

Keywords: acute kidney injury, cisplatin, myo-inositol, regulated cell death
Abstract
Regulated cell death (RCD), distinct from accidental cell death, refers to a process of well-controlled programmed cell death with well-defined pathological mechanisms. In the past few decades, various terms for RCDs were coined, and some of them have been implicated in the pathogenesis of various types of acute kidney injury (AKI). Cisplatin is widely used as a chemotherapeutic drug for a broad spectrum of cancers, but its usage was hampered because of being highly nephrotoxic. Cisplatin-induced AKI is commonly seen clinically, and it also serves as a well-established prototypic model for laboratory investigations relevant to acute nephropathy affecting especially the tubular compartment. Literature reports over a period of three decades have indicated that there are multiple types of RCDs, including apoptosis, necroptosis, pyroptosis, ferroptosis, and mitochondrial permeability transition-mediated necrosis, and some of them are pertinent to the pathogenesis of cisplatin-induced AKI. Interestingly, myo-inositol metabolism, a vital biological process that is largely restricted to the kidney, seems to be relevant to the pathogenesis of certain forms of RCDs. A comprehensive understanding of RCDs in cisplatin-induced AKI and their relevance to myo-inositol homeostasis may yield novel therapeutic targets for the amelioration of cisplatin-related nephropathy.
INTRODUCTION
Cisplatin, also known as cis-platinum, cis-diamminedichloroplatinum (II), and platinol, is a commonly used chemotherapeutic agent to treat various types of malignancies, including germline cancers, neoplasm of the prostate, bladder, and lung, and lymphoma (1–4). Interestingly, since its discovery, over a century’s trials and efforts were required to refine its clinical use. Cisplatin was initially discovered by an Italian chemist, M. Peyrone, in 1844 (described as Peyrone’ chloride), and its molecular configuration was thoroughly documented by a Swiss chemist, Alfred Werner, in 1893 (1). However, it took another seven decades to gain the recognition of its therapeutic implications. In 1965, Rosenberg et al. (5) noted that Escherichia coli cultured in the presence of an electrolysis product of platinum electrodes underwent morphological filamentation and growth inhibition, which led to its amenability for therapeutic usage afterward (6). Finally, cisplatin was approved for the treatment of ovarian and testicular neoplasms by the US Food and Drug Administration in 1978, and since then it has been recommended as a first line of chemotherapeutic agent to treat various malignancies.
The clinical use of cisplatin is largely limited by its high degree of nephrotoxicity. It is estimated that ∼30% of cisplatin-treated patients (a single dose at 50–100 mg/m2) exhibit notable nephrotoxic manifestations (7). In the laboratory settings, cisplatin nephropathy serves as a classical prototype for acute kidney injury (AKI)-related investigations due to the readily discernible severity of renal damage. However, it should be kept in perspective that the murine models of cisplatin-induced renal injury may not recapitulate the clinical events since the dose for mice (10–30 mg/kg) is much higher than what is used for its clinical usage (8). In this regard, repeated low-dose administration of cisplatin to mice is readily accepted to recapitulate the clinical settings (9, 10). Besides, mice used in laboratory investigations are usually healthy and young with good organ functions to handle other associated potential nephrotoxins, which may not be case in the clinical settings where patients may already be in a debilitated state (8). The selective organ (kidney) damage pattern of cisplatin may be partly related to its drug metabolism and pharmacokinetics. Cisplatin uptake experiments with kidney slices revealed that its concentration in renal tissues was five times higher than that of the culture medium following ∼4 h of exposure (11). Cisplatin, after freely traversing across the glomerular filter, is handled by certain efflux transporters localized at the apical surfaces of renal tubular epithelia (12, 13). Most cisplatin in the glomerular ultrafiltrate is reabsorbed by proximal tubules, among which the S3 segment tubules efficiently handle majority of the resorptive load, and such a localized buildup would likely contribute toward nephrotoxicity (14). Organic cation transporter 2 (OCT2) and copper transporter 1 (Ctr1) are the major transporters participating in the reabsorption of cisplatin in renal tubules, and genetic or pharmaceutical inhibition of these two transporters could provide protection against cisplatin-induced nephrotoxicity (15–17).
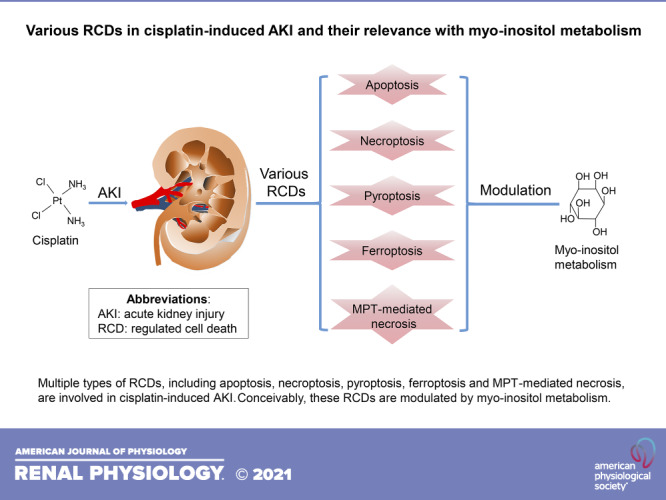
It is now well established that cisplatin [PtCl2(NH3)2] is a square planar inorganic complex (1). The molecular structure of cisplatin is characterized by a centralized platinum ion encircled by two amine groups on one side and two chloride radicals on the other side (Fig. 1A). Conceivably, the interactions between the platinum ion and two chloride radicals are unstable in an intracellular environment, which apparently allows the sequential replacement of chloride radicals by water molecules (Fig. 1A) (18). Hydrated cisplatin gets activated and crosslinks with nucleophilic N7 sites of the purine bases in the DNA, followed by the generation of DNA:DNA adducts, mostly via intrastrand crosslinking (Fig. 1B) (1, 19). These crosslinks lead to DNA damage by dampening DNA synthesis and replication, which ultimately results in cellular demise (20). This DNA damage has been implicated as the major cause of cisplatin being endowed with anticancer properties and unfortunately also the side effects, such as nephrotoxicity (20, 21). However, questions have been raised regarding its role in predominant DNA damage in cisplatin-induced AKI, since only ∼1% of intracellular cisplatin is translocated into the nuclear compartment (22). Besides, renal proximal tubular cells, the primary target of cisplatin-related nephrotoxicity, are the quiescent cells that are devoid of active DNA replication. Other mechanisms, such as oxidant stress, inflammation, vascular dysfunction, and mitochondrial damage, are probably of much greater significance in the pathogenesis of cisplatin-induced AKI (23–26). In recent years, various forms of regulated cell death (RCD) have been described in the literature, and some of them have been implicated in cisplatin-induced AKI (Fig. 2). In addition, the pathogenesis of RCDs has been reported to be conceivably modulated by the metabolism of myo-inositol, a unique carbocyclic sugar. Here, we discuss the available information in the previous investigations on various forms of RCDs in cisplatin-induced AKI and their relevance to myo-inositol metabolism, which may provide unique insights into the novel pathogenetic mechanisms, and of course would eventually facilitate in the development of therapies for cisplatin-induced AKI.
Figure 1.

The structure, hydration, and DNA binding of cisplatin. A: cisplatin is a square-planar complex with a platinum ion in the center; the two chloride ligands can be replaced by H2O in two successive steps. B: cisplatin binds with N7 sites of purine bases (A or G) in DNA, leading to DNA damage. [Partially adapted from Ref. (19).]
Figure 2.

Top: denomination. Bottom: timeline of the events for the five regulated cell deaths related to cisplatin-induced acute kidney injury (AKI). MPT, mitochondrial permeability transition.
APOPTOSIS: THE CLASSIC FORM OF RCD
The term apoptosis was first coined by Kerr et al. (27) in the early 1970s to differentiate it from necrosis (Fig. 2). It is the most extensively investigated RCD in various forms of AKI. Mechanistically, apoptosis is caspase dependent, and its executionary phase mainly relies on the activation of effector caspases, including caspase-3, caspase-6, and caspase-7 (Fig. 3) (28, 29). Apparently, the caspase-3, caspase-6, and caspase-7 complex, and especially caspase-3, then activates caspase-activated DNase (CAD) by proteolytically cleaving the inhibitor of CAD (ICAD) (30). Normally, CAD is bound to ICAD and remains detached with genomic DNA (30). However, in an apoptotic scenario, ICAD gets cleaved and is detached from CAD, which is followed by the dimerization and activation of CAD (30, 31). Activated CAD then binds to genomic double-stranded DNA, culminating in DNA fragmentation (30). In doing so, the apoptotic bodies are generated during the late stages of apoptosis, and they are phagocytosed by contiguous cells without causing obvious inflammation and damage to the adjacent cells (32), a process that is distinct from necrosis. Interestingly, the process of apoptosis in cisplatin-induced AKI was initially identified by two independent groups of investigators in 1996 (33, 34) (Fig. 2). Lieberthal et al. (33) reported cisplatin-induced apoptosis in proximal tubular epithelial cells with morphological evidence of cellular damage and DNA fragmentation, whereas Takeda et al. (34) presented evidence of considerable DNA fragmentation in the S3 segment of proximal tubules and outer medullary collecting tubular cells following cisplatin treatment. Overall, it seems that protein levels of activated caspase-3 are now considered as one of the major markers for apoptosis in cisplatin-induced AKI (35–37). However, the role of other executionary caspases, i.e., caspase-6/7, in cisplatin-induced AKI has not been adequately investigated.
Figure 3.

Cellular events leading to apoptosis. The initiation of apoptosis includes three pathways. Extrinsic pathway apoptosis is initiated by the binding between death receptor and death ligand, followed by sequential activation of Fas-associated death domain (FADD)/tumor necrosis factor receptor-associated death domain (TRADD), caspase-8/10, and caspase-3/6/7. Intrinsic pathway apoptosis is triggered by the mitochondrial leakage of cytochrome c (Cyt C), which binds to apoptotic protease activating factor 1 (APAF1) in the cytoplasm. APAF1 then leads to consequential activation of caspase-9 and caspase-3/6/7. The leakage of Cyt C is strictly controlled by proapoptotic proteins (BAX, BAK1, and P53) and antiapoptotic proteins (BCL-2 and BCL-XL). Endoplasmic reticulum (ER) stress pathway-related apoptosis is mediated by ER stress sensors. Inositol-requiring protein 1 (IRE1) promotes apoptosis by activating successively caspase-12/4, caspase-9, and caspase-3/6/7. Protein kinase R-like ER kinase (PERK) and activating transcription factor 6 (ATF6) drive apoptosis through C/EBP homologous protein (CHOP), which potentiates death receptor activation and mitochondrial permeability. Apoptosis is executed by caspase-3/6/7, among which caspase-3 cleaves inhibitor of caspase-activated DNase (ICAD) to revitalize caspase-activated DNase (CAD), followed by DNA damage.
Apoptosis can be induced using three different routes, i.e., the extrinsic pathway, the intrinsic pathway, and the endoplasmic reticulum (ER) stress pathway (Fig. 3). Extrinsic pathway apoptosis is initiated when death ligands (such as Fas ligand and TNF-α), secreted by immune cells or tubular cells, bind with the corresponding death receptors [such as Fas and TNF receptor (TNFR)] on the plasma membrane of the targeted cells (38–41). This interaction leads to the activation of two initiator caspases (caspase-8 and caspase-10) via Fas-associated death domain or TNFR-associated death domain, which then activates effector caspases (38). Earlier investigations indicated that there was an increased expression of Fas and Fas ligand following cisplatin treatment in both in vivo and in vitro systems (42). Interestingly, studies have shown that Fas mutation attenuates cisplatin-induced proximal tubular cell death and morphological cellular disruption (43). Also, in support of this notion are studies where TNF-α was reported to be upregulated in cisplatin-treated tubular cells, whereas its inhibition dampened the tubular damage (44). Specifically, genetic ablation of TNF-α conferred the mice with resistance to cisplatin-induced cellular injury (44). However, in the initial studies, it was not clear if the upregulated TNF-α by itself modulates cisplatin-induced tubular injury or in conjunction with the activation of a particular receptor, the interaction with which leads to a calamitous inflammatory response and ultimately acute renal failure (45). In this regard, Ramesh and Reeves (45, 46) contemporaneously identified that it is TNFR2 and not TNFR1 that dominantly mediates TNF-α-induced apoptosis. However, other investigators reported that TNFR1 deletion also alleviated cisplatin-induced AKI (43). Of note, one of the death receptors, i.e., death receptor 3 (DR3), and its ligand TNF-like ligand 1A have been reported to mitigate cisplatin-induced AKI, via alleviating proapoptotic signals (47). Other death receptors, such as TNF-related apoptosis inducing ligand (TRAIL)-R1 and TRAIL-R2, and their ligands have not been investigated in cisplatin-induced AKI. Similarly, the role of caspase-10 in such a cellular damage has not been examined, although the role of death receptors in the activation of caspase-8 leading to tubular damage has been well implicated in cisplatin-induced AKI (43).
