

Abstract
This paper proposes natural drug candidate compounds for the treatment of coronavirus disease 2019 (COVID-19). We investigated the binding properties between the compounds in the Moringa oleifera plant and the main protease (Mpro) of severe acute respiratory syndrome coronavirus 2 using molecular docking and ab initio fragment molecular orbital calculations. Among the 12 compounds, niaziminin was found to bind the strongest to Mpro. We furthermore proposed novel compounds based on niaziminin and investigated their binding properties to Mpro. The results reveal that the introduction of a hydroxyl group into niaziminin enhances its binding affinity to Mpro. These niaziminin derivatives can be promising candidate drugs for the treatment of COVID-19.
Keywords: COVID-19, SARS-CoV-2, Main protease, Moringa oleifera, Natural product, In silico drug design, Molecular simulation, Molecular docking, Fragment molecular orbital
Graphical abstract
The interacting structure between our proposed compound 9d and selected important residues of SARS-CoV-2 main protease. Red and blue lines indicate hydrogen bonding and electrostatic interactions, respectively.
1. Introduction
The outbreak of the novel coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has raised serious global health concerns. On March 11, 2020, the World Health Organization officially declared the SARS-CoV-2 outbreak a pandemic [1]. Coronaviruses are large RNA viruses belonging to the Coronaviridae family, which is further categorized into four genera, i.e., alpha, beta, gamma, and delta coronaviruses. Typically, the former two infect mammals, whereas the latter two infect birds [2,3]. SARS-CoV-2 is a beta coronavirus. To date, no specific antiviral drugs are available for the treatment of COVID-19 [4], although several clinical trials are currently underway to identify effective drugs. In the development of these drugs, it is essential to identify and evaluate novel drug lead compounds for treating COVID-19. In the present study, using molecular docking and ab initio molecular orbital calculations, we attempt to establish the specific compounds of the Moringa oleifera (M. oleifera) plant that can bind strongly to the SARS-CoV-2 main protease (Mpro) and inhibit its activity.
The Mpro plays a major role in mediating the replication and transcription of SARS-CoV-2. The mutagenesis rate is low in the Mpro, and it cleaves the pp1a and pp1b polyproteins, which release functional proteins, including RNA polymerase, exoribonuclease, and endoribonuclease [5,6]. Moreover, Mpro inhibitors are considered less cytotoxic because of the low similarity between Mpro and human proteases [[7], [8], [9]]. Therefore, the Mpro is a promising target for drugs with which to treat coronaviruses, and the inhibition of Mpro activity is vital to blocking coronavirus replication [5,9,10].
To date, several studies have focused on the inhibition of Mpro activity [[5], [6], [7]]. Pendyala et al. [11] conducted molecular docking studies to identify potential inhibitors for the Mpro and the RNA-dependent RNA polymerase in bioactive food compounds. Their results showed that phycocyanobilin, riboflavin, cyanidin, daidzein, and genistein were more potent as Mpro inhibitors than antiviral drugs (remdesivir, nelfinavir, and lopinavir). Similarly, on the base of molecular docking simulations, Das et al. proposed a natural compound, rutin, which had the highest inhibitor efficiency among the 33 compounds they studied [12]. Theaflavin digallate, which is an antioxidant natural phenol and a theaflavin derivative found in black tea, was shown to bind strongly to the target Mpro [13]. Furthermore, Gurung et al. reported that bonducellpin D, a furanoditerpenoid lactone isolated from Caesalpinia minax, exhibited a high binding affinity toward the Mpro [14].
Natural compounds play an important role in the treatment of a wide range of diseases [15]. For example, M. oleifera is a plant belongs to the Moringaceae family, originating in India. It is cultivated commercially in Africa, Mexico, the United States (including Hawaii), and across Asia. Its antifungal, antioxidant, antibacterial, anti-inflammatory, diuretic, and hepato-protective properties has been studied [16]. Several studies have reported the antiviral activity of M. oleifera, and the plant has been used in many traditional medicines. It has proved to be effective at fighting several viruses, such as the human immunodeficiency virus (HIV), the herpes simplex virus, hepatitis B virus, Epstein–Barr virus (EBV), foot-and-mouth disease virus, and Newcastle disease virus. Moreover, it has been used for the treatment of HIV-related diseases in African countries [17].
In a previous study [16], the antiviral properties of M. oleifera in Huh7 cells were evaluated. M. oleifera is known to contain several bioactive compounds that include phenolic acids, flavonoids, alkaloids, vitamins, tannins, saponins, and isothiocyanates. The different parts of M. oleifera such as root, leaf, seed, flower etc. exhibit various biological activities. Furthermore, M. oleifera was found to improve hepatic and renal functions, as well as the regulation of thyroid hormone status [[18], [19], [20], [21]].
