

Abstract
Macular telangiectasia type 2 (MacTel) is a progressive, late-onset retinal degenerative disease linked to decreased serum levels of serine that elevate circulating levels of a toxic ceramide species, deoxysphingolipids (deoxySLs); however, causal genetic variants that reduce serine levels in patients have not been identified. Here, we identify rare, functional variants in the gene encoding the rate-limiting serine biosynthetic enzyme, phosphoglycerate dehydrogenase (PHGDH), as the single locus accounting for a significant fraction of MacTel. Under a dominant collapsing analysis model of a genome-wide enrichment analysis of rare variants predicted to impact protein function in 793 MacTel cases and 17,610 matched controls, the PHGDH gene achieves genome-wide significance (p=1.2×10−13) with variants explaining ~3.2% of the affected individuals. We further show that the resulting functional defects in PHGDH cause decreased serine biosynthesis and accumulation of deoxySLs in retinal pigmented epithelial cells. PHGDH is the first significant locus for MacTel that explains the typical disease phenotype and suggests specific treatment options, including serine supplementation.
Introduction
Macular telangiectasia type 2 (MacTel) is a rare, late-onset macular degenerative disease with an estimated prevalence of 0.0045-0.06%1,2. MacTel has a definite, but complex, genetic basis supported by extended families with multiple affected individuals3, linkage, and genome-wide association studies4,5. Recent studies have associated MacTel with a deficiency in circulating levels of serine5,6. However, to date no disease-causing variants leading to a serine deficiency in MacTel have been identified. Serine is a non-essential amino acid that is central to mammalian cell metabolism acting not only as a proteinogenic amino acid, but contributing to central metabolic processes like nucleotide synthesis, redox homeostasis, and lipid biosynthesis. Recently, we determined that low serine levels in MacTel patients drive a concomitant increase in a neurotoxic atypical ceramide lipid species, deoxysphingolipids (deoxySL) that is causal in MacTel retinopathy6.
To date the only disease-causing variants linked to MacTel have been identified in serine palmitoyltransferase (SPT), the first enzyme in the ceramide/sphingolipid pathway6,7. These variants are gain-of-function mutations that directly elevate the synthesis of deoxySLs in patients8–10. However, these variants are extremely rare in MacTel patients, accounting for <0.1% of the disease load. Furthermore, these variants also cause a severe peripheral neuropathy, hereditary sensory and autonomic neuropathy type 1 (HSAN1), which is uncommon in most cases of MacTel. While the identification of causal SPT mutations has advanced our understanding of the disease mechanism underlying MacTel, causal genes and variants leading to reduced serine levels in MacTel patients have remained elusive. A genome-wide association study (GWAS) has identified several significant MacTel risk loci that lie within or near genes involved with serine and glycine synthesis, including PHGDH, the rate-limiting enzyme in de novo biosynthesis of serine5. Biallelic loss-of-function variants in PHGDH cause rare autosomal recessive disorders, PHGDH-deficiency and Neu-Laxova syndrome, characterized by severe neurological malformation and dysfunction 11–15. To date no disorders of the retina caused by PHGDH variants have been reported.
The increasing availability of whole exome sequencing (WES) data has aided the development of population-genetic approaches, such as case-control collapsing analysis, to detect disease associations in complex diseases with rare variants. Collapsing analyses test the association of the burden of rare, presumably deleterious variants between cases and controls on a gene, not a variant, level. This approach has been used to identify disease-associated variation in complex traits, such as amyotrophic lateral sclerosis (ALS) and epilepsy16,17. Recently, we have successfully applied collapsing method in rare, early-onset retinal diseases with variable phenotypes, as in cases resembling Stargardt/ABCA4 disease18. These advances suggest that a collapsing approach is useful for agnostically exploring associations in rare diseases, such as MacTel, where specific causal variants with variable penetrance are very rare in individual genes. This offers an objective measure of statistical enrichment of putatively causal variants in genes in which disease associations are suspected.
The goal of this study was to further our understanding of the genetic and functional basis of MacTel, specifically the prominent disease feature of low serine, by a combination of whole exome sequencing, case-control collapsing analysis, and functional studies including enzymatic assays, and CRISPR-edited patient iPS cell lines. We identified variants in the PHGDH locus, which result in deficiency of serine biosynthesis, as the major contributor to MacTel. This finding further solidifies the status of MacTel as a predominantly metabolic disease.
Results
Genetic analyses
To identify genes associated with MacTel, we used WES data from 793 cases and 17,610 controls. All affected individuals received diagnoses compatible with the clinical presentation of MacTel (see Materials and Methods). All controls were negative for any eye disease; in addition, those with history of neurological diseases (Alzheimer’s disease, ALS, etc.) were excluded for more stringent comparisons. These two groups were compared in a standard gene-level collapsing analysis where individuals were coded by the presence or absence of qualifying variants (QVs). We used several collapsing models defined by allele frequency, from ultra-rare, with minor allele frequency (MAF) <0.00005 and absent in gnomAD, to rare, MAF<0.001 in gnomAD; see Supplementary Table 1. In silico pathogenicity assessment by PolyPhen (“probably pathogenic”) or REVEL (>0.5) was applied to all models.
In the dominant model, the only gene where QVs reached study (genome)-wide significance (p = 1.33 x 10−6, see Materials and Methods) using two-sided Fisher’s exact test (FET) p-values, was PHGDH (Figure 1). The highest association was achieved in the model 3b with MAF<0.001 and REVEL >0.5 as the intolerance filters (Figure 1). In this model, we identified QVs in PHGDH in 3.15% of cases versus 0.56% of controls (Odds ratio [OR], 5.82; FET p-value=7.52×10−11). Application of additional filtering criteria by including loss-of-function (stop-gain and splicing) variants, “possibly pathogenic” or “deleterious” variants according to PolyPhen-2 HVAR, and functional data, resulted in 3.7% and 0.56% in cases and controls, respectively, OR=6.7, 95% CI [4.2;10.3]; FET p=1.2x10−13 (Table 1). PHGDH was the only gene, which reached study (genome)-wide significance in all models except for the ultra-rare (see below). The second gene, small integral membrane protein 8 (SMIM8), which was close to genome-wide significance (Figure 1), is a protein with unknown function and currently no suggested disease association. However, the excess of QVs in cases vs controls was close to 20x, so this gene/protein warrants further evaluation.
Figure 1. Collapsing analysis identifies PHGDH as a MacTel gene.
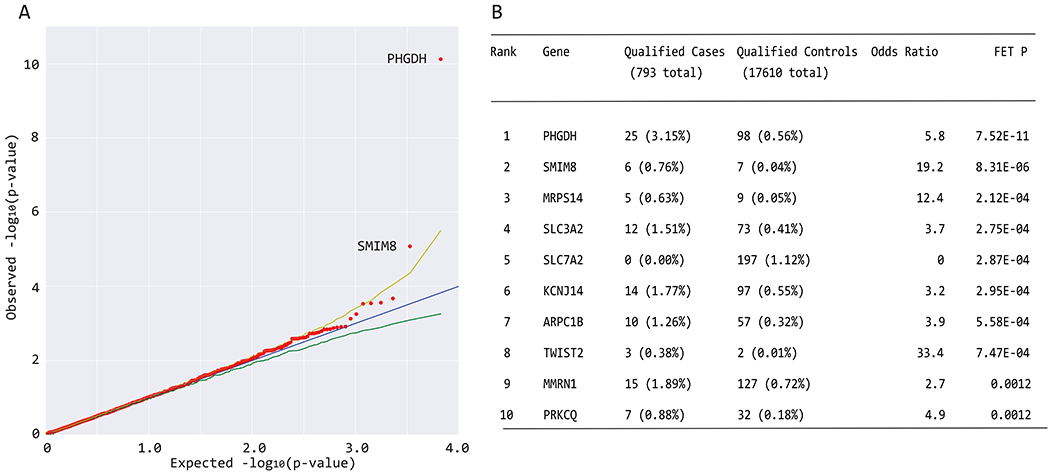
(a) Quantile-quantile (QQ) plot for exome-wide gene-based collapsing analysis under the dominant genetic model with MAF<0001 and REVEL>0.5 filters. The y-axis represents the −log10 of the observed two-sided Fisher’s exact test (FET) p-values (sorted). The x-axis represents the −log10 of the permutation-based expected FET p-values (sorted). The red dots represent the data points, while the blue line is the diagonal with slope 1. The green and yellow lines represent permutation-based 95% confidence intervals. Data points falling outside the 97.5th percentile bound are labeled with corresponding gene symbols. PHGDH reached study (genome)-wide significance, SMIM8 association was borderline. (b) The top 10 genes from collapsing analyses under the same model are shown, including the exact numbers of all qualifying cases and controls and statistical calculations of association (OR and FET P).
Table 1.
All qualified variants in PHGDH in 793 MacTel cases and 17610 controls under a dominant model with gnomAD MAF < 0.001.
| Variant ID Chr-Pos-Ref-Alt |
HGVS.c | HGVS.p | Alleles in 793 MacTel cases | Alleles in 17610 controls | Allele frequency in gnomAD (if available) | REVEL | Polyphen2 HVAR | CADD | Relative enzyme activity to wt protein |
|---|---|---|---|---|---|---|---|---|---|
| 1-120254666-GA-G | c.21_22G | p.Val9Cysfs*32 | 1 | ||||||
| 1-120254667-A-T | c.22A>T | p.Lys8Ter | 1 | 39.0 | |||||
| 1-120263820-G-A | c.166G>A | p.Ala56Thr | 1 | 0.000004 | 0.53 | B | 24.9 | ||
| 1-120263835-G-A | c.181G>A | p.Ala61Thr | 1 | 0.000046 | 0.53 | P | 27.5 | ||
| 1-120263880-G-T | c.226G>T | p.Ala76Ser | 2 | 0.000099 | 0.78 | D | 32.0 | ||
| 1-120263896-A-T | c.242A>T | p.Asp81Val | 1 | 0.000064 | 0.95 | D | 31.0 | ||
| 1-120263899-A-G | c.245A>G | p.Asn82Ser | 2 | 0.000004 | 0.78 | D | 27.0 | ||
| 1-120263904-G-T | c.250G>T | p.Asp84Tyr | 1 | 0.97 | D | 32.0 | |||
| 1-120263941-T-C | c.287T>C | p.Met96Thr | 1 | 0.85 | D | 25.6 | 0.17 | ||
| 1-120266027-G-A | c.319G>A | p.Ala107Thr | 1 | 2 | 0.000078 | 0.89 | D | 34.0 | |
| 1-120266052-T-G | c.344T>G | p.Met115Arg | 1 | 0.78 | B | 25.4 | −0.01 | ||
| 1-120266064-G-C | c.356G>C | p.Arg119Thr | 1 | 0.95 | D | 31.0 | |||
| 1-120269476-A-C | c.359A>C | p.Gln120Pro | 1 | 0.64 | B | 23.5 | |||
| 1-120269497-C-T | c.380C>T | p.Ser127Leu | 1 | 0.000016 | 0.74 | D | 35.0 | ||
| 1-120269654-A-C | c.439A>C | p.Thr147Pro | 1 | 0.84 | D | 27.1 | |||
| 1-120269667-T-C | c.452T>C | p.Leu151Pro | 1 | 0.93 | D | 28.0 | |||
| 1-120269682-T-C | c.467T>C | p.Ile156Thr | 1 | 0.000018 | 0.92 | D | 29.2 | ||
| 1-120269720-A-G | c.505A>G | p.Met169Val | 1 | 0.91 | D | 24.6 | |||
| 1-120277270-A-G | c.524A>G | p.Asp175Gly | 1 | 0.97 | D | 33.0 | |||
| 1-120277272-C-G | c.526C>G | p.Pro176Ala | 1 | 1 | 0.000008 | 0.85 | D | 29.3 | 1.34 |
| 1-120277282-C-G | c.536C>G | p.Ser179Cys | 1 | 0.60 | B | 28.3 | |||
| 1-120277308-G-T | c.562G>T | p.Val188Phe | 1 | 0.000004 | 0.80 | D | 32.0 | ||
| 1-120277318-T-A | c.572T>A | p.Leu191Gln | 1 | 0.000039 | 0.66 | P | 23.3 | ||
| 1-120277365-A-T | c.619A>T | p.Thr207Ser | 5 | 0.000052 | 0.78 | P | 26.6 | ||
| 1-120277384-C-T * | c.638C>T | p.Thr213Met | 1 | 0.000008 | 0.96 | D | 34.0 | 0.13 | |
| 1-120277956-G-C | c.682G>C | p.Gly228Arg | 1 | 0.000004 | 0.86 | D | 31.0 | ||
| 1-120277956-G-T | c.682G>T | p.Gly228Trp | 8 | 31 | 0.000843 | 0.88 | D | 33.0 | 0.17 |
| 1-120277980-C-T | c.706C>T | p.Arg236Cys | 1 | 3 | 0.000060 | 0.79 | D | 34.0 | 0.19 |