Compared with extrinsic apoptosis, intrinsic apoptosis has drawn much more attention in cisplatin-induced AKI. Intrinsic apoptosis is closely associated with outer mitochondrial membrane permeabilization and leakage of mitochondrial proteins, such as cytochrome c, apoptosis-inducing factor, somatic cytochrome c, Smac/DIABLO, endonuclease G, etc. (Fig. 3) (48, 49). This process is strictly modulated by proapoptotic factors (Bax and Bak1) and antiapoptotic factors (Bcl-2 and Bcl-xL) (50). Released cytochrome c binds to apoptogenic protease activating factor 1 in the cytoplasm, and this intricate interaction leads to the assembling of the multimeric apoptosome, where procaspase-9 is recruited, cleaved, and activated (51). Following the activation of caspase-9, there is activation of effector system caspases, i.e., caspase-3/6/7 (51). After the first report regarding BAX upregulation and membrane translocation in cisplatin-treated tubular cells (52), the process of intrinsic apoptosis was extensively investigated during the last two decades. Dong and coworker’s (53) laboratory reported that Bax expression levels were increased in cisplatin-treated mice, and mice with genetic ablation of Bax were partially resistant to cisplatin-induced tubular damage. Likewise, pharmacological inhibition of Bax by nutlin-3 alleviated cytochrome c leakage and cellular disruption following cisplatin treatment (54). Interestingly, both Bax and Bak were upregulated, and they were oligomerized and activated in mitochondria following cisplatin exposure (54). On the other hand, Bcl-2 and Bcl-xL have been reported to be downregulated in cisplatin-treated mice or cells, whereas overexpression of Bcl-2 renders renal tubular cells to be resistant to cisplatin (55–58). Also, p53, a tumor suppressor, exacerbates cisplatin-induced AKI via inhibition of antiapoptotic proteins (Bcl-2 and Bcl-xL) (39).
The third route, i.e, ER stress-related apoptosis, has been poorly investigated in cisplatin-induced AKI. Under basal conditions, the ER is the organelle where proteins are synthesized and processed, in other words, properly folded and packaged (59). However, in pathological states, excessive unfolded/misfolded proteins accumulate in the ER lumen due to its functional disruption, and that leads to an accentuated unfolded protein response (UPR) and thereby causes ER stress (60). When ER stress persists for a long time, glucose-regulated protein 78/binding protein is dissociated from the three key ER stress sensors, i.e., inositol-requiring protein 1 (IRE1), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6), and this process downstream facilitates apoptosis (Fig. 3) (59, 60). IRE1 accelerates apoptosis by activating murine caspase-12 or its human homolog caspase-4, followed by cleavage of procaspase-9 and procaspase-3 (60, 61). PERK and ATF6 drive apoptosis via increasing transcription of C/EBP homologous protein, which, in turn, participates in apoptosis via modulating mitochondrial permeability and death receptor activation (60). Overall, one can say that the literature reports, to a certain extent, support the view that these three ER stress sensors (IRE1, PERK, and ATF6) are conceivably activated and upregulated in cisplatin-induced AKI (58, 62, 63). Besides, heme oxygenase-1, an ER-resident enzyme, accelerates apoptosis in cisplatin-induced AKI (7). In addition, activation of downstream molecules, including cleaved caspase-12 and C/EBP homologous protein, were reported to be induced in cisplatin-treated kidney tissues (64, 65). However, all these publications provide somewhat weak evidence for the involvement of ER stress-related apoptosis in cisplatin-induced AKI due to the paucity of data regarding mice with relevant genetic deficiency or lack of specific pharmacological inhibitory agents for these key sensors. Further validation is needed to elucidate a definitive concrete role of the ER stress pathway and apoptosis in cisplatin-induced AKI.
NECROPTOSIS
The term “necroptosis” was initially coined by Yuan and coworkers (66) in 2005 while working on ischemic brain injury (Fig. 2). These authors characterized necroptosis as a process in which the Fas/TNFR family activates a nonapoptotic death pathway in the absence of apoptotic signaling. Later, several investigators noted that necroptosis is an essential process seen in many forms of AKI (67–72). The process of necroptosis includes an array of cascade of events, which culminate in the generation of the necrosome complex and phosphorylation of mixed lineage kinase domain-like (MLKL) pseudokinase (73–75). Phosphorylation of MLKL is mediated by receptor-interacting serine/threonine protein kinase (RIPK)1 and RIPK3 as well as death receptors (Fig. 4, top left) (73, 74). Upon activation of death receptors (notably TNFR1 and Fas), the kinase domain of RIPK1 is activated, which downstream leads to the phosphorylation of RIPK3 (73, 74). In this scenario, RIPK1 and RIPK3 are recruited and assembled into a necrosome complex, where RIPK1 and RIPK3 undergo auto- and transphosphorylation, followed by phosphorylation and activation of MLKL (73, 74). Phosphorylated MLKL gets homotrimerized and undergoes a conformational change, which results in the exposure of its NH2-terminal 4-helix bundle (NB) domain and central brace region (76–78). Subsequently, MLKL translocates into the plasma membrane, where the central brace region and NB domain interact with polyphosphoinositides, resulting in pore formation in the plasma membrane and ultimately leading to cellular rupture, i.e., necroptosis (76–78).
Figure 4.

Molecular mechanisms pertaining to four types of nonapoptotic regulated cell deaths. Top left: necroptosis is triggered by receptor-interacting serine/threonine-protein kinase (RIPK)1 activation upon binding between death receptor and death ligand. RIPK1 then phosphorylates RIPK3, leading to the assembly of necrosome, where mixed lineage kinase domain-like (MLKL) is recruited and activated. Activated MLKL gets oligomerized and disrupts the cytomembrane, ultimately culminating in cell death. Top right: the initiation of pyroptosis includes two ways. Canonical pyroptosis is induced by the binding of the pattern recognition receptor (PPR) and its corresponding adaptor, which leads to the activation of caspase-1. Caspase-1 then cleaves gasdermin D (GSDMD), IL-1β, and IL-18. Noncanonical pyroptosis is mediated by lipopolysaccharide (LPS)-activated caspase-4/5/11, which also cleaves GSDMD. NH2-terminal domain of GSDMD (GSDMD-N) gets oligomerized and generates pores in the cytomembrane, resulting in IL-1β and IL-18 leakage and cell death. Bottom left: ferroptosis is executed by overwhelming lipid peroxidation, which is catalyzed by ferritinophagy-induced free iron and lipoxygenase (LOX). The lethal lipid peroxidation can be inhibited by glutathione peroxidase 4 (GPX4) together with GSH. Bottom right: mitochondrial permeability transition (MPT)-mediated necrosis is characterized by generation of the MPT pore (MPTP), which disrupts the inner mitochondrial membrane (IMM) and allows the passage of small solutes [molecular weight (MW) < 1.5 kDa]. The formation of the MPTP is usually induced by increased calcium concentration or oxidant stress, and the MPTP can be inhibited by cyclosporine A (CsA), a potent antagonist of cyclophilin D (Cyp-D). NCOA4, nuclear receptor coactivator 4.
The role of necroptosis in cisplatin-induced proximal tubular injury was originally reported almost a decade ago by Monte and coworkers (79) (Fig. 2). These authors demonstrated that necrostatin-1 (Nec-1; an inhibitor of RIPK1) alleviated cell death following cisplatin exposure to HK-2 cells. Following this seminal observation, several groups of investigators confirmed that the administration of Nec-1 indeed attenuates aberrant renal pathological alterations induced by cisplatin (80–83). Interestingly, the expression of molecules relevant to necroptosis, i.e., RIPK1, RIPK3, and MLKL, was noted to be upregulated in kidneys of mice that received cisplatin (83–86). Other RIPK1 inhibitors, i.e., wogonin and Cpd-71, or gene disruption of the molecules mentioned above also had an ameliorative effect in cisplatin-induced nephropathy (82, 83, 85). However, certain concerns were raised regarding the investigations related to RIPK1 inhibition in necroptosis. First, although Nec-1 is commonly used as a necroptosis inhibitor, it should be noted that it is also involved in the modulation of certain aspects of the immunoregulatory system, which raises the question regarding the specificity of the inhibitory effect (87–89). Second, RIPK1 participates in other biological processes, including inflammation, immunity, and cell survival, besides the downstream activation of RIPK3 (90, 91). In this regard, genetic interference of the downstream molecules, i.e., RIPK3 and MLKL, would be good potential targets for the inhibition of necroptosis. Indeed, Han and coworkers (83) observed that RIPK3 or MLKL knockout mice had a notable alleviation of tubular damage following cisplatin exposure compared with wild-type mice. In line with these observations, Meng and coworkers (92) recently reported that cisplatin-induced HK-2 cell death can be attenuated by the microRNA hsa-miR-500a-3P; the latter binds to the 3′-untranslated region of MLKL and suppresses its phosphorylation and plasma membrane translocation. Other points worth considering in the process of necroptosis include the dose and duration of exposure of the offending agent or inhibitors. In vitro studies have indicated that necroptosis usually occurs following the administration of a high dose of cisplatin or its prolonged exposure at low dosages (83). Coneivably, the prevailing notion is that the first low dose of cisplatin sensitizes mouse kidneys to necroptosis whereas the second dose renders them susceptible to cisplatin-induced injury (93, 94). Likewise, a recovery pattern of injury in necroptosis was observed in the mouse model of folic acid-induced AKI. In folic acid-induced injury, where the injury is usually milder compared with cisplatin, no notable protective effect of Nec-1 administration was observed at day 2 (95). However, in the state of prolonged folic acid-induced renal injury at day 4, the administration of Nec-1/Nec-1s or genetic ablation of RIPK3/MLKL had significant protection against tubular injury (71).
PYROPTOSIS
Pyroptosis, recently redefined as gasdermin-dependent cell death, is a caspase-mediated nonapoptotic RCD, and it was originally described by Cookson and Brennan and other investigators (96–98) in monocyte/macrophage death induced by mounting of an overwhelming bacterial assault (Fig. 2). Strikingly different from apoptosis, the affected cells undergoing pyroptosis exhibit a diffuse pattern of DNA fragmentation, disruption of membrane integrity, and triggering of inflammation, and the process is characterized by the absence of traditional caspase-3 activation (96, 97, 99). The onset of pyroptosis can be triggered by two different routes, which include a canonical inflammasome pathway and the other noncanonical inflammasome pathway (100). In canonical pyroptosis, exposure to different stimuli leads to the generation of various oligomeric inflammasome complexes. They are usually formed following activation of a pattern recognition receptor (PRR) and a downstream adaptor (101, 102). The PRRs sense specific stimuli and transmit the signals to the adaptor, leading to the activation of caspase-1, which is followed by cleavage of IL-1β, IL-18, and gasdermin D (GSDMD; a pyroptosis effector) (Fig. 4, top right) (98, 102). Of note, the PPRs usually undergo oligomerization to increase efficiency of caspase-1 activation (98). Interestingly, multiple types of inflammasomes having their own unique potencies have been identified to recognize a wide variety of stimuli (103). Noncanonical pyroptosis is mediated by murine caspase-11 or its human homolog caspase-4/5 (104–106). Upon exposure of lipopolysaccharide (LPS), caspase-4/5/11 binds to the lipid A motif in LPS, thereafter culminating into its activation (Fig. 4, top right) (105). Subsequently, activated caspase-4/5/11 is oligomerized and exerts its proteolytic effect to cleave the common substrate GSDMD, without involving the inflammatory cytokine IL-1β or IL-18 (98, 104–106).
Here, a brief description of the common substrate, i.e., GSDMD, of canonical and noncanonical pathways may be worth discussion. GSDMD, the executor of pyroptosis, is a member of the gasdermin family, which is characterized by having a highly conserved NH2-terminal domain (98, 107, 108). To date, six members of gasdermin, i.e., GSDMA, GSDMB, GSDMC, GSDMD, GSDME (DFNA5), and DFNB59, have been identified in humans, whereas there are 12 members in mice (107, 108). GSDMD is the only member that harbors the cleavage site for caspase-1/4/5/11 (108). The NH2-terminal domain of GSDMD (GSDMD-N) gets oligomerized and binds to phosphoinositides and cardiolipin while being inserted into the plasma membrane (109, 110), which is followed by pore formation, membrane disruption, and cellular lysis. Interestingly, GSDMD only disrupts the plasma membrane from the inside since phosphoinositides are localized in the cytoplasmic leaflet, and not the exoplasmic leaflet, of the plasmalemma (109, 110). The membrane pore formed by oligomerized GSDMD has an inner diameter of 10–14 nm, which readily allows the secretion of IL-1β (4.5 nm) or IL-18 (7.5 nm) (98, 109). In this regard, IL-1β and IL-18, processed by caspase-1 and secreted through pores formed by GSDMD-N, are regarded as sensitive markers for pyroptosis (111–113). Other gasdermins also generate pyroptotic alterations after activation even if no caspase cleavage sites are present, suggesting that there are other potential routes for pyroptosis to ensue (109).
