

Abstract
Cancer treatment has been associated with accelerated aging that can lead to early-onset health complications typically experienced by older populations. In particular, cancer survivors have an increased risk of developing premature cardiovascular complications. In the last two decades, cellular senescence has been proposed as an important mechanism of premature cardiovascular diseases. Cancer treatments, specifically anthracyclines and radiation, have been shown to induce senescence in different types of cardiovascular cells. Additionally, clinical studies identified increased systemic markers of senescence in cancer survivors. Preclinical research has demonstrated the potential of several approaches to mitigate cancer therapy-induced senescence. However, strategies to prevent and/or treat therapy-induced cardiovascular senescence have not yet been translated to the clinic. In this review, we will discuss how therapy-induced senescence can contribute to cardiovascular complications. Thereafter, we will summarize the current in vitro, in vivo, and clinical evidence regarding cancer therapy-induced cardiovascular senescence. Then, we will discuss interventional strategies that have the potential to protect against therapy-induced cardiovascular senescence. To conclude, we will highlight challenges and future research directions to mitigate therapy-induced cardiovascular senescence in cancer survivors.
Keywords: Senescence, cancer therapy, cardiovascular diseases, radiation, cardio-oncology, doxorubicin, cardiotoxicity
1. Introduction
There are more than 15 million cancer survivors in the United States and this number is expected to increase due to the continued advance of diagnosis, therapy, and care models (“Study cancer survivors,” 2019). Nearly 85% of cancer survivors have a high risk of developing chronic adverse health conditions, age-related disorders, and frailty, mainly due to cancer and/or cancer treatment (Dowling, et al., 2013). Cancer treatment is associated with accelerated aging and declining body reserves in relatively young cancer survivors, which in turn lead to premature onset of frailty, chronic diseases, and geriatric syndromes (Cupit-Link, et al., 2017). Indeed, cancer survivors appear to be older than their stated age after the completion of chemotherapy by as much as 20 years, with the intensity of treatment correlated with the aging process (Hill, Sadda, LaBarge, & Hurria, 2020). In particular, cancer survivors have an increased risk of developing premature cardiovascular complications. A broad spectrum of anticancer agents has adverse effects on the cardiovascular system, including anthracyclines, trastuzumab, proteasome inhibitors (e.g., carfilzomib), tyrosine kinase inhibitors (e.g., sunitinib), and immune checkpoint inhibitors (e.g., nivolumab and ipilimumab) (Bansal, et al., 2020; Faber, et al., 2018; Foulkes, et al., 2020). Cardio-oncology aims to identify the mechanisms of and mitigate cardiovascular complications in cancer patients, and also to optimize cardiac surveillance and treatment of cardiac complications (Coviello, 2018; Tajiri, Aonuma, & Sekine, 2017).
The exact mechanisms of cancer therapy-induced cardiovascular complications remain incompletely understood. However, multiple potential mechanisms have been proposed including oxidative stress, mitochondrial dysfunction, altered myocardial energy metabolism, apoptotic cell death, inflammation, and recently cellular senescence (A. Ferreira, et al., 2017; Nakamura, et al., 2000; Takemura & Fujiwara, 2007; Ueno, et al., 2006). Cellular senescence, a state of permanent cell-cycle arrest, has been identified in the past two decades as an essential component of cell response to cancer treatment, including chemotherapy and radiation, as reviewed by several authors previously (T. Saleh, et al., 2020; B. Wang, Kohli, & Demaria, 2020; Wyld, et al., 2020). Additionally, accumulating evidence implicates senescent cardiovascular cells in the onset and/or exacerbation of multiple cardiovascular diseases (CVDs) (P. Song, Zhao, & Zou, 2020; Tang, Li, & Chen, 2020; C. M. Wu, Zheng, Wang, & Hu, 2020). The objective of this review is to summarize and critically evaluate the current knowledge about cancer therapy-induced cardiovascular senescence. We will first delineate the role of cardiovascular cellular senescence in CVDs. Thereafter, we will discuss the mechanisms by which cancer treatment, particularly anthracyclines and radiation, induces senescence in cardiovascular cells. Finally, we will discuss potential protective strategies that can mitigate senescence in cardiovascular cells and hence prevent premature cardiovascular complications in cancer survivors.
2. Cellular senescence and cardiovascular diseases (CVDs)
Senescence is a signaling event that occurs in response to a myriad of cellular stressors resulting in irreversible cell cycle arrest, i.e. the cells lose their replicative potential. Senescence was first described in 1961 when Hayflick and his team observed that human fibroblasts cease division after a certain number of passages, which was named thereafter as “Hayflick Limit” (Hayflick & Moorhead, 1961). Later, it became clear that senescent cells accumulate with aging in many tissues of most vertebrates (Dimri, et al., 1995; Terzibasi, Valenzano, & Cellerino, 2007; Yousefzadeh, et al., 2020). Senescent cells also have characteristic changes in structure, morphology, gene expression, and metabolism (van Deursen, 2014). Senescent cells are metabolically active, have a more flattened and irregular shape, and have enlarged nuclei that are sometimes multinucleated (Rattan, 2008). Other features include increased activity of senescence-associated ß-galactosidase (SA-ß-gal), upregulation of cell cycle inhibitors, e.g., p16Ink4a and p21Cip1, and accumulation of DNA damage foci and senescence-associated heterochromatin foci (SAHF) (Campisi & d’Adda di Fagagna, 2007; Dimri, et al., 1995). Another hallmark feature is the expression of the senescence-associated secretory phenotype (SASP) (Coppe, et al., 2008). SASP encompasses multiple soluble and insoluble components, including inflammatory cytokine interleukins [IL-1α, IL1β, IL-6, IL-8, IL-18, CCL-2, tumor necrosis factor-alpha (TNF-α)], chemokines, growth factors, matrix metalloproteinases (MMP-1, -2, -3, -7, -8, -9, -10), serine proteases, and extracellular matrix components. Physiologically, SASP allows senescent cells to interact with the microenvironment to recruit immune cells, macrophages and lymphocytes, to clear senescent cells and restore normal tissue functions (Greten & Eggert, 2017). SASP components can vary depending on the cell type and causes of senescence (van Deursen, 2014). It is also critical to note that there is no single marker of senescence that is specific. Thus, multiple endpoints must be measured to identify senescent cells (Gorgoulis, et al., 2019).
Cellular senescence has an essential role in embryonic development (Muñoz-Espín, et al., 2013) and wound healing (Telgenhoff & Shroot, 2005). Senescence also represents a primary tumor-suppression mechanism in response to oncogenic activation (Campisi, Kim, Lim, & Rubio, 2001), a process known as oncogene-induced senescence (OIS) (X. L. Liu, Ding, & Meng, 2018) and to prevent the replication of a damaged genome leading to mutagenesis and potentially carcinogenesis. Thus, senescence evolved as a protective mechanism necessary of organism health and homeostasis. However, there is now abundant genetic and pharmacologic data making it clear that too many persistent senescent cells disrupt tissue homeostasis and drive aging and age-related disease (Baker, et al., 2011; Marco Demaria, et al., 2014; Xu, et al., 2018). Indeed, senescent cells contribute to pathophysiology of chronic kidney disease (Knoppert, Valentijn, Nguyen, Goldschmeding, & Falke, 2019), type 2 diabetes (Palmer, Gustafson, Kirkland, & Smith, 2019), diabetic nephropathy (Xiong & Zhou, 2019), Alzheimer’s disease (Lyons & Bartolomucci, 2020; Walton, Begelman, Nguyen, & Andersen, 2020), osteoarthritis (Jeon, et al., 2017; Martin & Buckwalter, 2001), osteoporosis (Farr, et al., 2017; Khosla, Farr, & Kirkland, 2018), multiple sclerosis (Papadopoulos, Magliozzi, Mitsikostas, Gorgoulis, & Nicholas, 2020), and chronic lung diseases, e.g. chronic obstructive lung diseases (Barnes, Baker, & Donnelly, 2019) and asthma (Z.-N. Wang, et al., 2020).
Senescence can contribute to the loss of organ function through cell-autonomous events such as impaired intercellular communication, loss of contractility or cell function for example in immune cells (Fafián-Labora & O’Loghlen). However, cell non-autonomous effects of the SASP seems to dominate in disease processes, as clearing senescent cells can improve stem cell function (Chang, et al., 2016) and reverse frank tissue damage (Yousefzadeh, et al., 2018). SASP can induce senescence in neighboring non-senescent cells by paracrine signaling, which is described as a “bystander effect.” The bystander effect occurs both in vitro (Nelson, et al., 2012) and in vivo (Acosta, et al., 2013; da Silva, et al., 2019). Intriguingly, even a small number of senescent cells (10%) is enough to spread senescence in vitro (Pulakat & Chen, 2020) and shorten health and lifespan in vivo (Xu, et al., 2018). Co-culture of late passage senescent fibroblasts with early passage fibroblasts can cause an increase in DNA damage markers in the young bystander cells via gap junction-mediated cell-to-cell communication (Nelson, et al., 2012). Second, SASP promotes chronic low-grade inflammation, known as “inflammaging” (Franceschi, et al., 2000).
Cellular senescence driven by many types of cancer therapy, including chemotherapy and radiation, is called therapy-induced senescence (TIS) (Ewald, Desotelle, Wilding, & Jarrard, 2010; Roninson, 2003). TIS is extensively studied in tumor cells and is a desirable outcome since senescence impedes tumor growth (S. Lee & Lee, 2019; Nardella, Clohessy, Alimonti, & Pandolfi, 2011). However, recent studies show that TIS can provide alternative ways for cancer cells to escape the lethality by entering a transient dormant state, which can later lead to a more aggressive cancer relapse (Tareq Saleh, et al., 2020; Saleh, et al., 2019). Cancer therapy can also induce senescence in healthy non-tumor cells, leading to multiple adverse effects (T. Saleh, et al., 2020). Different types of cells are affected by therapy-induced senescence, including stem cells, bone marrow, and cardiovascular cells (M. Demaria, et al., 2017).
Cardiovascular aging is associated with cell death and structural remodeling, such as fibrosis, stiffness, circulatory impairment, and hypertrophy, which ultimately leads to heart failure (Pulakat & Chen, 2020). These changes occur with chronological and accelerated aging. Notably, cardiovascular senescence is linked to tissue remodeling and the predisposition to many CVDs, including coronary heart diseases (CHD), atrial fibrillation, congestive heart failure, atherosclerosis, and arterial diseases (Gorenne, Kavurma, Scott, & Bennett, 2006; Shimizu & Minamino, 2020; Stojanović, Fiedler, Bauersachs, Thum, & Sedding, 2020; Veronica & Esther, 2012). Recent studies demonstrate a significant accumulation of senescent vascular smooth muscle cells (VSMCs) and endothelial cells (ECs) in the walls of atherosclerotic vessels (Veronica & Esther, 2012). Using transgenic mice, Childs et al. found that in atherosclerosis, senescent foamy macrophages initially accumulate in the sub-endothelial space, then drive atherosclerosis pathology by overexpression of atherogenic and inflammatory cytokines and chemokines (Childs, et al., 2016). Indeed, SASP inflammatory components, such as IL-6 and TNF-α, contribute to the development of CVDs (Rea, et al., 2018). Additionally, senescence-mediated inflammaging boosts the risk of endothelial dysfunction, insulin resistance, and atherosclerosis (Soysal, Arik, Smith, Jackson, & Isik, 2020). Moreover, excessive matrix metalloproteinases (MMPs), a SASP component, in cardiomyocytes can induce sarcoplasmic proteins proteolysis, which eventually impairs cardiac contractile function (Chan, et al., 2020). Cellular senescence may also contribute to thrombosis. Administration of the chemotherapeutic agent doxorubicin (DOX) in p16–3MR transgenic mice induces senescence markers in the liver and the secretion of multiple homeostasis-related factors regulated by SASP, which potentiates blood clotting with shorter bleeding times relative to controls (Wiley, et al., 2019). Interestingly, clearance of senescent cells mitigates the activated clotting induced by DOX (Wiley, et al., 2019).
Since senescence occurs at the cellular level, it is important to discuss how senescence in specific populations of cardiovascular cells can contribute to the pathogenesis of CVDs. By understanding this, we can predict how senescence can contribute to cancer therapy-induced cardiovascular adverse effects.
2.1. Senescent Cardiomyocytes
It is hard to define the senescence of cardiomyocytes as cell cycle arrest because adult cardiomyocytes are generally considered terminally differentiated post-mitotic cells. Instead, senescent cardiomyocytes exhibit other functional changes that are characteristic of senescent cells, including SASP secretion, mitochondrial dysfunction, and DNA damage response (Tang, et al., 2020). Additionally, senescent cardiomyocytes have alterations in the cellular functions that can increase the risk of heart failure, including a decrease in the β-adrenergic response, marked prolonged relaxation, and impaired contractility (Boccardi & Mecocci, 2020). Moreover, the flattened and enlarged morphological senescence changes in cardiomyocytes can also contribute to age-related diastolic dysfunction (Boccardi & Mecocci, 2020). Aged cardiomyocytes demonstrate decreased levels of cardiac troponin I and telomerase activity (Maejima, Adachi, Ito, Hirao, & Isobe, 2008). Shorter cardiomyocyte telomeres are observed in a number of CVDs, including heart failure and hypertrophic cardiomyopathy (Sharifi-Sanjani, et al., 2017). Interestingly, growing evidence suggests that adult cardiomyocytes retain proliferative capacity with a turnover rate of less than 1% per year (Bergmann, et al., 2009); however, the role of cellular senescence in halting this proliferative capacity is still not completely defined.
2.2. Senescent Endothelial Cells (ECs)
Senescent endothelial cells (ECs) exhibit a lower activity of endothelial nitric oxide synthase (eNOS) (Minamino, et al., 2002), nitric oxide (NO) production (Hoffmann, et al., 2001), and prostacyclin (PGI2) secretion (Nakajima, et al., 1997). Moreover, significantly higher levels of IL-1α and TNF-α have been demonstrated in senescent ECs (Khan, et al., 2017). These changes contribute to endothelial dysfunction, including impairment of vascular homeostasis, altered angiogenic response, and decreased endothelium-dependent dilation (Lesniewski, et al., 2017). Consequently, senescent ECs can play an important role in the development of atherosclerosis. Indeed, the overproduction of SASP activates the initial invasion of monocytes into the vessel wall, the first step in plaque formation (Boccardi & Mecocci, 2020). Additionally, chemoattractant factors in the SASP may trigger plaque formation since it has pro-atherosclerotic properties. Furthermore, senescent ECs demonstrated higher levels of CD9, a tetraspanin membrane protein associated with cell adhesion regulation and contributing to atherosclerosis (Cho, et al., 2020). Senescent human aortic endothelial cells (HAECs) express increased levels of the anti-angiogenic vascular endothelial growth factor 165b isoform (VEGFA165b) (Latorre, et al., 2018). Interestingly, higher levels of VEGFA165b are reported in patients with CHD, which can suggest that EC senescence can contribute to CHD by this mechanism (Latorre, et al., 2018). Moreover, senescent ECs have changes in the microRNAs (miRNA), which regulates multiple processes, including inflammation, apoptosis, and eNOS production (Rippe, et al., 2012). This may lead to oxidative stress, which contributes to the development of mitochondrial dysfunction (Y. Wang, Boerma, & Zhou, 2016). Notably, although aged mice do not spontaneously develop atherosclerosis, a survey of tissues in aged wild-type mice and progeroid mice prematurely aged due to a DNA repair defect, revealed that expression of the senescence markers p16Ink4a and p21Cip1 were greatest in the aorta, relative to young mice as compared to thirteen other tissues (Yousefzadeh, et al., 2020).
2.3. Senescent Vascular Smooth Muscle Cells (VSMCs)
Senescence of vascular smooth muscle cells (VSMCs) has been shown to contribute to arterial stiffness by promoting vascular inflammation and matrix remodeling (Schellinger, Mattern, & Raaz, 2019). Senescent VSMCs have higher calcification levels as a result of trans-differentiation into osteoblasts. (Nakano-Kurimoto, et al., 2009). Indeed, several osteogenic pathways were shown to be activated in senescent VSMCs, including bone morphogenetic protein 2 (BMP-2), alkaline phosphatase (ALP), osteopontin (OPN), and osteoprotegerin (OPG), which contribute to plaque calcification. Almost one-fifth of the VSMCs in human carotid artery plaque stain positively for senescence markers, such as p16Ink4a, p21Cip1, and SA-β-gal (Matthews, et al., 2006), increased IL-6 (Gardner, Humphry, Bennett, & Clarke, 2015), and reduced levels telomeric repeat binding factors (TRF-2). Interestingly, activation of senescence in VSMCs augmented abdominal aortic aneurysm, and inhibition of senescence using SIRT1 activators prevents the disease (Chen, et al., 2016).
2.4. Senescent Cardiac Progenitor Cells (CPCs)
Adult cardiac progenitor cells (CPCs) have the stem cell properties of being self-renewing and multipotent, generating cardiomyocytes, ECs, and VSMCs. Senescence of CPCs plays an essential role in the onset and progression of heart failure (Chimenti, et al., 2003; Rota, et al., 2006). Endothelial progenitor cells isolated from CHD patients show telomere shortening and decreased telomerase activity (Satoh, et al., 2008). Recent studies revealed that endothelial progenitor cells are more sensitive to DOX-induced senescence than to apoptosis compared to other cell types, which results in dose-dependent upregulation of SASP markers, increased p16Ink4a, p21Cip1, p53, and SA-ß-gal activity (Jahn, et al., 2020).
2.5. Senescent Cardiac fibroblasts (CFs)
Cardiac fibroblasts (CFs) are a major cell population within the heart and play an essential role in maintaining mechanical, structural, and electrical homeostasis. The contribution of senescent CFs to heart disease is still unclear. On one hand, transient senescence in particular of myofibroblasts appears important for preventing fibrosis in a mechanical model of myocardial fibrosis (K. Meyer, Hodwin, Ramanujam, Engelhardt, & Sarikas, 2016). Conversely, accumulation of senescent cells is positively correlated with fibrotic lesions in atrial fibrillation patients (J. Xie, et al., 2017). In this study, the majority of senescent cells are cardiac fibroblasts with minor contribution of cardiomyocytes and endothelial cells (J. Xie, et al., 2017).
3. Cancer therapy-induced cardiovascular senescence
3.1. Anthracycline-induced cardiovascular senescence
Anthracyclines (e.g., DOX and daunorubicin) are among the most commonly used chemotherapeutic agents in a wide variety of human cancers, including leukemia, lymphoma, and multiple solid tumors such as breast cancer. Despite its broad spectrum of therapeutic efficacy, the clinical utility of DOX is hindered by dose-limiting and often life-threatening cardiovascular toxicity (van Dalen, van der Pal, Caron, & Kremer, 2009). Indeed, DOX treatment increases the risk of developing characteristic cardiomyopathy, including tachycardia, arrhythmia, and eventually congestive heart failure. The risk of DOX-induced cardiotoxicity increases with higher cumulative doses. DOX-induced heart failure can affect approximately 26% of the patients with cumulative doses exceeding 600 mg/m2 (Lefrak, Pitha, Rosenheim, & Gottlieb, 1973). Vascular toxicities are also associated with DOX, including induction of endothelial cell death, endothelial dysfunction, and premature vascular aging (Carlson, et al., 2018; H. He, et al., 2019). A previous clinical study reported endothelial dysfunction in children treated with anthracyclines (Jang, Choi, & Jeon, 2013). DOX-induced apoptosis is hypothesized to be the primary driver of DOX-induced cardiotoxicity. However, low and moderate doses of DOX are associated with subclinical cardiovascular toxicity (Drafts, et al., 2013), without the induction of significant apoptosis in cardiomyocytes, which suggests that other mechanisms, such as cellular senescence, may contribute to cardiovascular dysfunction, especially after chronic administration of low DOX doses.
Studies that demonstrate DOX-induced senescence in cardiovascular cells/tissues are summarized in Table 1. Multiple studies demonstrate DOX-induced senescence in different types of cardiovascular cells, including ventricular myocytes, ECs, VSMCs, endothelial progenitor cells, and cardiac progenitor cells. Importantly, the majority of these studies were in vitro studies with only a few in vivo or clinical studies. Based on these studies, low concentrations of DOX (≤ 0.5 μM) preferentially induce senescence of cardiovascular cells, with no induction of apoptosis (Table 1). Currently, low doses of DOX are commonly used to induce senescence in vitro. Three main mechanisms were identified by which DOX induces senescence in cardiovascular cells:
Table 1.
