

Abstract
Drug-target interaction (DTI) prediction has drawn increasing interest due to its substantial position in the drug discovery process. Many studies have introduced computational models to treat DTI prediction as a regression task, which directly predict the binding affinity of drug-target pairs. However, existing studies (i) ignore the essential correlations between atoms when encoding drug compounds and (ii) model the interaction of drug-target pairs simply by concatenation. Based on those observations, in this study, we propose an end-to-end model with multiple attention blocks to predict the binding affinity scores of drug-target pairs. Our proposed model offers the abilities to (i) encode the correlations between atoms by a relation-aware self-attention block and (ii) model the interaction of drug representations and target representations by the multi-head attention block. Experimental results of DTI prediction on two benchmark datasets show our approach outperforms existing methods, which are benefit from the correlation information encoded by the relation-aware self-attention block and the interaction information extracted by the multi-head attention block. Moreover, we conduct the experiments on the effects of max relative position length and find out the best max relative position length value  . Furthermore, we apply our model to predict the binding affinity of Corona Virus Disease 2019 (COVID-19)-related genome sequences and
. Furthermore, we apply our model to predict the binding affinity of Corona Virus Disease 2019 (COVID-19)-related genome sequences and 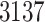 FDA-approved drugs.
FDA-approved drugs.
Keywords: deep learning, drug-target interaction, self-attention, COVID-19
Introduction
Drugs work by interacting with target proteins to activate or inhibit the biological process of the targets. Thus, identifing novel drug-target interactions (DTIs) is an essential step in the drug discovery field, like drug repurposing [12, 18, 26]. However, transitional costly experiments limit the process to identify new DTIs [26, 28, 31]. Thus, the computational approach for DTI prediction is urgent [37]. Recently, a large of studies proposed computational methods for DTI prediction. Parts of studies [6, 16, 29, 32] considered the DTI prediction task as a binary classification problem. They focused on the existence of DTI, while some other studies [9, 18, 24] treat it as a regression task to directly predict the binding affinity scores. Here, the binding affinity scores describe the strength of the interactions in drug-target pairs. In this study, we focus on the drug-target binding affinity prediction.
Recently, deep learning methods are utilized for DTI prediction. DeepDTA [18] proposed a convolutional neural networks (CNNs)-based model for drug representation learning, target representation learning and predicting interaction between them. As one of the widely used deep learning-based models for predicting the binding affinity values, it has achieved an acceptable result. However, it is limited due to that CNN cannot capture the long-distance relationship among atoms in drugs. Based on this, the study [24] introduced a self-attention mechanism-based model with position embedding to encode the relationship among all atoms in compounds. Nevertheless, firstly, it is far from enough to model the compounds since these existing methods just label each atom a corresponding integer according to a dictionary. During modeling the compounds, what is learned is an atom at a specific position. It ignores the correlation between atoms and separates each atom. For example, the compounds ’COC1=C(C=C2C(=C1)CCN=C2C3=CC(=C(C=C3)Cl)Cl)Cl’. In existing methods, given a dictionary  , the character ’C’ would be coded as ’1’ and ’l’ is labeled as ’2’. The existing methods separated the chloride atom ’Cl’ as two fake atoms since they cannot further learn the relative position information between character ’C’ and character ’l’. Moreover, the correlation between atoms not only depicts relative position information but also enhances the diversity of atoms. As the ’C’ in that example, ’C’ atoms would be in any position, but each one includes unique information since their connected atoms are different. Secondly, most existing methods always simply modeled the interaction between drugs and targets by concatenating their representations which is not sufficient to describe the interactions.
, the character ’C’ would be coded as ’1’ and ’l’ is labeled as ’2’. The existing methods separated the chloride atom ’Cl’ as two fake atoms since they cannot further learn the relative position information between character ’C’ and character ’l’. Moreover, the correlation between atoms not only depicts relative position information but also enhances the diversity of atoms. As the ’C’ in that example, ’C’ atoms would be in any position, but each one includes unique information since their connected atoms are different. Secondly, most existing methods always simply modeled the interaction between drugs and targets by concatenating their representations which is not sufficient to describe the interactions.
Based on these observations, we propose an end-to-end model with multiple attention blocks, named MATT_DTI, to predict the binding affinity scores of drug-target pairs. The protein sequences and SMILES (Simplified Molecular Input Line Entry System) of drugs are the inputs of our proposed model. Firstly, we propose a relation-aware self-attention block to model the drugs from SMILES data, considering the correlation between atoms. The relative self-attention block makes it possible to enhance the relative position information between atoms in compounds while considering the relationship of all elements at the same time. Secondly, two CNN models are utilized to learn the representations of drugs and targets, respectively. Finally, a multi-head attention block is built to model the similarity of drug-target pairs as the interaction information and fully connected networks (FNNs) are used to extract interaction features. Compared with the baseline DeepDTA [18], both of us are sequence representation methods and the protein representation learning part uses the same CNN model. The difference is that we employ a relation-aware self-attention block in drug representation learning to encode correlations of atoms, and a multi-head attention block to model the interaction information of DTIs.
In the experiments, we evaluate our proposed model on two public benchmark datasets, Davis [4] and KIBA [25] datasets, and compare our model with regression-based baselines, KronRLS [19], SimBoost [9], DeepDTA [18] and other recent sequence representation learning methods for DTI prediction. Our MATT_DTI model outperforms these baseline models on Concordance index (CI) and 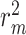 index metrics. Moreover, in order to further investigate the potential of our model, we apply our proposed model to Corona Virus Disease 2019 (COVID-19)-related proteins and list the FDA-approved drugs with high binding affinity scores predicted by our model.
index metrics. Moreover, in order to further investigate the potential of our model, we apply our proposed model to Corona Virus Disease 2019 (COVID-19)-related proteins and list the FDA-approved drugs with high binding affinity scores predicted by our model.
The main contributions of this paper can be summarized as follows.
(i) In order to model the drug compounds, a relation-aware self-attention block is built to enhance the relative position information between atoms in drugs and capture the long-distance relationship among all the atoms at the same time (section 3 Methods).