In 2012, Yu and coworkers (114) initially investigated the relevance of pyroptosis in renal injury in a murine model of unilateral ureteral obstructive nephropathy, where they observed increased expression of key pyroptosis markers, i.e., caspase-1 and IL-1β. Subsequently, the pathogenetic role of pyroptosis was implicated in a number of experimental renal injury models, including ischemia-reperfusion injury (IRI)-, contrast-, sepsis-, cadmium-, diabetes-, and calcium oxalate-induced nephropathy (115–120). However, the evidence presented in these reports was somewhat weak and indirect, since they lack the use of specific pyroptosis inhibitors and due to a paucity of pyroptosis-related gene disruption in in vivo studies in various experimental models. However, recently, two independent group of investigators demonstrated a direct pathogenetic role of pyroptosis in cisplatin-induced AKI using GSDMD knockout mice (121, 122). In this regard, Lu and coworkers (122) presented evidence that gene deletion of caspase-11 or GSDMD apparently protect mice against cisplatin- and IRI-induced AKI, suggesting the participation of noncanonical pyroptosis. In addition, Jia and coworkers (121) reported that GSDMD gene ablation is beneficial, whereas GSDMD-N overexpression is detrimental, to the kidney in cisplatin-induced nephropathy, as assessed by aberrant alterations in renal functional and morphological parameters. Also, the anomalous changes observed in IL-1β and IL-18 levels following cisplatin exposure were partially restored following GSDMD gene disruption (121, 122). Along these lines, Edelstein and coworkers (123) observed that caspase-1 knockout mice are resistant to cisplatin-induced tubular injury, suggesting the involvement of canonical pyroptosis, although the relevance of pyroptosis was not alluded to in this publication.
FERROPTOSIS
Almost a decade ago, ferroptosis was described as an erastin-triggered cell death by Dixon et al. (124). Incidentally, erastin is a small molecule that inhibits cystine-glutamate antiporter system Xc−. Ferroptosis is a unique type of iron-dependent nonapoptotic RCD (Fig. 2). Furthermore, Stockwell and coworkers (124, 125) suggested that the executionary phase of ferroptosis is characterized by iron catalyzed-lipid hydroperoxidation, indicating disruption in iron metabolism and associated with various redox perturbations. In fact, the process of ferroptosis encompasses an array of metabolic disorders involving lipids, iron, and amino acids (Fig. 4, bottom left) (125). Lipid peroxidate, the executor of ferroptosis, can be synthesized via Fenton reaction or the lipoxygenase (LOX) pathway (126, 127). Fenton reaction refers to a nonenzymatic catalysis between peroxides and ferrous iron, leading to the generation of highly reactive oxidant radicals (128). These radicals then target bis-allylic hydrogen atoms of polyunsaturated fatty acids (PUFAs), thus resulting in deleterious lipid peroxidation (126). Of note, PUFAs need to be esterified into membrane phospholipids before lipid peroxidation (125, 129). This may mean that a large number of cellular components, including plasmalemma, mitochondria, the ER, and lysosomes would be affected due to having PUFA in their membranes. LOX, a family of the iron-containing enzymes, mediates ferroptosis via catalyzing PUFA-containing phospholipids (130). Many of the LOXs, including LOX5, LOX12, and LOX15, are intimately associated with ferroptosis in a variety of pathobiological processes (131–133). The notion regarding the participation of iron metabolism is well supported by the fact that both Fenton reaction and LOX-mediated enzyme catalysis are iron dependent (126). Along these lines, it is worth mentioning that the onset of ferroptosis induced by various chemicals can be prevented by the administration of iron chelators (125, 134), whereas iron supplementation by ferric ammonium citrate facilitates the induction of ferroptosis, as alluded to in vitro studies (135). In addition, ferritinophagy, a selective autophagy of ferritin guided by its specific cargo receptor nuclear receptor coactivator 4, leads to an increased availability of free iron (136), which is essential for the Fenton reaction. Other iron metabolic sensors, including transferrin and transferrin receptor (iron import), ferroportin (iron export), ferritin and heme (iron storage), iron-responsive element-binding protein 2, heat shock protein-β1, and mitoNEET, iron metabolism regulator/transporter in mitochondria, have been also implicated in various pathobiological processes relevant to ferroptosis (137–143). The iron-catalyzed overwhelming lipid peroxidation damages the cells by membrane disruption and toxicity of lipid peroxides decomposition products (126). Nonetheless, the deleterious process of ferroptosis can be terminated by an enzyme, glutathione peroxidase 4 (GPX4), whereby noxious lipid peroxidates are converted into harmless lipid alcohols in the presence of glutathione (144). Here, it should be noted that two amino acids, i.e., cysteine and glutamate, of the antiporter system Xc− that are pertinent to glutathione synthesis, are relevant to the induction of ferroptosis (145). In fact, the inhibitors of antiporter system Xc− and GPX4 have been widely used as inducers of ferroptosis. Other agents, including NADPH, nuclear factor erythroid 2-related factor 2 (NRF2), heme oxygenase-1, p53, and coenzyme Q10, also have been reported to be relevant in the onset of ferroptosis (131, 144, 146).
In 2014, Linkermann et al. (147) described the role of ferroptosis in AKI in an excellent seminal report, and the authors indicated that ferroptosis, rather than necroptosis and apoptosis, mediated renal parenchymal cellular necrosis following ischemia-reperfusion-induced AKI. Also, other investigators discovered that ferroptosis is also operative in oxalate-, rhabdomyolysis-, and folic acid-induced AKI (95, 147, 148). In a recent report, with convincing in vivo and in vitro evidence, we described the role of ferroptosis in cisplatin-induced AKI (149). In this report, we also indicated that cisplatin treatment leads to excessive lipid peroxidation, ferritinophagy-mediated free iron release, glutathione depletion, and a decrease in the activity of GPX4 (149). Here, it is encouraging to note that our findings were immediately validated by other group of investigators (150, 151). The link between ferroptosis and cisplatin-induced AKI has been only recently appreciated; nevertheless, the investigators of decades-old earlier publications should be credited for pointing out this unique association, although the evidence was somewhat indirect. In 1991, two groups of investigators noted that cisplatin-induced lipid peroxidation in rat kidneys was iron dependent (152, 153). A few years later, Baliga et al. (154) described that cisplatin treatment leads to excessive free iron load in both LLC-PK1 cells (pig proximal tubular cells) and kidneys and that iron chelators protected the tubular cells against cisplatin-induced cellular damage. Afterward, a flurry of articles suggesting perturbations in the iron metabolism induced by cisplatin were reported in the literature by several investigators (155–157).
MITOCHONDRIAL PERMEABILITY TRANSITION-MEDIATED NECROSIS
Mitochondrial permeability transition (MPT)-mediated necrosis refers to a variant of RCD induced by the formation of the MPT pore (MPTP), which disrupts the inner mitochondrial membrane and facilitates the traffic of solutes (molecular weight < 1,500 Da) across the inner mitochondrial membrane (Fig. 4, bottom right) (158–160). These aberrant changes lead to perturbations in the mitochondrial membrane potential, mitochondrial swelling and dysfunction, and eventually cell death (161). Inexplicably, well-defined molecular components of the MPTP remain elusive. Previous publications indicated several candidates for MPTP components, including adenine nucleotide translocase, voltage-dependent anion channel, c subunit of FoATP synthase, phosphate carrier, and cyclophilin D (CypD) (162–165). However, some of the notions regarding these potential candidates were challenged by other investigators following the observations made in genetically modified mice (166–169). Nevertheless, it is well accepted that CypD, a mitochondrial molecular chaperone, is the key regulator participating in the functionality of the MPTP (170). The idea of possible involvement of the MPTP in necrosis was initially marshalled by the experiments in which cyclosporine A, a CypD inhibitor, was noted to inhibit Ca2+-dependent MPT and cell death (171–174). However, this has been somewhat controversial because of lack of specificity since cyclosporine A has the tendency to interact with other cyclophilins and calcineurin, and, moreover, overexpression of CypD has opposite effects on apoptosis and necrosis (172, 175). However, Li et al. (175) provided evidence that CypD-overexpressing cells were susceptible to sodium nitroprusside (nitric oxide donor)-induced necrosis and that their mitochondria were prone to undergo permeability transition (Fig. 2). Interestingly, convincing proof of the role of CypD was obtained by gene deletion approaches in mice by three different groups of investigators. Their elegant work indicated that CypD-deficient cells were more resistant to necrotic cell death induced by oxidant stress or Ca2+ overload (159, 176, 177). In addition, they noted that CypD knockout mice were less susceptible to neuronal or cardiac IRI. Of note, as mentioned above, CypD is considered as an essential regulator rather than an integral component of the MPTP, since the functionality of the MPTP was still preserved under higher Ca2+ concentration and oxidant stress even under conditions of CypD deficiency (160). MPT-mediated necrosis has been implicated in the pathogenesis of various injury models in several organ systems, including the pancreas, lung, kidney, and liver (87, 178–180). However, due to the limited knowledge, especially related to the constitutional component of the MPTP, the precise mechanisms of MPT-mediated necrosis remain still somewhat elusive.
The role of MPT-mediated necrosis in the kidney was initially implicated in renal IRI, which indicated beneficial effects of CypD deficiency in IRI-induced AKI (181, 182). The research literature related to MPT-mediated necrosis and cisplatin-induced AKI is limited. In this regard, a few years ago, Linkermann et al. (87) observed that CypD-deficient mice had prolonged survival following cisplatin treatment compared with wild-type mice, and these investigators documented that the pathogenesis of MPT-mediated necrosis is distinct from RIPK1/RIPK3-mediated necroptosis. Interestingly, some of the Chinese herbs, such as Panax notoginseng saponins and Astragalus polysaccharide, were noted to alleviate cisplatin-induced AKI by inhibition of the MPTP (183, 184). Of note, before MPT-mediated necrosis was fully recognized, cisplatin-induced AKI was reported to be relieved by cyclosporine A more than two decades ago (185), and the authors suggested a conceivable role of Ca2+-dependent MPT-mediated necrosis in this mouse model.
METABOLISM OF MYO-INOSITOL AND ITS RELATED DERIVATIVES AND THEIR RELEVANCE IN THE PATHOBIOLOGY OF RCDs
Myo-inositol (C6H12O6, a carbocyclic sugar) is the predominant isomer (among the nine isomers) of inositol present in mammalian cells, and it participates in various biological processes either as a free molecule or as inositol-containing phospholipids and inositol phosphate derivatives (186). Myo-inositol was once termed as vitamin B8, but this conception has been forsaken due to its extensive endogenous synthesis from glucose (186). It is estimated that the two kidneys, the dominant sites of myo-inositol metabolism in human, produce ∼4 g of myo-inositol daily, which substantially exceeds myo-inositol intake (∼1 g/day) from the regular diet (186, 187). Of note, other organs, including the brain, testis, and liver, are also involved in the endogenous synthesis of myo-inositol (188–190). The synthesis of myo-inositol involves three steps: the initial generation of glucose-6-phosphate via hexokinase-mediated phosphorylation of glucose, and following the conversion of glucose-6-phosphate to myo-inositol-1-phosphate by 1-d-myo-inositol phosphate synthase, it is then converted into myo-inositol by the action of inositol monophosphatase, which apparently dephosphorylates myo-inositol-1-phosphate (Fig. 5) (186). Dietary myo-inositol intake is absorbed from the gastrointestinal tract in free form or as phosphatidylinositol (PI) (186). Once inside the bloodstream, myo-inositol (∼30 μM in normal human plasma) (191) is transported into various organs via its specific transporters, including Na+/myo-inositol transporter 1/2 (SMIT1/2) and H+/myo-inositol transporter (HMIT) (Fig. 5) (192, 193). In addition, the dephosphorylation of inositol phosphates following the decomposition of inositol-containing membrane phospholipids also participates in the generation of free myo-inositol (Fig. 5) (194).
Figure 5.

The structure, source, and catabolism of myo-inositol. Intracellular myo-inositol is derived in three different ways: de novo synthesis, H+/myo-inositol transporter (HMIT)/Na+/myo-inositol transporter 1/2 (SMIT1/2)-mediated absorption, and dephosphorylation of inositol phosphates. De novo synthesis of myo-inositol from glucose is catalyzed by hexokinase, 1-d-myo-inositolphosphate synthase (MIPS), and inositol monophosphatase (IMPase) sequentially. Myo-inositol is catabolized by myo-inositol oxygenase (MIOX), a renal proximal tubule-specific enzyme, to generate d-glucuronic acid, which is further converted into xylitol via the glucuronate-xylulose (G-X) pathway. Xylitol is catabolized into d-xylulose-5-phosphate, which is involved in the pentose phosphate cycle. ALR1, aldehyde reductase; DGDC, dehydro-l-gulonate decarboxylase; DXR, d-xylulose reductase; GDH, l-gulonate dedydrogenase; LXR, l-xylulose reductase. [Partially adapted from Ref. (197).]
Unlike the multiple sites of myo-inositol synthesis, its catabolism is restricted only in the kidneys, and it is mediated by the proximal tubular specific enzyme, myo-inositol oxygenase (MIOX) (Fig. 5) (195). The pioneering work by Howard and Anderson revealed that bilateral nephrectomized rats were unable to degrade [2-14C]myo-inositol into 14CO2, indicating an essential role of the kidneys in myo-inositol catabolism (196). It is noteworthy that the urinary excretion of myo-inositol is marginal in normal states, which gets accelerated in the diabetic state with depletion of plasma myo-inositol levels (186). It was later on discovered that myo-inositol undergoes oxidative cleavage of the cyclic ring by MIOX in renal proximal tubular cells, yielding d-glucuronic acid (195, 197). Incidentally, it has been reported in the literature that genetic deletion of MIOX leads to elevated myo-inositol levels in murine serum and renal tissues (197). d-Glucuronic acid is then converted into xylitol via the glucuronate-xylulose pathway, and xylitol is further catabolized into d-xylulose-5-phosphate, which enters the pentose phosphate cycle (Fig. 5) (195). d-Chiro-inositol, an isomer of myo-inositol involved in insulin signaling transduction, is also a cleavage substrate of MIOX (195). Previous investigations have revealed that the catalytic activity of MIOX consumes NADPH and drives renal redox-related injuries in various pathological states (198–200). In addition, our recent work has implicated MIOX overexpression in the induction of apoptosis and ferroptosis in cisplatin-induced AKI (149, 198).