| 1-120277981-G-A | c.707G>A | p.Arg236His | 1 | 1 | 0.000036 | 0.81 | D | 32.0 | 0.14 |
| 1-120278004-G-A | c.730G>A | p.Ala244Thr | 1 | 0.000028 | 0.80 | D | 26.7 | 0.23 | |
| 1-120278016-G-A | c.742G>A | p.Ala248Thr | 2 | 0.000016 | 0.88 | D | 34.0 | ||
| 1-120278040-G-A | c.766G>A | p.Gly256Arg | 1 | 0.000012 | 0.94 | D | 34.0 | ||
| 1-120278046-G-T | c.772G>T | p.Ala258Ser | 1 | 0.000008 | 0.71 | P | 28.2 | ||
| 1-120278065-A-G | c.791A>G | p.Glu264Gly | 1 | 0.54 | B | 32.0 | |||
| 1-120279791-C-A | c.847C>A | p.His283Asn | 1 | 0.94 | D | 34.0 | |||
| 1-120279801-C-T * | c.857C>T | p.Ala286Val | 1 | 0.000004 | 0.92 | D | 34.0 | 0.11 | |
| 1-120279825-G-A | c.881G>A | p.Arg294His | 1 | 0.000095 | 0.71 | P | 31.0 | ||
| 1-120279833-G-T | c.889G>T | p.Glu297Ter | 1 | 0.000004 | 43.0 | −0.08 | |||
| 1-120279844-TG-T | c.900_901T | p.Val301Phefs*6 | 1 | ||||||
| 1-120283009-G-A | c.946G>A | p.Val316Met | 1 | 0.000052 | 0.80 | P | 26.5 | ||
| 1-120283054-C-A | c.991C>A | p.Pro331Thr | 1 | 0.000004 | 0.67 | B | 24.5 | ||
| 1-120283057-T-G | c.994T>G | p.Trp332Gly | 1 | 0.67 | P | 29.7 | |||
| 1-120283093-C-T * | c.1030C>T | p.Arg344Ter | 1 | 0.000017 | 37.0 | 0.47 | |||
| 1-120283117-G-T | c.1054G>T | p.Gly352Trp | 1 | 0.53 | P | 32.0 | 0.76 | ||
| 1-120284447-T-C | c.1136T>C | p.Leu379Pro | 1 | 0.95 | D | 28.1 | 0.23 | ||
| 1-120284516-T-G | c.1205T>G | p.Leu402Arg | 1 | 0.80 | D | 26.6 | 0.47 | ||
| 1-120285493-G-A * | c.1273G>A | p.Val425Met | 1 | 0.000004 | 0.74 | P | 28.7 | 0.45 | |
| 1-120285545-C-T | c.1325C>T | p.Thr442Met | 2 | 0.000004 | 0.55 | B | 12.3 | ||
| 1-120285569-G-T | c.1349G>T | p.Gly450Val | 1 | 0.71 | B | 25.1 | |||
| 1-120285625-C-T | c.1405C>T | p.Arg469Trp | 1 | 1 | 0.000016 | 0.61 | D | 24.0 | 0.36 |
| 1-120285626-G-A | c.1406G>A | p.Arg469Gln | 1 | 3 | 0.000032 | 0.40 | P | 17.6 | 0.63 |
| 1-120285672-G-A | c.1447+5G>A | 1 | 14.0 | ||||||
| 1-120286529-G-A | c.1468G>A | p.Val490Met | 1 | 5 | 0.000142 | 0.66 | B | 27.1 | 0.55 |
| 1-120286532-C-T | c.1471C>T | p.Arg491Trp | 1 | 7 | 0.000103 | 0.53 | B | 29.7 | 0.52 |
| 1-120286568-G-C | c.1507G>C | p.Gly503Arg | 2 | 0.000012 | 0.58 | D | 23.4 | ||
| 1-120286595-T-G | c.1534T>G | p.Ser512Ala | 1 | 0.000024 | 0.64 | B | 21.3 | ||
| 1-120286599-C-G | c.1538C>G | p.Ser513Cys | 1 | 0.50 | P | 24.0 | |||
| 1-120286599-C-T | c.1538C>T | p.Ser513Phe | 1 | 0.58 | P | 25.2 | 0.85 |
All qualified variants (QVs) with gnomAD MAF < 0.001 in case and control cohorts are listed. These satisfy at least one of the following additional criteria: REVEL>0.5, Polyphen2 = P or D, Loss-of-function (stop-gain, frameshift, splicing). CADD values are shown for reference purpose only and were not used for variant qualification. A total of 29/793 (3.7%) cases and 99/17,610 (0.56%) of controls carried a single QV, resulting in two-sided FET p=1.2x10−13, OR = 6.7, 95% CI [4.2;10.3]. No cases or controls carried more than one QV. Pathogenicity of all variants present in MacTel cases was assessed by PHGDH enzymatic activity assay; results are shown in the last column. No data indicates no assay was performed, resulting in a conservative overestimate of pathogenic variants in the control cohort.
indicates variants that are causal in Neu-Laxova syndrome.
All PHGDH variants identified in all 793 cases and 17610 controls under the most significant model with additional filtering criteria are detailed in Table 1. The QVs in both cases and controls were primarily missense with a few loss-of-function variants (2 stop-gain and 1 splice variant in 793 cases, 1 stop-gain and 2 indels in 17610 controls). Possible causality of the identified variants was initially estimated by a combination of in silico methods including previous knowledge of causality, MAF in the matched controls and the general population, analysis with predictive software, e.g., CADD, PolyPhen-2 HVAR, and REVEL. In total, 22 unique rare variants (MAF<0.001) were detected in 29 MacTel cases, all of which passed pathogenicity criteria (Table 1). Definitive data on the effect of variants were obtained by functional analyses (Table 1, and see below). Five of the 22 variants in cases, p.Thr213Met, p.Ala286Pro (p.Ala286Val in MacTel), p.Arg344Ter, p.Val425Met, and p.Val490Met, have been proven causal in recessive Neu-Laxova syndrome 1 (NLS1, MIM 256520) or recessive phosphoglycerate dehydrogenase deficiency (PHGDHD; MIM 601815), a severe neuropathological disorder with reduced plasma serine levels11,19. No NLS1-causing variants were present in any of the 17610 controls (Table 1). Since MacTel is a late onset, genetically heterogeneous disease (suggesting low penetrance of causal variants), the presence of many highly penetrant variants in any given gene in a substantial fraction of patients is not expected. We assessed the penetrance of identified variants by analyzing available family members. Of the 22 pathogenic PHGDH variants, based on both in silico and functional analyses, informative family members (parents, siblings, offspring) were available for only 3 variants in 5 families (Extended Data Figure 1), including a total of 17 family members with clinical and genetic data. In one family member (Extended Data Figure 1) the clinical diagnosis was ambiguous (“possibly affected”), further underlining the complications of genetic analyses in MacTel. PHGDH variants segregated with the disease in 13/17 cases; i.e., in 76.5% of the time, suggesting incomplete penetrance. The results from the ultra-rare model support this hypothesis, where plausible candidate genes have likely disease-associated variants in <1% of qualified cases (Table 2 and Supplementary Table 2).
Table 2.
Candidate genes for association with MacTel based on established co-morbidity with HSAN/CMT
| Genes | Qualified variants in MacTel cases [n=793] | Qualified variants in controls [n=17610] | Excess QV | FET P | OR | Collapsing Model | Disease association [dominant] [recessive] |
|---|---|---|---|---|---|---|---|
| SLC25A46 | 4 | 5 | 0.0024 | 3.6 x 10−4 | 17.7 | 1a | hereditary motor and sensory neuropathy, type VIB [rec] |
| SCN11A | 9 | 53 | 0.0042 | 0.001 | 3.8 | 1b | hereditary sensory and autonomic neuropathy, type VII [dom] |
| PHYH | 6 | 32 | 0.0029 | 0.005 | 4.2 | 2a | Refsum disease [rec] |
| DCTN1 | 6 | 39 | 0.0027 | 0.012 | 3.5 | 1a | distal hereditary motor neuropathy, type VIIB [dom] |
| MFN2 | 7 | 55 | 0.0029 | 0.016 | 2.9 | 2b | CMT, axonal, 2A2A [dom], hereditary motor and sensory neuropathy VIA [dom] |
| Combined | 0.015 | ||||||
| SPTLC1 | 1 | 14 | 0.0002 | 0.484 | 1.6 | 1a | hereditary sensory and autonomic neuropathy, type IA [dom] |
| SPTLC2 | 2 | 11 | 0.0009 | 0.106 | 4.0 | 1b | hereditary sensory and autonomic neuropathy, type IC [dom] |
Collapsing models and filtering criteria are specified in Supplementary Table 1. In short, Models 1a and 1b included only variants absent from gnomAD (“ultra rare”) and filtered for pathogenicity by PolyPhen as “probably pathogenic” (Model 1a) or REVEL >0.5 (Model 1b). Models 2a/b and 3a/b had the same pathogenicity filtering criteria and gnomAD minor allele frequencies <0.00005 (“very rare”) and <0.001 (“rare”), respectively.
We previously established a link between MacTel and low serine levels5 and recently identified two ultra-rare, highly penetrant variants in SPTLC1 and SPTLC2 genes that are causal for MacTel6. These variants also cause hereditary sensory and autonomic neuropathy (HSAN1), a rare dominant peripheral neuropathy8–10. Considering the co-morbidities we have identified in the MacTel population - neuropathy, deficiency in serine or sphingolipid metabolism - along with collapsing analysis, MAFs, and in silico predictions, we established a short list of candidate genes (Table 2). These include causal genes for HSAN and the phenotypically overlapping neurological disease Charcot-Marie-Tooth (CMT), SLC25A46 (OR, 17.85; FET p-value, 3.62×10−04), SCN11A (OR, 3.6; FET p-value, 0.0018), DCTN1, PHYH, and MFN2. Of these, variants in SCN11A, DCTN1 and MFN2 cause dominant neuropathies, while those in SLC25A46 and PHYH are responsible for recessive HSAN/CMT. It is likely that very rare QVs in dominant genes may be directly responsible for MacTel through a dominant negative mechanism, as is the case with SPTLC subunits6. Haploinsufficiency in recessive variants, as is the case in PHGDH, may also cause, or increase susceptibility to, MacTel.
Several systemic conditions have been reported at increased frequency in MacTel, of which the most prevalent is Type 2 Diabetes (T2D) where it is more common in MacTel patients than in age-matched controls (20-30% vs 5-10%, 20). We previously assessed the possible confounding effect of T2D in MacTel genetic analyses and found no evidence of association5. The chromosome 1p12 locus, where PHGDH resides, has been, as expected, associated with serine levels21; however, no robust evidence for association of the 1p21 locus with T2D has been found. We analyzed the incidence of T2D in our cohort of cases and, separately, in 29 cases with PHGDH variants as listed in Table 1. The incidence of T2D was 213 out of 793 (~27%) independent cases included in the collapsing analyses, in line with the previously published data. Interestingly, the incidence of T2D in cases carrying PHGDH variants was much lower, only one of 29 cases (~3.5%) carries a possible pathogenic PHGDH variant, a statistically significant difference (two-sided FET P=0.002) even below the lower end of general population frequencies. Because the fraction of carriers of PHGDH variants in our cohort is small, careful assessment and additional studies will be necessary to confirm the possible protective association of PHGDH variants with T2D.
The main reason for only one gene (PHGDH) reaching genome-wide significance is a substantial locus heterogeneity. For example, the two ultra-rare variants in SPTLC1 (p.Cys133Tyr) and SPTLC2 (p.Ser384Phe), which are known to cause MacTel as a co-morbidity with HSAN16, were not significant in collapsing analysis since only two cases harboring one of the two causal variants each were present in 793 samples (Table 2), further illustrating the extremely heterogeneous genetic structure of MacTel. The list of other promising candidate genes is given in Supplementary Table 2. In addition to having excess of QVs in MacTel vs controls, these genes were selected for their role in sphingosine and serine metabolism5,6. Finally, two plausible genes, which did not make the list, but have strong collapsing signals and/or intriguing disease association, include SLC12A1 (OR, 6.18; FET p-value, 1.46×10−04), and SIK1, heterozygous variants in which cause dominant early-onset severe neurodevelopmental disorder with epilepsy.
