

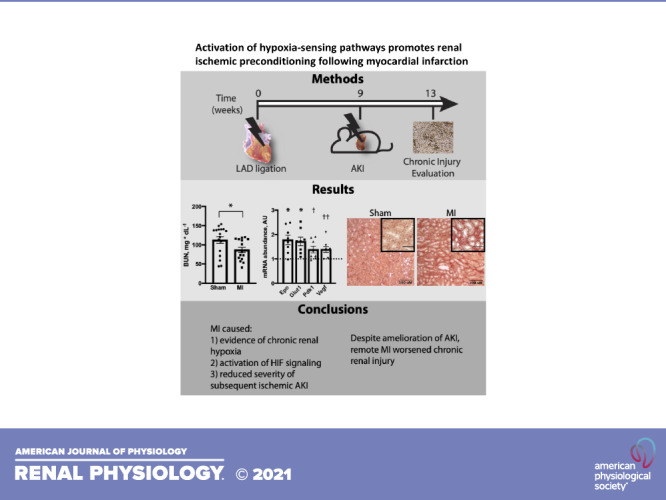
Keywords: acute kidney injury, cardiorenal syndrome, chronic kideny disease, hypoxia-inducible factor, myocardial infarction
Abstract
Ischemic heart disease is the leading cause of death worldwide and is frequently comorbid with chronic kidney disease. Physiological communication is known to occur between the heart and the kidney. Although primary dysfunction in either organ can induce dysfunction in the other, a clinical entity known as cardiorenal syndrome, mechanistic details are lacking. Here, we used a model of experimental myocardial infarction (MI) to test effects of chronic cardiac ischemia on acute and chronic kidney injury. Surprisingly, chronic cardiac damage protected animals from subsequent acute ischemic renal injury, an effect that was accompanied by evidence of chronic kidney hypoxia. The protection observed post-MI was similar to protection observed in a separate group of healthy animals housed in ambient hypoxic conditions prior to kidney injury, suggesting a common mechanism. There was evidence that chronic cardiac injury activates renal hypoxia-sensing pathways. Increased renal abundance of several glycolytic enzymes following MI suggested that a shift toward glycolysis may confer renal ischemic preconditioning. In contrast, effects on chronic renal injury followed a different pattern, with post-MI animals displaying worsened chronic renal injury and fibrosis. These data show that although chronic cardiac injury following MI protected against acute kidney injury via activation of hypoxia-sensing pathways, it worsened chronic kidney injury. The results further our understanding of cardiorenal signaling mechanisms and have implications for the treatment of heart failure patients with associated renal disease.
NEW & NOTEWORTHY Experimental myocardial infarction (MI) protects from subsequent ischemic acute kidney injury but worsens chronic kidney injury. Observed protection from ischemic acute kidney injury after MI was accompanied by chronic kidney hypoxia and increased renal abundance of hypoxia-inducible transcripts. These data support the idea that MI confers protection from renal ischemic injury via chronic renal hypoxia and activation of downstream hypoxia-inducible signaling pathways.
INTRODUCTION
Ischemic heart disease is the leading cause of death worldwide (1). Patients with chronic cardiac dysfunction often have multiple comorbidities, and kidney disease is among the most common and most serious (2). The clinical entity wherein cardiac dysfunction induces renal dysfunction is known as cardiorenal syndrome (3). With the rising prevalence of both cardiac and renal disease, clinicians encounter the cardiorenal syndrome more frequently in both inpatient and outpatient settings. Patients with this dual-organ dysfunction consume substantial financial and medical resources and have a high mortality risk (4–6). Despite the large burden cardiorenal syndrome places on medical systems, a clear understanding of the physiological communication between the heart and the kidney eludes us. Limited understanding of the basic physiology hinders our ability to treat these patients when this interorgan communication fails in disease states.
Physiological explanations for cardiorenal syndrome often cite hypoperfusion from reduced cardiac output and/or venous congestion from fluid overload as overarching mechanisms driving renal dysfunction (7). Although these are certainly contributing factors, they may more accurately reflect the phenotype observed in the setting of acute decompensated heart failure leading to acute kidney injury (AKI) rather than the subtler pathophysiology seen chronically in patients with heart failure. Indeed, evidence from animal models clearly supports a role for acute cardiac dysfunction affecting renal function. It was recently reported that a mouse model of cardiac arrest causes acute renal failure and ultimately leads to chronic kidney injury weeks later (8); however, factors driving renal injury in the setting of chronic cardiac dysfunction are likely different and remain poorly defined. Furthermore, a clear mechanistic model of renal dysfunction at the cellular and molecular levels of cardiorenal syndrome continues to elude us.
To gain a better understanding of cardiorenal signaling, we used a model of myocardial infarction to investigate effects of chronic heart damage on both acute and chronic renal injury. Unexpectedly, we found that chronic cardiac dysfunction conferred protection from subsequent ischemic AKI. Protection was accompanied by evidence of preischemic renal hypoxia and increased abundance of several important glycolytic enzymes, suggesting that a shift to anaerobic glycolysis may underlie ischemic preconditioning. Despite amelioration of AKI, transition to chronic renal injury and fibrosis was worse in the setting of cardiac injury, demonstrating that even modest cardiac injury drives renal dysfunction in the chronic setting.