Myo-inositol is present mainly in its free form or as myo-inositol phospholipid in the kidneys, among which free myo-inositol is the predominant form (187). It is worth mentioning here that the renal concentration of myo-inositol also increases in mice following dietary myo-inositol supplementation (201). Although the beneficial effects of myo-inositol supplementation have been reported in various disease states, its role in renal pathology has been poorly investigated. Previous in vitro studies have revealed that myo-inositol treatment alleviates high-glucose-induced collagen synthesis by renal proximal tubular cells (202), whereas diabetic glomerulopathy was unaffected by myo-inositol administration (203). Besides, myo-inositol treatment alleviates the acute renal failure induced by methylene-myo-inositol (a SMIT inhibitor), which may possibly be related to the perturbations in osmoregulation by renal medullary tubules (204). Along these lines, more recent studies have indicated that myo-inositol is protective against cadmium-induced AKI via attenuating oxidant stress and apoptosis (205), although the precise mechanism(s) in alleviation of renal injury remains elusive.
Compared with the limited research on free myo-inositol relevant to AKI, the role of myo-inositol phospholipids in AKI has been extensively investigated. The intricacies and interconversion of various inositol phospholipids have been alluded to earlier (186, 187), and thus only RCD and AKI-related mechanisms are discussed here. Among the various forms of inositol phospholipids, PI, localized to the ER, is the most common form of inositol in mammalian cells (187, 206). Synthesis of PI relies on catalysis by PI synthase, which allows the reaction between myo-inositol and cytidine diphosphate-diacylglycerol (DAG) to yield PI (Figs. 6 and 7) (186, 207). PI is readily phosphorylated into PI phosphate and PI 4,5-bisphosphate [PI(4,5)P2] by PI kinase and PI phosphate kinase, respectively, in various biological settings (Figs. 6 and 7) (186). Under the catalysis of phospholipase C, PI(4,5)P2 is further hydrolyzed into DAG and inositol 1,4,5-trisphosphate (IP3) (Figs. 6 and 7), and both participate in various biological processes as second messengers but initiate distinct downstream signaling (208). DAG remains on the plasma membrane and facilitates the plasmalemmal translocation of protein kinase C (PKC) and its subsequent activation (209). On the other hand, IP3 is diffused into the cytosol and binds to its receptor localized on the ER Ca2+ channel, culminating in an increased concentration of cytosolic Ca2+ (Fig. 6D), which also leads to PKC activation (210). Various PKCs have been implicated in the pathogenesis of AKI in different murine models (211–214). PKC inhibition by Go6976 alleviates cisplatin-induced apoptosis in renal proximal tubular cells, which is apparently related to the reduced mitochondrial cytochrome c release and caspase-3 activation (215). Of note, the role of PKC in apoptosis and downstream signaling may be cell specific, and it can decelerate apoptosis in certain states. Also, there are reports that have indicated that PKC can drive necroptosis, pyroptosis, ferroptosis, and MPT-mediated necrosis in other non-AKI settings (214, 216–218). Another inositol phospholipid PI 3,4,5-trisphosphate [PI(3,4,5)P3], which is synthesized by class I phosphoinositol 3-kinase (PI3K)-mediated phosphorylation of PI(4,5)P2, also modulates RCDs via the phosphoinositide-dependent kinase 1 (PDK1)/AKT pathway (Figs. 6 and 7) (186). PI(3,4,5)P3 resides on the plasma membrane and recruits PDK1 and AKT, and both contain PH domains that are essential for interactions (219). The interaction between PI(3,4,5)P3 and PDK1 leads to activation of PDK1, which then phosphorylates and actives AKT (Fig. 7) (219). PI3K inhibition promotes cisplatin-induced proximal tubular apoptosis via hindering AKT-mediated Bcl-2-associated death promoter phosphorylation, which restores the antiapoptosis capacity of Bcl-XL/Bcl-2 (220). Besides, PI3K/AKT signaling also protects against other forms of AKIs via activation of NRF2 (221, 222), with the latter being a vital antioxidant in the pathogenesis of ferroptosis. Also, the role of PI3K/AKT signaling has also been implicated in necroptosis, pyroptosis, and MPT-mediated necrosis in non-AKI states (217, 223, 224); however, the precise mechanism(s) in these processes has not been thoroughly investigated. Apart from IP3/DAG- and PI3K-mediated intracellular events, multiple types of polyphosphoinositides per se participate in necroptosis and pyroptosis in terminal stages via binding with phosphorylated MLKL and GSDMD-N, which apparently leads to membrane disruption and cell death (76, 78, 110).
Figure 6.

Key events of myo-inositol derivatives. A: synthesis of phosphatidylinositol (PI). B: synthesis of phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] and phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3]. C: decomposition of PI(4,5)P2 into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). D: IP3 binds with its receptor on the endoplasmic reticulum Ca2+ channel, resulting in endoplasmic retriculum Ca2+ release. CDP-DAG, cytidine diphosphate-diacylglycerol; CMP, cytidine monophosphate; PI3K, phosphatidylinositol 3-kinase; PIP, phosphatidylinositol phosphate; PLC, phospholipase C.
Figure 7.

Connections between myo-inositol metabolism and regulated cell deaths in acute kidney injury (AKI). Phosphatidylinositol (PI), synthesized in the endoplasmic reticulum (ER), is converted into phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] after a series of events. PI(4,5)P2 is hydrolyzed into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) by phospholipase C (PLC). DAG and IP3-mediated increased cytoplasmic Ca2+ bind with protein kinase C (PKC), leading to its activation. PKC promotes cisplatin-induced AKI by facilitating cytochrome c (Cyt C) release-mediated apoptosis. On the other hand, PI(4,5)P2 can be further phosphorylated by phosphatidylinositol 3-kinase (PI3K) into phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3], which binds with phosphoinositide-dependent kinase 1 (PDK1) to activate AKT. AKT attenuates apoptosis in cisplatin-induced AKI via BAD-restored BCL-2/BCL-XL potency. AKT also protects against other AKIs through nuclear factor erythroid 2-related factor 2 (NRF2), a potent antioxidant of ferroptosis. Of note, polyphosphoinositides present as the binding substrates of mixed lineage kinase domain-like (MLKL) and NH2-terminal domain of gasdermin D (GSDMD-N), indicating their involvement in necroptosis and pyroptosis. PIP, phosphatidylinositol phosphate.
PERSPECTIVES, CONFLICTS, AND CONCLUSIONS
Over a period of the past two decades, more than 10 variants of RCDs have been described, among which only five types (related to cisplatin-induced AKI) are discussed in this review. Nevertheless, it should be stressed here that other forms of RCDs may also be involved in cisplatin-induced AKI. Due to the insufficient period of investigation following their discovery, the pathological mechanisms of most of the RCDs are poorly investigated, and thus the research in cisplatin-induced AKI has not been fully explored. Limited past investigatory efforts and the diversity of RCDs provide ample research opportunities for AKI-related investigations, since some key factors or relevant mechanism(s) in other forms of RCDs have been investigated before their discoveries. For example, autosis, an autophagic machinery-mediated RCD (49), has never been investigated in cisplatin-induced AKI, even if the role of autophagy has been well documented in the literature (225). In addition, parthanatos, driven by poly(ADP-ribose) polymerase 1 hyperactivation, can be kindled by excessive oxidative stress, hypoxia, or inflammation (226), the very biological processes that are notably observed in cisplatin-induced AKI. Further research is needed to explore the full profile of various RCDs in cisplatin-related nephropathy. Due to the lack of specific markers, it is noteworthy that the evaluation of ferroptosis in vivo by pathological staining is somewhat limited, which is different from the detection of apoptosis by its specific indicators, i.e., cleaved caspase-3, BAX, Bcl-2, or others. More convincing investigations are required to uncover the unique indicators for ferroptosis. The interaction between RCD and myo-inositol metabolism also remains to be fully investigated. For instance, free myo-inositol supplement protects against AKI via alleviating apoptosis (205), while its role in other RCDs has not been investigated. Besides, the PI(4,5)P2-mediated PKC pathway and PI(3,4,5)P3-related PI3K/AKT signaling are involved in some of the nonapoptotic RCDs, but the exact mechanisms have been poorly explored.
It is noteworthy that the conclusions in different publications seem to be controversial in certain aspects. For instance, our recent investigation revealed that ferroptosis and apoptosis, rather than necroptosis, are essential in cisplatin-induced AKI (149), and these results are similar to the seminal observations made by Linkermann et al. (147). However, other investigations using genetic modification approaches revealed that “necroptosis” is a vital component in cisplatin-induced tubular cell death (79, 83). A comprehensive examination of these publications revealed that the cisplatin concentration in our experiments was 20 μM, far lower than that used by Monte and Han (100 − 200 μM) (79, 83, 149). These unique observations may suggest that various RCDs are initiated at different stages of injury following cisplatin treatment in in vitro studies. In another setting, the research work by Han and coworkers et al. (83) on MLKL or RIPK3 knockout mice provided convincing evidence for the pathogenetic role of necroptosis in cisplatin-induced AKI, whereas other investigators believed that necroptosis seems to modulate renal vascular effects rather than renal tubular injuries (147). It seems that caspase-11-mediated noncanonical pyroptosis is conceivably operative in cisplatin-induced AKI (122). The pathogenetic role of caspase-11 in this scenario may be somewhat debatable since no exogenous LPS was introduced into the kidneys to activate caspase-11, indicating that other potential mechanisms may be operational in the induction of pyroptosis.
Finally, it should be borne in mind that cisplatin-induced renal cell death is rather complex, involving various types of RCDs that are initiated at different stages of injury. Besides, various RCDs per se are interconnected, and myo-inositol metabolism may be participating in the pathogenesis of RCDs at different stages/levels. The involvement of RCDs at different stages/levels may yield the opportunity for exploration of novel drugs with differing properties for the inhibition of RCDs or small inhibitory molecules relevant to myo-inositol metabolism may provide potential avenues to battle against cisplatin-induced AKI.
GRANTS
The work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK60635, Visiting Scholarships from China Scholar Council Grant 201706370164, National Natural Science Foundation of China Grant 81470925, and Science and Technology program of Guangdong Scientific Committee Grant 2019A1515012116.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F.D., Y.D., and Y.S.K. conceived and designed research; X.Z. prepared figures; F.D. drafted manuscript; X.Z., I.S., Y.W., and Y.S.K. edited and revised manuscript; Y.D. and Y.S.K. approved final version of manuscript.
