

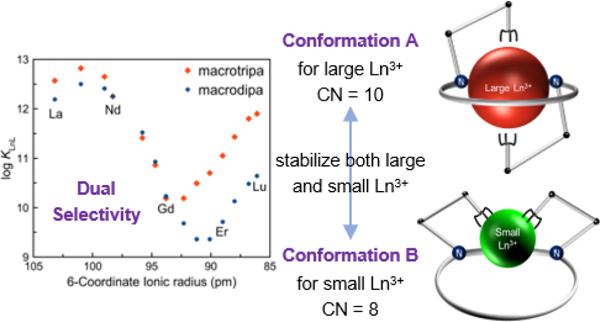
Abstract
Lanthanides (Ln3+) are critical materials used for many important applications, often in the form of coordination compounds. Tuning the thermodynamic stability of these compounds is a general concern, which is not readily achieved due to the similar coordination chemistry of lanthanides. Herein, we report two 18-membered macrocyclic ligands called macrodipa and macrotripa that show for the first time a dual selectivity towards both the light, large Ln3+ ions and the heavy, small Ln3+ ions, as determined by potentiometric titrations. The lanthanide complexes of these ligands were investigated by NMR spectroscopy and X-ray crystallography, revealing the occurrence of a significant conformational toggle between a 10-coordinate Conformation A and an 8-coordinate Conformation B that accommodates Ln3+ ions of different sizes. The origin of this selectivity pattern was further supported by density functional theory (DFT) calculations, which show the complementary effects of ligand strain energy and metal-ligand binding energy that contribute to this conformational switch. This work demonstrates how novel ligand design strategies can be applied to tune the selectivity pattern for the Ln3+ ions.
Graphical Abstract
Introduction
The unique physical properties of the lanthanides are critical for use in magnets, superconductors, catalysts, luminescent phosphors, and medicinal agents.1–6 Despite their diverse magnetic and electronic properties, the lanthanides possess similar chemical properties dictated by their strong preference for the +3 oxidation state and tendency to engage in ionic bonding.7,8 The major distinguishing feature among them is their ionic radius, which decreases across the series, a phenomenon known as the lanthanide contraction.9
For many applications of the lanthanides (Ln3+), a ligand is required to chelate these ions to control and modify their chemical properties. The availability of a range of chelators with different affinities and selectivity patterns for the Ln3+ ions, measured by the stability constant KLnL, is valuable for the implementation of these ions.10 Generally, most ligands possess a higher affinity for the heavier, smaller Ln3+ ions because the increased charge density on these smaller ions enhances metal-ligand electrostatic interactions.11 Recent ligand design efforts, however, have led to systems with other Ln3+-selectivity patterns. To date, three types of selectivity patterns have been identified (Figure 1). As noted above, the most common trend shows a systematic increase in KLnL across the lanthanide series (Type I). This Type I behavior is observed for many well-known ligands including EDTA,12 (Figure 1, Chart 1), 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA),13 and diethylenetriaminepentaacetic acid (DTPA).14,15
Figure 1.
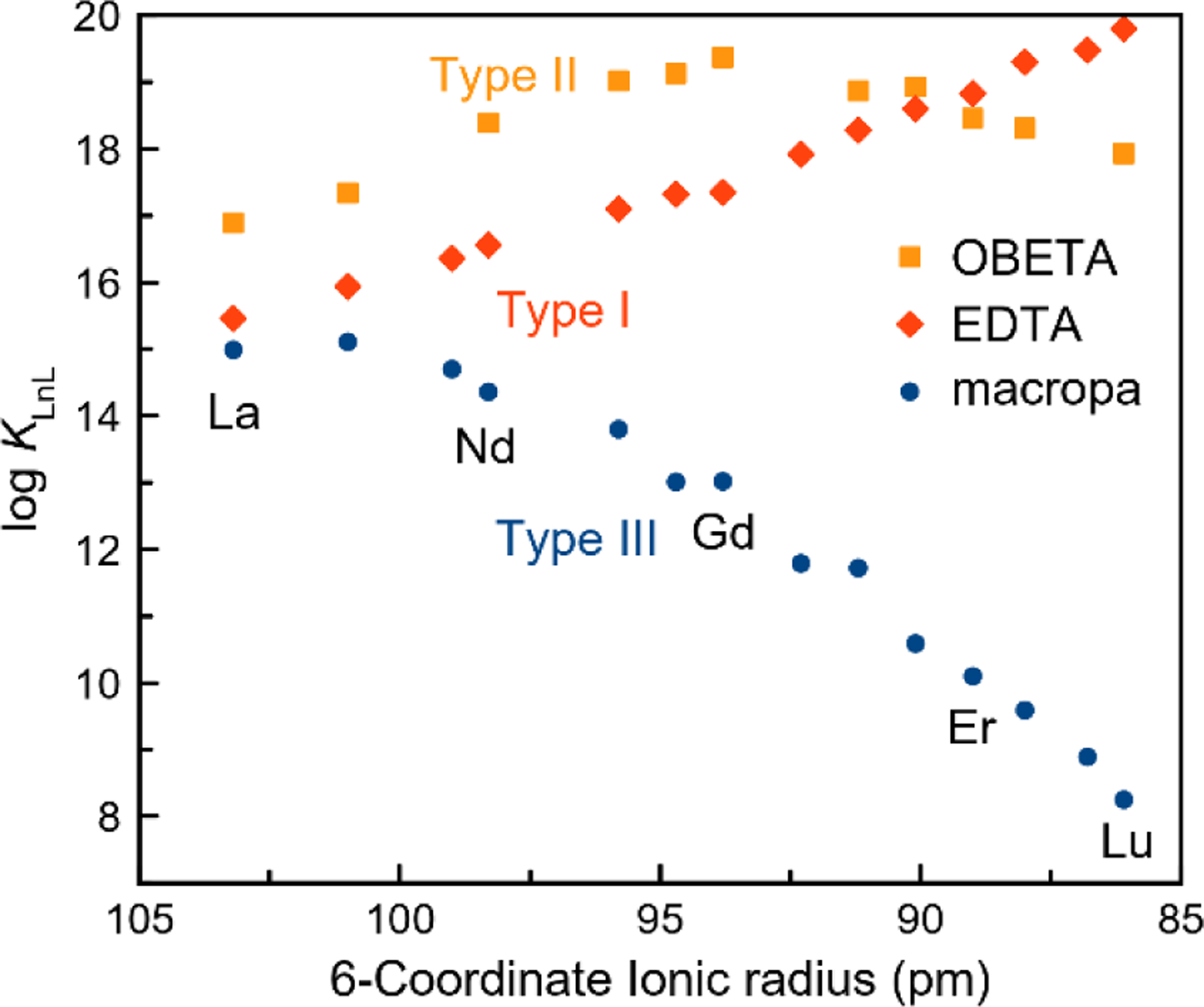
Stability constants of Ln3+ complexes formed with EDTA, OBETA, and macropa plotted versus ionic radii.
Chart 1.