METHODS
Animals
All animals used were male wild-type 8- to 10-wk-old C57Bl/6 mice purchased from The Jackson Laboratory (Bar Harbor, ME).
Ethical Approval
All animal experiments were performed in accordance with the guidelines and with the approval of the Institutional Animal Care and Use Committee of Vanderbilt University Medical Center.
Experimental Myocardial Infarction
Experimental mouse MI was induced by permanent ligation of the left anterior descending (LAD) coronary artery. Briefly, after being anesthetized with ketamine and xylazine (80/10 mg/kg ip), intubated, and ventilated with a positive-pressure ventilator (Harvard Apparatus, Holliston, MA), the heart was exposed via a left thoracotomy between the third and fourth ribs. The pericardium was opened, and the LAD artery was ligated using 8-0 silk suture (AD Surgical, Sunnyvale, CA). Occlusion of the LAD artery was visually confirmed by rapid myocardial blanching, as well as ST segment elevation on electrocardiogram (ECG) recorded by the PowerLab system (ADInstruments). Then, the ribcage and muscles were closed layer by layer with 7-0 absorbable suture (AD Surgical). The skin was closed with 6-0 silk suture (AD Surgical). Sham-operated mice underwent the same procedure without the LAD artery ligation. Immediately after surgery, mice were administered buprenorphine (0.1 mg/kg sc) for postsurgical analgesia and subsequently every 8–12 h for 72 h.
Renal Ischemia-Reperfusion Injury
Ischemia-reperfusion (IR) AKI was induced as described previously (9). Briefly, after being anesthetized with ketamine and xylazine (100/10 mg/kg ip), animals underwent right uninephrectomy followed immediately by left renal pedicle clamping for 30 min. Mice were warmed on a warming pad during the surgery, and reperfusion of the left kidney was confirmed after clamp release.
Hypoxia Model
Animals were exposed to either room air in our standard animal facility or to 10% oxygen in a hypoxia chamber for 10 days. Animals underwent IR injury as aforementioned immediately after exposure.
Echocardiography
Mouse cardiac function was evaluated with transthoracic echocardiography in the conscious state using a Vevo2100 Imaging System (VisualSonics). Prior to the acquisition of echo images, chest hair was removed by applying hair removal cream. For echocardiographic examination, the mice were gently held by their nape in the palm of one hand with the tail held between the last two fingers. Prewarmed echo transmission gel was applied to the hairless chest. Parasternal long- and short-axis view at the papillary muscle level and two-dimensional guided M-mode images were recorded. Qualitative and quantitative measurements were made offline using analytical software (VisualSonics). Ejection fraction and fractional shortening were measured in three consecutive beats according to the guidelines and standards of American Society of Echocardiography leading edge method (10).
Transdermal Glomerular Filtration Rate Measurement
Transdermal measurement of FITC-sinistrin clearance was performed to determine glomerular filtration rate (GFR) in conscious mice, as previously described (11). The FITC-sinistrin half-life was calculated using a three-compartment model with linear fit using MPD Studio software (MediBeacon, Mannheim, Germany). The FITC-sinistrin half-life (in min) was converted to GFR (in μL/min), as previously described (12), with correction for individual mouse body weight.
Blood Urea Nitrogen Measurement
Blood was collected from the tail vein in heparinized tubes on the indicated days post-kidney injury. Blood was centrifuged for 5 min at 10,000 g, and the plasma layer was removed and stored at −20°C until measurements were performed. Plasma blood urea nitrogen (BUN) was subsequently measured using a urea assay kit (BioAssay Systems, Hayward, CA).
Real-Time PCR
Total tissue RNA from kidneys was isolated using TRIzol reagent (Invitrogen). SuperScript IV First-Strand Synthesis System kit (Invitrogen) was used to synthesize cDNA from total RNA from each sample. Quantitative real-time PCR was performed using TaqMan real-time PCR (7900HT; Applied Biosystems). The master mix and all gene probes were also purchased from Applied Biosystems. The probes used in the experiments included S18 (Mm02601778), erythropoietin (Epo; Mm01202755), Slc2a1 (Mm00441480), pyruvate dehydrogenase kinase (Pdk1; Mm00554300), vascular endothelial growth factor (Vegf; Mm00437306), hexokinase 1 (Hk1; Mm00439344), hexokinase 2 (Hk2; Mm00443385), pyruvate kinase (Pkm1; Mm00834102), phosphoglucomutase (Pgm1; Mm00804141), and glyceraldehyde 3-phosphate dehydrogenase (Gapdh; Mm99999915).
Pimonidazole Staining
Staining was carried out as previously described (13) using a Hypoxyprobe kit (Burlington, MA). Briefly, 8–10 wk after MI, mice received 60 mg/kg pimonidazole (dissolved in 0.9% saline) via an intraperitoneal injection. Sixty minutes after injection, animals were euthanized, and kidneys were removed and immediately placed in fixative containing 3.7% formaldehyde, 10 mM sodium m-periodate, 40 mM phosphate buffer, and 1% acetic acid. Subsequent processing and immunohistochemical detection were performed using the manufacturer-supplied anti-pimonidazole monoclonal antibody (1:50) according to the following immunohistochemistry protocol. The M.O.M. immunodetection kit from Vector Laboratories was used to reduce endogenous mouse IgG staining.