In order to ascertain the proportion of disease burden in our cohort that could be explained by genetic variation in these genes, we calculated the excess of qualified individuals (cases with QVs) in PHGDH and each of the 5 other plausible candidate genes, which are involved in HSAN/CMT (Tables 1 and 2). We subtracted the frequency of qualified controls from the frequency of qualified cases to quantify the potential percent of cases explained by each gene.
Taken together, the excess of qualified cases in PHGDH alone explains ~3.2% of the affected individuals, while the excess across all five nominally significant, plausible candidate genes potentially explains only another ~1.5% of MacTel patients (Table 2).
Identified variants result in PHGDH with reduced enzymatic function
The PHGDH variants identified in MacTel patients result in amino acid changes that are located throughout the protein and found in all of the characterized and/or predicted domains (Figure 2a). PHGDH catalyzes the oxidation of the glycolytic intermediate 3-phosphoglycerate to 3-phosphohydroxypyruvate (Figure 2b, inset). To assess the impact of the variants on PHGDH function we used site-directed point mutagenesis and assayed enzymatic activity of the overexpressed mutated protein after partial purification from cell lysates for 20 variants found in patients, 9 of which were also present in controls at statistically significantly lower frequency (Table 1). In addition, enzyme activity data were also obtained for several variants found in both cases and controls, which did not reach the selection threshold by predictive methods, but could have been qualified as very rare variants significantly enriched in cases (e.g., p.Gln253Arg, p.Glu420Lys, p.Val508Ile) (Extended Data Figure 2). Enzyme activity was determined using 3-phosphoglycerate as a substrate and monitoring the reduction of the cofactor NAD+ to NADH. All of the variants were expressed at detectable levels and distinguishable from the endogenous protein by a size shift due to an N-terminal FLAG-HA tag (Extended Data Figure 2a–b). Enzymatic activity was corrected for variant protein abundance and normalized to the activity of the overexpressed wild type enzyme (Figure 2b, Extended Data Figure 2c). All but one tested variants reduced PHGDH activity, with 10 variants reducing activity more than 75% (Table 1, Figure 2b), indicating that a large portion of the MacTel variants could significantly affect enzyme function. Using an available structure of truncated PHGDH (PDB 6CWA, residues 6 – 30422), we find that many of the most detrimental variants are located near the substrate binding pockets or within the dimerization interface (Figure 2c–d). Detrimental variants are also found on exposed surfaces (for example, p.Gly228Trp), potentially interfering with function through allosteric structural changes or via interactions with domains not present in the truncated structure.
Figure 2. PHGDH variants identified in MacTel patients disrupted enzyme function.
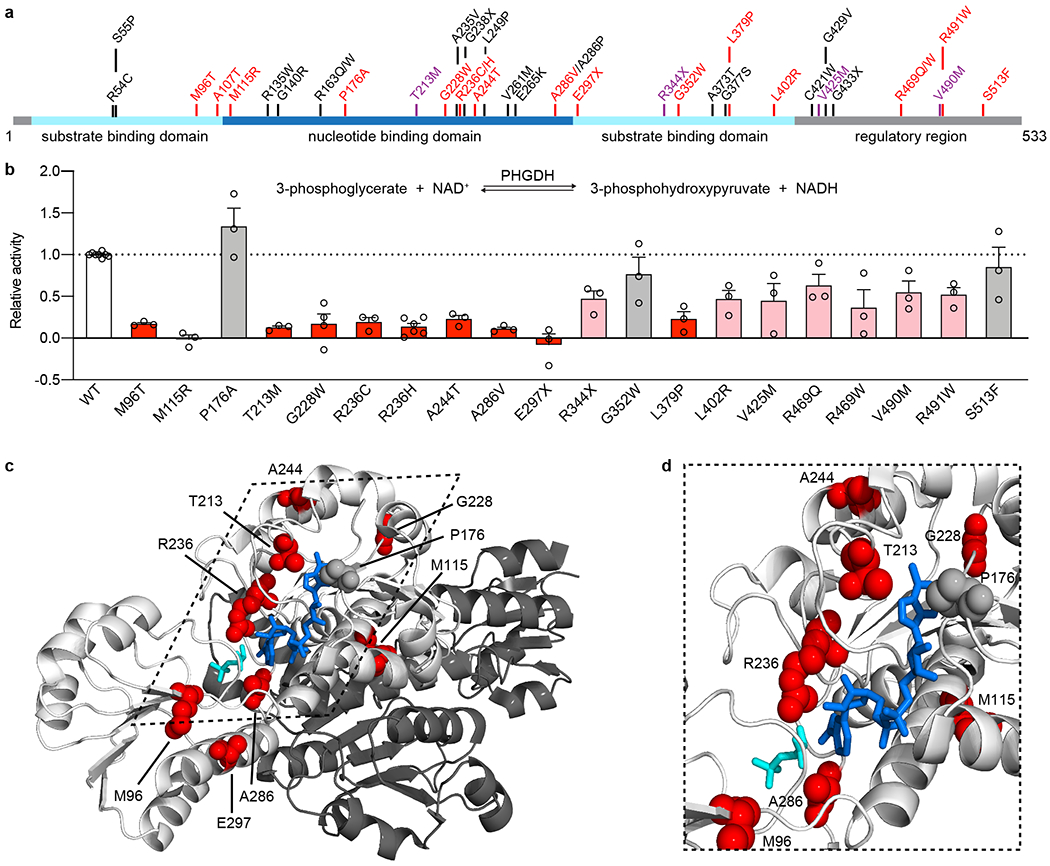
(a) Schematic of human PHGDH domain structure indicating identified MacTel variants (red), known PHGDH-deficiency/NLS1 variants (black) 11–14,19,23,38–42 and variants found in both groups (purple). (b) Relative activity of PHGDH variants compared to wild type (WT) PHGDH. Assayed activity was corrected for endogenous PHGDH by subtracting the activity of the empty vector and then normalized to the protein expression of the exogenous variant. Data for variants that retain less than 25% activity are shown in red, between 25-75% activity are shown in pink, and more than 75% activity are shown in grey. Data are shown as the mean +/− SEM, n = 3 independent experiments. A schematic of the PHGDH enzymatic reaction is shown as an inset at the top of the graph. (c) Structural representation of the location of MacTel variants on the available partial PHGDH structure. Chain A is shown in white and chain B is shown in dark grey, with substrates and variants only shown on chain A. The substrates NADH and 3PG are shown in dark blue and cyan. MacTel variants are shown in red or grey according to their activity in panel b. (d) Zoomed in and rotated view of the PHGDH active site with chain B omitted for clarity.
While many of the variants identified in MacTel patients are novel (Table 1), as described in the previous section, 5 of the 22 variants have previously been determined to cause NLS1 or PHGDHD when compound heterozygous or homozygous11,14,19,23 (Figure 2a, highlighted in purple). We found that the reduction in enzymatic activity by p.Val425Met and p.Val490Met was comparable to that previously reported11.
PHGDH p.Gly228Trp variant causes reduced serine biosynthesis in RPE
We have previously linked MacTel to a reduction in circulating serine levels causing an increase in circulating levels of deoxySLs, which are toxic to the retina6,24. In MacTel it is unclear if the retinal pathology is the result of reduced serine and elevated deoxySLs levels derived from circulation, or if serine deficiency and deoxySL accumulation are also occurring locally in retinal tissue. Retinal pigmented epithelium (RPE) are situated between the retinal blood supply in the choroid and the photoreceptors of the neural retina, and support the retina by forming a blood/retinal barrier, generating trophic factors and supplying nutrients to meet the high-energy demands of photoreceptors 25,26. Among the nutrients RPE supply to the retina are serine and glycine, which RPE synthesize from glucose27 (Extended Data Figure 3), and compared to the neural retina, serine synthesis enzymes and intermediate metabolites are enriched in the RPE28,29. (Supplementary Table 3). We sought to determine if a PHGDH haploinsufficiency in RPE is sufficient to cause disease-associated conditions directly in cells of the retina.
We introduced a heterozygous c.628G>T PHGDH substitution (p.Gly228Trp), using CRISPR editing, into an iPSC line derived from an individual without MacTel. This variant was selected because it occurs at the highest frequency in our MacTel cohort and significantly reduces enzyme function (Table 1 and Figure 2b). Three independent iPSC clones with a heterozygous substitution (p.Gly228TrpHet) and wild type (WT) PHGDH were differentiated into pigmented RPE (iPSC-RPE) and matured for 12 weeks. We functionally validated that iPSC-RPE synthesized serine and glycine from glucose and secreted these amino acids on their apical side (towards the retina), as previously reported in human fetal RPE27 (Extended Data Figure 3). The presence of a p.Gly228TrpHet variant in iPSC-RPE did not alter PHGDH transcript and protein levels compared to wild type cells, nor were expression levels of serine biosynthetic enzymes, their regulatory transcription factor ATF4, and serine transporters changed (Extended Data Figure 3a–d).
To measure the impact of p.Gly228TrpHet on serine biosynthesis in RPE we cultured iPSC-RPE in the presence of [U-13C6] glucose and quantified isotope enrichment in metabolites using mass spectrometry (Figure 3a, Extended Data Figure 4). The abundance of 13C in these amino acids measures the carbon contribution from [U-13C6] glucose through PHGDH to each, providing information on the relative flux of glucose to serine and glycine synthesis. 13C abundance of both serine and glycine was significantly reduced in p.Gly228TrpHet RPE, indicating decreased synthesis of serine/glycine via PHGDH (Figure 3b and Extended Data Figure 5a, b). Furthermore, total serine abundance was significantly lower in p.Gly228TrpHet RPE compared to WT RPE, which exhibited 35% decrease in serine abundance in RPE and a 58% decrease in the secretory flux of serine (Figure 3c, d). Glycine abundance was not significantly decreased (Figure 3c, d) however intracellular glycine pools in RPE are 10 fold greater than serine, potentially increasing glycine’s buffering capacity (Extended Data Figure 4e).
Figure 3. PHGDH p.Gly228Trp variant decreases serine synthesis in RPE.
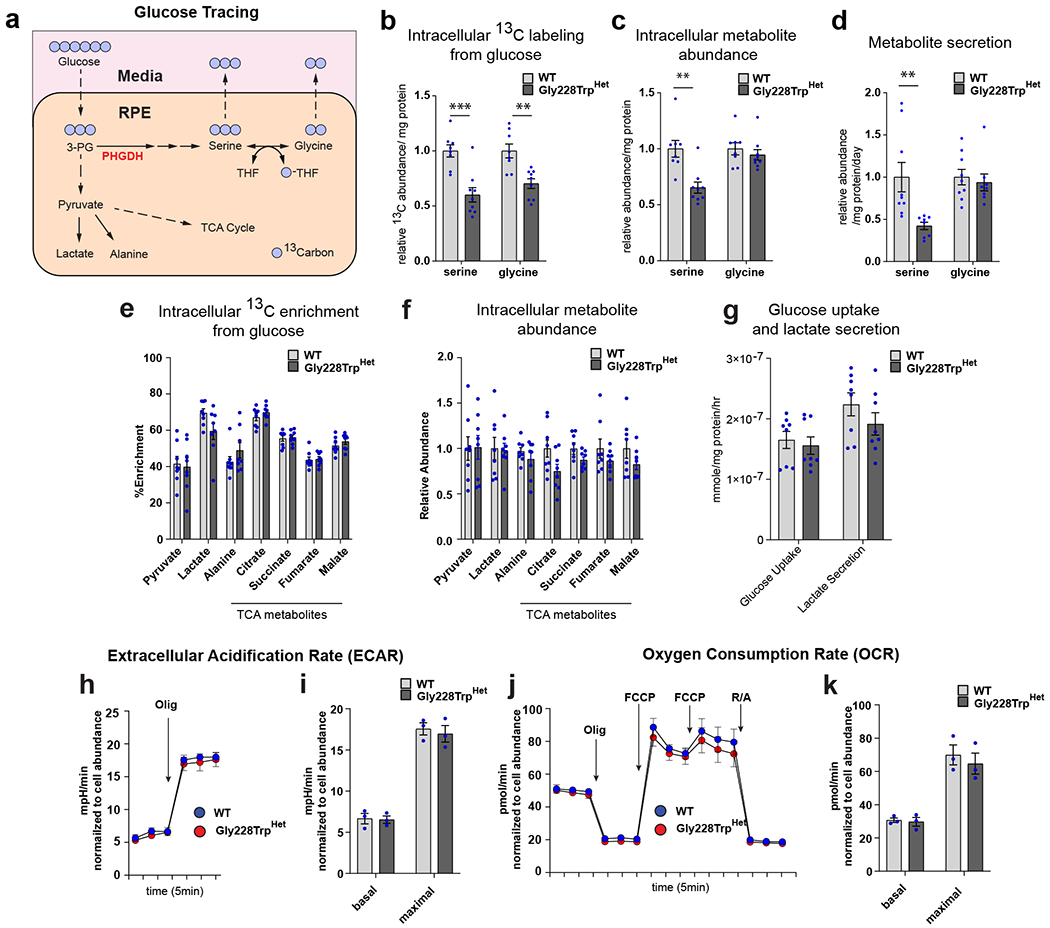
(a) Schematic illustrating [U-13C6] glucose tracing through central carbon metabolism, and the serine biosynthesis pathway in RPE. (b) Relative abundance of 13C isotope in fully labeled serine (M3) and glycine (M2) from [U-13C6] glucose in cell pellet (intracellular) between WT and PHGDH p.Gly228TrpHET iPSC-RPE (serine p=0.00035, glycine p=0.0013). (c) Relative intracellular metabolite abundance of total serine and glycine between WT and PHGDH p.Gly228Trp iPSC-RPE (serine p=0.0013). (d) Relative secretion flux of serine and glycine into media from RPE (serine p=0.0080). e) Intracellular percent labeling (% enrichment) of central carbon metabolites from [U-13C6] glucose of control and Gly228TrpHET iPSC-RPE. (f) Relative intracellular metabolite abundance of central carbon metabolites between WT and PHGDH p.Gly228Trp iPSC-RPE. (g) The rate of glucose uptake and lactate secretion in iPSC-RPE culture media during glucose tracing. (b-g) Data shown as the mean of at least 8 replicates from three independently derived WT or PHGDH p.Gly228Trp iPSC-RPE clones. Error bars +/− SEM. **p<0.01 ***p<0.001 unpaired two-tailed T-test. (h-k) Bioenergetic analysis of WT and PHGDH p.Gly228Trp iPSC-RPE in the absence of serine and glycine measuring glycolysis (h,i) and mitochondrial respiration (j,k). (h) ECAR measurement trace. (i) Basal ECAR represents the time point prior to oligo treatment, and maximal ECAR represents the third time point following Olig treatment. (j) OCR measurement trace. (k) Basal and maximal measurements are normalized to OCR output following R/A treatment. Basal OCR represents the time point prior to Olig treatment maximal OCR represents time point following FCCP treatment. Data shown as the mean of three independently derived clones of WT and PHGDH p.Gly228Trp iPSC-RPE. Each clone is the average of 12 independent measurements normalized to cell abundance +/− SEM.
