

Abstract
The Abelson-helper integration site 1 (AHI1) gene encodes for a ciliary transition zone localizing protein that when mutated causes the human ciliopathy, Joubert syndrome. We prepared and examined neuronal cultures derived from male and female embryonic Ahi1+/+ and Ahi1–/– mice (littermates) and found that the distribution of ciliary melanin-concentrating hormone receptor-1 (MchR1) was significantly reduced in Ahi1–/– neurons; however, the total and surface expression of MchR1 on Ahi1–/– neurons was similar to controls (Ahi1+/+). This indicates that a pathway for MchR1 trafficking to the surface plasma membrane is intact, but the process of targeting MchR1 into cilia is impaired in Ahi1-deficient mouse neurons, indicating a role for Ahi1 in localizing MchR1 to the cilium. Mouse Ahi1–/– neurons that fail to accumulate MchR1 in the ciliary membrane have significant decreases in two downstream MchR1 signaling pathways [cAMP and extracellular signal-regulated kinase (Erk)] on MCH stimulation. These results suggest that the ciliary localization of MchR1 is necessary and critical for MchR1 signaling, with Ahi1 participating in regulating MchR1 localization to cilia, and further supporting cilia as critical signaling centers in neurons.
SIGNIFICANCE STATEMENT Our work here demonstrates that neuronal primary cilia are powerful and focused signaling centers for the G-protein-coupled receptor (GPCR), melanin-concentrating hormone receptor-1 (MCHR1), with a role for the ciliary transition zone protein, Abelson-helper integration site 1 (AHI1), in mediating ciliary trafficking of MCHR1. Moreover, our manuscript further expands the repertoire of cilia functions on neurons, a cell type that has not received significant attention in the cilia field. Lastly, our work demonstrates the significant influence of ciliary GPCR signaling in the overall signaling of neurons.
Keywords: AHI1, cilia, Joubert syndrome, MchR1, neuron, signaling
Introduction
Primary cilia are microtubule-based, polarized cell structures that protrude from the apical surface of the plasma membrane into the extracellular space. These organelles are considered critical signaling centers important for vertebrate development (Goetz and Anderson, 2010; Pal and Mukhopadhyay, 2015). One of the unique features of primary cilia is the selective molecular composition of their membrane, which is enriched in signaling receptors and molecules, such as several G-protein-coupled receptors (GPCRs) and adenylyl cyclase, respectively (Mykytyn and Askwith, 2017; Nachury and Mick, 2019). When defects in cilia formation or function occur, abnormal embryonic development and cellular dysfunction result because of impaired signal transduction at primary cilia (Pazour and Rosenbaum, 2002; Huangfu et al., 2003; Huangfu and Anderson, 2005; Pal and Mukhopadhyay, 2015). This indicates that the correct localization of receptors at the ciliary membrane, and not just at the plasma membrane, is critical for initiating proper signaling, but this remains unclear particularly for terminally differentiated neurons.
Mutations in the Abelson-helper integration site 1 (AHI1) gene cause an inherited neurodevelopmental disorder called Joubert syndrome (JBTS; Dixon-Salazar et al., 2004; Ferland et al., 2004), which is defined by aplasia/hypoplasia of the cerebellar vermis and an abnormal configuration of the superior cerebellar peduncles, and is characterized by cognitive impairment, apnea/hyperpnea, abnormal eye movements and hypotonia. Ahi1 is a cytoplasmic and transition zone localizing protein required for the function of primary cilia (Doering et al., 2008; Eley et al., 2008; Hsiao et al., 2009; Lee et al., 2014), supporting JBTS as a ciliopathy (Hildebrandt et al., 2011). In addition, inactivation of Ahi1 hampers primary cilia function because of defective ciliary trafficking through an effect of Ahi1 in regulating Rab8a localization (Hsiao et al., 2009; Westfall et al., 2010). Thus, these studies indicate that Ahi1 at the ciliary transition zone may be an important regulator in trafficking receptors to the primary cilium (Hsiao et al., 2012; Muñoz-Estrada and Ferland, 2019).
The melanin-concentrating hormone receptor-1 (MchR1) is a GPCR (Chambers et al., 1999), and one of the GPCRs enriched at neuronal cilia (Berbari et al., 2008a,b; Tuz et al., 2013). In humans, rare MCHR1 missense mutations have been identified in individuals with obesity whereas loss of function MCHR1 mutations have been associated with underweight individuals (Gibson et al., 2004; Goldstein et al., 2010). Complementary findings in murine models implicate MchR1 in feeding behavior and metabolism as well as in anxiety and depression (Shimazaki et al., 2006; Macneil, 2013). Studies that used cell lines exogenously expressing MchR1, showed that stimulation of MchR1 by adding melanin-concentrating hormone (MCH) can inhibit forskolin (FSK)-induced cAMP production and increase intracellular calcium concentrations (Chambers et al., 1999; Saito et al., 1999; Hawes et al., 2000), in addition to activating the extracellular signal-regulated kinase (Erk) pathway (Pissios et al., 2003; Eberle et al., 2004). Therefore, we were interested in determining the extent of ciliary MchR1 localization and its stimulation in Ahi1 knock-out neurons with defective primary cilia function.
Here, we show that targeted deletion of Ahi1 in mice reduces the localization of ciliary markers at the neuronal primary cilium in different brain regions, including a dramatic reduction in ciliary MchR1 localization in Ahi1–/– neurons. Given that the total cell surface expression of MchR1 was normal in Ahi1–/– neurons, the decreased ciliary MchR1 distribution in Ahi1–/– neurons suggest defective MchR1 cilia trafficking that is regulated by the transition zone protein, Ahi1. Strikingly, Ahi1–/– neurons with normal MchR1 plasma membrane localization, but lacking MchR1 localization on primary cilia, have reduced signaling on MCH stimulation as indicated by an inhibition of FSK-induced cAMP production and decreased Erk pathway activation. This study suggests a critical role for neuronal primary cilia as a requisite site for MchR1 localization and ligand binding to activate downstream signaling pathways, and further supports the requirement of a functional ciliary transition zone for proper signal transduction.
Materials and Methods
Primary neuronal cultures
Ahi1+/+ and Ahi1–/– hypothalamic, hippocampal and cerebral cortical neuron cultures were prepared from embryonic day (E)18.5 mouse embryos harvested from anesthetized timed-pregnant female Ahi1+/− mice that were crossed with male Ahi1+/− mice. All mice used for breeding were backcrossed at least 10 generations onto a Balb/CJ genetic background (this strain has been deposited in The Jackson Laboratory/MMRRC repository, stock #033663/65561-JAX, designated C.Cg-Ahi1tm1Rujf/Mmjax). All experimental protocols were performed under approval of the Institutional Animal Care and Use Committee of Albany Medical College and complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Brains were dissected in ice-cold dissecting solution (Dulbecco's PBS containing 20% glucose and penicillin/streptomycin). Hypothalami, hippocampi, or cerebral cortices from each mouse embryo were isolated and rinsed three times with sterile, ice-cold dissecting solution. Tissues were then treated with 0.05% trypsin/EDTA for 10 min at 37°C and were incubated with 1 mg/ml trypsin inhibitor (Sigma) in Dulbecco's PBS for 5 min at room temperature (RT). Tissues were dissociated by gentle trituration in Neurobasal medium (Invitrogen) supplemented with 0.5% glucose, 0.4 mm L-glutamine, 0.04 mg/ml gentamicin, and 2% B27 supplement (Invitrogen). Cells were counted with a hemocytometer and seeded on poly-D-lysine (Sigma) coated glass coverslips at a density of 2 × 105 cells/cm2. Neuronal culture media was replaced every 3 d. Neuronal cultures were grown at 37°C in a humidified 5% CO2 incubator.
Immunolabeling and imaging
Cells were fixed with 4% paraformaldehyde/4% sucrose in Banker's PBS (Goslin and Banker, 1989), with methanol at −20°C (10 min), or with 4% paraformaldehyde/4% sucrose in Banker's PBS (18 min) followed by incubation with 100% iced-cold methanol at −20°C (5 min). For tissue immunolabeling, mice were perfused with 4% paraformaldehyde/PBS and postfixed for at least 3 d before being transferred to a 30% sucrose cryopreservation solution. Fixed brains were embedded in OCT and sectioned in the sagittal plane at a thickness of 20 µm.
Fixed cells or tissue sections were permeabilized in 0.04–0.1% Triton X-100/PBS (PBSTx) for 15 min at RT followed by incubation in 10% normal donkey serum in PBSTx or 1% bovine serum albumin (BSA) in PBS for 1 h at RT before primary antibody incubation. Cells or tissue sections were then incubated with primary antibodies diluted in 1% normal donkey serum/PBSTx at 4°C overnight. The following antibodies were used for immunolabeling: rabbit polyclonal anti-Ahi1 (1:1000; Doering et al., 2008), mouse monoclonal anti-acetylated α-tubulin (1:1000; Sigma), rabbit polyclonal anti-adenylyl cyclase III (AC3; 1:1000; Santa Cruz Biotechnology), goat polyclonal anti-AC3 (1:200; Santa Cruz Biotechnology), mouse monoclonal anti-Arl13b (1:1000; UC Davis/NIH NeuroMab Facility), rabbit monoclonal anti-Arl13b (1:2000; a gift from Caspary at Emory University), mouse monoclonal anti-γ-tubulin (1:2000; Sigma), goat polyclonal anti-MchR1 (1:200; Santa Cruz Biotechnology), or mouse monoclonal anti-NeuN (1:2000; Millipore). After extensive washing with PBS, tissue sections and cells were incubated with fluorescently conjugated secondary antibodies [Alexa Fluor 488 or 546 or Cy5 (Life Technologies) or Dylight 488, 546, or 649 (Jackson ImmunoResearch), all at 1:500–1:2000 dilutions] for 1 h at RT, followed by Hoechst 33258 DNA labeling (1 μg/ml). After labeling, cells and tissue sections were mounted on slides with Fluoromount-G antifade solution (Southern Biotechnology).