Immunohistochemistry
Following euthanasia, kidneys were removed and incubated at room temperature overnight in fixative containing 3.7% formaldehyde, 10 mM sodium m-periodate, 40 mM phosphate buffer, and 1% acetic acid. The fixed kidney was subsequently dehydrated through a graded series of ethanol, embedded in paraffin, sectioned (4 µm), and mounted on glass slides. Immunostaining was carried out as reported previously (14). Sections from both sham and MI animals were mounted on a single slide to minimize slide-to-slide and temporal variation. Three sections per animal were stained to ensure minimal section-to-section variation. Subsequent images were acquired at the same time under the same exposure conditions. Quantification was performed using ImageJ software. To do this, threshold parameters were set to differentiate immunopositive versus negative signal. These parameters were then applied to all sections being quantified, and the software calculated the percent area that was determined to be positive for each image. At least four images were quantified per animal. Antibodies used include mouse anti-α-smooth muscle actin (α-SMA; 1:4,000 A5228, Sigma), goat anti-kidney injury molecule (KIM)-1 (used for Fig. 2; 1:50 AF1817, R&D), and rat anti-KIM-1 (used for Fig. 6; 1:50 MAB1817-100, R&D). The M.O.M. immunodetection kit (Vector Laboratories) was used to reduce endogenous mouse IgG staining in combination with the anti-α-SMA antibody. Negative controls were performed without incubation in primary antibody to ensure the absence of nonspecific staining.
Figure 2.

Effects of myocardial infarction (MI) on ischemic acute kidney injury. A–D: peak blood urea nitrogen (BUN; A), kidney injury molecule (Kim)-1 immunostaining (both low-magnification and high-magnification images included; B), weight loss (C), and mortality (D) following ischemia reperfusion (IR) in animals that had undergone either a sham or MI surgery 9 wk prior. Kim-1 immunostaining was performed on kidneys 24 h following IR. *P < 0.05 by unpaired t test. For sham and MI groups, respectively, n = 17 and n = 16 in A, n = 5 and n = 6 in B, n = 6 and n = 8 in C, and n = 9 and n = 8 in D.
Figure 6.

Effects of myocardial infarction (MI) on kidney fibrosis. A–C: picrosirius red (A), α-smooth muscle actin (α-SMA; B), and kidney injury molecule-1 (KIM-1; C) staining 4 wk after acute renal ischemia in animals that had previously undergone MI and sham-operated controls. Each panel includes a low-magnification image and a high-magnification image in the inset. Scale bar length for the inset = 50 µM. *P < 0.05 by unpaired t test. For A, n = 3 for sham and 5 for MI. For B, n = 5 for sham and n = 7 for MI. For C, n = 5 for sham and n = 8 for MI.
Picrosirius Red Staining
Staining was performed according to the protocol provided by the manufacturer (Sigma, St. Louis, MO). Quantification was performed using ImageJ software.
Statistical Analyses
All values are expressed as means ± SE. Between-group comparisons were made using two-sided Student’s t test or two-way ANOVA, as indicated in the figure legends. A P value of <0.05 was considered significant. When sample size was five or fewer per group, nonparametric analyses were performed using the Mann–Whitney test.
RESULTS
To study effects of ischemic heart disease on renal function, MI was induced by permanent LAD artery ligation. Proximal ligations were avoided in an attempt to cause cardiac injury without a congestive heart failure phenotype (Fig. 1A). When compared with control mice that underwent a sham procedure, this resulted in a small, yet statistically significant, reduction in cardiac ejection fraction and fractional shortening as assessed by conscious echocardiography (Fig. 1, B and C). Mice that underwent MI did have slightly higher presurgery body weight, but there was no detectable difference in postsurgery weight gain or urine output between the two groups (Fig. 1, D–F). In a separate group of animals, GFR was not different between the sham and MI groups when measured 3 wk after surgery (Fig. 1G).
Figure 1.

Effects of myocardial infarction (MI) on cardiac and renal function. A: animals underwent either sham or left anterior descending artery (LAD) ligation at time 0. Nine weeks later, animals in both groups underwent ischemia-reperfusion acute kidney injury (IR AKI) with contralateral uninephrectomy (UNx). Chronic effects on renal injury and fibrosis were subsequently determined at 13 wk after the initial cardiac procedure. B: cardiac ejection fraction (EF) and C: fractional shortening (FS) 1 wk after sham or MI surgery. D–G: weight gain (D and E), 24-h urine output (F), and glomerular filtration rate (GFR) (G) after mice underwent sham or MI surgery. *P < 0.05 by unpaired t test. For B and C, n = 4 for sham and n = 13 for MI. For D and E, n = 5 for sham and n = 13 for MI. For F, n = 9 for sham and n = 5 for MI. For G, n = 4 for sham and n = 5 for MI.