Contrary to previous findings from PHGDH inhibition in cancer cell lines30 we found no decrease in the 13C labeling from glucose of glycolytic intermediates and TCA cycle metabolites, nor did we observe a significant decrease in their abundance (Figure 3e,f). The glucose uptake and lactate secretion were also unchanged in the p.Gly228TrpHet RPE compared to WT (Figure 3g). Furthermore, we found no difference in extracellular acidification rates (ECAR) or oxygen consumption rates (OCR) between WT and p.Gly228TrpHet RPE (Figure 3h–k). These data suggest that p.Gly228TrpHet does not broadly alter central carbon metabolism in RPE.
PHGDH p.Gly228Trp and SPTLC1 p.Cys133Tyr variants cause elevated deoxysphingolipids in RPE
MacTel can be caused by elevations in deoxySLs, which occur in the context of mutations in SPTLC1 or SPTLC28,10, or reduced serine availability6,31. We next determined if a reduction in serine synthesis in RPE results in an elevation in the synthesis of deoxySLs. The first step of the sphingolipid/ceramide pathway is the condensation of either serine or alanine with palmitoyl-CoA by the enzyme SPT to make sphinganine (SA) or deoxysphinganine (deoxySA), respectively (Figure 4a). An increase in the deoxySA to SA ratio is cytotoxic, and can be elevated by an imbalance in cellular serine to alanine ratios, as seen in MacTel, or by gain-of-function variants in SPT subunits, LC1 and LC2, that preferentially generate deoxySAs, as in HSAN1/MacTel6,8,31. In the retina, sphingolipid/ceramides synthesis is highly enriched in the RPE28,32. We demonstrate that the relative synthesis of deoxySA and SA in RPE is sensitive to perturbations in serine levels by culturing iPSC-RPE in custom media depleted of serine and glycine then measured hydrolyzed sphingolipids (i.e. total sphingoid bases) using mass spectrometry. Upon removal of serine and glycine from the culture media, iPSC-RPE rapidly undergo a 4 to 5-fold increase in deoxySA within two days (Figure 4b and Extended Data Figure 6a). To determine if altered PHGDH activity affects deoxySL synthesis, we measured deoxySA and SA in the p.Gly228TrpHet CRISPR modified RPE. The reduction in serine synthesis in these cells result in a two-fold increase in deoxySA compared to wild type PHGDH RPE controls (Figure 4c and Extended Data Figure 6b). We next generated iPSC-RPE from individuals with and without the HSAN1/MacTel-associated variant, STPLC1 p.Cys133Tyr, known to increase the synthesis of deoxySA (Supplementary Table 4)8. Similar to p.Gly228TrpHet iPSC-RPE, iPSC-RPE derived from HSAN1 patients have a two-fold increase in deoxySA compared to iPSC-RPE derived from control patients (Figure 4d and Extended Data Figure 6c), indicating that the p.Gly228TrpHet PHGDH variant is sufficient to raise RPE deoxySL levels similarly to that observed with the causative MacTel variant SPTLC1 p.Cys133Tyr.
Figure 4. HSAN1/MacTel linked SPTLC1 p.Cys133Tyr and PHGDH p.Gly228Trp variants elevate deoxySA in RPE.
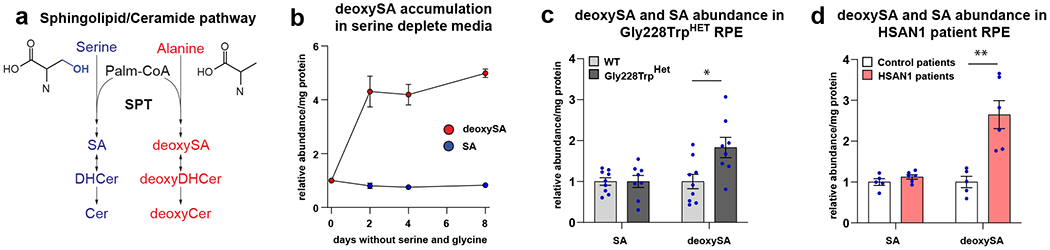
(a) Schematic illustrating the synthesis of sphingolipids and deoxysphingolipids from serine and alanine. SPT condenses palmitoyl-CoA (Palm-CoA) with either serine or alanine to generate the canonical or deoxy forms of sphinganine (SA), dihydroceramide (DHCer), and ceramide (Cer). (b) Relative sphinganine (SA, blue), and deoxysphinganine (deoxySA, red) abundance in control iPSC-RPE following removal of serine and glycine from culture media. Each time point represents the mean of triplicate WT samples. Error bars +/− SEM. (c) Relative SA and deoxySA abundance in iPSC-RPE following 2 days in serine and glycine free media between WT and PHGDH p.Gly228Trp iPSC-RPE. Data shown as the mean of at least 8 replicates from three independently derived clones of WT and PHGDH p.Gly228Trp iPSC-RPE. Error bars +/− SEM. deoxySA p=0.014 unpaired two-tailed T-test.*p<0.05. (d) Relative SA and deoxySA abundance in iPSC-RPE derived from 2 control patients and two HSAN1 patients with SPTLC1 p.Cys133Tyr following 2 days in serine and glycine free media. Data represents at least 5 replicates from clones derived from two control patients and two HSAN1 patients. Error bars +/−SEM. deoxySA p=0.0025 unpaired two-tailed T-test. **p<0.01
Discussion
We have used a case-control collapsing analysis to identify 22 rare variants in 29 MacTel patients in the gene encoding the rate-limiting serine biosynthetic enzyme PHGDH, accounting for ~3.2% of the population affected with MacTel. Using enzymatic assays, we showed that the vast majority of the variants encoded an enzyme with significantly reduced activity, including 10 variants by more than 75%. Using the most common missense mutation, p.Gly228Trp, we showed that reduced PHGDH function significantly affects serine synthesis in critical support cells of the retina, the RPE, leading to a selective decrease in serine synthesis and elevation of neurotoxic deoxySLs. These deoxySLs have been shown to be correlated with the extent of disease in MacTel6
This is the first report of variants in PHGDH causing, or significantly increasing susceptibility to, disease in the retina. Highly penetrant deleterious PHGDH variants have previously been shown to cause severe autosomal recessive neuropathological disorders, NLS1 (MIM 256520) and PHGDHD (MIM 601815) 11–15. We identified five of the same variants in 793 MacTel cases. We also identified more than 10 new variants in MacTel cases associated with an even greater loss of function. The same variants leading to multiple diseases suggests there is a genetic dose response to PHGDH, with biallelic inheritance of deleterious PHGDH variants causing severe developmental neuropathy, and monoallelic inheritance often leading to the late onset retinal disease, MacTel. This indicates the retina, and specifically the macula, might be highly sensitive to even partial loss of PHGDH activity. This is an important consideration since PHGDH inhibitors are currently in clinical trials as potential chemotherapies due to the central role of PHGDH in cancer metabolism where its genetic amplification, expression and enzymatic activity positively correlate with tumor survival and growth33. In light of our findings, macular function should be evaluated and closely monitored when treating patients with PHGDH inhibitors or serine-lowering treatments.
RPE are specialized retinal support cells critical for the health of the outer retina. They are situated at the interface between photoreceptors and the choriocapillaris where they form the outer blood-retinal barrier, and provide nutrient support for the neural retina25,26,34. Serine biosynthesis in the retina primarily occurs in the Müller glia and RPE, which then presumably provide serine for the photoreceptors 27,35. RPE also have a major role in cycling lipids with photoreceptors, as well as being enriched for sphingolipid/ceramides when compared to other retinal cell types25,26,28,32. Here, we demonstrate that MacTel-linked variants in PHGDH and SPTLC1 both elevate the synthesis of deoxySLs directly in the RPE.
Our previous work demonstrated that low levels of circulating serine, present in the typical form of MacTel, and mutations in SPT, present in HSAN1, both lead to increased circulating levels of neurotoxic deoxySLs and MacTel retinopathy6. Due to the unique role of RPE for serine and sphingolipid/ceramides synthesis in the retina, and that MacTel-linked variants in PHGDH and SPTLC1 directly elevate deoxySL synthesis in RPE, we speculate that elevated deoxySL synthesis in the RPE could play a direct role in MacTel pathogenesis25,26,28,32.
Our current and previous results define MacTel as a genetically heterogeneous disease. While we have determined that many MacTel patients have decreased serine levels, the specific genetic causes (i.e., penetrant variants in associated loci) of MacTel have remained elusive. Our earlier GWA study had identified five statistically significant or strongly suggestive loci associated with MacTel5. Here, we identified specific, rare, penetrant variants in only one of the five loci, the 46.4 kb locus on 1p12 that contains the PHGDH gene. It is accepted that rare, presumably (highly) penetrant variants are seldom found in genes in, or adjacent to, GWAS loci even in extensive sequencing-based follow-up studies36,37. One explanation is that these rare variants do not exist, especially in complex diseases, and that the associations result from subtle effects of frequent variants. Another possible explanation is that the disease-causing or -associated variants are in regulatory sequences, sometimes very distant from the causal genes36. The identification of specific, rare, and functionally validated variants in the PHGDH gene further confirms the role of coding variation in this gene as highly associated with a small but significant fraction of MacTel.
We have determined that the genetic architecture of MacTel is highly heterogeneous, where no gene except for PHGDH accounts for >0.5% of the disease in a global cohort of mostly Caucasian descent. The collapsing data and comorbidity of MacTel with other neurodegenerative diseases, (e.g., HSAN/CMT) allowed us to propose several additional candidate genes for MacTel, which contain possibly highly penetrant variants in a very small subset of patients. These include SLC25A46, SCN11A, DCTN1, PHYH, and MFN2 (Table 2). Considering the proven association of MacTel with serine and sphingolipid metabolism, we have also identified more nominally significant candidate genes (Supplementary Table 2). However, the fraction of each gene in MacTel is <0.5%, therefore, even when these genes are causal, their association with the disease will not reach genome-wide significance due to a limited sample size. Since MacTel is a rare disease with estimated prevalence of ~1:5000, low penetrance and expressivity, it is virtually impossible to increase the study cohort substantially for significant increase of the power of statistical analyses. The current results, however, coupled with the other accumulated knowledge of the disease etiology, will allow detailed analysis of the genetic basis of MacTel in extended families, which, together with functional analyses, will likely yield additional, albeit very rare, causal genes for the disease. This study demonstrates the power of collapsing analysis in a rare, genetically heterogeneous disease setting as an objective method to understand the heterogeneity and interplay of genetics and disease phenotypes. In disease phenotypes that lack specific genetic signature beyond GWAS, collapsing analyses offer a powerful and economical approach to expand our understanding of the genetic basis of the disease with limited sample sizes.