The present study aims to discover natural inhibitors of the SARS-CoV-2 Mpro. To identify novel drug lead compounds for the inhibitor, we first investigated the specific interactions between the Mpro and the 12 natural compounds found in M. oleifera at an electronic level, using molecular docking, classical molecular mechanics (MM), and ab initio fragment molecular orbital (FMO) simulations. Among the 12 compounds, we determined which could bind strongly to the Mpro and be potential lead compounds for Mpro inhibitors. Additionally, we proposed novel compounds based on the most promising lead compound and investigated their binding properties to the Mpro to derive novel natural Mpro inhibitors. These simulated results will be useful for designing novel natural drugs to target the Mpro of SARS-CoV-2.
2. Details of molecular simulations
2.1. Construction and optimization of the Mpro + compound complexes
Data on the compounds contained in the various parts of M. oleifera were collected from the literature [[18], [19], [20], [21]]. The initial dataset included 33 compounds. These were filtered using “Lipinski's rule of five” [22], which is important for the screening of drugs with pharmacological activity. To predict the chemical properties of the compounds, we used the SwissADME web tool [23]. As listed in Table S1 of the Supplementary Information (SI), only 12 of the 33 compounds satisfied Lipinski's rule. We used these 12 compounds [24] shown in Fig. 1 as lead candidates for the Mpro inhibitor. Their three-dimensional (3D) structures were obtained from a structure data file format in the PubChem database [25].
Fig. 1.

The chemical structures of the 12 compounds found in Moringa oleifera.
The structures of the 12 compounds were fully optimized in a vacuum using the B3LYP/6-31G(d,p) method of the ab initio molecular orbital calculation program Gaussian 09 (G09) [26]. The charge distributions of the optimized structures were evaluated using restrained electrostatic potential (RESP) analysis [27] of G09 using the HF/6-31G(d) method, and the RESP charges were employed as the charge parameters in the MM force fields of the compounds. These RESP charges were essential to the docking simulations, along with the MM optimizations of the Mpro + compound complexes, to precisely describe the electrostatic interactions between the Mpro and each of the compounds.
The 3D structure of the Mpro was downloaded from the Protein Data Bank (PDB ID: 6LU7) [28]. This Mpro has seven histidine (His) residues, each of which can have three types of protonation states (Hid, Hie, and Hip+), depending on the pKa values around them. When His residues have a pKa value greater than 6, they prefer a protonated Hip+ state, whereas other His residues can possess Hid or Hie states, depending on the structure around them. To predict the pKa values for the His residues, we used the PROPKA3.1 software [29,30]. Of the seven His residues, only His64 was assigned as Hip+. His41, His80, His163, and His172 were assigned to the Hid state, whereas His164 and His246 were assigned to the Hie state, considering the structures around these His residues. For the remaining ionizable amino acid residues in the Mpro, an ionized state was adopted.
Fig. 2 shows the ligand binding pocket of Mpro [28]. To find the binding site of the compound in the pocket, we conducted molecular docking studies using AutoDock4.2.6 [31]. In the docking simulations, the size of the grid box was set to 19.5 × 19.5 × 19.5 Å3, which is almost 1.5 times the size of the compound, and the center of the grid box was set to the center of the ligand in the PDB structure [28] of the Mpro + ligand complex. The number of created candidate poses was 256, and the threshold distance for clustering these poses was set as 2 Å. From the various clusters generated by AutoDock [31], we selected the three with the largest number of poses, and the representative structures of these clusters were used in the subsequent MM and FMO calculations.
Fig. 2.

Structure of Mpro and its ligand-binding pocket marked by a yellow ellipse. Charge distribution on Mpro is shown in red (negative), blue (positive), and green (neutral), respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To obtain stable structures for the Mpro + compound complexes, the representative structures of the clusters obtained from the docking simulations were fully optimized in water using the classical MM method. In the MM optimizations, approximately 1800 water molecules existing within 8 Å of the complex surface were explicitly considered. The MM and molecular dynamics simulation program AMBER 12 [32] was used. The AMBER FF99-SBLIN force field [33], TIP3P model [34], and general AMBER force field [35] were assigned to the Mpro, water molecules, and compounds, respectively. The criterion for the convergence of structure optimization was set as 0.0001 kcal/mol/Å.
2.2. Ab initio FMO calculations for the optimized structures of the Mpro + compound complexes
To clarify the specific interactions and binding affinity between the Mpro and the compound, we investigated the electronic properties of the Mpro + compound complexes in explicit waters using the ab initio FMO method [36]. This method has been applied to many biomolecules to obtain accurate results that are comparable with experimental results. Because water molecules can contribute to the specific interactions between the Mpro and the compound, the water molecules existing within 10 Å of the compound were explicitly considered. The number of water molecules used in the FMO calculations was approximately 150 for all clusters. To predict the binding affinity between the compound and the Mpro, we evaluated the total inter-fragment interaction energies (IFIEs) [37] between the compound and all the Mpro residues using the ab initio FMO method [36].
In this study, we did not consider the effect of entropy on the binding affinity because a vibrational analysis for the solvated Mpro + compound complex is not practical when using the ab initio FMO method. Furthermore, the entropic effect is unlikely to be markedly different for each of the compounds, as they have similar chemical structures and bind to the same Mpro site. We thus investigated the total IFIEs between the Mpro residues and the compounds using ab initio FMO calculations and estimated the binding affinity under the assumption that the entropic effect was the same for each of the compounds.