Using this model, we tested the hypothesis that chronic cardiac injury worsened renal function following acute ischemic kidney injury. Nine weeks after cardiac surgery, animals that had previously undergone the sham procedure or LAD ligation underwent acute IR injury via renal pedicle clamping for 30 min with contralateral uninephrectomy. Surprisingly, AKI was attenuated in animals that had previously received MI, as shown by decreased peak BUN and Kim-1 immunostaining following ischemia (Fig. 2, A and B). They also had decreased acute weight loss and mortality compared with sham-operated controls that underwent IR (Fig. 2, C and D).
These data suggested that chronic cardiac injury promoted ischemic preconditioning in the kidney. Previous reports have demonstrated that activation of hypoxia-sensing pathways induces renal ischemic preconditioning (15–17). Thus, we hypothesized that our cardiorenal preconditioning might be providing protection via similar mechanisms. To determine if our MI model caused chronic renal hypoxia, we performed pimonidazole staining on mouse kidneys 2 mo after LAD ligation. This revealed reduced renal oxygen tension in animals that underwent MI compared with sham controls (Fig. 3A and Supplemental Fig. S1; all Supplemental Material available online at https://doi.org/10.6084/m9.figshare.13519064.v1). As this was highly suggestive of activated HIF signaling, we measured mRNA levels of several HIF target genes. Indeed, mice that had undergone MI had increased transcript abundance of Epo, glucose transporter 1 (Slc2a1), and Pdk1. Vegf mRNA showed a statistical trend toward increased abundance (Fig. 3B).
Figure 3.

Evidence of renal hypoxia and hypoxia-inducible factor (HIF) activation 2 mo after myocardial infarction (MI). A: pimonidazole staining as a marker of renal hypoxia 8–10 wk after cardiac surgery, and prior to undergoing renal ischemia reperfusion (IR). B: total kidney mRNA abundance, as determined by quantitative real-time polymerase chain reaction (qPCR), of several HIF-regulated genes, including erythropoietin (Epo), glucose transporter 1 (Slc2a1), pyruvate dehydrogenase kinase (Pdk1), and vascular endothelial growth factor (Vegf) as evidence of HIF signaling. C: total kidney mRNA abundance of several glycolytic enzymes, including hexokinase 1 (Hk1), hexokinase 2 (Hk2), pyruvate kinase (Pkm1), phosphoglucomutase (Pgm1), and glyceraldehyde 3-phosphate dehydrogenase (Gapdh). The dotted line in B and C represents normalized sham control transcript abundance. *P < 0.05, †P = 0.05, and ††P = 0.07 all by unpaired t test. For A, n = 8 for sham and n = 7 for MI. For B and C, n = 8 per group.
Renal metabolic reprogramming has been shown to be an important determinant to modulate both acute and chronic renal injury (15, 16, 18). The HIF pathway is central in this process, by promoting a shift away from oxidative phosphorylation toward anaerobic glycolysis. To determine if this was occurring in the setting of chronic MI, we measured the mRNA abundance of key glycolytic enzymes 2 mo after MI, but before IR. Post-MI mRNA abundance of Hk1, Hk2, Pkm1, Pgm1, and Gapdh were all increased when compared with sham-operated controls (Fig. 3C).
These data provided evidence for chronic cardiac ischemia causing renal hypoxia and subsequent activation of hypoxia-sensing pathways to confer renal ischemic preconditioning. Although previous studies demonstrated a protective effect of HIF activation on ischemic kidney injury, these reports used pharmacological treatment with prolyl hydroxylase inhibitors (16, 17) and exposure to acute systemic hypoxia (15). As the current studies focused on chronic renal hypoxia, we sought to determine whether a similar effect would occur in the setting of chronic systemic hypoxic conditions. After being housed in 10% oxygen for 10 days, wild-type animals that had not undergone any cardiac procedures were protected from acute renal ischemic injury. Although the protective effects on BUN elevation and weight loss did not reach statistical significance until 7 and 14 days, respectively, overall, this was a similar effect, as was previously observed post-MI (Fig. 4, A and B). Interestingly, the differences in weight and BUN persisted for several weeks following acute injury, though BUN did not quite reach statistical significance at later time points (Fig. 4, C and D).
Figure 4.

Effects of chronic systemic hypoxia on ischemic acute kidney injury. A and B: peak blood urea nitrogen (BUN; A) and weight loss (B) as measured in healthy animals housed in either normal or hypoxic conditions (10% O2) for 10 days prior to renal ischemia. C and D: BUN (C) and weight loss (D) were monitored for several weeks following renal ischemia. *P < 0.05 by Mann–Whitney test, † indicates interaction between treatment and time is significant for two-way ANOVA with repeated measures; n = 4–5 per group.
Although the studies described thus far focused on AKI, effects on chronic renal injury in our model remained unclear. To determine long-term effects of MI on progression to chronic kidney injury after ischemic acute renal injury, we compared trends in body weight and BUN between sham and MI groups following IR. Despite worsened acute injury as already described, the sham group had recovery of body weight and BUN that was similar to the MI group (Fig. 5, A and B). There was also no detectable difference in GFR 4 wk after IR (Fig. 5C). Despite the observed protection from acute renal injury, animals that underwent MI had evidence of worsened chronic kidney fibrosis, as shown by increased picrosirius red and α-SMA staining (Fig. 6, A and B). In addition, the MI group had increased Kim-1 abundance (Fig. 6C).