The principal conundrum is whether MacTel is a polygenic, complex disease (similar to age-related macular degeneration (AMD)) or a collection of Mendelian diseases (similar to, for example, retinitis pigmentosa (RP) where variation in >100 genes result in generally the same phenotype. While some features are remarkably similar to AMD (e.g., strong association with specific loci, late onset, variable phenotype, etc.), MacTel is a rare disease while AMD is common. Conversely, while MacTel often presents as a dominantly inherited Mendelian disease in extended families with suggested penetrance up to 50%3, and >76% in this study, extensive research has not identified the same causal genes in even two distinct families, except for the SPTLC1/2 and PHGDH genes, arguing against simple Mendelian inheritance in most cases. However, elevated levels of deoxySLs and low levels of serine seen in MacTel appear to be common to the broader population and not driven by an outlying subpopulation of MacTel patients6. Serine and sphingolipid metabolism are impacted by many genetic/metabolic pathways and, as such, are vulnerable to variants in numerous genes. While the question remains as to the specific genetic causes of a large fraction of MacTel, we have now identified two underlying genetic defects and their functional consequences in a small but significant fraction of patients with the disease. These findings facilitate precise diagnosis and prognosis of the disease and, most importantly, provide a basis for specific treatment options.
Methods
Study Subjects:
All enrolled patients read and signed informed consent forms approved by local Institutional Review Boards, and all clinical research complied with the Declaration of Helsinki and HIPAA privacy regulations. Clinical diagnoses were made by experienced retina specialists and were subsequently confirmed by masked readers at the Moorfields Eye Hospital MacTel Reading Centre in London. Subjects’ best-corrected visual acuities were recorded using standard ETDRS protocols and retinal examinations were conducted after pupil dilation. All subjects were imaged with color fundus photography, fluorescein angiography, blue light reflectance, autofluorescence, and optical coherence tomography on a Heidelberg Spectralis confocal imaging system (Heidelberg, Germany). The initial analysis included 815 MacTel cases and 19,589 controls. Controls were selected from studies at the Institute for Genomic Medicine (IGM) known to have no comorbid phenotypes such as ophthalmic disease, metabolic disease, etc. Written informed consent was obtained from all study subjects at the time of recruitment. Patient collection and sharing of anonymized specimens for research was approved by site-specific Institutional Review Boards and ethics committees at 24 clinical centers in seven countries (Australia, Germany, France, UK, Switzerland, Israel and United States) as follows: (also see Reporting Summary) Protocols and records of consent were centrally managed by the EMMES Corporation. The following ethics boards granted approval for human subject enrollment: Quinze-Vingts, Paris, France: Comité de Protection des Personnes Hôpital Saint-Antonie; Centre for Eye Research, Victoria, Australia: The Royal Victorian Eye and Ear Hospital; QIMR Berghofer Institute of Medical Research, Queensland, Australia; Clinique Ophtalmolgie de Creteil, Paris, France: Comité de Protection des Personnes Hôpital Saint-Antonie; Hospital Lariboisiere, Paris, France: Comité de Protection des Personnes Hôpital Saint-Antonie; Jules Stein Eye Institute, UCLA, California, USA: The UCLA Institutional Review Board; Lions Eye Institute, Nedlands, Australia: Sire Charles Gairdner Group Human Research Ethics Committee; Manhattan Eye, Ear and Throat Hospital, New York, USA: Lenox Hill Hospital Institutional Review Board; Moorfields Eye Hospital, London, UK: National Research Ethics Service; Retina Associates of Cleveland, Inc., Cleveland, Ohio, USA: Sterling Institutional Review Board; Save Sight Institute, Sydney, Australia: South Eastern Sydney Illawarra Area Health Service Human Research Ethics Committee–Northern Hospital Network; Scripps Research Institute, La Jolla, California, USA: Scripps Institutional Review Board; St. Franziskus Hospital, Munster, Germany: Ethik-Kommission der Arztekammer Westfalen-Lippe und der Medizinishchen Fakultat der Westfallschen Wilhelms-Universitat; The Goldschleger Eye Institute, Tel Hashomer, Israel: Ethics Committee The Chaim Sheba Medical Center; The New York Eye and Ear Infirmary, New York, USA: The Institutional Review Board of the New York Eye and Ear Infirmary; The Retina Group of Washington, Olympia, Washington, USA: Western Institutional Review Board; University of Bonn, Bonn, Germany: Rheinische Friedrich-Wilhelms-Universität Ethik-Kommission; University of Chicago, Chicago, Illinois, USA: The University of Chicago Division of Biological Sciences–The Pritzker School Institutional Review Board; University of Michigan, Ann Arbor, Michigan, USA: Medical School Institutional Review Board (IRBMED); University of Wisconsin, Madison, Wisconsin, USA: Office of Clinical Trials University of Wisconsin School of Medicine and Public Health; The Wilmer Eye Institute of Johns Hopkins University, Baltimore, Maryland, USA: Johns Hopkins School of Medicine Office of Human Subjects Research; Scheie Eye Institute University of Pennsylvania, Philadelphia, Pennsylvania, USA: University of Pennsylvania Office of Regulatory Affairs; University of Bern, Bern, Switzerland: Kantonale Ethikkommission Bern; John Moran Eye University of Utah, Salt Lake City, Utah, USA: The University of Utah Institutional Review Board; Bascom Palmer Eye Institute University of Miami, Miami, Florida, USA: The University of Miami Human Subjects Research Office; Columbia University, New York, New York, USA: Columbia University Medical Center Institutional Review Board Category 4 waiver for research involving specimens obtained from de-identified subjects.
Sequencing and Collapsing Analysis:
All samples were sequenced at IGM with the IDT’s Exome Research Panel Version 1 (differing capture kits) or off-site using mostly AgilentV5 and V7 kits. All exome data were processed using the IGM alignment and annotation pipeline for standardized analysis outcome. Samples were sequenced on Illumina next-generation sequencing machines (San Diego, CA, USA) using DRAGEN Bio-IT Platform v. 2.5.1 (Illumina) to align reads to the Genome Reference Consortium Human Build 37, calling variants in accordance with the Genome Analysis Tool Kit (GATK, v.4.0.2.1) Best Practices Workflow, using ATAV (v.7.0.16), an IGM variant-calling pipeline.
We used the protocols previously laid out43 for cohort pruning and rare variant collapsing analyses. Specifically, we required samples to have below 8% contamination rate and to have at least 10X coverage in at least 90% of the CCDS regions. We removed any samples with second-degree or closer kinship evaluated using KING (1.4.2), preferentially retaining cases over controls, resulting in excluding 4 cases and 1,912 controls. To ensure accurate case-control comparisons, population substructures were corrected using the EIGENSTRAT (6.1.4) pruning algorithm44. We computed the principal components (PCs) of a matrix of 13,000 common variants in the cohort. We pruned samples further than 7 standard deviations away from the cohort mean in each of the 10 most informative PCs across 5 iterations, resulting in the exclusion of 18 cases and 67 control samples. While other methods to correct for population stratification, including those utilizing logistic regression, can perform similarly45, the EIGENSTRAT pruning method has been shown to perform well in rare variant collapsing analyses17,18,46–48. We only considered sites with equivalent rates of coverage between cases and controls, removing sites in CCDS (hg19, release 20) with an 11% or greater coverage difference between all cases and all controls (2MB sites removed).
We ran a variety of collapsing models (Supplementary Table 1), each of which considers specific properties for “qualifying variants” (QVs). Variants are filtered for varying rates of frequency, intolerance scores, and pathogenicity scores. We only keep variants that pass GATK’s recommended variant filters. For every gene, we computed a two-sided Fisher’s Exact Test (FET, SciPy module, Python 2.7.7) of the amount of cases with a QV versus the number of controls. We ensured the quality of our models by generating expected significance values, graphing them against the sorted observed p-values in quantile-quantile (QQ) plots, and confirming that the resulting regression line was not inflated. For specific hypotheses about potentially associated pathways and known gene sets, we collapsed onto specific gene sets. For each of the varying collapsing model filters, we checked the rates of cases with at least one QV in any of the genes in the genes set, versus the rates of controls. For all models, with 18,852 genes being tested, we used the Bonferroni multiple-test correction to set a study-wide significance threshold of p = 1.33×10−6.
Quantile-quantile plots and genomic inflation factor (λ):
We generated QQ plots using a permutation-based empirical expected probability distribution. For the dominant model’s qualification state matrix (genes × samples), we randomly permuted the case-control labels associated with each column of qualification states. After each permutation of the 793 case and 17,610 control labels, we recomputed the two-sided FET p-values for each gene and ordered the p-values from lowest to highest. We repeated this process 1,000 times and calculated the mean of each rank-ordered value across the 1,000 permutations, i.e. the average 1st rank p-value, the average 2nd rank p-value, etc. These averages represent the empirical estimates of the ordered expected p-values. This empirical expected p-value distribution does not depend on the assumption that the p-values are uniformly distributed under the null.
We estimated the λ inflation factor in a manner similar to the procedure defined in the ‘estlambda’ method in the genABEL R package (cran.r-project.org/web/packages/GenABEL/). Following the removal of data points corresponding to observed p-values of study-wide significance and data points where either the observed or the expected p-value was 1, we calculated the “inverse” of each p-value by subtracting it from 1. We then transformed these “inverse” observed and expected p-values using χ2 quantile function (with one degree of freedom). We performed a least-squares linear regression on the transformed observed and expected p-values. The λ factor was taken to be the slope of this regression line.
Cellular assays
Western Blot:
PHGDH protein levels were determined by western blotting using an anti-PHGDH antibody (Sigma-Aldrich HPA021241, 1:5000). For the enzymatic activity assay, a fluorescent secondary antibody was used (LI-COR 926-32213, 1:15000) and blots were visualized using LICOR. For the RPE cells, anti-β-actin was used as a loading control (Cell Signaling Technology 3700, 1:5000) with HRP secondary antibody (Sigma-Aldrich AP160P, 1:15000) and blots were visualized using enhanced chemiluminescence (Pierce). Protein abundance was quantified using Image J (version 1.50b). Unprocessed western blot available in Source Data.
PHGDH Activity Assay:
To test the activity of the PHGDH variants, a library of expression plasmids harboring each variant was generated. Wildtype (WT) human PHGDH (Addgene 83901) was cloned into the mammalian expression vector, pcDNA3.1-FLAG-HA (Addgene 52535). Variants were introduced using site-directed point mutagenesis according to the manufacturer’s protocol (QuikChange II, Agilent). HEK-293 cells were transfected using polyethyleneimine (1:3, DNA:PEI) with the constructs carrying the WT or variant PHGDH sequence or the empty pcDNA vector. After 48 hrs, cells were collected for western blot and PHGDH enzymatic activity analysis. Enzymatic activity of partially purified cell lysates was measured in the physiological direction (conversion of 3-phosphoglycerate to 3-phosphohydroxypyruvate) using a PHGDH colorimetric activity assay kit (BioVision) according to the manufacturer’s protocol. For the enzymatic assay, two million cells were lysed by a single freeze-thaw cycle and centrifuged at 17,000 x g for 10 minutes to remove cell debris. Two volumes of 4.32 M ammonium sulfate were added dropwise to the supernatant and the mixture was incubated on ice for 1 hour followed by centrifugation at 17,000 x g for 15 minutes. The resulting pellet was re-suspended in 200 μL assay buffer. The enzymatic activity of the overexpressed PHGDH variants was determined by subtracting the activity of the endogenous PHGDH determined from the empty vector controls. The corrected PHGDH activity was then normalized to the protein abundance of the overexpressed enzyme (upper band in western blots due to the additional FLAG-HA tag). The relative activity was then determined by normalizing the samples to the WT PHGDH activity.
Generation and maintenance of iPSCs:
The human induced pluripotent stem cell (hiPSC) lines were derived from peripheral blood mononuclear cells from patients. Reprogramming was performed by the Harvard and Salk iPS core facilities using sendai virus for reprogramming factor delivery. All cell lines used were verified for a normal karyotype (Cell Line Genetics Inc) and contamination-free. hiPSC were maintained on Matrigel (BD Biosciences) coated plates with mTeSR1 medium (STEMCELL Technologies). Cells were passaged every 3-4 days at approximately 80% confluency. Colonies containing clearly visible differentiated cells were marked and mechanically removed before passaging.