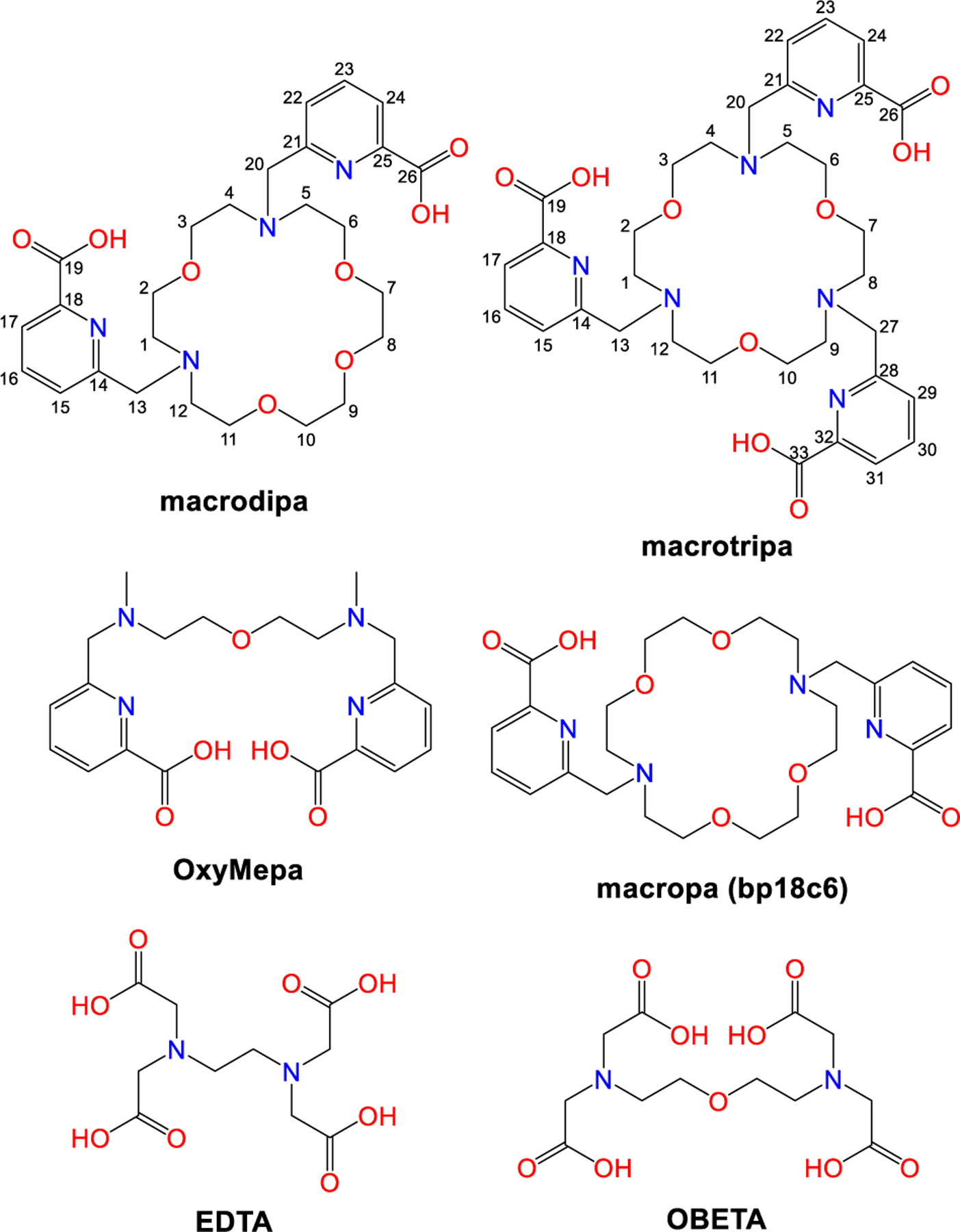
Structures of Ligands Discussed in this Work.
Another less frequently observed trend, Type II, has the stability reach a maximum prior to dropping again. The ligands OBETA16,17 (Figure 1, Chart 1) and 1,4,7,10-tetrakis(carbamoylmethyl)-1,4,7,10-tetraazacyclododecane (TCMC)18 follow this pattern. The rarely observed behavior, Type III, occurs with ligands having reverse-size selectivity, an unusual thermodynamic preference for large over small Ln3+ ions. This pattern has been observed in ligands containing the large diaza-18-crown-6 macrocycles, such as macropa19 (Figure 1, Chart 1) and 1,10-diaza-4,7,13,16-tetraoxacyclooctadecane-N,N′-diacetic acid (dacda).20 A common feature of these three selectivity patterns is that they all have the maximum affinity for only one Ln3+ ion.
Inspired by macropa and our ongoing research efforts on chelating agents for large metal ions of biomedical and industrial relevance,21–24 we sought to explore other ligands containing 18-membered macrocycles. Specifically, we investigated an isomer of macropa, called macrodipa, and a triaza-18-crown-6 ligand25,26 bearing three pendent picolinate donors, macrotripa (Chart 1). Notably, these macrocycles are cyclic analogues of the previously reported ligand OxyMepa27 (Chart 1) that provides a similar albeit truncated set of donor atoms. Over the course of our studies on their Ln3+ coordination chemistry, we found that macrodipa and macrotripa undergo dramatic conformational changes upon binding large versus small ions. These conformational changes manifest in a previously unreported Type IV selectivity pattern with one minimum and two maxima of stability across the Ln3+ series. This work demonstrates how novel ligand design strategies can be employed to differentiate these ions in new ways.
Results and Discussion
The syntheses of macrodipa and macrotripa (Schemes S1–S2) involve the assembly of the tosyl-protected 18-crown-6 macrocycles, deprotection of the tosyl groups, and subsequent alkylation of the picolinate donor arms. They were characterized by NMR spectroscopy, mass spectrometry, and HPLC (Figures S1–10).
Potentiometric titrations were performed to determine their protonation constants (Ki, Table S1). To probe the thermodynamic affinities of these ligands for Ln3+ ions, we conducted potentiometric titrations to obtain their stability constants (KLnL and KLnHL, Table 1). These protonation and stability constants are defined in Eqs 1–3, with the concentrations of all species at chemical equilibrium.
Table 1.
Stability Constants of the Lanthanide Complexes Formed with macrodipa, macrotripa, OxyMepa, macropa, EDTA, and OBETA.
