

Abstract
Traumatic brain injury (TBI) is a debilitating disorder associated with chronic progressive neurodegeneration and long-term neurological decline. Importantly, there is now substantial and increasing evidence that TBI can negatively impact systemic organs including the pulmonary, gastrointestinal, cardiovascular, renal, and immune system. Less well appreciated, until recently, is that such functional changes can affect both the response to subsequent insults or diseases, as well as contribute to chronic neurodegenerative processes and long-term neurological outcomes. In this review, we summarize evidence showing bi-directional interactions between the brain and systemic organs following TBI and critically assess potential underlying mechanisms.
Keywords: Traumatic brain injury, neuroinflammation, microglia, bi-directional, systemic
Systemic Changes Following Traumatic Brain Injury
It has long been recognized that traumatic brain injury (TBI) can induce substantial systemic alterations that impact morbidity and mortality. TBI related changes in systemic organ function can affect both the response to subsequent insults or diseases, as well as to potentially contribute to chronic neurodegeneration and related neuropsychiatric dysfunction [1, 2]. Experimental and/or clinical studies have reported posttraumatic changes involving the systemic immune system, gut, lung, spleen, thymus, liver, heart and kidney [1, 2]. Potential mechanisms include activation of the hypothalamic-pituitary-adrenal (HPA) axis, alterations of the autonomic nervous system, immune system, microbiome and release of extracellular vesicles, among others [2–6].
TBI is associated with a high rate of infections that exceeds that for other acute neurological conditions and that contributes to mortality. This includes both nosocomial infections in hospitalized patients, as well as subsequent increased susceptibility to infection, suggesting that TBI may compromise systemic immune function [7]. Experimental studies convincingly show considerable posttraumatic functional alterations in systemic immune cells, including those involved in both innate and adaptive immunity [5]. Posttraumatic immune changes continue chronically, contributing to neuroinflammation and associated neurodegeneration [5, 8–10].
Experimental reports demonstrate that posttraumatic infections and/or inflammatory challenges can increase chronic neuroinflammation and associated neurodegeneration, particularly in chronic neurodegenerative conditions, with exacerbation of neurological dysfunction [11–13]. These observations raise important therapeutic implications for the care of TBI patients.
Here we review brain-systemic interactions after TBI, both pre-clinical and clinical, addressing how they may influence recovery after injury. We underscore the potential mechanisms involved, the importance of systemic effects induced by TBI on outcome, and how such changes may require modifications in clinical care. Lastly, we suggest venues for possible therapeutic targeting.
Critical Assessment of Prior Work: Caveats
Although studies that address the systemic consequences of TBI have been accelerating and provide considerable evidence that such changes can critically affect outcome and future risk, there are important caveats regarding most published studies. Issues include: nature and clinical relevance of animal models used; injury severity and heterogeneity; potential sex differences; possible confounding effects of trauma; design issues for clinical research; and the general absence of conclusive evidence from human studies.
TBI is perhaps the most heterogeneous among central nervous system disorders. Critical confounding issues for interpretation of studies include injury severity, type of injury (focal, diffuse, penetrating), sex/gender, age, presence of co-morbidities, genetics/epigenetics, and prior injury. Concordantly, it is well known that the same clinical presentation may reflect highly different underlying pathology. Established animal models cannot fully reflect such diversity, but rather generally focus on selected aspects of clinical trauma such as contusion (controlled cortical impact, CCI), or more complex injury plus brain displacement (fluid-percussion, F-P), most often using only one level of severity. Even F-P models differ with regard to site of injury (lateral versus central), which cause very different physiological responses. Both CCI and F-P models require craniotomy but have the advantage of generating relatively consistent patterns of injury. In contrast, although certain closed injury models may be more clinically relevant, they induce more variable injury and require much higher animal numbers. Repeated head injury models usually utilize only mild injury and vary considerably as a function of numbers of insults and timing between injuries. Given constraints on use of most gyrencephalic animals, whose brain structure better parallels that of humans, most widely used models employ rodents, with mice often preferred because of size and availability of transgenic strains. Given model differences, it is often difficult to compare studies across laboratories. Moreover, the predominant use of relatively simplistic animal models in lissencephalic species markedly adds to potential translational difficulties. To partially address such issues, some groups have attempted to replicate experimental results across species and/or use pathobiologically different models, either within or across laboratories.
Although it is well recognized that both mechanisms and outcomes may differ as a function of sex, the majority of reported studies have utilized animals of a single sex; this important issue may also confound and limit clinical translation.
Most clinical studies that examine systemic changes after TBI are retrospective, often with a small patient number. The relatively few prospective studies generally have methodological and conceptual limitations. Specifically, further investigation of how direct systemic trauma may complicate TBI outcomes will be important at a physiological level and in the identification of optimal therapeutic options for patients. This is particularly relevant in cases of severe TBIs where direct systemic trauma is more likely to occur. Given these caveats one must be cautious in interpreting much of the published work to date. Yet some of the consistent data being generated across experimental models and laboratories, as well as clinically, suggest opportunities for future mechanistic studies and translation, as well as for future therapeutic targeting and clinical care.
Immune system
TBI causes complex time-dependent changes in systemic and brain immune systems, affecting both innate and adaptive immune responses. Posttraumatic immune changes can persist chronically and contribute to morbidity/mortality after TBI [5, 14–16]. Clinically, TBI is associated with both innate and adaptive immune changes. There is evidence of increased circulating neutrophils following TBI [17]. During the early phase, neutrophils have enhanced reactive oxygen species (ROS) production [18, 19], but display impaired ROS generation more chronically following TBI. Furthermore, there are posttraumatic reductions in circulating monocytes [19] and natural killer (NK) cells [17], with decreased circulating T lymphocytes beginning within the first 24 hours following more severely injured patients [20, 21].
Pre-clinical studies also report significant TBI induced systemic immune responses. Following CCI in mice with moderate injury, there is mobilization of newly created myeloid cells (particularly neutrophils) into the blood, a decrease in T lymphocytes, atrophy of the thymus and defective T cell maturation [5]. Importantly, the posttraumatic circulating leukocytes showed functional changes, including alterations in leukocyte-derived reactive oxygen species (ROS) production in monocytes and neutrophils, cytokine production, and phagocytic ability [5]. Similarly, in a closed head injury model, trauma caused a significant loss of thymocytes as early as 3 days following injury; blood monocytes were reduced at 1 day following injury and remained suppressed up to 1 month [22]. Overall, these clinical and preclinical findings show that TBI suppresses both innate and adaptive immune responses.