Differentiation of RPE:
For directed differentiation of RPE, 10 mM nicotinamide (Sigma-Aldrich) /62 ng/ml activin A (Peprotech) based protocols were used according to previously described protocols49. Confluent iPSCs cultures were switched to differentiation media containing: 78% Knockout DMEM (ThermoFisher, 10829018), 20% Knockout Serum Replacement (ThermoFisher, 10828028), 1% L-Glutamine (ThermoFisher, 25030081), 1% PenStrep (ThermoFisher, 15140-122), 800mg Nicotinamide (Sigma, N0636). Differentiating iPSCs are maintained in differentiation media for 3 weeks, then switched to differentiation media plus 140ng/mL Activin A (PeproTech, 120-14E) two weeks, then switched back to differentiation media. Following differentiation, pigmented colonies were manually excised and plated on growth factor reduced matrigel (corning) for expansion. RPE were passaged once at 1 to 6 for expansion before for being passaged and plated for assay conditions. For experiments on transwell inserts, we used 0.4uM pore sized coated with fibronectin (Corning 47743-654). RPE were plated in assay conditions at 50% confluency. In each assay RPE were plated at the same time and matured for a minimum of 12 weeks past the point of confluency before being assayed.
CRISPR genome editing:
To generate the c. 682G>T substitution in PHGDH we designed a guide RNA (gRNA) for a targeted double stranded break and a single-stranded oligo DNA nucleotide (ssODN) with the desired substitution for homologous recombination. We generated three independent heterozygous clones. Clones for three independent control iPSCs were selected from iPSCs that underwent the same protocol without obtaining a 682G>T substitution. The gRNA was designed using http:/tools.genome-engineering.org and cloned it into the px330-puro-eGFP plasmid using Fast digest BpiI restriction enzyme (ThermoFisher). The ssODN was designed with the 682G>T substitution and substitutions at the gRNA PAM site that disrupted the PAM site without altering amino acid sequence of PHGDH.
gRNA: TGGAGGGATCGTGGACGAA
ssODN: CCCCAGCAGGAAGATGCTTCGCTTTCTTCCAGGCTTGCTGAATGACAACACCTTTGCCCAG
TGCAAGAAGTGGGTGCGTGTGGTGAACTGTGCACGGGGAGGGATCGTGGACGAAGGCGCCCTGCTCCGGGCCCTGCAGTCTGGCCAGTGTGCCGGGGCTGCACTGGACGTGTTTACGGAAG
We transfected control iPSC line with the gRNA containing plasmid (1ug) and ssODN (10uM) using the Neon Transfection System (ThermoFisher) with two 30sec 850V pulses. Transfected iPSCs were selected using puromycin selection. Clones were established from single cell plating of puro selected iPSCs and manually excised from individual iPSC clusters following 10 days of expansion. Sequences were validated using sanger sequencing.
Sanger sequencing primers:
For 5’-3’-TGGCCCAGATCCATAACAGG
Rev 5’-3’-TGTAAAACGCCAGGCTTTCC
RNA isolation and quantitative RT-PCR:
Total RNA was purified from frozen tissues using Trizol Reagent (Life Technologies) according to the manufacturer’s instructions. First-strand cDNA was synthesized from 400ng of total RNA using High-Capacity Reverse Transcriptase kit (Applied Biosystems) according to the manufacturer’s instructions. Individual 10 μl SYBR Green Master Mix (Applied Biosystems) real-time PCR reactions consisted of 2 μl of diluted cDNA, 5 μl of Power Up SYBR Green(Applied Biosystems), and 1 μl of each 5 μM forward and reverse primers. The PCR was carried out on 384-well plates on a Quant Studio Real Time PCR system(Applied Biosystems) using a three-stage program: 95 °C for 10 min, 40 cycles of 95 °C for 20 s, 60 °C for 20 s and 72 C for 20 s. PCR data for intergene comparison were corrected for primer efficiency. Samples were normalized to internal loading control, 36B4. Primer sequences are provided in Supplementary Table 5.
Cell culture protocol for metabolite abundance measurements and glucose tracing:
To measure metabolite levels in RPE cell pellet and co-cultured media iPSC-RPE were switched to serine and glycine deplete custom RPE media substituted with custom Knock Out DMEM media and custom KSR (outlined below). iPSC-RPE were switched to custom media with 25 mM glucose and deplete of serine and glycine for 24 hours allowing for an acclimation to biosynthetic requirements. Following 24 hours cells were switched to fresh deplete media containing 25 mM glucose (for standard GC/MS metabolite abundance and sphingolipid abundance) or 25 mM U13C glucose (for glucose tracing) and cultured for 24 hours. Following 24 hours samples were spun down and supernatant was removed and snap frozen. Cell pellets were rinsed in ice cold saline, manually scrapped from the plate, and snap frozen. Metabolite measurements from GC/MS and LC/MS were normalized to protein content of RPE culture using BCA protein assay (Pierce, ThermoFisher).
GCMS analysis:
Polar metabolites from cell pellets or media were extracted using a MeOH:CHCL3:H2O extraction with norvaline as an internal standard and analyzed using GCMS as previously described50. Specifically, polar metabolites from cells were extracted with cold methanol:water:chloroform (1:1:2) solution with inclusion of norvaline as an internal standard. Polar metabolites from media were extracted from cold methanol:water (8:1) solution with inclusion of norvaline as internal standard. Samples were vortexed for 10 min and centrifuged at 16,000g for 10 min to allow phase separation. The upper aqueous layer was collected and dried under vacuum. Polar metabolites were derivatized in 2% (w/v) methoxyamine hydrochloride (Thermo Scientific) in pyridine and incubated at 37°C for 60 minutes. Samples were then silylated with N-tertbutyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA) with 1% tert-butyldimethylchlorosilane (tBDMS) (Regis Technologies) at 37°C for 30 minutes. The derivatized metabolites were analysed by an Agilent 7890B gas chromatograph equipped with a DB-35MS column (30 m (length) × 0.25 mm (inner diameter) Agilent J&W Scientific] and connected with an Agilent 5977C mass spectrometer. For metabolite separation, the gas chromatograph oven was held at 100 °C for 2 min followed by increasing the temperature to 300 °C at a ramp rate of 10 °C/min, and held for 3 min. The eluates from the gas chromatograph were subjected to electron impact ionization. Mass spectrometer scanning was performed over m/z range of 100–650. The quadrupole was held at 150 °C. The percentage isotopologue distribution of each metabolite was determined and corrected for natural abundance using in-house algorithms adapted from Fernandez et al46. Metabolite abundances were calculated by normalizing metabolite spectral counts to the norvaline internal standard and protein abundance determined by BCA protein assay (Pierce Thermo-Fisher). The percentage isotopologue distribution of each metabolite was determined and corrected for natural abundance using in-house algorithms adapted from Fernandez et al51. Metabolite abundances were calculated by normalizing metabolite spectral counts to the norvaline internal standard and protein abundance determined by BCA protein assay (Pierce Thermo-Fisher). Relative 13C abundance was calculated using M3 (serine) and M2 (glycine) fractional enrichment multiplied by the intracellular metabolite abundance.
Hydrolyzed sphingoid base determination:
Snap frozen RPE cell pellet was mixed with 0.5 mL of methanol and spiked with internal standards, sphinganine d7, sphingosine d7 and deoxysphinganine d3 (Avanti lipids). The samples were placed on a mixer for 1 h at 37 °C, centrifuged @ 2,800 x g and the supernatant collected and acid hydrolyzed overnight at 65 °C with 75 μL of methanolic HCl (1N HCl, 10M H2O in methanol). Next, 100 μL of 10 M KOH was added to neutralize. 625 μL of chloroform, 100 μL of 2N NH4OH and 500 μL of alkaline water were added, the sample vortex-mixed and centrifuged for 5 min at 16,000 x g. The lower organic phase was washed three times with alkaline water and dried under air. LCMS analysis was performed on an Agilent 6460 QQQ LC-MS/MS. Metabolite separation was achieved with a C18 column (Luna 100 x 2.1 mm, 3 μm, Phenomenex). Mobile phase A was composed of a 60:40 ratio of methanol:water and mobile phase B consisted of 100% methanol with 0.1% formic acid with 5 mM ammonium formate added to both mobile phases. The gradient elution program consisted of holding at 40% B for 0.5 min, linearly increasing to 100% B over 15 min, and maintaining it for 9 min, followed by re-equilibration to the initial condition for 10 min. The capillary voltage was set to 3.5 kV, the drying gas temperature was 350 °C, the drying gas flow rate was 10 L/min, and the nebulizer pressure was 60 psi. Sphingoid bases were analyzed by SRM of the transition from precursor to product ions at associated optimized collision energies and fragmentor voltages as per Gantner et al, 20196.
Custom Deplete RPE Media:
Custom Knock Out DMEM without glucose, pyruvate, serine, glycine, manufactured by Gibco with all additional components unchanged and proprietary. Custom KSR formulation provided in Supplementary Table 6.
Extracellular Metabolic Flux Analysis:
Oxygen consumption rate (OCR) and extracellular acidification rates (ECAR) were performed on an Agilent Seahorse XFe96 Analyzer system (Seahorse Biosciences) following instructions by the manufacturer. Cells were plated 24 hours prior to analysis at a density of 5x104 cells per well on to a Seahorse XF96 V3 PS cell culture microplate (catalog #101085) coated with synthemax (Corning, catalog #CLS3535). RPE culture media was changed to Custom Knock Out DMEM and incubated for 1 hour at 37°C without CO2 supply prior to assay. Custom Knock Out DMEM (Gibco) for assay was manufactured without phenol red, glucose, serine, glycine, or sodium bicarbonate and supplemented with 12 mM glucose, 10 mM HEPES, 5 mM sodium pyruvate, 2 mM L-glutamine at a pH of 7.4. Assay parameters were set at 3 minute mix, and 2 minute measure in four steps: (1) 2 μM oligomycin (Sigma), (2) 0.5 μM FCCP (Sigma), (3) repeat injection of 0.5 μM FCCP to ensure maximal OCR was achieved, (4) 1 μM rotenone (Sigma) and 1μM antimycin (Sigma). Following OCR and ECAR assay cells were quantified using CyQUANT Cell Proliferation Assay (catalog #C7026). Each OCR and ECAR measurement was normalized to the corresponding cell quantity in that well. Each clone of WT and PHGDH p.Gly228Trp iPSC-RPE was run in 12 separate wells and averaged.
Measurement of Glucose and Lactate in Culture Media
Glucose and lactate concentrations in the culture media were determined by using the YSI 2950 Biochemistry Analyzer. Media was assayed following 24 hours on cell culture. Media samples were centrifuged at 300 x g for 5 minutes before 100 uL of sample was analyzed on the instrument. Concentrations in medium were compared to concentrations measured in the initial (t=0) media and normalized to the protein concentration of cells in the corresponding well and experiment duration.
Extended Data
Extended Data Fig. 1. Pedigrees segregating possibly pathogenic PHGDH variants.
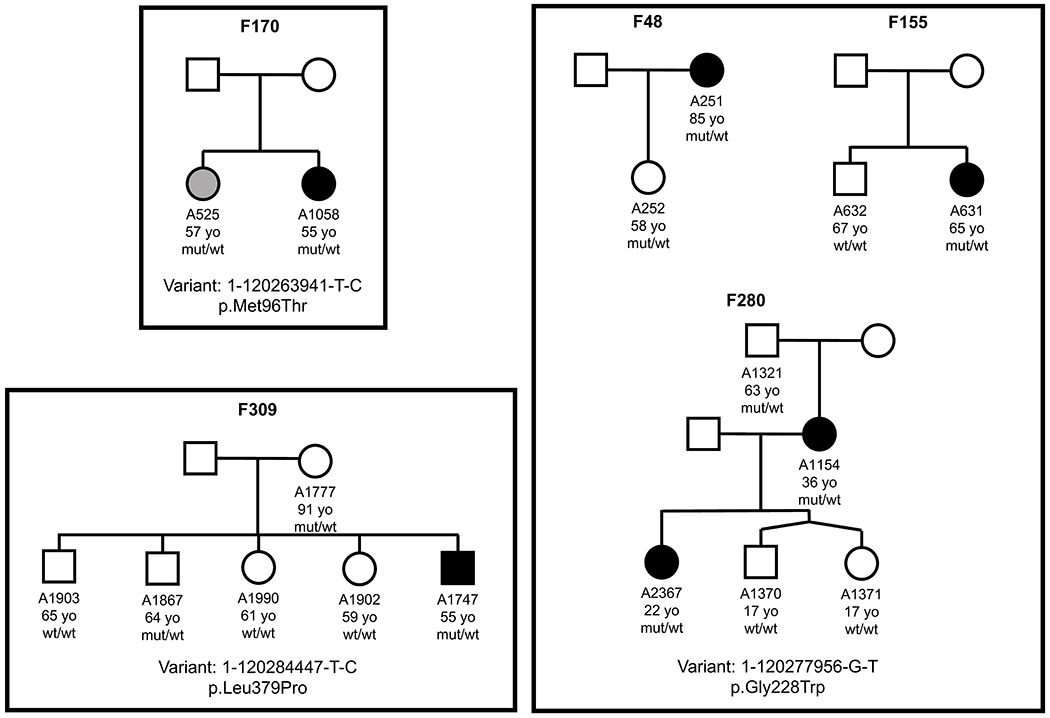
Three variants, given for each group, were analyzed for segregation with the disease in 5 families. The specific number, age, and result of genetic analysis is given for all family members who were available for clinical and genetic analyses. Filled, black symbols represent affected individuals, white symbols define unaffected family members and grey symbols depict family members with ambiguous diagnoses (maybe or maybe not affected). Ages of family members at the time of recruitment, when clinical diagnosis was determined, are given below each symbol. Wt, wild type allele; mut, the allele with the specific PHGDH variant.