| Ln3+ | macrodipaa | macrotripaa | OxyMepab | macropac | EDTAd | OBETAe | ||
|---|---|---|---|---|---|---|---|---|
| log KLnL | Log KLnL | log KLnHL | log KLnL | Log KLnL | log KLnHL | log KLnL | log KLnL | |
| La3+ | 12.19(2) | 12.57(1) | 3.67(2) | 9.93 | 14.99 | 2.28 | 15.46 | 16.89 |
| Ce3+ | 12.50(4) | 12.82(2) | 3.66(4) | 10.74 | 15.11 | 2.07 | 15.94 | 17.34 |
| Pr3+ | 12.41(2) | 12.65(1) | 3.65(2) | 11.25 | 14.70 | 2.96 | 16.36 | |
| Nd3+ | 12.25(3) | 12.25(2) | 3.88(1) | 11.49 | 14.36 | 2.08 | 16.56 | 18.39 |
| Sm3+ | 11.52(2) | 11.41(1) | 3.99(1) | 12.13 | 13.80 | 2.70 | 17.10 | 19.02 |
| Eu3+ | 10.93(1) | 10.86(1) | 4.24(2) | 12.15 | 13.01 | 1.97 | 17.32 | 19.13 |
| Gd3+ | 10.23(2) | 10.19(2) | 4.51(1) | 12.02 | 13.02 | 2.48 | 17.35 | 19.37 |
| Tb3+ | 9.68 (1) | 10.19(2) | 4.82(1) | 12.17 | 11.79 | 2.91 | 17.92 | |
| Dy3+ | 9.36(2) | 10.49(3) | 4.86(2) | 12.18 | 11.72 | 2.42 | 18.28 | 18.87 |
| Ho3+ | 9.36(1) | 10.70(3) | 4.90(1) | 12.10 | 10.59 | 18.60 | 18.93 | |
| Er3+ | 9.71(4) | 11.05(3) | 4.92(4) | 12.00 | 10.10 | 18.83 | 18.46 | |
| Tm3+ | 10.13(1) | 11.43(5) | 4.89(2) | 12.05 | 9.59 | 19.30 | ||
| Yb3+ | 10.48(1) | 11.80(3) | 4.88(1) | 12.14 | 8.89 | 19.48 | 18.31 | |
| Lu3+ | 10.64(4) | 11.90(1) | 4.93(2) | 12.21 | 8.25 | 19.80 | 17.93 | |
| (1) |
| (2) |
| (3) |
Figure 2 shows a plot of log KLnL versus the Ln3+ ionic radius28 for both macrodipa and macrotripa, revealing them to be the first known ligands that exhibit Type IV selectivity. In comparing the log KLnL values between Ln3+-macrodipa and Ln3+-macrotripa systems, they are similar for early lanthanides, La3+–Gd3+. Ln3+-macrodipa reaches a minimum for Dy3+ and Ho3+, whereas for macrotripa the minimum occurs earlier in the series, between Gd3+ and Tb3+. For late lanthanides, the macrotripa complexes are significantly more stable than those of macrodipa.
Figure 2.
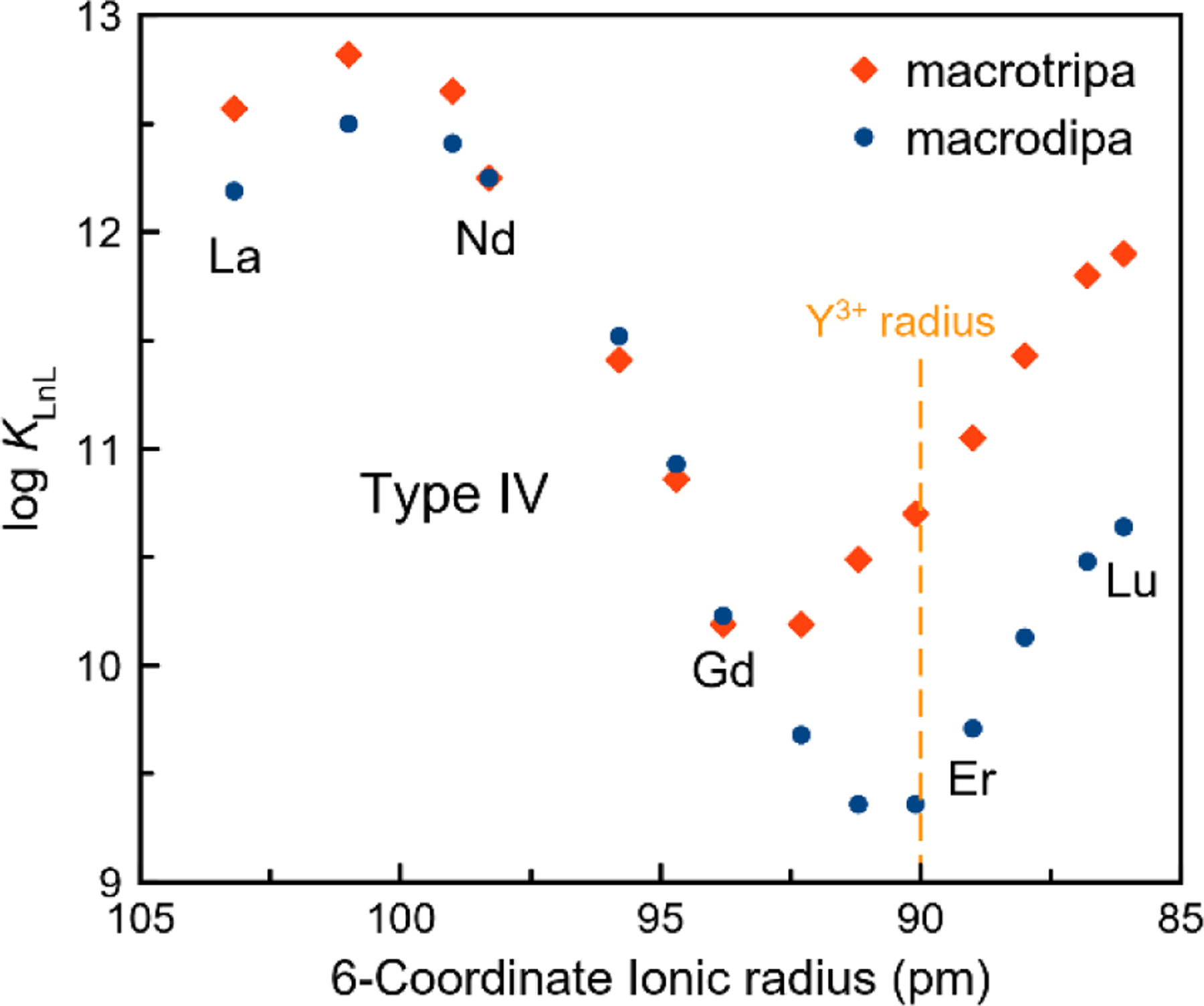
Stability constants of Ln3+ complexes formed with macrodipa and macrotripa plotted versus ionic radii.
The log KLnHL values, which represent the protonation of LnL complex, are also noteworthy. These values were not found for Ln3+-macrodipa complexes but were observed for Ln3+-macrotripa complexes. Comparing the chemical structures of macrodipa and macrotripa and their complexes (vide infra), it can be reasonably inferred that this protonation event occurs on the third picolinate arm in macrotripa. The KLnHL values of the macrotripa complexes remain steady from La3+ to Pr3+, but then increase abruptly from Nd3+ to Tb3+, before leveling off from Dy3+ to Lu3+. The sudden change in log KLnHL implies that a conformational change may be present for Ln3+-macrotripa complexs when crossing the Ln3+ series.
To gain insight on the Type IV selectivity of macrodipa and macrotripa, we analyzed their complexes of the largest and smallest lanthanides, La3+ and Lu3+, by NMR spectroscopy. These diamagnetic La3+ and Lu3+ complexes were characterized by 1H, 13C{1H}, and 2D (HSQC, HMBC, COSY, ROESY) NMR spectroscopy in D2O at pD = 7 (Figures 3, S41–S80). The 1H NMR spectra of all four complexes indicate that a single species is present in solution. However, significant differences are apparent in comparing the La3+ and Lu3+ complexes. For example, the La3+-macrodipa complex is 2-fold symmetric, indicated by half the number of 1H and 13C resonances relative to the asymmetric Lu3+-macrodipa complex.
Figure 3.