Gastrointestinal (GI) System
TBI patients often experience alterations in GI function including upper GI intolerance, intestinal barrier disruption and intestinal tract dysmotility (Figure 1, Key Figure), which can affect posttraumatic morbidity and mortality [23–26]. Experimental TBI models demonstrate motility and permeability changes occurring within several days following mild-severe injury [12, 27]. In drosophila, TBI also caused intestinal barrier dysfunction associated with increased mortality [28]. Some models show early effects, including transient changes in small bowel permeability, but increased colonic permeability was reported in mice beginning several days posttrauma and persisting through 28 days [12]. Increased colon permeability can allow access of pathogenic bacteria or bacterial toxins to the systemic circulation, potentially causing endotoxemia and/or systemic toxicity. Delayed intestinal permeability changes also occur clinically and correlate with injury severity [23]. Permeability changes are associated with systemic inflammatory response syndrome, multiple organ failure and infections [23].
Figure 1: Bi-directional effects of TBI and systemic organs.
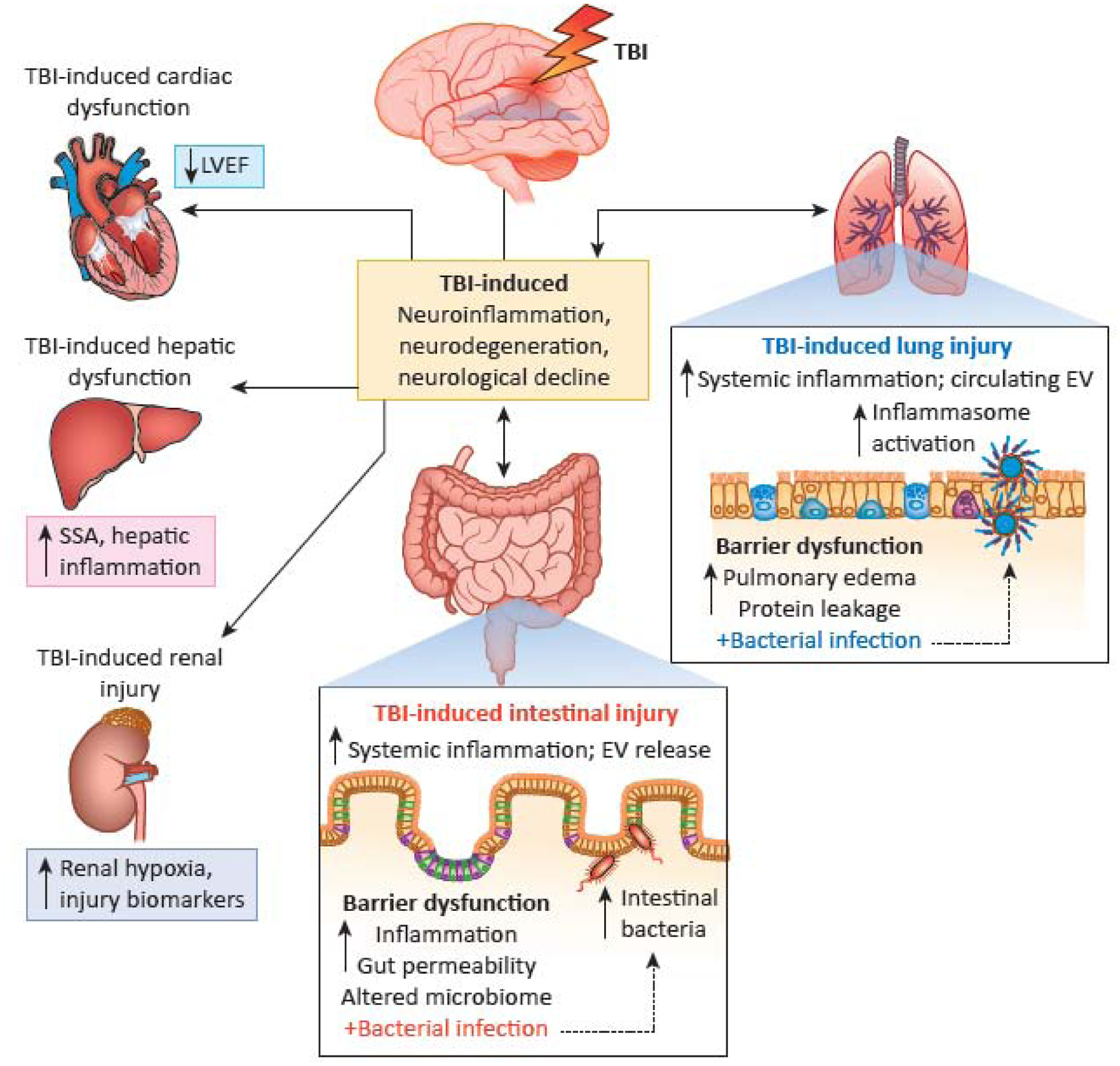
TBI can induce lung and intestinal injury (right-side and bottom insets, respectively). This in turn can lead to organ dysfunction and subsequent increases in systemic inflammatory responses including EV release. Subsequent bacterial insults to both the lung and gut may result in exacerbation of TBI-induced deficits including neuroinflammation, neurodegeneration and neurological decline. In addition, TBI can result in cardiac, hepatic and renal dysfunction which may increase mortality (left-side column); however, the subsequent effects on long-term neurological function remain to be determined. Abbreviations: Traumatic brain injury (TBI); Extracellular vesicles (EV); Left ventricular ejection fraction (LVEF); Serum amyloid A (SSA).
Mice subjected to moderate CCI show increased paracellular permeability at 28 days, associated with decreased expression of claudin-1 at both mRNA and protein levels [12]. CCI also induced pathological changes in the colon, with thickening of smooth muscle and greater mucosal depth, as well as increased expression of GFAP and Sox 10 in glial cells. When subsequently subjected to infectious colitis by Citrobacter rodentium at 6 weeks after injury, injured animals demonstrated greater progressive neuroinflammation and neurodegeneration changes as compared to non-infected TBI animals (Figure 1) [12]. Posttraumatic intestinal mucosal damage with permeability changes, as well as increased expression of inflammatory cytokines in the intestine and plasma endotoxin levels, have been observed in mice deficient for nuclear factor erythroid 2-related factor 2 (Nrf2), suggesting a potential protective role for Nrf2 [29]. Furthermore, delayed decreases of contractile activity in the ileum was found at 7 days after rat CCI, with reduced transit time; intestinal smooth muscle showed edema and increased expression of inflammatory cytokines [27].
