, Zhang B, Cui W, Gross ML, Angew. Chem. Int. Ed. Engl 2017, 56, 14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chorev DS, Tang H, Rouse SL, Bolla JR, von Kügelgen A, Baker LA, Wu D, Gault J, Grünewald K, Bharat TAM, Matthews SJ, Robinson CV, Nat. Protoc 2020, 15, 1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stathopulos PB, Scholz GA, Hwang Y-M, Rumfeldt JAO, Lepock JR, Meiering EM, Protein Sci 2004, 13, 3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fraczkiewicz R, Braun W, J. Comput. Chem 1998, 19, 319. [Google Scholar]
- [28].Hong H, Park S, Flores Jiménez RH, Rinehart D, Tamm LK, J. Am. Chem. Soc 2007, 129, 8320. [DOI] [PubMed] [Google Scholar]
- [29].Howard JAK, Hoy VJ, O’Hagan D, Smith GT, Tetrahedron 1996, 52, 12613. [Google Scholar]
- [30].Limpikirati P, Pan X, Vachet RW, Anal. Chem 2019, 91, 8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Giansanti P, Tsiatsiani L, Low TY, Heck AJR, Nat. Protoc 2016, 11, 993. [DOI] [PubMed] [Google Scholar]
- [32].Cheng M, Asuru A, Kiselar J, Mathai G, Chance MR, Gross ML, J. Am. Soc. Mass Spectrom 2020, 31, 1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bern M, Cai Y, Goldberg D, Anal. Chem 2007, 79, 1393. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.