Figure 5.
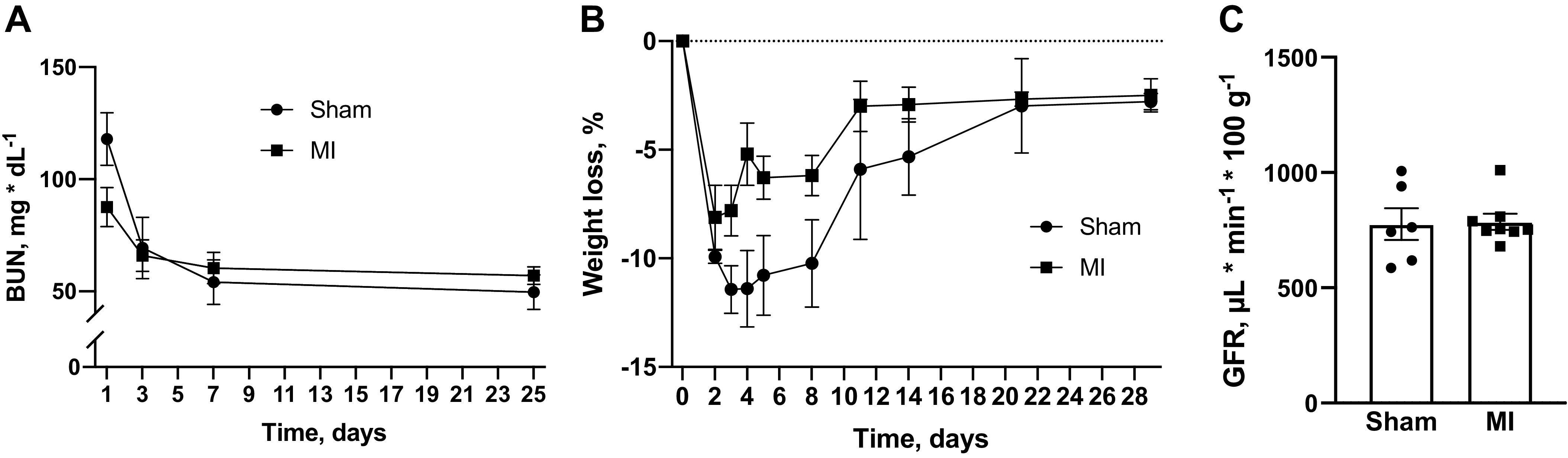
Effects of myocardial infarction (MI) on chronic renal function. A–C: blood urea nitrogen (BUN; A), weight loss (B), and glomerular filtration rate (GFR; C) as measured 4 wk after acute renal ischemia in animals that had previously undergone MI or sham procedure. Comparisons were made by unpaired t test. For A and B, n = 9 for sham and n = 8 for MI. For C, n = 6 for sham and n = 8 for MI.
DISCUSSION
Physiological evidence for communication between the heart and the kidneys has existed for nearly a century (19). Patients suffering from chronic heart failure serve as an important clinical example of the renal consequences endured when this interorgan communication fails. Indeed, heart and renal failure coexist frequently, and it is clear that the cardiorenal syndrome is bidirectional, with primary dysfunction in either organ capable of worsening function in the other. As the rising prevalence of both heart and kidney failure results in more hospitalizations and deaths, a critical understanding of the intricacies of this bidirectional communication is key to developing novel therapeutics. To date, there are no specific therapies approved for the treatment of this syndrome.
Here, we used a model of ischemic cardiomyopathy, the most common cause of death in the world, and determined effects on both acute and chronic renal injury. Importantly, we honed our model to cause ischemic cardiac injury without producing an overt heart failure phenotype at baseline. Given that we produced only a modest reduction in cardiac ejection fraction, this scenario is most akin to the clinical entity of heart failure with preserved ejection fraction. Our data show that even modest cardiac damage affects both acute and chronic kidney physiology following injury. Surprisingly, chronic heart damage conferred protection from ischemic AKI. Protection was accompanied by evidence of tissue hypoxia and activation of HIF signaling pathways in the kidney.
Ischemic preconditioning effects observed in this study are similar to previous reports of preconditioning observed with remote ischemia or caloric restriction (20–22). These reports focused on calorie restriction or ischemia in remote organs conferring renal preconditioning via mechanisms functioning at a distance (23, 24), which is in subtle contrast to the present study. In our model, it cannot be excluded that the remote cardiac injury may confer some renal protection, as has been postulated previously via release of endocrine factors (25). However, our data suggest that ischemic preconditioning via a mechanism that has effects directly on renal tissue, namely, chronic renal hypoxia leading to metabolic reprogramming via activation of hypoxia inducible factors. Although our mice that underwent MI demonstrate a similar phenotype to our chronically hypoxic mice following IR and we observed increased HIF-responsive genes, it should be noted these associations are not mechanistically definitive.