REFERENCES
- 1.Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 740: 364–378, 2014. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene 31: 1869–1883, 2012. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 3.Hermans TJN, Voskuilen CS, van der Heijden MS, Schmitz-Drager BJ, Kassouf W, Seiler R, Kamat AM, Grivas P, Kiltie AE, Black PC, van Rhijn BWG. Neoadjuvant treatment for muscle-invasive bladder cancer: the past, the present, and the future. Urol Oncol 36: 413–422, 2018. doi: 10.1016/j.urolonc.2017.10.014. [DOI] [PubMed] [Google Scholar]
- 4.Pichler R, Horninger W, Heidegger I. ASCO 2018: highlights of urothelial cancer and prostate cancer. Memo 11: 284–290, 2018. doi: 10.1007/s12254-018-0422-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg B, Vancamp L, Krigas T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 205: 698–699, 1965. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg B, VanCamp L. The successful regression of large solid sarcoma 180 tumors by platinum compounds. Cancer Res 30: 1799–1802, 1970. [PubMed] [Google Scholar]
- 7.Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, Nick HS, Agarwal A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol 278: F726–F736, 2000. doi: 10.1152/ajprenal.2000.278.5.F726. [DOI] [PubMed] [Google Scholar]
- 8.Sharp CN, Siskind LJ. Developing better mouse models to study cisplatin-induced kidney injury. Am J Physiol Renal Physiol 313: F835–F841, 2017. doi: 10.1152/ajprenal.00285.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu Y, Cai J, Li F, Liu Z, Shu S, Wang Y, Liu Y, Tang C, Dong Z. Chronic effects of repeated low-dose cisplatin treatment in mouse kidneys and renal tubular cells. Am J Physiol Renal Physiol 317: F1582–F1592, 2019. doi: 10.1152/ajprenal.00385.2019. [DOI] [PubMed] [Google Scholar]
- 10.Sharp CN, Doll MA, Dupre TV, Shah PP, Subathra M, Siow D, Arteel GE, Megyesi J, Beverly LJ, Siskind LJ. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am J Physiol Renal Physiol 310: F560–F568, 2016. doi: 10.1152/ajprenal.00512.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Safirstein R, Miller P, Guttenplan JB. Uptake and metabolism of cisplatin by rat kidney. Kidney Int 25: 753–758, 1984. doi: 10.1038/ki.1984.86. [DOI] [PubMed] [Google Scholar]
- 12.Estrela GR, Wasinski F, Felizardo RJF, Souza LL, Camara NOS, Bader M, Araujo RC. MATE-1 modulation by kinin B1 receptor enhances cisplatin efflux from renal cells. Mol Cell Biochem 428: 101–108, 2017. doi: 10.1007/s11010-016-2920-x. [DOI] [PubMed] [Google Scholar]
- 13.Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res Int 2014: 967826, 2014. doi: 10.1155/2014/967826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kroning R, Lichtenstein AK, Nagami GT. Sulfur-containing amino acids decrease cisplatin cytotoxicity and uptake in renal tubule epithelial cell lines. Cancer Chemother Pharmacol 45: 43–49, 2000. doi: 10.1007/PL00006741. [DOI] [PubMed] [Google Scholar]
- 15.Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther 86: 396–402, 2009. doi: 10.1038/clpt.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pabla N, Gibson AA, Buege M, Ong SS, Li L, Hu S, Du G, Sprowl JA, Vasilyeva A, Janke LJ, Schlatter E, Chen T, Ciarimboli G, Sparreboom A. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc Natl Acad Sci USA 112: 5231–5236, 2015. doi: 10.1073/pnas.1424313112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pabla N, Murphy RF, Liu K, Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am J Physiol Renal Physiol 296: F505–F511, 2009. doi: 10.1152/ajprenal.90545.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodsell DS. The molecular perspective: cisplatin. Stem Cells 24: 514–515, 2006. doi: 10.1634/stemcells.2006-CSC2. [DOI] [PubMed] [Google Scholar]
- 19.Cisplatin. In: Encyclopedia of Cancer, edited by Schwab M. Berlin, Heidelberg: Springer, 2011, p. 869–872. [Google Scholar]
- 20.Hu J, Lieb JD, Sancar A, Adar S. Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc Natl Acad Sci USA 113: 11507–11512, 2016. doi: 10.1073/pnas.1614430113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu S, Pabla N, Tang C, He L, Dong Z. DNA damage response in cisplatin-induced nephrotoxicity. Arch Toxicol 89: 2197–2205, 2015. doi: 10.1007/s00204-015-1633-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol 59: 657–663, 2001. doi: 10.1124/mol.59.4.657. [DOI] [PubMed] [Google Scholar]
- 23.Ichinomiya M, Shimada A, Ohta N, Inouchi E, Ogihara K, Naya Y, Nagane M, Morita T, Satoh M. Demonstration of mitochondrial damage and mitophagy in cisplatin-mediated nephrotoxicity. Tohoku J Exp Med 246: 1–8, 2018. doi: 10.1620/tjem.246.1. [DOI] [PubMed] [Google Scholar]
- 24.Soetikno V, Sari SDP, Ul Maknun L, Sumbung NK, Rahmi DNI, Pandhita BAW, Louisa M, Estuningtyas A. Pre-treatment with curcumin ameliorates cisplatin-induced kidney damage by suppressing kidney inflammation and apoptosis in rats. Drug Res (Stuttg) 69: 75–82, 2019. doi: 10.1055/a-0641-5148. [DOI] [PubMed] [Google Scholar]
- 25.Soni H, Kaminski D, Gangaraju R, Adebiyi A. Cisplatin-induced oxidative stress stimulates renal Fas ligand shedding. Ren Fail 40: 314–322, 2018. doi: 10.1080/0886022X.2018.1456938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soni H, Matthews AT, Pallikkuth S, Gangaraju R, Adebiyi A. Gamma-secretase inhibitor DAPT mitigates cisplatin-induced acute kidney injury by suppressing Notch1 signaling. J Cell Mol Med 23: 260–270, 2019. doi: 10.1111/jcmm.13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26: 239–257, 1972. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galluzzi L, Lopez-Soto A, Kumar S, Kroemer G. Caspases connect cell-death signaling to organismal homeostasis. Immunity 44: 221–231, 2016. doi: 10.1016/j.immuni.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 29.Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity 50: 1352–1364, 2019. doi: 10.1016/j.immuni.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larsen BD, Sorensen CS. The caspase-activated DNase: apoptosis and beyond. FEBS J 284: 1160–1170, 2017. doi: 10.1111/febs.13970. [DOI] [PubMed] [Google Scholar]
- 31.Samejima K, Ogawa H, Ageichik AV, Peterson KL, Kaufmann SH, Kanemaki MT, Earnshaw WC. Auxin-induced rapid degradation of inhibitor of caspase-activated DNase (ICAD) induces apoptotic DNA fragmentation, caspase activation, and cell death: a cell suicide module. J Biol Chem 289: 31617–31623, 2014. doi: 10.1074/jbc.M114.583542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pefanis A, Ierino FL, Murphy JM, Cowan PJ. Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int 96: 291–301, 2019. doi: 10.1016/j.kint.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 33.Lieberthal W, Triaca V, Levine J. Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: apoptosis vs. necrosis. Am J Physiol 270: F700–F708, 1996. doi: 10.1152/ajprenal.1996.270.4.F700. [DOI] [PubMed] [Google Scholar]
- 34.Takeda M, Fukuoka K, Endou H. Cisplatin-induced apoptosis in mouse proximal tubular cell line. Contrib Nephrol 118: 24–28, 1996. doi: 10.1159/000425072. [DOI] [PubMed] [Google Scholar]
- 35.Kim JY, Jo J, Kim K, An HJ, Gwon MG, Gu H, Kim HJ, Yang AY, Kim SW, Jeon EJ, Park JH, Leem J, Park KK. Pharmacological activation of Sirt1 ameliorates cisplatin-induced acute kidney injury by suppressing apoptosis, oxidative stress, and inflammation in mice. Antioxidants (Basel) 8: 322, 2019. doi: 10.3390/antiox8080322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh CJ, Ha CM, Choi YK, Park S, Choe MS, Jeoung NH, Huh YH, Kim HJ, Kweon HS, Lee JM, Lee SJ, Jeon JH, Harris RA, Park KG, Lee IK. Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int 91: 880–895, 2017. doi: 10.1016/j.kint.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 37.Tsogbadrakh B, Ryu H, Ju KD, Lee J, Yun S, Yu KS, Kim HJ, Ahn C, Oh KH. AICAR, an AMPK activator, protects against cisplatin-induced acute kidney injury through the JAK/STAT/SOCS pathway. Biochem Biophys Res Commun 509: 680–686, 2019. doi: 10.1016/j.bbrc.2018.12.159. [DOI] [PubMed] [Google Scholar]
- 38.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 35: 495–516, 2007. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holditch SJ, Brown CN, Lombardi AM, Nguyen KN, Edelstein CL. Recent advances in models, mechanisms, biomarkers, and interventions in cisplatin-induced acute kidney injury. Int J Mol Sci 20: 3011, 2019. doi: 10.3390/ijms20123011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu G, Yuan Y, Long M, Luo T, Bian J, Liu X, Gu J, Zou H, Song R, Wang Y, Wang L, Liu Z. Beclin-1-mediated autophagy protects against cadmium-activated apoptosis via the Fas/FasL pathway in primary rat proximal tubular cell culture. Sci Rep 7: 977, 2017. doi: 10.1038/s41598-017-00997-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Liu N, Xue X, Zhou S. The effect of overexpression of the enhancer of zeste homolog 1 (EZH1) gene on aristolochic acid-induced injury in HK-2 human kidney proximal tubule cells in vitro. Med Sci Monit 25: 801–810, 2019. doi: 10.12659/MSM.911611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Razzaque MS, Koji T, Kumatori A, Taguchi T. Cisplatin-induced apoptosis in human proximal tubular epithelial cells is associated with the activation of the Fas/Fas ligand system. Histochem Cell Biol 111: 359–365, 1999. doi: 10.1007/s004180050368. [DOI] [PubMed] [Google Scholar]
- 43.Tsuruya K, Ninomiya T, Tokumoto M, Hirakawa M, Masutani K, Taniguchi M, Fukuda K, Kanai H, Kishihara K, Hirakata H, Iida M. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int 63: 72–82, 2003. doi: 10.1046/j.1523-1755.2003.00709.x. [DOI] [PubMed] [Google Scholar]
- 44.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 110: 835–842, 2002. doi: 10.1172/JCI15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramesh G, Reeves WB. Inflammatory cytokines in acute renal failure. Kidney Int Suppl 66: S56–S61, 2004. doi: 10.1111/j.1523-1755.2004.09109.x. [DOI] [PubMed] [Google Scholar]
- 46.Ramesh G, Reeves WB. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am J Physiol Renal Physiol 285: F610–F618, 2003. doi: 10.1152/ajprenal.00101.2003. [DOI] [PubMed] [Google Scholar]
- 47.Al-Lamki RS, Lu W, Finlay S, Twohig JP, Wang EC, Tolkovsky AM, Bradley JR. DR3 signaling protects against cisplatin nephrotoxicity mediated by tumor necrosis factor. Am J Pathol 180: 1454–1464, 2012. doi: 10.1016/j.ajpath.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 48.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007, 2008. [DOI] [PubMed] [Google Scholar]
- 49.Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res 29: 347–364, 2019. doi: 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu S, Huang H, Deng G, Xie Z, Ye Y, Guo R, Cai X, Hong J, Qian D, Zhou X, Tao Z, Chen B, Li Q. miR-326 targets antiapoptotic Bcl-xL and mediates apoptosis in human platelets. PloS One 10: e0122784, 2015. doi: 10.1371/journal.pone.0122784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274: 11549–11556, 1999. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 52.Lee RH, Song JM, Park MY, Kang SK, Kim YK, Jung JS. Cisplatin-induced apoptosis by translocation of endogenous Bax in mouse collecting duct cells. Biochem Pharmacol 62: 1013–1023, 2001. [DOI] [PubMed] [Google Scholar]
- 53.Wei Q, Dong G, Franklin J, Dong Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int 72: 53–62, 2007. doi: 10.1038/sj.ki.5002256. [DOI] [PubMed] [Google Scholar]
- 54.Jiang M, Pabla N, Murphy RF, Yang T, Yin XM, Degenhardt K, White E, Dong Z. Nutlin-3 protects kidney cells during cisplatin therapy by suppressing Bax/Bak activation. J Biol Chem 282: 2636–2645, 2007. doi: 10.1074/jbc.M606928200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang M, Wei Q, Wang J, Du Q, Yu J, Zhang L, Dong Z. Regulation of PUMA-alpha by p53 in cisplatin-induced renal cell apoptosis. Oncogene 25: 4056–4066, 2006. doi: 10.1038/sj.onc.1209440. [DOI] [PubMed] [Google Scholar]
- 56.Kruidering M, van de Water B, Zhan Y, Baelde JJ, Heer E, Mulder GJ, Stevens JL, Nagelkerke JF. Cisplatin effects on F-actin and matrix proteins precede renal tubular cell detachment and apoptosis in vitro. Cell death Differ 5: 601–614, 1998. doi: 10.1038/sj.cdd.4400392. [DOI] [PubMed] [Google Scholar]
- 57.Li W, Yang Y, Li Y, Zhao Y, Jiang H. Sirt5 attenuates cisplatin-induced acute kidney injury through regulation of Nrf2/HO-1 and Bcl-2. BioMed Res Int 2019: 4745132, 2019. doi: 10.1155/2019/4745132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tan Z, Guo F, Huang Z, Xia Z, Liu J, Tao S, Li L, Feng Y, Du X, Ma L, Fu P. Pharmacological and genetic inhibition of fatty acid-binding protein 4 alleviated cisplatin-induced acute kidney injury. J Cell Mol Med 23: 6260–6270, 2019. doi: 10.1111/jcmm.14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maekawa H, Inagi R. Stress signal network between hypoxia and ER stress in chronic kidney disease. Front Physiol 8: 74, 2017. doi: 10.3389/fphys.2017.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim C, Kim B. Anti-cancer natural products and their bioactive compounds inducing ER stress-mediated apoptosis: a review. Nutrients 10: 1021, 2018. doi: 10.3390/nu10081021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem 276: 13935–13940, 2001. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 62.Hao Y, Guo F, Huang Z, Feng Y, Xia Z, Liu J, Li L, Huang R, Lin L, Ma L, Fu P. 2-Methylquinazoline derivative 23BB as a highly selective histone deacetylase 6 inhibitor alleviated cisplatin-induced acute kidney injury. Biosci Rep 40: BSR20191538, 2020. doi: 10.1042/BSR20191538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang Z, Guo F, Xia Z, Liang Y, Lei S, Tan Z, Ma L, Fu P. Activation of GPR120 by TUG891 ameliorated cisplatin-induced acute kidney injury via repressing ER stress and apoptosis. Biomed Pharmacother 126: 110056, 2020. doi: 10.1016/j.biopha.2020.110056. [DOI] [PubMed] [Google Scholar]