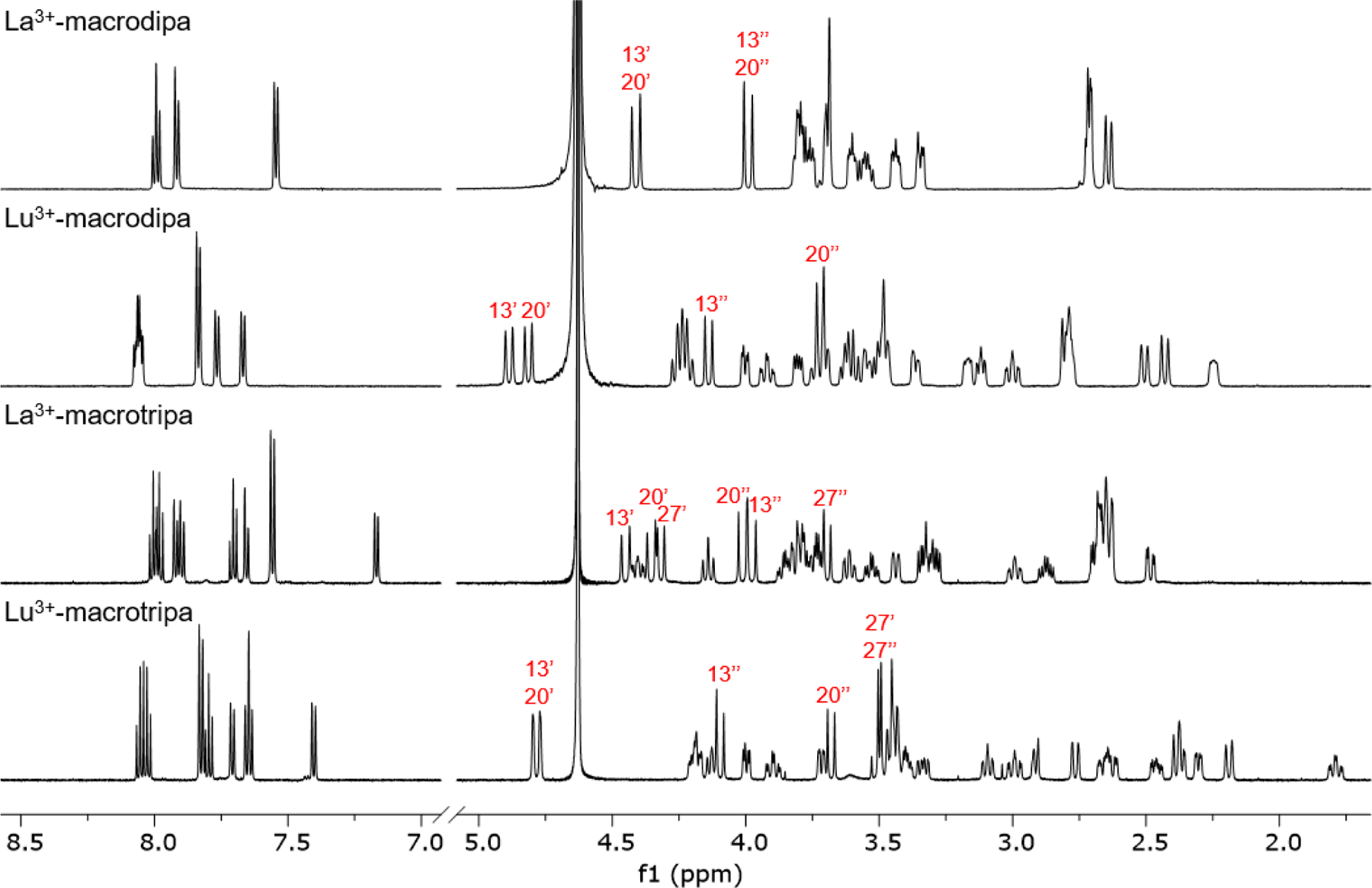
1H NMR spectra of macrodipa and macrotripa complexes formed with La3+ and Lu3+ (600 MHz, D2O, pD = 7, 25 °C). Selected peaks are labeled using the numbering scheme in Chart 1.
Additionally, the hydrogen resonances from the methylene groups linking the crown and picolinate donors (H-13, H-20 for macrodipa, and H-13, H-20, H-27 for macrotripa; Chart 1), which are informative due to their proximities to the picolinate donors, are significantly different between the La3+ and Lu3+ complexes. For example, the peaks for the diastereotopic H-27′ and H-27′′ in La3+-macrotripa case are well-separated, whereas for Lu3+-macrotripa they have near-identical chemical shifts. Collectively, these NMR data suggest that there is a significant conformational difference between La3+ and Lu3+ complexes of these ligands.
To further explore these different conformations, we characterized these complexes by X-ray crystallography. Crystal structures of [La(macrodipa)]+, [Lu(macrodipa)(OH2)]+, and [La(macrotripa)]+ are shown in Figure 4. A weakly diffracting and partially twinned crystal of [Lu(macrotripa)(OH2)] was also obtained, but the connectivity information was reliably ascertained (Figure S89). Confirming the NMR spectroscopic data, the La3+ complexes attain a significantly different conformation than the Lu3+ complexes. In both La3+ structures, this ion is encapsulated into the 18-membered macrocyclic core, interacting with all its six donor atoms. Moreover, the pendent picolinate groups bind to La3+ from two opposite faces of the macrocycle, resulting in 10-coordinate complexes. The third picolinate donor of macrotripa does not participate in coordination. Consistent with its NMR spectra, [La(macrodipa)]+ attains a slightly distorted C2 symmetry. By contrast, both Lu3+ structures show significantly different coordination environments. Specifically, only two tertiary nitrogens and one ethereal oxygen from the macrocycle act as donors. The coordination sphere is completed with the four donor atoms from two picolinate groups and an inner-sphere water molecule, yielding 8-coordinate Lu3+ centers. In [Lu(macrotripa)(OH2)], the third unbound picolinate donor is positioned to interact with the bound water molecule through hydrogen bonding. Except for the macrocycle, these Lu3+ structures are highly comparable to those found for the acyclic ligand OxyMepa,27 which displays Type I selectivity, indicating that the complete 18-crown-6 macrocycles of macrodipa and macrotripa are critical for their unique Ln3+-selectivity profiles.
Figure 4.