HIF effects are pleiotropic, with studies demonstrating its role in promoting metabolic reprograming toward anaerobic glycolysis, oxygen delivery, erythropoiesis, cellular proliferation, and cell survival (16, 26). Prior work in humans and animals has shown preischemic HIF activation via acute systemic hypoxia (15, 27), prior renal ischemia (28), or HIF stabilization through prolyl hydroxylase inhibition protects against AKI (17). Beneficial effects in those studies were accompanied by metabolic reprogramming toward anaerobic glycolysis. We observed a similar increase in glycolytic enzyme transcripts in animals that underwent MI, consistent with a glycolytic shift preconditioning the kidney to respond to ischemic injury. Evidence supporting an important role for HIF signaling in acute renal injury in endothelial (29), epithelial (30), and myeloid (31) cell types exists. Determining the cell type(s) mediating these effects in our model will be the subject of future studies.
Our proposed mechanism, although consistent with prior reports, may initially appear to conflict with clinical observations that have been made in individuals with ischemic cardiomyopathy. Clinical data strongly suggest that heart failure increases the risk of AKI, rather than protects from it (32). Although the differences between our experimental data and patient studies cannot entirely be elucidated at this time, several key differences should be noted.
First, our studies were performed on young mice with healthy kidneys at baseline, a substrate that is quite different from the encountered clinical scenario involving older individuals with multiple underlying comorbidities affecting renal function, including diabetes, hypertension, coronary artery disease, peripheral vascular disease, atherosclerosis, and baseline chronic kidney disease, among others. These baseline differences make it such that many patients that experience myocardial ischemia already have baseline kidney dysfunction, which may alter outcomes independent of the mechanism under investigation in this report. Investigating effects of MI in preinjured or aged kidneys would be interesting to study in the future.
Second, our model involves a permanent LAD ligation, which does not precisely mimic the corresponding clinical scenario, which frequently involves coronary reperfusion following percutaneous coronary artery interventions. This clinical scenario is associated with cardiac ischemia and reperfusion injury rather than permanent ischemia, which may alter subsequent effects on renal perfusion and injury.
Third, although clinical data indicate patients with heart failure have an increased risk of AKI, one of the more commonly observed forms of renal injury in this setting is type 1 cardiorenal syndrome where acute cardiac dysfunction results in acute renal dysfunction, which is not an ischemic renal insult. This is different from our model in that we are modeling effects of chronic cardiac dysfunction on acute renal ischemia.
Finally, the clinical data in this area arise from observational studies rather than randomized trials. This is an important distinction to note because the lack of randomization makes it more difficult to define cause-and-effect relationships. Specifically, although patients with heart failure experience AKI with increased frequency, making it a risk factor for AKI, we cannot clearly delineate the cause-and-effect relationship that exists between these two entities. Therefore, it remains possible that a significant proportion of the increased AKI risk experienced by patients with heart failure can be attributed to confounding variables that put them at increased risk of hospitalizations and results in a greater AKI incidence.
Although acute renal injury and weight loss were ameliorated post-MI, chronic effects were not. These data from the chronic phase of our model reflect our general understanding of cardiorenal syndrome, that is, cardiac dysfunction promoting renal dysfunction. They suggest that although chronic cardiac injury may, to an extent, prime the kidney to respond more favorably to an acute insult, it has a deleterious effect on renal function over time. As our model involved chronic cardiac dysfunction promoting renal dysfunction, it is most analogous to cardiorenal syndrome type 2. Although prior studies have focused on type 1 cardiorenal syndrome, our model provides an opportunity to define cardiorenal signaling in the chronic setting, outside of acute hospitalizations, in the physiological setting where patients spend the majority of their time. This information will have important implications for the development of novel therapeutics.
GRANTS
This work was supported by the National Institutes of Health Grants DK51265, DK95785, DK62794, DK7569, and P30DK114809 (to R.C.H. and M.Z.Z.), by Veterans Affairs Merit Award 00507969 (to R.C.H.), and by the Vanderbilt Center for Kidney Disease.
DISCLOSURES
A.S.T. has a consulting agreement with Ampio Pharmaceuticals. No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.S.T., M.-Z.Z., and R.C.H. conceived and designed research; A.S.T., K.S., A.N., S.W., X.F., Y.Z., and S.N. performed experiments; A.S.T., M.-Z.Z., and R.C.H analyzed data; A.S.T., K.S., J.P.A. M.-Z.Z., and R.C.H interpreted results of experiments; A.S.T. prepared figures; A.S.T. and R.C.H. drafted manuscript; A.S.T., M.-Z.Z., and R.C.H edited and revised manuscript; A.S.T., K.S., J.P.A., A.N., S.W., X.F., Y.Z., S.N., M.-Z.Z., and R.C.H approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Volker Haase and Hanako Kobayashi for the assistance with performing hypoxia chamber experiments. They also thank Lin Zhong for assistance with LAD ligations and echocardiography.
REFERENCES
- 1.WHO. The Top 10 Causes of Death. World Health Organization. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death. [2020. December 9].