More recently, our group has demonstrated that induction of a chemical colitis with dextran sodium sulfate (DSS) - a well characterized mouse model - beginning 6 weeks after CCI, also increases posttraumatic neuroinflammation, neurodegeneration and neurobehavioral dysfunction; this occurred both in mild, as well as moderate-severe injury [30]. Together, these observations appear consistent with studies of sickness behavior, in which systemic inflammation can lead to brain inflammation and functional alterations, particularly following a primary brain insult or disorder causing neurodegeneration [31]. Consistent with this concept, systemic immune challenge with LPS exacerbated neurological dysfunction in a midline fluid-percussion model (mFPI) [13].
Future studies should further delineate the time course of immune/inflammatory responses in both the gut and brain following TBI. Moreover, as pre-clinical investigations in these areas have often focused on male animals and given recognized sex-dependent differences in immune responses [32], further studies should address potential sex-dependent differences for development and progression of TBI-induced brain-gut interactions.
There has been growing interest in the influence of the microbiome on brain function and dysfunction, including after TBI. Following CCI in mice, bacterial changes (dysbiosis) were observed in the gut or feces in several studies [33–36]. Following repetitive mild TBI in mice, transient microbiome changes were reported [33]; however, repetitive mild TBI rats showed decreased gut microbial diversity from 6h to 30 days [34]. Given the methodological challenges in following posttraumatic microbiota changes in mice, such as the need to individually house mice because of confounding effects of coprophagia (with resulting stressor effects), as well as injury model, mouse strain and severity differences, comparison of results across studies must be interpreted cautiously.
Other work has sought to administer protective bacteria to alter the microbiome in order to improve outcomes [37, 38] or begun to examine the impact of aging on the microbiome and brain injury. The aged microbiome shows increased pro-inflammatory cytokines, and modifying the microbiome in young animals to that of an aged phenotype through fecal gavage exacerbated outcomes after experimental stroke [39]. This study has potential implications more generally for neurodegenerative disorders, including TBI, where poorer outcomes are observed with aging [40]. To what extent the microbiome may contribute to chronic neurotoxic inflammation - an important component of TBI and chronic neurodegenerative disorders - requires further study.
Lung
Hospitalized patients with TBI frequently show lung damage, with a prevalence greater than 20% that has increased in the past 20 years [41]. Moreover, there is a high incidence of infections in TBI patients that appears to correlate with injury severity [7, 42]. Among cases of patients with infection, most involve lung pathology, with pneumonia occurring in 30–50% of those who develop nosocomial infections [42]. Such individuals have increased mortality and disability. However, various risk factors may influence infections rates after head injury [42, 43]. For example, admission to an intensive care unit (ICU) and more prolonged ICU stays increase infection risk [44]. Several groups have examined the effects of experimental TBI on lung physiology and function. Closed head injury in rats caused pulmonary edema, protein leakage and increased inflammatory cytokines in alveolar fluid [45]. Acute lung injury was also reported following CCI in mice [46]. The latter study also demonstrated that extracellular vesicles (EV) containing inflammasome proteins were released after clinical TBI into the serum and caused lung injury; moreover, blocking EV uptake or using monoclonal antibodies to inhibit inflammasome activation protected against posttraumatic lung injury (Figure 1) [46]. More recently, the same group showed that treatment with a low molecular weight heparin limited the posttraumatic inflammasome response in both brain and lung, reducing lung injury [47]. A separate recent study reported that TBI increased lung permeability and monocyte infiltration at 24 hours after moderate mouse CCI [11]. Importantly, both clinical and experimental studies demonstrate that pathological pulmonary changes after TBI are associated with increased levels of high mobility group box 1 (HMGB1) in serum or plasma [46, 48]. Moreover, experimental evidence suggests that this may result from damage to neurons or endothelial cells in the blood-brain-barrier (BBB), with subsequent induction of inflammation [48].
Similar to GI studies, bacterial infection within the lung after experimental TBI exacerbates neuroinflammation, as well as neurological dysfunction (Figure 1) [11]. In one study, for instance, following moderate CCI, mice were infected with Streptococcus pneumoniae either early (3 days) or late (60 days) after injury. Infection at either time increased mortality, neuroinflammation and motor dysfunction [11]. Monocyte infiltrates in the lung early after injury showed evidence of immunosuppression, with reduced expression of inflammatory cytokines and reactive oxygen species (ROS). In contrast, delayed infection showed higher expression levels of both inflammatory cytokines and ROS as compared to sham infected animals. Intratracheal administration of LPS after CCI injury in mice can also exacerbate posttraumatic cognitive impairment [49]. These experimental models differ from ventilator-associated pneumonia, which is commonly found in severely injured TBI patients and is associated with a poor prognosis [50]. Collectively, these studies demonstrate important bi-directional effects between the lung and the brain that potentially impact TBI outcomes [11]. They also suggest that chronic TBI patients should be closely followed for evidence of systemic infections, and if present, treated aggressively. However, to better understand these interactions, future studies should delineate the effects of injury severity, infection model, pre-existing co-morbidities, and sex differences.
Heart
Cardiac complications appear to be relatively common after moderate to severe TBI. Clinical studies in this context have largely been retrospective. Specifically, two major trauma groups reported detectable or elevated serum cardiac troponin I (cTnI) levels in approximately 30% of TBI patients (420 and 580, respectively), which were associated with increased mortality [51, 52]. A more recent, prospective study found that nearly 60% of patients showed detectable cTnI levels; again increased levels were a predictor of mortality [53]. Although intriguing and potentially important, future studies should further delineate relationship to injury severity, presence of major polytrauma and be sufficiently powered to address issues such as age, sex/gender and presence of known co-morbidities.
A mouse CCI study reported significant reductions in posttraumatic cardiac function at 3 and 30 days, with decreased left ventricular ejection fraction (LVEF) (Figure 1) [54]. However, findings from clinical studies have been mixed. In one report, 5 of 15 patients with moderate-severe TBI showed reduced systolic function [55]. Another study examined 46 patients with moderate-severe TBI; the authors reported cardiac dysfunction in 6 of the patients (13%), with a mild to moderate reduction of LVEF, although cardiac changes were not associated with adverse outcome [56]. A larger study involving 139 patients with isolated TBI found 12% with reduced LVEF; an abnormal echocardiogram was associated with mortality [57]. Yet in a prospective study of 20 severe TBI patients, LVEF did not differ from controls, although diastolic function was slightly lower in the injury group [58].