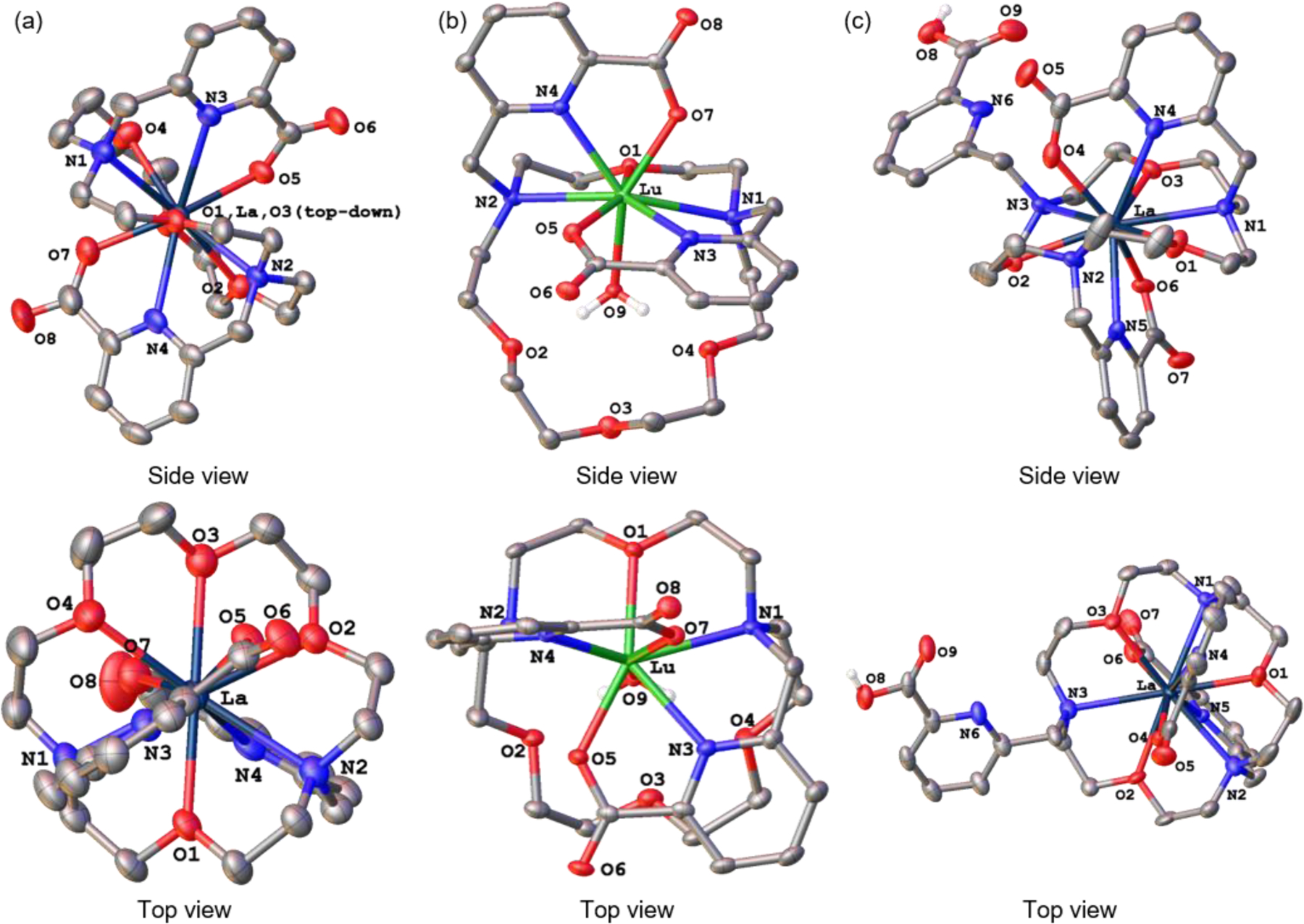
Crystal structures of (a) [La(macrodipa)]+, (b) [Lu(macrodipa)(OH2)]+, and (c) [La(macrotripa)]+ complexes. Thermal ellipsoids are drawn at the 50% probability level. Solvent, counterions, and non-acidic hydrogen atoms are omitted for clarity. Only one of the two [La(macrodipa)]+ units in the asymmetric cell is shown.
As a further validation on the proposed intramolecular hydrogen bond in [Lu(macrotripa)(OH2)], we optimized its structure (Figure S98) using validated DFT methods (vide infra), and carried out a topological analysis of the electron density using the quantum theory of atoms in molecules (QTAIM).29 Specifically, we found all bond critical points (BCP) with the Multiwfn30 program (Figure S99). A BCP was located between a hydrogen atom of the coordinated water molecule and the oxygen atom of the pendent picolinate group. At this BCP, the magnitude of its local electron density (ρ) is 0.079 a.u. and the Laplacian of the electron density (∇2ρ) is 0.16 a.u. The positive value for ∇2ρ reflects a closed-shell hydrogen bond, and the magnitude of ρ suggests that this interaction is strong, comparable to that found between OH– and H2O.31–33 This analysis supports the proposed intramolecular hydrogen-bonding interaction present in [Lu(macrotripa)(OH2)].
Based on the NMR and X-ray crystallographic data, it is clear that macrodipa and macrotripa attain distinct conformations depending on whether they bind large or small Ln3+ ions (Scheme 1). Large ions, like La3+, attain Conformation A, in which the ion is fully encapsulated by the macrocycle, whereas small ions, like Lu3+, sit in Conformation B, held by only part of the macrocycle. The ability of these ligands to drastically alter their conformations to match the sizes of metal ions accounts for the Type IV selectivity pattern. The structures may also explain the difference in thermodynamic stability of macrodipa and macrotripa for the late, but not early Ln3+ (Figure 2). Both ligands give rise to identical coordination spheres for the large early lanthanides, like La3+, and therefore exhibit only minor differences in their thermodynamic stabilities. However, for the small late lanthanides, like Lu3+, the inner coordination spheres are nearly identical between macrodipa and macrotripa, but the outer sphere differs due to the hydrogen-bonding interaction with the coordinated water molecule. Thus, the differences in thermodynamic stability between the macrodipa and macrotripa complexes of the late lanthanides is most likely a consequence of the hydrogen bonding of the pendent picolinate donor arm. This result highlights how modifying the outer coordination sphere of lanthanide complexes fine-tunes their thermodynamic properties.
Scheme 1.
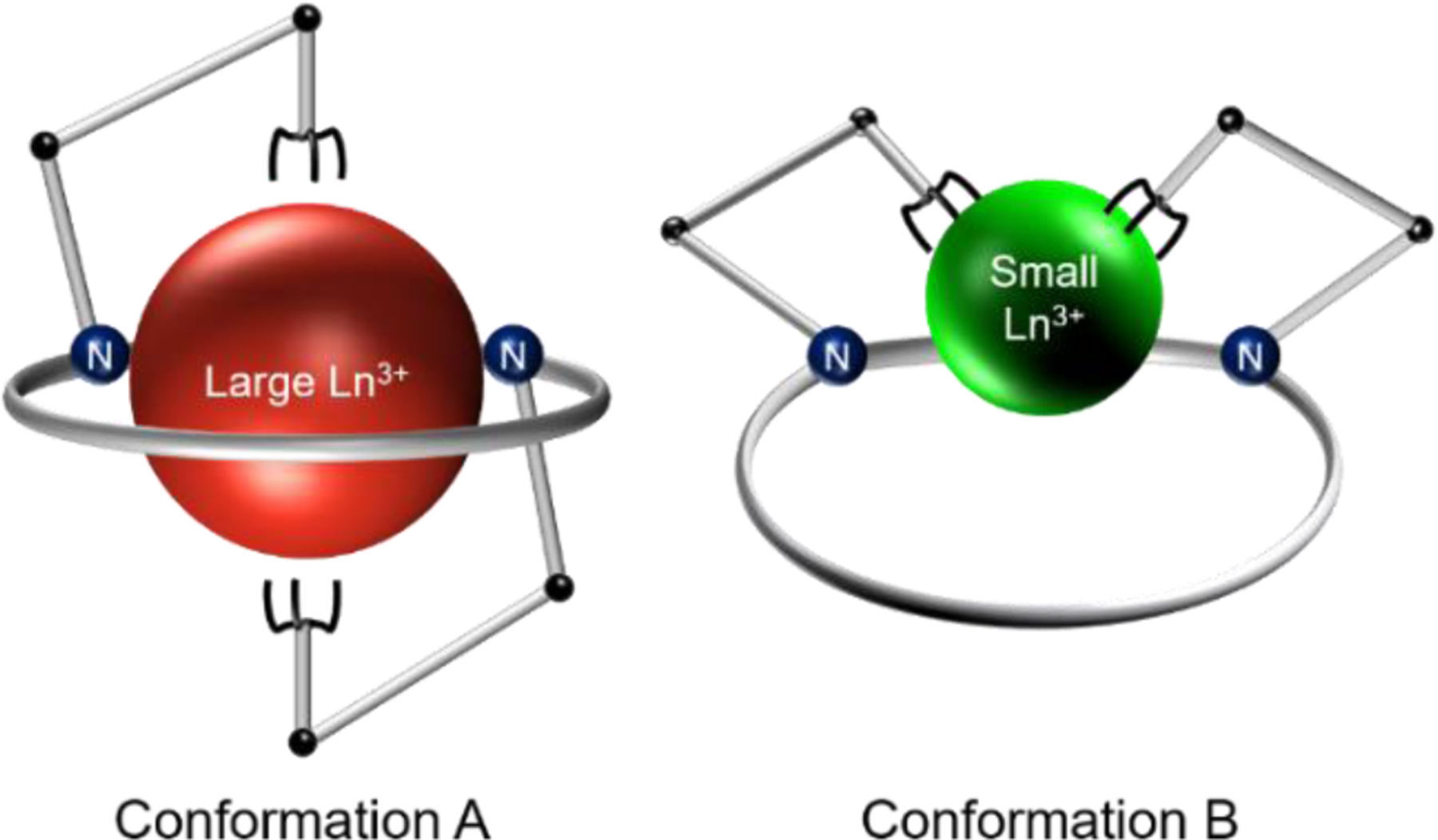
Depiction of the Conformational Toggle Present in Ln3+-macrodipa and Ln3+-macrotripa complex systems.