Studies demonstrating doxorubicin-induced cardiovascular senescence.
| Study | Cell type | Dox Treatment (Conc / Time) | Detection of Senescence | Finding | Proposed Mechanism |
|---|---|---|---|---|---|
| (Maejima, et al., 2008) | Neonatal rat cardiomyocy tes | 0.1 μM for 7 days | ↑SA-β-gal activity ↑ Acetylated p53/p21cip1 ↑p27kip1 ↓Telomere length ↓cTnI phosphorylation | Low concentrations of DOX induce senescence in cardiomyocytes | Oxidative stress Telomere dysfunction |
| (Spallarossa, et al., 2009) | Neonatal rat cardiomyocytes and H9c2 cells | 0.01, 0.05, or 0.1 μM for 3 h Different experiments at 6 h, 24, or 48 h following treatment | Cell cycle alterations Morphological changes ↑ SA-β-gal activity ↓Chk2 ↓ TRF2, TRF1 | Low doses of DOX induce a senescence-like phenotype in cardiomyocytes, which undergo late cardiac death by mitotic catastrophe | Telomere dysfunction through p53 and MAPKs |
| (Spallarossa, et al., 2010) | Endothelial progenitor cells (EPCs) | 0.01, 0.05, 0.25, 0.05, 0.1 μM for 3 h (further experiments with 0.25 μM at 24 or 48 h following treatment) | Cell cycle arrest at G2/M phase Morphological changes with cytoskeleton remodeling ↑ SA-β-gal activity ↑ Cytoplasmic p16INK4a ↓ TRF2 | Low concentrations of DOX induce senescence in EPCs Higher concentrations induce apoptosis | Oxidative stress Telomere dysfunction through p38-MAPK activation |
| (Altieri, et al., 2012) | Neonatal rat ventricular myocytes | 0.1 μM for 3 h Different experiments at 6h, 24, or 48 h following treatment | Cell cycle arrest at S phase Morphological changes with cytoskeleton remodeling ↑ SA-β-gal activity ↑ pi6Ink4a ↓ TRF2 | Pretreatment with the PPARδ agonist L-165041, protected against DOX- induced senescence | DOX activates PPARδ, which sequesters the anti-senescence protein BCL-6. PPARδ agonist L-165041 releases BCL-6 in MAPK/AKT dependent mechanism |
| (Hodjat, et al., 2013) | Human primary vascular smooth muscle cells (VSMCs) | 0.25, 0.5, and 1 μM for 3 h, experiments were done 3 days after treatment | ↑ SA-β-gal activity ↑ p53/ p21CIP1 No change p16INK4 ↓ TRF2 | Low doses of DOX induce senescence in VSMC that may initiate vascular damage | Telomere dysfunction (DOX upregulates uPAR-mediated TRF2 ubiquitination and proteasomal degradation) No oxidative stress was observed at these concentrations |
| (Piegari, et al., 2013) | Human cardiac progenitor cells (CPCs) | 0.1, 0.5, and 1.0 μM for 24 or 48 h | Morphological changes ↑ SA-β-gal activity ↑ p16INK4a No change in p21CIP1 ↑ p-p53 ↑ γ-H2AX ↓ Cyclin D1 ↓ phosphorylated RbSer798 | DOX exposure induces senescence in hCPCs which may mediate DOX-induced cardiomyopathy | DNA Damage |
| (Bielak-Zmijewska, et al., 2014) | Human VSMCs | 0.1 μM for 1, 3, or 7 days | Morphological changes Cell cycle arrest at G2/M ↑ SA-β-gal activity ↑ p53/p-p53 ↑ p16INK4a,↑ p21CIP1 ↑ SASP (IL-6, IL-8, VEGF) ↑Superoxide production | DOX-induced senescence in VSMCs with some differences to replicative senescence | DNA Damage |
| (Heo, et al., 2016) | Human pulmonary microvascular endothelial cells (HMVECs) | 1 μM for 4 h then incubated for 6, 24, or 48 h | Morphological changes ↑ SA-β-gal activity ↑ p53 | XAF1 may contribute to inducing senescence in HMVECs | Activation of XAF1 via a p53-dependent mechanism |
| (Altieri, et al., 2016) | H9c2 cells and neonatal mouse cardiomyocytes | 0.1 μM for 3 h +/− pretreatment with 0.01 μM testosterone or 0.001 μM 17β- estradiol for 15 minutes. Different experiments were carried out at 24 or 48 h | ↑ SA-β-gal activity ↑ SAHF ↑ p16INK4a ↑ p53 phosphorylation/p21cip1 ↓ TRF2 | Testosterone, but not 17β- estradiol, protects against DOX- induced senescence in Cardiomyocytes | Testosterone modulates TRF2 via PI3K/AKT/NO S3 mechanism |
| (Bent, Gilbert, & Hemann, 2016) | Human umbilical vein endothelial cells (HUVECs) | 0.225 μM for 24 or 120 h | Cell cycle arrest ↑ SA-β-gal activity ↑ p21CIP1 ↑ p16INK4a ↑ Acute IL-6 production | DOX induces endothelial cell senescence without the typical SASP but rather ASAP | Oxidative stress induces ASAP through p38 signaling and downregulation of PI3K/AKT/mT OR pathway |
| (De Falco, et al., 2016) | Endothelial progenitor cells (EPCs) | 0.25 μM for 3 h | ↑ SA-β-gal activity ↑ p21CIP1 ↑ IL-6 ↓ NO | DOX induces senescence in EPC. Nox2 Inhibition may protect against DOX-induced senescence | Oxidative stress via Nox2- dependent mechanism |
| (Przybylska, et al., 2016) | Human VSMCs | 1 μM for 2 h Experiments were done 6 days after | Morphological changes ↑ SA-β-gal activity ↑ p53/p-p53 ↑ p21CIP1 ↑ SASP (IL-6, IL-8, VEGF) | DOX induces senescence in VSMCs Both increased and diminished ROS levels can lead to senescence | DOX-induced DSB and ROS activates p53/p21 pathway NOX4 silencing activates p27 |
| (Xia & Hou, 2018) | H9c2 Rat BM-MSCs | 0.5 μM for 24 h | ↑ p53/ p16INK4a gene expression ↓ Telomere length ↓Telomerase activity | Co-culture with MSCs attenuated DOX-induced senescence and increased cells proliferation | MSCs induce anti-senescence effect by upregulating Sirt1 expression via miR-34a Inhibition |
| (Z. Xie, et al., 2018) | HL-1 murine cardiomyocytes | 5 μM for 24 h | ↑ p53/ p16INK4a gene expression ↓Telomere length ↓Telomerase activity ↓ Proliferation SOD activation | lincRNA-p21 silencing attenuated DOX-induced senescence in cardiomyocytes | DOX induces lincRNA-p21 which regulates oxidative stress via WNT/β-catenin signaling pathway (Decrease β-catenin) |
| (Fallah, et al., 2019) | Wistar rats (Heart tissues) | DOX: 0.75, 0.5, 0.1 mg/kg Liposomal DOX: 0.1, 0.025, 0.05 mg/kg. Both daily for 6 weeks | ↑ SA-β-gal activity ↑ p53 | Both DOX and liposomal DOX induce senescence and mild inflammation | Oxidative stress |
ASAP, Acute stress-associated phenotype; Akt, Protein Kinase B; Bcl-6, B cell lymphoma-6; Chk2, checkpoint kinase 2; CPCs, Cardiac progenitor cells; CTn, Cardiac troponin; DSB, double-strand breaks; HUVECs, Human umbilical vein endothelial cells; IL, Interleukin; linc, Long intergenic non-coding; MAPK, Mitogen-activated protein kinase; MSCs, Mesenchymal stem cells; NOX, NADPH oxidases; PI3K, Phosphatidylinositol 3-kinase; ROS, Reactive oxygen species; SA-β-gal, Senescence associated-β-galactosidase assay; SAHF, Senescence-associated heterochromatin foci; SASP, Senescence-associated secretory phenotype; TRF, Telomeric repeat binding factor; VSMCs, Vascular smooth muscle cells; VEGF, Vascular endothelial growth factor
3.1.1. DOX-induced DNA damage
Replication stress caused by DNA damage or frank double-strand breaks (DSBs) activates the DNA damage response (DDR), which in turn causes stabilization/upregulation of the p53/p21 pathway. Activation of p53 through ATM-dependent phosphorylation leads to increased expression of many effector genes, in particular p21Cip1, a cyclin-dependent kinase (CDK) inhibitor, and thereby growth arrest (Larsson, 2011). Additionally, DNA damage increases p16INK4a expression, which blocks cyclin-dependent kinase activity through the retinoblastoma (Rb) pathway (Rayess, Wang, & Srivatsan, 2012). The increase of CDK inhibitors, p16Ink4a and p21Cip1, is both dose and time-dependent, with p21Cip1 usually being induced first followed by a delayed upregulation in p16Ink4a. Prolonged activation of the PI3K/AKT/mTORC1 results in p53-mediated cellular senescence (Astle, et al., 2012). While most studies link mTORC1 to senescence, a recent study demonstrates increased mTORC2 in H2O2-induced senescence in human umbilical vein endothelial cells (HUVECs) (Yang, et al., 2018). In addition, DOX increases the expression of promyelocytic leukemia protein (PML), which induces acetylation of p53 by forming PML-acetylated p53 complex, leading to activation of p21Cip1 (Maejima, et al., 2008). Other studies demonstrate that DOX-induced activation of p53 is JNK-dependent (Spallarossa, et al., 2009).
3.1.2. DOX-induced oxidative stress
DOX causes an increased abundance of reactive oxygen species (ROS) through a variety of mechanisms. First, ROS is increased by the metabolism of DOX, mostly because of the unstable intermediate formed (Wallace, 2003). Moreover, DOX-induced mitochondrial damage can lead to increased ROS, which is mediated by mitochondrial NADPH oxidase (Asensio-Lopez, Soler, Pascual-Figal, Fernandez-Belda, & Lax, 2017). This can lead to increased lipid peroxidation, depletion of anti-oxidants, and a feed-forward cycle of ROS production contributing to the overall DOX-induced oxidative stress. Similar to genotoxic stress, oxidative stress can lead to p53 activation and triggering of senescence. Hydrogen peroxide (H2O2), like DOX is used to induce senescence in vitro. The transcription factor NF-κB plays an important role in regulating the cellular response to oxidative and genotoxic stress (Lingappan, 2018). Additionally, it plays an important role in regulating SASP and can increase the expression of pro-inflammatory cytokines, such as TNF-α and IL-1 (Chien, et al., 2011). A number of studies have shown that DOX significantly activates NF-κB in cultured cardiomyocytes (Guo, et al., 2013; Notarbartolo, et al., 2005), which leads to the generation of free radicals and activation of the DDR, which ultimately induces senescence (Tilstra, et al., 2012). Similarly, DOX administration to Wistar rats induces NF-κB activation and oxidative stress and upregulated p53 and SA-β-gal expression in heart tissues (Fallah, et al., 2019). In endothelial progenitor cells, DOX treatment induces the activity of NADPH oxidase isoform 2 (Nox2), which leads to superoxide generation resulting in oxidative stress-induced senescence (De Falco, et al., 2016). Low doses of DOX induce activation of AKT by phosphorylation at Ser473 in cardiac muscle cells (Altieri, et al., 2012). AKT activation induces phosphorylation of FOXO transcription factors, which leads to a decrease in superoxide dismutase-2 (SOD2) levels, a key antioxidant, eventually leading to increased oxidative stress (Bourgeois & Madl, 2018).
3.1.3. DOX-induced telomere dysfunction
Telomeres are repeated sequences at the ends of chromosomes and are essential for genome stability. Many cell stress stimuli, such as oxidative stress and mitochondrial dysfunction, can induce telomere damage, and repeated replication drives telomere attrition. When telomeres become critically short, this activates the DDR, and phosphorylation of histone variant H2AX (γH2AX), which marks sites of DNA DSBs including in telomeric DNA (d’Adda di Fagagna, et al., 2003). Telomere shortening is prevented by telomerase, which replaces telomeric repeat DNA lost during cell division. Telomere dysfunction contributes to senescence via activation of either p53 or p16Ink4a signaling pathways in human cells or via p53 only in mouse cells (Smogorzewska & de Lange, 2002). DOX decreases telomerase in different cardiovascular cells (Maejima, et al., 2008; Xia & Hou, 2018; Z. Xie, Xia, & Hou, 2018).
Telomeric repeat binding factors 1 and 2 (TRF-1, TRF-2) are important shelterin complex proteins, which prevent telomeric DNA from becoming damaged or eroding. Upregulation of TRF-2 is associated with suppression of senescence. Downregulation of TRF-2 is implicated in the progression of CHD (Satoh, et al., 2017). The knockout of TRF-2 in mice accelerates the progression of atherosclerosis (J. Wang, et al., 2015). Low doses of DOX downregulate the levels of both TRF-1 and TRF-2 via increased p38-MAPK and p53 phosphorylation (Altieri, et al., 2012; Spallarossa, et al., 2009), which contributes to DOX-induced senescence in neonatal cardiomyocytes and endothelial progenitor cells (Spallarossa, et al., 2010). DOX also induces senescence in VSMCs via TRF-2 downregulation (Hodjat, Haller, Dumler, & Kiyan, 2013). However, TRF-2 downregulation is induced by a different mechanism that is dependent on the urokinase receptor (uPAR) upregulation, which drives ubiquitination and proteasomal degradation of TRF-2 (Hodjat, et al., 2013). Interestingly, pretreatment of cardiomyocytes with testosterone protects against DOX-induced senescence (Altieri, et al., 2016). The protective effect of testosterone is mediated through TRF2 modulation via a pathway involving the PI3K/AKT/nitric oxide synthase 3 (NOS3)/ androgen receptor (Altieri, et al., 2016).
3.1.4. DOX-induced epigenetic alterations
The effects of DOX on epigenetic alterations were recently reviewed in (Kumari, Huang, & Chan, 2020). Previous studies show that DOX treatment downregulates DNMT1 (DNA Methyltransferase 1) and induces DNA hypomethylation both in vitro in H9c2 cells (L. L. Ferreira, et al., 2019) and in vivo in rat hearts (A. Ferreira, et al., 2017). Additionally, DOX caused fluctuation in DNMT1 level in a model of DOX-induced senescence in VSMCs (Bielak-Zmijewska, et al., 2014). Recently, DNMT1 was demonstrated to be suppressed before initiation of senescence in human fibroblasts (Jung, et al., 2017). Additionally, DOX treatment upregulates histone deacetylases (HDACs) levels in cardiomyocytes (R. Song, et al., 2018) and the heart of DOX-treated mice (Piotrowska, Isalan, & Mielcarek, 2017). Interestingly, the past few years have witnessed significant interest in HDAC inhibitors as anti-aging drugs to increase lifespan (McIntyre, Daniels, Molenaars, Houtkooper, & Janssens, 2019). To conclude, DOX induces dysregulation of multiple epigenetics pathways in cardiovascular cells both in vivo and in vitro. These alterations interplay with DOX-induced cellular senescence and may have an important role in DOX-induced premature cardiovascular aging. Taking into consideration that epigenetic alterations can be reversed (Freije & Lopez-Otin, 2012), epigenetic reprograming can be an important therapeutic strategy to mitigate cancer therapy-induced aging.
3.2. Radiation-induced Cardiovascular Senescence
Radiation-induced heart disease (RIHD) is a serious complication of radiotherapy that can affect the quality of life of cancer survivors (Nabialek-Trojanowska, et al., 2018). Radiation causes a number of cardiovascular complications, including atherosclerosis, CHD, myocardial fibrosis, pericarditis, pericardial fibrosis, valve dysfunction, and microvascular damage (Tapio, 2016). Although a plausible strategy to protect against radiation-induced cardiac complications is to limit cardiac exposure to radiation, this approach is not always feasible, especially with thoracic cancer patients, including breast, lung, and esophageal cancers. Several studies propose multiple mechanisms to mediate radiation-induced cardiovascular complications such as oxidative stress (Pradeep, et al., 2012), inflammation (Meeren, Bertho, Vandamme, & Gaugler, 1997), apoptosis (R. A. Panganiban, O. Mungunsukh, & R. M. Day, 2013), and cellular senescence (Table 2). The majority of these studies focus particularly on radiation-induced senescence (RIS) in endothelial cells, with few studies evaluating RIS in other cardiovascular cells, including cardiomyocytes. Additionally, most of these studies were conducted in vitro using different cell lines, with much fewer in vivo studies. Radiation-induced genotoxic and oxidative stress are the two major drivers of senescence in cardiovascular cells. The mechanisms of RIS in endothelial cells and its contribution to RIHD were recently reviewed (Y. Wang, et al., 2016). Additionally, while senescence is observed in most of these studies in the first weeks following radiation, others report persistent senescence for four weeks (Lafargue, et al., 2017) and even up to 20 weeks following radiation (Oh, Bump, Kim, Janigro, & Mayberg, 2001). Since RIHD can take more than a decade to manifest (Y. Wang, et al., 2016), chronic senescence could explain part of the delayed effects of radiation.
Table 2.
In vitro and In vivo studies demonstrating radiation-induced cardiovascular senescence
| Study | Cell type | Radiation Dose | Detection of Senescence | Finding | Proposed Mechanism |
|---|---|---|---|---|---|
| (Oh, et al., 2001) | BAECs | 5–15 Gy Different experiments at 0–8 weeks after | Morphological changes ↓ BrdU incorporation ↑ SA-β-gal activity ↑ p21cip1 | IR induces a senescence-like phenotype in endothelial cells (ECs) | DNA damage |
| (Igarashi, Sakimoto, Kataoka, Ohta, & Miura, 2007) | BAECs HUVECs | 2, 4, and 8 Gy | Morphological changes ↑ SA-β-gal activity ↓ BrdU incorporation ↑ SASP (IL-8, IL-1α, VEGF-B) ↑ VCAM-1, ICAM-1 | IR induces a senescence-like phenotype in ECs which could inhibit their angiogenic features | DNA damage |
| (Sermsathanasawadi, et al., 2009) | HUVECs | 8 Gy | Morphological changes ↑ SA-β-gal activity ↑ VCAM-1, ICAM-1, E-selectin | IR induces senescence-like phenotype in ECs with increased expression of adhesion molecules that may promote tumor neovascularization | DNA damage |
| (Mendonca, et al., 2011) | Endothelial colony-forming cells (ECFCs) isolated from adult peripheral blood | 3 and 10 Gy | ↑ SA-β-gal activity | While 10 Gy induces senescence in adults ECFCs, this response is not observed with 3 Gy radiation | Possible telomere dysfunction |
| (Panganiban & Day, 2013) | HPAECs | 10 Gy followed by 1, 2, or 3 days | Morphological changes ↑ SA-β-gal activity ↑ p53/ p21CIP1 ↑ Phosphorylation of IGF-1R and AKT | IR induces accelerated senescence in ECs | ROS which activates IGF-1R-mediated senescence via mTOR/PI3k pathway |
| (R. A. M. Panganiban, O. Mungunsukh, & R. M. Day, 2013) | Bovine PAECs | 2 – 50 Gy followed by 1, 3, or 5 days | Morphological changes ↑ SA-β-gal activity ↑ p21cip1 ↓ Sirt1 ↑ Bcl-2 | Low doses (< 10 Gy) of IR induces senescence with limited apoptosis in ECs | DNA damage independent of ER stress |
| (Ungvari, et al., 2013) | Rat primary CMVECs | 2–8 Gy for 1–14 days | ↑ SA-β-gal activity ↑ p53, p16INK4a gene expression No change in p21cip1 ↑ SASP (IL- 6, IL-1α,, MCP-1,GM- CSF) | Low doses of IR induces senescence and impaired angiogenic capacity in CMVECs | DNA Damage |
| (Yentrapalli, Azimzadeh, Barjaktarovic, et al., 2013) | HUVECs | Continual low dose gamma radiation (4.1 mGy/h), cells were harvested 1, 3, or 6 weeks after. | Morphological changes ↑ SA-β-gal activity Acute ↑ p53/p-p53 Acute ↑ p21CIP1 Delayed ↑ p16INK4a No change in AKT or p16INK4a | Chronic radiation inhibits the replicative potential of HUVECs and induces premature senescence | Activation of p53/p21 pathway due to radiation-induced oxidative stress and DNA damage |
| (Yentrapalli, Azimzadeh, Sriharshan, et al., 2013) | HUVECs | Two doses were compared: 1.4 mGy/h and 2.4 mGy/h | ↑ SA-β-gal activity ↑ p21CIP1 No change p53 or p16INK4a | The 2.4 mGy/h dose, but not 1.4, was able to induce senescence in ECs | DNA damage mediated by altered PI3K/Akt/mT OR pathway |
| (Lowe & Raj, 2014) | HCAECs | 10 Gy | ↑ SA-β-gal activity | IR induces premature aging in ECs which may contribute to atherosclerosis | DNA damage |
| (K. S. Kim, Kim, Choi, Bae, & Kim, 2014) | HUVECs | 4 Gy | Morphological changes ↑ SA-β-gal activity ↑ p53/ p21CIP1 ↑ γ-H2AX foci ↓ Cyclin A, Cyclin B1 | Using microarray analysis, IR altered the expression of senescence genes in ECs | DNA damage |
| (E. J. Kim, et al., 2015) | HL-1 and H9C2 cells | 8 and 15 Gy | ↑ SA-β-gal activity ↑ p21cip1 | Radiation induces senescence in cardiomyocytes by ROS generation via impairing corin function | Oxidative stress |
| (Azimzadeh, et al., 2015) | 10 week old male C57Bl/6 mice | Single Dose of 8 or 16 Gy Mice were sacrificed 16 weeks later Senescence markers are measured in cardiac microvascular ECs | ↑ ICAM-1,2, PECAM-1, VCAM-1 ↑ p21cip1 ↑ p16Ink4a ↑ p21, p16, and Igfbp3 gene expression No change p53/p-p53 No increase in SA-β-gal activity | Acute irradiation-induced endothelial dysfunction was mediated by senescence in cardiac microvascular ECs | Increased ROS and decrease NO bioavailability via deactivation of the insulin/IGF-PI3K-Akt |
| (Dong, et al., 2015) | HUVECs | 0–8 Gy | Morphological changes ↑ SA-β-gal activity ↑ IL-6 gene expression | IR induces senescence-like phenotype, but not apoptosis, in ECs | DNA damage via NF-κB activation through NEMO |
| (Heo, et al., 2016) | HMVECs | 4 Gy then incubated for 6, 24, or 48 h | Morphological changes ↑ SA-β-gal activity ↑ p53 | XAF1 may contribute to inducing senescence in HMVECs | Activation of XAF1 via p53-dependent mechanism |
| (Park, Kim, Jeong, Park, & Kim, 2016) | HAECs | 4 Gy for 24 or 48 h | Morphological changes ↑ SA-β-gal activity ↑ p-p53 ↑ p21CIP1 | GDF15 is involved in IR- induced senescence in ECs | DNA damage induces GDF15 which contributes to senescence via oxidative stress-mediated p16 pathway |
| (Lafargue, et al., 2017) | HMVECs | 0–15 Gy, cell cultures were maintained for 28 days | ↑ SA-β-gal activity ↑ p21CIP1 ↑ p16INK4a ↑ γ-H2AX foci ↑ IL-8 ↑ ATM phosphorylation | IR induces long term senescence in HMVEC | DNA damage Oxidative stress-mediated mitochondrial dysfunction |
| (Hu, et al., 2018) | Human cardiac myocytes (HCMs) | 5 Gy | ↑ SA-β-gal activity ↓Telomere length ↓ Telomerase activity ↓ Sirt1 ↑ p21CIP1 gene expression | IR induces senescence in cardiomyocytes via induction of miR-34a | Oxidative stress |
| (Casella, et al., 2019) | HUVEC, HAECS | 4 Gy Cells were harvested after 10 days | ↑ SA-β-gal activity ↑ p16INK4a gene expression | The study identified common transcriptomic signature in different senescence models | |
| (K. Wu, et al., 2019) | HUVECs | Two days following cell transfection with Ku86, continual IR was applied for 7 days with cumulative doses of 0, 0.1, 0.2, 0.3, and 0.5 Gy | ↑ SA-β-gal activity ↑ p16INK4a | Low doses of IR induces senescence, higher intensities were associated with apoptosis | DNA damage Ku86 activated Sirt1 and SOD2, abrogated IR-induced senescence |
AKT, Protein Kinase B; Bcl-2, B Cell Lymphoma-2; DDR, DNA damage response; BAECs,Bovine aortic endothelial cells; ECFCs, Endothelial colony-forming cells; ECs, Endothelial cells; GM-CSF, Granulocyte-macrophage colony-stimulating factor; HAECs, Human aortic endothelial cells; HCAECs, Human coronary artery endothelial cells; HMVEC-L, Human Cardiac Microvascular Endothelial Cells; HUVECs, Human umbilical vein endothelial cells; IR, Ionizing radiation; ICAM, Intercellular adhesion molecule; IGF-1R, Insulin-like growth factor type 1 receptor; IL-6, Interleukin-6; Sirt1, Sirtuin 1, silent information regulator 2 homolog 1; MCP, Monocyte chemoattractant protein; miR-34a, microRNA-34a; NEMO, NFkappa-B essential modulator; NO, Nitric oxide; PECAM, platelet endothelial cell adhesion molecule; ROS, Reactive Oxygen Species; SA-β-gal, SA-β-gal, Senescence associated-β-galactosidase; SOD2, Superoxide dismutase-2; VCAM, Vascular cell adhesion molecule; VEGF, Vascular endothelial growth factor; XAF1, X-linked inhibitor of apoptosis (XIAP)-associated factor 1
A recent study revealed that RIS in cardiomyocytes occurs via upregulating of microRNA-34a (miR-34a), which inhibits Sirt1 expression and further decreases cardiomyocytes tolerance to stress (Hu, Xia, & Hou, 2018). Pretreatment of cardiomyocytes with macrophage migration inhibitory factor (MIF) suppresses radiation-induced oxidative stress via inhibition of miR-34a and consequently alleviates RIS (Hu, et al., 2018). The role of miRNAs in senescence and aging were recently reviewed (Majidinia, et al., 2020; Williams, Smith, Kumar, Vijayan, & Reddy, 2017). Long non-coding RNA (lncRNA) dysregulation is also involved in senescence (Puvvula, 2019) A recent transcriptomic analysis reveals that exposure of endothelial cells to 4 Gy of radiation alters the expression of more than 50 RNAs, including protein-coding and non-coding RNAs (Casella, et al., 2019). Another proposed mechanism of RIS is that radiation-induced oxidative stress downregulates the expression of corin protein in cardiomyocyte-like cell lines HL-1 and H9c2 (E. J. Kim, et al., 2015). Corin is a cardiac protease that is responsible for cleavage of pro-atrial natriuretic peptide (pro-ANP) and pro-brain natriuretic peptide (pro-BNP) to generate the active forms. These natriuretic peptides are important regulators for myocardial function. Inhibition of corin function will inhibit ANP and BNP, which can contribute to the RIS in cardiomyocytes (E. J. Kim, et al., 2015).