In the FMO calculations, the MP2/6-31G(d) method [38,39] of the FMO calculation program ABINIT-MP ver.6.0 [40] was used. Each amino acid residue of the Mpro, compound, and individual water molecule were assigned to a fragment in the FMO calculations. This fragmentation enabled us to analyze the interactions between each Mpro residue and the compound affected by the solvating water molecules. In our previous study [41], the binding properties between the androgen receptor (AR) protein and its ligands were investigated using the same FMO calculations. The evaluated total IFIEs between the AR residues and the ligands were confirmed to correlate well with the binding affinities of these ligands obtained through experiments. The correlation coefficient (R2) was 0.94 for all the nine different ligands, confirming that our evaluated total IFIEs can explain the trend of the observed binding affinities between AR and these ligands. Therefore, the present FMO calculations are expected to obtain accurate binding properties between Mpro residues and the compounds.
3. Results and discussion
3.1. Binding properties between Mpro and compounds contained in Moringa oleifera
M. oleifera is effective at inhibiting Mpro activity and contains many types of natural compounds. However, it is unclear which of these compounds is the most effective. We identified 33 compounds from the literature [[18], [19], [20], [21]] and filtered them using “Lipinski's rule of five” [22] to obtain the 12 candidate compounds for the Mpro inhibitor. Their pharmacokinetic properties evaluated using the SwissADME web tool [23] are listed in Table S1 (see SI), and their chemical structures are shown in Fig. 1. Their binding properties to the Mpro were investigated using the present ab initio FMO calculations to elucidate which compound bonded the strongest to the Mpro.
Morphine (Fig. 1a) is an anti-ulcer and anti-inflammatory agent contained in Moringa root bark. Kaempferol (Fig. 1b) and quercetin (Fig. 1c) are flavonoids found in Moringa flowers [20]. The antiviral, anti-inflammatory, and anti-cancer properties of kaempferol have been reported in previous studies [[42], [43], [44], [45], [46], [47]], whereas quercetin has been shown to reduce the replication of several respiratory viruses [48]. Pterygospermin (Fig. 1d) is found in Moringa seeds and has antibacterial properties [20]. The dried leaves of M. oleifera are rich in polyphenols, of which phenolic acids and flavonoids are the main compounds. Gallic acid (Fig. 1f) is a phenolic acid with anti-tumor and antioxidant functions [49]. Niaziminin (Fig. 1i) is a thiocarbamate contained in M. oleifera leaves. Murakami et al. [50] reported that the presence of an acetoxy group at the 4′-position of niaziminin is important for the inhibition of EBV activation. It is noted that niaziminin, niazinin (Fig. 1j), and O-ethyl N-carbamothioate (Fig. 1k) possess significantly different binding properties to the Mpro, although they have similar chemical structures, as will be shown later in this study.
Using the AutoDock [31] program, each compound was docked to the ligand-binding pocket of the Mpro shown in Fig. 2. The created structures of the Mpro + compound complex were classified into several clusters according to their structural similarities, and the clusters were ranked on the basis of the lowest binding energy (BE) between the Mpro and the compound, as evaluated using AutoDock.
Table 1 lists the ranks, BEs, the number of poses, and Mpro residues involved in H-bonds with each compound for the three clusters with a larger number of poses obtained by AutoDock. In the present study, we selected these clusters because a larger number of poses indicate that the compound has a higher possibility of having one of the structures included in the cluster. In the last column of Table 1, the total IFIEs between each compound and all residues of Mpro evaluated using ab initio FMO method are listed. The representative structures included in the three clusters were employed as the candidate structures of the complexes in the subsequent molecular simulations.
Table 1.
Lowest binding energy (BE: kcal/mol), number of poses, and Mpro residues involved in H-bonds with each compound for the selected clusters obtained by AutoDock4.2.6 program [31].
