

Abstract
We describe an individual in whom clinical and radiographic features are typical for achondroplasia, but in whom the common variants of FGFR3 that result in achondroplasia are absent. Whole exome sequencing demonstrated a novel, de novo 6 base pair tandem duplication in FGFR3 that results in the insertion of Ser-Phe after position Leu324. in vitro studies showed that this variant results in aberrant dimerization, excessive spontaneous phosphorylation of FGFR3 dimers and excessive, ligand-independent tyrosine kinase activity. Together, these data suggest that this variant leads to constitutive disulfide bond-mediated dimerization, and that this, surprisingly, occurs to an extent similar to the neonatal lethal thanatophoric dysplasia type I Ser249Cys variant.
Keywords: achondroplasia, fibroblast growth factor receptor 3, in vitro functional studies
1 |. INTRODUCTION
Achondroplasia is the most common of the bone dysplasias resulting in small stature (Pauli, 2019). Typically, it arises from a recurrent variant in FGFR3 that results in a Gly380Arg substitution (Rousseau et al., 1994; Shiang et al., 1994). This mutational homogeneity is reflected in the relative phenotypic homogeneity seen in individuals with this disorder; indeed, that is such that, in most instances, diagnosis on the basis of clinical and radiologic criteria, without need for molecular assessment, is unequivocal (Pauli, 2019; Xue et al., 2014).
Rare instances of individuals with the achondroplasia phenotype, or a similar clinical presentation, with other variants of FGFR3 have been reported, but in none has there been in vitro demonstration that the variant resulted in alteration of FGFR3 function which could be predicted to cause achondroplasia. We report here a boy with clinical and radiologic characteristics of achondroplasia in whom a novel variant in FGFR3 was found. Subsequent in vitro studies present evidence of its effect on FGFR3 function.
2 |. CLINICAL REPORT
The now 13-year-old propositus was born at 38 weeks gestation to a 38 years of age mother and 41 years of age father. Pregnancy was generally uncomplicated. Ultrasound evaluation at 34 weeks gestation showed limb shortening. Delivery was by (repeat) caesarian section. Apgar scores were 8 and 9 at 1 and 5 min, respectively.
He was assessed by a geneticist in the newborn period and both clinical features and radiographic ones (Figure 1a) were thought to be completely consistent with a diagnosis of achondroplasia. However, initial clinical molecular testing failed to demonstrate the common achondroplasia variant of FGFR3 (Gly380Arg). This negative result was subsequently confirmed by repeat testing. Nonetheless, both radiologic and clinical characteristics remained fully consistent with a diagnosis of achondroplasia (Figure 1b–d).
FIGURE 1.
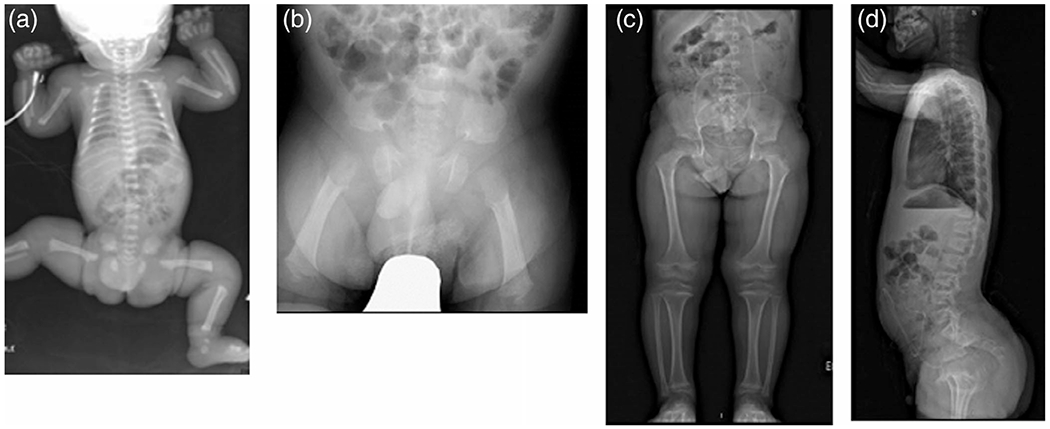
Radiographic features of the propositus. (a) Full-body radiograph in the first day of life showing typical features of achondroplasia including long bone shortening and characteristic pelvic configuration; (b) AP pelvis at 2 months age. The presence of a squared off ilium, a markedly narrowed sacrosciatic notch, characteristics proximal femoral radiolucency and femoral shortening are all well illustrated; (c) Standing AP view of the pelvis and legs at 10 years of age, in which, as in early radiographs, there are no features atypical for achondroplasia; (d) Lateral spine at 10 years of age–while there is mild anterior wedging, there is not marked or atypical platyspondyly evident
He experienced central hypopneas with desaturations, requiring oxygen supplementation. However, these resolved by 2 months of age.
Sequential assessment beginning at 2 months of age through the Midwest Regional Bone Dysplasia Clinics demonstrated the following.
Morphologic features (Figure 2) included rhizomesomelic shortening of the arms and rhizomelic shortening of the legs; small stature with linear growth initially at ~70%ile and subsequently ~50%ile compared with achondroplasia standards; macrocephaly, with head circumference initially at 60th-80th%ile on achondroplasia standards; midface flattening and retrusion; mild, reversible kyphosis; trident configuration and moderate brachydactyly.
FIGURE 2.