Immunofluorescent images were either acquired with a Zeiss AxioImager-Z1 microscope (Zeiss EX Plan-NeoFluar 40×/0.75, ∞/0.17 lens or a Zeiss Plan-Apochromat 63×/1.4 oil DIC, ∞/0.17 lens) through a Zeiss AxioCam MRm monochrome camera using AxioVision Rel 4.6 (Zeiss) software or a Zeiss confocal microscope LSM800 with Airyscan (63×/1.4 NA oil objective) processed with Zen black 2.1 or Zen blue lite 2.3 (Zeiss). The immersion medium for the 63× lens was Zeiss Immersol 518F oil. All images comparing Ahi1+/+ and Ahi1–/– cells or tissues were acquired with the same exposure times. Contrast and brightness of images were adjusted through linear level adjustments, as needed, to optimize the intensity ranges of the images using Adobe Photoshop CS2 (version 9.0.2; Adobe Systems).
Quantitative immunofluorescence was performed using ImageJ, with fluorescent images acquired with identical microscope settings. To determine MchR1 intensity in the primary cilium of Ahi1+/+ and Ahi1–/– neurons, individual cilia were defined with Arl13b immunolabeling and integrated intensity was measured in the MchR1 channel, with background correction performed by subtracting the integrated density of an adjacent area of identical dimensions to the region of interest (ROI). Note that integrated density (IntDen) in the ROI represents the product of area and mean gray value, IntDen = [area in µm2 (sum of the gray values of all pixels ÷ number of pixels)].
Western blot analysis
Cells and brain tissue lysates were prepared for western blot analysis as described previously (Hsiao et al., 2009). For membrane protein solubilization, tissues or cells were homogenized in normal radioimmunoprecipitation assay (RIPA) buffer with 2% SDS followed by sonication. Cleared lysates were collected after 16,000 × g centrifugation at 4°C for 5 min. Membranes prepared for western blot analysis were probed with primary antibodies diluted in 5% skim milk/Tris-buffered saline/0.01% Triton X-100 at 4°C overnight. The following antibodies were used for western blot analysis: rabbit polyclonal anti-Ahi1 (1:1000; Doering et al., 2008), mouse monoclonal anti-Gapdh (1:5000; Abcam), goat polyclonal anti-MchR1 (1:200; Santa Cruz Biotechnology), mouse monoclonal anti-Arl13b (1:1000; UC Davis/NIH NeuroMab Facility), or chicken polyclonal anti-βIII tubulin (1:2000; Millipore). Primary antibodies were detected with either the SuperSignal West Femto Maximum Sensitivity Substrate Chemiluminescence kit (ThermoFisher) with horseradish peroxidase (HRP)-conjugated secondary antibodies (Invitrogen; 32230 and 32260) or with fluorescently labeled secondary antibodies (Alexa Fluor 488, Alexa Fluor 546, or Cy5). Signals were detected with a G:Box iChemi XT imaging system using GeneSnap software and analyzed with GeneTool analysis software (SynGene). In order to quantify and compare the signal intensities between each sample, all detected signals were unsaturated and in the linear range of detection.
Surface protein isolation
Surface proteins on cells were isolated with the Pierce Cell Surface Protein Isolation kit (ThermoFisher) according to the manufacturer's instructions. In brief, neuronal cultures were grown in vitro for 9–14 d [day in vitro (DIV)9–DIV14] in 12-well culture plates and then rinsed with PBS supplemented with Ca2+ and Mg2+ followed by incubation with 1 ml of 0.5 mg/ml EZ-link sulfo-NH-SS-Biotin in PBS for 15 min at 4°C. The reaction was stopped by adding 50 µl of quench solution followed by three rinses with 1 ml of 1 mg/ml BSA. Cell lysates were collected in 100 µl of RIPA lysis buffer containing protease inhibitors [20 µl of the lysate (20% of the total lysate) was kept as a sample for assessing total protein levels]. The remaining lysate (80 µl, 80% of the total lysate) was incubated with 200 µl of NeutrAvidin agarose resin (50% slurry) and shaken on a Nutator for 2 h at 4°C to pull-down biotin-labeled proteins. Resins were washed three times with RIPA buffer containing protease inhibitors, and were incubated with 1× SDS-PAGE sample buffer containing 0.05 m dithiothreitol for 1 h at RT to release biotin-labeled proteins. Levels of MchR1 and Gapdh were evaluated from total lysates and from surface protein fractions using western blot analysis. Gapdh was used as an indicator for any contamination of cytoplasmic proteins in the surface protein fractions.
cAMP assay
Ahi1+/+ and Ahi1–/– neuronal cells, cultured in 96-well plates for 9–14 d, were used for the determination of cAMP concentrations ([cAMP]) using the LANCE cAMP 384 kit (PerkinElmer) following the manufacturer's instructions. Briefly, attached neuronal cells were rinsed with HBSS (Life Technologies) three times and then incubated with Alexa Fluor 647-conjugated anti-cAMP antibodies diluted in stimulation buffer (5 mm HEPES, 5 mm IBMX, and 0.1% BSA in HBSS) in the presence of 1 μm FSK (Sigma) or 1 μm FSK and 100 nm MCH (Bachem Americas). Each treatment was run in triplicate for each embryo. After 15 min of MCH stimulation at 37°C, the detection mix containing biotin-labeled cAMP and Europium-conjugated anti-cAMP antibodies was added to each sample and incubated for 240 min at RT. Signals (time-resolved fluorescence resonance energy transfer) were detected at 665 nm with an excitation of 340 nm using a Victor3 1420 multilabel plate reader (PerkinElmer). Measured values were then calibrated to known [cAMP] for further statistical analyses.
Erk signaling experiments
Cells were grown on poly-D-lysine coated 12-well cultured plates and after 10 d in culture, cells were stimulated with 100 nm MCH (Bachem Americas) for 3 min. Cells were then washed with ice-cold PBS and harvested with RIPA buffer supplemented with protease and phosphatase inhibitors. Cell lysates were then incubated for 20 min on a nutating mixer and centrifuged at 16,000 × g for 20 min at 4°C. The supernatant was transferred to a clean tube and mixed with loading buffer, resolved by 10% TGX stain-free polyacrylamide gels (Bio-Rad) and transferred to PVDF membranes (Millipore). Membranes were incubated in blocking buffer consisting of either 5% (w/v) nonfat dry milk or 5% BSA in Tris-buffered saline with 0.1% Tween (TBST) for 1 h at RT followed by overnight incubation of primary antibodies, diluted in blocking buffer, at 4°C. Primary antibodies were as follows: mouse monoclonal anti-α-tubulin (1:5000; Abcam), rabbit monoclonal anti-Erk1/2 (1:1000; Cell Signaling), or mouse monoclonal anti-pErk1/2 (1:1000; Santa Cruz Biotechnology). After extensive washing with TBST, membranes were incubated for 1 h at RT with the corresponding HRP secondary antibodies (1:10,000 dilution; Invitrogen) in TBST. Chemiluminescent detection was performed using the SuperSignal kit (Thermo) and quantified using a ChemiDoc MP imaging system with Image Lab software (Bio-Rad).
Statistics
Data were assessed as to whether they were normally distributed. For experiments with a small sample size (n = 3 or 4), normality was tested using the Shapiro–Wilk test. Depending on the distribution, data were analyzed by unpaired two-tailed t tests or Dunnett's multiple comparison tests, or using the Mann–Whitney test (for data not having a normal distribution). The tests used are indicated in the corresponding figure legend. Prism 7 (v7.0c) was used for all statistical analyses. Statistical significance was set to p < 0.05. Significance is marked with asterisks in figures.
Results
Ahi1 is localized to the ciliary transition zone in neurons
Our previous work has shown that Ahi1 is highly expressed in the ventral telencephalon, ventral diencephalon (hypothalamus), midbrain, and hindbrain. However, Ahi1 immunohistochemistry in other brain regions, such as the cortex, hippocampus, and cerebellum, revealed either low or no detectable Ahi1 protein levels in these areas (Doering et al., 2008), despite the presence of detectable levels of Ahi1 mRNA (Ferland et al., 2004; Doering et al., 2008). This suggested the possibility that Ahi1 was localized in very discrete subcellular regions in cortical, hippocampal, and cerebellar neurons (neurons with low Ahi1 expression). To assess this, we performed immunolabeling on cultured neurons to provide higher sensitivity and resolution of the subcellular distribution of Ahi1. Neuronal cells from cerebral cortex, hippocampus, and cerebellum were isolated from wild-type mouse embryos at day 18.5 gestation and were cultured in vitro for at least one week. Ahi1 immunolabeling in these cultured neuronal populations was much less than that observed in cultured hypothalamic neurons (cells with high Ahi1 expression; Doering et al., 2008). However, distinct punctate Ahi1 immunolabeling was observed in most neurons (Fig. 1A,B). Since the localization of Ahi1 at the base of primary cilia has been demonstrated previously in non-CNS cells (Eley et al., 2008; Hsiao et al., 2009; Lee et al., 2014), it prompted us to determine whether this neuronal punctate Ahi1 labeling was at the ciliary base. Use of the ciliary markers, AC3 and ADP ribosylation factor like GTPase 13b (Arl13b), confirmed Ahi1 localization at the base of AC3-positive and Arl13b-positive primary cilia in cultured neurons (Fig. 1A,B), with this Ahi1 immunolabeling being absent in Ahi1–/– knock-out neurons demonstrating our Ahi1 antibody specificity (Fig. 1B, lower panel). Co-immunolabeling of Ahi1 with γ-tubulin (a centrosome marker) in neurons confirmed Ahi1 localization in close proximity with the centrosome, a domain recognized as the ciliary transition zone (Lee et al., 2014) in neuronal primary cilia (Fig. 1C). The low expression of Ahi1 protein in the cerebral cortex, hippocampus, and cerebellum (Doering et al., 2008) was further confirmed by co-immunoprecipitation of Ahi1 (Fig. 1D, right, IP lanes), and not by whole lysate western blotting (Fig. 1D, left, input lanes), indicating a low expression level for Ahi1 protein in these particular neuronal populations. Overall, these results indicate that Ahi1 protein expression is very low in some regions of the brain, but with highly localized levels of Ahi1 occurring at the ciliary transition zone. These results are in stark comparison to the high expression levels of Ahi1 observed throughout the cell body (and ciliary base) in ventral forebrain and hindbrain regions (Doering et al., 2008).