- 2.Ezekowitz J, McAlister FA, Humphries KH, Norris CM, Tonelli M, Ghali WA, Knudtson ML; APPROACH Investigators. The association among renal insufficiency, pharmacotherapy, and outcomes in 6,427 patients with heart failure and coronary artery disease. J Am Coll Cardiol 44: 1587–1592, 2004. doi: 10.1016/j.jacc.2004.06.072. [DOI] [PubMed] [Google Scholar]
- 3.Ronco C, House AA, Haapio M. Cardiorenal syndrome: refining the definition of a complex symbiosis gone wrong. Intensive care Med 34: 957–962, 2008. doi: 10.1007/s00134-008-1017-8. [DOI] [PubMed] [Google Scholar]
- 4.Damman K, Valente MA, Voors AA, O'Connor CM, van Veldhuisen DJ, Hillege HL. Renal impairment, worsening renal function, and outcome in patients with heart failure: an updated meta-analysis. Eur Heart J 35: 455–469, 2014. doi: 10.1093/eurheartj/eht386. [DOI] [PubMed] [Google Scholar]
- 5.Heywood JT, Fonarow GC, Costanzo MR, Mathur VS, Wigneswaran JR, Wynne J; ADHERE Scientific Advisory Committee and Investigators. High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail 13: 422–430, 2007. doi: 10.1016/j.cardfail.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez RP, Comba PC, Esteban MR, Sanchez JJ, Afonso JH, Perez MD, Rodriguez IM, Diaz BB, Elosua R, Cabrera de LA. Incidence, mortality and positive predictive value of type 1 cardiorenal syndrome in acute coronary syndrome. PLoS One 11: e0167166, 2016. doi: 10.1371/journal.pone.0167166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shamseddin MK, Parfrey PS. Mechanisms of the cardiorenal syndromes. Nat Rev Nephrol 5: 641–649, 2009. doi: 10.1038/nrneph.2009.156. [DOI] [PubMed] [Google Scholar]
- 8.Matsushita K, Saritas T, Eiwaz MB, McClellan N, Coe I, Zhu W, Ferdaus MZ, Sakai LY, McCormick JA, Hutchens MP. The acute kidney injury to chronic kidney disease transition in a mouse model of acute cardiorenal syndrome emphasizes the role of inflammation. Kidney Int 97: 95–105, 2020. doi: 10.1016/j.kint.2019.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, Yin H, Wong K, Miyazawa T, Chen J, Chang I, Singh A, Harris RC. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest 122: 4519–4532, 2012. doi: 10.1172/JCI60363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ; Chamber Quantification Writing Group, American Society of Echocardiography's Guidelines and Standards Committee, European Association of Echicardiography. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 18: 1440–1463, 2005. doi: 10.1161/JAHA.117.008181. [DOI] [PubMed] [Google Scholar]
- 11.Scarfe L, Schock-Kusch D, Ressel L, Friedemann J, Shulhevich Y, Murray P, Wilm B, de Caestecker M. Transdermal measurement of glomerular filtration rate in mice. J Vis Exp 58520, 2018. doi: 10.3791/58520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schreiber A, Shulhevich Y, Geraci S, Hesser J, Stsepankou D, Neudecker S, Koenig S, Heinrich R, Hoecklin F, Pill J, Friedemann J, Schweda F, Gretz N, Schock-Kusch D. Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol 303: F783–F788, 2012. doi: 10.1152/ajprenal.00279.2012. [DOI] [PubMed] [Google Scholar]
- 13.Ow CPC, Ullah MM, Ngo JP, Sayakkarage A, Evans RG. Detection of cellular hypoxia by pimonidazole adduct immunohistochemistry in kidney disease: methodological pitfalls and their solution. Am J Physiol Renal Physiol 317: F322–F332, 2019. doi: 10.1152/ajprenal.00219.2019. [DOI] [PubMed] [Google Scholar]
- 14.Zhang MZ, Yao B, Wang S, Fan X, Wu G, Yang H, Yin H, Yang S, Harris RC. Intrarenal dopamine deficiency leads to hypertension and decreased longevity in mice. J Clin Invest 121: 2845–2854, 2011. doi: 10.1172/JCI57324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnsen M, Kubacki T, Yeroslaviz A, Spath MR, Morsdorf J, Gobel H, Bohl K, Ignarski M, Meharg C, Habermann B, Altmuller J, Beyer A, Benzing T, Schermer B, Burst V, Muller RU. The integrated RNA landscape of renal preconditioning against ischemia-reperfusion injury. J Am Soc Nephrol 31: 716–730, 2020. doi: 10.1681/ASN.2019050534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kapitsinou PP, Haase VH. Molecular mechanisms of ischemic preconditioning in the kidney. Am J Physiol Renal Physiol 309: F821–F834, 2015. doi: 10.1152/ajprenal.00224.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapitsinou PP, Jaffe J, Michael M, Swan CE, Duffy KJ, Erickson-Miller CL, Haase VH. Preischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am J Physiol Renal Physiol 302: F1172–F1179, 2012. doi: 10.1152/ajprenal.00667.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46, 2015. doi: 10.1038/nm.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merrill AJ, Morrison JL, Branno ES. Concentration of renin in renal venous blood in patients with chronic heart failure. Am J Med 1: 468, 1946. doi: 10.1016/0002-9343(46)90067-8. [DOI] [PubMed] [Google Scholar]