- 64.Kong D, Zhuo L, Gao C, Shi S, Wang N, Huang Z, Li W, Hao L. Erythropoietin protects against cisplatin-induced nephrotoxicity by attenuating endoplasmic reticulum stress-induced apoptosis. J Nephrol 26: 219–227, 2013. doi: 10.5301/jn.5000177. [DOI] [PubMed] [Google Scholar]
- 65.Peyrou M, Hanna PE, Cribb AE. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol Sci 99: 346–353, 2007. doi: 10.1093/toxsci/kfm152. [DOI] [PubMed] [Google Scholar]
- 66.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1: 112–119, 2005. [Erratum in Nat Chem Biol 1: 234, 2005] doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 67.Abd-Ellatif RN, Hegab II, Atef MM, Sadek MT, Hafez YM. Diacerein protects against glycerol-induced acute kidney injury: Modulating oxidative stress, inflammation, apoptosis and necroptosis. Chem Biol Interact 306: 47–53, 2019. doi: 10.1016/j.cbi.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 68.Homsi E, Andreazzi DD, Faria JB, Janino P. TNF-alpha-mediated cardiorenal injury after rhabdomyolysis in rats. Am J Physiol Renal Physiol 308: F1259–F1267, 2015. doi: 10.1152/ajprenal.00311.2014. [DOI] [PubMed] [Google Scholar]
- 69.Linkermann A, Heller JO, Prokai A, Weinberg JM, De Zen F, Himmerkus N, Szabo AJ, Brasen JH, Kunzendorf U, Krautwald S. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J Am Soc Nephrol 24: 1545–1557, 2013. [Erratum in J Am Soc Nephrol 25: 2942, 2014] doi: 10.1681/ASN.2012121169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu W, Chen B, Wang Y, Meng C, Huang H, Huang XR, Qin J, Mulay SR, Anders HJ, Qiu A, Yang B, Freeman GJ, Lu HJ, Lin HY, Zheng ZH, Lan HY, Huang Y, Xia Y. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc Natl Acad Sci USA 115: E1475–E1484, 2018. doi: 10.1073/pnas.1716959115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martin-Sanchez D, Fontecha-Barriuso M, Carrasco S, Sanchez-Nino MD, Massenhausen AV, Linkermann A, Cannata-Ortiz P, Ruiz-Ortega M, Egido J, Ortiz A, Sanz AB. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc Natl Acad Sci USA 115: 4182–4187, 2018. [Erratum in Proc Natl Acad Sci USA 115: E4731, 2018] doi: 10.1073/pnas.1716578115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mulay SR, Desai J, Kumar SV, Eberhard JN, Thomasova D, Romoli S, Grigorescu M, Kulkarni OP, Popper B, Vielhauer V, Zuchtriegel G, Reichel C, Brasen JH, Romagnani P, Bilyy R, Munoz LE, Herrmann M, Liapis H, Krautwald S, Linkermann A, Anders HJ. Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat Commun 7: 10274, 2016. doi: 10.1038/ncomms10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anders HJ. Necroptosis in acute kidney injury. Nephron 139: 342–348, 2018. doi: 10.1159/000489940. [DOI] [PubMed] [Google Scholar]
- 74.Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol 12: 103–130, 2017. doi: 10.1146/annurev-pathol-052016-100247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muller T, Dewitz C, Schmitz J, Schroder AS, Brasen JH, Stockwell BR, Murphy JM, Kunzendorf U, Krautwald S. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci 74: 3631–3645, 2017. doi: 10.1007/s00018-017-2547-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, Bertin J, Gough PJ, Savvides S, Martinou JC, Bertrand MJ, Vandenabeele P. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7: 971–981, 2014. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 77.Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RC, Webb AI, Tripaydonis A, Babon JJ, Mulcair MD, Scanlon MJ, Alexander WS, Wilks AF, Czabotar PE, Lessene G, Murphy JM, Silke J. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA 111: 15072–15077, 2014. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T, Green DR. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol Cell 61: 589–601, 2016. doi: 10.1016/j.molcel.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tristão VR, Gonçalves PF, Dalboni MA, Batista MC, Durão M. D S, Monte JCM. Nec-1 protects against nonapoptotic cell death in cisplatin-induced kidney injury. Ren Fail 34: 373–377, 2012. doi: 10.3109/0886022X.2011.647343. [DOI] [PubMed] [Google Scholar]
- 80.Ning Y, Shi Y, Chen J, Song N, Cai J, Fang Y, Yu X, Ji J, Ding X. Necrostatin-1 attenuates cisplatin-induced nephrotoxicity through suppression of apoptosis and oxidative stress and retains klotho expression. Front Pharmacol 9: 384, 2018. doi: 10.3389/fphar.2018.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tristao VR, Pessoa EA, Nakamichi R, Reis LA, Batista MC, Durao Junior Mde S, Monte JC. Synergistic effect of apoptosis and necroptosis inhibitors in cisplatin-induced nephrotoxicity. Apoptosis 21: 51–59, 2016. doi: 10.1007/s10495-015-1190-5. [DOI] [PubMed] [Google Scholar]
- 82.Wang JN, Liu MM, Wang F, Wei B, Yang Q, Cai YT, Chen X, Liu XQ, Jiang L, Li C, Hu XW, Yu JT, Ma TT, Jin J, Wu YG, Li J, Meng XM. RIPK1 inhibitor Cpd-71 attenuates renal dysfunction in cisplatin-treated mice via attenuating necroptosis, inflammation and oxidative stress. Clin Sci (Lond) 133: 1609–1627, 2019. doi: 10.1042/CS20190599. [DOI] [PubMed] [Google Scholar]
- 83.Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, Wang Y, Huang Z, Ren J, Liu S, Chen X, Han J. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol 26: 2647–2658, 2015. doi: 10.1681/ASN.2014080741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim JW, Jo J, Kim JY, Choe M, Leem J, Park JH. Melatonin attenuates cisplatin-induced acute kidney injury through dual suppression of apoptosis and necroptosis. Biology (Basel) 8: 64, 2019. doi: 10.3390/biology8030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meng XM, Li HD, Wu WF, Ming-Kuen Tang P, Ren GL, Gao L, Li XF, Yang Y, Xu T, Ma TT, Li Z, Huang C, Zhang L, Lv XW, Li J. Wogonin protects against cisplatin-induced acute kidney injury by targeting RIPK1-mediated necroptosis. Lab Invest 98: 79–94, 2018. doi: 10.1038/labinvest.2017.115. [DOI] [PubMed] [Google Scholar]
- 86.Zang H, Yang Q, Li J. Eleutheroside B protects against acute kidney injury by activating IGF pathway. Molecules 24: 3876, 2019. doi: 10.3390/molecules24213876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA 110: 12024–12029, 2013. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis 3: e437, 2012. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vandenabeele P, Grootjans S, Callewaert N, Takahashi N. Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ 20: 185–187, 2013. doi: 10.1038/cdd.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, Lee TH, Chan FKM, Pasparakis M, Kelliher MA. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol 193: 1539–1543, 2014. doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol 16: 689–697, 2015. [Erratum in Nat Immunol 16: 889, 2015] doi: 10.1038/ni.3206. [DOI] [PubMed] [Google Scholar]
- 92.Jiang L, Liu XQ, Ma Q, Yang Q, Gao L, Li HD, Wang JN, Wei B, Wen J, Li J, Wu YG, Meng XM. hsa-miR-500a-3P alleviates kidney injury by targeting MLKL-mediated necroptosis in renal epithelial cells. FASEB J 33: 3523–3535, 2019. doi: 10.1096/fj.201801711R. [DOI] [PubMed] [Google Scholar]
- 93.Landau SI, Guo X, Velazquez H, Torres R, Olson E, Garcia-Milian R, Moeckel GW, Desir GV, Safirstein R. Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int 95: 797–814, 2019. doi: 10.1016/j.kint.2018.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tonnus W, Belavgeni A, Xu Y, Linkermann A. Don't trick me twice! Kidney Int 95: 736–738, 2019. doi: 10.1016/j.kint.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 95.Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Nino MD, Ruiz Ortega M, Egido J, Linkermann A, Ortiz A, Sanz AB. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol 28: 218–229, 2017. doi: 10.1681/ASN.2015121376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol 38: 31–40, 2000. doi: 10.1046/j.1365-2958.2000.02103.x. [DOI] [PubMed] [Google Scholar]
- 97.Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol 9: 113–114, 2001. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 98.Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci 42: 245–254, 2017. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 99.Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, Yu T, Wu X, Shi Y, Ma P, Shu Y. Pyroptosis: a new frontier in cancer. Biomed Pharmacother 121: 109595, 2020. doi: 10.1016/j.biopha.2019.109595. [DOI] [PubMed] [Google Scholar]
- 100.Kayagaki N, Lee BL, Stowe IB, Kornfeld OS, O'Rourke K, Mirrashidi KM, Haley B, Watanabe C, Roose-Girma M, Modrusan Z, Kummerfeld S, Reja R, Zhang Y, Cho V, Andrews TD, Morris LX, Goodnow CC, Bertram EM, Dixit VM. IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci Signal 12: eaax4917, 2019. doi: 10.1126/scisignal.aax4917. [DOI] [PubMed] [Google Scholar]
- 101.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16: 407–420, 2016. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 102.Mamik MK, Power C. Inflammasomes in neurological diseases: emerging pathogenic and therapeutic concepts. Brain 140: 2273–2285, 2017. doi: 10.1093/brain/awx133. [DOI] [PubMed] [Google Scholar]
- 103.Marchesan JT, Girnary MS, Moss K, Monaghan ET, Egnatz GJ, Jiao Y, Zhang S, Beck J, Swanson KV. Role of inflammasomes in the pathogenesis of periodontal disease and therapeutics. Periodontol 82: 93–114, 2020. doi: 10.1111/prd.12269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bierschenk D, Boucher D, Schroder K. Salmonella-induced inflammasome activation in humans. Mol Immunol 86: 38–43, 2017. doi: 10.1016/j.molimm.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 105.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514: 187–192, 2014. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 106.Yi YS. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology 152: 207–217, 2017. doi: 10.1111/imm.12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fujii T, Tamura M, Tanaka S, Kato Y, Yamamoto H, Mizushina Y, Shiroishi T. Gasdermin D (GSDMD) is dispensable for mouse intestinal epithelium development. Genesis 46: 418–423, 2008. doi: 10.1002/dvg.20412. [DOI] [PubMed] [Google Scholar]
- 108.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665, 2015. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 109.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111–116, 2016. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 110.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: 153–158, 2016. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li D, Ren W, Jiang Z, Zhu L. Regulation of the NLRP3 inflammasome and macrophage pyroptosis by the p38 MAPK signaling pathway in a mouse model of acute lung injury. Mol Med Rep 18: 4399–4409, 2018. doi: 10.3892/mmr.2018.9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liao J, Yang F, Tang Z, Yu W, Han Q, Hu L, Li Y, Guo J, Pan J, Ma F, Ma X, Lin Y. Inhibition of Caspase-1-dependent pyroptosis attenuates copper-induced apoptosis in chicken hepatocytes. Ecotoxicol Environ Saf 174: 110–119, 2019. doi: 10.1016/j.ecoenv.2019.02.069. [DOI] [PubMed] [Google Scholar]
- 113.Zhang L, Liu H, Jia L, Lyu J, Sun Y, Yu H, Li H, Liu W, Weng Y, Yu W. Exosomes mediate hippocampal and cortical neuronal injury induced by hepatic ischemia-reperfusion injury through activating pyroptosis in rats. Oxid Med Cell Longev 2019: 3753485, 2019. doi: 10.1155/2019/3753485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chung SD, Lai TY, Chien CT, Yu HJ. Activating Nrf-2 signaling depresses unilateral ureteral obstruction-evoked mitochondrial stress-related autophagy, apoptosis and pyroptosis in kidney. PLoS One 7: e47299, 2012. doi: 10.1371/journal.pone.0047299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chou X, Ding F, Zhang X, Ding X, Gao H, Wu Q. Sirtuin-1 ameliorates cadmium-induced endoplasmic reticulum stress and pyroptosis through XBP-1s deacetylation in human renal tubular epithelial cells. Arch Toxicol 93: 965–986, 2019. doi: 10.1007/s00204-019-02415-8. [DOI] [PubMed] [Google Scholar]
- 116.Li X, Zeng L, Cao C, Lu C, Lian W, Han J, Zhang X, Zhang J, Tang T, Li M. Long noncoding RNA MALAT1 regulates renal tubular epithelial pyroptosis by modulated miR-23c targeting of ELAVL1 in diabetic nephropathy. Exp Cell Res 350: 327–335, 2017. doi: 10.1016/j.yexcr.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 117.Song Z, Zhang Y, Gong B, Xu H, Hao Z, Liang C. Long noncoding RNA LINC00339 promotes renal tubular epithelial pyroptosis by regulating the miR-22-3p/NLRP3 axis in calcium oxalate-induced kidney stone. J Cell Biochem 120: 10452–10462, 2019. doi: 10.1002/jcb.28330. [DOI] [PubMed] [Google Scholar]