RIS also triggers pro-atherosclerotic events (Lowe & Raj, 2014). Radiation increases monocyte adhesion to senescent endothelial cells observed as a higher number of monocyte clusters forming following primary endothelial cells irradiation with 10 Gy (Lowe & Raj, 2014). Interestingly, this was time-dependent, with the number of monocyte clusters increasing up to 15 days post-irradiation. RIS is also seen in fibroblasts (Casella, et al., 2019; Cmielova, et al., 2011; de Magalhaes, Chainiaux, Remacle, & Toussaint, 2002; Gorbunova, Seluanov, & Pereira-Smith, 2002; Studencka & Schaber, 2017; Suzuki, et al., 2001) and alveolar epithelial cells (AECs) (Citrin, et al., 2013), which may contribute to radiation-induced pulmonary fibrosis (RIPF) (Y. He, et al., 2019); astrocytes, which may mediate radiation-induced brain injury (Turnquist, et al., 2019); salivary glands, which may contribute to radiation-induced dry-mouth syndrome (Marmary, et al., 2016); and bone marrow stem cells, dental pulp stem cells (Muthna, et al., 2010), and bone resident cells which may drive radiation-induced bone loss (Yao, et al., 2020). Transplanting relatively small numbers of senescent cells into young mice is sufficient to cause detrimental long-lasting systemic effects and reduce lifespan (Xu, et al., 2018); therefore, it is possible that RIS of non-cardiovascular tissues could also have detrimental cardiovascular effects.
3.3. Other Cancer Treatments
Anthracyclines and radiation therapy are the most studied cancer treatments with regard to their effects on cellular senescence in cardiovascular cells and organs. Nevertheless, other chemotherapeutic agents have been shown in a few studies to induce senescence in endothelial cells (Table 3). When there is a paucity of research regarding the senescence-inducing effects of other chemotherapeutic agents in cardiovascular cells, we will briefly discuss their effects in non-cardiovascular cells.
Table 3.
Studies demonstrating other chemotherapy-induced cardiovascular cellular senescence.
| Study | Cell type | Chemotherapy | Cell Treatment (Conc./Time) | Detection of Senescence | Finding | Proposed Mechanism |
|---|---|---|---|---|---|---|
| (Focaccetti, et al., 2015) | HUVECs HCMs | 5-Fluorouracil (5-FU) | Cells were treated with 5-FU up to 72 h | ↑ SA-β-gal activity t SASP | 5-FU induces endothelial senescence leading to vascular collapse or vasospasm | ROS production resulting in senescence induction |
| (Altieri, et al., 2017) | EA.hy926 cells | 5-Fluorouracil (5-FU), sera from capecitabine-treated patients | 100 μgmL−1 5-FU for 4 h or 10% human serum from patients receiving capecitabine | ↑ SA-β-gal activity ↑ p16INK4a Morphological changes ↓ eNOS | Both 5-FU and sera from capecitabine-treated patients induce endothelial cell senescence | Activation of p38 and JNK Downregulation of Sirt-1 and eNOS |
| (Yin, et al., 2017) | HUVECs | Bleomycin | HUVECs were exposed to bleomycin (5 – 20 μM) for 2 – 6 days | ↑ p53 and p21 ↑ SA-β-gal activity ↑ IL-1 Sirt1 mRNA level remained unchanged | Bleomycin significantly induces HUVECs senescence in a dose- and time-dependent manner | ROS-dependent activation of the NLRP3 inflammasome |
| (Mongiar di, et al., 2019) | HUVECs | Axitinib | Axitinib 25 μM for 1 h then HUVECs were cultured in drug free medium for 4 days | ↑ SA-β-gal activity ↓ ki67 expression ↓ CDKN1B (p27) ↓ LMNB1, LMNB2, LBR ↑ SASP (CCL2, CX3CL1) ↑ BTG2 expression No H2AX Phosphorylation No p53 activation | Axitinib-induced endothelial cells senescence phenotype was quite different from DOX-induced senescence Both antioxidants as GSH, NAC; and ATM inhibitors as KU-60019,KU-55933, abrogated Axitinib-induced senescence |
ATM activation (phosphorylation) through increased ROS without DDR |
| (Merolle, et al., 2020) | HUVECs co-cultured with GBM tumor cells in transwell plates | Axitinib | 25 μM for 1 h then GBM cells were removed and HUVECs were cultured another four days | ↑ SA-β-gal activity ↓ ki67 expression ↓ CDKN1B (p27) ↓ LMNB1 downregulation ↑ SASP (CCL2, CX3CL1) ↑BTG2 expression | Co-culture with GBM did not prevent Axitinib-induced HUVEC senescence, rather it modified transcriptomic profile | ROS-dependent ATM activation |
ATM, Ataxia Telangectasia Mutated; BTG2, B-cell translocation gene 2; DOX, Doxorubicin; DDR, DNA damage response; eNOS, Endothelial nitric oxide synthase; GBM, glioblastoma; GSH, Glutathione; HCMs, Human cardiac myocytes; HUVECs, Human umbilical vein endothelial cells; IL-1, interleukin-1; JNK, N-terminal kinase; LMNB1, Lamin B1; NAC, N-acetyl cysteine; NLRP3, NOD-like receptor family pyrin domain-containing3; PAI-1, Plasminogen activator inhibitor-1; ROS, reactive oxygen species; SA-β-gal, senescence-associated β-galactosidase; SASP, senescence-associated secretory phenotype
3.3.1. Fluoropyrimidines
The antimetabolite 5-fluorouracil (5-FU) and its prodrug capecitabine are used for the treatment of multiple solid tumors. Focaccetti et al. found that 5-FU induces senescence as indicated by increased activity of SA-β-Gal in HUVECs and human cardiac myocytes (Focaccetti, et al., 2015). Senescence could be due to replication stress or increased ROS abundance. These results were further confirmed by Altieri et al. in the human endothelial-derived EA.hy926 cells treated with 5-FU and sera from capecitabine treated patients (Altieri, et al., 2017).
3.3.2. Axitinib
Axitinib is an oral tyrosine kinase inhibitor with selectivity to vascular endothelial growth factors 1, 2, and 3 (Hu-Lowe, et al., 2008). It is used as a second-line treatment for advanced renal cell carcinoma. In HUVECs, axitinib triggers senescence and SASP by inducing oxidative stress and ATM activation in a way that does not depend on DNA damage or p53 phosphorylation (Mongiardi, et al., 2019). Axitinib-mediated senescence is not affected by the presence of glioblastoma tumor cells (GBM) (Merolle, Mongiardi, Piras, Levi, & Falchetti, 2020).
3.3.3. Bleomycin
Bleomycin is a chemotherapeutic drug that is used to induce DNA damage and senescence in multiple cell lines. Bleomycin induces senescence in HUVECs in a dose- and time-dependent manner (Yin, et al., 2017). ROS-mediated interaction of thioredoxin-interacting protein (TXNIP) with NOD-like receptor family pyrin domain-containing 3 (NLRP3) in senescent endothelial cells activates NLRP3 inflammasome and caspase-1, which triggers IL-1 secretion.
3.3.4. Pegylated interferons (pIFN-α)
Peg IFN-α is added as adjuvant therapy in the treatment of melanoma (Agha & Tarhini, 2017). IFN-α acts by inducing interferon regulatory factor-1 (IRF-1), activating its tumor suppressor function resulting in an antiangiogenic agent (J. H. Lee, Chun, Park, & Rho, 2008). Peg IFN-α induces senescence in endothelial-derived EA.hy926 cells (Upreti, Koonce, Hennings, Chambers, & Griffin, 2010). The combination of pegylated IFN-α with the chemotherapeutic vinblastine (VBL), induces cell death of melanoma cells via IRF-1-mediated signaling and senescence of endothelial cells reducing angiogenesis to act as a further tumor suppressor.
3.3.5. Chemotherapeutic agents causing senescence in non-cardiovascular cell lines/tissues
Alkylating agents such as cyclophosphamide, busulfan, and temozolomide induce senescence in multiple cell lines and mice. Cyclophosphamide activates the MAPK pathway in response to oxidative stress resulting in senescence in human fetal lung fibroblasts (TIG-7) (Palaniyappan, 2009). Moreover, cyclophosphamide is capable of inducing senescence in mouse ovarian granulosa cells by activating the lncRNA-Meg3-p53-p66Shc pathway leading to premature ovarian failure (Xiong, et al., 2017). Busulfan, an anticancer medication that causes DNA damage, induces senescence in human fibroblasts (WI-38) via Erk/MAPK activation in a p53-independent mechanism (Probin, Wang, Bai, & Zhou, 2006; Probin, Wang, & Zhou, 2007). In addition, busulfan induces senescence in bone-marrow-derived mesenchymal stem cells (BMSCs) and adipose tissue-derived mesenchymal stem cells (ADSCs) (Qi, et al., 2012). The adverse hematopoietic effects of busulfan could be explained by its ability to induce senescence in murine bone marrow cells (BM-MNCs) (Papaconstantinou, 2019). Temozolomide increases the expression of p16Ink4a in mice (M. Demaria, et al., 2017). Cisplatin induces senescence in several human cell lines including human lung fibroblasts, human dental pulp stem cells, human dermal fibroblasts, and primary human oral fibroblasts (C. Liu, Ma, Zhuang, Liu, & Sun, 2020; Seifrtova, et al., 2012; Tasnuva D. Kabir1, Eric K. Parkinson5, & McCall, 2016). Moreover, cisplatin induces senescence in rat renal tubule epithelial cells (NRK-52E) and senescence in renal tissues which may explain the chronic kidney disease caused by cisplatin (Li, et al., 2019). Cisplatin-induced peripheral neuropathy (CIPN) is the most common dose-limiting adverse effect of cisplatin (Cioroiu & Weimer, 2017; Kandula, et al., 2017). This could be because cisplatin causes the accumulation of senescent-like neuronal cells in primary culture and in mouse dorsal root ganglion (Acklin, et al., 2020). The cisplatin-induced DDR activation and p21Cip1 upregulation cause senescence, not apoptosis, in mouse dorsal root ganglion sensory neurons (Calls, et al., 2020). Paclitaxel, a potent microtubule inhibitor, induces senescence in primary mouse cells and in vivo (M. Demaria, et al., 2017). Furthermore, paclitaxel induces senescence in mesenchymal stem cells (MSCs) which may help to explain the severe myelosuppression caused by taxane-based anticancer treatments (Munz, et al., 2018). Etoposide is commonly used to induce senescence in vitro. Low dose etoposide induces senescence in human diploid fibroblasts (WI-38) in a p53-dependent mechanism (Probin, et al., 2006). Etoposide also induces senescence in mouse embryonic fibroblasts (MEFs), normal human skin fibroblasts (BJ cells), retinal pigment epithelial cells (RPE), NRK-52E rat renal tubular epithelial cells, and normal human lung fibroblasts (IMR-90) (Biran, et al., 2017; Blagosklonny, 2010; L. Gu & Kitamura, 2012; Yosef, et al., 2017). Actinomycin D is an antimetabolite that induces senescence in human foreskin fibroblasts (HDF-2, NHF-3), human lung fibroblasts (MRC-5), and hMSCs (Minieri, et al., 2015; Steven J Robles & Adami, 1998). The topoisomerase inhibitor, irinotecan, induces senescence in normal human colonic fibroblasts (NCF), and normal human colonic mucosa cells (NCM) (Rudolf, John, & Cervinka, 2012). Low concentrations of mitoxantrone, another topoisomerase inhibitor, induces senescence in hDPSCs and HDFs (Seifrtova, et al., 2013).
In summary, multiple anticancer agents induce senescence in non-cardiovascular cells and tissues, accounting for some of their adverse effects. Taking into account that several chemotherapeutic agents cause cardiovascular adverse effects (Minami, Matsumoto, & Horiuchi, 2010; Yeh & Bickford, 2009), further investigation is warranted to better understand the mechanisms by which chemotherapy-induced senescence may contribute to these adverse cardiovascular effects.
4. Clinical Evidence for Premature Aging in Cancer Survivors
4.1. Cellular Senescence
Numerous in vitro and animal studies provide evidence for senescence in cardiac and non-cardiac cells after exposure to chemotherapy or radiation (Tables 1–3). Cancer treatment effectively damages malignant cells but also causes unintended injury to nonmalignant cells. Treatment-induced DNA damage causes cell cycle arrest resulting in cellular senescence, telomere shortening, and is associated with a sterile pro-inflammatory state. Table 4 summarizes the clinical studies that demonstrate increased senescence in cancer survivors.
Table 4:
Clinical studies demonstrating elevation of cellular senescence biomarkers in cancer survivors
| Study | Study type | Cellular Senescence Biomarkers | Population of interest | Study groups | Methods | Findings |
|---|---|---|---|---|---|---|
| (Piegari, et al., 2013) | Case control | p16INK4a | Heart autopsies to examine cardiac progenitor cells among cancer patients | Cases: 6 heart autopsies from DOX- treated cancer patients who died from cardiomyopat hy, 2 who died from other causes Controls: 6 heart autopsies from cancer free individuals | p16INKa levels were collected from cardiac progenitor cells obtained from autopsy samples | p16INK4a levels were higher in cardiac progenitor cells from individuals who were exposed to DOX and deceased from cardiomyopathy |
| (Marcoux, et al., 2013) | Case control | p16INK4a | Survivors of childhood ALL who received chemotherapy and cranial radiation | Cases: 10 survivors of childhood ALL who received chemotherapy and cranial radiation Controls: 11 sibling controls without cancer | p16INKa levels were collected from skin biopsies of the scalp (exposed) and buttocks (unexposed) in the cases, and from the buttocks only in the controls | p16INK4a levels were higher in skin biopsies from the scalp compared to skin biopsies from the buttocks in ALL survivors at a mean of 12 years post-diagnosis. There was no difference between p16INK4a levels from biopsies of the buttocks in the cases compared to the controls |
| (Sanoff, et al., 2014) | Cohort | p16INK4a, p14ARF Telomere length Senescence-associated cytokines (VEGFA and MCP1) | Women treated for stage I-III breast cancer | Prospective cohort = 33 women who were treated with adjuvant chemotherapy Cross-sectional cohort = 176 women, 39% received adjuvant chemotherapy, 61% did not | Serum samples were drawn prior to chemotherapy exposure, immediately after exposure, 3 months, and 12 months later in the prospective cohort. A single sample obtained in cross-sectional cohort | Women who received chemotherapy had elevated p16INK4a ARF mRNA, and VEGFA and MCP1 expression immediately after and at 12 months after chemotherapy exposure. Telomere length was not affected |
| (Ariffin, et al., 2017) | Case control | Inflammatory cytokines (IL-2, IL-10, IL-17a) Telomere length High-sensitivity CRP | Survivors of childhood ALL | Cases: 87 young adult childhood ALL survivors with a median of 18 years off therapy Controls: 87 age and sex- matched volunteers without history of cancer |
Serum biomarkers measured | Survivors have significantly higher levels of inflammatory cytokines and shorter leukocyte telomere lengths compared to controls. Telomere lengths in survivors were similar to that of healthy individuals aged 20 years older |
| (Alfano, et al., 2017) | Cohort | Inflammatory cytokines (TNF-α, IL-6) | Women treated for stage I-III breast cancer | Survivor cohort: 209 women treated with multimodal therapy for breast cancer Controls = 106 women worked up and found to not have breast cancer | Baseline questionnaire, interview, and blood draw at work-up for both groups. Post-treatment assessments were performed at 6 and 12 months off- therapy for cases | Breast cancer survivors had significantly elevated inflammatory cytokines and higher burder of comorbid conditions compared to controls |
| (Uziel, et al., 2020) | Case control | DNA methylation status Telomere length | HSCT survivors | Cases: 26 survivors of allogenic- HSCT for a hematologic malignancy Controls: matched sibling donors | Blood samples collected from survivors and their matched sibling donors. Buccal swabs collected in survivors | WBC methylation and buccal cells predicted accelerated aging in survivors compared to controls. No difference in telomere length |
| (Sehl, et al., 2020) | Prospective cohort study | DNA methylation and epigenetic biomarkers | Stage 0- IIIA breast cancer patients | 72 women treated for breast cancer with surgery followed by adjuvant radiation alone (n=37) or chemoradiation (n=35) | Blood samples with epigenetic analysis collected pre- and posttreatment | Epigenetic markers of accelerated aging were most significant in patients treated with radiation compared to those treated with chemotherapy and radiation. |
| (Shachar, et al., 2020) | Prospective cohort study | P16INK4a | Stage I-III breast cancer patients | 146 women treated for breast cancer; 47.9% treated with anthracyclines, 34.9% treated without anthracyclines | Serum p16INK4a levels drawn prior to chemotherapy initiation and >/=60 days after completion of chemotherapy | P16INK4a expression was significantly elevated to levels equivalent to 23 to 26 years of accelerated aging in patients treated with anthracycline |
ALL, Acute lymphoblastic leukemia; CRP, C-reactive protein; DOX, Doxorubicin; HSCT, Hematopoietic stem cell transplant; IL-6, Interlukin-6; MCP1, Monocyte chemoattractant protein-1; TNF-α, Tumor Necrosis Factor alpha; VEGFA, Vascular endothelial growth factor A; WBC, White blood cell
In humans, p16INK4a is a measurable biomarker of cellular senescence utilized in many clinical studies. Among survivors of childhood acute lymphoblastic leukemia (ALL) treated with cranial radiation, a significantly higher level of p16INK4a is detected in skin biopsies of radiation-exposed tissue from the scalp compared to unexposed tissue from the buttocks (Marcoux, et al., 2013). Women who had undergone chemotherapy for breast cancer express higher levels of p16INK4a than those without cancer (Sanoff, et al., 2014). Breast cancer survivors treated with anthracycline-based regimens demonstrated significant increases in p16INK4a expression, equivalent to a 23- to 26- year acceleration in aging, compared to a more modest increase equivalent to 17 years of accelerated aging in those treated with non-anthracycline based treatments (Shachar, et al., 2020). Likewise, cardiac progenitor cells procured from heart biopsies taken during autopsies of DOX-treated cancer patients who died from cardiomyopathy have elevated levels of p16INK4a compared to age-matched unexposed controls (Piegari, et al., 2013). RNA sequencing of T cells in breast cancer patients demonstrates higher expressions of genes associated with cellular senescence. e.g., p16IKN4a, IL8, HMGA2, and CCL4 following chemotherapy with DOX and cyclophosphamide (Wood, et al., 2016).
Telomere length is another surrogate marker for cellular aging. Ariffin et al. did a case-control study examining telomere length in 87 long term young adult childhood ALL survivors compared to 87 age and sex-matched cancer-free controls (Ariffin, et al., 2017). Telomere length amongst survivors was shorter than the controls and similar to that predicted of healthy individuals 20 years older (Ariffin, et al., 2017). Shortened telomere length is also associated with increased risk for age-related diseases that are characterized by chronic inflammation (Kordinas, Ioannidis, & Chatzipanagiotou, 2016), such as insulin resistance and metabolic syndrome (Armstrong, et al., 2014; W. A. Smith, et al., 2014). Multiple studies show elevated inflammatory markers in survivors compared to their age and sex-matched controls without a history of cancer (Alfano, et al., 2017; Ariffin, et al., 2017; Sanoff, et al., 2014).