| Compound | Cluster | BE | Poses | Residues involved in H-bonds | IFIE |
|---|---|---|---|---|---|
| 1 | 2 | −5.18 | 141 | Glu166 | −41.4 |
| 3 | −5.13 | 77 | Gln189 | −67.0 | |
| 5 | −4.82 | 33 | Thr24, Thr26 | −66.3 | |
| 2 | 1 | −4.65 | 52 | Thr24, Thr26, Asn142, His163 | −55.0 |
| 2 | −4.58 | 131 | Thr26, Leu141,Gly143, Ser144, His164 | −64.6 | |
| 3 | −4.56 | 42 | Leu141, Gly143, Ser144, Cys145, Glu166 | −94.6 | |
| 3 | 2 | −3.95 | 31 | Thr25, Thr26, Thr45, Ser144 | −86.8 |
| 7 | −3.87 | 60 | Thr26, Leu141, Gly143, Ser144, Glu166 | −83.3 | |
| 10 | −3.61 | 37 | Thr24, Thr26, Leu141, Asn142, Gly143 | −58.7 | |
| 4 | 1 | −6.64 | 193 | No H-bonds | −72.9 |
| 2 | −6.46 | 25 | No H-bonds | −73.1 | |
| 4 | −5.74 | 18 | Glu166 | −83.9 | |
| 5 | 1 | −3.49 | 85 | Glu166 | −25.9 |
| 2 | −3.46 | 94 | Leu141, Gly143, Ser144 | −50.5 | |
| 3 | −3.38 | 77 | Glu166, Gln189 | −19.5 | |
| 6 | 1 | −3.12 | 62 | Leu141, Gly143, His163, Glu166 | −112.0 |
| 2 | −2.81 | 41 | Asn142, Glu166 | −95.0 | |
| 3 | −2.76 | 69 | Thr24, Thr26, Cys44, Thr45, Ser46 | −57.4 | |
| 7 | 1 | −4.25 | 181 | No H-bonds | −35.7 |
| 2 | −4.17 | 50 | No H-bonds | −37.1 | |
| 3 | −3.99 | 25 | No H-bonds | −30.6 | |
| 8 | 2 | −4.39 | 28 | Glu166, His172 | −105.1 |
| 3 | −4.11 | 22 | Glu166 | −94.7 | |
| 10 | −3.83 | 57 | Thr26, Phe140, Gly143, Glu166 | −66.5 | |
| 9 | 1 | −4.95 | 28 | Thr26, Gln189 | −108.2 |
| 3 | −4.78 | 24 | His41, Asn142, His164, Glu166, Gln189 | −92.3 | |
| 6 | −4.02 | 30 | Gly143, Glu166, Gln189 | −136.5 | |
| 10 | 2 | −4.58 | 67 | Asn142, His164, Glu166, Gln189 | −106.1 |
| 9 | −3.85 | 25 | Thr26, Gly143, Gln189 | −77.3 | |
| 14 | −3.67 | 24 | Thr26, Gly143, His164 | −75.1 | |
| 11 | 1 | −4.33 | 36 | His164, Glu166 | −97.0 |
| 2 | −4.28 | 40 | Asn142, His164, Glu166, Gln189 | −63.7 | |
| 5 | −3.81 | 40 | Thr26, Gly143, Gln189 | −72.5 | |
| 12 | 1 | −5.41 | 37 | Gly143, Glu166, Gln189, Thr190 | −111.0 |
| 2 | −5.11 | 42 | Asn142, Glu166 | −119.0 | |
| 3 | −4.87 | 29 | Asn142, Gly143, Glu166, Gln189 | −107.1 |
The created 256 poses were clustered based on their structural similarity, and each cluster was ranked in the order of BE between Mpro and each compound from Moringa oleifera. We selected three clusters with the largest number of poses, and the total inter fragment interaction energy (IFIE: kcal/mol) between each compound and all Mpro residues was evaluated using the FMO method. These values are listed in the last column.
To clarify the difference in conformation of the compound in the Mpro + compound complex for the three clusters, we compared the conformations in Fig. S1 (see SI). Small-sized compounds (compounds 5–7) were able to bind to different sites in the ligand-binding pocket of the Mpro, because the size of the pocket was larger than the size of these compounds. In contrast, as compounds 9–12 were large and had similar structures, they bonded to similar sites on the Mpro, although their conformations differed.
We optimized the candidate structures in explicit water molecules using the AMBER 12 [32] MM method [[33], [34], [35]]. For the optimized structures, the total IFIEs between the compound and all the Mpro residues were precisely evaluated using the ab initio FMO method [36,40] to determine which compound bonded the strongest to the Mpro. As indicated in Table 1, compound 9 had the largest total IFIEs (−136.5 kcal/mol) among the 12 compounds. The size of this value was at least 17.5 kcal/mol larger than that of the other compounds. Accordingly, it was expected that compound 9 would bond the strongest to the Mpro. Additionally, compounds 6, 8, 10, and 12 had relatively large total IFIEs.
To understand why compound 9 bonded strongly to the Mpro, we compared the conformation of compound 9 in the ligand-binding pocket of the Mpro for the three clusters obtained using AutoDock. As shown in Fig. S1(i) (see SI), in the third ranked cluster (in pink), compound 9 exists near the negatively charged Glu166 residue of the Mpro and is stabilized by the electrostatic attractive interactions between it and this residue. As a result, compound 9 of the cluster 6 has the largest total IFIEs. Furthermore, we analyzed the IFIEs between the Mpro residues and compound 9 in the structures of the three clusters. As shown in Fig. 3 , Asn142, Glu166, and Gln189 residues primarily contribute to the interactions with compound 9 in all the clusters. These residues were thus considered to be important for the strong binding of compound 9 to the Mpro. In the cluster 6 structure (Fig. 3c), compound 9 interacted strongly (−45 kcal/mol) with Glu166; it also interacted with Cys145. As a result, compound 9 had the largest total IFIEs in the cluster 6 structure.
Fig. 3.