- 118.Yang JR, Yao FH, Zhang JG, Ji ZY, Li KL, Zhan J, Tong YN, Lin LR, He YN. Ischemia-reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am J Physiol Renal Physiol 306: F75–F84, 2014. doi: 10.1152/ajprenal.00117.2013. [DOI] [PubMed] [Google Scholar]
- 119.Ye Z, Zhang L, Li R, Dong W, Liu S, Li Z, Liang H, Wang L, Shi W, Malik AB, Cheng KT, Liang X. Caspase-11 mediates pyroptosis of tubular epithelial cells and septic acute kidney injury. Kidney Blood Press Res 44: 465–478, 2019. doi: 10.1159/000499685. [DOI] [PubMed] [Google Scholar]
- 120.Zhang Z, Shao X, Jiang N, Mou S, Gu L, Li S, Lin Q, He Y, Zhang M, Zhou W, Ni Z. Caspase-11-mediated tubular epithelial pyroptosis underlies contrast-induced acute kidney injury. Cell Death Dis 9: 983, 2018. doi: 10.1038/s41419-018-1023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li Y, Xia W, Wu M, Yin J, Wang Q, Li S, Zhang A, Huang S, Zhang Y, Jia Z. Activation of GSDMD contributes to acute kidney injury induced by cisplatin. Am J Physiol Renal Physiol 318: F96–F106, 2020. doi: 10.1152/ajprenal.00351.2019. [DOI] [PubMed] [Google Scholar]
- 122.Miao N, Yin F, Xie H, Wang Y, Xu Y, Shen Y, Xu D, Yin J, Wang B, Zhou Z, Cheng Q, Chen P, Xue H, Zhou L, Liu J, Wang X, Zhang W, Lu L. The cleavage of gasdermin D by caspase-11 promotes tubular epithelial cell pyroptosis and urinary IL-18 excretion in acute kidney injury. Kidney Int 96: 1105–1120, 2019. doi: 10.1016/j.kint.2019.04.035. [DOI] [PubMed] [Google Scholar]
- 123.Faubel S, Ljubanovic D, Reznikov L, Somerset H, Dinarello CA, Edelstein CL. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int 66: 2202–2213, 2004. doi: 10.1111/j.1523-1755.2004.66010.x. [DOI] [PubMed] [Google Scholar]
- 124.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B III, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149: 1060–1072, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stockwell BR, Friedmann AJ, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA, Zhang DD. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171: 273–285, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Feng H, Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol 16: e2006203, 2018. doi: 10.1371/journal.pbio.2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hu Z, Zhang H, Yang SK, Wu X, He D, Cao K, Zhang W. Emerging role of ferroptosis in acute kidney injury. Oxid Med Cell Longev 2019: 8010614, 2019. doi: 10.1155/2019/8010614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett 82–83: 969–974, 1995. [DOI] [PubMed] [Google Scholar]
- 129.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayir H. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13: 81–90, 2017. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev 111: 5866–5898, 2011. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 131.Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, Song S, Tavana O, Gu W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol 21: 579–591, 2019. doi: 10.1038/s41556-019-0305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Karuppagounder SS, Alin L, Chen Y, Brand D, Bourassa MW, Dietrich K, Wilkinson CM, Nadeau CA, Kumar A, Perry S, Pinto JT, Darley-Usmar V, Sanchez S, Milne GL, Pratico D, Holman TR, Carmichael ST, Coppola G, Colbourne F, Ratan RR. N-Acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol 84: 854–872, 2018. doi: 10.1002/ana.25356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA, Mikulska-Ruminska K, Shrivastava IH, Kenny EM, Yang Q, Rosenbaum JC, Sparvero LJ, Emlet DR, Wen X, Minami Y, Qu F, Watkins SC, Holman TR, VanDemark AP, Kellum JA, Bahar I, Bayir H, Kagan VE. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171: 628–641.e626, 2017. doi: 10.1016/j.cell.2017.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Meng X, Deng J, Liu F, Guo T, Liu M, Dai P, Fan A, Wang Z, Zhao Y. Triggered all-active metal organic framework: ferroptosis machinery contributes to the apoptotic photodynamic antitumor therapy. Nano Lett 19: 7866–7876, 2019. doi: 10.1021/acs.nanolett.9b02904. [DOI] [PubMed] [Google Scholar]
- 135.Fang S, Yu X, Ding H, Han J, Feng J. Effects of intracellular iron overload on cell death and identification of potent cell death inhibitors. Biochem Biophys Res Commun 503: 297–303, 2018. doi: 10.1016/j.bbrc.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 136.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509: 105–109, 2014. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 59: 298–308, 2015. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, Zhou H, Cai JH. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci 22: 3826–3836, 2018. doi: 10.26355/eurrev_201806_15267. [DOI] [PubMed] [Google Scholar]
- 139.Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ III, Kang R, Tang D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12: 1425–1428, 2016. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.NaveenKumar SK, SharathBabu BN, Hemshekhar M, Kemparaju K, Girish KS, Mugesh G. The role of reactive oxygen species and ferroptosis in heme-mediated activation of human platelets. ACS Chem Biol 13: 1996–2002, 2018. doi: 10.1021/acschembio.8b00458. [DOI] [PubMed] [Google Scholar]
- 141.Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, Wang H, Cao L, Tang D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 34: 5617–5625, 2015. doi: 10.1038/onc.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, Li B, Fan BY, Wang X, Li WX, Fu XH, Hu Y, Liu C, Kong XH, Feng SQ. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen Res 14: 532–541, 2019. [Erratum in Neural Regen Res 14: 1068, 2019] doi: 10.4103/1673-5374.245480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun 478: 838–844, 2016. doi: 10.1016/j.bbrc.2016.08.034. [DOI] [PubMed] [Google Scholar]
- 144.Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, Bassik MC, Nomura DK, Dixon SJ, Olzmann JA. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575: 688–692, 2019. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, Xia H, Zhou J, Li G, Li J, Li W, Wei S, Vatan L, Zhang H, Szeliga W, Gu W, Liu R, Lawrence TS, Lamb C, Tanno Y, Cieslik M, Stone E, Georgiou G, Chan TA, Chinnaiyan A, Zou W. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569: 270–274, 2019. doi: 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Adedoyin O, Boddu R, Traylor A, Lever JM, Bolisetty S, George JF, Agarwal A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol Renal Physiol 314: F702–F714, 2018. doi: 10.1152/ajprenal.00044.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, Weinlich R, Vanden Berghe T, Vandenabeele P, Pasparakis M, Bleich M, Weinberg JM, Reichel CA, Brasen JH, Kunzendorf U, Anders HJ, Stockwell BR, Green DR, Krautwald S. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA 111: 16836–16841, 2014. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Guerrero-Hue M, Garcia-Caballero C, Palomino-Antolin A, Rubio-Navarro A, Vazquez-Carballo C, Herencia C, Martin-Sanchez D, Farre-Alins V, Egea J, Cannata P, Praga M, Ortiz A, Egido J, Sanz AB, Moreno JA. Curcumin reduces renal damage associated with rhabdomyolysis by decreasing ferroptosis-mediated cell death. FASEB J 33: 8961–8975, 2019. doi: 10.1096/fj.201900077R. [DOI] [PubMed] [Google Scholar]
- 149.Deng F, Sharma I, Dai Y, Yang M, Kanwar YS. Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J Clin Invest 129: 5033–5049, 2019. doi: 10.1172/JCI129903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hu Z, Zhang H, Yi B, Yang S, Liu J, Hu J, Wang J, Cao K, Zhang W. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis 11: 73, 2020. doi: 10.1038/s41419-020-2256-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mishima E, Sato E, Ito J, Yamada KI, Suzuki C, Oikawa Y, Matsuhashi T, Kikuchi K, Toyohara T, Suzuki T, Ito S, Nakagawa K, Abe T. Drugs repurposed as antiferroptosis agents suppress organ damage, including aki, by functioning as lipid peroxyl radical scavengers. J Am Soc Nephrol 31: 280–296, 2020. doi: 10.1681/ASN.2019060570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hannemann J, Duwe J, Baumann K. Iron- and ascorbic acid-induced lipid peroxidation in renal microsomes isolated from rats treated with platinum compounds. Cancer Chemother Pharmacol 28: 427–433, 1991. doi: 10.1007/BF00685818. [DOI] [PubMed] [Google Scholar]
- 153.Kameyama Y, Gemba M. The iron chelator deferoxamine prevents cisplatin-induced lipid peroxidation in rat kidney cortical slices. Jpn J Pharmacol 57: 259–262, 1991. doi: 10.1254/jjp.57.259. [DOI] [PubMed] [Google Scholar]
- 154.Baliga R, Zhang Z, Baliga M, Ueda N, Shah SV. In vitro and in vivo evidence suggesting a role for iron in cisplatin-induced nephrotoxicity. Kidney Int 53: 394–401, 1998. doi: 10.1046/j.1523-1755.1998.00767.x. [DOI] [PubMed] [Google Scholar]
- 155.Liu H, Baliga R. Cytochrome P450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney Int 63: 1687–1696, 2003. doi: 10.1046/j.1523-1755.2003.00908.x. [DOI] [PubMed] [Google Scholar]
- 156.Muselin F, Dumitrescu E, Berbecea A, Doma AO, Brezovan D, Savici J, Trif A, Cristina RT. The effect of cisplatin administration on certain trace elements homeostasis in rats and the protective effect of silver birch (Betula pendula) sap. J Trace Elem Med Biol 50: 474–481, 2018. doi: 10.1016/j.jtemb.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 157.Zarjou A, Bolisetty S, Joseph R, Traylor A, Apostolov EO, Arosio P, Balla J, Verlander J, Darshan D, Kuhn LC, Agarwal A. Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J Clin Invest 123: 4423–4434, 2013. doi: 10.1172/JCI67867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol 25: 2689–2701, 2014. doi: 10.1681/ASN.2014030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA 102: 12005–12010, 2005. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ying Y, Padanilam BJ. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cell Mol Life Sci 73: 2309–2324, 2016. doi: 10.1007/s00018-016-2202-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J 77: 1111–1122, 2013. doi: 10.1253/circj.cj-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12: 674–683, 2013. doi: 10.4161/cc.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem 258: 729–735, 1998. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- 164.Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol 152: 237–250, 2001. doi: 10.1083/jcb.152.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Varanyuwatana P, Halestrap AP. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 12: 120–125, 2012. doi: 10.1016/j.mito.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9: 550–555, 2007. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chinopoulos C, Konrad C, Kiss G, Metelkin E, Torocsik B, Zhang SF, Starkov AA. Modulation of F0F1-ATP synthase activity by cyclophilin D regulates matrix adenine nucleotide levels. FEBS J 278: 1112–1125, 2011. doi: 10.1111/j.1742-4658.2011.08026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427: 461–465, 2004. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.McGee AM, Baines CP. Phosphate is not an absolute requirement for the inhibitory effects of cyclosporin A or cyclophilin D deletion on mitochondrial permeability transition. Biochem J 443: 185–191, 2012. doi: 10.1042/BJ20111881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lindblom RSJ, Higgins GC, Nguyen TV, Arnstein M, Henstridge DC, Granata C, Snelson M, Thallas-Bonke V, Cooper ME, Forbes JM, Coughlan MT. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clin Sci (Lond) 134: 239–259, 2020. doi: 10.1042/CS20190787. [DOI] [PubMed] [Google Scholar]
- 171.Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J 255: 357–360, 1988. [PMC free article] [PubMed] [Google Scholar]
- 172.Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun 304: 463–470, 2003. doi: 10.1016/s0006-291x(03)00618-1. [DOI] [PubMed] [Google Scholar]
- 173.Qian T, Nieminen AL, Herman B, Lemasters JJ. Mitochondrial permeability transition in pH-dependent reperfusion injury to rat hepatocytes. Am J Physiol 273: C1783–C1792, 1997. doi: 10.1152/ajpcell.1997.273.6.C1783. [DOI] [PubMed] [Google Scholar]
- 174.Tanveer A, Virji S, Andreeva L, Totty NF, Hsuan JJ, Ward JM, Crompton M. Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. Eur J Biochem 238: 166–172, 1996. doi: 10.1111/j.1432-1033.1996.0166q.x. [DOI] [PubMed] [Google Scholar]