Figure 1.
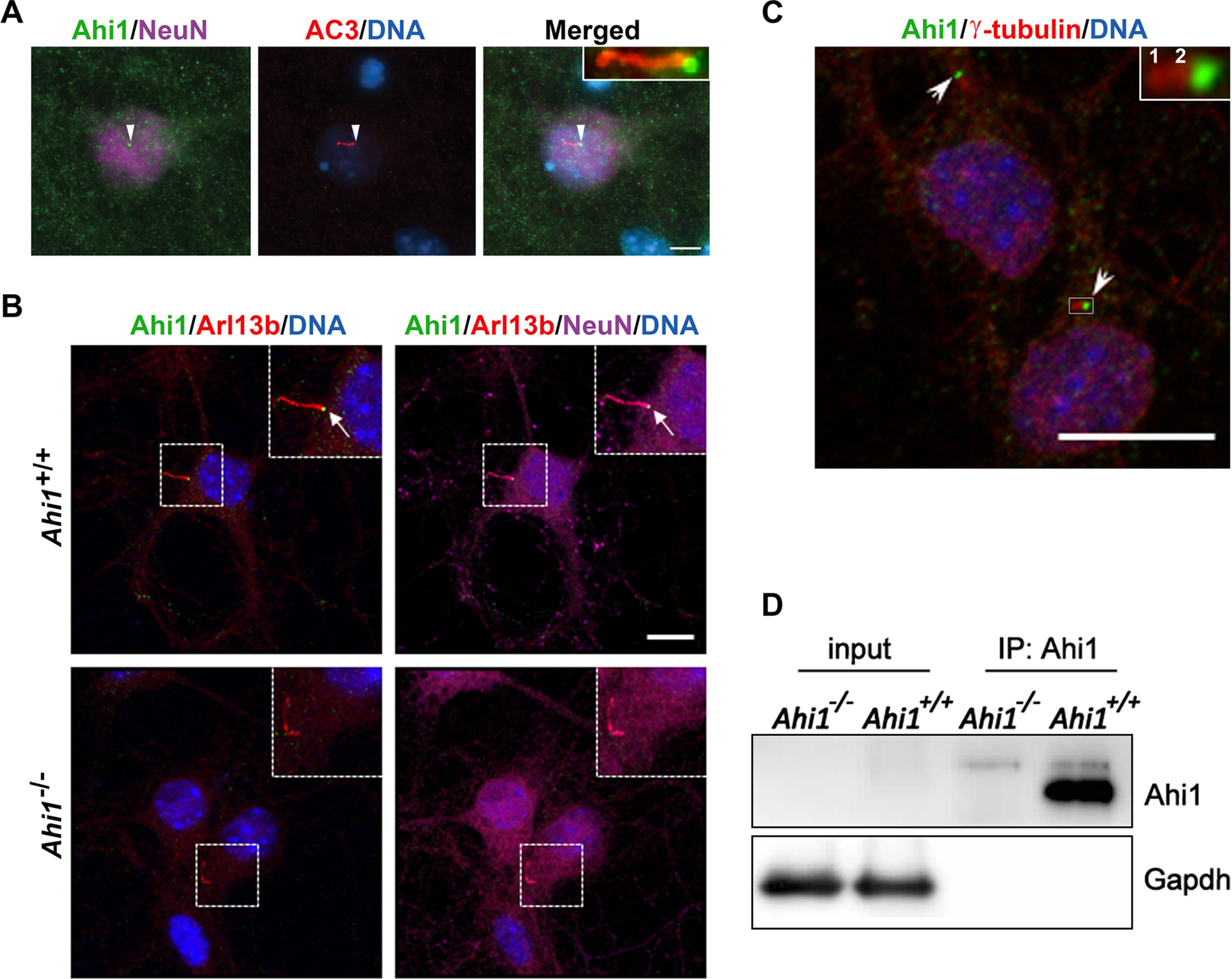
Ahi1 localizes at the transition zone of neuronal primary cilia. Subcellular distribution of Ahi1 was evaluated in primary hippocampal neuron cultures by immunofluorescence. A, Neurons isolated from Ahi1+/+ mouse embryos were co-labeled for Ahi1 (green) and the ciliary marker, AC3 (red). B, Ahi1+/+ and Ahi1–/– hippocampal cells were co-labeled with Ahi1 (green) and the ciliary marker, ADP ribosylation factor like GTPase 13B (Arl13b, red). In A, B, neurons were identified by the neuron-specific nuclear protein, NeuN (magenta), and DNA was visualized with Hoechst 33258 (blue). White arrowheads (A) and white arrows (B) point to Ahi1 localization at the base of the cilium at the transition zone. Scale bars: 5 µm (A) and 10 µm (B). Note in B the lack of Ahi1 immunoreactivity at the base of cilia in Ahi1–/– neurons. C, Co-immunolabeling of Ahi1 (green) and the centrosome marker γ-tubulin (red labels 1 and 2, which are the centrioles). DNA was visualized with Hoechst 33258 (blue). White arrows indicate Ahi1 localization in close proximity with the two centrioles, a domain recognized as the ciliary transition zone. Scale bar: 10 µm. In A–C, insets show magnified images of the boxed region. D, Co-immunoprecipitation of Ahi1 from hippocampal culture lysates (a neuronal population with low Ahi1 expression) from Ahi1+/+ and Ahi1–/– mice. Ahi1 was not able to be detected in the input control but was detected in immunoprecipitated samples, demonstrating reduced and discrete ciliary expression in hippocampal neurons (note the absence of Ahi1 signal in the Ahi1–/– lane). Gapdh was used as a loading control.
Loss of Ahi1 results in defective primary cilia in neurons
Given that Ahi1 is expressed in neurons, including neurons with very low levels of Ahi1 protein expression, and that it is localized at the ciliary transition zone, we sought to determine whether Ahi1 loss could affect primary cilia in brain. We observed a reduction of Arl13b-positive primary cilia immunolabeling in brain sections from the cerebral cortex, hippocampus, and cerebellum in Ahi1–/– mice as compared with littermate controls (Fig. 2A). Western blot analysis showed no significant differences in Arl13b protein levels in Ahi1–/– mouse brain compared with wild-type animals (Fig. 2B,C), indicating defective trafficking of ciliary Arl13b in the absence of Ahi1. In support of this finding, previous studies in several cell lines have demonstrated that Ahi1 promotes ciliary trafficking of Arl13b (Lee et al., 2014; Muñoz-Estrada and Ferland, 2019).
Figure 2.

Ahi1 promotes ciliary trafficking of Arl13b in adult brain tissue. A, Arl13b immunostaining in brain sections from Ahi1+/+ and Ahi1–/– mice. Arrows indicate some Arl13b-positive cilia in brain sections. Reductions in Arl13b ciliary labeling were noted in cerebral cortex, hippocampus, and cerebellum of Ahi1–/– mice. Scale bar: 20 μm. Roman numerals denote the cerebral cortical layer shown. DG, dentate gyrus; H, hilus; ML, molecular layer; P, Purkinje cell layer; GL, granular layer. B, Western blot analysis of brain tissue lysates from Ahi1+/+ and Ahi1–/– mice probed with Arl13b and actin antibodies. C, Quantification of Arl13b levels normalized to Ahi1+/+ values using actin as a loading control, n = 3/genotype. Arl13b levels were analyzed in high Ahi1 expressing brain regions (brainstem and diencephalon) and low Ahi1 expressing brain regions (telencephalon). Error bars represent SEM. Significance was determined by unpaired two-tailed t tests; ns, not significant.
Because of difficulties in quantifying these small organelles in brain sections, we counted cilia in neuronal cultures from Ahi1+/+ and Ahi1–/– mice in three brain regions: hypothalamus (region of highest Ahi1 expression), hippocampus, and cerebral cortex. Significant differences in primary cilia were noted in these three brain regions with Ahi1–/– neurons having fewer AC3-positive cilia as well (Fig. 3A,B). Interestingly, Ahi1–/– hypothalamic and hippocampal neurons had significantly longer cilia as compared with Ahi1+/+ neurons (Fig. 3A,C,D). Similar observations have also been found in mouse embryonic fibroblasts (MEFs) from Ahi1–/– embryos (Muñoz-Estrada and Ferland, 2019). Overall, these results suggest that loss of Ahi1 results in a decreased recruitment of ciliary markers as well as defects in primary cilia length in neurons. Also, although the hippocampus and cerebral cortex have very low expression of Ahi1 protein, the deletion of Ahi1 still has a significant effect in reducing the number of AC3-positive cilia observed on these neurons (similar to Arl13b-positive cilia).
Figure 3.
