

Abstract
Current coronavirus disease-2019 (COVID-19) pandemic has caused massive loss of lives. Clinical trials of vaccines and drugs are currently being conducted around the world; however, till now no effective drug is available for COVID-19. Identification of key genes and perturbed pathways in COVID-19 may uncover potential drug targets and biomarkers. We aimed to identify key gene modules and hub targets involved in COVID-19. We have analyzed SARS-CoV-2 infected peripheral blood mononuclear cell (PBMC) transcriptomic data through gene coexpression analysis. We identified 1520 and 1733 differentially expressed genes (DEGs) from the GSE152418 and CRA002390 PBMC datasets, respectively (FDR < 0.05). We found four key gene modules and hub gene signature based on module membership (MMhub) statistics and protein–protein interaction (PPI) networks (PPIhub). Functional annotation by enrichment analysis of the genes of these modules demonstrated immune and inflammatory response biological processes enriched by the DEGs. The pathway analysis revealed the hub genes were enriched with the IL-17 signaling pathway, cytokine–cytokine receptor interaction pathways. Then, we demonstrated the classification performance of hub genes (PLK1, AURKB, AURKA, CDK1, CDC20, KIF11, CCNB1, KIF2C, DTL and CDC6) with accuracy >0.90 suggesting the biomarker potential of the hub genes. The regulatory network analysis showed transcription factors and microRNAs that target these hub genes. Finally, drug–gene interactions analysis suggests amsacrine, BRD-K68548958, naproxol, palbociclib and teniposide as the top-scored repurposed drugs. The identified biomarkers and pathways might be therapeutic targets to the COVID-19.
Keywords: COVID-19, differentially expressed genes, gene coexpression network, systems biology, protein–protein interaction, machine learning
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a newly evolved virus, first identified in Wuhan, China in December 2019. Pneumonia caused by the SARS-CoV-2 was referred to as coronavirus disease (COVID-19), which was declared as the COVID-19 as a pandemic by the World Health Organization (WHO) [1]. The main symptoms of COVID-19 are fever, cough, pneumonia and shortness of breath [2]. The SARS-CoV-2 has infected almost 65 943 003 peoples with more than 1 519 137 deaths globally (as of 5 December 2020) [3].
The spread of the virus infection may be controlled through early detection of COVID-19 patients. However, the currently used methods including real-time polymerase chain reaction (RT-PCR) are subject to limited sensitivity and specificity as well as time-consuming. Moreover, careful sample collections and preparations, and skilled manpower are required, which are a tremendous drawback for developing countries. Thus, transcriptomic analysis of SARS-CoV-2 PBMC may provide candidate biomarkers. Past studies have conducted a transcriptomic analysis of various organs including lung epithelial cell [4–6], PBMC [7, 8]. Most of the previous reports detected the hub genes in COVID-19 from each module either via the PPI network or module membership criterion [5, 9–13]. Extensive gene expression analysis to identify differentially expressed genes (DEGs) and associated gene ontologies have been proposed by previous reports [5, 9–13]. Detection of specific gene modules has not been performed in COVID-19, and identification of key gene module hubs and targeting those critical genes for drugs repurposing is crucial to combat COVID-19. However, despite important findings from those studies, integrative analysis is needed to detect novel dysregulated genes and pathways for the pathogenesis of COVID-19.
The weighted gene coexpression network analysis (WGCNA) identifies significant modules (clusters) of highly correlated genes [14]. It explains the correlation patterns within genes and samples and biologically interprets the function of gene modules. The hub genes of differentially coexpressed modules provide more significance than the usual hub DEGs. WGCNA is widely used for biomarkers identification in various diseases [15–17], it has a great prospect in COVID-19. Identification of repurposable drug candidates for COVID-19 may reverse the DEGs of COVID-19, we have decided to utilize the anti-signature approach [5]. Identification of this in silico based approach offer opportunities to identify potential candidate drugs that might be considered for drug repositioning for COVID-19 treatment.
In this study, we implemented a system biology approach to key gene modules (identified via WGCNA) that were DEGs in COVID-19 PBMC. The hub genes were then identified from the key gene modules based on gene module membership (MMhub) and protein–protein interaction network (PPIhub), respectively. Then, we employed machine learning methods to determine the validity of these hub genes. Finally, we identified several candidate drugs considering these hub genes as therapeutic targets. Our results may provide novel insights into the pathogenesis of COVID-19 and the potential molecular targets.
Materials and methods
RNA-sequencing datasets
In this study, we used two RNA-Sequencing PBMC datasets of SARS-CoV-2 (COVID-19). One of the COVID-19 gene expression raw counts dataset was obtained with the accession number GSE152418 under the platform GPL24676 from the NCBI Gene Expression Omnibus (GEO) [18]. Recently this dataset was deposited by Arunachalam et al. [7] that contained 34 samples (17 COVID-19 samples and 17 healthy control samples). The other dataset was obtained from the Chinese Academy of Science with the accession number CRA002390 that contained PBMC samples from three COVID-19 infected patients and three healthy donors [6]. In this study, the GSE152418 discovery dataset was used to analyze WGCNA, and the CRA002390 dataset was used for independent validation.
Data preprocessing and differential gene expression analysis
The transcriptomics dataset, GSE152418, of COVID-19 contained a large number of genes (60 683 genes). For data preprocessing, the low expressed genes (sum of the gene counts for all samples <100) were excluded from this dataset. Then the differential gene expression analysis of the dataset was carried out through the DESeq2 package in the R language [19]. For the CRA002390 normalized count dataset, we used the limma package in R [20] to identify the DEGs. We considered adjusted P-value with Benjamini-Hochberg FDR correction techniques (FDR < 0.05) and 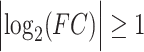 statistical threshold parameters for DEGs identification.
statistical threshold parameters for DEGs identification.
Weighted gene coexpression networks construction
The gene coexpression network construction was executed after removing the outlier samples (if there exist). The cluster dendrogram of the samples was constructed to check the outliers through the hclust function in R. We used the WGCNA package in R [25] to construct the weighted gene coexpression network. For finding numerous soft-thresholding powers β over R2, we used the pickSoftThreshold function. Then we picked the value of β for which the value of R2 maximum. The adjacency matrix and Topological Overlap Matrix (TOM) were then constructed using this soft threshold power with the transformed gene expression matrix. Then the dissimilarity of TOM (dissTOM) was computed to construct a network heatmap plot and for further analyses.
For the detection of the module, the dendrogram of genes was constructed with a dissTOM matrix using hclust function with different colors. The Automerged technique was used to get modules using the parameters: deepSplit = 2 and minClusterSize = 30 for avoiding the generation of small or large modules. Here MEDissThres = 0.25 was taken for merging similar modules [15].
Preservation analysis for the key module selection
To find the key modules, we used module preservation analysis. The modulePreservation function [21] was used to evaluate each of the modules whether it was robust and reproducible across datasets or not. If preservation statistics Z summary>10, then the module is considered as preserved [15]. It is apparent that the module preservation and preservation statistics-median Rank are negatively correlated and there is present a positive correlation between Z summary statistic and module preservation.
Gene ontology and pathway enrichment analysis
The high connectivity of genes inside the coexpression modules may represent crucial information about the similar biological roles within the same module. The functional enrichment analysis of the genes was studied in each selected key modules via Gene Ontology (GO) and pathway analysis [22, 23]. The GO and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis was executed via the Database for Annotation, Visualization, and Integrated Discovery and visualized (DAVID) tools [24] and the result was visualized through clusterProfiler package in R [25]. A statistical threshold criterion with an adjusted P-value <0.05 was used to select significant GO terms and KEGG pathways.