(ii) In order to further extract the interaction information of drug-target pairs, a multi-head attention block is proposed to model the similarity between drugs and target (section 3 Methods).
(iii) To the best our knowledge, our results are the state-of-the-art on the two datasets in sequence presentation learning methods for drug-target binding affinity prediction (section 4 Experiments).
(iv) We apply our model to COVID-19-related proteins and provide a reference to medical experts to find related drugs (section 5 Discussion).
Preliminaries
In this section, we introduce existing approaches for DTI prediction, the background knowledge of attention mechanism and the motivation of this work.
The related studies on DTI prediction
Many studies [6, 16, 29, 32] regarded DTI prediction as a binary classification problem. The proposed models to determine whether the interactions exist between drugs and targets. However, those methods simplified the DTI problem as with chosen binding affinity threshold values [18]. They overlooked the information for the binding affinity value, which describes the strength of the interaction between a drug-target pair. Therefore, the exact way for DTI prediction is directly to predict the binding affinity value based on a regression model.
In recent years, many efforts have been conducted on regression-based models for DTI prediction. The approaches based on random forest algorithm [11, 22] have been successful to predict the binding affinities of drugs and targets. Moreover, similarity-based methods were one option for regression-based DTI prediction, which utilized the similarity information of drugs and targets, such as SimBoost [9] and KronRLS [19].
With the significant success of deep neural networks in the computer version, speech recognition and natural language processing (NLP), many deep learning-based models were proposed to predict DTIs based on regression motivation. In recent works, deep models for DTI prediction mainly include two branches, graph representation method-based approaches with structure information as inputs [15, 30] and sequence representation-based approaches considering sequence information of DTI [18, 34, 35]. In this work, we focus on sequence representation learning approaches DeepDTA [18] and OnionNet [36] proposed CNN-based models for DTI predicting. Especially, DeepDTA focused on the sequence information of both drugs and targets and then used two CNN models for drugs (the SMILES input) and targets (the protein sequence input) as representation learning parts. Then, an information fusion part was connected to predict the binding affinities of drugs and targets. There, three FNN layers were regarded as the information fusion part. Since it could not capture the long-distance relationship between atoms, the study [24] applied a self-attention network (SAN) with position embedding to extract drug representation for DTI prediction.
The background of attention mechanism
Most neural sequence transaction models have an encoder-decoder structure. The transformer [27] is a typical encoder-decoder model based on an attention mechanism. It is widely used in the field of NLP, which has proven the strong ability of transformer in processing text data. The main block of the transformer is the attention function. The attention function can be described as mapping a query (Q) and a set of key-value (K-V) pairs to an output.
Scaled dot-product attention is defined as the generalized attention with Q, K and V. Let the dimension of Q and K be 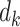 and the dimension of
and the dimension of 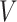 be
be 
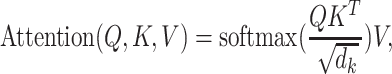 |
(1) |
where 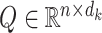 ,
,  and
and 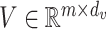 . Attention describes the similarity between the query and each value. The similarity can be measured by inner product of the softmax results and the value. The factor
. Attention describes the similarity between the query and each value. The similarity can be measured by inner product of the softmax results and the value. The factor 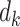 plays a regulatory role so that the inner product is not too large. When Q, K and V are projections from the same inputs, the attention function is the self-attention.
plays a regulatory role so that the inner product is not too large. When Q, K and V are projections from the same inputs, the attention function is the self-attention.
Multi-head Attention is an improved attention mechanism. Firstly, before scaled dot-product attention, the 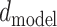 -dimensional Q, K and V should be linearly projected
-dimensional Q, K and V should be linearly projected  times with learned linear projections to
times with learned linear projections to 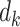 ,
,  and
and  , respectively. Specifically,
, respectively. Specifically,
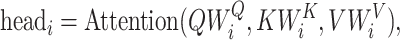 |
(2) |
where 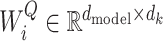 ,
, 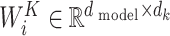 and
and 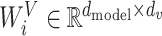 . Then, concatenate the results of attention
. Then, concatenate the results of attention
 |
(3) |
where  . Here, for each of those Q, K and V, a number of different ’heads’ are obtained through linear projections and attention with different weights. Thus, multi-head can be regarded as multiple the same operation in parallel, while parameters are not shared.
. Here, for each of those Q, K and V, a number of different ’heads’ are obtained through linear projections and attention with different weights. Thus, multi-head can be regarded as multiple the same operation in parallel, while parameters are not shared.
Motivation
As seen, when SANs are used on DTI prediction, an atom will conduct an attention operation with all atoms. It leads to SANs disperse the attention distribution to all elements and then overlook the essential correlation between atoms. In addition, most deep models for DTI prediction simply concatenate drug and protein representations to model the interaction between them. The way ignores the interaction features between drug and protein representations. In this study, we propose a deep model on DTI binding affinity prediction, in which the correlation between atoms and the interaction information between drugs and targets are considered.
Methods
In this work, we introduce a multiple attention blocks-based model—MATT_DTI, to predict the binding affinity scores of drug-target pairs, as shown in Figure 1. Like most deep learning-based DTI models, our model consists of three parts: drug representation learning, protein representation learning and interaction learning. Specifically, we propose a relation-aware self-attention block in the drug representation learning process. The relation-aware self-attention block is to encode correlations by enhancing the relative position information between atoms. Then, two CNN models are utilized to extract features from drugs and proteins in drug representation learning and protein representation learning processes, respectively. Finally, the interaction learning model is exploited to combine and extract interaction features from both drug representations and protein representations by multi-head attention.
Figure 1.

Illustration of our proposed MATT_DTI which considers drug SMILES and protein sequence as input to predict the binding affinity of drug-target pairs, which use a relation-aware self-attention block to strengthen the relative position information when encoding drug compounds and employ multi-head attention to model the interaction of drug representations and protein representations.