As a further test of this conformational toggle, we investigated the complexes of Y3+, a diamagnetic Ln3+ analogue with a comparable ionic radius to that of Ho3+,7,28 by NMR spectroscopy. The 1H and 13C{1H} NMR spectra of Y3+-macrodipa and Y3+-macrotripa were acquired in D2O at pD = 7 (Figure S81–S88). Both Conformations A and B are detected for Y3+-macrodipa, in a molar ratio of 1:15. For the Y3+-macrotripa complex, only Conformation B is observed. The 90-pm ionic radius of Y3+ places its macrodipa complex near the local minimum of log KLnL, but its macrotripa complex rather far from the minimum (Figure 2). Thus, these NMR data show that the conformational switch occurs for complexes of ions with their radii near the minimum of stability; larger and smaller ions show preferences for Conformations A and B, respectively.
DFT has been extensively used to investigate the properties of Ln3+ coordination compounds.34–36 In this study, we took advantage of this powerful tool to help understand the origin of the Type IV selectivity pattern of these ligands. We focused exclusively on the Ln3+-macrodipa system. These complexes lack the third non-coordinated picolinate arm of the Ln3+-macroptripa, and therefore provide a straightforward system to model the inner coordination spheres of these complexes. DFT calculations were executed using Gaussian 0937 with the ωB97XD functional.38,39 This functional, which is long-range corrected and includes dispersion corrections, has been shown to give accurate geometries of Ln3+ complexes.40 Because of the importance of relativistic effects in Ln3+ ions,41 we used the large-core relativistic effective core potential (LCRECP) by Dolg42 to account for these effects in a computationally efficient manner. For light atoms, the 6–31G(d,p) basis set43,44 was applied. The SMD solvation model45,46 was implemented to take the solvent effects into consideration. The ΔG° for the conformational equilibrium
| (4) |
was calculated for Ln3+-macrodipa complexes. The ΔG° (Figure 5) is positive for light Ln3+ and negative for heavy Ln3+. This observation is consistent with the experimental results with La3+-macrodipa and Lu3+-macrodipa complexes attaining Conformations A and B, respectively. Additionally, ΔG° changes its sign between Gd3+ and Tb3+, indicating the switch of favored conformation. This crossover suggests that the Type IV behavior of macrodipa is a consequence of the significant conformational changes that occur when binding Ln3+ ions of different sizes.
Figure 5.
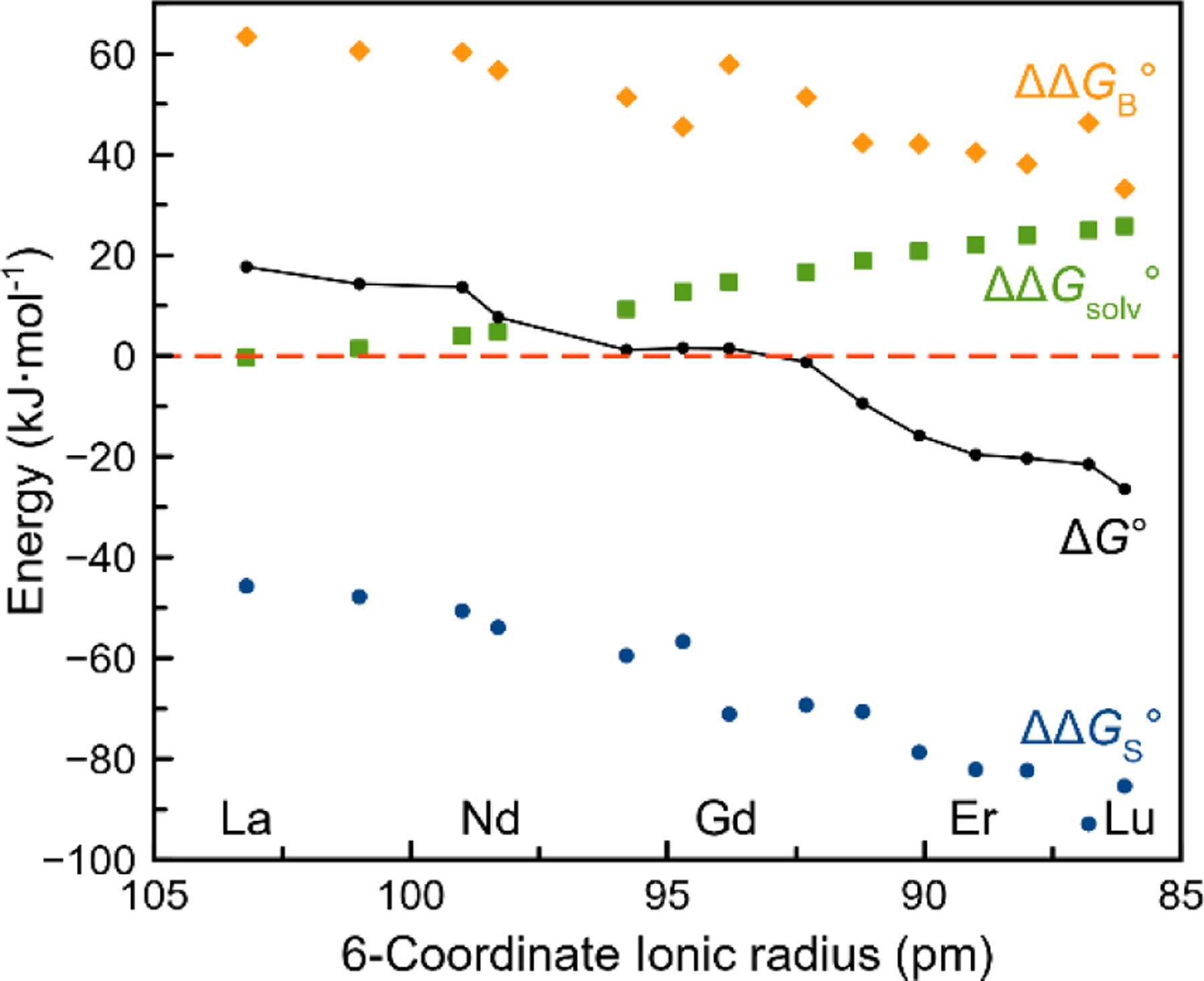
DFT-computed free energies for Ln3+-macrodipa complexes.