The inter fragment interaction energies (IFIEs) between compound 9 and each Mpro residue for the structures of (a) cluster 1, (b) cluster 3, and (c) cluster 6. The total IFIEs between compound 9 and all Mpro residues are also shown for each cluster. The red bars indicate the residues with attractive IFIE, the size of which is larger than 10 kcal/mol. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Additionally, we attempted to clarify which part of compound 9 was important for its strong binding to the Mpro, to propose novel potent Mpro inhibitors based on this compound. As shown in Fig. 4a, the NH group and the oxygen atom connecting the two rings of compound 9 contribute to the strong electrostatic interactions with the backbone and the side chain of Glu166. These interactions are the main reason for compound 9 having the largest total IFIEs. Additionally, Fig. 4b reveals that Asn142, Gly143, and Gln189 of the Mpro form hydrogen bonds with the hydroxide (OH) and carbonyl groups of the cyclohexane ring of compound 9. Therefore, these groups, as well as the oxygen atom of compound 9, are considered essential for maintaining the strong binding between the compound and the Mpro residues. In contrast, as the other sites of compound 9 make no significant contribution to its binding to the Mpro, it is expected that the replacement of these sites will enhance the binding affinity of compound 9 to the Mpro.
Fig. 4.

The interacting structures between compound 9 (ball-and-stick model) and selected important Mpro residues (stick model) in the optimized structure of the Mpro + compound 9 complex for cluster 6. (a) Compound 9 and Glu166, and (b) compound 9 and Asn142, Gly143, Cys145, and Glu189. Hydrogen bonding and electrostatic interactions are indicated by red and blue lines, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The binding properties to the Mpro of the other compounds included in M. oleifera were investigated in the same manner. Their docking conformations to the Mpro, IFIEs with the Mpro residues, and their interacting structures with Mpro residues are shown in Figs. S1–S13 (see SI). As listed in the last column of Table 1, the compound 12 has the second largest total IFIE among the 12 compounds. Its IFIEs with the Mpro residues and the interacting structure between compound 12 and some important residues are shown in Fig. 5 . Compound 12 interacts strongly with Glu166 and Asn142, however, it has no significant interaction with Gln189 and Cys145 as for the compound 9. As a result, the size of total IFIE of compound 12 is remarkably smaller than that for the top ranked compound 9.
Fig. 5.

(a) The inter fragment interaction energies (IFIEs) between compound 12 and each Mpro residue for the structure of cluster 2. The total IFIE between compound 12 and all Mpro residues is also shown. The red bars indicate the residues with attractive IFIE, the size of which is larger than 10 kcal/mol. (b) The interacting structures between compound 12 (ball-and-stick model) and selected important Mpro residues (stick model) in the optimized structure of the Mpro + compound 12 complex for cluster 2. Hydrogen bonding, electrostatic, and NH-π interactions are indicated by red, blue, and orange lines, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. Proposal of novel potent inhibitors and their binding properties to the Mpro
As compound 9 was found to bind the strongest to the Mpro, we adopted it as a lead compound for proposing novel potent Mpro inhibitors. Fig. 4 shows that the OH groups of compound 9 contribute to its strong interactions with the Mpro residues. We thus considered the introduction of an OH group to selected sites of compound 9. The seven sites shown in Table 2 were considered because the lower part of compound 9 already contributed to the interactions with the Mpro, as shown in Fig. 4b. The proposed compounds are defined as compounds 9a − 9 g, based on the site to be introduced by the OH group indicated in Table 2.
Table 2.
The total inter fragment interaction energy (IFIE; kcal/mol) between each compound and all Mpro residues evaluated using the FMO method.
| Compound | Total IFIE |
|---|---|
| 9 | −136.5 |
| 9a | −149.9 |
| 9b | −139.1 |
| 9c | −160.6 |
| 9d | −175.4 |
| 9e | −145.1 |
| 9f | −139.6 |
| 9g | −153.9 |
The proposed compounds are defined as compounds 9a–9g, based on the site to be replaced by a hydroxyl group. For example, in compound 9a, the hydrogen atom at the a-site of compound 9 shown as follows is replaced by a hydroxyl group.
The total IFIEs between the Mpro residues and our proposed compounds, as well as compound 9, are listed in Table 2. The compounds 9d and 9c had larger total IFIEs compared with the other compounds. The size of their total IFIEs was at least 24 kcal/mol larger than that of compound 9, indicating that the introduction of an OH group enhanced the interaction between compound 9 and the Mpro residues.
To establish why compounds 9d and 9c had larger total IFIEs, we compared the IFIEs for our proposed compounds. As shown in Fig. 6 , the Glu166 residue of Mpro has the strongest attractive interaction with all the compounds, indicating its importance for ligand binding of the Mpro. Specifically, compound 9d interacted strongly with Glu166. In addition, Phe140, Asn142, Cys145, and Gln189 contributed to interactions between the Mpro and compounds 9d/9c.
Fig. 6.