Identification and validation of hub genes
In a gene module, a series of genes with the greatest degree of connectivity was detected as hub genes that investigate the characteristics of a module. Also, a module’s connectivity defining the hub genes was calculated using the absolute value of the Pearson’s correlation (|cor.geneModuleMembership| > 0.8) [15]. Furthermore, we uploaded all genes of the key modules into the STRING database, choosing confidence score cutoff >900 to construct protein–protein interaction (PPI). In the PPI network, genes with a connectivity degree of ≥8 were also defined as hub genes [26]. The present study further analyzed the CRA002390 validation dataset to confirm the role of these hub genes as molecular signature genes for COVID-19. The common genes among the MMhub, PPIhub and the DEGs of CRA002390 will be considered for further network analyses.
Performance evaluation of the hub genes with classification algorithms
To check the validity of the identified common hub genes in two different analyses results (MMhub and PPIhub), the five popular classification algorithms, support vector machine (SVM) [27] radial basis kernel function, random forest (RF) [28], Poisson linear discriminant analysis (PLDA) [29], negative binomial linear discriminant analysis (NBLDA) [30] and voom-based diagonal linear discriminant analysis (voomDLDA) [31] were conducted through MLSeq package in R [32]. For the SVM and RF classifiers, we used DESeq normalization and VST transformation on the count dataset. Here, we considered the COVID-19 GSE152418 transcriptomic dataset for the classification analysis separately with MMhub genes and PPIhub genes, respectively. We computed four performance measures namely, accuracy, area under the ROC curve (AUC), sensitivity and specificity based on the data with MMhub and PPIhub genes, respectively.
PPI network analysis
The common genes among MMhub, PPIhub and DEGs of CRA002390 were considered for further network analysis. The PPI network for these genes was constructed via the STRING web tools [33]. STRING provides the PPI network that shows how the identified hub genes (proteins) interrelate functionally and physically with each other through encoding the gene list as input. Through the STRING information, the PPI network was constructed via Cytoscape [34] which is an open-source platform. The hub genes from this network were chosen based on degree connectivity via Cytohubba in Cytoscape. These hub genes were considered for final biomarkers of COVID-19.
Transcription factor and miRNAs identification
The significant transcription factors (TFs) were identified through a freely accessible database of TFs repository-JASPAR [35] by executing the interaction of the TFs-target genes via NetworkAnalyst [36]. The significant miRNAs were identified from miRNAs-target gene interaction analysis through the Tarbase [37] and mirTarbase [38] database via NetworkAnalyst [36]. These networks were visualized with Cytoscape and the significant hub TFs and miRNAs were selected via the CytoHubba plugin in Cytoscape based on the degree connectivity.
Drug–gene interaction analysis
To predict potential drugs for the treatment of COVID-19, we performed a transcriptomic anti-signature approach [5, 39] using the L1000FWD web-based tool [40], which measures the similarity score between input DEGs and expression signature and LINCS-L1000 data to detect drugs that may reverse the input gene signature. LINCS-L1000 contained drugs induced gene signature of about 50 human cell lines in response to 20 000 compounds. The significant drugs were chosen based on the criterion, q-value < 0.05.
Proposed bioinformatics pipeline
The workflow of the proposed bioinformatics methods to identify significant pathway and drug targets were illustrated in Figure 1 with followings:
Figure 1.
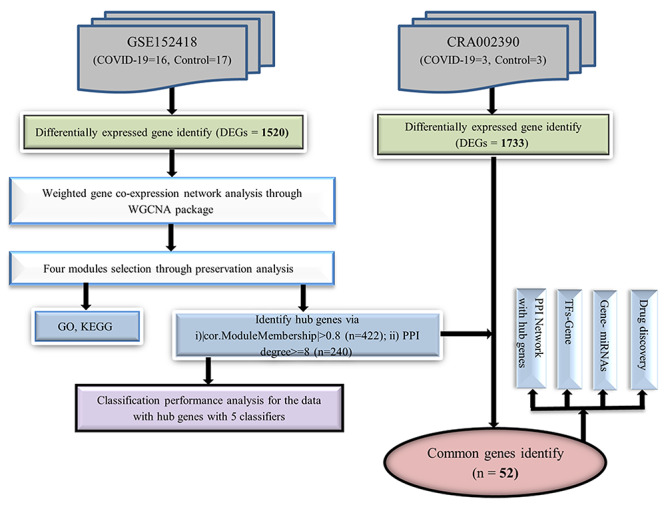
Flowchart of this study.
RNA-sequencing datasets: Differential gene expression analysis was achieved using RNA-seq data. One dataset was used to analyze the weighted gene coexpression network and the other independent dataset was used for validation.
Coexpression network reconstruction: The weighted gene coexpression network was constructed through the WGCNA package in R.
Key module selection: The Module Preservation function was used to identify robust and reproducible modules.
Enrichment analysis: Biological insights of key module genes were determined via the DAVID tools.
Hub genes identification: Hub gene signature was identified based on module membership (MMhub) statistics which calculated using the absolute value of the Pearson’s correlation and PPI networks (PPIhub) by using degree metrics. The common genes as hub signature were selected among the MMhub, PPIhub and the DEGs of CRA002390..
Validation analysis with machine learning methods: For validation of hub signatures, the five popular classification algorithms, SVM-radial basis kernel function, RF, PLDA, NBLDA and voomDLDA were conducted through MLSeq package in R.
Performance evaluation of hub genes: The performance measures including accuracy, AUC, sensitivity and specificity of MMhub and PPIhub genes were calculated by using an independent dataset.
Transcriptional regulator identification: Potential regulators (i.e.: TF and miRNA) of hub gene signature were determined using JASPAR, Tarbase and mirTarbase databases via NetworkAnalyst.
Candidate drug identification: LINCS-L1000 data were used to detect significant putative drugs that may reverse the input hub gene signature.
Results
DEGs identification
The discovery dataset (GSE152418) contained 60 683 genes with 17 COVID-19 and 17 health control samples. After excluding the lowly expressed genes, we selected 20 251 genes for DEGs identification. We identified a total of 1520 DEGs (1299 upregulated and 221 downregulated) from this dataset (FDR < 0.05) that were considered for further weighted gene coexpression network analysis. We identified 1733 DEGs (1139 upregulated and 594 downregulated) from the validation dataset (CRA002390) in the COVID-19 samples. Table 1 presented the information regarding the datasets used in this study.
Table 1.
Overview of the COVID-19 datasets used in this study
| Data accession No. | Tissue sources | # of identified DEGs | # of Samples | # of Control | # of Case |
|---|---|---|---|---|---|
| GSE152418 | PBMC | 1520 | 34 | 17 | 17 (one outlier sample) |
| CRA002390 | PBMC | 1733 | 6 | 3 | 3 |
Weighted gene coexpression networks and module preservation analysis
The gene coexpression network analyses were performed with identified 1520 DEGs of 16 COVID-19 samples in the GSE152418 dataset. The cluster dendrogram of these samples was visualized in Figure 2A that revealed, no outlying samples presented in this dataset. We selected the optimized soft-thresholding power, β = 6 with R2 = 0.90 as the scale-free topology criteria (Figure 2B). The coexpression networks were then constructed with this soft threshold power β = 6 and obtained 10 coexpressed modules namely, black, blue, brown, green, gray, magenta, pink, red, turquoise and yellow. The cluster dendrogram of these 10 modules presented in Figure 2D. We observed 63, 273, 259, 95, 46, 58, 77, 426 and 198 genes for black, blue, brown, green, magenta, pink, red, turquoise and yellow modules, respectively. The gray module contained 25 genes that were tied up as noncoexpressed. Figure 2C showed the network heatmap of all genes with these nine modules. The interactions among these coexpressed modules were presented with the module eigengene dendrogram and eigengene network heatmap (Figure 2E).
Figure 2.