- 20.Grundmann F, Muller RU, Reppenhorst A, Hulswitt L, Spath MR, Kubacki T, Scherner M, Faust M, Becker I, Wahlers T, Schermer B, Benzing T, Burst V. Preoperative short-term calorie restriction for prevention of acute kidney injury after cardiac surgery: a randomized, controlled, open-label, pilot trial. J Am Heart Assoc 7: e008181, 2018. doi: 10.1161/JAHA.117.008181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitchell JR, Verweij M, Brand K, van de Ven M, Goemaere N, van den Engel S, Chu T, Forrer F, Muller C, de Jong M, van IW, Jn IJ, Hoeijmakers JH, de Bruin RW. Short-term dietary restriction and fasting precondition against ischemia reperfusion injury in mice. Aging Cell 9: 40–53, 2010. doi: 10.1111/j.1474-9726.2009.00532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spath MR, Bartram MP, Palacio-Escat N, Hoyer KJR, Debes C, Demir F, Schroeter CB, Mandel AM, Grundmann F, Ciarimboli G, Beyer A, Kizhakkedathu JN, Brodesser S, Gobel H, Becker JU, Benzing T, Schermer B, Hohne M, Burst V, Saez-Rodriguez J, Huesgen PF, Muller RU, Rinschen MM. The proteome microenvironment determines the protective effect of preconditioning in cisplatin-induced acute kidney injury. Kidney Int 95: 333–349, 2019. doi: 10.1016/j.kint.2018.08.037. [DOI] [PubMed] [Google Scholar]
- 23.Gigliotti JC, Huang L, Ye H, Bajwa A, Chattrabhuti K, Lee S, Klibanov AL, Kalantari K, Rosin DL, Okusa MD. Ultrasound prevents renal ischemia-reperfusion injury by stimulating the splenic cholinergic anti-inflammatory pathway. J Am Soc Nephrol 24: 1451–1460, 2013. doi: 10.1681/ASN.2013010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zarbock A, Schmidt C, Van Aken H, Wempe C, Martens S, Zahn PK, Wolf B, Goebel U, Schwer CI, Rosenberger P, Haeberle H, Gorlich D, Kellum JA, Meersch M; RenalRIPC Investigators. Effect of remote ischemic preconditioning on kidney injury among high-risk patients undergoing cardiac surgery: a randomized clinical trial. JAMA 313: 2133–2141, 2015. doi: 10.1001/jama.2015.4189. [DOI] [PubMed] [Google Scholar]
- 25.Wakasaki R, Matsushita K, Golgotiu K, Anderson S, Eiwaz MB, Orton DJ, Han SJ, Lee HT, Smith RD, Rodland KD, Piehowski PD, Hutchens MP. Glomerular filtrate proteins in acute cardiorenal syndrome. JCI Insight 4: e122130, 2019. doi: 10.1172/jci.insight.122130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haase VH. The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int 69: 1302–1307, 2006. doi: 10.1038/sj.ki.5000221. [DOI] [PubMed] [Google Scholar]
- 27.Vesnina ZV, Lishmanov YB, Alexandrova EA, Nesterov EA. Evaluation of nephroprotective efficacy of hypoxic preconditioning in patients undergoing coronary artery bypass surgery. Cardiorenal Med 6: 328–336, 2016. doi: 10.1159/000446571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinsey GR, Huang L, Vergis AL, Li L, Okusa MD. Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney. Kidney Int 77: 771–780, 2010. doi: 10.1038/ki.2010.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapitsinou PP, Sano H, Michael M, Kobayashi H, Davidoff O, Bian A, Yao B, Zhang MZ, Harris RC, Duffy KJ, Erickson-Miller CL, Sutton TA, Haase VH. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J Clin Invest 124: 2396–2409, 2014. doi: 10.1172/JCI69073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schley G, Klanke B, Schodel J, Forstreuter F, Shukla D, Kurtz A, Amann K, Wiesener MS, Rosen S, Eckardt KU, Maxwell PH, Willam C. Hypoxia-inducible transcription factors stabilization in the thick ascending limb protects against ischemic acute kidney injury. J Am Soc Nephrol 22: 2004–2015, 2011. doi: 10.1681/ASN.2010121249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobayashi H, Gilbert V, Liu Q, Kapitsinou PP, Unger TL, Rha J, Rivella S, Schlondorff D, Haase VH. Myeloid cell-derived hypoxia-inducible factor attenuates inflammation in unilateral ureteral obstruction-induced kidney injury. J Immunol 188: 5106–5115, 2012. doi: 10.4049/jimmunol.1103377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jentzer JC, Bihorac A, Brusca SB, Del R-PG, Kashani K, Kazory A, Kellum JA, Mao M, Moriyama B, Morrow DA, Patel HN, Rali AS, van Diepen S, Solomon MA; Critical Care Cardiology Working Group of the Heart Failure and Transplant Section Leadership Council. Contemporary management of severe acute kidney injury and refractory cardiorenal syndrome: JACC council perspectives. J Am Coll Cardiol 76: 1084–1101, 2020. doi: 10.1016/j.jacc.2020.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]