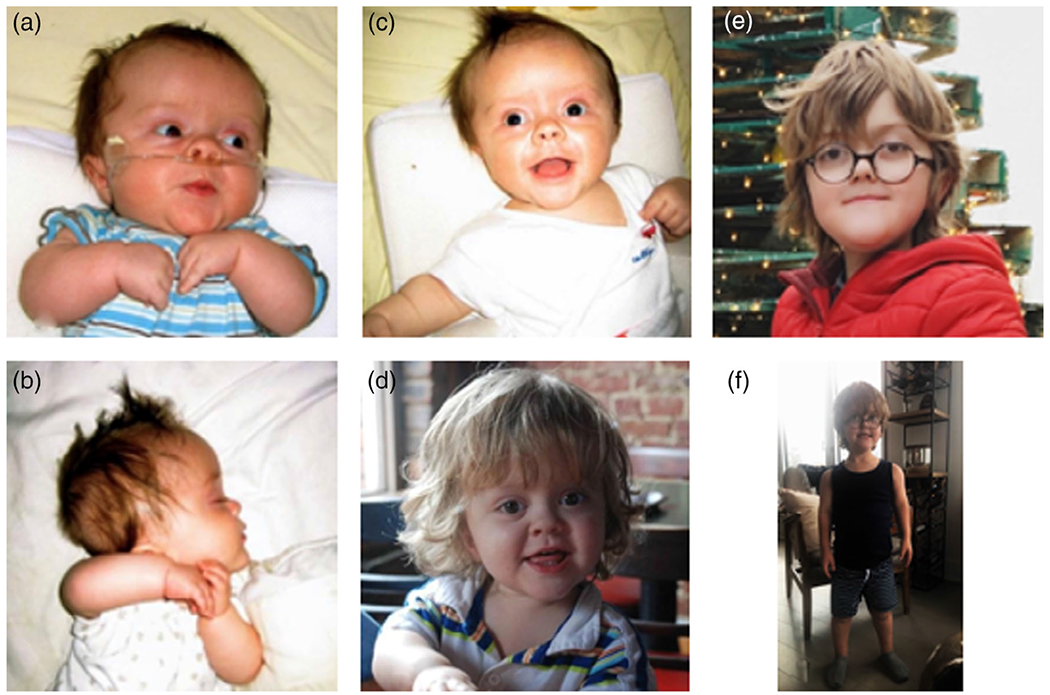
Clinical characteristics of the propositus. (a) Facial features are typical and unremarkable for an individual with achondroplasia, (b) 6 weeks of age, (c) 4 months of age, (d) 24 months of age, (e) 11 years of age, and (f) shows overall body proportions at 11 years of age
Radiographs both in infancy and subsequently showed characteristic findings of achondroplasia (Figure 1b–d). There were no features unexpected for that diagnosis.
Early development was typical for children with achondroplasia. He is cognitively normal but does have both learning disabilities and a complicated form of attention deficit–hyperactivity disorder.
Calvarial asymmetry was noted from birth. Both posterior plagiocephaly and asymmetric skull prominence just superior to the left ear was present by 2 months of age. Abnormal head shape persisted. Computerized tomography confirmed the presence of left lambdoidal craniosynostosis at 15 months of age (Legare et al., 2020). No surgery has been deemed necessary.
Increasing ventriculomegaly without clinical symptoms but with apparent papilledema resulted in ventriculoperitoneal shunt placement at age 13 m. No subsequent complications have arisen.
There were ongoing concerns related to obstructive sleep apnea, which was confirmed by polysomnography at 55 months of age. Tonsillectomy and adenoidectomy were completed. He also was found to have mild tracheomalacia. Obstructive apnea was a recurring concern leading to repeat adenoidectomy, turbinate shaving and current consideration of initiation of CPAP.
Other medical issues have included middle ear dysfunction requiring pressure equalizing tubes and marked malocclusions treated with palatal expansion and orthodontic manipulation. No skin abnormalities are present.
Whole exome sequencing testing was undertaken due to the clear clinical and radiologic diagnosis of achondroplasia and lack of identification of typical relevant variants known to cause achondroplasia and other FGFR3-associated skeletal dysplasias.
3 |. METHODS
3.1 |. Whole exome sequencing
Exome sequencing was performed utilizing the Agilent SureSelect HumanAllExonV4_51MbKit_S03723314 for library preparation. Libraries were sequenced on the HiSeq2500 platform using 100 bp paired end runs and sequencing chemistry kits TruSeq Rapid PE Cluster Kit-HS and TruSeq Rapid SBS-HS. Fastq files were aligned with BWA (Li & Durbin, 2009) version 0.5.10-tpx to the 1,000 genomes phase 2 GRCh37/hg19 human genome reference. Duplicate molecules were flagged with Picard version 1.74. Local realignment around indels and base call quality score recalibration was performed using the Genome Analysis Toolkit (GATK) (McKenna et al., 2010) version 2.3-9-ge5ebf34. Variant filtering was done using the Variant Quality Score Recalibration (VQSR) method (DePristo et al., 2011). Analysis of variant call format (vcf) files were performed using the PhenoDB analysis tool (Sobreira, Schiettecatte, Boehm, Valle, & Hamosh, 2011) to identify rare (MAF <0.01 in the 1,000 Genomes project, Exome Variant server, gnomAD, and our in-house database), functional variants (missense, nonsense, frameshift, splicing, or stoploss) with recessive or dominant inheritance patterns.
3.2 |. Functional studies
3.2.1 |. Plasmid constructs, cell culture and transfections
FGFR3-WT, FGFR3-Ser249Cys, FGFR3-Gly380Arg and FGFR3-Lys650Glu plasmids have been previously described (d’Avis et al., 1998; Webster, D’Avis, Robertson, & Donoghue, 1996). FGFR3-Asn540Lys was generated by QuikChange site-directed mutagenesis (Agilent) using FGFR3-WT as the template. FGFR3-patient variant was generated via an insertion PCR protocol (Liu & Naismith, 2008). Mutagenic Oligos D3624 (5′-GCTAGAGGTTCTCTCCTTCTCCTTGCACAACGTCACCTTTG-3′) and D3625 (5′-GAGAGAACCTCTAGCTCCTTGTCGGTGGTGTTAGC-3′) were used to insert the six bases, identified in the patient variant, resulting in the insertion of residues Ser and Phe between Leu324 and Ser325 of FGFR3-WT (Uniprot P22607-1). All clones are in the pcDNA3 expression plasmid.