Input embedding
The inputs of our model are SMILES sequences for drugs and FASTA sequences for proteins. According to the work [18], the SMILES sequence is comprised of characters representing atoms and structure indicators. Mathematically, a drug is
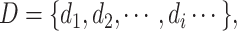 |
(1) |
where 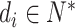 and the sequence length is varied, which depends on a compound. In this study, we define a hyperparameter
and the sequence length is varied, which depends on a compound. In this study, we define a hyperparameter 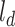 to restrict the max input length for drugs. Inspired by the token embedding and position embedding in transformer [27], the input of drug representation learning is the sum of token embedding and position embedding of SMILE sequences. The token embedding
to restrict the max input length for drugs. Inspired by the token embedding and position embedding in transformer [27], the input of drug representation learning is the sum of token embedding and position embedding of SMILE sequences. The token embedding 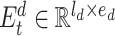 has a trainable weight
has a trainable weight 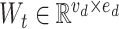 , where
, where 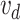 is the vocabulary size of drugs and
is the vocabulary size of drugs and 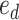 is the embedding size of drugs. The position embedding
is the embedding size of drugs. The position embedding 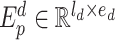 has a trainable weight
has a trainable weight  . The output of the embedding operations is
. The output of the embedding operations is
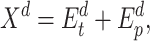 |
(2) |
where 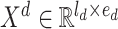 .
.
The same as the mathematical expression of drugs, a protein sequence is mathematically expressed as,
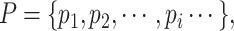 |
(3) |
where 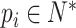 and the length of
and the length of 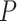 depends on proteins. We also define a hyperparameter
depends on proteins. We also define a hyperparameter  as the fixed protein input length to ensure the same size of inputs. Different from drug sequence, the trainable embedding layer of protein sequence is similar to DeepDTA [18] as
as the fixed protein input length to ensure the same size of inputs. Different from drug sequence, the trainable embedding layer of protein sequence is similar to DeepDTA [18] as
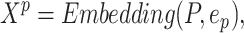 |
(4) |
where  is the embedding size of protein sequences and
is the embedding size of protein sequences and 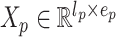 .
.
Protein representation learning model
As for protein representation leaning process, our proposed model is developed from DeepDTA [18] and the learning model for protein sequences also utilizes three convolutional layers as the feature extractor, followed by a max pooling layer. As for the CNN model in presentation learning model, suppose there exist 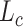 convolutional layer
convolutional layer
 |
(5) |
where  is the output of
is the output of 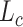 th convolutional layer and the
th convolutional layer and the  th
th 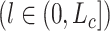 layer can be formally expressed as
layer can be formally expressed as
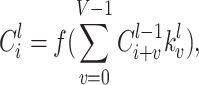 |
(6) |
where  indicates the trainable filters in
indicates the trainable filters in 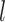 th convolutional layer, the size of it is
th convolutional layer, the size of it is 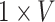 ,
, 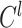 is the output of
is the output of 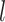 th layer and
th layer and  is the activation function. Based on this, when
is the activation function. Based on this, when 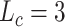 in protein representation learning, the output of protein representation learning could be calculated as
in protein representation learning, the output of protein representation learning could be calculated as
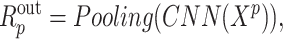 |
(7) |
where 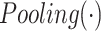 is the max pooling function.
is the max pooling function.
Drug representation learning model
SANs have drawn increasing interest, especially in the NLP field. SANs have the ability to capture long-distance dependencies by explicitly attending to all the elements, regardless of distance [33]. It contributes to representing a drug because SANs capture the long-distance relation between all atoms in a compound. However, SANs have a major limitation that is it disperses the attention distribution and thus overlooks relative information of elements [7, 23]. The relative information of atoms in drugs describes the essential correlation between atoms. This leads to that SAN is not sufficient to model SMILES data. Thus, a relation-aware self-attention block is proposed in drug representation learning.
The relation-aware self-attention block
In the field of NLP, self-attention with relative position representations [23] has already considered the pairwise relationship between words. It considers the relative distance information to model the distance between the words during conducting the self-attention. According to work [23], it can be simplified as
 |
(8) |
where 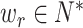 is the relative distance length between words.
is the relative distance length between words.
Inspired by this, we developed it to encode the correlation between atoms. We first define 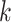 kinds of relative relationships between atoms, which are embedded into learnable parameters
kinds of relative relationships between atoms, which are embedded into learnable parameters 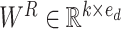 . Taken
. Taken 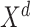 as the input, the output of a self-attention with relative position representations [23] layer can be formally expressed as
as the input, the output of a self-attention with relative position representations [23] layer can be formally expressed as
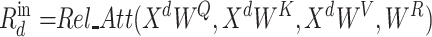 |
(9) |
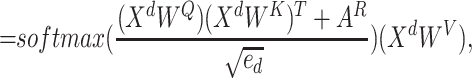 |
(10) |
where 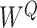 ,
, 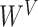 ,
, 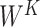
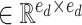 denote parameter matrices of attention layer.
denote parameter matrices of attention layer.  indicates the relationship matrix, of which,
indicates the relationship matrix, of which, 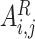 represents the correlation between the
represents the correlation between the  th and the
th and the  th elements
th elements
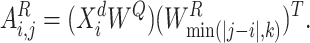 |
(11) |
Here, 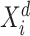 is the
is the 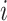 th vector in
th vector in 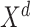 .
. 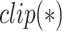 is employed to select corresponding embedding in
is employed to select corresponding embedding in  . Thus,
. Thus, 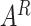 can be served as inductive biases to revise the attention distribution.
can be served as inductive biases to revise the attention distribution.