Overall, these clinical and pre-clinical studies provide some support for the conclusion that more severe TBI can negatively impact cardiac function. When present, increased cardiac troponin I levels appear to predict worse outcome. Although no mechanism has been established, the excessive sympathetic activity observed in these patients may be a key factor.
Other Organs
More limited information is available regarding TBI’s impact on other systemic organ systems, such as the kidney or liver (Figure 1). One retrospective clinical review of 207 TBI patients with moderate-severe injury reported acute kidney injury (AKI) in 9% of the patients; higher rates were found in older patients and those with more severe trauma [59]. Another study reported that plasma obtained from patients with severe TBI and evidence of subclinical kidney injury could induce cellular dysfunction and apoptosis in cultured human renal proximal tubular epithelial cells [60]. Several retrospective studies have identified potential biomarkers for assessment of TBI severity and the occurrence of AKI including: liver-type-fatty acid-binding-protein (L-FABP) [61]; procalcitonin (PCT) [62]; and urinary neutrophil gelatinase-associated lipocalin (NGAL) [63]. Additionally, diffuse brain injury in rats is associated with renal hypoxia [64].
TBI can activate acute phase responses in the liver early after injury [65, 66]. Liver expression of serum amyloid A (SSA) peaked 1 day following injury in both male and female mice; SSA mRNA and protein expression co-localized with astrocytes and macrophage/microglia in discrete brain regions in a sex-dependent manner [67]. Fluid percussion injury in rats induced hepatic inflammation and oxidative stress at 24h following injury [68]. However, the impact of changes in these organs on long term behavioral outcomes or neurodegeneration requires further study.
Bidirectional Interactions and TBI: Potential Mechanisms
A number of potential mechanisms have been suggested or implicated in TBI-associated systemic alterations. Historically, most emphasis has been placed on the role of the hypothalamic-pituitary- adrenal (HPA) axis, the autonomic nervous system (ANS) and the immune system.
Acute activation of the HPA can alter cortisol and multiple endocrine factors; resultant physiological changes can affect, among others, immune function and metabolism [1, 2].
Glucocorticoids (cortisol in humans and corticosterone (CORT) in rodents) are the main effectors of the HPA axis and exert potent immunosuppressive effects through activation of the glucocorticoid receptor [17, 69]. Both clinical and preclinical studies show alterations in HPA function following TBI [70–72]. Patients with mild- to moderate-level TBI have increased circulating cortisol for 2 days following injury, whereas severely injured patients have decreased circulating cortisol at 1–3 days [73]. Such differential responses have also been shown experimentally. Rodents subjected to mild-level CCI show heightened restraint-induced CORT at 34 and 70 days post-injury [74]; in contrast, restraint-induced CORT responses were significantly more blunted in moderate CCI versus mild level CCI at 7 and 35 days following injury [75]. In addition, sex-dependent effects have been reported in HPA axis responses; male mice exposed to mild blast TBI had increased restraint-induced CORT at 7 and 10 days post-injury, whereas females displayed the opposite response [76]. Increased microglia activation in the paraventricular nucleus (PVN) was reported in male, but not female animals, following midline F-P [77]. Such sex-dependent effects on TBI-induced alterations of HPA activation may have important implications for outcomes.
Adrenal insufficiency (AI) occurs in about a quarter of TBI patients, likely due to HPA axis dysfunction [78, 79]. In addition, many patients with severe TBI demonstrate abnormal circulating levels of glucocorticoids in response to pharmacologically-induced stress within 10 days following injury [79]. Experimentally, in a closed head injury model, there was a decreased number of circulating lymphocytes (particularly T cells) with changes inversely correlated with plasma CORT [80]. Elevated CORT has also been associated with lymphocytopenia after experimental stroke [81]. Given that brain-induced immunosuppression makes patients more vulnerable to infections [82] and the known effects of HPA axis dysfunction on immune and endocrine responses, additional mechanistic studies should be performed to address implications for therapeutic interventions and patient care.
Paroxysmal sympathetic hyperactivity (PSH, sympathetic storm) has been implicated in diverse peripheral pathophysiologic effects after acute brain injuries [3]. Beta blockers have been examined clinically to modulate systemic organ pathological changes after TBI, with limited evidence for improvement in mortality but no data relating to impact on functional outcomes [83–85]; overall, because of study design issues the level of evidence supporting therapeutic benefit has been considered low [82]. A randomized study [84] purported to demonstrate reduced mortality and better longer-term clinical outcomes in TBI patients following administration of the beta blocker Propranolol, but it is important to note that the study was unblinded, lacked placebo controls and actually showed no significant benefit on primary outcomes. Collectively, these studies suggest that ANS mediated effects are more complex and nuanced than may initially seem. Although activation of the sympathetic nervous system has been implicated in inflammation, such as through effects in the spleen and liver [86], it can also control resolution of inflammation [87]. In addition, the parasympathetic nervous system may have a role in modulating inflammation [88].
TBI results in both early and late systemic immune changes, which appears to contribute to chronic neuroinflammation and neurodegeneration (Figure 2). Expression of toll-like receptor (TLR)4 on myeloid cells that enter the brain in the early period after TBI may affect longer-term adaptive immune responses [89]. Immune cell changes after brain injury substantially parallel those observed with normal aging and suggest that TBI may accelerate immune aging or senescence [5, 40]. A possible mechanistic role for circulating monocytes in posttraumatic toxic neuroinflammation after TBI has been supported by the work of several groups [14, 90–92]. This mechanism appears to be exaggerated in aged animals [92]. Global depletion of monocytes, as well as knock-out or inhibition of peripheral C-C chemokine receptor type 2 (CCR2(+) monocytes, reduce posttraumatic neuroinflammation, and neurobehavioral responses in both juvenile and aged mice [14, 90, 92].
Figure 2: TBI-induced alterations in systemic and central immune function.