Extended Data Fig. 2. Example of western blots.
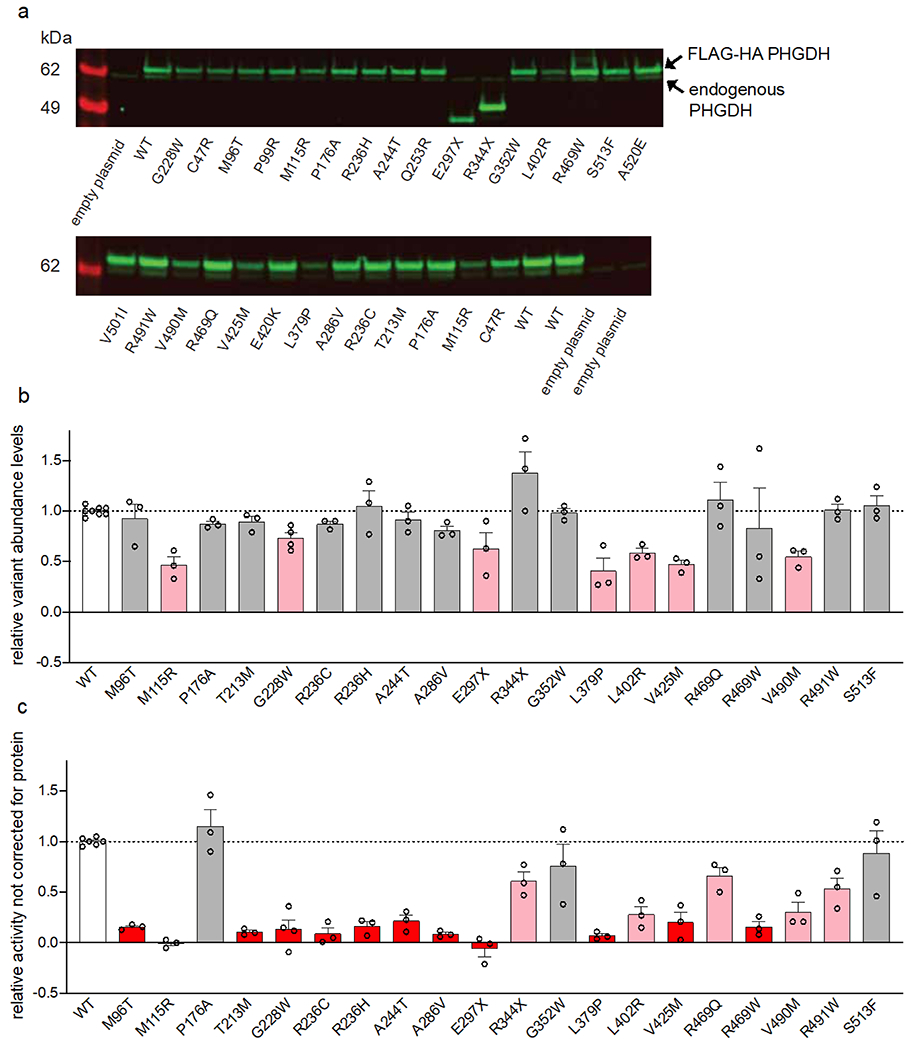
a) Example western blots of overexpressed variants. Arrows indicated overexpressed protein (upper band, shifted from the FLAG-HA tag) and endogenous PHGDH protein (lower band). b) Relative expression of each PHGDH variant calculated from three independent experiments and normalized to corresponding WT expression in each blot. c) PHGDH enzymatic activity after correcting for endogenous activity and without normalizing for overexpressed variant protein abundance. Data for variants that retain less than 25% activity or expression are shown in red, between 25-75% activity or expression are shown in pink, and more than 75% activity or expression are shown in grey. Data are shown as the mean +/− SEM, n ≥ 3 independent experiments.
Extended Data Fig. 3. Relative gene expression.
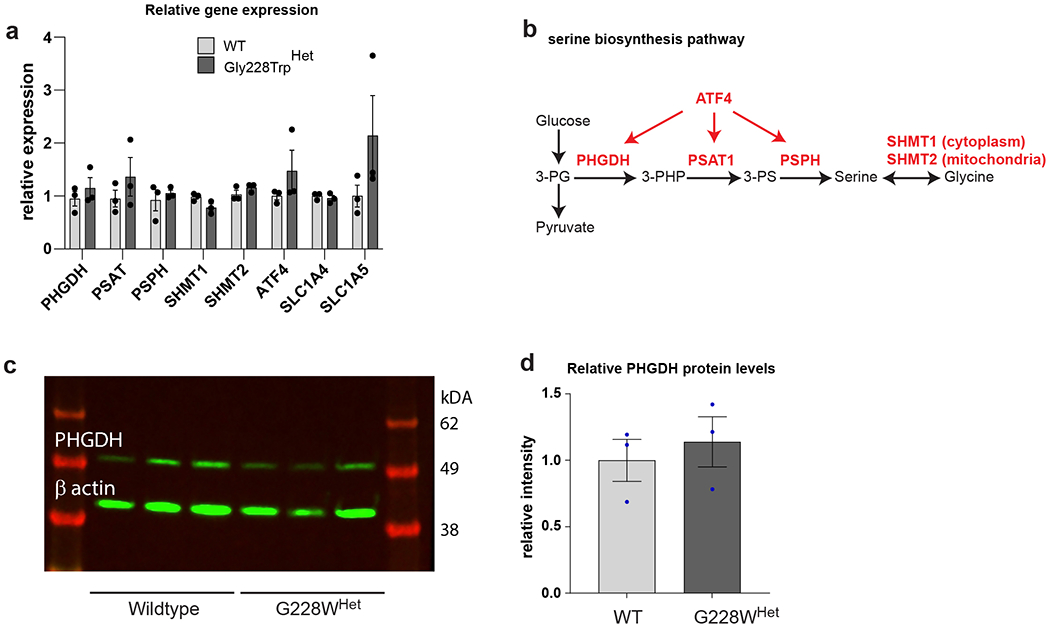
a) qPCR showing relative gene expression of enzymes from serine biosynthesis pathway. b) Schematic of the serine biosynthesis pathway from glucose showing metabolites (black) and enzymes/regulators (red). Data shown as the mean of three independently derived clones of wildtype (WT) and PHGDH p.Gly228TrpHET iPSC-RPE assayed in triplicate. Error bars +/− SEM. *p<0.05, **p<0.01 with unpaired two tailed t-test. c) Western blot of PHGDH and beta-actin loading control in WT and Gly228TrpHET iPSC-RPE clones. d) Relative protein levels of PHGDH normalized to beta-actin. Data shown as mean of 3 clones. Error bars +/− SEM. Unpaired two tailed t-test shows no difference.
Extended Data Fig. 4. Metabolite tracing.
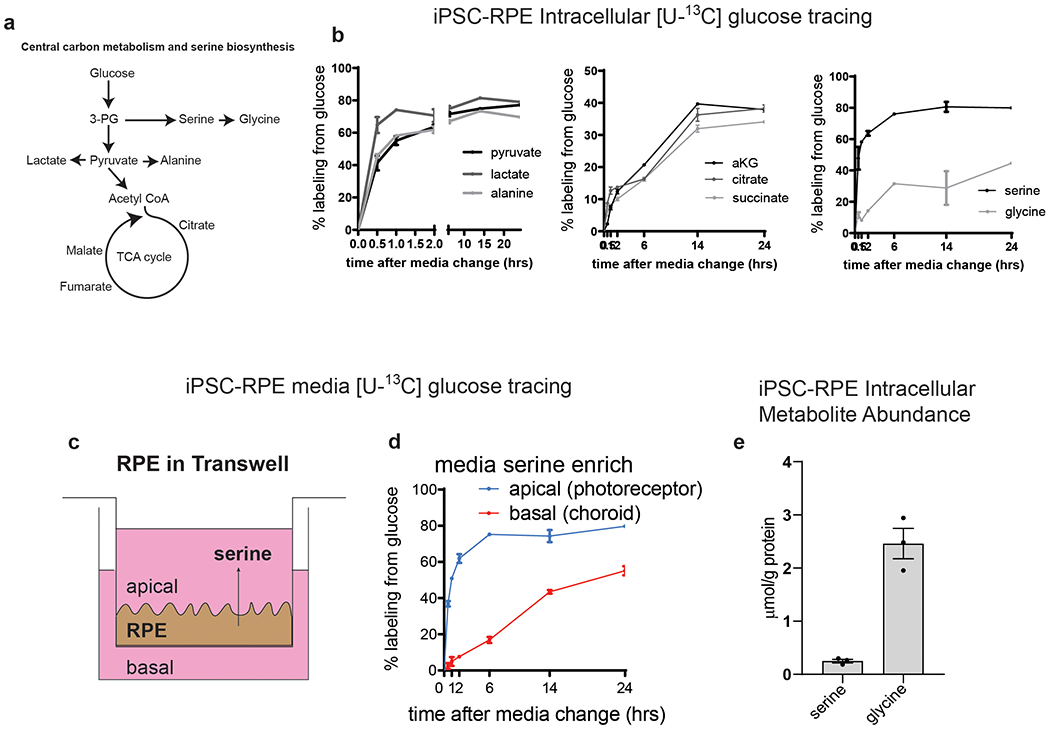
a) Schematic illustrating key metabolites in central carbon metabolism. b) % labeling of central carbon metabolites from [U-13C] glucose in cell pellet of iPSC-RPE. Points are the mean of three separately run wildtype iPSC-RPE samples. Error bars are +/− SEM. c) Schematic showing basal and apical secretion of metabolites from iPSC-RPE in transwells. d) Apical (blue) and basal (red) media measurements of serine from iPSC-RPE. e) Mean intracellular abundance of serine and glycine in wildtype iPSC-RPE. n=3 independent clones of wildtype iPSC-RPE. Error bars are +/− SEM.
Extended Data Fig. 5. Isotopologue distribution.
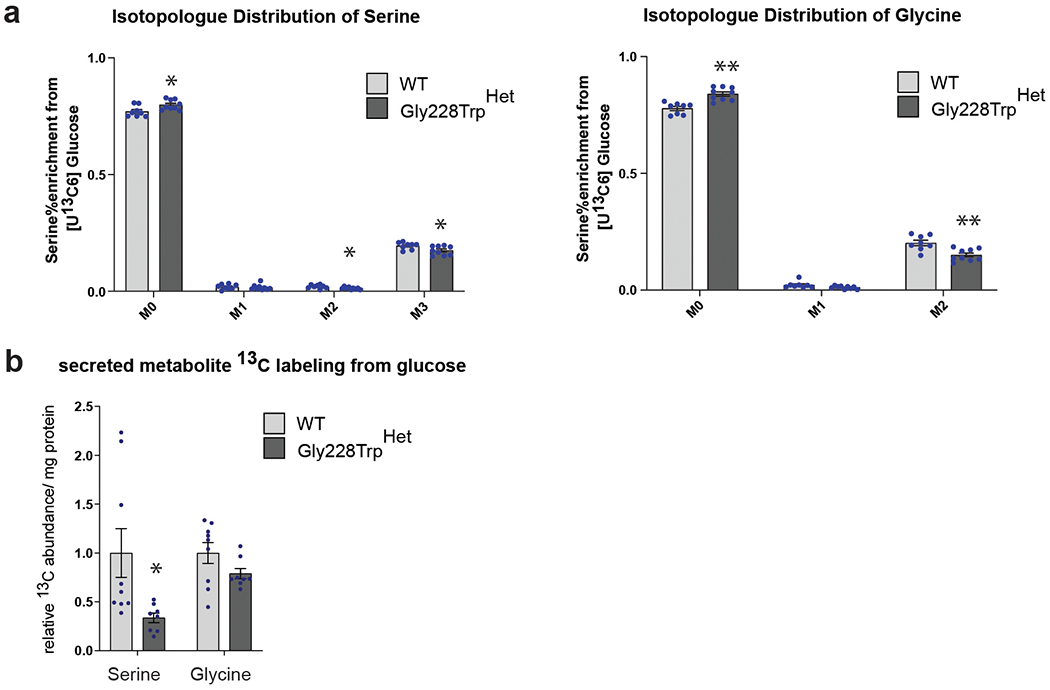
a) Isotopologue Distribution of U-13C from glucose in serine and glycine. b) Relative abundance of 13C isotope in fully labeled serine (M3) and glycine (M2) from [U-13C6] glucose in cell culture media (secreted) between WT and PHGDH p.Gly228TrpHET iPSC-RPE over a period of 24 hours. (a,b) Data shown as the mean of nine WT and eight PHGDH p.Gly228Trp replicates from three independently derived clones. Error bars +/− SEM. *p>0.05, **p>0.01 with unpaired two-tailed T-test. (a) serine: M0 p=0.02, M2 p=0.04, M3 p=0.03; glycine: M0 p=0.0002, M1 p=0.052, M2 p=0.002. (b) serine p=0.03.