HEK293 and HEK293T cells were cultured in 10% FBS DMEM plus penicillin/streptomycin in 10% CO2 at 37°C. Twenty-four hour prior to transfection by calcium phosphate, cells were seeded at 1 × 106 cells/100 mm plate (Nelson et al., 2016). Approximately 24 hr after transfection, cells were starved for 16–18 hr in media with no serum prior to lysis.
3.2.2 |. Dimerization assays, immunoblotting and kinase assays
For dimerization assays, cells were washed twice with 1×PBS + 10 mM iodoacetamide and then lysed in RIPA+ Buffer [10 mM sodium phosphate pH 7.0, 150 mM sodium chloride, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate, 2 mM EDTA, 50 mM sodium fluoride, 10 mM iodoacetamide, 10 μg/ml aprotinin, 1 mM sodium orthovanadate, 0.1 mM PMSF]. Lysates were boiled in 2X non-reducing sample buffer [4% SDS, 10 mM sodium phosphate pH 7.0, 20% glycerol, 0.08% bromophenol blue] then half of the sample was transferred to a new tube and reduced with 5% β-mercaptoethanol and 20 mM DTT (Chen, Webster, Meyer, & Donoghue, 1997). Samples were resolved on 4–12% SDS-PAGE gradient gels, transferred to Immobilon-P membrane and immunoblotted with FGFR3 (C-15), FGFR3 (B-9) (Santa Cruz Biotechnology) or phospho-FGF Receptor (3471) (Cell Signaling). Membranes were blocked in 5% nonfat milk/TBS/0.05% Tween 20 for anti-FGFR3 or 3% bovine serum albumin/TBS/0.05% Tween 20 for anti-P-FGFR. Immunoblotting was performed as described (Meyer, McAndrew, & Donoghue, 2008).
For kinase assays, cells were washed twice with PBS prior to lysing in 1% NP-40 Lysis Buffer [20 mM Tris pH 7.5, 137 mM sodium chloride, 1% IGEPAL (octylphenoxypolyethoxyethanol), 5 mM EDTA, 10% glycerol, 10 μg/ml aprotinin, 1 mM sodium orthovanadate] and immunoprecipitated with FGFR3 (B-9) antiserum overnight at 4°C. Immune complexes were collected with Protein A sepharose, washed twice with 1% NP-40 Lysis Buffer and twice with 20 mM Tris pH 7.5, then incubated with 40 μl kinase buffer [20 mM Tris pH 7.5, 10 mM MnCl2, 5 mM MgCl2, 10 μCi of γ-32P-ATP] at 37°C for 15 min. Samples were washed extensively with 1% NP-40 Lysis Buffer, boiled in 2X reducing sample buffer and resolved by 7.5% SDS-PAGE and autoradiography (Manickam, Donoghue, Meyer, Snyder, & Prior, 2014).
4 |. RESULTS
4.1 |. Sequencing
Whole exome sequencing identified in FGFR3 a de novo 6 base pair tandem duplication (g.1805458 insertion of TCTCCT) that results in the insertion of Ser-Phe after position Leu324 (NM_000142:exon8: c.970_971insTCTCCT:p.L324delinsLSF).
4.2 |. In vitro protein studies
In order to examine the effect of this atypical achondroplasia variant of FGFR3, we wished to compare it with other known skeletal dysplasia variants of FGFR3, ranging from the relatively mild hypochondroplasia to the very severe thanatophoric dysplasia, types I and II. The FGFR3 derivatives used for comparison all contain point mutations in the various domains of FGFR3 and have been found to lead to activation of the receptor in the absence of ligand (Bellus et al., 1995; d’Avis et al., 1998; Webster et al., 1996; Webster & Donoghue, 1996).
We examined the novel patient variant described here for dimerization and discovered that the 2-residue insertion between Leu324 and Ser325 does, indeed, result in aberrant dimerization of the receptor (Figure 3a, lane 3). The ratio of dimer to monomer was quantitated in the nonreducing samples and is shown in Figure 3b. Interestingly, the amount of FGFR3 dimer detected for the two amino acid insert patient variant is similar to the Ser249Cys, thanatophoric dysplasia type I (TD I) variant, which is typically lethal in the newborn period (Figure 3b, lanes 3 and 4).
FIGURE 3.