- 175.Li Y, Johnson N, Capano M, Edwards M, Crompton M. Cyclophilin-D promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis. Biochem J 383: 101–109, 2004. doi: 10.1042/BJ20040669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658–662, 2005. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 177.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652–658, 2005. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 178.Armstrong JA, Cash NJ, Ouyang Y, Morton JC, Chvanov M, Latawiec D, Awais M, Tepikin AV, Sutton R, Criddle DN. Oxidative stress alters mitochondrial bioenergetics and modifies pancreatic cell death independently of cyclophilin D, resulting in an apoptosis-to-necrosis shift. J Biol Chem 293: 8032–8047, 2018. doi: 10.1074/jbc.RA118.003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, Peixoto PM, Molkentin JD. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci Adv 5: eaaw4597, 2019. doi: 10.1126/sciadv.aaw4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kim H, Zhao J, Zhang Q, Wang Y, Lee D, Bai X, Turrell L, Chen M, Gao W, Keshavjee S, Liu M. deltaV1-1 reduces pulmonary ischemia reperfusion-induced lung injury by inhibiting necrosis and mitochondrial localization of PKCdelta and p53. Am J Transplant 16: 83–98, 2016. doi: 10.1111/ajt.13445. [DOI] [PubMed] [Google Scholar]
- 181.Devalaraja-Narashimha K, Diener AM, Padanilam BJ. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol 297: F749–F759, 2009. doi: 10.1152/ajprenal.00239.2009. [DOI] [PubMed] [Google Scholar]
- 182.Park JS, Pasupulati R, Feldkamp T, Roeser NF, Weinberg JM. Cyclophilin D and the mitochondrial permeability transition in kidney proximal tubules after hypoxic and ischemic injury. Am J Physiol Renal Physiol 301: F134–F150, 2011. doi: 10.1152/ajprenal.00033.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Li Q, Liang X, Yang Y, Zeng X, Zhong X, Huang C. Panax notoginseng saponins ameliorate cisplatin-induced mitochondrial injury via the HIF-1alpha/mitochondria/ROS pathway. FEBS Open Bio 10: 118–126, 2020. doi: 10.1002/2211-5463.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ma Q, Xu Y, Tang L, Yang X, Chen Z, Wei Y, Shao X, Shao X, Xin Z, Cai B, Wang Q, Mou S. Astragalus polysaccharide attenuates cisplatin-induced acute kidney injury by suppressing oxidative damage and mitochondrial dysfunction. BioMed Res Int 2020: 2851349, 2020. doi: 10.1155/2020/2851349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Campbell PI, Al-Nasser IA. Renal insufficiency induced by cisplatin in rats is ameliorated by cyclosporin A. Toxicology 114: 11–17, 1996. doi: 10.1016/s0300-483x(96)03411-7. [DOI] [PubMed] [Google Scholar]
- 186.Croze ML, Soulage CO. Potential role and therapeutic interests of myo-inositol in metabolic diseases. Biochimie 95: 1811–1827, 2013. doi: 10.1016/j.biochi.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 187.Holub BJ. Metabolism and function of myo-inositol and inositol phospholipids. Annu Rev Nutr 6: 563–597, 1986. doi: 10.1146/annurev.nu.06.070186.003023. [DOI] [PubMed] [Google Scholar]
- 188.Parthasarathy L, Parthasarathy R, Vadnal R. Molecular characterization of coding and untranslated regions of rat cortex lithium-sensitive myo-inositol monophosphatase cDNA. Gene 191: 81–87, 1997. doi: 10.1016/s0378-1119(97)00045-0. [DOI] [PubMed] [Google Scholar]
- 189.Ragan CI, Watling KJ, Gee NS, Aspley S, Jackson RG, Reid GG, Baker R, Billington DC, Barnaby RJ, Leeson PD. The dephosphorylation of inositol 1,4-bisphosphate to inositol in liver and brain involves two distinct Li+-sensitive enzymes and proceeds via inositol 4-phosphate. Biochem J 249: 143–148, 1988. doi: 10.1042/bj2490143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sherman WR, Rasheed A, Mauck LA, Wiecko J. Incubations of testis myo-inositol-1-phosphate synthase with d-(5-18O)glucose 6-phosphate and with H218O show no evidence of Schiff base formation. J Biol Chem 252: 5672–5676, 1977. doi: 10.1016/S0021-9258(17)40075-5. [DOI] [PubMed] [Google Scholar]
- 191.Holub BJ. 1982 Borden Award lecture. Nutritional, biochemical, and clinical aspects of inositol and phosphatidylinositol metabolism. Can J Physiol Pharmacol 62: 1–8, 1984. doi: 10.1139/y84-001. [DOI] [PubMed] [Google Scholar]
- 192.Farias VX, Uchoa PN, Aquino CP, Britto LRG, Fonteles MC, Leal-Cardoso JH, Silva-Alves KS, Havt A, Prata MMG, Heimark DB, Nascimento NRF, Santos CF. Expression of myo-inositol cotransporters in the sciatic nerve and dorsal root ganglia in experimental diabetes. Braz J Med Biol Res 52: e8589, 2019. doi: 10.1590/1414-431x20198589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Fu H, Li B, Hertz L, Peng L. Contributions in astrocytes of SMIT1/2 and HMIT to myo-inositol uptake at different concentrations and pH. Neurochem Int 61: 187–194, 2012. doi: 10.1016/j.neuint.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 194.Deranieh RM, Greenberg ML. Cellular consequences of inositol depletion. Biochem Soc Trans 37: 1099–1103, 2009. doi: 10.1042/BST0371099. [DOI] [PubMed] [Google Scholar]
- 195.Prabhu KS, Arner RJ, Vunta H, Reddy CC. Up-regulation of human myo-inositol oxygenase by hyperosmotic stress in renal proximal tubular epithelial cells. J Biol Chem 280: 19895–19901, 2005. doi: 10.1074/jbc.M502621200. [DOI] [PubMed] [Google Scholar]
- 196.Howard CF Jr, Anderson L. Metabolism of myo-inositol in animals. II. Complete catabolism of myo-inositol-14C by rat kidney slices. Arch Biochem Biophys 118: 332–339, 1967. doi: 10.1016/0003-9861(67)90357-8. [DOI] [PubMed] [Google Scholar]
- 197.Sharma I, Deng F, Liao Y, Kanwar YS. Myo-inositol oxygenase (MIOX) overexpression drives the progression of renal tubulo-interstitial injury in diabetes. Diabetes 69: 1248–1263, 2020. doi: 10.2337/db19-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Dutta RK, Kondeti VK, Sharma I, Chandel NS, Quaggin SE, Kanwar YS. Beneficial effects of myo-inositol oxygenase deficiency in cisplatin-induced AKI. J Am Soc Nephrol 28: 1421–1436, 2017. doi: 10.1681/ASN.2016070744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sharma I, Tupe RS, Wallner AK, Kanwar YS. Contribution of myo-inositol oxygenase in AGE:RAGE-mediated renal tubulointerstitial injury in the context of diabetic nephropathy. Am J Physiol Renal Physiol 314: F107–F121, 2018. doi: 10.1152/ajprenal.00434.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tominaga T, Sharma I, Fujita Y, Doi T, Wallner AK, Kanwar YS. Myo-inositol oxygenase accentuates renal tubular injury initiated by endoplasmic reticulum stress. Am J Physiol Renal Physiol 316: F301–F315, 2019. doi: 10.1152/ajprenal.00534.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Burton LE, Wells WW. myo-Inositol metabolism during lactation and development in the rat. The prevention of lactation-induced fatty liver by dietary myo-inositol. J Nutr 106: 1617–1628, 1976. doi: 10.1093/jn/106.11.1617. [DOI] [PubMed] [Google Scholar]
- 202.Ziyadeh FN, Simmons DA, Snipes ER, Goldfarb S. Effect of myo-inositol on cell proliferation and collagen transcription and secretion in proximal tubule cells cultured in elevated glucose. J Am Soc Nephrol 1: 1220–1229, 1991. [DOI] [PubMed] [Google Scholar]
- 203.Cohen AM, Wald H, Popovtzer M, Rosenmann E. Effect of myo-inositol supplementation on the development of renal pathological changes in the Cohen diabetic (type 2) rat. Diabetologia 38: 899–905, 1995. doi: 10.1007/BF00400577. [DOI] [PubMed] [Google Scholar]
- 204.Kitamura H, Yamauchi A, Sugiura T, Matsuoka Y, Horio M, Tohyama M, Shimada S, Imai E, Hori M. Inhibition of myo-inositol transport causes acute renal failure with selective medullary injury in the rat. Kidney Int 53: 146–153, 1998. doi: 10.1046/j.1523-1755.1998.00747.x. [DOI] [PubMed] [Google Scholar]
- 205.Pallio G, Micali A, Benvenga S, Antonelli A, Marini HR, Puzzolo D, Macaione V, Trichilo V, Santoro G, Irrera N, Squadrito F, Altavilla D, Minutoli L. Myo-inositol in the protection from cadmium-induced toxicity in mice kidney: an emerging nutraceutical challenge. Food Chem Toxicol 132: 110675, 2019. doi: 10.1016/j.fct.2019.110675. [DOI] [PubMed] [Google Scholar]
- 206.Bozelli JC Jr, Epand RM. Role of membrane shape in regulating the phosphatidylinositol cycle at contact sites. Chem Phys Lipids 221: 24–29, 2019. doi: 10.1016/j.chemphyslip.2019.03.002. [DOI] [PubMed] [Google Scholar]
- 207.Blunsom NJ, Cockcroft S. CDP-diacylglycerol synthases (CDS): gateway to phosphatidylinositol and cardiolipin synthesis. Front Cell Dev Biol 8: 63, 2020. doi: 10.3389/fcell.2020.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yuan WF, Chen Q, Gao XT, Zheng ZM, Jia H, Zhu HF, Xin T, Sui XK, Li M, Hou SH, Guo XY. Phospholipase C signaling is involved in porcine reproductive and respiratory syndrome virus infection in cell cultures. Acta Virol 63: 117–120, 2019. doi: 10.4149/av_2019_115. [DOI] [PubMed] [Google Scholar]
- 209.Lucic I, Truebestein L, Leonard TA. Novel features of DAG-activated PKC isozymes reveal a conserved 3-D architecture. J Mol Biol 428: 121–141, 2016. doi: 10.1016/j.jmb.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 210.Finley J. Oocyte activation and latent HIV-1 reactivation: AMPK as a common mechanism of action linking the beginnings of life and the potential eradication of HIV-1. Med Hypotheses 93: 34–47, 2016. doi: 10.1016/j.mehy.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 211.Aguilar A. Acute kidney injury: loss of PKC-varepsilon protects against IRI. Nat Rev Nephrol 12: 714, 2016. doi: 10.1038/nrneph.2016.162. [DOI] [PubMed] [Google Scholar]
- 212.Rong S, Hueper K, Kirsch T, Greite R, Klemann C, Mengel M, Meier M, Menne J, Leitges M, Susnik N, Meier M, Haller H, Shushakova N, Gueler F. Renal PKC-epsilon deficiency attenuates acute kidney injury and ischemic allograft injury via TNF-alpha-dependent inhibition of apoptosis and inflammation. Am J Physiol Renal Physiol 307: F718–F726, 2014. doi: 10.1152/ajprenal.00372.2013. [DOI] [PubMed] [Google Scholar]
- 213.Xu X, Pan J, Li H, Li X, Fang F, Wu D, Zhou Y, Zheng P, Xiong L, Zhang D. Atg7 mediates renal tubular cell apoptosis in vancomycin nephrotoxicity through activation of PKC-delta. FASEB J 33: 4513–4524, 2019. [Erratum in FASEB J 33: 9695, 2019] doi: 10.1096/fj.201801515R. [DOI] [PubMed] [Google Scholar]
- 214.Yang CC, Yao CA, Yang JC, Chien CT. Sialic acid rescues repurified lipopolysaccharide-induced acute renal failure via inhibiting TLR4/PKC/gp91-mediated endoplasmic reticulum stress, apoptosis, autophagy, and pyroptosis signaling. Toxicol Sci 141: 155–165, 2014. doi: 10.1093/toxsci/kfu121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Nowak G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J Biol Chem 277: 43377–43388, 2002. doi: 10.1074/jbc.M206373200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Alharbi RA, Pandha HS, Simpson GR, Pettengell R, Poterlowicz K, Thompson A, Harrington K, El-Tanani M, Morgan R. Inhibition of HOX/PBX dimer formation leads to necroptosis in acute myeloid leukemia cells. Oncotarget 8: 89566–89579, 2017. doi: 10.18632/oncotarget.20023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Cheng Y, Xia Z, Han Y, Rong J. Plant natural product formononetin protects rat cardiomyocyte H9c2 cells against oxygen glucose deprivation and reoxygenation via inhibiting ROS formation and promoting GSK-3beta phosphorylation. Oxid Med Cell Longev 2016: 2060874, 2016. doi: 10.1155/2016/2060874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Dachert J, Ehrenfeld V, Habermann K, Dolgikh N, Fulda S. Targeting ferroptosis in rhabdomyosarcoma cells. Int J Cancer 146: 510–520, 2020. doi: 10.1002/ijc.32496. [DOI] [PubMed] [Google Scholar]
- 219.Chakraborty A. The inositol pyrophosphate pathway in health and diseases. Biol Rev Camb Philos Soc 93: 1203–1227, 2018. doi: 10.1111/brv.12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kaushal GP, Kaushal V, Hong X, Shah SV. Role and regulation of activation of caspases in cisplatin-induced injury to renal tubular epithelial cells. Kidney Int 60: 1726–1736, 2001. doi: 10.1046/j.1523-1755.2001.00026.x. [DOI] [PubMed] [Google Scholar]
- 221.Tongqiang L, Shaopeng L, Xiaofang Y, Nana S, Xialian X, Jiachang H, Ting Z, Xiaoqiang D. Salvianolic acid B prevents iodinated contrast media-induced acute renal injury in rats via the PI3K/Akt/Nrf2 pathway. Oxid Med Cell Longev 2016: 7079487, 2016. doi: 10.1155/2016/7079487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Zhang G, Wang Q, Zhou Q, Wang R, Xu M, Wang H, Wang L, Wilcox CS, Liu R, Lai EY. Protective effect of tempol on acute kidney injury through PI3K/Akt/Nrf2 signaling pathway. Kidney Blood Press Res 41: 129–138, 2016. doi: 10.1159/000443414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Diao MY, Zhu Y, Yang J, Xi SS, Wen X, Gu Q, Hu W. Hypothermia protects neurons against ischemia/reperfusion-induced pyroptosis via m6A-mediated activation of PTEN and the PI3K/Akt/GSK-3beta signaling pathway. Brain Res Bull 159: 25–31, 2020. doi: 10.1016/j.brainresbull.2020.03.011. [DOI] [PubMed] [Google Scholar]
- 224.Xin C, Guangliang S, Qing Z, Qingqing L, Hang Y, Yiming Z, Shu L. Astilbin protects chicken peripheral blood lymphocytes from cadmium-induced necroptosis via oxidative stress and the PI3K/Akt pathway. Ecotoxicol Environ Saf 190: 110064, 2020. doi: 10.1016/j.ecoenv.2019.110064. [DOI] [PubMed] [Google Scholar]
- 225.Liu J, Livingston MJ, Dong G, Tang C, Su Y, Wu G, Yin XM, Dong Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell death Dis 9: 322, 2018. doi: 10.1038/s41419-018-0374-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, , et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell death Differ 25: 486–541, 2018. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]