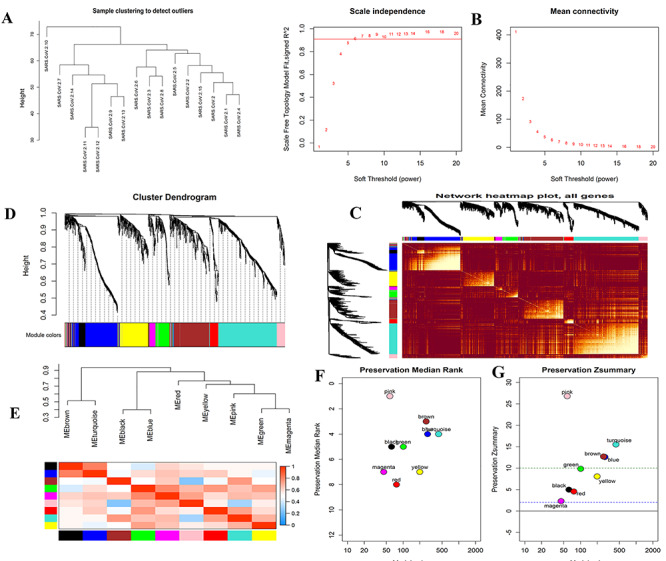
Construction of WGCNA coexpression modules and hub modules selection. (A) The cluster dendrogram of COVID-19 infected samples. (B) Analysis of the scale-free fit index (left) and the mean connectivity (right) for various soft-thresholding powers. (C) Heatmap plot of all genes. (D) Dendrogram of all differentially expressed genes clustered based on a dissimilarity measure (1-TOM). (E) Module eigengene dendrogram and eigengene network heatmap summarize the modules yielded in the clustering analysis. (F) The median rank of the modules; the rank value close to zero indicates a high degree of module preservation. (G) The Z summary statistics plot over each module; the blue and green dashed lines indicate the thresholds Z = 2 and Z = 10, indicate moderate and strong preservation thresholds, respectively.
In the module preservation analysis, we used the GSE152418 dataset with 17 health control samples as the test dataset. We identified turquoise, blue, brown and pink modules as the most stable through preservation analysis. The remaining modules were considered nonstable since their Z summary statistic <10 (Figure 2G). Figure 2F presented that the turquoise, blue, brown and pink modules were the best-preserved among all modules since their medianRank statistic were minimum than other modules.
Biological insights of the four-module genes
To obtain further biological insight into the genes of the selected four modules, the GO and KEGG pathway analysis was conducted in this study. The significant biological process (BP) mainly enriched in the immune response, division and fission related BP (Figure 3A). The significant molecular function (MF) mainly enriched in the binding related functions (Figure 3B). The most significant cellular components (CC) for the four modules are enriched in several cell compartments (Figure 3C).
Figure 3.

GO and KEGG enrichment analysis for four key modules. (A) biological process, (B) molecular function, (C) cellular components and (D) KEGG enrichment analysis.
The KEGG pathways for the genes of four modules significantly enriched in several pathways such as infection-related pathways (i.e: herpes simplex virus 1 infection, human papillomavirus infection), autoimmune diseases related pathways (i.e.: systemic lupus erythematosus, rheumatoid arthritis, type I diabetes mellitus), ECM-receptor interaction, IL-17 signaling pathway and p53 signaling pathway (Figure 3D and Table 2). Interestingly, alcoholism and systemic lupus erythematosus were significantly enriched for the genes of these four modules.
Table 2.
KEGG pathway enrichment results of four modules (top ten)
| Modules | ID | Description | Adjusted P-value | Related genes | Count |
|---|---|---|---|---|---|
| Turquoise | hsa05034 | Alcoholism | 4.28E-10 | HIST1H2AE;HIST1H3H;HIST1H4I;GNG11;HIST1H2AG;HIST1H2AI;HIST2H2BE HIST1H2BD;HIST1H4K;HIST1H2BJ;SLC18A2;HIST1H4H;HIST1H2AC;HIST2H4A; HIST1H2BG;HIST1H2BL;HIST1H2BK;HIST1H2BH;HIST1H2BC;HIST1H2BO; MAOB |
21 |
| hsa05322 | Systemic lupus erythematosus | 5.59E-10 | MYLK;GP9;GUCY1B1;ITGA2;COL1A2;PTGS1;ARHGEF12;PRKG1;VWF;GP1BA; ITGA2B;ITGB3;PLCB4;F2RL3;GP6 |
18 | |
| hsa04611 | Platelet activation | 5.66E-08 | MYLK;GP9;GUCY1B1;ITGA2;COL1A2;PTGS1;ARHGEF12;PRKG1;VWF;GP1BA; ITGA2B;ITGB3;PLCB4;F2RL3;GP6 |
15 | |
| hsa04512 | ECM-receptor interaction | 0.000108 | ITGB5;GP9;ITGA2;COL1A2;VWF;GP1BA;ITGA2B;ITGB3;GP6 | 9 | |
| hsa04540 | Gap junction | 0.000108 | EGF;GUCY1B1;PDGFA;LPAR1;PRKG1;TJP1;PLCB4;TUBB1;TUBA8 | 9 | |
| hsa05203 | Viral carcinogenesis | 0.000123 | H4C14;H2BC21;H2BC4;H2BC5;H2BC8;H2BC9;H4C8;H2BC11;H4C9;H2BC12; H2BC13;H4C12;H2BC17;CDKN2A |
14 | |
| hsa04022 | cGMP-PKG signaling pathway | 0.000247 | OPRD1;MYLK;PDE5A;GUCY1B1;PDE2A;TRPC6;PRKG1;ADRA2A;PDE3A; SLC8A3;PLCB4;MYL9 |
12 | |
| hsa04510 | Focal adhesion | 0.000385 | MYLK;ITGB5;EGF;VEGFC;ITGA2;PDGFA;COL1A2;CAV2;VWF;BCAR1; ITGA2B;ITGB3;MYL9 |
13 | |
| hsa04270 | Vascular smooth muscle contraction | 0.002487 | PLA2G2C;MYLK;GUCY1B1;CALD1;ARHGEF12;PRKG1;PLCB4;MYL9;PPP1R14A | 9 | |
| hsa04810 | Regulation of actin cytoskeleton | 0.002582 | IQGAP3;MYLK;ITGB5;EGF;ITGA2;PDGFA;LPAR1;ARHGEF12;BCAR1;ITGA2B; ITGB3;MYL9 |
12 | |
| Blue | hsa04110 | Cell cycle | 3.14E-24 | CDC20;CDKN2C;ORC1;BUB1;MCM6;CDC25A;MCM2;MAD2L1;CCNA2;CCNB1; CDC25C;PTTG1;TTK;MCM4;CCNE2;CHEK1;CDK1;ESPL1;BUB1B;CCNB2;PKMYT1; PLK1;ORC6;CDC6;CCNE1;CDC45 |