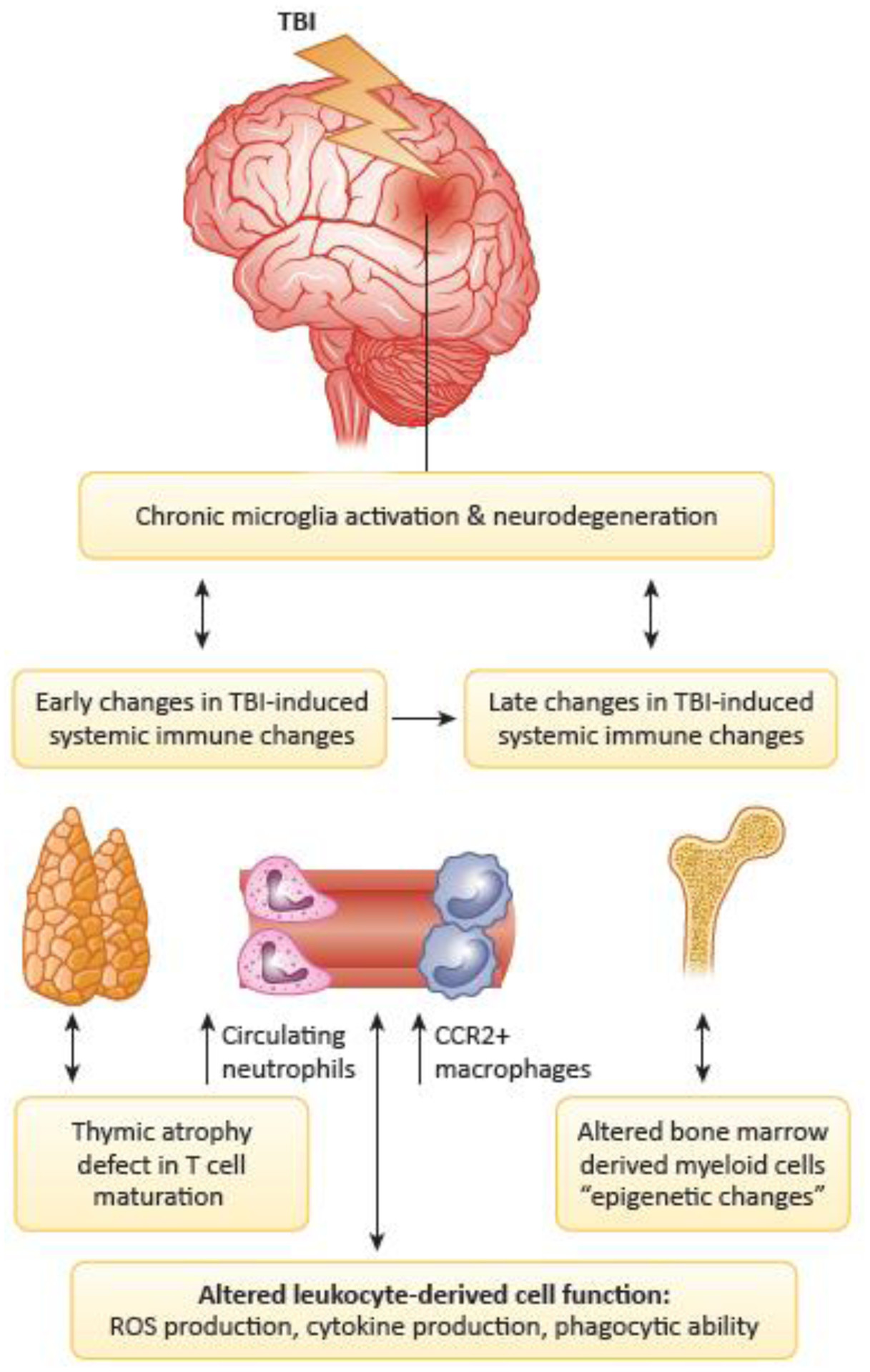
During the early stages of TBI, alterations can emerge in the systemic innate and adaptive immune responses that can in turn alter TBI-induced microglia responses and subsequent neurodegenerative processes. In addition, TBI can induce late changes in systemic immune responses including altered bone-marrow-derived myeloid cell function, which may in turn alter chronic microglial responses and neurodegenerative processes. Abbreviations: Traumatic brain injury (TBI); Reactive oxygen species (ROS); C-C chemokine receptor 2 (CCR2).
Clinically, TBI increases plasma levels of inflammatory factors such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, and C-reactive protein (CRP). TBI substantially elevates both numbers of circulating leukocytes and their expression of TNF-α and inducible nitric oxide synthase (iNOS); induces greater free radical production in leukocyte homogenates; and elevates expression of iNOS, cyclooxygenase (COX)-2 and NADPH oxidase (gp91(phox)) in circulating leukocytes [19]. Moreover, after injury circulating neutrophils show markedly up-regulated oxidative activity and suppressed phagocytic ability [19]. In a recent clinical study, patients with mild TBI had altered plasma cytokine levels that persisted up to one year post injury [93].
Microglial activation, as well as infiltration of myeloid cells after experimental TBI, are implicated in posttraumatic neurotoxic neuroinflammation [15, 16, 91, 94]. In mice, these responses are sexually dimorphic, with males showing greater and earlier myeloid infiltration than females, as well as dissimilar cytokine responses [95, 96]. These differences are associated with less pathological changes in female animals during the early period after injury [95]. Moreover, posttraumatic brain injury responses are exacerbated in aged mice, with evidence of greater oxidative stress responses and greater microglial functional changes - including altered phagocytosis and increased senescent markers [40, 97]. Pro-inflammatory microglial activation persists chronically, both clinically and in experimental brain trauma models over months to years [16, 94, 98, 99]. Chronic changes in TBI-induced systemic immune responses include alterations in bone-marrow derived myeloid cells. These effects, as well as chronic alterations in microglia, may reflect in part epigenetic changes that may be modifiable (Figure 2) [5, 94]. Further research is needed to better characterize sex and age dependent differences in immune responses to TBI, both peripheral and central, as well as their potential bi-directional interactions across experimental models.