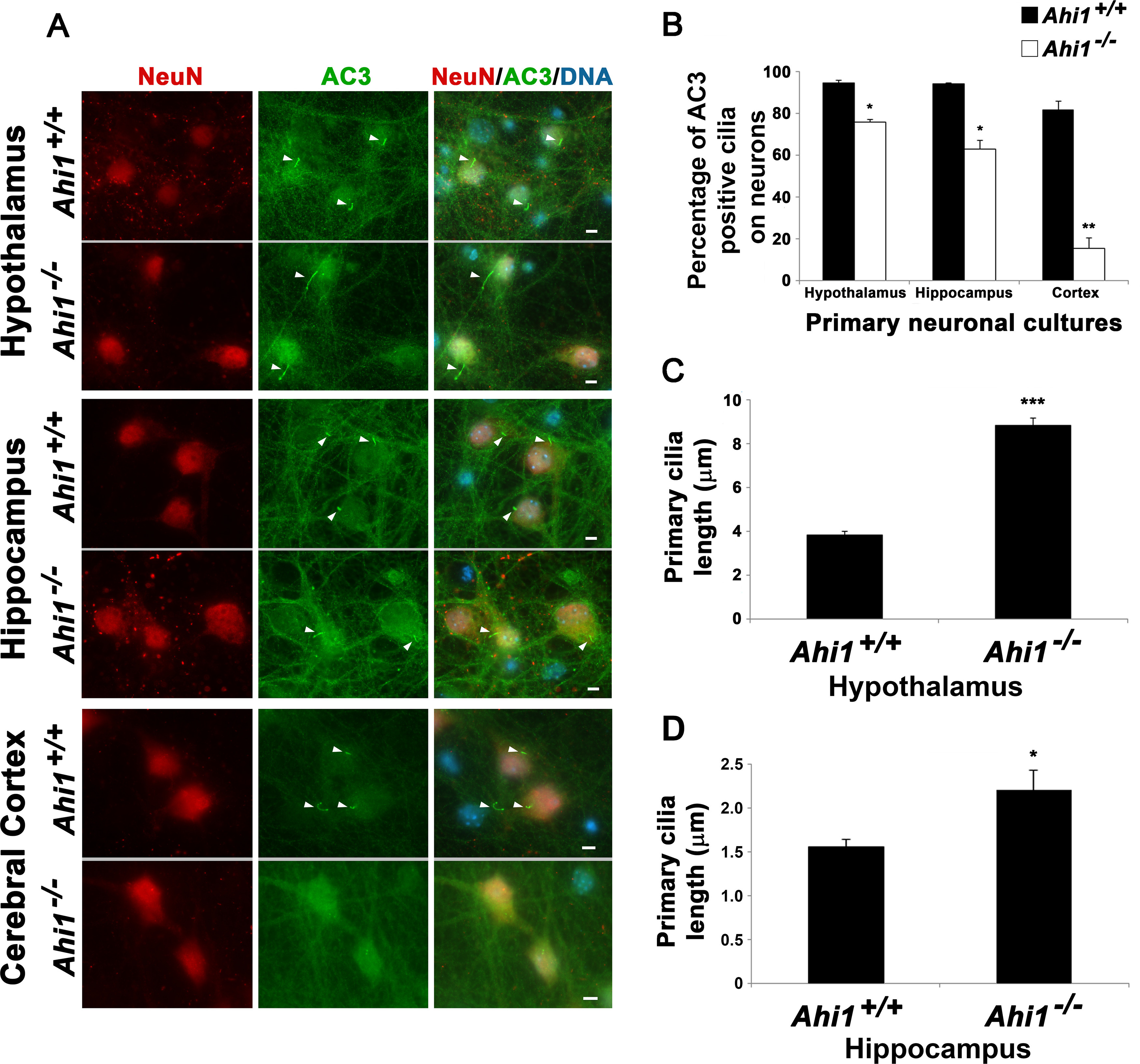
Loss of Ahi1 results in reduced AC3-positive cilia number and cilia elongation in neurons. A, Primary cilia on Ahi1+/+ and Ahi1–/– neurons were identified by performing co-immunolabeling of AC3 (ciliary marker) and NeuN (neuronal marker). DNA was visualized with Hoechst 33258 (blue). Analysis was performed in neurons isolated from three different brain areas: hypothalamus, hippocampus, and cerebral cortex. White arrowheads point to primary cilia. Scale bar: 5 µm. B, Percentage of neurons with AC3-positive cilia in the three different brain areas; n ≥ 100 neuronal cells from each embryo (n = 3 per tissue/genotype). Error bars represent SEM. Significance was determined by χ2 tests (*p < 0.05, **p < 0.005). C, D, Cilia length analysis from Ahi1+/+ and Ahi1–/– hypothalamic (C) and hippocampal (D) neurons. Primary cilia lengths were measured using AxioVison software, n ≥ 100 (C) and n ≥ 20 (D) neuronal cells/group. Error bars represent SEM. Significance was determined by unpaired two-tailed t tests (*p < 0.03, ***p < 0.0005).
Ciliary localization of the GPCR, MchR1, is reduced in Ahi1–/– neurons
Although AC3-positive and Arl13b-positive cilia were decreased in Ahi1–/– neurons, immunoreactivity of these cilia markers could still be found on Ahi1-null neurons. We therefore sought to determine whether these primary cilia on Ahi1–/– neurons have normal ciliary function through assessing the localization and function of the GPCR, MchR1 (AC3 and Arl13b are not GPCRs). Given that MchR1 is localized at primary cilia (Berbari et al., 2008b), we examined whether the ciliary distribution of MchR1 is altered in Ahi1–/– neurons by performing MchR1 immunolabeling on both cultured hypothalamic and hippocampal neurons (both brain regions with high MchR1 expression and activity; Saito et al., 1999; Berbari et al., 2008b; Domire and Mykytyn, 2009; Chee et al., 2013; Engle et al., 2018). Primary cilia, identified by Arl13b immunoreactivity, were present on ∼90% and ∼80% of wild-type cultured hypothalamic and hippocampal neurons, respectively (Fig. 4A–D). Of these wild-type neurons, ∼70% of the hypothalamic and ∼55% of the hippocampal neurons had MchR1 localized to the primary cilium (Fig. 4A–D). However, a significant loss of ciliary localization of MchR1 was observed in Ahi1–/– neurons with ∼20% of hypothalamic knock-out neurons and ∼10% of hippocampal knockout neurons having ciliary MchR1 localization (Fig. 4A–D). These data suggest that deletion of Ahi1 in neurons, not only reduces the localization of Arl13b and AC3 at cilia, but also significantly disrupts the localization of MchR1 to the primary cilium. Lastly, we have shown that disruption of Ahi1 at the ciliary transition zone in fibroblasts causes defects in ciliary protein composition, despite increases in cilia length (Muñoz-Estrada and Ferland, 2019). Similarly, we observed significant decreases in MchR1 immunolabeling in primary cilia from Ahi1–/– neurons although increased cilia lengths were observed (Figs. 3C,D, 4E). This further illustrates that there is significant loss of MchR1 localization at primary cilia in Ahi1–/– neurons, although elongated cilia are observed with loss of Ahi1, supporting the potential for decreased ciliary MchR1 signaling in Ahi1–/– neurons.
Figure 4.
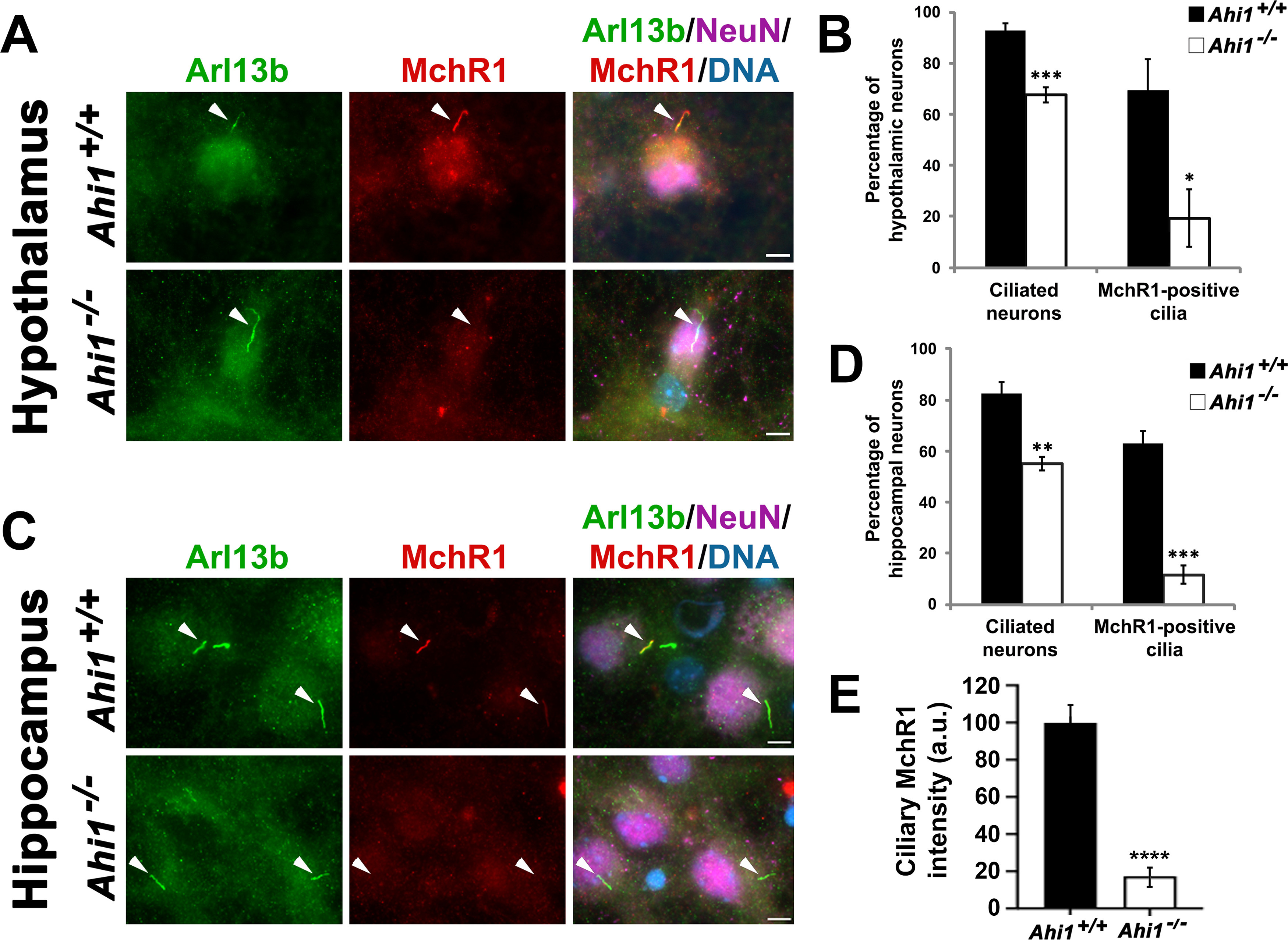
Lack of Ahi1 reduces ciliary trafficking of MchR1. Subcellular distribution of the MchR1 in hypothalamic (A) and hippocampal (C) neuronal cultures isolated from Ahi1+/+ and Ahi1–/– mice. Cells were co-stained with MchR1 (red) and NeuN (magenta), and primary cilia were immunolabeled by Arl13b (green). DNA was visualized with Hoechst 33258 (blue). White arrowheads indicate primary cilia presence. Co-localization of Arl13b and MchR1 labeling was observed in Ahi1+/+ neurons. However, most Ahi1–/– neurons lacked MchR1 immunoreactivity at primary cilia that were Arl13b-positive. Scale bar: 5 μm. Percentage of hypothalamic (B) and hippocampal (D) neurons with Arl13b-positive and MchR1-positive cilia from Ahi1+/+ and Ahi1–/– cultures; n > 100 NeuN positive cells/genotype (n ≥ 4 per tissue/genotype). E, Graph showing MchR1 integrated intensities at primary cilia (which were longer in Ahi1–/– neurons) normalized to Ahi1+/+ values in both Ahi1+/+ and Ahi1–/– neurons (n > 12 neural cells, across 4 litters). Error bars represent SEM. Significance was determined by unpaired two-tailed t tests (*p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.00005).