DNA methylation is a method used to measure the cellular age of leukocytes (Horvath, 2013; Weidner, et al., 2014). In a study examining 26 survivors of allogeneic hematopoietic stem cell transplant for hematologic malignancies, peripheral blood was collected from the recipients and their matched sibling donors. DNA methylation predicted a cellular age that was significantly higher in 62% of the transplanted recipients compared to the predicted cellular age in the donor (Uziel, et al., 2020). Another study demonstrated an increase in the epigenetic age acceleration following breast cancer treatment. Interestingly, those treated with radiation alone had more significant increases in age acceleration than those treated with chemotherapy and radiation (Sehl, Carroll, Horvath, & Bower, 2020). Taken together, these biomarkers are evidence for accelerated aging in survivors after exposure to cancer treatments.
4.2. Frailty
In addition to evidence for accelerated aging in cardiac and non-cardiac cells, cancer survivors display clinical signs of premature aging that manifest as frailty. Fried et al. were the first to describe a frailty or aging phenotype, defined as individuals who are vulnerable to adverse health outcomes, which often precedes the onset of chronic disease, and is a predictor of early mortality (Fried, et al., 2001). Fried developed clinical criteria for frailty, consisting of 5 components: 1) low muscle mass, 2) self-reported exhaustion, 3) low energy expenditure, 4) slow walking speed, and 5) weakness. Individuals who fulfill two of the five criteria are considered “pre-frail” and those who fulfill ≥ three criteria are “frail.” Table 5 summarizes literature examining premature functional aging in cancer survivors measured by “frailty.”
Table 5.
Clinical studies demonstrating increased frailty in cancer survivors
| Study | Study type | Frailty Measurement | Population of interest | Study groups | Methods | Findings |
|---|---|---|---|---|---|---|
| (K. K. Ness, et al., 2010) | Case control | Muscle strength Fitness Physical performance Participation | Survivors of childhood brain tumors treated at St. Jude or University of Minnesota | Cases: 78 survivors of childhood brain tumors Controls: 78 age, sex, and zip codematched population-based controls | In-home evaluations for muscle strength, fitness, physical performance, and an interview | Survivors with a median age of 22 demonstrated muscle strength and fitness similar to that expected of an individual in their 60’s. |
| (K. K. Ness, et al., 2012) | Cohort | Neuromuscular impairment | Survivors of childhood ALL enrolled in the St. Jude Lifetime Cohort Study | Participants : 415 survivors of childhood ALL Non-participants: 285 controls | Chart abstraction and tests for neuromuscular function | Survivors in their 30’s demonstrated neuromuscular impairments that limit physical performance similar to what is observed in individuals in their 60’s. This effect correlated with higher cumulative doses of vincristine and/or intrathecal methotrexate. |
| (Kirsten K. Ness, et al., 2013) | Cohort | Prefrailty Frailty Morbidity Mortality | Survivors of childhood cancer from the St. Jude Lifetime Cohort Study | Survivors: 1922 adult childhood cancer survivors Controls: 341 individuals without history of cancer | Chart extraction for medical records, questionnaires for frailty, and in-clinic assessments at follow up visits | Prevalence of prefrailty and frailty were higher in survivors compared to controls, particularly in women. Frailty was also associated with higher risk of chronic condition onset and with risk of death. |
| (Vatanen, et al., 2017) | Case control | Frailty Cardiovascular function Inflammatory markers Telomere length | Survivors of high-risk neuroblastoma who underwent high dose chemotherapy followed by autologous stem cell rescue | Cases: 19 survivors of high risk neuroblastoma Controls: 20 age and sex- matched volunteers | Assessed frailty using tests for muscle mass, energy expenditure, running, and weakness | Survivors were more likely to be “frail” and to report physical health limitations in vigorous activities compared to controls. Survivors also had higher CRP and shorter telomere length than controls. |
| (Smitherman, et al., 2018) | Cross sectional | Prefrailty Frailty Comorbid conditions | Adolescent- young adult cancer survivors treated at University of North Carolina | 271 survivors who were diagnosed between ages 15–39 | Frailty questionnaire to assess frailty status and comorbid conditions | Prevalence of prefrailty and frailty were high in AYA survivors. Frailty was associated with higher prevalence of comorbidities. |
| (Blair, et al., 2019) | Case control | Deficiencies in geriatric assessment domains All-cause mortality | Female survivors of any cancer in participants from the Iowa Women’s Health Study | Cases = 1723 female survivors of cancer Controls = 11,145 age matched cancer free women | Questionnaire to assess for Geriatric assessment domains and for all-cause mortality | Cancer survivors were more likely than controls to have deficits in multiple geriatric domains. Predicted 10- year mortality was higher in survivors than in controls. |
| (Hayek, et al., 2020) | Cohort | Prefrailty Frailty | Survivors of childhood cancer in the Childhood Cancer Survivor Study | Survivors: 10,899 survivors Controls: 2,097 Sibling controls | Baseline and follow up questionnaire | Demonstrated that prefrailty and frailty are higher in survivors compared to controls, and higher among females than in males. Exposure to cranial, abdominal/pelvic radiation, lung surgery, and comorbidities were also with risk of frailty. Findings suggest cancer therapies are a risk factor for the premature aging. |
ALL, Acute lymphoblastic leukemia; AYA, Adolescent and young adult
Several studies examined the prevalence of the prefrailty and frailty phenotypes in childhood cancer survivors (CCSs). In an analysis from the St. Jude Lifetime Cohort Study, 1,922 adult CCSs were assessed for prefrailty and frailty and compared them to 341 individuals without a history of cancer (Kirsten K. Ness, et al., 2013). The mean age of the survivors was 33.6 years, yet the prevalence of frailty was similar to that of persons aged 65 or older (Collard, Boter, Schoevers, & Oude Voshaar, 2012). Prefrailty was identified in 31.5% of female and 12.9% of male survivors compared to 7.8% of female and 4.6% of male controls. Additionally, frailty was observed in 13.1% of female and 2.7% of male survivors compared to no individuals in the age-matched control group fulfilling this criterion. Importantly, frailty was associated with an increased risk of chronic health conditions (RR 2.2, 95% CI 1.2–4.2) and a heightened risk for death (HR 2.6, 95% CI 1.2–6.2). In another large study comprised of 10,899 survivors in the Childhood Cancer Survivorship Study (CCSS), 6.4% of survivors were frail at a mean age of 37.6 years, compared to 2.2% in the sibling controls with a higher prevalence for frailty among females compared to males (Hayek, et al., 2020). Others examined smaller cohorts of survivors of disease-specific childhood cancers and also reported higher rates of frailty in those treated for brain tumors (K. K. Ness, et al., 2010), ALL (K. K. Ness, et al., 2012), and high-risk neuroblastoma (Vatanen, et al., 2017). Frailty and comorbid conditions were found to be more prevalent in survivors of adolescent/young adult-onset cancers as well (Smitherman, et al., 2018).
In a study examining frailty in long-term survivors of adult-onset cancers in women, a geriatric domain assessment tool was utilized to distinguish functional age from chronological age. This assessed physical function, comorbidities, nutritional status, mental health, and cognition. Cancer survivors with greater deficits had a higher risk of 10-year all-cause mortality. Cancer survivors without deficits still had a 1.3 to 1.4-fold excess risk of death compared to cancer-free controls (Blair, et al., 2019). Additionally, Hayek et al. demonstrated that cranial radiation, pelvic radiation ≥ 34 Gy, and lung surgery, all cancer-directed therapies, were associated with a higher prevalence of frailty even after adjusting for chronic diseases and modifiable lifestyle factors such as physical activity, smoking, and obesity (Hayek, et al., 2020). This evidence suggests that cancer survivors experience premature functional aging in excess of their chronological age due to exposures to cancer therapies, and this is associated with accelerated morbidity and mortality. Thus, there is a need for interventions to delay the onset of chronic disease and to promote healthy lifestyle behaviors in cancer survivors.
5. Prevention/Treatment Strategies Against Cancer Therapy-induced Cardiovascular Senescence
Cardiovascular complications are the second leading cause of death in cancer survivors. Therefore, mitigation of cancer therapy-induced cardiovascular complications will improve the quality and quantity of survivors’ lives. As discussed earlier, cardiovascular senescence emerges as an important mechanism in mediating these cardiovascular complications (Figure 1). Therefore, it is pivotal to develop effective protective strategies that can prevent or treat cancer therapy-induced cardiovascular senescence. Multiple strategies have been demonstrated to modulate senescence and prevent adverse effects of cellular senescence, called as “senotherapy”. These strategies can be divided into two broad categories: senomorphics and senolytics. While senomorphics modulate function and morphology of senescent cells without induction of death of senescent cells, senolytics can selectively induce death of senescent cells as illustrated in Figure 2 (H. Fuhrmann-Stroissnigg, et al., 2019; E. C. Kim & Kim, 2019).
Figure 1. Cancer therapy-induced senescence in cardiovascular cells.
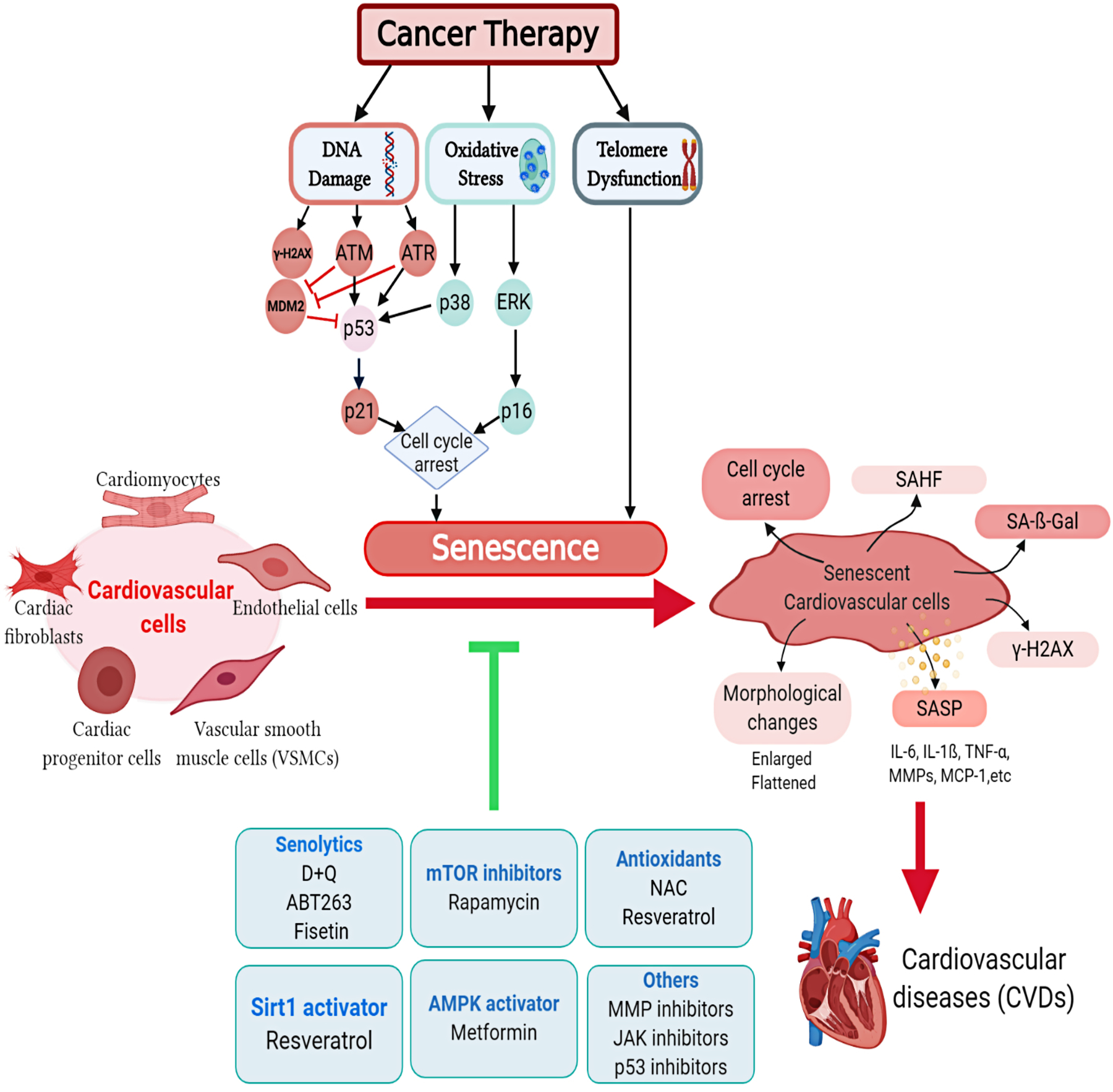
Cancer therapy induces senescence in different cardiovascular cells through a number of molecular mechanisms. Accumulation of senescent cells contributes to premature cardiovascular diseases in cancer survivors. Multiple interventions are proposed to mitigate cancer therapy-induced cardiovascular senescence. D+Q, Dasatinib and quercetin; IL-6, Interleukin-6; MMP, Matrix metalloproteinase inhibitor; NAC, N-acetyl cysteine; SA-β-gal, Senescence associated-β-galactosidase assay; SAHF, Senescence-associated heterochromatin foci; SASP, Senescence-associated secretory phenotype; TNF-α, Tumor Necrosis Factor-alpha.
Figure 2. Senotherapeutics as a strategy to counteract cancer therapy-induced cardiovascular senescence.
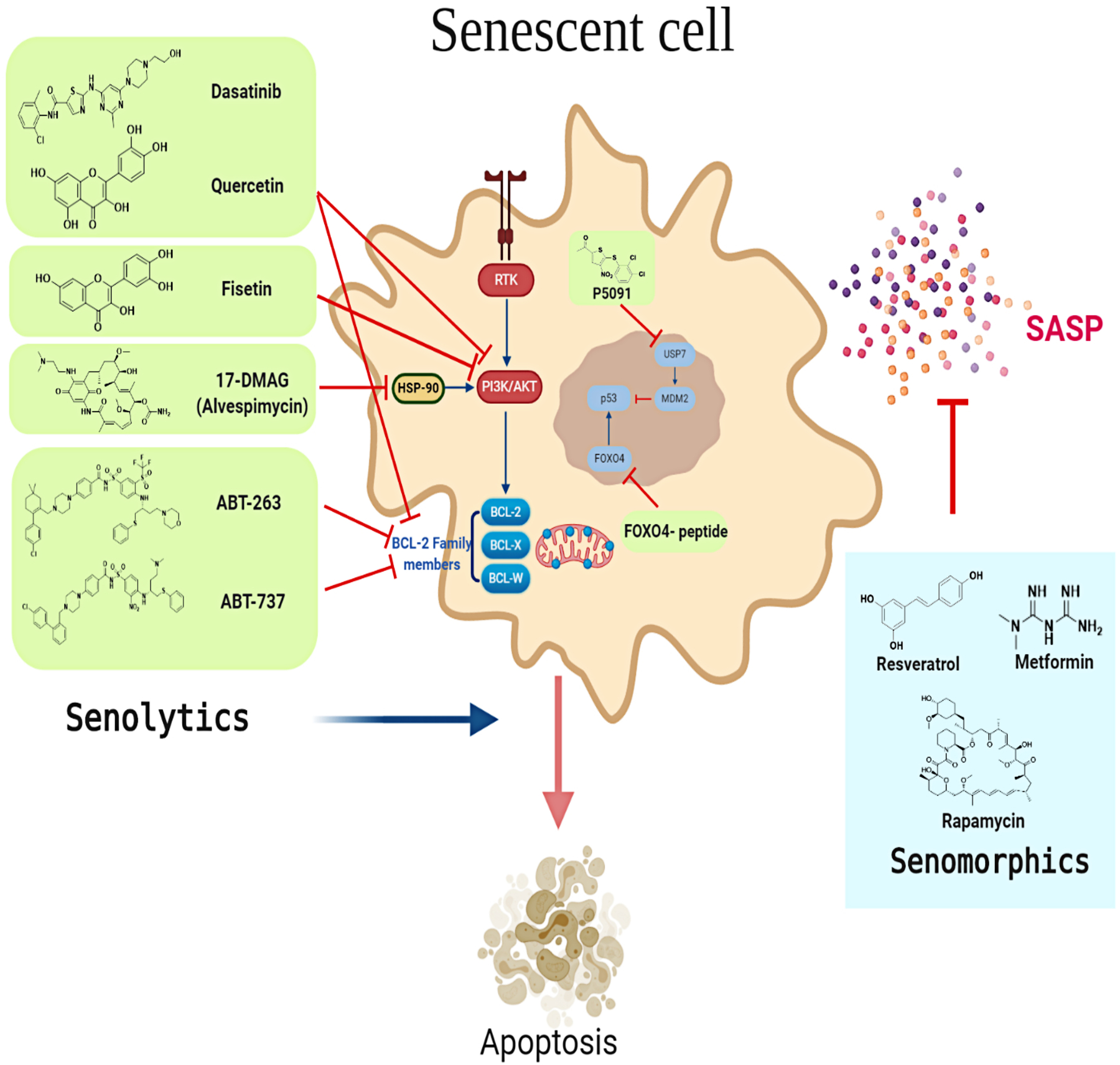
Several senotherapeutics have been developed to target and modulate the senescence phenotype. Senotherapeutics can be divided into senolytics (shown in light green rectangles) and senomorphics (shown in light blue rectangles). Senolytics target signaling pathways leading to apoptosis of senescent cells. Senomorphics modulate the senescence phenotype e.g. SASP without inducing death in senescent cells. SASP, Senescence-associated secretory phenotype; RTK, Receptor tyrosine kinase.
5.1. AMPK/mTOR/SIRT1 Pathway
Several longevity studies have suggested interventions targeting the AMPK/mTOR pathway to mitigate senescence and age-related diseases. These strategies include calorie restriction (Gelino, et al., 2016), and calorie restriction mimetics such as rapamycin, metformin, and resveratrol (D. L. Smith, Jr., Nagy, & Allison, 2010). The mechanisms of these treatments converge in autophagy induction mediated by AMPK and SIRT1 activation or mTOR inhibition (Kucheryavenko, Nelson, von Zglinicki, Korolchuk, & Carroll, 2019). Autophagy can facilitate the removal of senescent cells and hence decrease the spread of senescence to other cells (Pattison & Korolchuk, 2018). Unfortunately, cardiac autophagy levels are reduced with aging, which can precipitate cardiac diseases (Shirakabe, Ikeda, Sciarretta, Zablocki, & Sadoshima, 2016). Rapamycin is an FDA approved drug that inhibits mTORC1 and extends lifespan in mice (Evangelisti, Cenni, & Lattanzi, 2016). A recent study demonstrates that rapamycin supplementation in diet decreases arterial senescence markers and improves endothelial dysfunction in old mice (Lesniewski, et al., 2017). Rapamycin also activates nuclear factor erythroid 2-related factor 2 (NRF2) a regulator of the response to oxidative stress and suppresses SASP production via an NRF2-independent mechanism (R. Wang, et al., 2017). Other mTOR inhibitors, such as Torin 1 and PP242, are more potent than rapamycin (Khor & Wong, 2020), and newer rapalogs are currently being studied in clinical trials to improve age-related immunosenescence (Mannick, et al., 2018). Metformin, an FDA approved drug used to treat Type II diabetes, appears to attenuate multiple age-related diseases including CVD, and is thought to inhibit complex 1 of the mitochondria and AMPK, while activating SIRT1 (Longo, et al., 2015). Interestingly, metformin, like rapamycin attenuates SASP by suppressing NF-κB activation (Moiseeva, et al., 2013). Metformin abrogates the inflammaging state in T-cells isolated from old subjects (Bharath, et al., 2020). The protective effect of metformin is mediated via autophagy activation, suppressing STAT3 (regulator of age-dependent alterations in mitochondrial function), and improving mitochondrial function (Bharath, et al., 2020). A large clinical study, Targeting Aging with Metformin (TAME) trial, is designed to determine if metformin suppresses co-morbidities associated with old age (Barzilai, Crandall, Kritchevsky, & Espeland, 2016). Having a good safety profile with some studies demonstrating anticancer effects (Aljofan & Riethmacher, 2019; Zordoky, Bark, Soltys, Sung, & Dyck, 2014), would be key points for successful repurposing of metformin as an adjunct therapy to ameliorate TIS in cancer survivors, in case of demonstrating significant anti-aging effects in clinical trials.
SIRT1 can be activated using sirtuin-activating compounds (STACs) or by increasing NAD+ by using NAD+ precursors or inhibition of NAD+ hydrolase inhibitors (Escande, et al., 2013). Resveratrol, a SIRT1 agonist, has anti-aging effects (Sedding & Haendeler, 2007). Resveratrol abrogates oxidative stress-induced senescence in keratinocytes through AMPK-FOXO activation (Ido, et al., 2015). Recently, we demonstrate that the combined treatment of DOX followed by angiotensin II (Ang-II) increases the expression of multiple senescence-associated genes, including p21Cip1, growth arrest and DNA damage-inducible gamma (Gadd45g), and insulin-like growth factor-binding protein 3 (Igfbp3) (Matsumura, et al., 2018). Interestingly, co-administration of resveratrol with DOX corrects this upregulation and protects from the delayed DOX-induced detrimental cardiovascular effects (Matsumura, et al., 2018). In another study, co-treatment of neonatal cardiomyocytes with resveratrol and DOX suppresses the acetylation of p53 and decreases p21Cip1 levels (Maejima, et al., 2008). Despite promising anti-aging effects, the clinical utility of resveratrol is limited because of its poor bioavailability due to its first-pass intestinal/hepatic metabolism (Abdelgawad, Grant, & Zordoky, 2019). Spermidine, a natural product, has been shown to have cardio-protection and increase the lifespan of mice, which is thought to be mediated by autophagy activation (T. Eisenberg, et al., 2016). Spermidine also increases the lifespan in yeast through epigenetic modulation by inhibition of histone acetyltransferases (HATs) in aging yeast which results in hypoacetylation of chromatin and inhibition of oxidative stress and necrosis (Tobias Eisenberg, et al., 2009). IGF1-PI3k-AKT/mTOR pathway activation was shown to be involved in radiation-induced endothelial cell senescence (Panganiban & Day, 2013). Insulin-like growth factor-binding protein-7 (IGFBP7) inhibits cell proliferation via cell cycle arrest in the G1 phase. Overexpression of IGFBP7 is associated with tissue aging and poor prognosis in heart failure patients with preserved ejection fraction (Gandhi, et al., 2017). Specific inhibitors of this pathway, including the IGF-1R inhibitor (AG1024); the PI3k inhibitor (LY294002); and mTOR inhibitor (rapamycin), inhibit senescence of human pulmonary artery endothelial cells (HPAECs) following radiation (Panganiban & Day, 2013).