Then, a residual connection [8] and an FNN layer are following. As for the FNN layers in this study, 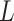 fully connected layers can be calculated by
fully connected layers can be calculated by
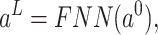 |
(12) |
where 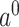 is the input of FNN layers, the
is the input of FNN layers, the 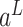 is the output of
is the output of 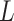 th FNN layer and the
th FNN layer and the  fully connected layer can be described as
fully connected layer can be described as
 |
(13) |
where 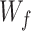 is the trainable wight and
is the trainable wight and 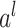 is the output of
is the output of 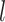 th fully connected layer. Therefore, the output of the residual connection and the FNN layer in the relation-aware self-attention block is
th fully connected layer. Therefore, the output of the residual connection and the FNN layer in the relation-aware self-attention block is
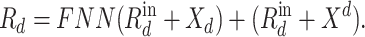 |
(14) |
Since the protein representation is learned by the CNN model, the three convolutional layers are also used to exploited drug information. We insist the two CNN models could ensure that the drug and protein representations are projected to the same space. Thus, the output is
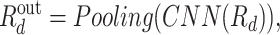 |
(15) |
while 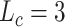 in drug representation learning. In this process, layer normalization [2] and dropout [10] are used.
in drug representation learning. In this process, layer normalization [2] and dropout [10] are used.
Interaction learning model
The existing way of interaction learning is to concatenate the representations of drugs and proteins. It overlooks the interaction information of drugs and proteins. In similarity-based DTI prediction models [9, 19], the similarity information of drug-protein pairs was used as the interaction information in them. Inspired by this, a multi-head attention block is exploited to model the similarity of drug-protein pairs as the interaction information of them. Here, the drug representations are regarded as the query, while the protein representations are the key and value in the attention mechanism. Mathematically, the output is
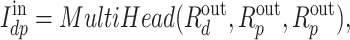 |
(16) |
with three heads in this study. Then, a residual connection [8] is used as
 |
(17) |
where 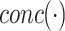 is a concatenation function and
is a concatenation function and 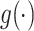 is global average pooling operation. Then, a 3-layered FCN (
is global average pooling operation. Then, a 3-layered FCN (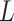 =3) is employed to learn the interaction information from
=3) is employed to learn the interaction information from 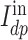 and the last layer of the network has only one neuron as the output of our model
and the last layer of the network has only one neuron as the output of our model
 |
(18) |
where  is the predicted binding affinity value of the drug-target pair. The weights of our proposed MATT_DTI model are optimized by the mean square error (MSE) between the network output
is the predicted binding affinity value of the drug-target pair. The weights of our proposed MATT_DTI model are optimized by the mean square error (MSE) between the network output 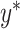 and the actual binding affinity value
and the actual binding affinity value 
 |
(19) |
Experiments
We proposed a novel drug-target binding affinity prediction method based on multiple attention blocks with sequence information of drugs (compounds) and proteins as inputs. In this section, we conducted experiments with our proposed model (MATT_DTI) on two benchmark datasets: Davis [4] and KIBA [25] datasets. The CI and 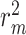 metrics were used to measure the performance of the proposed model and the baseline models.
metrics were used to measure the performance of the proposed model and the baseline models.
Benchmark datasets
We evaluated our proposed model on two benchmark datasets, Davis [4] and KIBA [25] datasets. The Davis dataset contains the 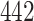 kinase proteins and their relevant inhibitors (
kinase proteins and their relevant inhibitors (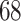 ligands) with respective dissociation constant (
ligands) with respective dissociation constant (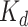 ) value. The
) value. The 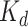 values were transformed into log space, as [9, 18],
values were transformed into log space, as [9, 18],  , as the binding affinity values, which is explained in 20,
, as the binding affinity values, which is explained in 20,
 |
(20) |
The KIBA dataset was developed from the KIBA approach, which comprised 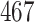 proteins,
proteins, 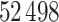 drugs and their binding affinity scores originally. Here, the KIAB scores measure the kinase inhibitor bioactivities and are regarded as the binding affinity values. SimBoost [9] filtered it to contain
drugs and their binding affinity scores originally. Here, the KIAB scores measure the kinase inhibitor bioactivities and are regarded as the binding affinity values. SimBoost [9] filtered it to contain 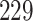 unique proteins and
unique proteins and 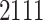 unique drugs for a fair comparison. As for the input of proteins and drugs in the Davis and KIBA dataset, we followed the DeepDTA method [18] in which the SMILES of drugs and protein sequences were digitized to a fixed maximum length by a dictionary. Table 1 summarizes the details of the Davis and KIBA dataset.
unique drugs for a fair comparison. As for the input of proteins and drugs in the Davis and KIBA dataset, we followed the DeepDTA method [18] in which the SMILES of drugs and protein sequences were digitized to a fixed maximum length by a dictionary. Table 1 summarizes the details of the Davis and KIBA dataset.
Table 1.
Summary of the benchmark datasets
| Davis | KIBA | |
|---|---|---|
| Proteins | 442 | 229 |
| Compounds | 68 | 2111 |
| Interactions | 30056 | 118254 |
| Training data | 25046 | 98545 |
| Test data | 5010 | 19709 |
Experiments setup
We evaluated the performance of our MATT_DTI on the benchmark datasets. Like the study DeepDTA [18], we firstly clipped the training data as training set and validation set to find the optimal settings of our model, like number of filters, filter length, hidden size, dropout rate and number of epochs. The final results given in this section were the average results on the test set with 5 times training. Table 2 gives the parameter settings in experiments depending on datasets. All models were trained on  NVIDIA 2080Ti GPU.
NVIDIA 2080Ti GPU.
Table 2.