The inter fragment interaction energies (IFIEs) between our proposed compound 9 derivatives and the Mpro residues. The red bars indicate the residues with attractive IFIE, the size of which is larger than 10 kcal/mol. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
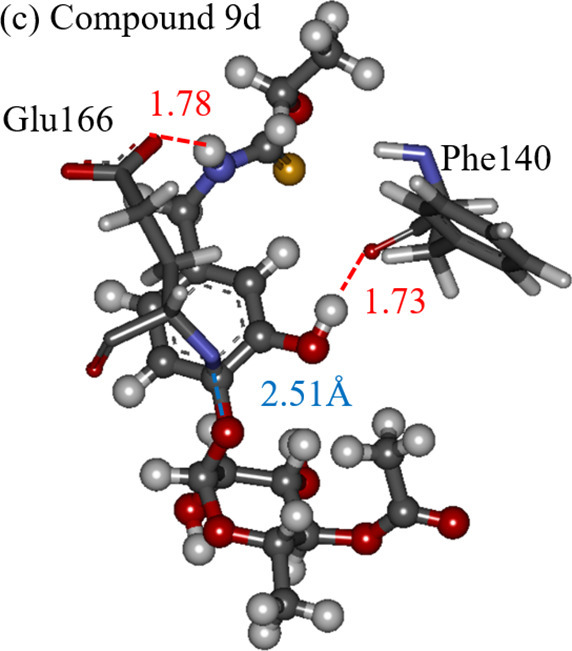
To elucidate the effect of the OH introduction on the IFIEs between the Mpro and compound 9, we investigated the difference in IFIEs for compounds 9, 9c, and 9d. As indicated in Fig. 7a, the effect of the OH introduction was not significant (smaller than 10 kcal/mol) for compound 9c. In contrast, Fig. 7b reveals that the interactions between compound 9 and Phe140/Glu166 of the Mpro were significantly enhanced by the introduction of OH at the d-site of compound 9.
Fig. 7.

The difference in inter fragment interaction energies (IFIEs) between the Mpro residues and (a) compounds 9c and 9 and (b) compounds 9d and 9. The red bars indicate the residues with IFIE difference, the size of which is larger than 5 kcal/mol. These residues interact more strongly with compound 9c/9d compared with compound 9. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We furthermore compared the interacting structures between these residues and compounds 9, 9c, and 9d to clarify the difference in the effect of the OH group, based on the introduced site. As shown in Fig. 8a, compound 9 interacted electrostatically with Glu166 and Phe140. Particularly, the Glu166 side chain interacted strongly with the NH group of compound 9. By introducing an OH group at the c-site of compound 9, a hydrogen bond was formed between the OH group and the oxygen atom of the Phe140 backbone (Fig. 8b). However, the interactions of the other parts of compound 9 with the Mpro residues did not change significantly due to the introduction of the OH group. In contrast, Fig. 8c elucidates that the introduction of an OH group at the d-site of compound 9 significantly enhanced the interactions between compound 9 and selected Mpro residues. The introduced OH group formed a hydrogen bond with Phe140. Additionally, the interaction between the Glu166 side chain and the NH group of compound 9 was enhanced to form a hydrogen bond at 1.78-Å distance. As a result, the IFIEs between compound 9d and Phe140/Glu166 were significantly enhanced by the OH group introduction at the d-site, leading to a larger amount of total IFIEs between the Mpro residues and compound 9d. Regarding the other proposed compounds shown in Table 2, the introduction of an OH group into compound 9 did not cause a significant change in the IFIEs and the interacting structures between compound 9 and the Mpro residues. Consequently, it is revealed that the site of the OH group replacement is important for enhancing the interactions between compound 9 and the Mpro residues, and that compound 9d binds more strongly to the Mpro than compound 9.
Fig. 8.

The interacting structures between our proposed compounds (ball-and-stick model) and selected important Mpro residues (stick model): (a) compound 9, (b) compound 9c, and (c) compound 9d. The red and blue lines indicate hydrogen bonding and electrostatic interactions, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In order to validate the improvement of the binding affinity between the Mpro and compound 9 by the addition of OH group, we additionally considered a methyl group and introduced it into the d-site of compound 9. The structure of its complex with the Mpro was optimized by the MM method, and the IFIEs between the Mpro and the methyl replaced compound 9 were investigated using FMO method. The total IFIE was evaluated −144.6 kcal/mol, whose size is about 30 kcal/mol smaller than that (−175.4 kcal/mol) of the OH replaced compound 9d. As shown in Fig. 8c, the OH group of compound 9d forms a hydrogen bond with the oxygen atom of Phe140 backbone. By replacing the OH group with a CH3 group, this hydrogen bond disappears, resulting in a weaker interaction between the Mpro and the CH3 replaced compound 9. Therefore, it is revealed that the introduction of OH group is more effective for improving the binding affinity between the Mpro and compound 9.