26 |
| hsa04114 | Oocyte meiosis | 1.11E-13 | CDC20;SPDYA;BUB1;SGO1;MAD2L1;CCNB1;CDC25C;PTTG1;FBXO5;CCNE2; FBXO43;CDK1;ESPL1;CCNB2;PKMYT1;PLK1;AURKA;CCNE1 |
18 | |
| hsa04914 | Progesterone-mediated oocyte maturation | 1.05E-08 | SPDYA;BUB1;CDC25A;MAD2L1;CCNA2;CCNB1;CDC25C;CDK1;CCNB2; PKMYT1;PLK1;AURKA |
12 | |
| hsa05322 | Systemic lupus erythematosus | 2.54E-06 | H3C2;H2AC4;H3C3;H4C4;H4C6;H3C7;H3C8;H2AC12;H2AC14;H4C11;H3C11 | 11 | |
| hsa04115 | p53 signaling pathway | 6.76E-06 | RRM2;CCNB1;CCNE2;CHEK1;CDK1;CCNB2;CCNE1;GTSE1 | 8 | |
| hsa05034 | Alcoholism | 9.77E-06 | GNG4;H3C2;H2AC4;H3C3;H4C4;H4C6;H3C7;H3C8;H2AC12;H2AC14;H4C11;H3C11 | 12 | |
| hsa05166 | Human T-cell leukemia virus 1 infection | 4.74E-05 | CDC20;CDKN2C;MAD2L1;CCNA2;TERT;PTTG1;CCNE2;CHEK1;ESPL1; BUB1B;CCNB2;CCNE1 |
12 | |
| hsa04218 | Cellular senescence | 5.66E-05 | CDC25A;CCNA2;CCNB1;CCNE2;CHEK1;CDK1;FOXM1;CCNB2;MYBL2;CCNE1 | 10 | |
| hsa03460 | Fanconi anemia pathway | 9.35E-05 | UBE2T;RAD51;FANCI;RMI2;BRCA1;EME1 | 6 | |
| hsa05203 | Viral carcinogenesis | 0.000115 | CDC20;CCR3;CCNA2;H4C4;H4C6;H4C11;SCIN;CCNE2;CHEK1;CDK1;CCNE1 | 11 | |
| Brown | hsa05322 | Systemic lupus erythematosus | 5.01E-06 | C1QA;C1QC;C1QB;FCGR1A;IL10;H2BC7;H2AC16;H3C12;H2AJ;ELANE;C3 | 11 |
| hsa05150 | Staphylococcus aureus infection | 0.000536 | C1QA;C1QC;C1QB;FCGR1A;IL10;CAMP;C3 | 7 | |
| hsa05142 | Chagas disease | 0.000772 | C1QA;C1QC;C1QB;FASLG;IL10;TGFBR1;C3 | 7 | |
| hsa05133 | Pertussis | 0.000889 | C1QA;C1QC;C1QB;IL10;IL23A;C3 | 6 | |
| hsa04610 | Complement and coagulation cascades | 0.001597 | C1QA;C1QC;C1QB;VSIG4;CLU;C3 | 6 | |
| hsa05168 | Herpes simplex virus 1 infection | 0.002736 | FASLG;PILRB;ZNF10;ZNF597;C3;ZNF563;ZNF540;ZNF571;ZNF607;ZNF284; ZNF600;ZNF543;ZNF304;ZNF547;ZNF419;ZNF132 |
16 | |
| hsa05165 | Human papillomavirus infection | 0.003638 | FZD5;FN1;WNT7A;SPP1;ITGA1;CREB3L1;WNT11;ITGA7;CCNA1;THBS1;NOTCH3 | 12 | |
| hsa05030 | Cocaine addiction | 0.005867 | MAOA;SLC18A1;CREB3L1;GRIN3B; | 4 | |
| hsa05034 | Alcoholism | 0.006577 | H2BC7;H2AC16;H3C12;MAOA;SLC18A1;CREB3L1;H2AJ;GRIN3B | 8 | |
| hsa04512 | ECM-receptor interaction | 0.009794 | FN1;SPP1;ITGA1;ITGA7;THBS1 | 5 | |
| Pink | hsa04657 | IL-17 signaling pathway | 2.17E-05 | IL1B;CCL20;TNF;TNFAIP3;FOSB | 5 |
| hsa05323 | Rheumatoid arthritis | 0.000359 | IL1A;IL1B;CCL20;TNF | 4 | |
| hsa05332 | Graft-versus-host disease | 0.000482 | IL1A;IL1B;TNF | 3 | |
| hsa04940 | Type I diabetes mellitus | 0.000516 | IL1A;IL1B;TNF | 3 | |
| hsa04060 | Cytokine–cytokine receptor interaction | 0.000639 | IL1A;IL1B;CXCR4;CCL20;TNF;OSM | 6 | |
| hsa04668 | TNF signaling pathway | 0.000727 | IL1B;CCL20;TNF;TNFAIP3 | 4 | |
| hsa04380 | Osteoclast differentiation | 0.001199 | IL1A;IL1B;TNF;FOSB | 4 | |
| hsa05162 | Measles | 0.001627 | IL1A;IL1B;TNFAIP3;CD209 | 4 | |
| hsa05418 | Fluid shear stress and atherosclerosis | 0.001627 | IL1A;IL1B;NFE2L2;TNF | 4 | |
| hsa05321 | Inflammatory bowel disease | 0.00173 | IL1A;IL1B;TNF | 3 |
Identification and validation of hub genes
We identified 422 hub genes (MMhub) in turquoise, blue, brown and pink modules with high connectivity using the module connectivity threshold criterion |cor.geneModuleMembership| > 0.8. Additionally, we identified 240 hub genes (PPIhub) for the four modules from the PPI through the STRING database with a connectivity degree ≥8. The present study further analyzed the CRA002390 validation set to confirm the role of these hub genes as candidate biomarker genes for COVID-19. We identified 52 common genes among MMhub, PPIhub and the DEGs of CRA002390 (Figure 5A). The summary of these 52 genes was described in Table 4. The expression values of these common genes over the COVID-19 and health control samples were presented in Figure 5C. The heatmap showed the two main clusters of these 52 hub genes in Figure 5B in terms of COVID-19 infected samples and control samples.
Figure 5.
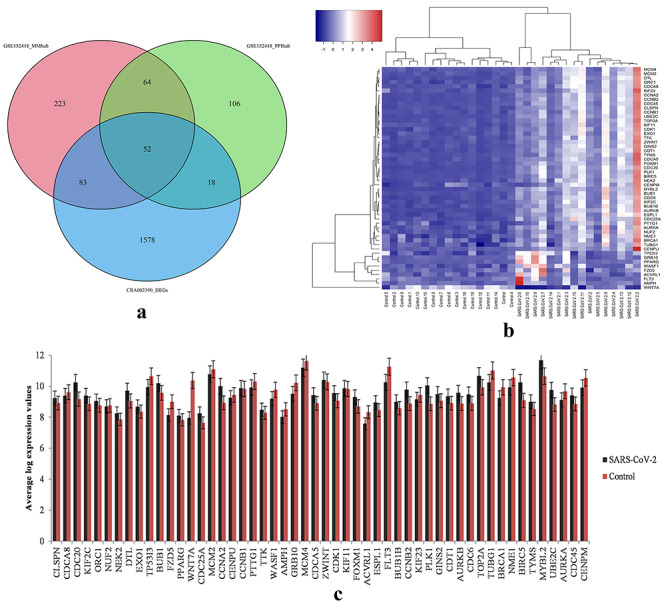
Hub gene expression profiles. (A) Venn diagram of common hub genes identified among the hub genes of GSE152418 identified via MM scores and PPI and the DEGs of CRA002390 data. (B) Heatmap of hub genes of GSE152418 dataset. (C) Bar chart of the log expression values of 52 common hub-genes in the GSE152418 dataset.
Table 4.