Summary of parameter settings for MATT_DTI
| parameter | KIBA | Davis |
|---|---|---|
| max length (drug) | 100 | 85 |
| max length (protein) | 1000 | 1200 |
| embedding size | 128 | |
| number of filters in CNNs | 32 64 96 | 16 32 64 |
| filter size (drug) | 8 | |
| filter size (protein) | 12 | 16 |
| hidden size in FNNs | 1024 1024 512 1 | |
| batch size | 256 | 128 |
| epoch | 300 | |
| dropout | 0.1 | |
| optimizer | Adam | |
| learning rate | 0.001 | |
| activation function | ReLU[14] | |
Metrics
To evaluate the performance of our model, firstly, the CI was used as the evaluation metrics
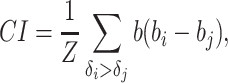 |
(21) |
where 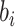 is the prediction value with larger affinity
is the prediction value with larger affinity 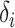 ,
, 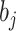 is the prediction value for smaller affinity
is the prediction value for smaller affinity 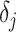 and
and 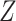 is a normalization constant. Moreover, the
is a normalization constant. Moreover, the 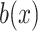 is the step function [19]
is the step function [19]
 |
(22) |
Then, in order to better evaluate our model,  [20, 21], which is widely used in this filed, is the another metric in this work. Mathematically,
[20, 21], which is widely used in this filed, is the another metric in this work. Mathematically,
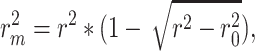 |
(23) |
where 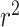 and
and 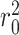 are the squared correlation coefficient values between the observed and predicted values with and without intercept, respectively. Only
are the squared correlation coefficient values between the observed and predicted values with and without intercept, respectively. Only 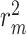 value of a model on test set is larger than
value of a model on test set is larger than 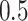 , the model is an acceptable model.
, the model is an acceptable model.
Experiment 1: relation-aware self-attention-based representation learning for drug compounds
Table 3 lists the average results on the drug-target binding affinity prediction tasks. As seen, MATT_DTIs improve the prediction quality in both two datasets, reconfirming the necessity of modeling the long-distance relationship and the relative position information of compounds. Besides, our models outperform all the baseline works in all metrics, indicating the superiority of the proposed approaches. In particular, The MATT_DTI with the self-attention block achieves better performance than DeepDTA, revealing the contribution of self-attention that modeling the long-distance relation of all elements in drugs. Moreover, the MATT_DTI with a relation-ware self-attention block (Rel_sa:CNN) outperforms the MATT_DTI model with a self-attention layer (sa:CNN), indicating that modeling the relative information can raise the ability of the self-attention model on capturing the atoms’ information.
Table 3.
Test results on KIBA and Davis dataset. The proposed MATT_DTI model includes a relative self-attention block in drug representation learning. The max relative position length 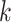 in ’Rel_sa:CNN’ is set to
in ’Rel_sa:CNN’ is set to 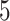 . In this table, ’sa:CNN’ denotes that the model has a self-attention block before CNN layers, while ’Rel_sa:CNN’ has a relative self-attention block in front of CNN layers
. In this table, ’sa:CNN’ denotes that the model has a self-attention block before CNN layers, while ’Rel_sa:CNN’ has a relative self-attention block in front of CNN layers
| Dataset | Methods | Compounds | Proteins | Interaction | CI (std) | MSE |
 (std) (std) |
|---|---|---|---|---|---|---|---|
| KIBA | KronRLS [19] | Pubchem Sim | S-W | – | 0.782 (0.001) | 0.411 | 0.342 (0.001) |
| SimBoost [9] | Pubchem Sim | S-W | – | 0.836 (0.001) | 0.222 | 0.629 (0.007) | |
| DeepDTA [18] | CNN | CNN | FNN | 0.863 (0.002) | 0.194 | 0.673 (0.009) | |
| MATT_DTI | sa:CNN | CNN | FNN | 0.881 (0.000) | 0.162 | 0.734 (0.002) | |
| MATT_DTI | Rel_sa:CNN | CNN | FNN | 0.889 (0.001) | 0.151 | 0.745 (0.008) | |
| Davis | KronRLS [19] | Pubchem Sim | S-W | – | 0.871 (0.001) | 0.379 | 0.407 (0.005) |
| SimBoost [9] | Pubchem Sim | S-W | – | 0.872 (0.001) | 0.282 | 0.644 (0.006) | |
| DeepDTA [18] | CNN | CNN | FNN | 0.878 (0.004) | 0.261 | 0.630 (0.017) | |
| MATT_DTI | sa:CNN | CNN | FNN | 0.880 (0.003) | 0.261 | 0.632 (0.015) | |
| MATT_DTI | Rel_sa:CNN | CNN | FNN | 0.884 (0.004) | 0.254 | 0.649 (0.009) |
Experiment 2: interaction learning with multi-head attention
In this section, we conducted the experiment about the interaction learning part based on multi-head attention and compared it with the existing way based on concatenation way. Table 4 gives the average test results on both KIBA and Davis datasets. One intuition of our approach is to capture interaction features via modeling the similarity between drug-protein pairs by a multi-head attention block. To evaluate it, we implemented models with a multi-head attention block in the interaction learning process. As shown in Table 4, the DeepDTA model and MATT_DTI model with a multi-head attention block (MulH_attention + FNN) achieve higher results than the models without it (Concatenation + FNN), revealing that extracting interaction features with multi-head attention is superior to concatenation.
Table 4.
Test results on KIBA and Davis dataset. The interaction model of the proposed MATT_DTI is based on multi-head attention. The max relative position length  in ’Rel_sa:CNN’ is
in ’Rel_sa:CNN’ is 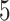
| Dataset | Methods | Compounds | Proteins | Interaction | CI (std) | MSE |
 (std) (std) |
|---|---|---|---|---|---|---|---|
| KIBA | DeepDTA[18] | CNN | CNN | Concatenation + FNN | 0.863 (0.002) | 0.194 | 0.673 (0.009) |
| DeepDTA-Attention | CNN | CNN | MulH_Attention + FNN | 0.875 (0.003) | 0.173 | 0.724 (0.012) | |
| MATT_DTI | Rel_sa:CNN | CNN | Concatenation+ FNN | 0.889 (0.001) | 0.151 | 0.745 (0.008) | |
| MATT_DTI | Rel_sa:CNN | CNN | MulH_Attention + FNN | 0.889 (0.001) | 0.150 | 0.756 (0.011) | |
| Davis | DeepDTA[18] | CNN | CNN | Concatenation + FNN | 0.878 (0.004) | 0.261 | 0.630 (0.017) |
| DeepDTA-Attention | CNN | CNN | MulH_Attention + FNN | 0.877 (0.002) | 0.252 | 0.648 (0.014) | |
| MATT_DTI | Rel_sa:CNN | CNN | Concatenation + FNN | 0.884 (0.004) | 0.254 | 0.649 (0.009) | |
| MATT_DTI | Rel_sa:CNN | CNN | MulH_Attention + FNN | 0.890 (0.003) | 0.229 | 0.682 (0.009) |
Experiment 3: effects of max relative position length
We finally investigated the effects of relation position length in the relation-aware self-attention block on the drug-target binding affinity prediction task. As shown in Figures 2 and 3, MATT_DTI with max relative position length 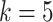 has the best performance in the KIBA dataset. As plotted in Figures 4 and 5, we believe the max relative position length with
has the best performance in the KIBA dataset. As plotted in Figures 4 and 5, we believe the max relative position length with  is superior to other setting for Davis dataset. The different distribution of KIBA and Davis dataset may lead to the slight difference of best max relative position length. As seen, MATT_DTI with max relative position length
is superior to other setting for Davis dataset. The different distribution of KIBA and Davis dataset may lead to the slight difference of best max relative position length. As seen, MATT_DTI with max relative position length  improves the prediction performance, indicating that the correlation between atoms is better modeled with max relative position length
improves the prediction performance, indicating that the correlation between atoms is better modeled with max relative position length 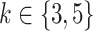 .