4. Conclusions
To propose novel natural compounds as potent inhibitors of the SARS-CoV-2 Mpro, we investigated the binding properties between the Mpro and the 12 natural compounds found in the M. oleifera plant, using molecular simulations based on protein−ligand docking, MM optimizations, and ab initio FMO calculations. The FMO results revealed that niaziminin bonded the strongest to the Mpro. Furthermore, to enhance the binding affinity of niaziminin to the Mpro, we introduced an OH group at different niaziminin sites and investigated the binding properties between these derivatives and the Mpro. Our proposed compound, which includes an OH group introduced into the phenyl ring of niaziminin, formed hydrogen bonds with the Glu166 and Phe140 residues of the Mpro to bind more strongly to the Mpro. It is thus expected that this niaziminin derivative (compound 9d in Table 2) can be a potent inhibitor of the Mpro.
Author statement
The authors have no statement.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bpc.2021.106608.
Appendix A. Supplementary data
Supplementary material
Data availability
Data will be made available on request.
References
- 1.Pachetti M., Marini B., Benedetti F., et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020;18:1–9. doi: 10.1186/s12967-020-02344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmad J., Ikram S., Ahmad F., et al. SARS-CoV-2 RNA dependent RNA polymerase (RdRp)–a drug repurposing study. Heliyon. 2020;6 doi: 10.1016/j.heliyon.2020.e04502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pal M., Kerorsa G.B., Kandi V.A. Knowledge update on SARS-Coronavirus-2 (SARS-CoV-2)/COVID-19 and its global public health implications. Am. J. Clin. Med. Res. 2020;8:23–27. [Google Scholar]
- 4.Saxena A. Drug targets for COVID-19 therapeutics: ongoing global efforts. J. Biosci. 2020;45:1–24. doi: 10.1007/s12038-020-00067-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motiwale M., Yadav N.S., Kumar S., et al. Finding potent inhibitors for COVID-19 main protease (Mpro): an in silico approach using SARS-CoV-3CL protease inhibitors for combating CORONA. J. Biomol. Struct. Dyn. 2020;7:1–12. doi: 10.1080/07391102.2020.1829501. [DOI] [PubMed] [Google Scholar]
- 6.Tachoua W., Kabrine M., Mushtaq M., et al. An in-silico evaluation of COVID-19 main protease with clinically approved drugs. J. Mol. Graph. Model. 2020;101:107758. doi: 10.1016/j.jmgm.2020.107758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joshi R.S., Jagdale S.S., Bansode S.B., et al. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J. Mol. Graph. Model. 2020;5:1–6. doi: 10.1080/07391102.2020.1760137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anand K., Ziebuhr J., Wadhwani P., et al. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 9.Zhang L., Lin D., Sun X., et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin Z., Du X., Xu Y., et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2019;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 11.Pendyala B., Patras A. 2020. In silico Screening of Food Bioactive Compounds to Predict Potential Inhibitors of COVID-19 Main protease (Mpro) and RNA-dependent RNA polymerase (RdRp) [DOI] [Google Scholar]
- 12.Das S., Sarmah S., Lyndem S., et al. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J. Biomol. Struct. Dyn. 2020:1–8. doi: 10.1080/07391102.2020.1763201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manish M. 2020. Studies on Computational Molecular Interaction between SARS-CoV-2 Main Protease and Natural Products. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurung A.B., Ali M.A., Lee J., et al. Unravelling lead antiviral phytochemicals for the inhibition of SARS-CoV-2 Mpro enzyme through in silico approach. Life Sci. 2020;255:117831. doi: 10.1016/j.lfs.2020.117831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Da Silva Antonio A., Wiedemann L.S., Veiga-Junior V.F. Natural products’ role against COVID-19. RSC Adv. 2020;10:23379–23393. doi: 10.1039/d0ra03774e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vergara-Jimenez M., Almatrafi M.M., Fernandez M.L. Bioactive components in Moringa Oleifera leaves protect against chronic disease. Antioxidants. 2017;6:91. doi: 10.3390/antiox6040091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biswas D., Nandy S., Mukherjee A., et al. Moringa oleifera lam. And derived phytochemicals as promising antiviral agents: a review. S. Afr. J. Bot. 2020;129:272–282. [Google Scholar]
- 18.Ragasa C.Y., Medecilo M.P., Shen C.C. Chemical constituents of Moringa oleifera Lam. Leaves. Der Pharma Chem. 2015;7:395–399. [Google Scholar]
- 19.Mehra M., Jakhar N., Joshi S., et al. Phytotherapaeutic functionality of Moringa oleifera Lam. for health. Int. J. Cell Sci. Mol. Biol. 2017;3:1–4. [Google Scholar]
- 20.Ragasa C.Y., Ng V.A., Shen C.C. Chemical constituents of Moringa oleifera lam. Seeds. Int. J. Pharmacogn. Phytochem. Res. 2016;8:495–498. [Google Scholar]
- 21.Karadi R.V., Palkar M.B., Gaviraj E.N., et al. Antiurolithiatic property of Moringa oleifera root bark. Pharm. Biol. 2008;46:861–865. [Google Scholar]
- 22.Lipinski C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 23.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaji D. 2020. Computational Identification of Drug Lead Compounds for COVID-19 from Moringa oleifera. [DOI] [Google Scholar]