Summary of the identified 52 genes in case of GSE152418 and CRA002390 datasets
| GSE152418 | CRA002390 | GSE152418 | CRA002390 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene name | Log2(FC) | Adj. P-value | Log2(FC) | Adj. P-value | Gene Name | Log2(FC) | Adj. P-value | Log2(FC) | Adj. P-value |
| PLK1 | 3.883 | 7.03E-56 | 1.99 | 0.000146 | KIF11 | 2.31 | 8.36E-26 | 2 | 3.24E-08 |
| AURKB | 3.22 | 1.57E-54 | 1.9 | 0.00184 | NEK2 | 2.841 | 2.21E-25 | 1.61 | 0.000793 |
| CCNA2 | 3.759 | 1.57E-54 | 1.98 | 7.74E-08 | MCM2 | 1.772 | 4.33E-25 | 1.43 | 0.00632 |
| UBE2C | 3.571 | 4.50E-54 | 3.02 | 4.31E-05 | CDCA8 | 1.874 | 1.02E-23 | 1.48 | 0.00108 |
| FOXM1 | 3.084 | 5.82E-53 | 1.35 | 0.0104 | TTK | 2.522 | 8.83E-23 | 2.3 | 9.53E-05 |
| CDC20 | 3.663 | 3.21E-46 | 3.65 | 2.91E-12 | ORC1 | 2.579 | 1.06E-21 | 1.23 | 0.00494 |
| BIRC5 | 3.849 | 6.59E-44 | 3.56 | 2.48E-12 | EXO1 | 2.687 | 1.71E-21 | 2.57 | 2.58E-07 |
| CDT1 | 2.791 | 1.21E-42 | 1.7 | 0.0116 | CDK1 | 2.736 | 6.64E-21 | 1.86 | 0.00141 |
| DTL | 3.164 | 8.78E-42 | 3.18 | 3.19E-05 | NME1 | 1.207 | 6.67E-20 | 1.29 | 0.00863 |
| BUB1 | 3.125 | 1.12E-38 | 2.96 | 1.97E-11 | MCM4 | 1.638 | 2.22E-17 | 1.29 | 0.0173 |
| ZWINT | 2.375 | 3.09E-38 | 2.49 | 7.28E-05 | CENPU | 1.955 | 1.25E-15 | 1.57 | 3.28E-05 |
| GINS2 | 2.812 | 9.82E-37 | 1.74 | 0.011 | KIF23 | 1.797 | 5.57E-15 | 2.47 | 5.05E-11 |
| CCNB2 | 3.433 | 2.69E-36 | 2.89 | 1.43E-09 | AURKA | 1.396 | 5.91E-15 | 2.02 | 2.43E-05 |
| CLSPN | 2.652 | 5.78E-35 | 1.42 | 0.00244 | PTTG1 | 1.577 | 7.69E-14 | 1.04 | 0.0383 |
| TYMS | 2.98 | 4.83E-33 | 2.53 | 3.05E-05 | TUBG1 | 1.007 | 2.35E-13 | 1.54 | 0.000929 |
| MYBL2 | 3.442 | 1.22E-32 | 3.45 | 2.91E-12 | PPARG | 3.169 | 4.61E-11 | 5.52 | 3.68E-07 |
| CDC45 | 3.039 | 2.51E-32 | 2.95 | 3.67E-06 | CENPM | 1.212 | 8.67E-11 | 1.67 | 0.0139 |
| CDC6 | 2.948 | 2.51E-32 | 2.32 | 2.85E-06 | BRCA1 | 1.128 | 1.95E-10 | 1.08 | 0.0261 |
| TOP2A | 3.174 | 6.24E-31 | 2.58 | 5.48E-10 | WASF1 | 1.503 | 2.10E-08 | 2.44 | 0.00218 |
| CDCA5 | 2.869 | 8.03E-31 | 3.38 | 2.37E-11 | TP53I3 | 1.175 | 1.19E-06 | 2.05 | 0.00108 |
| ESPL1 | 2.805 | 1.38E-30 | 2.68 | 3.48E-07 | GRB10 | 1.636 | 2.22E-06 | 3.82 | 4.48E-05 |
| BUB1B | 2.79 | 6.29E-30 | 2.45 | 2.34E-09 | AMPH | 1.853 | 4.06E-06 | 4.1 | 0.000303 |
| KIF2C | 2.909 | 8.87E-30 | 2.01 | 4.81E-05 | FZD5 | 1.205 | 6.73E-05 | 2.57 | 0.000514 |
| CCNB1 | 2.292 | 4.88E-29 | 2.06 | 3.27E-07 | ACVRL1 | 1.38 | 0.00033 | 3.89 | 0.000105 |
| CDC25A | 3.101 | 1.13E-28 | 4.05 | 4.44E-14 | WNT7A | −1.036 | 0.002901 | −2.27 | 0.0445 |
| NUF2 | 2.131 | 4.41E-26 | 1.55 | 9.15E-05 | FLT3 | 1.33 | 0.003703 | 4.7 | 4.36E-05 |
Performance evaluation of the hub genes with a classification algorithm
To investigate the validity of the identified hub DEGs in two different analyses (i.e.: MMhub and PPIhub), the five popular classification algorithms were executed in this study. The performance measures have been computed based on the datasets with MMhub and PPIhub genes, respectively. We executed these calculations 20 times using 5-fold cross-validation and the average performance measurement values were computed and summarized in Table 3. The boxplot of the five machine learning classifiers based on test accuracies and AUC were presented in Figure 4. In Table 3 and Figure 4, we observed that the SVM provides greater accuracy of 0.996 than the other four classifiers (RF 0.955, PLDA 0.821, NBLDA 0.956 and voomDLDA 0.988) for the dataset with MMhub genes. We also observed that the voomDLDA provides greater accuracy of 0.999 than the other four classifiers (SVM 0.986, RF 0.981, PLDA 0.768 and NBLDA 0.976) for the dataset with PPIhub genes.
Table 3.
Classification performance for the COVID-19 data with hub genes
| Classifier | Data with MMhub genes | Data with PPIhub genes | ||||||
|---|---|---|---|---|---|---|---|---|
| Accuracy | AUC | Sensitivity | Specificity | Accuracy | AUC | Sensitivity | Specificity | |
| SVM | 0.996 | 0.997 | 0.996 | 0.998 | 0.986 | 0.989 | 0.996 | 0.981 |
| RF | 0.955 | 0.959 | 0.923 | 0.994 | 0.981 | 0.983 | 0.973 | 0.992 |
| PLDA | 0.821 | 0.848 | 0.902 | 0.794 | 0.768 | 0.815 | 0.930 | 0.700 |
| NBLDA | 0.956 | 0.961 | 0.922 | 0.999 | 0.976 | 0.978 | 0.957 | 0.999 |
| voomDLDA | 0.988 | 0.990 | 0.980 | 0.999 | 0.999 | 0.999 | 0.997 | 0.999 |
Figure 4.
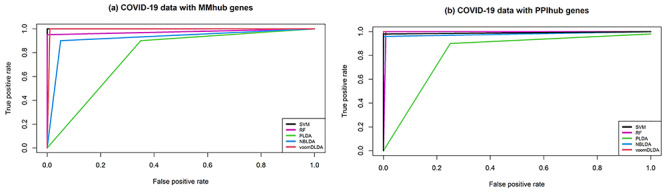
Receiver operating curve (ROC) plot of the five classifier performance based on (A) accuracies, (B) AUC.
PPI network analysis with identified common genes
The PPI networks for the 52 common genes were constructed via the STRING in Cytoscape. Figure 6A presented the network interaction among these genes and identified 10 hub genes (PLK1, AURKB, AURKA, CDK1, CDC20, KIF11, CCNB1, KIF2C, DTL and CDC6) based on a higher degree of connectivity.
Figure 6.