Total levels of MchR1 and trafficking of MchR1 to the plasma cell membrane are not affected in Ahi1–/– neurons
Loss of MchR1 localization at primary cilia in Ahi1–/– neurons could be the result of decreased protein expression or impaired global protein trafficking. We first evaluated the total protein levels of MchR1 from neuronal cultures and mouse brain tissues by western blot analyses. No significant differences were found in total levels of MchR1 in Ahi1–/– hippocampal or hypothalamic neurons as compared with Ahi1+/+ neurons (Fig. 5A–C). We also examined areas of the brain with either low Ahi1 expression (cortex, hippocampus, thalamus, and cerebellum) or high Ahi1 expression (ventral forebrain, midbrain, and brainstem) and found that there were no differences in MchR1 expression between Ahi1–/– or wild-type lysates (Fig. 5D–F). These results, in combination with no observed Arl13b protein reductions in Ahi1–/– neurons (Fig. 2C), suggest that reduction of MchR1 at primary cilia in Ahi1–/– neurons is not caused by decreased protein expression. In support, we previously reported that depletion of Ahi1 in MEFs does not alter protein expression of the ciliary proteins, Arl13b and IFT88, but did alter their localization to cilia instead. Furthermore, ectopic overexpression of the neuronal GPCR serotonin 6 receptor (Htr6) tagged with GFP showed a significant reduction in the fluorescent signal in Ahi1–/– MEFs compared with wild-type controls (Muñoz-Estrada and Ferland, 2019).
Figure 5.

Total and surface levels of MchR1 are intact in the absence of Ahi1. Western blot analysis of the MchR1 in primary neuronal cultures (A, B) and brain tissues (D, E) obtained from Ahi1+/+ and Ahi1–/– mice. Representative blots for total levels of MchR1 from hippocampal cultures (A), hypothalamic cultures (B), low Ahi1 expressing brain regions (cortex, hippocampus, striatum; D), and high Ahi1 expressing brain regions (midbrain and hindbrain; E). Quantification of total MchR1 levels in Ahi1+/+ and Ahi1–/– neuronal cultures (C) and brain lysates (F). MchR1 fold change was normalized to Ahi1+/+ values (=1; β-III tubulin levels), n ≥ 3 per tissue per genotype. Error bars represent SEM. Significance was determined by unpaired two-tailed t tests. G, Representative western blotting of MchR1 after cell-surface biotinylation assays and quantification of MchR1 surface levels normalized to Ahi1+/+ values (=1; H). Gapdh was used as an intracellular protein control as well as a loading control, n ≥ 3 per tissue/genotype. Error bars represent SEM. Significance was determined by unpaired two-tailed t tests; ns, not significant.
Although several receptor proteins have been identified at the primary cilium, mechanisms of transporting these receptors to the ciliary membrane are still unclear. A body of evidence indicates that ciliary receptors are transported to the cell surface first and then are targeted to the ciliary membrane by either diffusion or intraflagellar transport (IFT; Jin et al., 2010; Nachury et al., 2010). Therefore, defective protein trafficking to the cell surface could also result in a reduction of ciliary MchR1 in Ahi1–/– neurons. To test this possibility, surface proteins on neuronal cultures were isolated through biotinylation assays and evaluated by western blot analysis. These experiments showed that surface MchR1 expression in Ahi1–/– neurons was similar to control cultures (Fig. 5G,H). We also evaluated whether any accumulation of MchR1 occurred that could have resulted in a reduction of ciliary MchR1 in Ahi1–/– neurons, since defective GPCR trafficking could cause receptor accumulation in the endoplasmic reticulum (ER) or the Golgi (Fan et al., 2005; Dong et al., 2007). However, we did not observe any abnormal MchR1 distribution or accumulation in Ahi1–/– neurons (Fig. 4A,C). Taken together, the pathways for trafficking MchR1 to the cell surface from the ER or Golgi are not affected in Ahi1–/– neurons indicating that a loss of ciliary MchR1 in Ahi1-deficient cells is not a result of incorrect surface protein targeting. Given a normal expression and surface targeting of MchR1 in Ahi1–/– neurons, and a lack of MchR1 localization at Ahi1–/– neuronal primary cilia, this indicates a role of Ahi1 in MchR1 targeting to the primary cilium.
Ahi1 deletion inhibits MchR1-mediated FSK-induced cAMP production and downregulates Erk pathway activation on MCH administration
Loss of ciliary MchR1 in Ahi1–/– neurons could be the result of disrupted trafficking in or to primary cilia, since total and surface levels of MchR1 were normal in Ahi1–/– neurons. Therefore, we first determined whether ciliary MchR1 in Ahi1–/– neurons can respond to MCH stimulation. The use of Ahi1–/– neurons to test ciliary function for MchR1 signaling is ideal (as compared with other genetic models such as MchR1 knock-outs), since Ahi1–/– neurons have normal levels of endogenous MchR1 expression and normal levels of MchR1 surface localization (at the cell membrane; Fig. 5) but are simply lacking ciliary MchR1 (Fig. 4). Wild-type hypothalamic neurons treated with 1 μm FSK had significant increases in the production of cAMP, whereas cAMP production was significantly inhibited in these cells when exposed to both FSK and MCH (Fig. 6A). These data confirm the effect of MCH on the inhibition of FSK-induced cAMP production is also present in primary neuronal cells. Next, the same assays were also performed on Ahi1–/– hypothalamic neurons that lack ciliary MchR1. Interestingly, Ahi1–/– neurons showed no significant reduction in cAMP production when treated with FSK and MCH as compared with the FSK alone treatment (Fig. 6A). These effects were not because of an inability to produce cAMP, since Ahi1–/– neurons could be induced by FSK to similar levels as compared with wild-type cells (Fig. 6A). Therefore, these data indicate that the loss of MCH-induced decreases in cAMP production in Ahi1–/– neurons is because of the loss of MchR1s at primary cilia.
Figure 6.

Ahi1 depletion blocks inhibition of FSK-induced cAMP production and downregulates Erk pathway activation mediated by the binding of MCH to MchR1. A, Ahi1+/+ and Ahi1–/– hypothalamic neurons were stimulated with either FSK or FSK and MCH (FSK + MCH) for 15 min, and cAMP levels from each treatment were evaluated by cAMP assays. Each treatment was performed in triplicate, and the levels of cAMP obtained from different treatments were compared with the average cAMP levels from FSK-treated samples; n = 8/genotype. Error bars represent SEM. Significance was determined by Dunnett's multiple comparison test (*p < 0.05). B, Immunoblot analysis of Ahi1+/+ and Ahi1–/– hippocampal cultures treated with either vehicle or 100 nm MCH for 3 min. After the treatment, cells lysates were probed for pErk1/2, Erk1/2, and tubulin. C, Quantification of pErk1/2 in Ahi1+/+ and Ahi1–/– cultures. Graph displays pErk1/2-to-Erk1/2 band ratios normalized to Ahi1+/+ values of non-stimulated cells (set to =1). Tubulin was used as a loading control, n = 3/genotype. Error bars represent SEM. Significance was determined by unpaired two-tailed t tests (*p < 0.05). D, Ahi1 localization is unaltered at the transition zone of primary cilia after MCH treatment. Co-immunolabeling for Ahi1 (green) and AC3 (red) were performed on Ahi1+/+ neurons in: control conditions (no MCH added), stimulated with 100 nm MCH for 10 min (MCH 10'), or treated with 100 nm MCH for 10 min followed by a 30-min recovery in normal culture medium without MCH (MCH 10', recovery 30'). White arrows point to Ahi1 localization at the base of AC3-positive cilia. Of note, in the three experimental conditions, Ahi1 is present at the ciliary base. Scale bar: 5 μm.