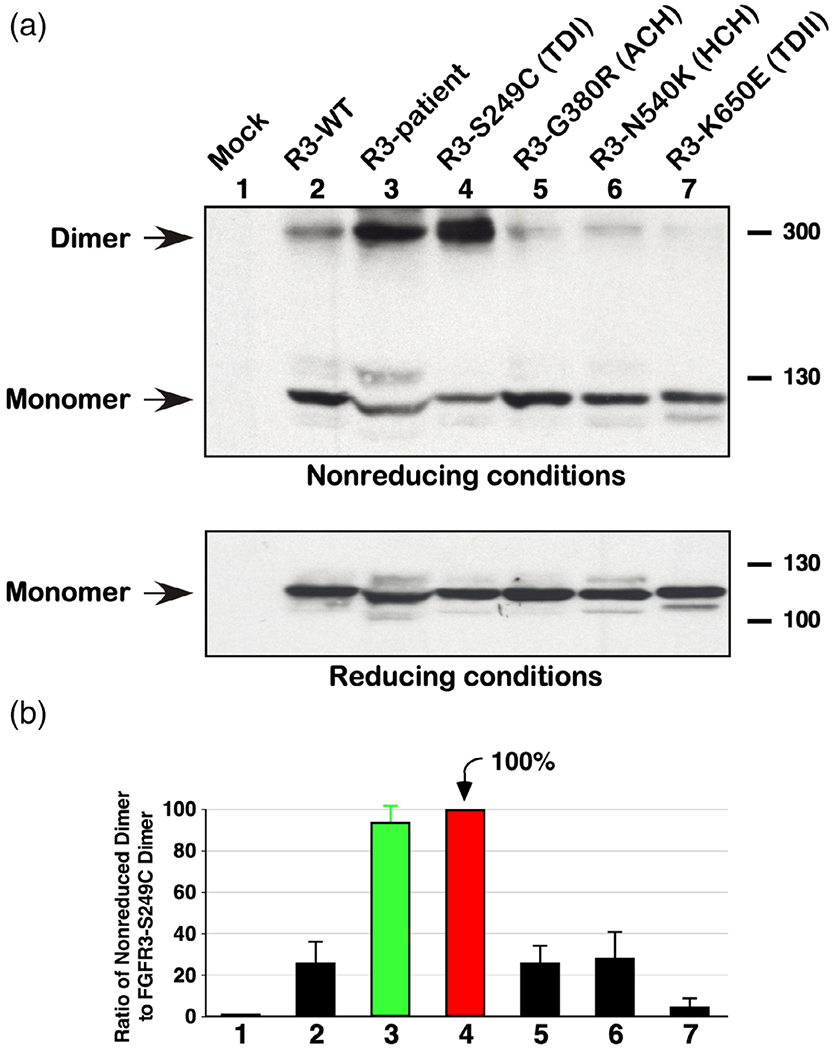
Ligand-independent dimerization. (a) Lysates from HEK293 cells expressing FGFR3 derivatives were resolved by 4–12% gradient SDS-PAGE under nonreducing conditions and immunoblotted for FGFR3 expression. Dimer and monomer FGFR3 is indicated (top panel). Duplicate samples were analyzed under reducing conditions and immunoblotted for FGFR3 showing the receptors only as monomers (bottom panel). (b) The ratio of dimer to monomer in the nonreducing samples was quantitated from four independent experiments with FGFR3-Ser249Cys set to 100%. Standard error of the mean is shown
In order to examine the phosphorylation on the FGFR3 receptor dimers, antisera specific for tyrosine phosphorylation of the activation loop tyrosine residues was used. As shown in Figure 4, top panel, the Ser249Cys (TD I) variant leads to a large phospho-tyrosine signal detected on the dimers which is also seen for the R3-patient dimers (lanes 3 and 4).
FIGURE 4.
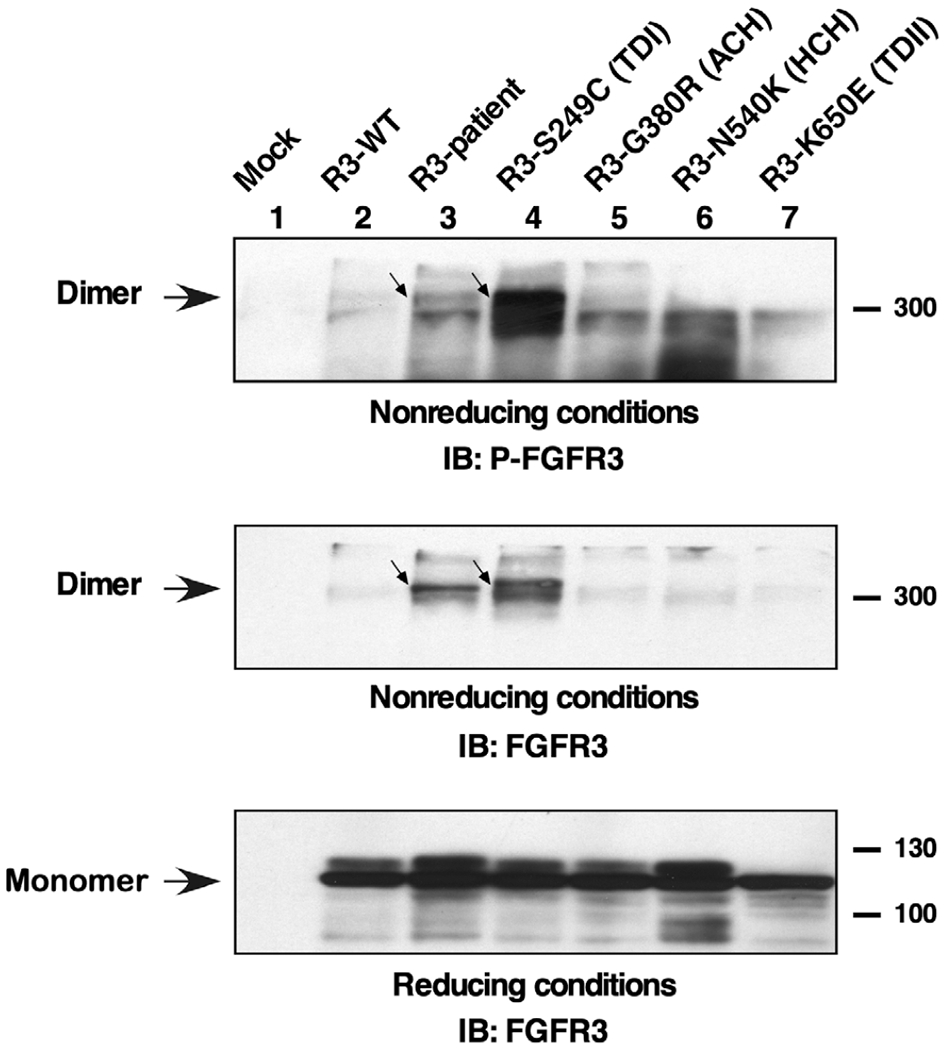
Tyrosine phosphorylation of FGFR3 dimers. Lysates from HEK293 cells expressing FGFR3 derivatives were resolved by 4–12% gradient SDS-PAGE under nonreducing conditions and immunoblotted with antisera specific for phosphorylation of the activation loop tyrosine residues of FGFR3. Phosphorylated dimers are indicated (top panel). The same membrane was reprobed for total FGFR3 (middle panel). Duplicate samples were analyzed under reducing conditions and immunoblotted for FGFR3 showing the total of monomer receptors (bottom panel)
Next, the activity of the receptors was determined by in vitro kinase assays (Figure 5). The receptors were immunoprecipitated with FGFR3 antisera and incubated with [γ-32P]-ATP. The autophosphorylation of the receptors is seen in Figure 5, second panel. As expected, the R3-Lys650Glu receptor has the most activity as has been well characterized. (Manickam et al., 2014; Webster et al., 1996). Nonetheless, the R3-patient receptor has elevated autophosphorylation compared to the wild type sample (lanes 2 and 3, second panel).