.
Figure 2.

CI results on KIBA dataset. Effects of relative position length in proposed MATT_DTI with a relation-aware self-attention block for drug representation and a multi-head self-attention in interaction learning.
Figure 3.

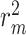 results on KIBA dataset. Effects of relative position length in proposed MATT_DTI with relative self-attention for drug representation and a multi-head self-attention in interaction learning.
results on KIBA dataset. Effects of relative position length in proposed MATT_DTI with relative self-attention for drug representation and a multi-head self-attention in interaction learning.
Figure 4.

CI results on Davis dataset. Effects of relative position length in proposed MATT_DTI with a relation-aware self-attention block for drug representation and a multi-head self-attention in interaction learning.
Figure 5.

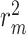 results on Davis dataset. Effects of relative position length in proposed MATT_DTI with a relation-aware self-attention block for drug representation and a multi-head self-attention in interaction learning.
results on Davis dataset. Effects of relative position length in proposed MATT_DTI with a relation-aware self-attention block for drug representation and a multi-head self-attention in interaction learning.
Moreover, we compare our proposed model with other sequence representation-based approaches in Table 5. As seen, our final results are higher 0.026 than DeepDTA on KIBA and higher 0.012 on Davis for CI metric. The  also higher 0.083 than DeepDTA on KIBA and higher 0.052 on Davis. In general, our model performs better than all other sequence representation learning approaches.
also higher 0.083 than DeepDTA on KIBA and higher 0.052 on Davis. In general, our model performs better than all other sequence representation learning approaches.
Table 5.
Results on KIBA and Davis dataset of our proposed model and the existing baseline methods
| Dataset | Methods | CI (std) | MSE |
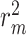 (std) (std) |
|---|---|---|---|---|
| KIBA | DeepDTA [18] | 0.863 (0.002) | 0.194 | 0.673 (0.009) |
| MT-DTI [24] | 0.882 (0.001) | 0.152 | 0.738 (0.006) | |
| WideDTA[17] | 0.875 (0.001) | 0.179 | – | |
| GANsDTA [34] | 0.866 (–) | 0.224 | 0.675 (–) | |
| MATT_DTI | 0.889 (0.001) | 0.150 | 0.756 (0.011) | |
| Davis | DeepDTA [18] | 0.878 (0.004) | 0.261 | 0.630 (0.017) |
| MT-DTI [24] | 0.887 (0.003) | 0.245 | 0.665 (0.014) | |
| WideDTA[17] | 0.886 (0.003) | 0.262 | – | |
| GANsDTA [34] | 0.881 (–) | 0.276 | 0.653 (–) | |
| MATT_DTI | 0.891 (0.002) | 0.227 | 0.683 (0.017) |
Discussion
Recently, the new coronavirus (SARS-CoV-2) infection is spreading rapidly, and the daily incidence rate is increasing worldwide. It is urgent to find out a valid drug for the patient. Drug repurposing is one of the computational efforts that re-utilize FDA approved drugs, or compound succeeded in phase one clinical trials, for a new indication, to take advantage of the proved toxicity [5, 13]. Drug repurposing is regarded as one potential way for finding new coronavirus treatment [3]. Therefore, in this section, we apply our trained model to predict the binding affinity scores between existing drugs and the genome sequences of COVID-19-related severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). We believe that the discussion could provide an example to apply our model in real-life situations and hope our results can provide scientists with an assistant to learn the coronavirus.
Based on studies [1, 3], we extract the genome sequences, 3C-like proteinase, RNA-dependent RNA polymerase, helicase, 3’-to-5’ exonuclease, endoRNAse and 2’-O-ribose methyltransferase of SARS-CoV-2 from the National Center for Biotechnology Information database; 3137 FDA-approved drugs are included in this section. Table 6 lists parts of the FDA-approval antiviral drugs with top binding affinity values predicted by our MATT_DTI with weights trained by KIBA dataset and existing approaches [1]. The full lists of the 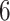 genome sequences could be found at supplementary data, see Supplementary Data available online at http://bib.oxfordjournals.org/.
genome sequences could be found at supplementary data, see Supplementary Data available online at http://bib.oxfordjournals.org/.
Table 6.
Parts of the FDA-approval antiviral drugs with top affinity scores of 3 genome sequences of SARS-CoV-2 predicted by our model
| The genome | Drug | Rank of |
|---|---|---|
| sequences | 3137 drugs | |
| 3C-like proteinase | Peramivir | 25 |
| Lopinavir | 45 | |
| Saquinavir | 54 | |
| Zanamivir | 73 | |
| Danoprevir | 83 | |
| Ritonavir | 89 | |
| RNA-dependent RNA polymerase | Saquinavir | 58 |
| Peramivir | 120 | |
| Lopinavir | 148 | |
| Danoprevir | 183 | |
| Daclatasvir (BMS-790052) | 202 | |
| MK-5172 | 243 | |
| helicase | Peramivir | 51 |
| Lopinavir | 55 | |
| Saquinavir | 102 | |
| Elvitegravir (GS-9137) | 118 | |
| Danoprevir | 137 | |
| Daclatasvir (BMS-790052) | 215 |
Nowadays, the effective drugs to cure COVID-19 have not been found; thus, we cannot verify our results. It is only a theoretical result on drug repurposing task. We just hope that the experiment will reflect the way to use our model in practical applications. Moreover, like studies [1, 3], we hope our work can provide scientists with some ideas for new drugs.