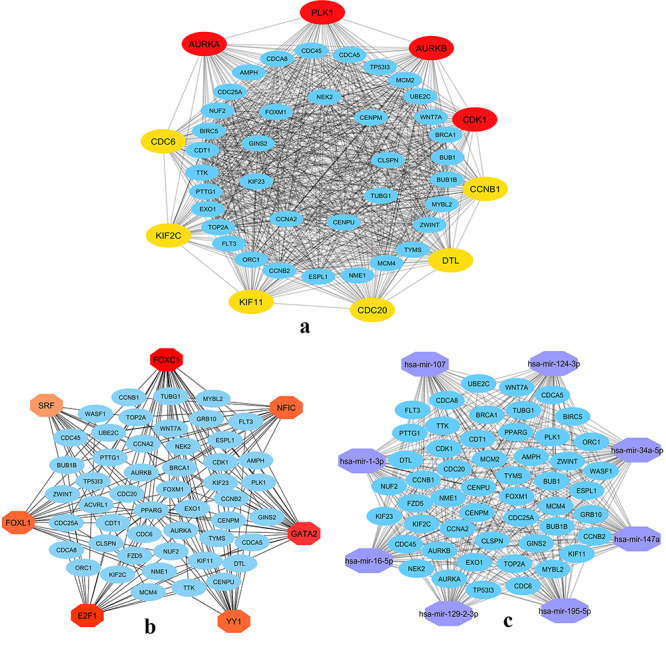
Network construction. (A) PPI network of the 52 common hub genes of COVID-19 data, (B) TFs-Gene interaction network of the 52 common hub genes, (C) gene-miRNAs interaction for the common hub genes of COVID-19.
Transcriptional regulators of identified common genes
We identified ‘FOXC1’, ‘GATA2’, ‘YY1’, ‘E2F1’, ‘NFIC’, ‘FOXL1’ and ‘SRF’ hub TFs from the TFs-gene interaction network (Figure 6B). The significant hub miRNAs were detected from the miRNAs-gene interaction network namely, ‘mir-16-5p’, ‘mir-124-3p’, ‘mir-34a-5p’, ‘mir-147a’, ‘mir-1-3p’, ‘mir-129-2-3p’, ‘mir-107’ and ‘mir-195-5p’ (Figure 6C).
Drug repositioning based on drug–gene overrepresentation analysis
We have identified 50 candidate drugs by reversal gene signature-based approach with q-value<0.05. Among them, the top 20 drugs have been summarized in Table 5. According to our analysis, ‘amsacrine’, ‘BRD-K68548958’, ‘naproxol’, ‘palbociclib’ and ‘teniposide’ were the top significantly identified drugs among others. These candidate drugs or components may be used for therapeutic applications in COVID-19.
Table 5.
Candidate drugs (top twenty) identified from gene–drug interaction enrichment analysis
| Drug | Similarity score | P-value | q-value | z-score | Combined score |
|---|---|---|---|---|---|
| Amsacrine | −0.7143 | 1.23E-48 | 1.32E-44 | 1.69 | −80.79 |
| BRD-K68548958 | −0.7143 | 5.98E-50 | 1.28E-45 | 1.82 | −89.76 |
| Naproxol | −0.6939 | 8.02E-47 | 5.72E-43 | 1.69 | −77.84 |
| Palbociclib | −0.6939 | 3.47E-46 | 1.65E-42 | 1.61 | −72.96 |
| SIB-1893 | −0.6939 | 7.00E-47 | 5.72E-43 | 1.67 | −77.09 |
| ZK-164015 | −0.6939 | 5.46E-46 | 2.12E-42 | 1.67 | −75.62 |
| Tanespimycin | −0.6939 | 4.22E-46 | 1.81E-42 | 1.63 | −74.15 |
| Emodic-acid | −0.6939 | 9.83E-47 | 6.01E-43 | 1.7 | −78 |
| BRD-K29506255 | −0.6735 | 4.27E-45 | 1.52E-41 | 1.78 | −78.99 |
| Teniposide | −0.6531 | 8.98E-43 | 2.13E-39 | 1.68 | −70.64 |
| Diphenyleneiodonium | −0.6531 | 3.33E-42 | 7.14E-39 | 1.64 | −67.86 |
| Homosalate | −0.6531 | 2.30E-42 | 5.19E-39 | 1.68 | −70.07 |
| SIB-1893 | −0.6531 | 6.11E-43 | 1.54E-39 | 1.68 | −70.75 |
| Ingenol | −0.6327 | 3.52E-40 | 5.79E-37 | 1.75 | −69.13 |
| FCCP | −0.6327 | 5.36E-41 | 9.98E-38 | 1.83 | −73.81 |
| Tremulacin | −0.6122 | 3.84E-39 | 4.98E-36 | 1.74 | −66.81 |
| BRD-K30836161 | −0.6122 | 4.89E-39 | 6.16E-36 | 1.76 | −67.42 |
| Idarubicin | −0.6122 | 2.67E-38 | 3.09E-35 | 1.59 | −59.71 |
| Wortmannin | −0.6122 | 9.75E-40 | 1.49E-36 | 1.84 | −71.72 |
| Devazepide | −0.6122 | 4.20E-38 | 4.73E-35 | 1.64 | −61.18 |
Discussion
The COVID-19 is affecting severely millions of people and taking thousands of precious lives every day over the globe due to its pandemic behavior. Though there are several candidate drugs and vaccines that were studied and proposed to treat the disease, no solid cure is available yet. The current study employed a gene coexpression network analysis to decode the critical genes and pathways of COVID-19. We identified 1520 and 1733 DEGs for the GSE152418 and CRA002390 RNA-sequencing PBMC based datasets, respectively. The four key modules were identified in GSE152418 COVID-19 data via WGCNA and module preservation analysis. The GO and pathway enrichment analysis were conducted for these key modules. We checked the validity of these identified hub genes (MMhub and PPIhub) with machine learning classifiers. We found 52 common genes in MMhub, PPIhub and the DEGs of CRA002390. The PPI networks, transcriptional regulatory networks of the common hub genes were constructed. We found 10 hub genes from the PPI of the identified common genes, and those were considered as the final candidate molecular blood signatures of COVID-19. These findings may provide new insights into the COVID-19 pathogenesis.
Our employed approach is significantly different from previous bioinformatics reports in COVID-19 research [5, 9, 12, 13, 41, 42] which relied on the identification of genes by differential analysis. However, in order to provide systems biology insights, we have implemented methodologies particularly gene coexpression module analysis that provides key gene modules rather than finding DEGs. Firstly it clustered genes in a specific condition followed by detection of hub genes which are highly connected nodes in each module network. There is a possibility of detecting some inappropriate genes as the hub if we use only one approach. To detect the hub genes from each module more accurately, we performed joint-analysis which revealed common hub genes by module membership significance analysis and PPI analysis, which is a robust approach in selecting hub genes in COVID-19. Our analysis focused on PBMC gene expression analysis, to obtain further insights regarding the potential utilization of the identified hubs in diagnostic development for COVID-19, we decided to perform classification by widely used machine learning classifiers. The performance of state-of-the-art methods was evaluated the classification performances in which obtained a higher score in accuracy, AUC, specificity and sensitivity.
To elucidate the roles of the identified DEGs, the GO and KEGG pathway analysis were executed in this study. Among the identified GO terms, the immune response, response to cytokines, cytokine-mediated signaling and response to external stimuli play crucial roles in restricting viral infections. Among the identified pathways of four modules the cytokine–cytokine receptor interaction, the IL-17 signaling pathway was highly enriched in COVID-19. Viral infectious diseases such as herpes simplex virus 1 infection, human papillomavirus infection and viral carcinogenesis pathways were detected. Human papillomavirus infection pathways are significantly related to colorectal cancer. An autoimmune disease like rheumatoid arthritis and type 1 diabetes mellitus were also expressively enriched. These pathways show massive significance to drug repurposing chances in COVID-19.
The application of machine learning classifiers have been widely used in different bioinformatics tasks [43–45]. We executed the machine learning classification algorithms based on MMhub and PPIhub genes data respectively to check their validity. We observed the satisfactory sample classification performance (accuracy >0.90; except PLDA) between COVID-19 and health control samples for both datasets (MMhub, PPIhub). The results indicate the validation of our identified hub genes through module membership and PPI networks.