Recently, it has been shown that posttraumatic microglial activation leads to the release of extracellular vesicles (EVs) in the blood, both at early periods and more chronically. These EVs contain pro-inflammatory factors, including micro RNAs (miRs), that can potentially affect systemic organs [4, 100–102]. EVs appear to play a role in the propagation of neuroinflammation and in promoting neurodegeneration at more distant sites [101]. They may also serve as a mechanism for both physiological and pathophysiological interactions between the brain and other organ systems [100–102]. In mice, CCI increased EVs (microparticles) in blood at 24h [101]. Enriched EVs from posttraumatic blood samples activated microglia in vitro, and like LPS- activated BV2 cells (which upregulate pro-inflammatory factors including miRs) caused propagation of neuroinflammation in the brain of uninjured mice after a stereotaxic cortical injection [101]. Another group also reported increased plasma EVs after CCI injury and demonstrated that enriched EVs from LPS stimulated macrophages, injected systemically, increased microglial activation and lesion volume after TBI [4]. Exosomal proteins were measured from blood samples of 21 patients with moderate-severe TBI; observed changes suggested potential prognostic value [102]. Another clinical study reported significantly increased serum EVs after severe TBI, with even larger changes in patients showing evidence of lung injury [100]. Using an epithelial cell line in vitro, administration of these EVs led to inflammasome activation and pyroptosis, implicating EVs in posttraumatic lung injury [100]. A study of patients with mild or repeated mild TBI showed dysregulated exosomal miRs in blood long after injury, potentially providing a method to help understand persisting neurological symptoms in such patients [103]. Whether EVs can serve as a therapeutic target or treatment has also begun to be addressed in pre-clinical models. Treatment with a neutral sphingomyelinase inhibitor, reduced EV release and downregulated pro-inflammatory factors induced by LPS treatment of BV2 microglia in vitro. Treatment with sphingomyelinase inhibitors reduced gene expression of multiple pro-inflammatory markers associated with microglial activation in brain after CCI injury [104]. Potential for therapeutic use of exosomes was also recently demonstrated. Exosomes from bone marrow mesenchymal stem cells administered retro-orbitally 15 minutes after CCI in mice decreased the inflammatory response, lesion size and functional outcomes [105].
Concluding Remarks
Although it has long been recognized that TBI patients have unusually high infection rates and evidence of changes in various systemic organs, more recent clinical and pre-clinical research has underscored the importance of brain-systemic interactions for TBI outcomes. Experimental work shows that such interactions have bi-directional implications with regard to secondary posttraumatic neuroinflammation and progressive neurodegeneration. TBI causes both acute and chronic systemic immune changes, which mimic, in part, those observed during aging, and can increase potential susceptibility to infection. TBI-related pathological changes occur in multiple systems and organs - including the immune system, intestine, lung and heart. Posttraumatic infections in the pulmonary or GI systems, or non-infectious inflammation in the lung, gut or systemic circulation appear to exacerbate chronic tissue damage and neurobehavioral deficits after head injury.
Mechanistic studies have begun to identify potential therapeutic targets to limit the consequences of these pathogenic brain-systemic changes. However, many questions remain to be addressed (see also “Outstanding Questions”). Among the pathological consequences of TBI-mediated effects in systemic organs, which are the most important with regard to clinical outcomes, and can they be successfully targeted to improve mortality and to facilitate recovery? Are the various changes in systemic organ function after injury additive or synergistic with regard to detrimental impact? Given the multiple proposed mechanisms for brain-systemic interactions that have some experimental support, which are most likely to be modifiable to alter outcome? Can systemic effects of TBI be used as biomarkers for mortality or functional recovery, and if so, can concurrent use of multiple systemic markers enhance predictive accuracy? As TBI appears to accelerate markers of aging, especially with regard to immune changes, can strategies used to modify “inflammaging” provide therapeutic benefit for TBI? Finally, in this emerging research area, as for many others, important sex differences have been observed, and if these translate to humans, it is possible in clinical settings, male and female patients may necessitate different evaluation or treatment approaches.
Outstanding Questions Box.
Among the pathological consequences of TBI-mediated effects in systemic organs, which are most important with regard to clinical outcomes?
Are the various changes in systemic organ function after injury additive or synergistic with regard to detrimental impact?
Given the multiple proposed mechanisms for brain-systemic interactions that have some experimental support, which are most likely to be modifiable to alter outcome?
Can systemic effects of TBI be used as biomarkers for mortality or functional recovery? And if so, can concurrent use of multiple systemic markers enhance predictive accuracy?
As TBI appears to accelerate markers of aging, especially with regard to immune changes, can strategies used to modify “inflammaging” provide therapeutic benefit for TBI?
Are there sex differences in the pathological consequences of TBI that may necessitate different evaluation or treatment approaches for male and female patients?
There is growing recognition, then, that brain trauma results in important systemic changes, many of which may influence outcome. From a clinical perspective, these insights should ultimately impact how such patients are followed and treated. Although animal TBI models, taken individually, may have relatively limited translational relevance, research findings - especially if confirmed across models and species may suggest novel therapeutic and biomarker targeting, as well as modify how head injured patients are monitored and treated.
Highlights.
Traumatic brain injury (TBI) can cause significant changes in systemic organ function
Peripheral immune challenges following TBI can increase chronic neuroinflammation and exacerbate neurological dysfunction
Recognition of brain-systemic interactions after TBI provides important insights relating to the pathobiology and treatment of head injury
Targeting brain systemic changes after TBI may influence long-term morbidity and mortality
Acknowledgements
This work was supported by National Institutes of Health grants R01NS037313 (A.I.F), and R01NS110756 (A.I.F/B.A.S)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing financial interests in relation to this work.