To assess an additional downstream MchR1-mediated pathway in neurons lacking Mchr1 ciliary localization, we examined the activation of the Erk pathway on MCH stimulation in neurons. Following addition of MCH (100 nm) or vehicle to Ahi1+/+ and Ahi1–/– hippocampal neuronal cultures for 3 min, lysates were probed with antibodies to pErk1/2, Erk1/2 and tubulin. As expected, the activation of the Erk pathway was observed in wild-type neurons exposed to MCH as determined by an increase in pErk/Erk levels (Fig. 6B,C). However, no significant increase in Erk activation (no change in pErk/Erk levels) were observed in Ahi1–/– neurons treated with MCH (Fig. 6B,C). Overall, given that Ahi1–/– neurons have normal MchR1 localization at the neuronal membrane, but not at the ciliary membrane, and the failure to observe changes in two downstream MchR1-mediated signaling pathways, this demonstrates the importance of MchR1 at the primary cilium for proper neuronal signal transduction.
Previous studies have reported that Ahi1 exhibits little to no turnover at the ciliary transition zone, suggesting that Ahi1, in conjunction with other proteins in the ciliary domain, defines a stable gating structure at the base of cilia (Takao et al., 2017). Therefore, wild-type hippocampal neurons, treated with MCH, were evaluated by immunolabeling for changes in Ahi1 subcellular distribution. Hippocampal neurons were tested here, instead of hypothalamic cultures which have high Ahi1 expression, because the diffuse cytoplasmic labeling of Ahi1 in hypothalamic neurons makes it difficult to examine the subcellular distribution of Ahi1. Ahi1 was localized at the base of cilia in cells with primary cilia during MCH treatment (Fig. 6D). Moreover, that Ahi1 localization does not change with MCH stimulation of MchR1 in neurons, also demonstrates that MCH ligand binding to MchR1 does not remove Ahi1 from the ciliary transition zone. Therefore, deletion of Ahi1, removing Ahi1 from the ciliary transition zone, thereby allows for decreased trafficking of MchR1 to cilia and a consequent decrease in ciliary MchR1 signaling. Consistent with previous findings, our results also indicate that Ahi1, at the transition zone, is a stable component that facilitates ciliary transport of transmembrane proteins. Overall, these results further support the concept that primary cilia serve as focused regions on the neuron/cell for rapid and potent signal amplification. Lastly, our results demonstrate the importance of Ahi1 in mediating the proper localization of MchR1 at primary cilia.
Discussion
Previously, we have shown that the transition zone localizing protein, Ahi1, participates in ciliary function based on the following findings: (1) defective rhodopsin trafficking through the connecting cilium of Ahi1–/– mouse photoreceptors (Westfall et al., 2010); (2) loss of Rab8a at the basal body in Ahi1 shRNAi knock-down kidney cell lines (Hsiao et al., 2009); (3) reduction of ARL13B ciliary localization in human fibroblasts carrying JBTS-causing mutations in AHI1, and in Ahi1–/– mouse fibroblasts (Tuz et al., 2013; Muñoz-Estrada and Ferland, 2019); and (4) decreased sonic hedgehog signaling in Ahi1–/– mouse fibroblasts (Muñoz-Estrada and Ferland, 2019). Here, we found that defective Arl13b ciliary trafficking is not limited to Ahi1-deficient cell lines but can also be observed in Ahi1–/– mouse brain tissue. Notably, Arl13b is currently considered the most upstream master regulator for factors targeted to cilia (Nozaki et al., 2017; Nachury and Mick, 2019). Consistent with this and our former and current findings, Ahi1–/– primary neuronal cultures showed decreased ciliary localization of MchR1, without noticeable alterations of the receptor on the cell body surface. Moreover, Ahi1–/– ciliated neurons with less or no MchR1 localization also display abnormally elongated axonemes. Abnormalities in ciliary length in the absence of transition zone proteins have been associated with ciliary signaling defects important for development of the CNS (Dowdle et al., 2011; Garcia-Gonzalo et al., 2011; Muñoz-Estrada and Ferland, 2019). Abnormally elongated cilia in the absence of Ahi1 has been associated with an increased concentration of ciliary IFT88 (Muñoz-Estrada and Ferland, 2019). IFT88 belongs to the IFT-B protein complex, which serves as a tubulin module that transports tubulin within cilia (Bhogaraju et al., 2013). We also report that Ahi1–/– neurons present a reduction in their ability to respond to MCH in inhibiting cAMP production and activating Erk signaling. Since the MCH/MchR1 system regulates feeding behavior and processes such as anxiety and depression (Antal-Zimanyi and Khawaja, 2009), our findings implicate a novel involvement of Ahi1 in neuromodulatory functions mediated by primary cilia.
Two main pathways have been described for sorting and targeting receptors to the primary cilium, the vesicular trafficking and lateral membrane transport pathway (Nachury et al., 2010). In the former, membrane proteins reach the base of the cilium through vesicles from the Golgi apparatus or recycling endosomes (Hoffmeister et al., 2011; Monis et al., 2017). The latter involves first the delivery of receptors to the plasma membrane and then these are targeted to ciliary membranes by crossing the transition zone (Garcia-Gonzalo and Reiter, 2012). Of note, in the vesicular trafficking pathway, cells capable of forming cilia under non-ciliating conditions confined receptors at the ER, but after cilia induction, receptors were not trafficked to the plasma membrane (Geng et al., 2006). Here, we show that Ahi1 at the transition zone promotes ciliary trafficking of MchR1 without affecting cell trafficking of the receptor to the plasma membrane. In addition, MchR1 subcellular distribution does not indicate accumulation of this GPCR in the Golgi/ER or intracellular vesicles in neurons lacking Ahi1. Therefore, our findings strongly suggest a role of Ahi1 in mediating lateral membrane transport of MchR1. This scenario has been also proposed for another conventional GPCR localized to cilia, the D1-type dopamine receptor (D1R; Leaf and Von Zastrow, 2015). The dynamic importing of receptors to cilia includes not only a functional transition zone, but also involves numerous other proteins, such as small GTPases and members of the IFT complex A and B families (Omori et al., 2008; Mukhopadhyay et al., 2010; Leaf and Von Zastrow, 2015; Madugula and Lu, 2016). Thus, the reduction in ciliary MchR1 observed in Ahi1–/– neurons may include intricate molecular mechanisms involving Rab8, Arl13b and IFT-B proteins, and alterations in the molecular composition of the transition zone (Hsiao et al., 2009; Tuz et al., 2013; Muñoz-Estrada and Ferland, 2019).
In addition to MchR1, other GPCRs are concentrated in neuronal cilia, including the somatostatin receptor 3 (Sstr3) and Htr6, with these GPCRs all having a common ciliary targeting sequence (CTS) in their third intracellular loop. This suggests similar mechanisms of ciliary trafficking for these GPCRs (Berbari et al., 2008b). Previous observations indicated that Ahi1 also facilitates Htr6 and Sstr3 ciliary trafficking (Takao et al., 2017; Muñoz-Estrada and Ferland, 2019). This links Ahi1 in trafficking of several rhodopsin-like GPCRs to the ciliary membrane and highlights the importance of the transition zone in different physiological and pathologic processes, including JBTS. In fact, mutations in AHI1 have also been associated with neuropsychiatric disorders, such as schizophrenia (Amann-Zalcenstein et al., 2006; Alvarez Retuerto et al., 2008; Porcelli et al., 2015). In addition to the relevance of AHI1 in the context of primary cilia, additional studies are necessary to better understand the contribution of extraciliary AHI1 in physiology and disease (Doering et al., 2008; Hua and Ferland, 2018a,b).
Neuronal GPCR signaling pathways have been mainly studied by using heterologous expression in non-neuronal cells. A previous study in kidney collecting duct-derived cells (IMCD3) overexpressing Flag-D1Rs showed that the D1R-mediated cAMP response on agonist addition does not require the primary cilium (Marley et al., 2013). Here, studying neurons from Ahi1–/– mice, which (1) endogenously express MchR1; (2) present a reduction in MchR1-positive cilia; and (3) express and localize similar levels of MchR1s at the plasma membrane, we found abnormalities in cAMP and Erk signaling pathways elicited by MCH. In contrast to the previous observations made for the D1R in cells that do not endogenously express this receptor (Marley et al., 2013), our results indicate that cilia are required for efficiently activating MchR1 intracellular pathways in neurons. Further studies are required to dichotomize extraciliary and ciliary membrane involvement of D1R signaling using a D1R native cell type to rule out potential similarities with MchR1 signaling.
In humans, MCHR1 missense mutations that failed to respond to MCH ligand have been associated with underweight individuals (Goldstein et al., 2010). In addition, Ahi1–/– mice have been reported to be smaller and thinner than their wild-type littermates (Bourgeois and Ferland, 2019). Given this phenotype of Ahi1–/– mice and an enriched expression of Ahi1 in the hypothalamus, this implicates a possible role for Ahi1 in energy homeostasis. Studies on Ahi1 expression in rats suggest a possible function for Ahi1 in regulating feeding behavior (Han et al., 2009; Niu et al., 2011). Ahi1 expression is increased in fasting rats; whereas Ahi1 levels are downregulated once rats are re-fed (Han et al., 2009). Here, we showed that Ahi1–/– mice have reduced MchR1 expression at primary cilia. Mice with both Ahi1 and MchR1 deficiency display the similar phenotype of lower body weight, implicating that Ahi1 and MchR1 work in a similar pathway for regulation of energy homeostasis. MCH and its receptor MchR1 are key regulators of feeding and energy balance (Naufahu et al., 2013). MCH is expressed increasingly in food-deprived animals (Qu et al., 1996). Moreover, MCH-deficient mice present reduced body weight, are lean and have higher energy consumption than control mice. Conversely, overexpression of MCH in transgenic mice leads to obesity. Similarly, MchR1 antagonist administration reduced the body weight in mice (Shimada et al., 1998; Ludwig et al., 2001; Borowsky et al., 2002; Marsh et al., 2002). These observations and the present results support a role of an Ahi1/MchR1 pathway, likely orchestrated in primary cilia, being involved in energy homeostasis that when inhibited, results in a lean phenotype.