- 25.Kim S., Thiessen P.A., Bolton E.E., et al. PubChem substance and compound databases. Nucleic Acids Res. 2015;44:D1202–D1213. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frisch M.J., Trucks G.W., Schlegel H.B., et al. Inc Wallingford CT; 2009. Gaussian 09, Revision D. 01, Gaussian. [Google Scholar]
- 27.Besler B.H., Merz K.M., Jr., Kollman P.A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990;11:431–439. [Google Scholar]
- 28.Jin Z., Du X., Xu Y., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 29.Søndergaard C.R., Mats Olsson H.M., Rostkowski J.H., et al. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comp. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 30.Mats Olsson H.M., Søndergaard C.R., Rostkowski J.H., et al. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comp. 2011;2:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 31.Morris G.M., Huey R., Lindstrom W., et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Case D.A., Darden T.A., Cheatham T.E., III, et al. Vol. 2012. University of California; San Francisco: 2010. AMBER 12; pp. 1–826. [Google Scholar]
- 33.Lindorff-Larsen K., Piana S., Palmo K., et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jorgensen W.L., Chandrasekhar J., Madura J.D., et al. Compatison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 35.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., et al. Development and testing of a general Amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 36.Kitaura K., Ikeo E., Asada T., et al. Fragment molecular orbital method: an approximate computational method for large molecules. Chem. Phys. Lett. 1999;313:701–706. [Google Scholar]
- 37.Fukuzawa K., Komeiji Y., Mochizuki Y., et al. Intra- and inter-molecular interactions between cyclic-AMP receptor protein and DNA: Ab initio fragment molecular orbital study. J. Comput. Chem. 2006;27:948–960. doi: 10.1002/jcc.20399. [DOI] [PubMed] [Google Scholar]
- 38.Mochizuki Y., Nakano T., Koikegami S., et al. A parallelized integral-direct second-order Mφller-Plesset perturbation theory method with a fragment molecular orbital scheme. Theor. Chem. Accounts. 2004;112:442–452. [Google Scholar]
- 39.Mochizuki Y., Koikegami S., Nakano T., et al. Large scale MP2 calculations with fragment molecular orbital scheme. Chem. Phys. Lett. 2004;396:473–479. [Google Scholar]
- 40.Mochizuki Y., Yamashita K., Nakano T., et al. Higher-order correlated calculations based on fragment molecular orbital scheme. Theor. Chem. Accounts. 2011;130:515–530. [Google Scholar]
- 41.Kobayashi I., Takeda R., Shimamura K., et al. Specific interactions between androgen receptor and its ligand: ab initio molecular orbital calculations in water. J. Mole Graph Model. 2017;75:383–389. doi: 10.1016/j.jmgm.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 42.Ren J., Lu Y., Qian Y., et al. Recent progress regarding kaempferol for the treatment of various diseases. Exp. Ther. Med. 2019;18:2759–2776. doi: 10.3892/etm.2019.7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imran M., Rauf A., Shah Z.A., et al. Chemopreventive and therapeutic effect of the dietary flavonoid kaempferol: a comprehensive review. Phytother. Res. 2019;33:263–275. doi: 10.1002/ptr.6227. [DOI] [PubMed] [Google Scholar]
- 44.Li J., Huang H., Feng M., et al. In vitro and in vivo anti-hepatitis B virus activities of a plant extract from Geranium carolinianum L. Antivir. Res. 2008;79:114–120. doi: 10.1016/j.antiviral.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 45.Jeong H.J., Ryu Y.B., Park S.J., et al. Neuraminidase inhibitory activities of flavonols isolated from Rhodiola rosea roots and their in vitro anti-influenza viral activities. Bioorg. Med. Chem. 2009;17:6816–6823. doi: 10.1016/j.bmc.2009.08.036. [DOI] [PubMed] [Google Scholar]
- 46.Zhang T., Wu Z., Du J., et al. Anti-Japanese-encephalitis-viral effects of kaempferol and daidzein and their RNA-binding characteristics. PLoS One. 2012;7 doi: 10.1371/journal.pone.0030259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zakaryan H., Arabyan E., Oo A., Zandi K. Flavonoids: promising natural compounds against viral infections. Arch. Virol. 2017;162:2539–2551. doi: 10.1007/s00705-017-3417-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu W., Li R., Li X., et al. Quercetin as an antiviral agent inhibits influenza A virus (IAV) entry. Viruses. 2016;8:6. doi: 10.3390/v8010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu Y., Yin Q., Yang Y. Comprehensive investigation of Moringa oleifera from different regions by simultaneous determination of 11 polyphenols using UPLC-ESI-MS/MS. Molecules. 2020;25:676. doi: 10.3390/molecules25030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murakami A., Kitazono Y., Jiwajinda S., et al. Niaziminin, a thiocarbamate from the leaves of Moringa oleifera, holds a strict structural requirement for inhibition of tumor-promotor-induced Epstein-Barr virus activation. Planta Med. 1998;64:319–323. doi: 10.1055/s-2006-957442. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Data Availability Statement
Data will be made available on request.