References
- 1.McDonald SJ, et al. (2020) Beyond the Brain: Peripheral Interactions after Traumatic Brain Injury. J Neurotrauma 37, 770–781 [DOI] [PubMed] [Google Scholar]
- 2.Royes LFF and Gomez-Pinilla F (2019) Making sense of gut feelings in the traumatic brain injury pathogenesis. Neurosci Biobehav Rev 102, 345–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(2018) Corrections. Lancet Neurol 17, 203. [DOI] [PubMed] [Google Scholar]
- 4.Hazelton I, et al. (2018) Exacerbation of Acute Traumatic Brain Injury by Circulating Extracellular Vesicles. J Neurotrauma 35, 639–651 [DOI] [PubMed] [Google Scholar]
- 5.Ritzel RM, et al. (2018) Chronic Alterations in Systemic Immune Function after Traumatic Brain Injury. J Neurotrauma 35, 1419–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sundman MH, et al. (2017) The bidirectional gut-brain-microbiota axis as a potential nexus between traumatic brain injury, inflammation, and disease. Brain Behav Immun 66, 31–44 [DOI] [PubMed] [Google Scholar]
- 7.Hu PJ, et al. (2017) Acute brain trauma, lung injury, and pneumonia: more than just altered mental status and decreased airway protection. Am J Physiol Lung Cell Mol Physiol 313, L1–L15 [DOI] [PubMed] [Google Scholar]
- 8.Needham EJ, et al. (2019) The immunological response to traumatic brain injury. J Neuroimmunol 332, 112–125 [DOI] [PubMed] [Google Scholar]
- 9.Puntambekar SS, et al. (2018) Cellular players that shape evolving pathology and neurodegeneration following traumatic brain injury. Brain Behav Immun 71, 9–17 [DOI] [PubMed] [Google Scholar]
- 10.Sharma R, et al. (2019) Infections after a traumatic brain injury: The complex interplay between the immune and neurological systems. Brain Behav Immun 79, 63–74 [DOI] [PubMed] [Google Scholar]
- 11.Doran SJ, et al. (2020) Early or Late Bacterial Lung Infection Increases Mortality After Traumatic Brain Injury in Male Mice and Chronically Impairs Monocyte Innate Immune Function. Crit Care Med 48, e418–e428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma EL, et al. (2017) Bidirectional brain-gut interactions and chronic pathological changes after traumatic brain injury in mice. Brain Behav Immun 66, 56–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muccigrosso MM, et al. (2016) Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun 54, 95–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chou A, et al. (2018) Persistent Infiltration and Impaired Response of Peripherally-Derived Monocytes after Traumatic Brain Injury in the Aged Brain. Int J Mol Sci 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar A, et al. (2016) Microglial/Macrophage Polarization Dynamics following Traumatic Brain Injury. J Neurotrauma 33, 1732–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loane DJ, et al. (2014) Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol 73, 14–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hazeldine J, et al. (2015) Traumatic Brain Injury and Peripheral Immune Suppression: Primer and Prospectus. Front Neurol 6, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Junger WG, et al. (2013) Prehospital hypertonic saline resuscitation attenuates the activation and promotes apoptosis of neutrophils in patients with severe traumatic brain injury. Shock 40, 366–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao Y, et al. (2013) Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS One 8, e68963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mrakovcic-Sutic I, et al. (2010) Early changes in frequency of peripheral blood lymphocyte subpopulations in severe traumatic brain-injured patients. Scand J Immunol 72, 57–65 [DOI] [PubMed] [Google Scholar]
- 21.Smrcka M, et al. (2005) Immune system status in the patients after severe brain injury. Bratisl Lek Listy 106, 144–146 [PubMed] [Google Scholar]
- 22.Schwulst SJ, et al. (2013) Traumatic brain injury-induced alterations in peripheral immunity. J Trauma Acute Care Surg 75, 780–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faries PL, et al. (1998) Intestinal permeability correlates with severity of injury in trauma patients. J Trauma 44, 1031–1035; discussion 1035–1036 [DOI] [PubMed] [Google Scholar]
- 24.Krakau K, et al. (2006) Metabolism and nutrition in patients with moderate and severe traumatic brain injury: A systematic review. Brain Inj 20, 345–367 [DOI] [PubMed] [Google Scholar]
- 25.Norton JA, et al. (1988) Intolerance to enteral feeding in the brain-injured patient. J Neurosurg 68, 62–66 [DOI] [PubMed] [Google Scholar]
- 26.Tan M, et al. (2011) Enteral nutrition in patients with severe traumatic brain injury: reasons for intolerance and medical management. Br J Neurosurg 25, 2–8 [DOI] [PubMed] [Google Scholar]
- 27.Bansal V, et al. (2009) Traumatic brain injury and intestinal dysfunction: uncovering the neuro-enteric axis. J Neurotrauma 26, 1353–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katzenberger RJ, et al. (2015) Death following traumatic brain injury in Drosophila is associated with intestinal barrier dysfunction. Elife 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin W, et al. (2008) Increased intestinal inflammatory response and gut barrier dysfunction in Nrf2-deficient mice after traumatic brain injury. Cytokine 44, 135–140 [DOI] [PubMed] [Google Scholar]
- 30.Hanscom M, et al. (2020) Acute Colitis During Chronic Experimental Traumatic Brain Injury in Mice Induces Dysautonomia and Persistent Extraintestinal, Systemic, and CNS Inflammation with Exacerbated Neurological Deficits. J Neuroinflammation, in press [DOI] [PMC free article] [PubMed]
- 31.Cunningham C, et al. (2009) Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry 65, 304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klein SL and Flanagan KL (2016) Sex differences in immune responses. Nat Rev Immunol 16, 626–638 [DOI] [PubMed] [Google Scholar]
- 33.Angoa-Perez M, et al. (2020) Repetitive, mild traumatic brain injury results in a progressive white matter pathology, cognitive deterioration, and a transient gut microbiota dysbiosis. Sci Rep 10, 8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matharu D, et al. (2019) Repeated mild traumatic brain injury affects microbial diversity in rat jejunum. J Biosci 44 [PubMed] [Google Scholar]
- 35.Nicholson SE, et al. (2019) Moderate Traumatic Brain Injury Alters the Gastrointestinal Microbiome in a Time-Dependent Manner. Shock 52, 240–248 [DOI] [PubMed] [Google Scholar]
- 36.Treangen TJ, et al. (2018) Traumatic Brain Injury in Mice Induces Acute Bacterial Dysbiosis Within the Fecal Microbiome. Front Immunol 9, 2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, et al. (2018) Clostridium butyricum exerts a neuroprotective effect in a mouse model of traumatic brain injury via the gut-brain axis. Neurogastroenterol Motil 30, e13260. [DOI] [PubMed] [Google Scholar]
- 38.Ma Y, et al. (2019) Lactobacillus acidophilus Exerts Neuroprotective Effects in Mice with Traumatic Brain Injury. J Nutr 149, 1543–1552 [DOI] [PubMed] [Google Scholar]
- 39.Spychala MS, et al. (2018) Age-related changes in the gut microbiota influence systemic inflammation and stroke outcome. Ann Neurol 84, 23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ritzel RM, et al. (2019) Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiol Aging 77, 194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rincon F, et al. (2012) Impact of acute lung injury and acute respiratory distress syndrome after traumatic brain injury in the United States. Neurosurgery 71, 795–803 [DOI] [PubMed] [Google Scholar]