We identified PLK1, AURKB, AURKA, CDK1, CDC20, KIF11, CCNB1, KIF2C, DTL and CDC6 hub genes representing that, they had a high association with clinical trait along with vital BPs and some of them were detected in COVID-19. Among them, the Polo-like kinase 1 (PLK1) gene was detected as down-regulates in Parainfluenza Virus 5 [46]. The Aurora Kinase B (AURKB) and Aurora Kinase A (AURKA) were found SARS-CoV-2 as DEGs in Caco-2 cells [47]. The Cyclin-Dependent Kinase 1 (CDK1) genes interact with the thrombocytopenia syndrome virus which initiates the cells into the M phase [48]. The Cell Division Cycle 20 (CDC20) and CDK1 were also found as potential biomarkers for hepatocellular carcinoma [49]. The identified hub TFs and miRNAs are also significantly associated with viral infectious diseases.
Finally, we detected the candidate drugs using the reversal gene signature-based approach [5, 9]. Among them, Naproxen is a nonsteroidal anti-inflammatory drug that was studied to use for the treatment of critically COVID-19 infected patients and to limit the spread of the virus [50, 51]. The drug Teniposide was suggested to evaluate the treatment of SARS-CoV-2 infected patients [52]. We proposed to send these candidate drugs for biological and clinical experimentation for the possible use in COVID-19 treatment.
Our analysis focused on PBMC gene expression analysis, to obtain further insights regarding the potential utilization of the identified hubs in diagnostic development for COVID-19, we decided to perform classification by widely used machine learning classifiers. Our analysis showed a good level of specificity in classification performances.
However, several limitations of the study should be noted as findings of this study relied on bioinformatics analysis without functional studies in wet-lab, thus caution should be taken in interpreting the results. Moreover, the transcriptomic analysis and candidate drugs were identified by reversal of PBMC gene expression in COVID-19 but the primary affected organ by SARS-CoV-2 is lung tissues, thus further research is now proposed to explore biological insights in COVID-19.
Conclusions
The present research aimed to identify key genes and molecular pathways altered in response to SARS-CoV-2 in blood cells compared to normal blood cells. We detected four key modules through module preservation analysis. The 52 common genes were identified from resultant 422 and 240 hub genes based on module membership statistics and PPI networks and from 1733 detected DEGs of CRA002390. The 10 hub genes (PLK1, AURKB, AURKA, CDK1, CDC20, KIF11, CCNB1, KIF2C, DTL and CDC6) were identified from the PPI networks of these 52 genes. The TFs (FOXC1, GATA2, YY1, E2F1, NFIC, FOXL1 and SRF) were also found as potential regulators of the hub genes. The naproxol, teniposide, amsacrine, BRD-K68548958, palbociclib were identified as the top-scored repurposed drugs for COVID-19 pathogenesis. The identified drugs should be judged with wet-lab experiments before clinical studies. Our results may provide novel insights into the pathogenesis of COVID-19 and the potential molecular targets for novel interventional approaches.
Key Points
COVID-19 pandemic has emerged as a massive threat to humankind limited by the unavailability of effective drugs.
This study has performed a comprehensive clinical bioinformatics and systems biology analysis of all available SARS-CoV-2 infected peripheral blood mononuclear cell (PBMC) transcriptomic datasets to identify gene modules by gene coexpression analysis.
A robust four key gene modules and hub gene signature were detected based on gene module membership statistics and protein–protein interaction networks and machine learning methods.
Functional annotation by enrichment analysis of the genes of these modules demonstrated immune and inflammatory response biological processes enriched by the gene signature.
Several potential candidate drugs based on the reversal of transcriptomic signature were also detected that may be effective treatment candidate for COVID-19.
Acknowledgments
Not applicable.
Md. Rabiul Auwul received his MSc in 2015 and BSc (honors) in 2014 in Statistics from Begum Rokeya University, Rangpur, Bangladesh. He is currently studying toward a PhD degree in Statistics at Guangzhou University, China. His research interests include data mining, robust statistics, supervised and unsupervised machine learning, system biology, transcriptomics study and bioinformatics.
Md Rezanur Rahman is a lecturer (on study leave) at Khwaja Yunus Ali University, Bangladesh. He is a Biotechnologist and Bioinformatician by training. He received MSc and a BSc in Biotechnology and Genetic Engineering with outstanding academic results holding the First Position in the Faculty in both degrees from Islamic University, Bangladesh. For the highest CGPA in the Faculty, he received the most prestigious award ‘Prime Minister Gold Medal 2017’ from Prime Minister of Bangladesh. He has published many research articles in internationally reputed journals. His research focuses on understanding the molecular mechanism of complex diseases utilizing systems biology and bioinformatics.
Dr Esra Gov received her Bachelor’s degree in Bioengineering from Ege University. Dr Gov received a PhD in Bioengineering from Marmara University, Istanbul. She is specialized in systems biology, bioinformatics and computational biology. Her interest lies in mostly biomarker identification of diseases via holistic systems medicine perspective. Dr GOV is now an assistant professor at Bioengineering Department at Adana Alparslan Turkes Science and Technology University.
Dr Md. Shahjaman has been working as an associate professor, Department of Statistics, Begum Rokeya University, Rangpur-5400, Bangladesh since 2010. He completed his PhD in Bioinformatics in 2018 from the University of Rajshahi, Bangladesh. He completed his BSc (honors) and MScin Statistics from the same institution. His research interest lies in theoretical and computational statistics, robust statistics, supervised and unsupervised machine learning, statistical inference, data science and deep learning with their application to big biological datasets such as biomarker identification from transcriptomics, metabolomics, and other OMICS datasets
Dr Mohammad Ali Moni is a senior research fellow and conjoint lecturer at WHO Collaborating Centre on eHealth and UNSW Digital Health of the University of New South Wales, Australia. Before joining the UNSW, he was a vice-chancellor fellow of the University of Sydney, Australia. He received his PhD in clinical Bioinformatics and Machine Learning from the University of Cambridge, UK. He has published over 70 peer-reviewed top tire journal articles during the last 5 years. His research interests encompass artificial intelligence, machine learning, data science, medical imaging and clinical bioinformatics.
Contributor Information
Md Rabiul Auwul, School of Economics and Statistics, Guangzhou University, Guangzhou 510006, China.
Md Rezanur Rahman, Department of Biochemistry and Biotechnology, School of Biomedical Science, Khwaja Yunus Ali University, Sirajgonj-6751, Bangladesh.
Esra Gov, Department of Bioengineering, Adana Alparslan Turkes Science and Technology University, Adana-01250, Turkey.
Md Shahjaman, Department of Statistics, Begum Rokeya University, Rangpur-5400, Bangladesh.
Mohammad Ali Moni, WHO Collaborating Centre on eHealth, UNSW Digital Health, School of Public Health and Community Medicine, Faculty of Medicine, UNSW Sydney, Australia.
Funding
No funding received for this study.
Data availability
All data utilized in this manuscript are available online from their respective database.