Conclusion
In this work, we propose a multiple attention blocks-based model to (i) enhance relative position information between atoms when encoding drugs and (ii) model the interaction between drug representations and target representations. Empirical results of the drug-target binding affinity prediction task on two benchmark datasets demonstrate the effectiveness of our proposed methods. The extensive analyses suggest that (i) encoding the relative position information is beneficial to drug representations, (ii) modeling the interaction can further improve the performance of predicting the binding affinity of DITs and (iii) the best max relative position length to encode drugs is in 3–5 for the KIBA and Davis dataset. Furthermore, we apply our trained model to predict the binding affinity scores of SARS-CoV-2-related genome sequences and 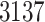 FDA-approved drugs to provide some reference for COVID-19-related scientists.
FDA-approved drugs to provide some reference for COVID-19-related scientists.
Key Points
MATT_DTI is a deep learning-based model for drug-target binding affinity score prediction.
In order to encode the correlation between atoms of drugs, MATT_DTI employs a relation-aware self-attention block to enhance the relative information between atoms when encoding drug compounds.
In order to extract interaction feature of drug-target pairs, a multi-head attention block is proposed to model the similarity between drugs and target in MATT_DTI.
Experimental results of DTI prediction on two benchmark datasets show our MATT_DTI outperforms existing models, which is benefit from the correlation and interaction information.
We further apply our model to FAD-approved drugs and COVID-19-related proteins, which could provide a reference to medical expert.
Supplementary Material
Funding
National Natural Science Foundation of China (Grants No. 61971296, U19A2078); Sichuan Science and Technology Planning Project (Grants No. 2020YFG0319, 2020YFH0186).
Yuni Zeng received her BS degree in the College of Computer Science, Sichuan University, Chengdu, at 2016, where she is currently pursuing his PhD degree. Her current research interests include neural networks and deep learning.
Xiangru Chen is currently pursuing the PhD degree with the College of Computer Science, Sichuan University. He received the BEng degree in computer science and technology from Sichuan University, China, in 2018. His current research interests include neural networks and deep learning.
Yujie Luo received his BEng degree in polymer science and technology from Sichuan University, Chengdu, China, in 2018. He is now a graduate student in the College of Computer Science, Sichuan University. His research interests include natural language processing and deep learning.
Xuedong Li is currently pursuing the PhD degree with the College of Computer Science, Sichuan University. His current research interests include machine learning and knowledge graph.
Dezhong Peng is a Professor at Sichuan University, Chengdu, China, currently. He received his BSc degree (1998) in applied mathematics and ME degree (2001) and PhD degree (2006) in computer science and engineering from the University of Electronic Science and Technology of China (UESTC), Chengdu, China. He was an Assistant Lecturer (2001) and a Lecturer (2003) at the School of Applied Mathematics, UESTC. He was also a Postdoctoral Research Fellow (2007.07–2009.09) at the School of Engineering, Deakin University, Melbourne, Australia.
Contributor Information
Yuni Zeng, College of Computer Science, Sichuan University, Chengdu, Sichuan,610065, China.
Xiangru Chen, College of Computer Science, Sichuan University, Chengdu, Sichuan,610065, China.
Yujie Luo, Shenzhen Peng Cheng Laboratory, Shenzhen, 518052, China.
Xuedong Li, Chengdu Sobey Digital Technology Co., Ltd, Chengdu, 610041,China.
Dezhong Peng, College of Computer Science, Sichuan University, Chengdu, Sichuan,610065, China.
References
- 1. Abdel-Basset M, Hawash H, Elhoseny M, et al. Deep learning for predicting drug-target interactions: A case study of COVID-19 drug repurposing. IEEE Access 2020; 8:170433–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ba LJ, Kiros JR, Hinton GE. Layer normalization. CoRR, abs/1607 2016;06450. [Google Scholar]
- 3. Bo RB, Shin B, Choi Y, et al. Predicting commercially available antiviral drugs that may act on the novel coronavirus (sars-cov-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J 2020; 18:784–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davis DMI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 2011; 29:1046–51. [DOI] [PubMed] [Google Scholar]
- 5. Dudley J, Deshpande T, Butte AJ. Exploiting drug-disease relationships for computational drug repositioning. Briefings Bioinform 2011; 12(4): 303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gao KY, Fokoue A, Luo H, et al. Interpretable drug target prediction using deep neural representation. In: Proceedings of the Twenty-Seventh International Joint Conference on Artificial Intelligence, IJCAI, July 13–19, Stockholm, Sweden, 2018, 3371–7. ijcai.org.
- 7. Guo M, Zhang Y, transformer TLG. A lightweight approach for natural language inference. In: The Thirty-Third AAAI Conference on Artificial Intelligence, AAAI 2019, Honolulu, Hawaii, USA, January 27–February 1, 2019, 2019, 6489–96.