FIGURE 5.
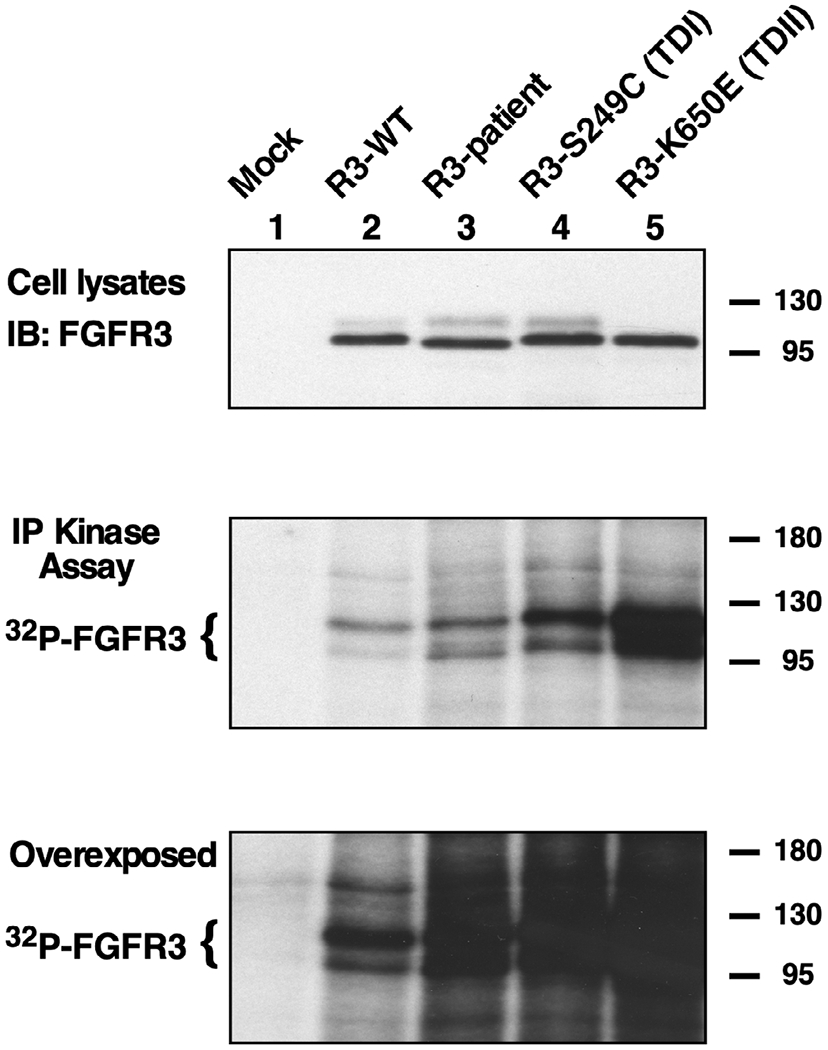
In vitro tyrosine kinase activity. Total expression of FGFR3 derivatives from HEK293T cells is shown by immunoblotting (top panel). FGFR3 immunoprecipitates were subjected to in vitro kinase assay with γ-32P-ATP. Samples were resolved by 7.5% SDS-PAGE and autoradiography (middle and bottom panels). The overexposure shown (bottom panel) is included to demonstrate the relative similarity of kinase activity in Lanes 3–5, showing the patient mutation together with a TD I and a TD II mutant, in comparison with FGFR3-WT in Lane 2
5 |. DISCUSSION
The patient described here has clinical and radiologic features that are only compatible with a diagnosis of achondroplasia. Each of the sequelae which he has experienced–middle ear dysfunction, obstructive apnea, increased intracranial pressure, etc. are well-described concomitants of this diagnosis (Pauli, 2019). Note that the presence of craniosynostosis also is a recently recognized complication seen in a minority of children with achondroplasia (Legare et al., 2020). Furthermore, he is cognitively normal, has growth comparable to those with the more common achondroplasia-causing variants, and has no unexpected features. A diagnosis of achondroplasia is fully appropriate unless, of course, one adheres to a strictly molecular method of defining disorders. In our view, such an approach is misguided, since the clinical phenotype is what is critical in providing ongoing, anticipatory care (Robin & Biesecker, 2001). We rather would choose to define the pro-band as having achondroplasia secondary to a rare FGFR3 variant.