Association of cilia with energy homeostasis control has been observed in several ciliopathy animal models (Mykytyn et al., 2004; Davenport et al., 2007; Seo et al., 2009; Mukhopadhyay et al., 2010). Bbs null mice recapitulate the obesity phenotype observed in human BBS (Mykytyn et al., 2004), although MchR1 fails to localize to cilia in neurons of Bbs2−/− and Bbs4−/− mice (Berbari et al., 2008b), MchR1 surface levels could be increased in these neurons explaining the obese phenotype. Alternatively, this phenotype could be because of an increase in adipogenesis as inhibition of Bbs10 and Bbs12 results in a reduction in ciliogenesis and activation of adipogenic pathways (Marion et al., 2011). Therefore, the BBS obesity phenotype may have a central nervous system origin and a peripheral origin. Interestingly, two extremely opposite phenotypes in body weight regulation are observed among mice with defective cilia, implicating that two (or more) independent pathways are involved in energy homeostasis regulation at cilia. Although neurons in the hypothalamus are master regulators for controlling feeding behavior and body metabolism, cells in peripheral tissues (pancreatic islets and adipocytes) are also crucial in the network for controlling energy homeostasis (Schwartz et al., 2000; Gao and Horvath, 2007). The phenotype of body weight displayed in mice with ciliary defects could be a result of impaired signaling combined from both central and peripheral tissues. Taken together, cilia are required for effective signal transduction, and proper trafficking of receptors to cilia is also essential for signaling. This study provides an insight into the regulatory mechanism for ciliary GPCR trafficking, which could be useful for future therapeutic application on cilium-associated disorders, such as obesity.
Footnotes
This work was supported in part by National Institutes of Health Grants K01MH71801 and R01NS092062 (to R.J.F.) and the March of Dimes Foundation Grant 5-FY09-29 (to R.J.F.).
The authors declare no competing financial interests.
References
- Alvarez Retuerto AI, Cantor RM, Gleeson JG, Ustaszewska A, Schackwitz WS, Pennacchio LA, Geschwind DH (2008) Association of common variants in the Joubert syndrome gene (AHI1) with autism. Hum Mol Genet 17:3887–3896. 10.1093/hmg/ddn291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann-Zalcenstein D, Avidan N, Kanyas K, Ebstein RP, Kohn Y, Hamdan A, Ben-Asher E, Karni O, Mujaheed M, Segman RH, Maier W, Macciardi F, Beckmann JS, Lancet D, Lerer B (2006) AHI1, a pivotal neurodevelopmental gene, and C6orf217 are associated with susceptibility to schizophrenia. Eur J Hum Genet 14:1111–1119. 10.1038/sj.ejhg.5201675 [DOI] [PubMed] [Google Scholar]
- Antal-Zimanyi I, Khawaja X (2009) The role of melanin-concentrating hormone in energy homeostasis and mood disorders. J Mol Neurosci 39:86–98. 10.1007/s12031-009-9207-6 [DOI] [PubMed] [Google Scholar]
- Berbari NF, Johnson AD, Lewis JS, Askwith CC, Mykytyn K (2008a) Identification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Mol Biol Cell 19:1540–1547. 10.1091/mbc.e07-09-0942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K (2008b) Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci USA 105:4242–4246. 10.1073/pnas.0711027105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogaraju S, Cajanek L, Fort C, Blisnick T, Weber K, Taschner M, Mizuno N, Lamla S, Bastin P, Nigg EA, Lorentzen E (2013) Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science 341:1009–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowsky B, Durkin MM, Ogozalek K, Marzabadi MR, DeLeon J, Lagu B, Heurich R, Lichtblau H, Shaposhnik Z, Daniewska I, Blackburn TP, Branchek TA, Gerald C, Vaysse PJ, Forray C (2002) Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nat Med 8:825–830. 10.1038/nm741 [DOI] [PubMed] [Google Scholar]
- Bourgeois JR, Ferland RJ (2019) Loss of the neurodevelopmental Joubert syndrome causing protein, Ahi1, causes motor and muscle development delays independent of central nervous system involvement. Dev Biol 448:36–47. 10.1016/j.ydbio.2019.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, Dytko GM, Foley JJ, Martin J, Liu WS, Park J, Ellis C, Ganguly S, Konchar S, Cluderay J, Leslie R, Wilson S, Sarau HM (1999) Melanin-concentrating hormone is the cognate ligand for the orphan G-protein-coupled receptor SLC-1. Nature 400:261–265. 10.1038/22313 [DOI] [PubMed] [Google Scholar]
- Chee MJ, Pissios P, Maratos-Flier E (2013) Neurochemical characterization of neurons expressing melanin-concentrating hormone receptor 1 in the mouse hypothalamus. J Comp Neurol 521:2208–2234. 10.1002/cne.23273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, Yoder BK (2007) Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol 17:1586–1594. 10.1016/j.cub.2007.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, Gleeson JG (2004) Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet 75:979–987. 10.1086/425985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering JE, Kane K, Hsiao YC, Yao C, Shi B, Slowik AD, Dhagat B, Scott DD, Ault JG, Page-McCaw PS, Ferland RJ (2008) Species differences in the expression of Ahi1, a protein implicated in the neurodevelopmental disorder Joubert syndrome, with preferential accumulation to stigmoid bodies. J Comp Neurol 511:238–256. 10.1002/cne.21824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domire JS, Mykytyn K (2009) Markers for neuronal cilia. Methods Cell Biol 91:111–121. 10.1016/S0091-679X(08)91006-2 [DOI] [PubMed] [Google Scholar]
- Dong C, Filipeanu CM, Duvernay MT, Wu G (2007) Regulation of G protein-coupled receptor export trafficking. Biochim Biophys Acta 1768:853–870. 10.1016/j.bbamem.2006.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdle WE, Robinson JF, Kneist A, Sirerol-Piquer MS, Frints SGM, Corbit KC, Zaghloul NA, Zaghloul NA, van Lijnschoten G, Mulders L, Verver DE, Zerres K, Reed RR, Attié-Bitach T, Johnson CA, García-Verdugo JM, Katsanis N, Bergmann C, Reiter JF (2011) Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am J Hum Genet 89:94–110. 10.1016/j.ajhg.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle AN, Mild G, Schlumberger S, Drozdz R, Hintermann E, Zumsteg U (2004) Expression and characterization of melanin-concentrating hormone receptors on mammalian cell lines. Peptides 25:1585–1595. 10.1016/j.peptides.2004.06.022 [DOI] [PubMed] [Google Scholar]
- Eley L, Gabrielides C, Adams M, Johnson CA, Hildebrandt F, Sayer JA (2008) Jouberin localizes to collecting ducts and interacts with nephrocystin-1. Kidney Int 74:1139–1149. 10.1038/ki.2008.377 [DOI] [PubMed] [Google Scholar]
- Engle SE, Antonellis PJ, Whitehouse LS, Bansal R, Emond MR, Jontes JD, Kesterson RA, Mykytyn K, Berbari NF (2018) A CreER mouse to study melanin concentrating hormone signaling in the developing brain. Genesis 56:e23217. 10.1002/dvg.23217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Perry SJ, Gao Y, Schwarz DA, Maki RA (2005) A point mutation in the human melanin concentrating hormone receptor 1 reveals an important domain for cellular trafficking. Mol Endocrinol 19:2579–2590. 10.1210/me.2004-0301 [DOI] [PubMed] [Google Scholar]
- Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, Shugart YY, Ruvolo M, Walsh CA (2004) Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet 36:1008–1013. 10.1038/ng1419 [DOI] [PubMed] [Google Scholar]
- Gao Q, Horvath TL (2007) Neurobiology of feeding and energy expenditure. Annu Rev Neurosci 30:367–398. 10.1146/annurev.neuro.30.051606.094324 [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Reiter JF (2012) Scoring a backstage pass: mechanisms of ciliogenesis and ciliary access. J Cell Biol 197:697–709. 10.1083/jcb.201111146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, Garcia-Verdugo JM, Katsanis N, Hildebrandt F, Reiter JF (2011) A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet 43:776–784. 10.1038/ng.891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L, Okuhara D, Yu Z, Tian X, Cai Y, Shibazaki S, Somlo S (2006) Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J Cell Sci 119:1383–1395. 10.1242/jcs.02818 [DOI] [PubMed] [Google Scholar]
- Gibson WT, Pissios P, Trombly DJ, Luan J, Keogh J, Wareham NJ, Maratos-Flier E, O'Rahilly S, Farooqi IS (2004) Melanin-concentrating hormone receptor mutations and human obesity: functional analysis. Obes Res 12:743–749. 10.1038/oby.2004.89 [DOI] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11:331–344. 10.1038/nrg2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein C, Schroeder JC, Fortin JP, Goss JM, Schaus SE, Beinborn M, Kopin AS (2010) Two naturally occurring mutations in the type 1 melanin-concentrating hormone receptor abolish agonist-induced signaling. J Pharmacol Exp Ther 335:799–806. 10.1124/jpet.110.174029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin K, Banker G (1989) Experimental observations on the development of polarity by hippocampal neurons in culture. J Cell Biol 108:1507–1516. 10.1083/jcb.108.4.1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SB, Choi BI, Lee D, Kee SH, Kim HS, Sun W, Kim H (2009) Regulation of AHI1 expression in adult rat brain: implication in hypothalamic feeding control. Biochem Biophys Res Commun 390:535–540. 10.1016/j.bbrc.2009.09.133 [DOI] [PubMed] [Google Scholar]
- Hawes BE, Kil E, Green B, O'Neill K, Fried S, Graziano MP (2000) The melanin-concentrating hormone receptor couples to multiple G proteins to activate diverse intracellular signaling pathways. Endocrinology 141:4524–4532. 10.1210/endo.141.12.7833 [DOI] [PubMed] [Google Scholar]