References
- 1. Cucinotta D, Vanelli M. WHO declares COVID-19 a pandemic. Acta Biomed 2020;90:157–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 2020;395:507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahamad MM, Aktar S, Rashed-Al-Mahfuz M, et al. A machine learning model to identify early stage symptoms of SARS-Cov-2 infected patients. Expert systems with applications 2020;160:113661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blanco-Melo D, Nilsson-Payant B, Liu W-C, et al. SARS-CoV-2 launches a unique transcriptional signature from in vitro, ex vivo, and in vivo systems. bioRxiv 2020. March 24, 2020. doi: 10.1101/2020.03.24.004655 Arxiv biorxiv;2020.03.24.004655v1, preprint: not peer reviewed. [DOI] [Google Scholar]
- 5. Islam T, Rahman MR, Aydin B, et al. Integrative transcriptomics analysis of lung epithelial cells and identification of repurposable drug candidates for COVID-19. Eur J Pharmacol 2020;887:173594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xiong Y, Liu Y, Cao L, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect 2020;9:761–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arunachalam PS, Wimmers F, Mok CKP, et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science (80-) 2020;369:1210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ong EZ, Fu Y, Chan Z, et al. A dynamic immune response shapes COVID-19 progression. Cell Host Microbe 2020;27:879–882.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fagone P, Ciurleo R, Lombardo SD, et al. Transcriptional landscape of SARS-CoV-2 infection dismantles pathogenic pathways activated by the virus, proposes unique sex-specific differences and predicts tailored therapeutic strategies. Autoimmun Rev 2020;19:102571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dolan M, Hill D, Mukherjee G, et al. Investigation of COVID-19 comorbidities reveals genes and pathways coincident with the SARS-CoV-2 viral disease. Sci Rep 2020;10:20848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Satu MS, Khan MI, Rahman MR, et al. Diseasome and comorbidities complexities of SARS-CoV-2 infection with common malignant diseases. Brief Bioinform 2021;22:1415–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moni MA, Quinn JMW, Sinmaz N, et al. Gene expression profiling of SARS-CoV-2 infections reveal distinct primary lung cell and systemic immune infection responses that identify pathways relevant in COVID-19 disease. Brief Bioinform 2020;22:1324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nashiry A, Sarmin Sumi S, Islam S, et al. Bioinformatics and system biology approach to identify the influences of COVID-19 on cardiovascular and hypertensive comorbidities. Brief Bioinform 2021;22:1387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jin X, Li J, Li W, et al. Weighted gene co-expression network analysis reveals specific modules and biomarkers in Parkinson ’ s disease. Neurosci Lett 2020;728:134950. [DOI] [PubMed] [Google Scholar]
- 16. Iu R, Zhang W, Liu Z, et al. Associating transcriptional modules with colon cancer survival through weighted gene co-expression network analysis. BMC Genomics 2017;18:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feng T, Li K, Zheng P, et al. Weighted gene coexpression network analysis identified MicroRNA coexpression modules and related pathways in type 2 diabetes mellitus. Oxid Med Cell Longev 2019;2019:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 2013;41:991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Love MI, Huber W, Anders S. Oderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Geneome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Langfelder P, Luo R, Oldham MC, et al. Is my network module preserved and reproducible? PLoS Comput Biol 2011; 7:e1001057 e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rahman MH, Rana HK, Peng S, et al. Bioinformatics and machine learning methodologies to identify the effects of central nervous system disorders on glioblastoma progression. Brief Bioinform 2021;bbaa365. [DOI] [PubMed] [Google Scholar]
- 23. Rahman MR, Islam T, Nicoletti F, et al. Identification of common pathogenetic processes between schizophrenia and diabetes mellitus by systems biology analysis. Genes (Basel) 2021;12:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang d W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 25. Yu G, Wang L, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. Omi A J Integr Biol 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yuan L, Chen L, Qian K, et al. Co-expression network analysis identi fi ed six hub genes in association with progression and prognosis in human clear cell renal cell carcinoma (ccRCC). Genomics Data 2017;14:132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boser B, Guyon I, Vapnik V. A training algorithm for optimal margin classes. Proc. 5th Annu. Work. Comput. Learn. theory 1992; 144–52
- 28. Ho TK. Random decision forests. Proc. Int. Conf. Doc. Anal. Recognition, ICDAR 1995; 1:278–82 [Google Scholar]
- 29. Witten DM. Classification and clustering of sequencing data using a Poisson model. Annals of Applied Statistics 2011;5:2493–518. [Google Scholar]
- 30. Dong K, Zhao H, Tong T, et al. NBLDA: negative binomial linear discriminant analysis for RNA-Seq data. BMC Bioinf 2016;17:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dudoit S, Fridlyand J, Speed TP. Comparison of discrimination methods for the classification of tumors using gene expression data. J Am Stat Assoc 2002;97:77–87. [Google Scholar]
- 32. Goksuluk D, Zararsiz G, Korkmaz S, et al. MLSeq: machine learning interface for RNA-sequencing data. Comput Methods Programs Biomed 2019;175:223–31. [DOI] [PubMed] [Google Scholar]
- 33. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 2017;45:D362–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smoot ME, Ono K, Ruscheinski J, et al. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 2011;27:431–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khan A, Fornes O, Stigliani A, et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 2018;46:D260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xia J, Gill EE, Hancock REW. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat Protoc 2015;10:823–44. [DOI] [PubMed] [Google Scholar]
- 37. Sethupathy P, Corda B, Hatzigeorgiou AG. TarBase: a comprehensive database of experimentally supported animal microRNA targets. RNA 2006;12:192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hsu S-D, Lin F-M, Wu W-Y, et al. miRTarBase: a database curates experimentally validated microRNA–target interactions. Nucleic Acids Res 2011;39:D163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rahman MR, Islam T, Gov E, et al. Identification of prognostic biomarker signatures and candidate drugs in colorectal cancer: insights from systems biology analysis. Medicina (Kaunas) 2019;55:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Z, Lachmann A, Keenan AB, et al. L1000FWD: fireworks visualization of drug-induced transcriptomic signatures. Bioinformatics 2018;34:2150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li X, Yu J, Zhang Z, et al. Network bioinformatics analysis provides insight into drug repurposing for COVID-2019 Preprints. 2019. [DOI] [PMC free article] [PubMed]
- 42. Feng Z, Chen M, Liang T, et al. Virus-CKB: an integrated bioinformatics platform and analysis resource for COVID-19 research. Brief Bioinform 2020;22:882–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hasan MM, Schaduangrat N, Basith S, et al. HLPpred-fuse: improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 2020;36:3350–6. [DOI] [PubMed] [Google Scholar]
- 44. Hasan MM, Khatun MS, Kurata H. iLBE for computational identification of linear B-cell epitopes by integrating sequence and evolutionary features. Genomics Proteomics Bioinformatics 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hasan MM, Basith S, Khatun MS, et al. Meta-i6mA: an interspecies predictor for identifying DNA N6-methyladenine sites of plant genomes by exploiting informative features in an integrative machine-learning framework. Brief Bioinform 2020;bbaa202. [DOI] [PubMed] [Google Scholar]
- 46. Sun D, Luthra P, Li Z, et al. PLK1 down-regulates parainfluenza virus 5 gene expression. PLoS One 2009;5:e1000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bock J-O, Ortea I. Re-analysis of SARS-CoV-2-infected host cell proteomics time-course data by impact pathway analysis and network analysis: a potential link with inflammatory response. Aging (Albany NY) 2020;12:11277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Su M, Chen Y, Qi S, et al. A mini-review on cell cycle regulation of coronavirus infection. Front Vet Sci 2020;7:943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang W-X, Pan Y-Y, You C-G. CDK1, CCNB1, CDC20, BUB1, MAD2L1, MCM3, BUB1B, MCM2, and RFC4 may be potential therapeutic targets for hepatocellular carcinoma using integrated bioinformatic analysis. Biomed Res Int 2019;2019:16 pp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Efficacy of addition of naproxen in the treatment of critically ill patients hospitalized for COVID-19 infection (ENACOVID). 2020.
- 51. Oany AR, Mia M, Pervin T, et al. Design of novel viral attachment inhibitors of the spike glycoprotein (S) of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) through virtual screening and dynamics. International journal of antimicrobial agents 2020;56:106177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bharadwaj S, Azhar EI, Amjad Kamal M, et al. SARS-CoV-2 Mpro inhibitors: identification of anti-SARS-CoV-2 Mpro compounds from FDA approved drugs. J Biomol Struct Dyn 2020;38:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data utilized in this manuscript are available online from their respective database.