Extended Data Fig. 6. deoxySA/SA ratios.
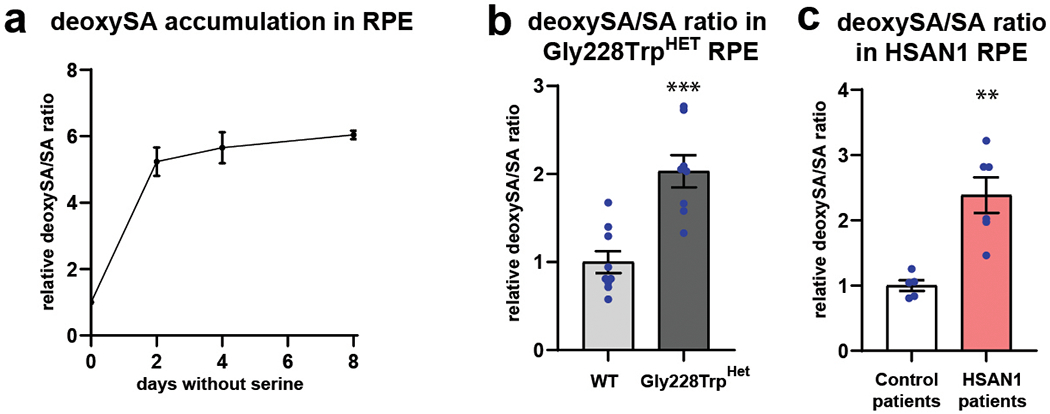
a) DeoxySA/SA ratios following 2, 4, and 8 days of culturing control iPSC-RPE in serine and glycine free media. Each time point run in triplicate. Error bars SEM. (b) Relative intracellular deoxySA/SA ratios in WT and PHGDH p.Gly228Trp iPSC-RPE following 2 days in serine and glycine free media. Data represented as mean of nine WT and eight PHGDH p.Gly228Trp replicates from three independently derived clones. (c) Relative intracellular deoxySA/SA ratios in control patient and HSAN1 patient iPSC-RPE following 2 days in serine and glycine free media. Data represented as the mean of five independently derived iPSC-RPE clones from two control patients and six independently derived iPSC-RPE clones from two HSAN1 patietns. Error bars SEM. **p<0.01 unpaired two-tailed T-test. (b) p=0.0002. (c) p=0.0015.
Supplementary Material
Acknowledgements:
We would like to thank the Lowy Family for their funding support of the MacTel project, the Lowy Medical Research Institute, and this study. Funding for the LC-MS used to measure lipids was provided by the NIH grant R01CA234245 to CM. Some genetic studies were supported, in part, by R01EY028203, R01EY029315, and P30EY019007 to RA, and the unrestricted funds from the Research to Prevent Blindness to the Department of Ophthalmology, Columbia University. We would like to thank the Moorfields Eye Hospital Reading Centre for their evaluation of clinical images. For discussions and expertise on measuring PHGDH activity we would like to thank K. Mattaini. We would like to thank M.L. Moon, and J. Orozco for administrative assistance, and J. Trombley for clinical oversight. We would like to express our appreciation of B. Hart and P. Bernstein at the Moran Eye Center for supplying us with patient monocytes from which we generated iPSCs. We would like to thank P. Westenskow for his assistance in establishing iPSC-RPE differentiation protocols, and C. Bautista, and J. Gleeson for their plasmids and assistance in establishing CRISPR editing protocols. We would also like to thank T. Cherry for processing the differential expression analysis for select genes in Extended Data Figure 3.
Footnotes
Declaration of competing interests: All authors declare no competing interests.
Data availability: The datasets generated during and/or analyzed during the current study, if not presented in the manuscript, are available at GitHub (https://github.com/igm-team/MacTel), and/or from the corresponding author on reasonable request.
References
- 1.Aung KZ, Wickremasinghe SS, Makeyeva G, Robman L & Guymer RH The prevalence estimates of macular telangiectasia type 2: the Melbourne Collaborative Cohort Study. Retina 30, 473–8 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Klein R et al. The prevalence of macular telangiectasia type 2 in the Beaver Dam eye study. Am J Ophthalmol 150, 55–62 e2 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ronquillo CC, Wegner K, Calvo CM & Bernstein PS Genetic Penetrance of Macular Telangiectasia Type 2. JAMA Ophthalmol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parmalee NL et al. Identification of a potential susceptibility locus for macular telangiectasia type 2. PLoS One 7, e24268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scerri TS et al. Genome-wide analyses identify common variants associated with macular telangiectasia type 2. Nat Genet 49, 559–567 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Gantner ML et al. Serine and Lipid Metabolism in Macular Disease and Peripheral Neuropathy. N Engl J Med 381, 1422–1433 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duan J & Merrill AH Jr. 1-Deoxysphingolipids Encountered Exogenously and Made de Novo: Dangerous Mysteries inside an Enigma. J Biol Chem 290, 15380–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Penno A et al. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem 285, 11178–87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rotthier A et al. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am J Hum Genet 87, 513–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eichler FS et al. Overexpression of the wild-type SPT1 subunit lowers desoxysphingolipid levels and rescues the phenotype of HSAN1. J Neurosci 29, 14646–51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klomp LW et al. Molecular characterization of 3-phosphoglycerate dehydrogenase deficiency--a neurometabolic disorder associated with reduced L-serine biosynthesis. Am J Hum Genet 67, 1389–99 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tabatabaie L et al. Novel mutations in 3-phosphoglycerate dehydrogenase (PHGDH) are distributed throughout the protein and result in altered enzyme kinetics. Hum Mutat 30, 749–56 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Shaheen R et al. Neu-Laxova syndrome, an inborn error of serine metabolism, is caused by mutations in PHGDH. Am J Hum Genet 94, 898–904 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Acuna-Hidalgo R et al. Neu-Laxova syndrome is a heterogeneous metabolic disorder caused by defects in enzymes of the L-serine biosynthesis pathway. Am J Hum Genet 95, 285–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaeken J et al. 3-Phosphoglycerate dehydrogenase deficiency: an inborn error of serine biosynthesis. Arch Dis Child 74, 542–5 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cirulli ET et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu X et al. A case-control collapsing analysis identifies epilepsy genes implicated in trio sequencing studies focused on de novo mutations. PLoS Genet 13, e1007104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolock CJ et al. A case-control collapsing analysis identifies retinal dystrophy genes associated with ophthalmic disease in patients with no pathogenic ABCA4 variants. Genet Med (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdelfattah F et al. Expanding the genotypic and phenotypic spectrum of severe serine biosynthesis disorders. Hum Mutat (2020). [DOI] [PubMed] [Google Scholar]
- 20.Clemons TE et al. Medical characteristics of patients with macular telangiectasia type 2 (MacTel Type 2) MacTel project report no. 3. Ophthalmic Epidemiol 20, 109–13 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Xie W et al. Genetic variants associated with glycine metabolism and their role in insulin sensitivity and type 2 diabetes. Diabetes 62, 2141–50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinstabl H et al. Intracellular Trapping of the Selective Phosphoglycerate Dehydrogenase (PHGDH) Inhibitor BI-4924 Disrupts Serine Biosynthesis. J Med Chem 62, 7976–7997 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Meneret A et al. A serine synthesis defect presenting with a Charcot-Marie-Tooth-like polyneuropathy. Arch Neurol 69, 908–11 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Scerri TS et al. Genome-wide analyses identify common variants associated with macular telangiectasia type 2. Nat Genet 49, 559–567 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Lehmann GL, Benedicto I, Philp NJ & Rodriguez-Boulan E Plasma membrane protein polarity and trafficking in RPE cells: past, present and future. Exp Eye Res 126, 5–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simo R, Villarroel M, Corraliza L, Hernandez C & Garcia-Ramirez M The retinal pigment epithelium: something more than a constituent of the blood-retinal barrier--implications for the pathogenesis of diabetic retinopathy. J Biomed Biotechnol 2010, 190724 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chao JR et al. Human retinal pigment epithelial cells prefer proline as a nutrient and transport metabolic intermediates to the retinal side. J Biol Chem 292, 12895–12905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinha T, Naash MI & Al-Ubaidi MR The Symbiotic Relationship between the Neural Retina and Retinal Pigment Epithelium Is Supported by Utilizing Differential Metabolic Pathways. iScience 23, 101004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cherry TJ et al. Mapping the cis-regulatory architecture of the human retina reveals noncoding genetic variation in disease. Proc Natl Acad Sci U S A 117, 9001–9012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reid MA et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat Commun 9, 5442 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esaki K et al. L-Serine Deficiency Elicits Intracellular Accumulation of Cytotoxic Deoxysphingolipids and Lipid Body Formation. J Biol Chem 290, 14595–609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zemski Berry KA, Gordon WC, Murphy RC & Bazan NG Spatial organization of lipids in the human retina and optic nerve by MALDI imaging mass spectrometry. J Lipid Res 55, 504–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Locasale JW et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43, 869–74 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinha T, Ikelle L, Naash MI & Al-Ubaidi MR The Intersection of Serine Metabolism and Cellular Dysfunction in Retinal Degeneration. Cells 9(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang T et al. Human macular Muller cells rely more on serine biosynthesis to combat oxidative stress than those from the periphery. Elife 8(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Price AL, Spencer CC & Donnelly P Progress and promise in understanding the genetic basis of common diseases. Proc Biol Sci 282, 20151684 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visscher PM et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet 101, 5–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mattos EP et al. Identification of a premature stop codon mutation in the PHGDH gene in severe Neu-Laxova syndrome-evidence for phenotypic variability. Am J Med Genet A 167, 1323–9 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Poli A et al. Phosphoglycerate dehydrogenase (PHGDH) deficiency without epilepsy mimicking primary microcephaly. Am J Med Genet A 173, 1936–1942 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Brassier A et al. Two new cases of serine deficiency disorders treated with l-serine. Eur J Paediatr Neurol 20, 53–60 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Cavole TR et al. Clinical, molecular, and pathological findings in a Neu-Laxova syndrome stillborn: A Brazilian case report. Am J Med Genet A 182, 1473–1476 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Ni C et al. Novel and recurrent PHGDH and PSAT1 mutations in Chinese patients with Neu-Laxova syndrome. Eur J Dermatol 29, 641–646 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Povysil G et al. Rare-variant collapsing analyses for complex traits: guidelines and applications. Nat Rev Genet 20, 747–759 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Price AL et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904–9 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Wu C, DeWan A, Hoh J & Wang Z A comparison of association methods correcting for population stratification in case-control studies. Ann Hum Genet 75, 418–27 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stanley KE et al. Causal Genetic Variants in Stillbirth. N Engl J Med 383, 1107–1116 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cameron-Christie S et al. Exome-Based Rare-Variant Analyses in CKD. J Am Soc Nephrol 30, 1109–1122 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gelfman S et al. A new approach for rare variation collapsing on functional protein domains implicates specific genic regions in ALS. Genome Res 29, 809–818 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Idelson M et al. Directed differentiation of human embryonic stem cells into functional retinal pigment epithelium cells. Cell Stem Cell 5, 396–408 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Wallace M et al. Enzyme promiscuity drives branched-chain fatty acid synthesis in adipose tissues. Nat Chem Biol 14, 1021–1031 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernandez CA, Des Rosiers C, Previs SF, David F & Brunengraber H Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J Mass Spectrom 31, 255–62 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.