- 8. He K, Zhang X, Ren S, et al. Deep residual learning for image recognition. In: 2016 IEEE Conference on Computer Vision and Pattern Recognition, CVPR 2016, Las Vegas, NV, USA, June 27-30, 2016. IEEE Computer Society, 2016, 770–8. [Google Scholar]
- 9. He T, Heidemeyer M, Ban F, et al. Simboost: a read-across approach for predicting drug-target binding affinities using gradient boosting machines. J. Cheminformatics 2017; 9(1): 24:1–24:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hinton GE, Srivastava N, Krizhevsky A, et al. Improving neural networks by preventing co-adaptation of feature detectors. CoRR, abs/1207 2012;0580. [Google Scholar]
- 11. Li H, Leung K-S, Wong M-H, et al. Low-quality structural and interaction data improves binding affinity prediction via random forest. Molecules 2015; 20(6): 10947–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maryam B, Elyas S, Kai W, et al. Machine learning approaches and databases for prediction of drug-target interaction: a survey paper. Brief Bioinform 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moriaud F, Richard SB, Adcock SA, et al. Identify drug repurposing candidates by mining the protein data bank. Briefings Bioinform. 2011; 12(4): 336–40. [DOI] [PubMed] [Google Scholar]
- 14. Nair V, Hinton GE. Rectified linear units improve restricted boltzmann machines. In: Proceedings of the 27th International Conference on Machine Learning (ICML-10), June 21–24, 2010, Haifa, Israel, 2010, 807–14.
- 15. Nguyen T, Le H, Venkatesh S. Graphdta: prediction of drug–target binding affinity using graph convolutional networks. BioRxiv, page 2019; 684662. [Google Scholar]
- 16. Öztürk H, Olmez EO, Özgür A. A comparative study of smiles-based compound similarity functions for drug-target interaction prediction. BMC Bioinform 2016; 17:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Öztürk H, Olmez EO, Özgür A. Widedta: prediction of drug-target binding affinity. CoRR, abs/1902 2019;04166. [Google Scholar]
- 18. Öztürk H, Özgür A, Olmez EO. Deepdta: deep drug-target binding affinity prediction. Bioinformatics 2018; 34(17): i821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pahikkala T, Airola A, Pietilä S, et al. Toward more realistic drug-target interaction predictions. Briefings Bioinform. 2015; 16(2): 325–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
20.
Roy K, Chakraborty P, Mitra I, et al. Some case studies on application of
 metrics for judging quality of quantitative structure-activity relationship predictions: Emphasis on scaling of response data. J. Comput. Chem 2013; 34(12): 1071–82. [DOI] [PubMed] [Google Scholar]
metrics for judging quality of quantitative structure-activity relationship predictions: Emphasis on scaling of response data. J. Comput. Chem 2013; 34(12): 1071–82. [DOI] [PubMed] [Google Scholar] - 21. Roy PP, Paul S, Mitra I, et al. On two novel parameters for validation of predictive qsar models. Molecules 2009; 14(5): 1660–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shar PA, Tao W, Gao S, et al. Pred-binding: large-scale protein-ligand binding affinity prediction. Journal of Enzyme Inhibition & Medicinal Chemistry 2016; 31(6): 1443–50. [DOI] [PubMed] [Google Scholar]
- 23. Shaw P, Uszkoreit J, Vaswani A. Self-attention with relative position representations. In: Proceedings of the 2018 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, NAACL-HLT, New Orleans, Louisiana, USA, June 1–6, 2018, 464–8.
- 24. Shin B, Park S, Kang K, et al. Self-attention based molecule representation for predicting drug-target interaction. In: Proceedings of the Machine Learning for Healthcare Conference, MLHC, Ann Arbor, Michigan, USA, volume 106. PMLR, 2019, 230–48. [Google Scholar]
- 25. Tang J, Szwajda A, Shakyawar S, et al. Making sense of large-scale kinase inhibitor bioactivity data sets: A comparative and integrative analysis. J Chem Inf Model 2014; 54(3): 735–43. [DOI] [PubMed] [Google Scholar]
- 26. Thafar MA, Olayan RS, Ashoor H, et al. Dtigems+: drug-target interaction prediction using graph embedding, graph mining, and similarity-based techniques. J Chem 2020; 12(1): 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vaswani A, Shazeer N, Parmar N, et al. Attention is all you need. In: Advances in Neural Information Processing Systems, NIPS, 2017, 5998–6008.
- 28. Wan F, Hong L, An X, et al. Neodti: neural integration of neighbor information from a heterogeneous network for discovering new drug-target interactions. Bioinformatics 2019; 35(1): 104–11. [DOI] [PubMed] [Google Scholar]
- 29. Wang L, You Z-H, Chen X, et al. A computational-based method for predicting drug-target interactions by using stacked autoencoder deep neural network. J Comput Biol 2018; 25(3): 361–73. [DOI] [PubMed] [Google Scholar]
- 30. Wang X, Liu Y, Lu F, et al. Dipeptide frequency of word frequency and graph convolutional networks for dta prediction. Front Bioeng Biotechnol 2020; 8:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Y, Zeng J. Predicting drug-target interactions using restricted boltzmann machines. Bioinformatics 2013; 29(13): 126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ming Wen, Zhimin Zhang, Shaoyu Niu, Haozhi Sha, Ruihan Yang, Yonghuan Yun,, and Hongmei Lu. Deep-learning-based drug-target interaction prediction. J Proteome Res, 16(4): 1401–9, 2017. [DOI] [PubMed] [Google Scholar]
- 33. Yang B, Wang L, Wong DF, et al. Convolutional self-attention networks. In: Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, NAACL-HLT 2019, Minneapolis, MN, USA, June 2–7, 2019, 2019, 4040–5.
- 34. Lingling Zhao, Junjie Wang, Long Pang, Yang Liu, and Jun Zhang. Gansdta: Predicting drug-target binding affinity using gans. Frontiers in Genetics, 10:1243–, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao Q, Xiao F, Yang M, et al. Attentiondta: prediction of drug-target binding affinity using attention model. In: 2019 IEEE International Conference on Bioinformatics and Biomedicine, BIBM 2019, San Diego, CA, USA, November 18–21, 2019, 2019, 64–9.
- 36. Zheng L, Fan J, Mu Y. Onionnet: a multiple-layer intermolecular-contact-based convolutional neural network for protein-ligand binding affinity prediction. ACS Omega 2019; 4(14): 15956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou M, Zheng C, Rong X. Combining phenome-driven drug-target interaction prediction with patients’ electronic health records-based clinical corroboration toward drug discovery. Bioinformatics 2020; 36(Supplement-1): i436–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.