- Hildebrandt F, Benzing T, Katsanis N (2011) Ciliopathies. N Engl J Med 364:1533–1543. 10.1056/NEJMra1010172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmeister H, Babinger K, Gürster S, Cedzich A, Meese C, Schadendorf K, Osten L, de Vries U, Rascle A, Witzgall R (2011) Polycystin-2 takes different routes to the somatic and ciliary plasma membrane. J Cell Biol 192:631–645. 10.1083/jcb.201007050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao YC, Tong ZJ, Westfall JE, Ault JG, Page-McCaw PS, Ferland RJ (2009) Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum Mol Genet 18:3926–3941. 10.1093/hmg/ddp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao YC, Tuz K, Ferland RJ (2012) Trafficking in and to the primary cilium. Cilia 1:4. 10.1186/2046-2530-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua K, Ferland RJ (2018a) Primary cilia reconsidered in the context of ciliopathies: extraciliary and ciliary functions of cilia proteins converge on a polarity theme? Bioessays 40:e1700132. 10.1002/bies.201700132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua K, Ferland RJ (2018b) Primary cilia proteins: ciliary and extraciliary sites and functions. Cell Mol Life Sci 75:1521–1540. 10.1007/s00018-017-2740-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV (2005) Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA 102:11325–11330. 10.1073/pnas.0505328102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV (2003) Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426:83–87. 10.1038/nature02061 [DOI] [PubMed] [Google Scholar]
- Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, Nachury MV (2010) The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141:1208–1219. 10.1016/j.cell.2010.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaf A, Von Zastrow M (2015) Dopamine receptors reveal an essential role of IFT-B, KIF17, and Rab23 in delivering specific receptors to primary cilia. Elife 4:e06996. 10.7554/eLife.06996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YL, Santé J, Comerci CJ, Cyge B, Menezes LF, Li FQ, Germino GG, Moerner WE, Takemaru K, Stearns T (2014) Cby1 promotes Ahi1 recruitment to a ring-shaped domain at the centriole-cilium interface and facilitates proper cilium formation and function. Mol Biol Cell 25:2919–2933. 10.1091/mbc.E14-02-0735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig DS, Tritos NA, Mastaitis JW, Kulkarni R, Kokkotou E, Elmquist J, Lowell B, Flier JS, Maratos-Flier E (2001) Melanin-concentrating hormone overexpression in transgenic mice leads to obesity and insulin resistance. J Clin Invest 107:379–386. 10.1172/JCI10660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macneil DJ (2013) The role of melanin-concentrating hormone and its receptors in energy homeostasis. Front Endocrinol (Lausanne) 4:49. 10.3389/fendo.2013.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madugula V, Lu L (2016) A ternary complex comprising transportin1, Rab8 and the ciliary targeting signal directs proteins to ciliary membranes. J Cell Sci 129:3922–3934. 10.1242/jcs.194019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion V, Schlicht D, Mockel A, Caillard S, Imhoff O, Stoetzel C, van Dijk P, Brandt C, Moulin B, Dollfus H (2011) Bardet-Biedl syndrome highlights the major role of the primary cilium in efficient water reabsorption. Kidney Int 79:1013–1025. 10.1038/ki.2010.538 [DOI] [PubMed] [Google Scholar]
- Marley A, Choy RW, von Zastrow M (2013) GPR88 reveals a discrete function of primary cilia as selective insulators of GPCR cross-talk. PLoS One 8:e70857. 10.1371/journal.pone.0070857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, Guan XM, Jiang MM, Feng Y, Camacho RE, Shen Z, Frazier EG, Yu H, Metzger JM, Kuca SJ, Shearman LP, Gopal-Truter S, MacNeil DJ, Strack AM, MacIntyre DE, et al. (2002) Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci USA 99:3240–3245. 10.1073/pnas.052706899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monis WJ, Faundez V, Pazour GJ (2017) BLOC-1 is required for selective membrane protein trafficking from endosomes to primary cilia. J Cell Biol 216:2131–2150. 10.1083/jcb.201611138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, Jackson PK (2010) TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev 24:2180–2193. 10.1101/gad.1966210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Estrada J, Ferland RJ (2019) Ahi1 promotes Arl13b ciliary recruitment, regulates Arl13b stability and is required for normal cell migration. J Cell Sci 132:jcs230680. 10.1242/jcs.230680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mykytyn K, Askwith C (2017) G-protein-coupled receptor signaling in cilia. Cold Spring Harb Perspect Biol 9:a028183. 10.1101/cshperspect.a028183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mykytyn K, Mullins RF, Andrews M, Chiang AP, Swiderski RE, Yang B, Braun T, Casavant T, Stone EM, Sheffield VC (2004) Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci USA 101:8664–8669. 10.1073/pnas.0402354101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Mick DU (2019) Establishing and regulating the composition of cilia for signal transduction. Nat Rev Mol Cell Biol 20:389–405. 10.1038/s41580-019-0116-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Seeley ES, Jin H (2010) Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol 26:59–87. 10.1146/annurev.cellbio.042308.113337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naufahu J, Cunliffe AD, Murray JF (2013) The roles of melanin-concentrating hormone in energy balance and reproductive function: are they connected? Reproduction 146:R141–R150. 10.1530/REP-12-0385 [DOI] [PubMed] [Google Scholar]
- Niu SN, Huang ZB, Wang H, Rao XR, Kong H, Xu J, Li XJ, Yang C, Sheng GQ (2011) Brainstem Hap1-Ahi1 is involved in insulin-mediated feeding control. FEBS Lett 585:85–91. 10.1016/j.febslet.2010.11.059 [DOI] [PubMed] [Google Scholar]
- Nozaki S, Katoh Y, Terada M, Michisaka S, Funabashi T, Takahashi S, Kontani K, Nakayama K (2017) Regulation of ciliary retrograde protein trafficking by the Joubert syndrome proteins ARL13B and INPP5E. J Cell Sci 130:563–576. 10.1242/jcs.197004 [DOI] [PubMed] [Google Scholar]
- Omori Y, Zhao C, Saras A, Mukhopadhyay S, Kim W, Furukawa T, Sengupta P, Veraksa A, Malicki J (2008) Elipsa is an early determinant of ciliogenesis that links the IFT particle to membrane-associated small GTPase Rab8. Nat Cell Biol 10:437–444. 10.1038/ncb1706 [DOI] [PubMed] [Google Scholar]
- Pal K, Mukhopadhyay S (2015) Primary cilium and sonic hedgehog signaling during neural tube patterning: role of GPCRs and second messengers. Dev Neurobiol 75:337–348. 10.1002/dneu.22193 [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Rosenbaum JL (2002) Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol 12:551–555. 10.1016/s0962-8924(02)02410-8 [DOI] [PubMed] [Google Scholar]
- Pissios P, Trombly DJ, Tzameli I, Maratos-Flier E (2003) Melanin-concentrating hormone receptor 1 activates extracellular signal-regulated kinase and synergizes with G(s)-coupled pathways. Endocrinology 144:3514–3523. 10.1210/en.2002-0004 [DOI] [PubMed] [Google Scholar]
- Porcelli S, Pae CU, Han C, Lee SJ, Patkar AA, Masand PS, Balzarro B, Alberti S, De Ronchi D, Serretti A (2015) The influence of AHI1 variants on the diagnosis and treatment outcome in schizophrenia. Int J Mol Sci 16:2517–2529. 10.3390/ijms16022517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek R, Kanarek R, Maratos-Flier E (1996) A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature 380:243–247. 10.1038/380243a0 [DOI] [PubMed] [Google Scholar]
- Saito Y, Nothacker HP, Wang Z, Lin SH, Leslie F, Civelli O (1999) Molecular characterization of the melanin-concentrating-hormone receptor. Nature 400:265–269. 10.1038/22321 [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404:661–671. 10.1038/35007534 [DOI] [PubMed] [Google Scholar]
- Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC (2009) Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet 18:1323–1331. 10.1093/hmg/ddp031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E (1998) Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature 396:670–674. 10.1038/25341 [DOI] [PubMed] [Google Scholar]
- Shimazaki T, Yoshimizu T, Chaki S (2006) Melanin-concentrating hormone MCH1 receptor antagonists: a potential new approach to the treatment of depression and anxiety disorders. CNS Drugs 20:801–811. 10.2165/00023210-200620100-00002 [DOI] [PubMed] [Google Scholar]
- Takao D, Wang L, Boss A, Verhey KJ (2017) Protein interaction analysis provides a map of the spatial and temporal organization of the ciliary gating zone. Curr Biol 27:2296–2306.e3. 10.1016/j.cub.2017.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuz K, Hsiao YC, Juárez O, Shi B, Harmon EY, Phelps IG, Lennartz MR, Glass IA, Doherty D, Ferland RJ (2013) The Joubert syndrome-associated missense mutation (V443D) in the Abelson-helper integration site 1 (AHI1) protein alters its localization and protein-protein interactions. J Biol Chem 288:13676–13694. 10.1074/jbc.M112.420786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfall JE, Hoyt C, Liu Q, Hsiao YC, Pierce EA, Page-McCaw PS, Ferland RJ (2010) Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. J Neurosci 30:8759–8768. 10.1523/JNEUROSCI.5229-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]