Furthermore, ΔG° for this conformation change can be broken up into three contributors. Specifically, it can be expressed as the sum of the relative ligand strain energies (ΔΔGS°), relative metal-ligand binding energies (ΔΔGB°), and relative solvation energies (ΔΔGsolv°) between Conformations A and B, as described in the Supporting Information. As shown in Figure 5, ΔΔGsolv° is positive for all Ln3+ complexes, revealing that Conformer A and the non-coordinated water ligand are better solvated in aqueous solution than Conformer B. Likewise, ΔΔGB° is positive for all Ln3+, indicating that Conformation A is better suited to neutralize the electrostatic charges of these ions than Conformation B. This observation can be rationalized by the fact that Conformation A interacts with the Ln3+ with two more donor atoms. However, ΔΔGB° decreases as the Ln3+ gets smaller, suggesting that Conformation A is less effective at binding the smaller ions. By contrast, ΔΔGS° is negative across the entire series, showing that Conformation B requires less ligand strain than Conformation A. Among the three values, ΔΔGS° shows the most significant changes as a function of the Ln3+ ionic radius, becoming more negative for smaller ions. Importantly, the strain energy is the only exothermic term for the switch from Conformation A to B, and thus it is the driving factor in the conformational switch of macrodipa. This result suggests that modifications of this ligand scaffold to alter the strain energy term could lead to a significant shift in the Ln3+-stability pattern for this ligand class.
Conclusion
In summary, this work describes the first ligands that display an unprecedented Type IV selectivity pattern with one minimum and two maxima of KLnL across the Ln3+ series. This novel selectivity pattern may have key applications in Ln3+ separations and nuclear medicine, where it is often desirable to stably chelate a range of metal ions with disparate ionic radii. Our structural data by NMR spectroscopy and X-ray crystallography show that both macrodipa and macrotripa undergo a significant conformational shift in moving from large to small Ln3+ ion, which allows for high thermodynamic stability for both early and late Ln3+. In comparing macrodipa and macrotripa, we have shown that one can modify these ligands to tune the position of the minimum and the overall magnitude of thermodynamic stability. Our DFT calculations further support the presence of this conformational toggle, and show that the ligand strain and metal-ligand binding energies are complementary factors contributing to the conformational switch. Between them, we believe that the ligand strain energy is more easily modified, such as including groups in the macrocyclic backbone that prevent conformational flexibility, thus presenting a path for tuning these ligands for different applications. This work demonstrates how the incorporation of conformational flexibility in ligands can be used to satisfy the coordination environments of different metal ions, a principle of great value for chelator design efforts.
Supplementary Material
ACKNOWLEDGMENT
This research was supported in part by seed funding from the Academic Integration grant program at Cornell University and from the College of Arts and Sciences at Cornell University and by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under Award Number R21EB027282. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This research made use of the NMR Facility at Cornell University, which is supported, in part, by the U.S. National Science Foundation under Award Number CHE-1531632.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and supplementary data for chemical synthesis, potentiometric titrations, NMR spectroscopy studies, X-ray crystallography, and DFT calculations (PDF)
Crystallographic data for La3+-macrodipa, Lu3+-macrodipa, and La3+-macrotripa complexes (CIF)
Geometry outputs for all DFT-optimized structures (ZIP)
The authors declare no competing financial interests.
REFERENCES
- (1).Cheisson T; Schelter EJ Rare Earth Elements: Mendeleev’s Bane, Modern Marvels. Science 2019, 363, 489–493. [DOI] [PubMed] [Google Scholar]
- (2).Woodruff DN; Winpenny REP; Layfield RA Lanthanide Single-Molecule Magnets. Chem. Rev 2013, 113, 5110–5148. [DOI] [PubMed] [Google Scholar]
- (3).Dos santos-García AJ; Alario-Franco MÁ; Sáez-Puche R The Rare Earth Elements: Superconducting Materials. In Encyclopedia of Inorganic and Bioinorganic Chemistry. John Wiley & Sons, Ltd.: Chichester, U.K., 2012. [Google Scholar]
- (4).Pellissier H Recent Developments in Enantioselective Lanthanide-Catalyzed Transformations. Coord. Chem. Rev 2017, 336, 96–151. [Google Scholar]
- (5).Qin X; Liu X; Huang W; Bettinelli M; Liu X Lanthanide-Activated Phosphors Based on 4f-5d Optical Transitions: Theoretical and Experimental Aspects. Chem. Rev 2017, 117, 4488–4527. [DOI] [PubMed] [Google Scholar]
- (6).Teo RD; Termini J; Gray HB Lanthanides: Applications in Cancer Diagnosis and Therapy. J. Med. Chem 2016, 59, 6012–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cotton SA Scandium, Yttrium & the Lanthanides: Inorganic & Coordination Chemistry. In Encyclopedia of Inorganic Chemistry; John Wiley & Sons, Ltd.: Chichester, U.K., 2006. [Google Scholar]
- (8).Huang C; Bian Z Introduction. In Rare Earth Coordination Chemistry: Fundamentals and Applications; Huang C, Eds.; John Wiley & Sons (Asia) Pte Ltd: Singapore, 2010; pp 1–39. [Google Scholar]
- (9).Seitz M; Oliver AG; Raymond KN The Lanthanide Contraction Revisited. J. Am. Chem. Soc 2007, 129, 11153–11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Peters JA; Djanashvili K; Geraldes CFGC; Platas-Iglesias C The Chemical Consequences of the Gradual Decrease of the Ionic Radius along the Ln-Series. Coord. Chem. Rev 2020, 406, 213146. [Google Scholar]
- (11).Piguet C; Bünzli J-CG Mono- and Polymetallic Lanthanide-Containing Functional Assemblies: A Field between Tradition and Novelty. Chem. Soc. Rev 1999, 28, 347–358. [Google Scholar]
- (12).Martell AE; Smith RM Critical Stability Constants; Plenum Press: New York, 1974; Vol. 1. [Google Scholar]
- (13).Cacheris WP; Nickle SK; Sherry AD Thermodynamic study of Lanthanide Complexes of 1,4,7-Triazacyclononane-N,N′,N″-triacetic Acid and 1,4,7,10-Tetraazacyclododecane-N,N′,N″,N‴-tetraacetic Acid. Inorg. Chem 1987, 26, 958–960. [Google Scholar]