5.2. Antioxidants
Since oxidative stress plays a crucial role in the induction of senescence, antioxidants have been proposed as a promising protective strategy against cardiovascular senescence (Papaconstantinou, 2019). Pretreatment of endothelial progenitor cells with N-acetyl cysteine (NAC) attenuates DOX-induced senescence and decreases SA-β-gal activity (Spallarossa, et al., 2010). In addition, pretreatment of cardiomyocytes with the antioxidant NAC abrogates ROS and inhibits RIS (E. J. Kim, et al., 2015). Similarly, chronic treatment of microvascular endothelial cells with NAC significantly decreases RIS (Lafargue, et al., 2017). In the same study, post-radiation treatment of microvascular endothelial cells with a superoxide dismutase (SOD) mimetic, manganese metalloporphyrin (MnTBAP), decreases SA-β-gal positive cells. Transfection of HUVECs with KU86, a protein critical for DSB repair, inhibits low dose radiation-induced cellular senescence via SIRT1 and SOD2 activation (K. Wu, et al., 2019). Supplementation of L-citrulline and L-arginine, in high glucose-induced senescence model in HUVECs, reduces p16Ink4a expression and SA-β-gal activity, and enhances telomerase function (Tsuboi, Maeda, & Hayashi, 2018). This protective effect against high glucose-induced senescence is suggested to be mediated through inhibition of ROS production.
It is noteworthy to mention that antioxidants that interfere with ROS homeostasis may reduce the effectiveness of some cancer treatments that depend on oxidative stress in their mechanism of action (Fernando, Rupasinghe, & Hoskin, 2019). Multiple pro-oxidant cancer treatments, including some tyrosine kinase inhibitors and monoclonal antibodies (Teppo, Soini, & Karihtala, 2017), procarbazine (Renschler, 2004), and cisplatin (Berndtsson, et al., 2007) require activation of oxidative stress and accumulation of ROS in tumors at high levels that overwhelm the antioxidant capacity, ultimately inducing death of cancer cells via multiple mechanisms, including DNA damage, disrupting cell membrane, calcium channels activity, protein functions and signaling pathways, and epigenetic alterations (Perillo, et al., 2020). However, elevated ROS levels in tumors can also contribute to cancer treatment resistance (Diehn, et al., 2009; H. Gu, et al., 2018). Therefore, maintaining an optimal balance between ROS production and scavenging is required to optimize the efficacy of cancer therapy. Previous studies using combinations of anti-oxidants and cancer therapies reported conflicting results about their effects on the efficacy of cancer therapy. The effects of anti-oxidants on chemotherapeutic efficacy were systematically reviewed in (Block, et al., 2007). Multiple studies demonstrate anti-oxidants augment the anticancer effects through a number of mechanisms including reduction of P-gp expression and increase chemo/radiosensitivity of cancer cells (Ahmad, et al., 2010; Ma, et al., 2014; Tak, Lee, & Park, 2012; Wartenberg, et al., 2005). On the other hand, some studies report the reduction of anticancer effects following the combination with anti-oxidants (D’Andrea, 2005; Meng, et al., 2020). Therefore, it is necessary to evaluate the potential of anti-oxidants to prevent cancer therapy-induced senescence without undermining their anticancer effects.
5.3. Anti-inflammatory agents
Using anti-inflammatory agents can be another means to antagonize the pro-inflammatory SASP. NF-κB is a critical regulator of age-related gene expression and SASP (Adler, et al., 2007). Accordingly, inhibition of NF-κB suppresses senescence in a murine model of lymphoma (Chien, et al., 2011). This can be a valid approach since DOX is a potent activator of NF-κB. NF-κB Essential Modulator (NEMO) has been suggested as a potential target of senescence. Inhibition of these pathways using specific inhibitors or using knockout in vitro model has been shown to abrogate radiation-induced in endothelial cells and DNA-damage-induced senescence in murine dermal fibroblasts (Dong, et al., 2015; P. Meyer, et al., 2017). Likewise, inhibition of NF-κB activation in mice with premature onset senescence due to increased genotoxic stress prevents senescence and slows aging (Robinson, et al., 2018; Tilstra, et al., 2012; Yousefzadeh, et al., 2020). Finally, inhibition of p38-MAPK antagonizes SASP effects (Cosgrove, et al., 2014) and ameliorates low doses DOX-induced senescence of ventricular myocytes (Spallarossa, et al., 2009).
5.4. Senolytics
Senolytics are promising pharmacological compounds that selectively induce apoptotic cell death in senescent cells, which normally demonstrate resistance to apoptosis (Chang, et al., 2016; Kirkland, Tchkonia, Zhu, Niedernhofer, & Robbins, 2017; Zhu, et al., 2017; Zhu, et al., 2015). Numerous studies show that senolytics have beneficial effects in age-related disease models. One advantage of the use of senolytics is that they have the potential to reverse premature aging following cancer treatment. Unlike other strategies that should be administered before or concurrently with cancer treatment exposure, senolytics can be used by cancer survivors months to years following exposure. Up to ten compounds have been identified to have senolytic properties with dasatinib and quercetin (D+Q) and ABT263 (navitoclax) being the most studied senolytics.
D+Q protective effects have been studied in diabetic kidney disease (Hickson, et al., 2019), idiopathic pulmonary fibrosis (Schafer, et al., 2017), hepatic steatosis (Ogrodnik, et al., 2017), among several in vivo aging models. Indeed, D+Q treatment improves vasomotor function in aged mice with hypercholesterolemia (Roos, et al., 2016). Moreover, D+Q administration mitigates systolic cardiac dysfunction and abrogated end-systolic left ventricle dilation in 24-month-old mice (Zhu, et al., 2015). ABT263 is a selective inhibitor of the anti-apoptotic proteins BCL-2 and BCL-xL, which can selectively induce apoptotic cell death in senescent cells. ABT263 clears p16Ink4a-positive senescent cells in bone marrow following irradiation of mice (Chang, et al., 2016). Additionally, ABT263 administration in an aged murine model of myocardial infarction improves cardiac remodeling and overall survival (Walaszczyk, et al., 2019). Fisetin, a quercetin-related flavonoid, increases the lifespan of mice and ameliorates tissue damage after administration in aged animal models (Yousefzadeh, et al., 2018). Interestingly, fisetin demonstrates both senolytic and senomorphic properties depending on the cell type (Zhu, et al., 2017). Recently, targeted inhibition of ubiquitin-specific peptidase 7 (USP7) is proposed as a novel senolytic strategy (He, et al., 2020). Inhibition of USP7 is associated with an increase in the degradation of MDM2, which increases p53 and selectively induces apoptosis of senescent cells. In some cell types, the levels of p53 are observed to decrease after senescence initiation to maintain senescence, so the hypothesis is the sustained increase in p53 level induces apoptosis and selectively eliminate senescent cells. Interestingly, in vivo administration of USP7 inhibitor, P5091, successfully removes senescent cells and abrogated SASP production in DOX-treated p16Ink4a -3MR mice (He, et al., 2020). A recent screening of small library of compounds identified heat shock protein (HSP90) as a novel target for senolytics (Heike Fuhrmann-Stroissnigg, et al., 2017). Inhibition of HSP90 using 17-DMAG extends the healthspan and decreases senescence markers expression in progeroid mice (Heike Fuhrmann-Stroissnigg, et al., 2017). Additionally, blocking p53-FOXO4 interaction using cell-permeable peptide induces apoptosis of senescent cells leading to improvement of fitness and renal function in both progeroid and naturally aged mice (Baar, et al., 2017).
Senolytics are also demonstrated to protect against radiation-induced cardiovascular senescence. Both fisetin and BCL-XL inhibitors selectively induce cell death in senescent HUVECs following exposure to radiation (Zhu, et al., 2017). Additionally, recent studies demonstrate the protective effects of senolytics against other models of RIS. For instance, ABT263 decreases senescent cells and reverses radiation-induced pulmonary fibrosis in C57BL/6 mice (Pan, et al., 2017). In another study, genetic or pharmacologic (using ABT263) depletion of senescent astrocytes improves cognitive function in mice following whole-brain irradiation (WBI) (Yabluchanskiy, et al., 2020). Similarly, ABT263 oral administration in C57BL/6 mice mitigates total body irradiation-induced premature aging of the hematopoietic system and depletes senescent hematopoietic stem cells (HSCs) and muscle stem cells (MuSCs) (Chang, et al., 2016).
Despite the effectiveness of senolytics, potential toxicity is considered the most critical limitation and a major concern for their clinical utility. For instance, ABT263which was initially developed as anticancer drugs causes several side effects, such as nausea, vomiting, diarrhea, and thrombocytopenia because it can induce apoptosis of non-senescent cells including platelets (Rudin, et al., 2012). Therefore, optimizing local administration of senolytics and the development of novel senolytics with a good safety profile is a priority in the next few years. Another challenging aspect is to identify the optimum time during the post-cancer treatment period to administer senolytic drugs. Based on this evidence, senolytics administration should be considered as an attractive protective approach for the elimination of senescent cardiovascular cells following cancer therapy treatment. Additionally, senolytics should not interrupt cell cycle pathways as this can exaggerate cancer or hinder the anticancer effects of chemotherapy (Robbins, et al., 2020). Further in vitro and in vivo studies are warranted to identify the efficacy of senolytics against cancer therapy-induced senescence.
5.5. Other potential strategies to protect from therapy-induced senescence (TIS)
Oral matrix metalloproteinases (MMPs) inhibitors, such as doxycycline (sub-antimicrobial dosing) or ONO-481, attenuate DOX-induced cardiotoxicity and improve diastolic and systolic function and extracellular matrix remodeling in C57BL/6J mice (Chan, et al., 2020). Improving chromosomal segregation delays cellular senescence and decreases the SASP in human dermal fibroblasts cultures (Barroso-Vilares, et al., 2020). Pioglitazone mitigates endothelial cell senescence through telomerase activation-mediated mechanism (Werner, et al., 2011). Rivaroxaban inhibits signaling cascades between factor Xa and Insulin-like growth factor binding protein 5 (IGFBP5), and hence decreases Factor Xa-induced VSMCs senescence (Sanada, et al., 2017). The ATM-TRAF6-TAK1 axis plays an important role in SASP. Furthermore, ATM signaling drives NF-kB-dependent senescence, stem cell dysfunction and premature aging in response to genotoxic stress and ATM inhibitors block that (Zhao, et al., 2020). Interestingly, high-throughput screening identified ATM inhibitor, KU-60019, to have anti-senescence effects (Kang, et al., 2017). Inhibition of the JAK/STAT pathway using JAK inhibitor 1 alleviates senescence in endothelial cells and preadipocytes by targeting SASP (Xu, et al., 2015). Cardiomyocytes treated with DOX and Pifithrin-α, a p53 inhibitor, exhibit a marked reduction of p53 and p21Cip1 protein expression and reduce the number of SA-ß-gal positive cells (Maejima, et al., 2008). Moreover, downregulation of X-linked inhibitor of apoptosis (XIAP)-associated factor 1 (XAF1) attenuates DOX-induced endothelial cell senescence (Heo, et al., 2016). Pretreatment with peroxisome proliferator-activated receptor delta (PPARδ) agonist, L-165041, prevents low dose DOX-induced senescence in ventricular myocytes and H9c2 cells (Altieri, et al., 2012). PPARδ is the most abundant subtype in the heart. It plays an important role in cardiomyocytes survival through the regulation of cell cycle progression via BCL-6 -mediated mechanism. JNK and p-38 activation is necessary for PPARδ protective effect (Altieri, et al., 2012). In the same manner, another PPARδ agonist, GW501516, prevents Ang-II-induced senescence of VSMCs (H. J. Kim, et al., 2011).
6. Conclusions and Future Directions
It has become increasingly clear that most if not all cancer survivors experience some form of accelerated aging. Every clinician caring for cancer survivors has heard repeatedly from patients, “I just feel like I’m 10 years older…” or “They keep telling me it’s all just in my head…” when referencing the symptoms of frailty or accelerated aging. The ultimate proof of this not being the case lies in identifying interventions and designing clinical trials to halt or reverse the chronic consequences of cancer therapy-induced senescence. Indeed, interventions are desperately needed to arm clinicians with the tools necessary to help cancer survivors attain the highest quality of life possible.
Several challenges may hinder the development of the interventions that target senescence. First, a comprehensive understanding of the complicated interplay between senescence and other cell death mechanisms is necessary, so we can have a clearer view of the role of senescence in mediating cancer therapy-induced cardiotoxicity. Second, the lack of specific markers of senescence can impede the detection of senescence and proof of efficacy in preclinical and clinical studies. Additionally, developing more quantitative tools to estimate senescent cell burden will enable selective treatment of cancer survivors with high senescence burden following cancer treatment. Detection of local senescence, for example, in specific cell types, can provide a better tool to target these particular cells since the efficacy of anti-senescence strategies has been shown to be cell-type dependent (Sikora, Bielak-Zmijewska, & Mosieniak, 2019).
Addressing these challenges will ultimately open the door to a new era of interventional research and clinical care that improves the lives of cancer survivors across the lifespan. Moreover, this can be a fundamental first step toward precision medicine approach following cancer treatment. Rigorous clinical research steeped in team science with expertise in the biology of aging will be needed to properly vet such interventions, but the reward will be exponential for cancer survivors in desperate need for ways to combat accelerated aging.
Acknowledgments:
This research is supported by the National Heart, Lung, and Blood Institute, grant 1R01HL151740 (B.N.Z. and K.T.S.), the St. Baldrick’s Foundation for Childhood Cancer (Award ID 638335, B.N.Z.); and the National Institutes of Health’s National Center for Advancing Translational Sciences, grant UL1TR002494 (B.N.Z.). L.J.N. is supported by ES029603, AG063543 U19 AG056278, R01063543, and P01AG062413.
List of abbreviations:
- 5-FU
5-Fluoro-uracil
- ALL
Acute lymphoblastic leukemia
- AMPK
AMP-activated protein kinase
- BAECs
Bovine aortic endothelial cells
- CCSs
Childhood cancer survivors
- CDK
Cyclin-dependent kinase
- CF
Cardiac fibroblasts
- CHD
Coronary heart disease
- CPCs
Cardiac progenitor cells
- CVD
Cardiovascular diseases
- D+Q
Dasatinib + Quercetin
- DDR
DNA damage response
- DOX
Doxorubicin
- DSB
Double-strand break
- ECFCs
Endothelial colony-forming cells
- ECs
Endothelial cells
- eNOS
Endothelial nitric oxide synthase
- GBM
Glioblastoma
- HMVECs
Human microvascular endothelial cells
- HSCT
Hematopoietic stem cell transplant
- HUVECs
Human umbilical vein endothelial cells
- IL
Interleukin
- MAPK
Mitogen-activated protein kinase
- MMP
Matrix metalloproteinase
- mTOR
Mammalian target of rapamycin
- NAC
N-acetyl cysteine
- OIS
Oncogene-induced senescence
- RIHD
Radiation-induced heart disease
- RIS
Radiation-induced senescence
- ROS
Reactive oxygen species
- SAHF
Senescence-associated heterochromatic foci
- SASP
Senescence-associated secretory pattern
- SA-β-Gal
Senescence-associated beta-galactosidase
- SOD
Superoxide dismutase
- TIS
Therapy-induced senescence
- TNF-α
Tumor necrosis factor-alpha
- VEGF
Vascular endothelial cell growth factor
- VSMCs
Vascular smooth muscle cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest Statement: The authors declare no conflict of interest.
References
- Abdelgawad IY, Grant MK, & Zordoky BN (2019). Leveraging the cardio-protective and anticancer properties of resveratrol in cardio-oncology. Nutrients, 11, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acklin S, Zhang M, Du W, Zhao X, Plotkin M, Chang J, Campisi J, Zhou D, & Xia F (2020). Depletion of senescent-like neuronal cells alleviates cisplatin-induced peripheral neuropathy in mice. Sci Rep, 10, 14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, & Gil J (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol, 15, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, & Chang HY (2007). Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev, 21, 3244–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agha A, & Tarhini AA (2017). Adjuvant Therapy for Melanoma. Curr Oncol Rep, 19, 36. [DOI] [PubMed] [Google Scholar]
- Ahmad IU, Forman JD, Sarkar FH, Hillman GG, Heath E, Vaishampayan U, Cher ML, Andic F, Rossi PJ, & Kucuk O (2010). Soy isoflavones in conjunction with radiation therapy in patients with prostate cancer. Nutr Cancer, 62, 996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfano CM, Peng J, Andridge RR, Lindgren ME, Povoski SP, Lipari AM, Agnese DM, Farrar WB, Yee LD, Carson WE 3rd, & Kiecolt-Glaser JK (2017). Inflammatory Cytokines and Comorbidity Development in Breast Cancer Survivors Versus Noncancer Controls: Evidence for Accelerated Aging? J Clin Oncol, 35, 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljofan M, & Riethmacher D (2019). Anticancer activity of metformin: a systematic review of the literature. Future Sci OA, 5, FSO410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri P, Barisione C, Lazzarini E, Garuti A, Bezante GP, Canepa M, Spallarossa P, Tocchetti CG, Bollini S, Brunelli C, & Ameri P (2016). Testosterone Antagonizes Doxorubicin-Induced Senescence of Cardiomyocytes. J Am Heart Assoc, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri P, Murialdo R, Barisione C, Lazzarini E, Garibaldi S, Fabbi P, Ruggeri C, Borile S, Carbone F, Armirotti A, Canepa M, Ballestrero A, Brunelli C, Montecucco F, Ameri P, & Spallarossa P (2017). 5-fluorouracil causes endothelial cell senescence: potential protective role of glucagon-like peptide 1. Br J Pharmacol, 174, 3713–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri P, Spallarossa P, Barisione C, Garibaldi S, Garuti A, Fabbi P, Ghigliotti G, & Brunelli C (2012). Inhibition of doxorubicin-induced senescence by PPARdelta activation agonists in cardiac muscle cells: cooperation between PPARdelta and Bcl6. Plos One, 7, e46126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariffin H, Azanan MS, Abd Ghafar SS, Oh L, Lau KH, Thirunavakarasu T, Sedan A, Ibrahim K, Chan A, Chin TF, Liew FF, Jeyamogan S, Rosli ES, Baharudin R, Yap TY, Skinner R, Lum SH, & Hainaut P (2017). Young adult survivors of childhood acute lymphoblastic leukemia show evidence of chronic inflammation and cellular aging. Cancer, 123, 4207–4214. [DOI] [PubMed] [Google Scholar]
- Armstrong GT, Kawashima T, Leisenring W, Stratton K, Stovall M, Hudson MM, Sklar CA, Robison LL, & Oeffinger KC (2014). Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. J Clin Oncol, 32, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asensio-Lopez MC, Soler F, Pascual-Figal D, Fernandez-Belda F, & Lax A (2017). Doxorubicin-induced oxidative stress: The protective effect of nicorandil on HL-1 cardiomyocytes. Plos One, 12, e0172803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astle MV, Hannan KM, Ng PY, Lee RS, George AJ, Hsu AK, Haupt Y, Hannan RD, & Pearson RB (2012). AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene, 31, 1949–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimzadeh O, Sievert W, Sarioglu H, Merl-Pham J, Yentrapalli R, Bakshi MV, Janik D, Ueffing M, Atkinson MJ, & Multhoff G (2015). Integrative proteomics and targeted transcriptomics analyses in cardiac endothelial cells unravel mechanisms of long-term radiation-induced vascular dysfunction. Journal of proteome research, 14, 1203–1219. [DOI] [PubMed] [Google Scholar]
- Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, van der Pluijm I, Essers J, van Cappellen WA, van IWF, Houtsmuller AB, Pothof J, de Bruin RWF, Madl T, Hoeijmakers JHJ, Campisi J, & de Keizer PLJ (2017). Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell, 169, 132–147 e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, Van De Sluis B, Kirkland JL, & van Deursen JM (2011). Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders. Nature, 479, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal N, Blanco JG, Sharma UC, Pokharel S, Shisler S, & Lipshultz SE (2020). Cardiovascular diseases in survivors of childhood cancer. Cancer Metastasis Rev, 39, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ, Baker J, & Donnelly LE (2019). Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am J Respir Crit Care Med, 200, 556–564. [DOI] [PubMed] [Google Scholar]
- Barroso-Vilares M, Macedo JC, Reis M, Warren JD, Compton D, & Logarinho E (2020). Small-molecule inhibition of aging-associated chromosomal instability delays cellular senescence. EMBO reports, 21, e49248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai N, Crandall JP, Kritchevsky SB, & Espeland MA (2016). Metformin as a Tool to Target Aging. Cell Metab, 23, 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bent EH, Gilbert LA, & Hemann MT (2016). A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes & development, 30, 1811–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, & Frisen J (2009). Evidence for cardiomyocyte renewal in humans. Science, 324, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndtsson M, Hagg M, Panaretakis T, Havelka AM, Shoshan MC, & Linder S (2007). Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int J Cancer, 120, 175–180. [DOI] [PubMed] [Google Scholar]
- Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, Liu J, Jiang K, Liu R, Guo Z, Deeney J, Apovian CM, Snyder-Cappione J, Hawk GS, Fleeman RM, Pihl RMF, Thompson K, Belkina AC, Cui L, Proctor EA, Kern PA, & Nikolajczyk BS (2020). Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielak-Zmijewska A, Wnuk M, Przybylska D, Grabowska W, Lewinska A, Alster O, Korwek Z, Cmoch A, Myszka A, Pikula S, Mosieniak G, & Sikora E (2014). A comparison of replicative senescence and doxorubicin-induced premature senescence of vascular smooth muscle cells isolated from human aorta. Biogerontology, 15, 47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biran A, Zada L, Abou Karam P, Vadai E, Roitman L, Ovadya Y, Porat Z, & Krizhanovsky V (2017). Quantitative identification of senescent cells in aging and disease. Aging Cell, 16, 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny O. V. L. a. M. V. (2010). DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging, 2, 924–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair CK, Jacobs DR Jr, Demark-Wahnefried W, Cohen HJ, Morey MC, Robien K, & Lazovich D (2019). Effects of cancer history on functional age and mortality. Cancer, 125, 4303–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block KI, Koch AC, Mead MN, Tothy PK, Newman RA, & Gyllenhaal C (2007). Impact of antioxidant supplementation on chemotherapeutic efficacy: a systematic review of the evidence from randomized controlled trials. Cancer Treat Rev, 33, 407–418. [DOI] [PubMed] [Google Scholar]
- Boccardi V, & Mecocci P (2020). The Importance of Cellular Senescence in Frailty and Cardiovascular Diseases. Adv Exp Med Biol, 1216, 79–86. [DOI] [PubMed] [Google Scholar]
- Bourgeois B, & Madl T (2018). Regulation of cellular senescence via the FOXO4-p53 axis. FEBS letters, 592, 2083–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calls A, Torres-Espin A, Navarro X, Yuste VJ, Udina E, & Bruna J (2020). Cisplatin-induced peripheral neuropathy is associated to neuronal senescence-like response. Neuro Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, & d’Adda di Fagagna F (2007). Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol, 8, 729–740. [DOI] [PubMed] [Google Scholar]
- Campisi J, Kim SH, Lim CS, & Rubio M (2001). Cellular senescence, cancer and aging: the telomere connection. Exp Gerontol, 36, 1619–1637. [DOI] [PubMed] [Google Scholar]
- Carlson BW, Craft MA, Carlson JR, Razaq W, Deardeuff KK, & Benbrook DM (2018). Accelerated vascular aging and persistent cognitive impairment in older female breast cancer survivors. Geroscience, 40, 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella G, Munk R, Kim KM, Piao Y, De S, Abdelmohsen K, & Gorospe M (2019). Transcriptome signature of cellular senescence. Nucleic acids research, 47, 7294–7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan BYH, Roczkowsky A, Cho WJ, Poirier M, Sergi C, Keschrumrus V, Churko JM, Granzier H, & Schulz R (2020). MMP inhibitors attenuate doxorubicin cardiotoxicity by preventing intracellular and extracellular matrix remodelling. Cardiovascular Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, & Zhou D (2016). Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med, 22, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HZ, Wang F, Gao P, Pei JF, Liu Y, Xu TT, Tang X, Fu WY, Lu J, Yan YF, Wang XM, Han L, Zhang ZQ, Zhang R, Zou MH, & Liu DP (2016). Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ Res, 119, 1076–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, & Lowe SW (2011). Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev, 25, 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, & van Deursen JM (2016). Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science, 354, 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimenti C, Kajstura J, Torella D, Urbanek K, Heleniak H, Colussi C, Di Meglio F, Nadal-Ginard B, Frustaci A, Leri A, Maseri A, & Anversa P (2003). Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res, 93, 604–613. [DOI] [PubMed] [Google Scholar]
- Cho JH, Kim E-C, Son Y, Lee D-W, Park YS, Choi JH, Cho K-H, Kwon K-S, & Kim J-R (2020). CD9 induces cellular senescence and aggravates atherosclerotic plaque formation. Cell Death & Differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioroiu C, & Weimer LH (2017). Update on Chemotherapy-Induced Peripheral Neuropathy. Curr Neurol Neurosci Rep, 17, 47. [DOI] [PubMed] [Google Scholar]
- Citrin DE, Shankavaram U, Horton JA, Shield W 3rd, Zhao S, Asano H, White A, Sowers A, Thetford A, & Chung EJ (2013). Role of type II pneumocyte senescence in radiation-induced lung fibrosis. J Natl Cancer Inst, 105, 1474–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cmielova J, Havelek R, Jiroutova A, Kohlerova R, Seifrtova M, Muthna D, Vavrova J, & Rezacova M (2011). DNA damage caused by ionizing radiation in embryonic diploid fibroblasts WI-38 induces both apoptosis and senescence. Physiol Res, 60, 667–677. [DOI] [PubMed] [Google Scholar]
- Collard RM, Boter H, Schoevers RA, & Oude Voshaar RC (2012). Prevalence of frailty in community-dwelling older persons: a systematic review. J Am Geriatr Soc, 60, 1487–1492. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, & Campisi J (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol, 6, 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, & Blau HM (2014). Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med, 20, 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coviello JS (2018). Cardio-oncology: A Subspecialty in its Infancy. J Adv Pract Oncol, 9, 154–155. [PMC free article] [PubMed] [Google Scholar]
- Cupit-Link MC, Kirkland JL, Ness KK, Armstrong GT, Tchkonia T, LeBrasseur NK, Armenian SH, Ruddy KJ, & Hashmi SK (2017). Biology of premature ageing in survivors of cancer. ESMO Open, 2, e000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, & Jackson SP (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature, 426, 194–198. [DOI] [PubMed] [Google Scholar]
- D’Andrea GM (2005). Use of antioxidants during chemotherapy and radiotherapy should be avoided. CA Cancer J Clin, 55, 319–321. [DOI] [PubMed] [Google Scholar]
- da Silva PFL, Ogrodnik M, Kucheryavenko O, Glibert J, Miwa S, Cameron K, Ishaq A, Saretzki G, Nagaraja-Grellscheid S, Nelson G, & von Zglinicki T (2019). The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell, 18, e12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Falco E, Carnevale R, Pagano F, Chimenti I, Fianchini L, Bordin A, Siciliano C, Monticolo R, Equitani F, Carrizzo A, Peruzzi M, Vecchione C, Rubattu S, Sciarretta S, & Frati G (2016). Role of NOX2 in mediating doxorubicin-induced senescence in human endothelial progenitor cells. Mech Ageing Dev, 159, 37–43. [DOI] [PubMed] [Google Scholar]
- de Magalhaes JP, Chainiaux F, Remacle J, & Toussaint O (2002). Stress-induced premature senescence in BJ and hTERT-BJ1 human foreskin fibroblasts. FEBS Lett, 523, 157–162. [DOI] [PubMed] [Google Scholar]
- Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, Alston S, Academia EC, Kilmarx S, Valdovinos A, Wang B, de Bruin A, Kennedy BK, Melov S, Zhou D, Sharpless NE, Muss H, & Campisi J (2017). Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov, 7, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge R-M, Vijg J, Van Steeg H, & Dollé ME (2014). An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Developmental cell, 31, 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL, & Clarke MF (2009). Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature, 458, 780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, & et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A, 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Tong F, Qian C, Zhang R, Dong J, Wu G, & Hu Y (2015). NEMO modulates radiation-induced endothelial senescence of human umbilical veins through NF-kappaB signal pathway. Radiat Res, 183, 82–93. [DOI] [PubMed] [Google Scholar]
- Dowling EC, Chawla N, Forsythe LP, de Moor J, McNeel T, Rozjabek HM, Ekwueme DU, & Yabroff KR (2013). Lost productivity and burden of illness in cancer survivors with and without other chronic conditions. Cancer, 119, 3393–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drafts BC, Twomley KM, D’Agostino R Jr., Lawrence J, Avis N, Ellis LR, Thohan V, Jordan J, Melin SA, Torti FM, Little WC, Hamilton CA, & Hundley WG (2013). Low to moderate dose anthracycline-based chemotherapy is associated with early noninvasive imaging evidence of subclinical cardiovascular disease. JACC Cardiovasc Imaging, 6, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A, Tong M, Ruckenstuhl C, Dammbrueck C, Gross AS, Herbst V, Magnes C, Trausinger G, Narath S, Meinitzer A, Hu Z, Kirsch A, Eller K, Carmona-Gutierrez D, Buttner S, Pietrocola F, Knittelfelder O, Schrepfer E, Rockenfeller P, Simonini C, Rahn A, Horsch M, Moreth K, Beckers J, Fuchs H, Gailus-Durner V, Neff F, Janik D, Rathkolb B, Rozman J, de Angelis MH, Moustafa T, Haemmerle G, Mayr M, Willeit P, von Frieling-Salewsky M, Pieske B, Scorrano L, Pieber T, Pechlaner R, Willeit J, Sigrist SJ, Linke WA, Muhlfeld C, Sadoshima J, Dengjel J, Kiechl S, Kroemer G, Sedej S, & Madeo F (2016). Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med, 22, 1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L, Hartl R, Schraml E, Criollo A, Megalou E, Weiskopf D, Laun P, Heeren G, Breitenbach M, Grubeck-Loebenstein B, Herker E, Fahrenkrog B, Fröhlich K-U, Sinner F, Tavernarakis N, Minois N, Kroemer G, & Madeo F (2009). Induction of autophagy by spermidine promotes longevity. Nature cell biology, 11, 1305–1314. [DOI] [PubMed] [Google Scholar]
- Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, O’Neil L, White TA, Sinclair DA, & Chini EN (2013). Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes, 62, 1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelisti C, Cenni V, & Lattanzi G (2016). Potential therapeutic effects of the MTOR inhibitors for preventing ageing and progeria-related disorders. Br J Clin Pharmacol, 82, 1229–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald JA, Desotelle JA, Wilding G, & Jarrard DF (2010). Therapy-induced senescence in cancer. J Natl Cancer Inst, 102, 1536–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber J, Wingerter A, Neu MA, Henninger N, Eckerle S, Munzel T, Lackner KJ, Beutel ME, Blettner M, Rathmann W, Peters A, Meisinger C, Linkohr B, Neuhauser H, Kaatsch P, Spix C, Schneider A, Merzenich H, Panova-Noeva M, Prochaska JH, & Wild PS (2018). Burden of cardiovascular risk factors and cardiovascular disease in childhood cancer survivors: data from the German CVSS-study. Eur Heart J, 39, 1555–1562. [DOI] [PubMed] [Google Scholar]
- Fafián-Labora JA, & O’Loghlen A Classical and Nonclassical Intercellular Communication in Senescence and Ageing. Trends in Cell Biology. [DOI] [PubMed] [Google Scholar]
- Fallah M, Mohammadi H, Shaki F, Hosseini-Khah Z, Moloudizargari M, Dashti A, Ziar A, Mohammadpour A, Mirshafa A, Modanloo M, & Shokrzadeh M (2019). Doxorubicin and liposomal doxorubicin induce senescence by enhancing nuclear factor kappa B and mitochondrial membrane potential. Life Sci, 232, 116677. [DOI] [PubMed] [Google Scholar]
- Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, LeBrasseur NK, Drake MT, Pignolo RJ, Pirtskhalava T, Tchkonia T, Oursler MJ, Kirkland JL, & Khosla S (2017). Targeting cellular senescence prevents age-related bone loss in mice. Nat Med, 23, 1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando W, Rupasinghe HPV, & Hoskin DW (2019). Dietary phytochemicals with anti-oxidant and pro-oxidant activities: A double-edged sword in relation to adjuvant chemotherapy and radiotherapy? Cancer Lett, 452, 168–177. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Cunha-Oliveira T, Simoes RF, Carvalho FS, Burgeiro A, Nordgren K, Wallace KB, & Oliveira PJ (2017). Altered mitochondrial epigenetics associated with subchronic doxorubicin cardiotoxicity. Toxicology, 390, 63–73. [DOI] [PubMed] [Google Scholar]
- Ferreira LL, Cunha-Oliveira T, Veloso CD, Costa CF, Wallace KB, & Oliveira PJ (2019). Single nanomolar doxorubicin exposure triggers compensatory mitochondrial responses in H9c2 cardiomyoblasts. Food Chem Toxicol, 124, 450–461. [DOI] [PubMed] [Google Scholar]
- Focaccetti C, Bruno A, Magnani E, Bartolini D, Principi E, Dallaglio K, Bucci EO, Finzi G, Sessa F, Noonan DM, & Albini A (2015). Effects of 5-fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ROS production in endothelial cells and cardiomyocytes. Plos One, 10, e0115686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes S, Claessen G, Howden EJ, Daly RM, Fraser SF, & La Gerche A (2020). The Utility of Cardiac Reserve for the Early Detection of Cancer Treatment-Related Cardiac Dysfunction: A Comprehensive Overview. Front Cardiovasc Med, 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, & De Benedictis G (2000). Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci, 908, 244–254. [DOI] [PubMed] [Google Scholar]
- Freije JM, & Lopez-Otin C (2012). Reprogramming aging and progeria. Curr Opin Cell Biol, 24, 757–764. [DOI] [PubMed] [Google Scholar]
- Fried LP, Tangen CM, Walston J, Newman AB, Hirsch C, Gottdiener J, Seeman T, Tracy R, Kop WJ, Burke G, McBurnie MA, & Cardiovascular Health Study Collaborative Research, G. (2001). Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci, 56, M146–156. [DOI] [PubMed] [Google Scholar]
- Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, Grassi D, Gregg SQ, Stripay JL, Dorronsoro A, Corbo L, Tang P, Bukata C, Ring N, Giacca M, Li X, Tchkonia T, Kirkland JL, Niedernhofer LJ, & Robbins PD (2017). Identification of HSP90 inhibitors as a novel class of senolytics. Nature Communications, 8, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann-Stroissnigg H, Santiago FE, Grassi D, Ling Y, Niedernhofer LJ, & Robbins PD (2019). SA-beta-Galactosidase-Based Screening Assay for the Identification of Senotherapeutic Drugs. J Vis Exp. [DOI] [PubMed] [Google Scholar]
- Gandhi PU, Chow SL, Rector TS, Krum H, Gaggin HK, McMurray JJ, Zile MR, Komajda M, McKelvie RS, Carson PE, Januzzi JL Jr., & Anand IS (2017). Prognostic Value of Insulin-Like Growth Factor-Binding Protein 7 in Patients with Heart Failure and Preserved Ejection Fraction. J Card Fail, 23, 20–28. [DOI] [PubMed] [Google Scholar]
- Gardner SE, Humphry M, Bennett MR, & Clarke MC (2015). Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1alpha-Dependent Senescence-Associated Secretory Phenotype. Arterioscler Thromb Vasc Biol, 35, 1963–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelino S, Chang JT, Kumsta C, She X, Davis A, Nguyen C, Panowski S, & Hansen M (2016). Intestinal Autophagy Improves Healthspan and Longevity in C. elegans during Dietary Restriction. PLOS Genetics, 12, e1006135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbunova V, Seluanov A, & Pereira-Smith OM (2002). Expression of human telomerase (hTERT) does not prevent stress-induced senescence in normal human fibroblasts but protects the cells from stress-induced apoptosis and necrosis. J Biol Chem, 277, 38540–38549. [DOI] [PubMed] [Google Scholar]
- Gorenne I, Kavurma M, Scott S, & Bennett M (2006). Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc Res, 72, 9–17. [DOI] [PubMed] [Google Scholar]
- Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, Jurk D, Maier AB, Narita M, Niedernhofer L, Passos JF, Robbins PD, Schmitt CA, Sedivy J, Vougas K, von Zglinicki T, Zhou D, Serrano M, & Demaria M (2019). Cellular Senescence: Defining a Path Forward. Cell, 179, 813–827. [DOI] [PubMed] [Google Scholar]
- Greten TF, & Eggert T (2017). Cellular senescence associated immune responses in liver cancer. Hepat Oncol, 4, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Huang T, Shen Y, Liu Y, Zhou F, Jin Y, Sattar H, & Wei Y (2018). Reactive oxygen species-mediated tumor microenvironment transformation: the mechanism of radioresistant gastric cancer. Oxidative Medicine and Cellular Longevity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, & Kitamura M (2012). Sensitive detection and monitoring of senescence-associated secretory phenotype by SASP-RAP assay. PLoS One, 7, e52305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo RM, Xu WM, Lin JC, Mo LQ, Hua XX, Chen PX, Wu K, Zheng DD, & Feng JQ (2013). Activation of the p38 MAPK/NF-kappaB pathway contributes to doxorubicin-induced inflammation and cytotoxicity in H9c2 cardiac cells. Mol Med Rep, 8, 603–608. [DOI] [PubMed] [Google Scholar]
- Hayek S, Gibson TM, Leisenring WM, Guida JL, Gramatges MM, Lupo PJ, Howell RM, Oeffinger KC, Bhatia S, Edelstein K, Hudson MM, Robison LL, Nathan PC, Yasui Y, Krull KR, Armstrong GT, & Ness KK (2020). Prevalence and Predictors of Frailty in Childhood Cancer Survivors and Siblings: A Report From the Childhood Cancer Survivor Study. J Clin Oncol, 38, 232–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L, & Moorhead PS (1961). The serial cultivation of human diploid cell strains. Experimental Cell Research, 25, 585–621. [DOI] [PubMed] [Google Scholar]
- He H, Wang L, Qiao Y, Zhou Q, Li H, Chen S, Yin D, Huang Q, & He M (2019). Doxorubicin Induces Endotheliotoxicity and Mitochondrial Dysfunction via ROS/eNOS/NO Pathway. Front Pharmacol, 10, 1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Li W, Lv D, Zhang X, Zhang X, Ortiz YT, Budamagunta V, Campisi J, Zheng G, & Zhou D (2020). Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell, e13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Thummuri D, Zheng G, Okunieff P, Citrin DE, Vujaskovic Z, & Zhou D (2019). Cellular senescence and radiation-induced pulmonary fibrosis. Transl Res, 209, 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JI, Kim W, Choi KJ, Bae S, Jeong JH, & Kim KS (2016). XIAP-associating factor 1, a transcriptional target of BRD7, contributes to endothelial cell senescence. Oncotarget, 7, 5118–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL, Kellogg TA, Khosla S, Koerber DM, Lagnado AB, Lawson DK, LeBrasseur NK, Lerman LO, McDonald KM, McKenzie TJ, Passos JF, Pignolo RJ, Pirtskhalava T, Saadiq IM, Schaefer KK, Textor SC, Victorelli SG, Volkman TL, Xue A, Wentworth MA, Wissler Gerdes EO, Zhu Y, Tchkonia T, & Kirkland JL (2019). Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine, 47, 446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A, Sadda J, LaBarge MA, & Hurria A (2020). How cancer therapeutics cause accelerated aging: insights from the hallmarks of aging. Journal of Geriatric Oncology, 11, 191–193. [DOI] [PubMed] [Google Scholar]
- Hodjat M, Haller H, Dumler I, & Kiyan Y (2013). Urokinase receptor mediates doxorubicin-induced vascular smooth muscle cell senescence via proteasomal degradation of TRF2. J Vasc Res, 50, 109–123. [DOI] [PubMed] [Google Scholar]
- Hoffmann J, Haendeler J, Aicher A, Rossig L, Vasa M, Zeiher AM, & Dimmeler S (2001). Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: important role of nitric oxide. Circ Res, 89, 709–715. [DOI] [PubMed] [Google Scholar]
- Horvath S (2013). DNA methylation age of human tissues and cell types. Genome Biology, 14, 3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu-Lowe DD, Zou HY, Grazzini ML, Hallin ME, Wickman GR, Amundson K, Chen JH, Rewolinski DA, Yamazaki S, Wu EY, McTigue MA, Murray BW, Kania RS, O’Connor P, Shalinsky DR, & Bender SL (2008). Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res, 14, 7272–7283. [DOI] [PubMed] [Google Scholar]
- Hu Y, Xia W, & Hou M (2018). Macrophage migration inhibitory factor serves a pivotal role in the regulation of radiation-induced cardiac senescencethrough rebalancing the microRNA-34a/sirtuin 1 signaling pathway. Int J Mol Med, 42, 2849–2858. [DOI] [PubMed] [Google Scholar]
- Ido Y, Duranton A, Lan F, Weikel KA, Breton L, & Ruderman NB (2015). Resveratrol prevents oxidative stress-induced senescence and proliferative dysfunction by activating the AMPK-FOXO3 cascade in cultured primary human keratinocytes. Plos One, 10, e0115341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi K, Sakimoto I, Kataoka K, Ohta K, & Miura M (2007). Radiation-induced senescence-like phenotype in proliferating and plateau-phase vascular endothelial cells. Experimental Cell Research, 313, 3326–3336. [DOI] [PubMed] [Google Scholar]
- Jahn SK, Hennicke T, Kassack M, Drews L, Reichert AS, & Fritz G (2020). Distinct influence of the anthracycline derivative doxorubicin on the differentiation efficacy of mESC-derived endothelial progenitor cells. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 118711. [DOI] [PubMed] [Google Scholar]
- Jang WJ, Choi DY, & Jeon IS (2013). Vascular endothelial dysfunction after anthracyclines treatment in children with acute lymphoblastic leukemia. Korean J Pediatr, 56, 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, Baker DJ, van Deursen JM, Campisi J, & Elisseeff JH (2017). Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med, 23, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HJ, Byun HO, Jee BA, Min S, Jeoun UW, Lee YK, Seo Y, Woo HG, & Yoon G (2017). The Ubiquitin-like with PHD and Ring Finger Domains 1 (UHRF1)/DNA Methyltransferase 1 (DNMT1) Axis Is a Primary Regulator of Cell Senescence. J Biol Chem, 292, 3729–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandula T, Farrar MA, Kiernan MC, Krishnan AV, Goldstein D, Horvath L, Grimison P, Boyle F, Baron-Hay S, & Park SB (2017). Neurophysiological and clinical outcomes in chemotherapy-induced neuropathy in cancer. Clin Neurophysiol, 128, 1166–1175. [DOI] [PubMed] [Google Scholar]
- Kang HT, Park JT, Choi K, Kim Y, Choi HJC, Jung CW, Lee YS, & Park SC (2017). Chemical screening identifies ATM as a target for alleviating senescence. Nat Chem Biol, 13, 616–623. [DOI] [PubMed] [Google Scholar]
- Khan SY, Awad EM, Oszwald A, Mayr M, Yin X, Waltenberger B, Stuppner H, Lipovac M, Uhrin P, & Breuss JM (2017). Premature senescence of endothelial cells upon chronic exposure to TNFalpha can be prevented by N-acetyl cysteine and plumericin. Sci Rep, 7, 39501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor E-S, & Wong P-F (2020). The roles of MTOR and miRNAs in endothelial cell senescence. Biogerontology. [DOI] [PubMed] [Google Scholar]