A number of instances in which rare variants of FGFR3 have caused achondroplasia or something similar to it have been reported previously. The most common of these is one that results in a Ser348Cys substitution. First reported by Hasegawa et al. (2016), there have been two additional instances described subsequently (Bengur, Ekmekci, Karaarslan, Gunoz, & Alanay, 2020; Couser et al., 2017). Although in two instances, the authors suggest that a diagnosis of “mild achondroplasia/severe hypochondroplasia” is appropriate (Bengur et al., 2020; Couser et al., 2017), review of the published materials does not suggest anything atypical for achondroplasia, which diagnosis seems appropriate in all three instances. None of these studies performed in vitro or in vivo studies to determine the functional impact of the variants.
Another variant that has been recurrent results in a Gly375Cys substitution. One family was described by Nishimura, Fukushima, Ohashi, and Ikegawa (1995) and Ikegawa, Fukushima, Takada, and Nakamura (1995). While reported as having achondroplasia, the radiologic characteristics documented seem not to be typical for this diagnosis, including considerable platyspondyly and some unusual metaphyseal changes. Similarly, Superti-Furga et al. (1995) describe one individual with this variant in whom there is quite considerable platyspondyly (seemingly incompatible with a diagnosis of achondroplasia) as well as mild thoracic butterfly vertebrae. Addor, Gudinchet, Truttmann, and Schorderet (2000) illustrated another sporadic occurrence of this variant that resulted in remarkably similar features–including atypical platyspondyly, butterfly vertebrae, severe femoral bowing and bowed clavicles; this patien’s growth also was atypically slow compared with achondroplasia standards. Lastly, Barton, Sweeney, Roberts, and McPartland (2010) reported a single instance in which termination was carried out at 33 weeks gestation. Clinical features seem consistent with achondroplasia, and radiographs, while quite severe, are probably within the range of those seen in achondroplasia. The authors reported that the histopathology was intermediate between achondroplasia and thanatophoric dysplasia. On balance, it appears that this variant might represent a rare, additional allelic disorder rather than another cause for classic achondroplasia.
Other rare variants that have been reported to cause achondroplasia include the following:
Ser279Cys (Heuretz et al., 2006), in whom the phenotype as described seems consistent with a diagnosis of achondroplasia;
Ser217Cys (Zhang et al., 2007), published with limited clinical documentation and no radiologic documentation;
Thr394Ser (Zhu et al., 2003 cited in Zhang et al., 2007)
Ser344Cys (Takagi et al., 2015), in a patient both clinically and radiologically atypical for achondroplasia and who probably should be considered to have a distinguishable allelic variant.
In summary, in addition to the novel variant described here, atypical mutations resulting in a disorder wholly compatible with a clinical diagnosis of achondroplasia include only those resulting in Ser348Cys and Ser279Cys substitutions. Note that in none of those previously reported instances of atypical mutations purported to cause achondroplasia has any functional analysis been completed, although arguments for their causal relevance are succinctly summarized by Superti-Furga et al. (1995). Other rare variants of FGFR3 that have been reported to cause achondroplasia in fact result in distinguishable phenotypes. These illustrate the utility of molecular analysis particularly in individuals with unusual achondroplasia-like manifestations.
The variant identified is unlike any previously described as causing achondroplasia or other members of the FGFR3 family of disorders. As noted above, virtually all previous pathogenic variants that result in FGFR3 related bone dysplasia are point mutations. Although single nucleotide variants are much more common in human evolution, 25–100 base pair duplications are not that rare. The genome is littered with them, and they are most likely caused by random slippage during DNA replication giving rise to duplications that may be relatively proximal, within 100 base pairs, or at distances up to ≈1,000 base pairs apart (Thomas et al., 2004). Conversely, the majority of short (1–100 base pair) DNA insertions in the human genome are tandem duplications of directly adjacent sequences with conserved polarity (Messer & Arndt, 2007). These authors note that these indels are preferentially generated in the male germ line, and most likely arise through a mechanism that is not recombination mediated. The apparent preferential occurrence of indels in the male germ line may arise from the higher number of germ cell divisions in males making them more prone to double-strand breaks as well as replication errors. Many FGFR disorders, including achondroplasia (Wilkin et al., 1998), arise secondary to variants that are of exclusive paternal origin, which may be because of “selfish spermatogonial selection” (Goriely & Wilkie, 2012). The increased basal activation of FGFR3 exhibited by the mutation described here would certainly be consistent with such a model, although at present there is no direct evidence for this. The variant described here is a 6 base pair insertion that has appeared de novo, and is otherwise likely to reoccur at extremely low frequency.
We have compared the in vitro consequences of this insertional variant with other, previously well characterized FGFR3 variants that result in constitutive activation of the FGFR3 receptor. The mechanism of constitutive activation, in some cases largely ligand-independent, has been characterized for many of these mutations. Variants within the tyrosine protein kinase domain, such as Asn540Lys and Lys650Glu, lead to activation by inducing conformational changes. In contrast, extracellular variants often create a novel cysteine or perturb the normal cysteine residues, resulting in abnormal dimerization of receptor monomers. Variants which occur in the extracellular domain of FGFR3 include many of the TD I mutations, such as those originally described, Arg248Cys and Ser371Cys (Tavormina et al., 1995). This eventually led to the description of other similar variants, including Ser249Cys, Tyr373Cys, and Gly370Cys (Rousseau et al., 1996; Tavormina et al., 1995). Some of the TD I variants were examined biochemically and were shown to directly induce aberrant receptor dimerization (d’Avis et al., 1998).