- (14).Moeller T; Thompson LC Observations on the Rare Earths—LXXV: The Stabilities of Diethylenetriaminepentaacetic Acid Chelates. J. Inorg. Nucl. Chem 1962, 24, 499–510. [Google Scholar]
- (15).Grimes TS; Nash KL Acid Dissociation Constants and Rare Earth Stability Constants for DTPA. J. Solution Chem 2014, 43, 298–313. [Google Scholar]
- (16).Baranyai Z; Botta M; Fekete M; Giovenzana GB; Negri R; Tei L; Platas-Iglesias C Lower Ligand Denticity Leading to Improved Thermodynamic and Kinetic Stability of the Gd3+ Complex: The Strange Case of OBETA. Chem. - Eur. J 2012, 18, 7680–7685. [DOI] [PubMed] [Google Scholar]
- (17).Negri R; Baranyai Z; Tei L; Giovenzana GB; Platas-Iglesias C; Bényei AC; Bodnár J; Vágner A; Botta M Lower Denticity Leading to Higher Stability: Structural and Solution Studies of Ln(III)–OBETA Complexes. Inorg. Chem 2014, 53, 12499–12511. [DOI] [PubMed] [Google Scholar]
- (18).Voss DA Jr.; Farquhar ER; Horrocks WD Jr.; Morrow JR Lanthanide(III) Complexes of Amide Derivatives of DOTA Exhibit an Unusual Variation in Stability across the Lanthanide Series. Inorg. Chim. Acta 2004, 357, 859–863. [Google Scholar]
- (19).Roca-Sabio A; Mato-Iglesias M; Esteban-Gómez D; Tóth É; de Blas A; Platas-Iglesias C; Rodríguez-Blas T Macrocyclic Receptor Exhibiting Unprecedented Selectivity for Light Lanthanides. J. Am. Chem. Soc 2009, 131, 3331–3341. [DOI] [PubMed] [Google Scholar]
- (20).Chang CA; Rowland ME Metal Complex Formation with 1,10-Diaza-4,7,13,16-tetraoxacyclooctadecane-N,N′-diacetic Acid. An Approach to Potential Lanthanide Ion Selective Reagents. Inorg. Chem 1983, 22, 3866–3869. [Google Scholar]
- (21).Thiele NA; Brown V; Kelly JM; Amor-Coarasa A; Jermilova U; MacMillan SN; Nikolopoulou A; Ponnala S; Ramogida CF; Robertson AKH; Rodríguez-Rodríguez C; Schaffer P; Williams C Jr.; Babich JW; Radchenko V; Wilson JJ An Eighteen-Membered Macrocyclic Ligand for Actinium-225 Targeted Alpha Therapy. Angew. Chem. Int. Ed 2017, 56, 14712–14717. [DOI] [PubMed] [Google Scholar]
- (22).Thiele NA; MacMillan SN; Wilson JJ Rapid Dissolution of BaSO4 by Macropa, an 18-Membered Macrocycle with High Affinity for Ba2+. J. Am. Chem. Soc 2018, 140, 17071–17078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Thiele NA; Woods JJ; Wilson JJ Implementing f-Block Metal Ions in Medicine: Tuning the Size Selectivity of Expanded Macrocycles. Inorg. Chem 2019, 58, 10483–10500. [DOI] [PubMed] [Google Scholar]
- (24).Aluicio-Sarduy E; Thiele NA; Martin KE; Vaughn BA; Devaraj J; Olson AP; Barnhart TE; Wilson JJ; Boros E; Engle JW Establishing Radiolanthanum Chemistry for Targeted Nuclear Medicine Applications. Chem. - Eur. J 2020, 26, 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen D; Squattrito PJ; Martell AE; Clearfield A Synthesis and Crystal Structure of a Nine-Coordinate Gadolinium(III) Complex of 1,7,13-Triaza-4,10,16-trioxacyclooctadecane-N,N′,N′′-triacetic Acid. Inorg. Chem 1990, 29, 4366–4368. [Google Scholar]
- (26).Delgado R; Sun Y; Motekaitis RJ; Martell AE Stabilities of Divalent and Trivalent Metal Ion Complexes of Macrocyclic Triazatriacetic Acids. Inorg. Chem 1993, 32, 3320–3326. [Google Scholar]
- (27).Hu A; Keresztes I; MacMillan SN; Yang Y; Ding E; Zipfel WR; DiStasio RA Jr.; Babich JW; Wilson JJ Oxyaapa: A Picolinate-Based Ligand with Five Oxygen Donors that Strongly Chelates Lanthanides. Inorg. Chem 2020, 59, 5116–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shannon RD Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr., Sect. A: Found. Adv 1976, 32, 751–767. [Google Scholar]
- (29).Bader RFW Atoms in Molecules - A Quantum Theory; Oxford University Press: Oxford, 1990. [Google Scholar]
- (30).Lu T; Chen F Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem 2012, 33, 580–592. [DOI] [PubMed] [Google Scholar]
- (31).Bader RFW A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev 1991, 91, 893–928. [Google Scholar]
- (32).Grabowski SJ; Robinson TL; Leszczynski J Strong Dihydrogen Bonds – ab initio and Atoms in Molecules Study. Chem. Phys. Lett 2004, 386, 44–48. [Google Scholar]
- (33).Parthasarathi R; Subramanian V; Sathyamurthy N Hydrogen Bonding without Borders: An Atoms-in-Molecules Perspective. J. Phys. Chem. A 2006, 110, 3349–3351. [DOI] [PubMed] [Google Scholar]
- (34).Tsipis AC DFT Flavor of Coordination Chemistry. Coord. Chem. Rev 2014, 272, 1–29. [Google Scholar]
- (35).Platas-Iglesias C; Roca-Sabio A; Regueiro-Figueroa M; Esteban-Gómez D; de Blas A; Rodriguez-Blas T Applications of Density Functional Theory (DFT) to Investigate the Structural, Spectroscopic and Magnetic Properties of Lanthanide(III) Complexes. Curr. Inorg. Chem 2011, 1, 91–116. [Google Scholar]
- (36).Platas-Iglesias C The Solution Structure and Dynamics of MRI Probes Based on Lanthanide(III) DOTA as Investigated by DFT and NMR Spectroscopy. Eur. J. Inorg. Chem 2012, 2023–2033.
- (37).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr., Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, 2009. [Google Scholar]
- (38).Chai J-D; Head-Gordon M Systematic Optimization of Long-Range Corrected Hybrid Density Functionals. J. Chem. Phys 2008, 128, 084106. [DOI] [PubMed] [Google Scholar]
- (39).Chai J-D; Head-Gordon M Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- (40).Roca-Sabio A; Regueiro-Figueroa M; Esteban-Gómez D; de Blas A; Rodríguez-Blas T; Platas-Iglesias C Density Functional Dependence of Molecular Geometries in Lanthanide(III) Complexes Relevant to Bioanalytical and Biomedical Applications. Comput. Theor. Chem 2012, 999, 93–104. [Google Scholar]
- (41).Barysz M; Ishikawa Y Relativistic Methods for Chemists; Springer Science+Business Media B.V.: Dordrecht, 2010. [Google Scholar]
- (42).Dolg M; Stoll H; Savin A; Preuss H Energy-Adjusted Pseudopotentials for the Rare Earth Elements. Theor. Chim. Acta 1989, 75, 173–194. [Google Scholar]
- (43).Hehre WJ; Ditchfield R; Pople JA Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys 1972, 56, 2257–2261. [Google Scholar]
- (44).Hariharan PC; Pople JA The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- (45).Marenich AV; Cramer CJ; Truhlar DG Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- (46).Regueiro-Figueroa M; Esteban-Gómez D; de Blas A; Rodríguez-Blas T; Platas-Iglesias C Understanding Stability Trends along the Lanthanide Series. Chem. - Eur. J 2014, 20, 3974–3981. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.