- Khosla S, Farr JN, & Kirkland JL (2018). Inhibiting Cellular Senescence: A New Therapeutic Paradigm for Age-Related Osteoporosis. J Clin Endocrinol Metab, 103, 1282–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EC, & Kim JR (2019). Senotherapeutics: emerging strategy for healthy aging and age-related disease. BMB Rep, 52, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Lee J, Jung YR, Park JJ, Park MJ, Lee JS, Kim CH, Lee YJ, & Lee M (2015). Involvement of corin downregulation in ionizing radiation-induced senescence of myocardial cells. Int J Mol Med, 35, 731–738. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Ham SA, Paek KS, Hwang JS, Jung SY, Kim MY, Jin H, Kang ES, Woo IS, Kim HJ, Lee JH, Chang KC, Han CW, & Seo HG (2011). Transcriptional up-regulation of antioxidant genes by PPARdelta inhibits angiotensin II-induced premature senescence in vascular smooth muscle cells. Biochem Biophys Res Commun, 406, 564–569. [DOI] [PubMed] [Google Scholar]
- Kim KS, Kim JE, Choi KJ, Bae S, & Kim DH (2014). Characterization of DNA damage-induced cellular senescence by ionizing radiation in endothelial cells. Int J Radiat Biol, 90, 71–80. [DOI] [PubMed] [Google Scholar]
- Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, & Robbins PD (2017). The Clinical Potential of Senolytic Drugs. J Am Geriatr Soc, 65, 2297–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoppert SN, Valentijn FA, Nguyen TQ, Goldschmeding R, & Falke LL (2019). Cellular Senescence and the Kidney: Potential Therapeutic Targets and Tools. Front Pharmacol, 10, 770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordinas V, Ioannidis A, & Chatzipanagiotou S (2016). The Telomere/Telomerase System in Chronic Inflammatory Diseases. Cause or Effect? Genes (Basel), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucheryavenko O, Nelson G, von Zglinicki T, Korolchuk VI, & Carroll B (2019). The mTORC1-autophagy pathway is a target for senescent cell elimination. Biogerontology, 20, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari H, Huang WH, & Chan MWY (2020). Review on the Role of Epigenetic Modifications in Doxorubicin-Induced Cardiotoxicity. Front Cardiovasc Med, 7, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafargue A, Degorre C, Corre I, Alves-Guerra MC, Gaugler MH, Vallette F, Pecqueur C, & Paris F (2017). Ionizing radiation induces long-term senescence in endothelial cells through mitochondrial respiratory complex II dysfunction and superoxide generation. Free Radic Biol Med, 108, 750–759. [DOI] [PubMed] [Google Scholar]
- Larsson LG (2011). Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin Cancer Biol, 21, 367–376. [DOI] [PubMed] [Google Scholar]
- Latorre E, Pilling LC, Lee BP, Bandinelli S, Melzer D, Ferrucci L, & Harries LW (2018). The VEGFA156b isoform is dysregulated in senescent endothelial cells and may be associated with prevalent and incident coronary heart disease. Clin Sci (Lond), 132, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Chun T, Park SY, & Rho SB (2008). Interferon regulatory factor-1 (IRF-1) regulates VEGF-induced angiogenesis in HUVECs. Biochim Biophys Acta, 1783, 1654–1662. [DOI] [PubMed] [Google Scholar]
- Lee S, & Lee JS (2019). Cellular senescence: a promising strategy for cancer therapy. BMB Rep, 52, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrak EA, Pitha J, Rosenheim S, & Gottlieb JA (1973). A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer, 32, 302–314. [DOI] [PubMed] [Google Scholar]
- Lesniewski LA, Seals DR, Walker AE, Henson GD, Blimline MW, Trott DW, Bosshardt GC, LaRocca TJ, Lawson BR, Zigler MC, & Donato AJ (2017). Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell, 16, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xie N, Li Y, Liu C, Hou FF, & Wang J (2019). N-acetylcysteine ameliorates cisplatin-induced renal senescence and renal interstitial fibrosis through sirtuin1 activation and p53 deacetylation. Free Radic Biol Med, 130, 512–527. [DOI] [PubMed] [Google Scholar]
- Lingappan K (2018). NF-kappaB in Oxidative Stress. Curr Opin Toxicol, 7, 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Ma X, Zhuang J, Liu L, & Sun C (2020). Cardiotoxicity of doxorubicin-based cancer treatment: What is the protective cognition that phytochemicals provide us? Pharmacol Res, 160, 105062. [DOI] [PubMed] [Google Scholar]
- Liu XL, Ding J, & Meng LH (2018). Oncogene-induced senescence: a double edged sword in cancer. Acta Pharmacol Sin, 39, 1553–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Antebi A, Bartke A, Barzilai N, Brown-Borg HM, Caruso C, Curiel TJ, de Cabo R, Franceschi C, Gems D, Ingram DK, Johnson TE, Kennedy BK, Kenyon C, Klein S, Kopchick JJ, Lepperdinger G, Madeo F, Mirisola MG, Mitchell JR, Passarino G, Rudolph KL, Sedivy JM, Shadel GS, Sinclair DA, Spindler SR, Suh Y, Vijg J, Vinciguerra M, & Fontana L (2015). Interventions to Slow Aging in Humans: Are We Ready? Aging Cell, 14, 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe D, & Raj K (2014). Premature aging induced by radiation exhibits pro-atherosclerotic effects mediated by epigenetic activation of CD44 expression. Aging Cell, 13, 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons CE, & Bartolomucci A (2020). Stress and Alzheimer’s Disease: A Senescence Link? Neuroscience & Biobehavioral Reviews. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Yang J, Wang C, Zhang N, Dong Y, Wang C, Wang Y, & Lin X (2014). Emodin augments cisplatin cytotoxicity in platinum-resistant ovarian cancer cells via ROS-dependent MRP1 downregulation. Biomed Res Int, 2014, 107671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima Y, Adachi S, Ito H, Hirao K, & Isobe M (2008). Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell, 7, 125–136. [DOI] [PubMed] [Google Scholar]
- Majidinia M, Mir SM, Mirza-Aghazadeh-Attari M, Asghari R, Kafil HS, Safa A, Mahmoodpoor A, & Yousefi B (2020). MicroRNAs, DNA damage response and ageing. Biogerontology, 21, 275–291. [DOI] [PubMed] [Google Scholar]
- Mannick JB, Morris M, Hockey HP, Roma G, Beibel M, Kulmatycki K, Watkins M, Shavlakadze T, Zhou W, Quinn D, Glass DJ, & Klickstein LB (2018). TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med, 10. [DOI] [PubMed] [Google Scholar]
- Marcoux S, Le ON, Langlois-Pelletier C, Laverdiere C, Hatami A, Robaey P, & Beausejour CM (2013). Expression of the senescence marker p16INK4a in skin biopsies of acute lymphoblastic leukemia survivors: a pilot study. Radiat Oncol, 8, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmary Y, Adar R, Gaska S, Wygoda A, Maly A, Cohen J, Eliashar R, Mizrachi L, Orfaig-Geva C, Baum BJ, Rose-John S, Galun E, & Axelrod JH (2016). Radiation-Induced Loss of Salivary Gland Function Is Driven by Cellular Senescence and Prevented by IL6 Modulation. Cancer Res, 76, 1170–1180. [DOI] [PubMed] [Google Scholar]
- Martin JA, & Buckwalter JA (2001). Roles of articular cartilage aging and chondrocyte senescence in the pathogenesis of osteoarthritis. The Iowa orthopaedic journal, 21, 1. [PMC free article] [PubMed] [Google Scholar]
- Matsumura N, Zordoky BN, Robertson IM, Hamza SM, Parajuli N, Soltys C-LM, Beker DL, Grant MK, Razzoli M, Bartolomucci A, & Dyck JRB (2018). Co-administration of resveratrol with doxorubicin in young mice attenuates detrimental late-occurring cardiovascular changes. Cardiovascular Research, 114, 1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, & Bennett M (2006). Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res, 99, 156–164. [DOI] [PubMed] [Google Scholar]
- McIntyre RL, Daniels EG, Molenaars M, Houtkooper RH, & Janssens GE (2019). From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol Med, 11, e9854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeren AV, Bertho JM, Vandamme M, & Gaugler MH (1997). Ionizing radiation enhances IL-6 and IL-8 production by human endothelial cells. Mediators Inflamm, 6, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendonca MS, Chin-Sinex H, Dhaemers R, Mead LE, Yoder MC, & Ingram DA (2011). Differential mechanisms of x-ray-induced cell death in human endothelial progenitor cells isolated from cord blood and adults. Radiat Res, 176, 208–216. [DOI] [PubMed] [Google Scholar]
- Meng D-F, Guo L-L, Peng L-X, Zheng L-S, Xie P, Mei Y, Li C-Z, Peng X-S, Lang Y-H, Liu Z-J, Wang M-D, Xie D-H, Shu D-T, Hu H, Lin S-T, Li H-F, Luo F-F, Sun R, Huang B-J, & Qian C-N (2020). Antioxidants suppress radiation-induced apoptosis via inhibiting MAPK pathway in nasopharyngeal carcinoma cells. Biochemical and Biophysical Research Communications, 527, 770–777. [DOI] [PubMed] [Google Scholar]
- Merolle M, Mongiardi MP, Piras M, Levi A, & Falchetti ML (2020). Glioblastoma Cells Do Not Affect Axitinib-Dependent Senescence of HUVECs in a Transwell Coculture Model. International Journal of Molecular Sciences, 21, 1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Hodwin B, Ramanujam D, Engelhardt S, & Sarikas A (2016). Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J Am Coll Cardiol, 67, 2018–2028. [DOI] [PubMed] [Google Scholar]
- Meyer P, Maity P, Burkovski A, Schwab J, Mussel C, Singh K, Ferreira FF, Krug L, Maier HJ, Wlaschek M, Wirth T, Kestler HA, & Scharffetter-Kochanek K (2017). A model of the onset of the senescence associated secretory phenotype after DNA damage induced senescence. PLoS Comput Biol, 13, e1005741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami M, Matsumoto S, & Horiuchi H (2010). Cardiovascular side-effects of modern cancer therapy. Circ J, 74, 1779–1786. [DOI] [PubMed] [Google Scholar]
- Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, & Komuro I (2002). Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation, 105, 1541–1544. [DOI] [PubMed] [Google Scholar]
- Minieri V, Saviozzi S, Gambarotta G, Lo Iacono M, Accomasso L, Cibrario Rocchietti E, Gallina C, Turinetto V, & Giachino C (2015). Persistent DNA damage-induced premature senescence alters the functional features of human bone marrow mesenchymal stem cells. J Cell Mol Med, 19, 734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva O, Deschenes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, Bourdeau V, Pollak MN, & Ferbeyre G (2013). Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell, 12, 489–498. [DOI] [PubMed] [Google Scholar]
- Mongiardi MP, Radice G, Piras M, Stagni V, Pacioni S, Re A, Putti S, Ferre F, Farsetti A, Pallini R, Barila D, Levi A, & Falchetti ML (2019). Axitinib exposure triggers endothelial cells senescence through ROS accumulation and ATM activation. Oncogene, 38, 5413–5424. [DOI] [PubMed] [Google Scholar]
- Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A, Varela-Nieto I, Ruberte J, & Collado M (2013). Programmed cell senescence during mammalian embryonic development. Cell, 155, 1104–1118. [DOI] [PubMed] [Google Scholar]
- Munz F, Lopez Perez R, Trinh T, Sisombath S, Weber KJ, Wuchter P, Debus J, Saffrich R, Huber PE, & Nicolay NH (2018). Human mesenchymal stem cells lose their functional properties after paclitaxel treatment. Sci Rep, 8, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthna D, Soukup T, Vavrova J, Mokry J, Cmielova J, Visek B, Jiroutova A, Havelek R, Suchanek J, & Filip S (2010). Irradiation of adult human dental pulp stem cells provokes activation of p53, cell cycle arrest, and senescence but not apoptosis. Stem Cells and Development, 19, 1855–1862. [DOI] [PubMed] [Google Scholar]
- Nabialek-Trojanowska I, Lewicka E, Wrona A, Kaleta AM, Lewicka-Potocka Z, Raczak G, & Dziadziuszko R (2018). Cardiovascular complications after radiotherapy. Cardiol J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M, Hashimoto M, Wang F, Yamanaga K, Nakamura N, Uchida T, & Yamanouchi K (1997). Aging decreases the production of PGI2 in rat aortic endothelial cells. Experimental Gerontology, 32, 685–693. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ueda Y, Juan Y, Katsuda S, Takahashi H, & Koh E (2000). Fas-mediated apoptosis in adriamycin-induced cardiomyopathy in rats: In vivo study. Circulation, 102, 572–578. [DOI] [PubMed] [Google Scholar]
- Nakano-Kurimoto R, Ikeda K, Uraoka M, Nakagawa Y, Yutaka K, Koide M, Takahashi T, Matoba S, Yamada H, Okigaki M, & Matsubara H (2009). Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol, 297, H1673–1684. [DOI] [PubMed] [Google Scholar]
- Nardella C, Clohessy JG, Alimonti A, & Pandolfi PP (2011). Pro-senescence therapy for cancer treatment. Nat Rev Cancer, 11, 503–511. [DOI] [PubMed] [Google Scholar]
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, & von Zglinicki T (2012). A senescent cell bystander effect: senescence-induced senescence. Aging Cell, 11, 345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness KK, Hudson MM, Pui CH, Green DM, Krull KR, Huang TT, Robison LL, & Morris EB (2012). Neuromuscular impairments in adult survivors of childhood acute lymphoblastic leukemia: associations with physical performance and chemotherapy doses. Cancer, 118, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness KK, Krull KR, Jones KE, Mulrooney DA, Armstrong GT, Green DM, Chemaitilly W, Smith WA, Wilson CL, Sklar CA, Shelton K, Srivastava DK, Ali S, Robison LL, & Hudson MM (2013). Physiologic Frailty As a Sign of Accelerated Aging Among Adult Survivors of Childhood Cancer: A Report From the St Jude Lifetime Cohort Study. Journal of Clinical Oncology, 31, 4496–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness KK, Morris EB, Nolan VG, Howell CR, Gilchrist LS, Stovall M, Cox CL, Klosky JL, Gajjar A, & Neglia JP (2010). Physical performance limitations among adult survivors of childhood brain tumors. Cancer, 116, 3034–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notarbartolo M, Poma P, Perri D, Dusonchet L, Cervello M, & D’Alessandro N (2005). Antitumor effects of curcumin, alone or in combination with cisplatin or doxorubicin, on human hepatic cancer cells. Analysis of their possible relationship to changes in NF-kB activation levels and in IAP gene expression. Cancer Lett, 224, 53–65. [DOI] [PubMed] [Google Scholar]
- Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM, Grellscheid SN, Hoeijmakers JHJ, Barnhoorn S, Mann DA, Bird TG, Vermeij WP, Kirkland JL, Passos JF, von Zglinicki T, & Jurk D (2017). Cellular senescence drives age-dependent hepatic steatosis. Nature Communications, 8, 15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh CW, Bump EA, Kim JS, Janigro D, & Mayberg MR (2001). Induction of a senescence-like phenotype in bovine aortic endothelial cells by ionizing radiation. Radiat Res, 156, 232–240. [DOI] [PubMed] [Google Scholar]
- Palaniyappan A (2009). Cyclophosphamide induces premature senescence in normal human fibroblasts by activating MAP kinases. Biogerontology, 10, 677–682. [DOI] [PubMed] [Google Scholar]
- Palmer AK, Gustafson B, Kirkland JL, & Smith U (2019). Cellular senescence: at the nexus between ageing and diabetes. Diabetologia, 62, 1835–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M, Lin S, Huang L, Chung EJ, Citrin DE, Wang Y, Hauer-Jensen M, Zhou D, & Meng A (2017). Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int J Radiat Oncol Biol Phys, 99, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panganiban RA, & Day RM (2013). Inhibition of IGF-1R prevents ionizing radiation-induced primary endothelial cell senescence. Plos One, 8, e78589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panganiban RA, Mungunsukh O, & Day RM (2013). X-irradiation induces ER stress, apoptosis, and senescence in pulmonary artery endothelial cells. Int J Radiat Biol, 89, 656–667. [DOI] [PubMed] [Google Scholar]
- Panganiban RAM, Mungunsukh O, & Day RM (2013). X-irradiation induces ER stress, apoptosis, and senescence in pulmonary artery endothelial cells. International journal of radiation biology, 89, 656–667. [DOI] [PubMed] [Google Scholar]
- Papaconstantinou J (2019). The Role of Signaling Pathways of Inflammation and Oxidative Stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos D, Magliozzi R, Mitsikostas DD, Gorgoulis VG, & Nicholas RS (2020). Aging, Cellular Senescence, and Progressive Multiple Sclerosis. Frontiers in Cellular Neuroscience, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Kim CH, Jeong JH, Park M, & Kim KS (2016). GDF15 contributes to radiation-induced senescence through the ROS-mediated p16 pathway in human endothelial cells. Oncotarget, 7, 9634–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison CJ, & Korolchuk VI (2018). Autophagy: ‘Self-Eating’ Your Way to Longevity. Subcell Biochem, 90, 25–47. [DOI] [PubMed] [Google Scholar]
- Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, Castoria G, & Migliaccio A (2020). ROS in cancer therapy: the bright side of the moon. Experimental & Molecular Medicine, 52, 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piegari E, De Angelis A, Cappetta D, Russo R, Esposito G, Costantino S, Graiani G, Frati C, Prezioso L, Berrino L, Urbanek K, Quaini F, & Rossi F (2013). Doxorubicin induces senescence and impairs function of human cardiac progenitor cells. Basic Res Cardiol, 108, 334. [DOI] [PubMed] [Google Scholar]
- Piotrowska I, Isalan M, & Mielcarek M (2017). Early transcriptional alteration of histone deacetylases in a murine model of doxorubicin-induced cardiomyopathy. Plos One, 12, e0180571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeep K, Ko KC, Choi MH, Kang JA, Chung YJ, & Park SH (2012). Protective effect of hesperidin, a citrus flavanoglycone, against gamma-radiation-induced tissue damage in Sprague-Dawley rats. J Med Food, 15, 419–427. [DOI] [PubMed] [Google Scholar]
- Probin V, Wang Y, Bai A, & Zhou D (2006). Busulfan selectively induces cellular senescence but not apoptosis in WI38 fibroblasts via a p53-independent but extracellular signal-regulated kinase-p38 mitogen-activated protein kinase-dependent mechanism. J Pharmacol Exp Ther, 319, 551–560. [DOI] [PubMed] [Google Scholar]
- Probin V, Wang Y, & Zhou D (2007). Busulfan-induced senescence is dependent on ROS production upstream of the MAPK pathway. Free Radic Biol Med, 42, 1858–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybylska D, Janiszewska D, Goździk A, Bielak-Zmijewska A, Sunderland P, Sikora E, & Mosieniak G (2016). NOX4 downregulation leads to senescence of human vascular smooth muscle cells. Oncotarget, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulakat L, & Chen HH (2020). Pro-Senescence and Anti-Senescence Mechanisms of Cardiovascular Aging: Cardiac MicroRNA Regulation of Longevity Drug-Induced Autophagy. Frontiers in pharmacology, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puvvula PK (2019). LncRNAs Regulatory Networks in Cellular Senescence. Int J Mol Sci, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Zhang Y, Liu L, Guo X, Qin J, & Cui G (2012). Mesenchymal stem cells derived from different origins have unique sensitivities to different chemotherapeutic agents. Cell Biol Int, 36, 857–862. [DOI] [PubMed] [Google Scholar]
- Rattan SI (2008). Cell Senescence In Vitro. eLS. [Google Scholar]
- Rayess H, Wang MB, & Srivatsan ES (2012). Cellular senescence and tumor suppressor gene p16. Int J Cancer, 130, 1715–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, & Ross OA (2018). Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Frontiers in immunology, 9, 586–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renschler MF (2004). The emerging role of reactive oxygen species in cancer therapy. Eur J Cancer, 40, 1934–1940. [DOI] [PubMed] [Google Scholar]
- Rippe C, Blimline M, Magerko KA, Lawson BR, LaRocca TJ, Donato AJ, & Seals DR (2012). MicroRNA changes in human arterial endothelial cells with senescence: relation to apoptosis, eNOS and inflammation. Exp Gerontol, 47, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PD, Jurk D, Khosla S, Kirkland JL, LeBrasseur NK, Miller JD, Passos JF, Pignolo RJ, Tchkonia T, & Niedernhofer LJ (2020). Senolytic Drugs: Reducing Senescent Cell Viability to Extend Health Span. Annu Rev Pharmacol Toxicol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson AR, Yousefzadeh MJ, Rozgaja TA, Wang J, Li X, Tilstra JS, Feldman CH, Gregg SQ, Johnson CH, Skoda EM, Frantz MC, Bell-Temin H, Pope-Varsalona H, Gurkar AU, Nasto LA, Robinson RAS, Fuhrmann-Stroissnigg H, Czerwinska J, McGowan SJ, Cantu-Medellin N, Harris JB, Maniar S, Ross MA, Trussoni CE, LaRusso NF, Cifuentes-Pagano E, Pagano PJ, Tudek B, Vo NV, Rigatti LH, Opresko PL, Stolz DB, Watkins SC, Burd CE, Croix CMS, Siuzdak G, Yates NA, Robbins PD, Wang Y, Wipf P, Kelley EE, & Niedernhofer LJ (2018). Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox Biol, 17, 259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roninson IB (2003). Tumor cell senescence in cancer treatment. Cancer Res, 63, 2705–2715. [PubMed] [Google Scholar]
- Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, Kirkland JL, & Miller JD (2016). Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell, 15, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota M, LeCapitaine N, Hosoda T, Boni A, De Angelis A, Padin-Iruegas ME, Esposito G, Vitale S, Urbanek K, Casarsa C, Giorgio M, Luscher TF, Pelicci PG, Anversa P, Leri A, & Kajstura J (2006). Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ Res, 99, 42–52. [DOI] [PubMed] [Google Scholar]
- Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D, Ramalingam SS, Ranson MR, Dive C, McKeegan EM, Chyla BJ, Dowell BL, Chakravartty A, Nolan CE, Rudersdorf N, Busman TA, Mabry MH, Krivoshik AP, Humerickhouse RA, Shapiro GI, & Gandhi L (2012). Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res, 18, 3163–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf E, John S, & Cervinka M (2012). Irinotecan induces senescence and apoptosis in colonic cells in vitro. Toxicol Lett, 214, 1–8. [DOI] [PubMed] [Google Scholar]