For the variant in this propositus, abnormal dimerization is clearly demonstrable (Figure 3). What is unexpected is that the amount of dimerization is comparable to that seen in a variant that results in TD I. Furthermore, excessive spontaneous phosphorylation of FGFR3 resulting from the propositus’ variant was clearly demonstrated, although the phosphorylation signal is not so large as that seen in the TD I (R3-Ser249Cys) sample (Figure 4). Lastly, the in vitro kinase assay clearly demonstrates abnormal ligand-independent activity (Figure 5). Together, these data indicate that the novel variant found in the patient leads to constitutive disulfide bond-mediated dimerization, and that this occurs to an extent similar to the neonatal lethal TD I mutation (Ser249Cys). This striking dimerization is notable given the relatively mild clinical phenotype observed. The difference may be the result of incomplete engagement of the receptor with downstream signaling pathways. Further biophysical analysis of the effects of this variant may be warranted to provide a complete understanding of the effects of this two-residue insertion upon FGFR3 conformation and activity.
ACKNOWLEDGMENTS
Whole exome sequence studies were completed with support of the Baylor-Hopkins Center for Mendelian Genomics (NHGRI UM1 HG006542). This work was also supported by generous support from the UC San Diego Foundation. We thank Marvin Miller, M.D., Dennis Weiner M.D. Michael Bober M.D., Ph.D. and Elaine Spector, Ph.D. for their contributions. None of the authors declare a conflict of interest. The data upon which this article is based are not publicly available due to privacy restrictions.
Funding information
National Human Genome Research Institute, Grant/Award Number: UM1 HG006542; UC San Diego Foundation
Footnotes
DATA AVAILABILITY STATEMENT
The data are not publicly available due to privacy restrictions.
REFERENCES
- Addor MC, Gudinchet F, Truttmann A, & Schorderet DF (2000). An uncommon G375C substitution in a newborn with achondroplasia. Genetic Counseling, 11, 169–174. [PubMed] [Google Scholar]
- Barton C, Sweeney E, Roberts D, & McPartland J (2010). Fibroblast growth receptor-3 (FGFR3) G375C mutation in a case of achondroplasia and thanatophoric dysplasia phenotypic overlap. Clinical Dysmorphology, 19, 146–149. 10.1097/MCD.0b013e328337586b [DOI] [PubMed] [Google Scholar]
- Bellus GA, McIntosh I, Smith EA, Aylsworth AS, Kaitila I, Horton WA, … Francomano CA (1995). A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nature Genetics, 10, 357–359. 10.1038/ng0795-357 [DOI] [PubMed] [Google Scholar]
- Bengur FB, Ekmekci CG, Karaarslan E, Gunoz H, & Alanay Y (2020). P.Ser348Cys mutation in FGFR3 gene leads to “mild ACH /severe HCH” phenotype. European Journal of Medical Genetics, 63, 103659. 10.1016/j.ejmg.2019.04.016 [DOI] [PubMed] [Google Scholar]
- Chen LI, Webster MK, Meyer AN, & Donoghue DJ (1997). Transmembrane domain sequence requirements for activation of the p185c-neu receptor tyrosine kinase. Journal of Cell Biology, 137, 619–631. 10.1083/jcb.137.3.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couser NL, Pande CK, Turcott CM, Spector EB, Aylsworth AS, & Powell CM (2017). Mild achondroplasia/hypochondroplasia with acanthosis nigricans, normal development, and a p.Ser348Cys FGFR3 mutation. American Journal of Medical Genetics A, 173, 1097–1101. 10.1002/ajmg.a.38141 [DOI] [PubMed] [Google Scholar]
- d’Avis PY, Robertson SC, Meyer AN, Bardwell WM, Webster MK, & Donoghue DJ (1998). Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type I. Cell Growth & Differentiation, 9, 71–78. [PubMed] [Google Scholar]
- DePristo M, Banks E, Poplin R, Garimella K, Maguire J, Hartl C, … Daly M (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics, 43, 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriely A, & Wilkie AO (2012). Paternal age effect mutations and selfish spermatogonial selection: Causes and consequences for human disease. American Journal of Human Genetics, 90, 175–200. 10.1016/j.ajhg.2011.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Fukuhara R, Moriwake T, Tanaka H, Higuchi Y, Yamashita M, & Tsukahara H (2016). A novel mutation p.Ser348Cys in FGFR3 causes achondroplasia. American Journal of Medical Genetics A, 170A, 1370–1372. 10.1002/ajmg.a.37557 [DOI] [PubMed] [Google Scholar]
- Heuretz S, Le Merrer M, Zabel B, Wright M, Legeai-Mallet L, Cormier-Daire V, … Bonaventure J (2006). Novel FGFR3 mutations creating cysteine residues in the extracellular domain of the receptor cause achondroplasia or severe forms of hypochondroplasia. European Journal of Human Genetics, 14, 1240–1247. 10.1038/sj.ejhg.5201700 [DOI] [PubMed] [Google Scholar]
- Ikegawa S, Fukushima Y, Takada MIF, & Nakamura Y (1995). Mutations of the fibroblast growth factor receptor-3 gene in one familial and six sporadic cases of achondroplasia in Japanese patients. Human Genetics, 96, 309–311. 10.1007/BF00210413 [DOI] [PubMed] [Google Scholar]
- Legare JM, Pauli RM, Hecht JT, Bober MB, Smid C, Modaff P, Little ME, Rodriguez-Buritica DF, Serna ME, Alade AY, Liu C, Hoover-Fong JE, & Hashmi SS (2020). CLARITY: Unrecognized or under-recognized co-occurrences in achondroplasia with special emphasis on craniosynostosis. Submitted for publication. [Google Scholar]