- Saleh T, Bloukh S, Carpenter VJ, Alwohoush E, Bakeer J, Darwish S, Azab B, & Gewirtz DA (2020). Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers (Basel), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh T, Bloukh S, Carpenter VJ, Alwohoush E, Bakeer J, Darwish S, Azab B, & Gewirtz DA (2020). Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers, 12, 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh T, Tyutyunyk-Massey L, Murray GF, Alotaibi MR, Kawale AS, Elsayed Z, Henderson SC, Yakovlev V, Elmore LW, Toor A, Harada H, Reed J, Landry JW, & Gewirtz DA (2019). Tumor cell escape from therapy-induced senescence. Biochem Pharmacol, 162, 202–212. [DOI] [PubMed] [Google Scholar]
- Sanada F, Muratsu J, Otsu R, Shimizu H, Koibuchi N, Uchida K, Taniyama Y, Yoshimura S, Rakugi H, & Morishita R (2017). Local Production of Activated Factor X in Atherosclerotic Plaque Induced Vascular Smooth Muscle Cell Senescence. Sci Rep, 7, 17172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanoff HK, Deal AM, Krishnamurthy J, Torrice C, Dillon P, Sorrentino J, Ibrahim JG, Jolly TA, Williams G, Carey LA, Drobish A, Gordon BB, Alston S, Hurria A, Kleinhans K, Rudolph KL, Sharpless NE, & Muss HB (2014). Effect of cytotoxic chemotherapy on markers of molecular age in patients with breast cancer. J Natl Cancer Inst, 106, dju057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Ishikawa Y, Takahashi Y, Itoh T, Minami Y, & Nakamura M (2008). Association between oxidative DNA damage and telomere shortening in circulating endothelial progenitor cells obtained from metabolic syndrome patients with coronary artery disease. Atherosclerosis, 198, 347–353. [DOI] [PubMed] [Google Scholar]
- Satoh M, Nasu T, Takahashi Y, Osaki T, Hitomi S, Morino Y, & Nakamura M (2017). Expression of miR-23a induces telomere shortening and is associated with poor clinical outcomes in patients with coronary artery disease. Clin Sci (Lond), 131, 2007–2017. [DOI] [PubMed] [Google Scholar]
- Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, & LeBrasseur NK (2017). Cellular senescence mediates fibrotic pulmonary disease. Nat Commun, 8, 14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellinger IN, Mattern K, & Raaz U (2019). The Hardest Part: Arterial Stiffness in the Context of Healthy Aging. Arteriosclerosis, Thrombosis, and Vascular Biology, 39, 1301–1306. [DOI] [PubMed] [Google Scholar]
- Sedding D, & Haendeler J (2007). Do we age on Sirt1 expression? Circ Res, 100, 1396–1398. [DOI] [PubMed] [Google Scholar]
- Sehl ME, Carroll JE, Horvath S, & Bower JE (2020). The acute effects of adjuvant radiation and chemotherapy on peripheral blood epigenetic age in early stage breast cancer patients. NPJ Breast Cancer, 6, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifrtova M, Havelek R, Cmielova J, Jiroutova A, Soukup T, Bruckova L, Mokry J, English D, & Rezacova M (2012). The response of human ectomesenchymal dental pulp stem cells to cisplatin treatment. Int Endod J, 45, 401–412. [DOI] [PubMed] [Google Scholar]
- Seifrtova M, Havelek R, Soukup T, Filipova A, Mokry J, & Rezacova M (2013). Mitoxantrone ability to induce premature senescence in human dental pulp stem cells and human dermal fibroblasts. J Physiol Pharmacol, 64, 255–266. [PubMed] [Google Scholar]
- Sermsathanasawadi N, Ishii H, Igarashi K, Miura M, Yoshida M, Inoue Y, & Iwai T (2009). Enhanced adhesion of early endothelial progenitor cells to radiation-induced senescence-like vascular endothelial cells in vitro. J Radiat Res, 50, 469–475. [DOI] [PubMed] [Google Scholar]
- Shachar SS, Deal AM, Reeder-Hayes KE, Nyrop KA, Mitin N, Anders CK, Carey LA, Dees EC, Jolly TA, Kimmick GG, Karuturi MS, Reinbolt RE, Speca JC, & Muss HB (2020). Effects of Breast Cancer Adjuvant Chemotherapy Regimens on Expression of the Aging Biomarker, p16INK4a. JNCI Cancer Spectrum. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi-Sanjani M, Oyster NM, Tichy ED, Bedi KC Jr, Harel O, Margulies KB, & Mourkioti F (2017). Cardiomyocyte-Specific Telomere Shortening is a Distinct Signature of Heart Failure in Humans. Journal of the American Heart Association, 6, e005086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu I, & Minamino T (2020). Cellular Senescence in Arterial Diseases. J Lipid Atheroscler, 9, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, & Sadoshima J (2016). Aging and Autophagy in the Heart. Circ Res, 118, 1563–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora E, Bielak-Zmijewska A, & Mosieniak G (2019). Targeting normal and cancer senescent cells as a strategy of senotherapy. Ageing Res Rev, 55, 100941. [DOI] [PubMed] [Google Scholar]
- Smith DL Jr., Nagy TR, & Allison DB (2010). Calorie restriction: what recent results suggest for the future of ageing research. Eur J Clin Invest, 40, 440–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WA, Li C, Nottage KA, Mulrooney DA, Armstrong GT, Lanctot JQ, Chemaitilly W, Laver JH, Srivastava DK, Robison LL, Hudson MM, & Ness KK (2014). Lifestyle and metabolic syndrome in adult survivors of childhood cancer: a report from the St. Jude Lifetime Cohort Study. Cancer, 120, 2742–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smitherman AB, Anderson C, Lund JL, Bensen JT, Rosenstein DL, & Nichols HB (2018). Frailty and Comorbidities Among Survivors of Adolescent and Young Adult Cancer: A Cross-Sectional Examination of a Hospital-Based Survivorship Cohort. J Adolesc Young Adult Oncol, 7, 374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, & de Lange T (2002). Different telomere damage signaling pathways in human and mouse cells. EMBO J, 21, 4338–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Zhao Q, & Zou MH (2020). Targeting senescent cells to attenuate cardiovascular disease progression. Ageing Res Rev, 60, 101072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R, Yang Y, Lei H, Wang G, Huang Y, Xue W, Wang Y, Yao L, & Zhu Y (2018). HDAC6 inhibition protects cardiomyocytes against doxorubicin-induced acute damage by improving alpha-tubulin acetylation. J Mol Cell Cardiol, 124, 58–69. [DOI] [PubMed] [Google Scholar]
- Soysal P, Arik F, Smith L, Jackson SE, & Isik AT (2020). Inflammation, Frailty and Cardiovascular Disease. In Veronese N (Ed.), Frailty and Cardiovascular Diseases : Research into an Elderly Population (pp. 55–64). Cham: Springer International Publishing. [Google Scholar]
- Spallarossa P, Altieri P, Aloi C, Garibaldi S, Barisione C, Ghigliotti G, Fugazza G, Barsotti A, & Brunelli C (2009). Doxorubicin induces senescence or apoptosis in rat neonatal cardiomyocytes by regulating the expression levels of the telomere binding factors 1 and 2. Am J Physiol Heart Circ Physiol, 297, H2169–2181. [DOI] [PubMed] [Google Scholar]
- Spallarossa P, Altieri P, Barisione C, Passalacqua M, Aloi C, Fugazza G, Frassoni F, Podesta M, Canepa M, Ghigliotti G, & Brunelli C (2010). p38 MAPK and JNK antagonistically control senescence and cytoplasmic p16INK4A expression in doxorubicin-treated endothelial progenitor cells. Plos One, 5, e15583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles Steven J, & Adami GR (1998). Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene, 16, 1113–1123. [DOI] [PubMed] [Google Scholar]
- Stojanović SD, Fiedler J, Bauersachs J, Thum T, & Sedding DG (2020). Senescence-induced inflammation: an important player and key therapeutic target in atherosclerosis. European heart journal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studencka M, & Schaber J (2017). Senoptosis: non-lethal DNA cleavage as a route to deep senescence. Oncotarget, 8, 30656–30671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Study cancer survivors. (2019). Nature, 568, 143. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Mori I, Nakayama Y, Miyakoda M, Kodama S, & Watanabe M (2001). Radiation-induced senescence-like growth arrest requires TP53 function but not telomere shortening. Radiat Res, 155, 248–253. [DOI] [PubMed] [Google Scholar]
- Tajiri K, Aonuma K, & Sekine I (2017). Cardio-oncology: a multidisciplinary approach for detection, prevention and management of cardiac dysfunction in cancer patients. Jpn J Clin Oncol, 47, 678–682. [DOI] [PubMed] [Google Scholar]
- Tak JK, Lee JH, & Park JW (2012). Resveratrol and piperine enhance radiosensitivity of tumor cells. BMB Rep, 45, 242–246. [DOI] [PubMed] [Google Scholar]
- Takemura G, & Fujiwara H (2007). Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis, 49, 330–352. [DOI] [PubMed] [Google Scholar]
- Tang X, Li PH, & Chen HZ (2020). Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. Front Endocrinol (Lausanne), 11, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapio S (2016). Pathology and biology of radiation-induced cardiac disease. J Radiat Res, 57, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir1 Tasnuva D., Leigh Ross J., Tasena Hataitip, Mellone Massimiliano, Coletta Ricardo D.,, Parkinson5 Eric K., P. SS, Thomas Gareth J., Paterson Ian C., Zhou Donghui, McCall John, S. PM, and Lambert Daniel W.. (2016). A miR-335/COX-2/PTEN axis regulates the secretory phenotype of senescent cancer-associated fibroblasts. In Aging (Vol. 8, pp. 1608–1624). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telgenhoff D, & Shroot B (2005). Cellular senescence mechanisms in chronic wound healing. Cell Death Differ, 12, 695–698. [DOI] [PubMed] [Google Scholar]
- Teppo H-R, Soini Y, & Karihtala P (2017). Reactive oxygen species-mediated mechanisms of action of targeted cancer therapy. Oxidative Medicine and Cellular Longevity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzibasi E, Valenzano DR, & Cellerino A (2007). The short-lived fish Nothobranchius furzeri as a new model system for aging studies. Exp Gerontol, 42, 81–89. [DOI] [PubMed] [Google Scholar]
- Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N, Huard J, Clemens PR, Stolz DB, Guttridge DC, Watkins SC, Garinis GA, Wang Y, Niedernhofer LJ, & Robbins PD (2012). NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest, 122, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi T, Maeda M, & Hayashi T (2018). Administration of L-arginine plus L-citrulline or L-citrulline alone successfully retarded endothelial senescence. Plos One, 13, e0192252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnquist C, Beck JA, Horikawa I, Obiorah IE, Von Muhlinen N, Vojtesek B, Lane DP, Grunseich C, Chahine JJ, Ames HM, Smart DD, Harris BT, & Harris CC (2019). Radiation-induced astrocyte senescence is rescued by Delta133p53. Neuro Oncol, 21, 474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Kakinuma Y, Yuhki K, Murakoshi N, Iemitsu M, Miyauchi T, & Yamaguchi I (2006). Doxorubicin induces apoptosis by activation of caspase-3 in cultured cardiomyocytes in vitro and rat cardiac ventricles in vivo. J Pharmacol Sci, 101, 151–158. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Podlutsky A, Sosnowska D, Tucsek Z, Toth P, Deak F, Gautam T, Csiszar A, & Sonntag WE (2013). Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J Gerontol A Biol Sci Med Sci, 68, 1443–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upreti M, Koonce NA, Hennings L, Chambers TC, & Griffin RJ (2010). Pegylated IFN-alpha sensitizes melanoma cells to chemotherapy and causes premature senescence in endothelial cells by IRF-1 mediated signaling. Cell Death Dis, 1, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel O, Lahav M, Shargian L, Beery E, Pasvolsky O, Rozovski U, Raanani P, & Yeshurun M (2020). Premature ageing following allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant, 55, 1438–1446. [DOI] [PubMed] [Google Scholar]
- van Dalen EC, van der Pal HJ, Caron HN, & Kremer LC (2009). Different dosage schedules for reducing cardiotoxicity in cancer patients receiving anthracycline chemotherapy. Cochrane Database Syst Rev, CD005008. [DOI] [PubMed] [Google Scholar]
- van Deursen JM (2014). The role of senescent cells in ageing. Nature, 509, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatanen A, Hou M, Huang T, Söder O, Jahnukainen T, Kurimo M, Ojala TH, Sarkola T, Turanlahti M, Saarinen-Pihkala UM, & Jahnukainen K (2017). Clinical and biological markers of premature aging after autologous SCT in childhood cancer. Bone Marrow Transplantation, 52, 600–605. [DOI] [PubMed] [Google Scholar]
- Veronica G, & Esther R-RM (2012). Aging, metabolic syndrome and the heart. Aging and disease, 3, 269–279. [PMC free article] [PubMed] [Google Scholar]
- Walaszczyk A, Dookun E, Redgrave R, Tual-Chalot S, Victorelli S, Spyridopoulos I, Owens A, Arthur HM, Passos JF, & Richardson GD (2019). Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell, 18, e12945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace KB (2003). Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol, 93, 105–115. [DOI] [PubMed] [Google Scholar]
- Walton CC, Begelman D, Nguyen W, & Andersen JK (2020). Senescence as an Amyloid Cascade: The Amyloid Senescence Hypothesis. Front Cell Neurosci, 14, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Kohli J, & Demaria M (2020). Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer. [DOI] [PubMed] [Google Scholar]
- Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, Gray K, Kumar S, Clarke M, & Bennett M (2015). Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation, 132, 1909–1919. [DOI] [PubMed] [Google Scholar]
- Wang R, Yu Z, Sunchu B, Shoaf J, Dang I, Zhao S, Caples K, Bradley L, Beaver LM, Ho E, Lohr CV, & Perez VI (2017). Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell, 16, 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Boerma M, & Zhou D (2016). Ionizing radiation-induced endothelial cell senescence and cardiovascular diseases. Radiation research, 186, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z-N, Su R-N, Yang B-Y, Yang K-X, Yang L-F, Yan Y, & Chen Z-G (2020). Potential Role of Cellular Senescence in Asthma. Frontiers in Cell and Developmental Biology, 8, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartenberg M, Gronczynska S, Bekhite MM, Saric T, Niedermeier W, Hescheler J, & Sauer H (2005). Regulation of the multidrug resistance transporter P-glycoprotein in multicellular prostate tumor spheroids by hyperthermia and reactive oxygen species. Int J Cancer, 113, 229–240. [DOI] [PubMed] [Google Scholar]
- Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jöckel K-H, Erbel R, Mühleisen TW, Zenke M, Brümmendorf TH, & Wagner W (2014). Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biology, 15, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner C, Gensch C, Poss J, Haendeler J, Bohm M, & Laufs U (2011). Pioglitazone activates aortic telomerase and prevents stress-induced endothelial apoptosis. Atherosclerosis, 216, 23–34. [DOI] [PubMed] [Google Scholar]
- Wiley CD, Liu S, Limbad C, Zawadzka AM, Beck J, Demaria M, Artwood R, Alimirah F, Lopez-Dominguez JA, Kuehnemann C, Danielson SR, Basisty N, Kasler HG, Oron TR, Desprez PY, Mooney SD, Gibson BW, Schilling B, Campisi J, & Kapahi P (2019). SILAC Analysis Reveals Increased Secretion of Hemostasis-Related Factors by Senescent Cells. Cell Rep, 28, 3329–3337 e3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J, Smith F, Kumar S, Vijayan M, & Reddy PH (2017). Are microRNAs true sensors of ageing and cellular senescence? Ageing Res Rev, 35, 350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood WA, Krishnamurthy J, Mitin N, Torrice C, Parker JS, Snavely AC, Shea TC, Serody JS, & Sharpless NE (2016). Chemotherapy and Stem Cell Transplantation Increase p16INK4a Expression, a Biomarker of T-cell Aging. EBioMedicine, 11, 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CM, Zheng L, Wang Q, & Hu YW (2020). The emerging role of cell senescence in atherosclerosis. Clin Chem Lab Med. [DOI] [PubMed] [Google Scholar]
- Wu K, Chen Z, Peng Q, Chen G, Yan W, & Chen X (2019). Ku86 alleviates human umbilical vein endothelial cellular apoptosis and senescence induced by a low dose of ionizing radiation. J Int Med Res, 47, 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyld L, Bellantuono I, Tchkonia T, Morgan J, Turner O, Foss F, George J, Danson S, & Kirkland JL (2020). Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers (Basel), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, & Hou M (2018). Mesenchymal stem cells confer resistance to doxorubicin-induced cardiac senescence by inhibiting microRNA-34a. Oncol Lett, 15, 10037–10046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Chen Y, Hu C, Pan Q, Wang B, Li X, Geng J, & Xu B (2017). Premature senescence of cardiac fibroblasts and atrial fibrosis in patients with atrial fibrillation. Oncotarget, 8, 57981–57990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Xia W, & Hou M (2018). Long intergenic noncoding RNAp21 mediates cardiac senescence via the Wnt/betacatenin signaling pathway in doxorubicin-induced cardiotoxicity. Mol Med Rep, 17, 2695–2704. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Liu T, Wang S, Chi H, Chen C, & Zheng J (2017). Cyclophosphamide promotes the proliferation inhibition of mouse ovarian granulosa cells and premature ovarian failure by activating the lncRNA-Meg3-p53-p66Shc pathway. Gene, 596, 1–8. [DOI] [PubMed] [Google Scholar]
- Xiong Y, & Zhou L (2019). The Signaling of Cellular Senescence in Diabetic Nephropathy. Oxid Med Cell Longev, 2019, 7495629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, Langhi LGP, Weigl M, Giorgadze N, LeBrasseur NK, Miller JD, Jurk D, Singh RJ, Allison DB, Ejima K, Hubbard GB, Ikeno Y, Cubro H, Garovic VD, Hou X, Weroha SJ, Robbins PD, Niedernhofer LJ, Khosla S, Tchkonia T, & Kirkland JL (2018). Senolytics improve physical function and increase lifespan in old age. Nat Med, 24, 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, White TA, Johnson KO, Stout MB, Mezera V, Giorgadze N, Jensen MD, LeBrasseur NK, & Kirkland JL (2015). JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A, 112, E6301–6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabluchanskiy A, Tarantini S, Balasubramanian P, Kiss T, Csipo T, Fulop GA, Lipecz A, Ahire C, DelFavero J, Nyul-Toth A, Sonntag WE, Schwartzman ML, Campisi J, Csiszar A, & Ungvari Z (2020). Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation-induced impairment of neurovascular coupling responses protecting cognitive function in mice. Geroscience, 42, 409–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Hong HL, Luo WW, Dai CM, Chen XY, Wang LP, Li Q, Li ZQ, Liu PQ, & Li ZM (2018). mTORC2 facilitates endothelial cell senescence by suppressing Nrf2 expression via the Akt/GSK-3beta/C/EBPalpha signaling pathway. Acta Pharmacol Sin, 39, 1837–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Murali B, Ren Q, Luo X, Faget DV, Cole T, Ricci B, Thotala D, Monahan J, van Deursen JM, Baker D, Faccio R, Schwarz JK, & Stewart SA (2020). Therapy-Induced Senescence Drives Bone Loss. Cancer research, 80, 1171–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh ET, & Bickford CL (2009). Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol, 53, 2231–2247. [DOI] [PubMed] [Google Scholar]
- Yentrapalli R, Azimzadeh O, Barjaktarovic Z, Sarioglu H, Wojcik A, Harms-Ringdahl M, Atkinson MJ, Haghdoost S, & Tapio S (2013). Quantitative proteomic analysis reveals induction of premature senescence in human umbilical vein endothelial cells exposed to chronic low-dose rate gamma radiation. Proteomics, 13, 1096–1107. [DOI] [PubMed] [Google Scholar]
- Yentrapalli R, Azimzadeh O, Sriharshan A, Malinowsky K, Merl J, Wojcik A, Harms-Ringdahl M, Atkinson MJ, Becker K-F, & Haghdoost S (2013). The PI3K/Akt/mTOR pathway is implicated in the premature senescence of primary human endothelial cells exposed to chronic radiation. Plos One, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Zhou Z, Liu W, Chang Q, Sun G, & Dai Y (2017). Vascular endothelial cells senescence is associated with NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation via reactive oxygen species (ROS)/thioredoxin-interacting protein (TXNIP) pathway. Int J Biochem Cell Biol, 84, 22–34. [DOI] [PubMed] [Google Scholar]
- Yosef R, Pilpel N, Papismadov N, Gal H, Ovadya Y, Vadai E, Miller S, Porat Z, Ben-Dor S, & Krizhanovsky V (2017). p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J, 36, 2280–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Zhao J, Bukata C, Wade EA, McGowan SJ, Angelini LA, Bank MP, Gurkar AU, McGuckian CA, Calubag MF, Kato JI, Burd CE, Robbins PD, & Niedernhofer LJ (2020). Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell, 19, e13094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, McGuckian C, Wade EA, Kato JI, Grassi D, Wentworth M, Burd CE, Arriaga EA, Ladiges WL, Tchkonia T, Kirkland JL, Robbins PD, & Niedernhofer LJ (2018). Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine, 36, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhang L, Lu A, Han Y, Colangelo D, Bukata C, Scibetta A, Yousefzadeh MJ, Li X, Gurkar AU, McGowan SJ, Angelini L, O’Kelly R, Li H, Corbo L, Sano T, Nick H, Pola E, Pilla SPS, Ladiges WC, Vo N, Huard J, Niedernhofer LJ, & Robbins PD (2020). ATM is a key driver of NF-kappaB-dependent DNA-damage-induced senescence, stem cell dysfunction and aging. Aging (Albany NY), 12, 4688–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, Niedernhofer LJ, Robbins PD, Tchkonia T, & Kirkland JL (2017). New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY), 9, 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, O’Hara SP, LaRusso NF, Miller JD, Roos CM, Verzosa GC, LeBrasseur NK, Wren JD, Farr JN, Khosla S, Stout MB, McGowan SJ, Fuhrmann-Stroissnigg H, Gurkar AU, Zhao J, Colangelo D, Dorronsoro A, Ling YY, Barghouthy AS, Navarro DC, Sano T, Robbins PD, Niedernhofer LJ, & Kirkland JL (2015). The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell, 14, 644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordoky BN, Bark D, Soltys CL, Sung MM, & Dyck JR (2014). The anti-proliferative effect of metformin in triple-negative MDA-MB-231 breast cancer cells is highly dependent on glucose concentration: implications for cancer therapy and prevention. Biochim Biophys Acta, 1840, 1943–1957. [DOI] [PubMed] [Google Scholar]