- Li H, & Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, & Naismith JH (2008). An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnology, 8, 91. 10.1186/1472-6750-8-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manickam K, Donoghue DJ, Meyer AN, Snyder PJ, & Prior TW (2014). Suppression of severe achondroplasia with developmental delay and acanthosis nigricans by the p.Thr651Pro mutation. American Journal of Medical Genetics A, 164A, 243–250. 10.1002/ajmg.a.36236 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, … DePristo MA (2010). The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 20, 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer PW, & Arndt PF (2007). The majority of recent short DNA insertions in the human genome are tandem duplications. Molecular Biology and Evolution, 24, 1190–1197. 10.1093/molbev/msm035 [DOI] [PubMed] [Google Scholar]
- Meyer AN, McAndrew CW, & Donoghue DJ (2008). Nor-dihydroguaiaretic acid inhibits an activated fibroblast growth factor receptor 3 mutant and blocks downstream signaling in multiple myeloma cells. Cancer Research, 68, 7362–7370. 10.1158/0008-5472.CAN-08-0575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KN, Meyer AN, Siari A, Campos AR, Motamedchaboki K, & Donoghue DJ (2016). Oncogenic gene fusion FGFR3-TACC3 is regulated by tyrosine phosphorylation. Molecular Cancer Research, 14, 458–469. 10.1158/1541-7786.MCR-15-0497 [DOI] [PubMed] [Google Scholar]
- Nishimura G, Fukushima Y, Ohashi H, & Ikegawa S (1995). Atypical radiological findings in achondroplasia with uncommon mutation of the fibroblast growth factor receptor-3 (FGFR-3) gene (Gly to Cys transition at codon 375). American Journal of Medical Genetics, 59, 393–395. 10.1002/ajmg.1320590325 [DOI] [PubMed] [Google Scholar]
- Pauli RM (2019). Achondroplasia: A comprehensive clinical review. Orphanet Journal of Rare Diseases, 14, 1–49. 10.1186/s13023-018-0972-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin NH, & Biesecker LG (2001). Considerations for a multiaxis nomenclature system for medical genetics. Genetics in Medicine, 3, 290–293. 10.1097/00125817-200107000-00004 [DOI] [PubMed] [Google Scholar]
- Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, … Munnich A (1994). Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature, 371, 252–254. 10.1038/371252a0 [DOI] [PubMed] [Google Scholar]
- Rousseau F, el Ghouzzi V, Delezoide AL, Legeai-Mallet L, Le Merrer M, Munnich A, & Bonaventure J (1996). Missense FGFR3 mutations create cysteine residues in thanatophoric dwarfism type I (TD1). Human Molecular Genetics, 5, 509–512. 10.1093/hmg/5.4.509 [DOI] [PubMed] [Google Scholar]
- Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, Bocian M, … Wasmuth JJ (1994). Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell, 78, 335–342. 10.1016/0092-8674(94)90302-6 [DOI] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Boehm C, Valle D, & Hamosh A (2011). New tools for Mendelian disease gene identification: PhenoDB variant analysis module; and GeneMatcher, a web-based tool for linking investigators with an interest in the same gene. Human Mutation, 36, 425–431. 10.1002/humu.22769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Superti-Furga A, Eich G, Bucher HU, Wisser J, Giedion A, Gitzelmann R, & Steinmann B (1995). A glycine 375-to-cysteine substitution in the transmembrane domain of the fibroblast growth factor receptor-3 in a newborn with achondroplasia. European Journal of Pediatrics, 154, 215–219. 10.1007/BF01954274 [DOI] [PubMed] [Google Scholar]
- Takagi M, Kouwaki M, Kawase K, Shinohara H, Hasegawa Y, Yamada T, … Hasegawa T (2015). A novel mutation Ser344Cys in FGFR3 causes achondroplasia with severe platyspondyly. American Journal of Medical Genetics A, 167A, 2851–2854. 10.1002/ajmg.a.37231 [DOI] [PubMed] [Google Scholar]
- Tavormina PL, Rimoin DL, Cohn DH, Zhu YZ, Shiang R, & Wasmuth JJ (1995). Another mutation that results in the substitution of an unpaired cysteine residue in the extracellular domain of FGFR3 in thanatophoric dysplasia type I. Human Molecular Genetics, 4, 2175–2177. 10.1093/hmg/4.11.2175 [DOI] [PubMed] [Google Scholar]
- Tavormina PL, Shiang R, Thompson LM, Zhu YZ, Wilkin DJ, Lachman RS, … Wasmuth JJ (1995). Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nature Genetics, 9, 321–328. 10.1038/ng0395-321 [DOI] [PubMed] [Google Scholar]
- Thomas EE, Srebro N, Sebat J, Navin N, Healy J, Mishra B, & Wigler M (2004). Distribution of short paired duplications in mammalian genomes. Proceedings of the National Academy of Sciences USA, 101, 10349–10354. 10.1073/pnas.0403727101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MK, D’Avis PY, Robertson SC, & Donoghue DJ (1996). Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Molecular and Cellular Biology, 16, 4081–4087. 10.1128/mcb.16.8.4081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MK, & Donoghue DJ (1996). Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO Journal, 15, 520–527. [PMC free article] [PubMed] [Google Scholar]
- Wilkin DJ, Szabo JK, Cameron R, Henderson S, Bellus GA, Mack ML, … Francomano CA (1998). Mutations in fibroblast growth-factor receptor 3 in sporadic cases of achondroplasia occur exclusively on the paternally derived chromosome. American Journal of Human Genetics, 63, 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Sun A, Mekikian PB, Martin J, Rimoin DL, Lachman RS, & Wilcox WR (2014). FGFR3 mutation frequency in 324 cases from the international skeletal dysplasia registry. Molecular Genetics and Genomic Medicine, 2, 497–503. 10.1002/mgg3.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhou X, Ren X, Wang T, Yuan M, Wang Q, …Liu M (2007). Ser217Cys mutation in the Ig II domain of FGFR3 in a Chinese family with autosomal dominant achondroplasia. Chinese Medical Journal, 120, 1017–1019. [PubMed] [Google Scholar]