- 42.Scott BN, et al. (2013) Incidence, prevalence, and occurrence rate of infection among adults hospitalized after traumatic brain injury: study protocol for a systematic review and meta-analysis. Syst Rev 2, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vincent JL (2003) Nosocomial infections in adult intensive-care units. Lancet 361, 2068–2077 [DOI] [PubMed] [Google Scholar]
- 44.Kourbeti IS, et al. (2012) Infections in traumatic brain injury patients. Clin Microbiol Infect 18, 359–364 [DOI] [PubMed] [Google Scholar]
- 45.Vermeij JD, et al. (2013) Traumatic brain injury in rats induces lung injury and systemic immune suppression. J Neurotrauma 30, 2073–2079 [DOI] [PubMed] [Google Scholar]
- 46.Kerr NA, et al. (2018) Traumatic Brain Injury-Induced Acute Lung Injury: Evidence for Activation and Inhibition of a Neural-Respiratory-Inflammasome Axis. J Neurotrauma 35, 2067–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kerr NA, et al. (2020) Enoxaparin Attenuates Acute Lung Injury and Inflammasome Activation after Traumatic Brain Injury. J Neurotrauma [DOI] [PMC free article] [PubMed]
- 48.Parker TM, et al. (2017) The danger zone: Systematic review of the role of HMGB1 danger signalling in traumatic brain injury. Brain Inj 31, 2–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jacovides CL, et al. (2019) An inflammatory pulmonary insult post-traumatic brain injury worsens subsequent spatial learning and neurological outcomes. J Trauma Acute Care Surg 87, 552–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, et al. (2020) Incidence, Risk Factors, and Outcomes of Ventilator-Associated Pneumonia in Traumatic Brain Injury: A Meta-analysis. Neurocrit Care 32, 272–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai SS, et al. (2016) The role of cardiac troponin I in prognostication of patients with isolated severe traumatic brain injury. J Trauma Acute Care Surg 80, 477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salim A, et al. (2008) Significance of troponin elevation after severe traumatic brain injury. J Trauma 64, 46–52 [DOI] [PubMed] [Google Scholar]
- 53.Rimaz S, et al. (2019) Significance of Cardiac Troponin I Elevation in Traumatic Brain Injury Patients. Anesth Pain Med 9, e90858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao Q, et al. (2019) Immune Response Mediates Cardiac Dysfunction after Traumatic Brain Injury. J Neurotrauma 36, 619–629 [DOI] [PubMed] [Google Scholar]
- 55.Chaikittisilpa N, et al. (2019) Early cardiovascular function and associated hemodynamics in adults with isolated moderate-severe traumatic brain injury: A pilot study. J Clin Neurosci 69, 97–103 [DOI] [PubMed] [Google Scholar]
- 56.Venkata C and Kasal J (2018) Cardiac Dysfunction in Adult Patients with Traumatic Brain Injury: A Prospective Cohort Study. Clin Med Res 16, 57–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prathep S, et al. (2014) Preliminary report on cardiac dysfunction after isolated traumatic brain injury. Crit Care Med 42, 142–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cuisinier A, et al. (2016) Myocardial function at the early phase of traumatic brain injury: a prospective controlled study. Scand J Trauma Resusc Emerg Med 24, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore EM, et al. (2010) The incidence of acute kidney injury in patients with traumatic brain injury. Ren Fail 32, 1060–1065 [DOI] [PubMed] [Google Scholar]
- 60.Civiletti F, et al. (2019) Acute Tubular Injury is Associated With Severe Traumatic Brain Injury: in Vitro Study on Human Tubular Epithelial Cells. Sci Rep 9, 6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu GY, et al. (2016) [Value of urinary liver fatty acid-binding protein in assessing severity of brain trauma and predicting acute kidney injury]. Nan Fang Yi Ke Da Xue Xue Bao 36, 1527–1530 [PubMed] [Google Scholar]
- 62.Wang R, et al. (2020) Serum Procalcitonin Level Predicts Acute Kidney Injury After Traumatic Brain Injury. World Neurosurg [DOI] [PubMed]
- 63.Li N, et al. (2013) Neutrophil gelatinase-associated lipocalin as an early marker of acute kidney injury in patients with traumatic brain injury. J Nephrol 26, 1083–1088 [DOI] [PubMed] [Google Scholar]
- 64.Mrozek S, et al. (2016) Cerebral and extracerebral vulnerability to hypoxic insults after diffuse traumatic brain injury in rats. Brain Res 1646, 334–341 [DOI] [PubMed] [Google Scholar]
- 65.Nizamutdinov D, et al. (2017) Hepatic alterations are accompanied by changes to bile acid transporter-expressing neurons in the hypothalamus after traumatic brain injury. Sci Rep 7, 40112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Villapol S, et al. (2015) Hepatic expression of serum amyloid A1 is induced by traumatic brain injury and modulated by telmisartan. Am J Pathol 185, 2641–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Soriano S, et al. (2020) Serum Amyloid A is Expressed in the Brain After Traumatic Brain Injury in a Sex-Dependent Manner. Cell Mol Neurobiol 40, 1199–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de Castro MRT, et al. (2017) Previous physical exercise alters the hepatic profile of oxidative-inflammatory status and limits the secondary brain damage induced by severe traumatic brain injury in rats. J Physiol 595, 6023–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coutinho AE and Chapman KE (2011) The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol 335, 2–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Agha A, et al. (2007) Hypopituitarism following traumatic brain injury (TBI). Br J Neurosurg 21, 210–216 [DOI] [PubMed] [Google Scholar]
- 71.Lieberman SA, et al. (2001) Prevalence of neuroendocrine dysfunction in patients recovering from traumatic brain injury. J Clin Endocrinol Metab 86, 2752–2756 [DOI] [PubMed] [Google Scholar]
- 72.Rowe RK, et al. (2016) Diffuse traumatic brain injury affects chronic corticosterone function in the rat. Endocr Connect 5, 152–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cernak I, et al. (1999) Neuroendocrine responses following graded traumatic brain injury in male adults. Brain Inj 13, 1005–1015 [DOI] [PubMed] [Google Scholar]
- 74.Taylor AN, et al. (2008) Injury severity differentially affects short- and long-term neuroendocrine outcomes of traumatic brain injury. J Neurotrauma 25, 311–323 [DOI] [PubMed] [Google Scholar]
- 75.Taylor AN, et al. (2010) Injury severity differentially alters sensitivity to dexamethasone after traumatic brain injury. J Neurotrauma 27, 1081–1089 [DOI] [PubMed] [Google Scholar]
- 76.Russell AL, et al. (2018) Differential Responses of the HPA Axis to Mild Blast Traumatic Brain Injury in Male and Female Mice. Endocrinology 159, 2363–2375 [DOI] [PubMed] [Google Scholar]
- 77.Bromberg CE, et al. (2020) Sex-Dependent Pathology in the HPA Axis at a Sub-acute Period After Experimental Traumatic Brain Injury. Front Neurol 11, 946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Powner DJ and Boccalandro C (2008) Adrenal insufficiency following traumatic brain injury in adults. Curr Opin Crit Care 14, 163–166 [DOI] [PubMed] [Google Scholar]
- 79.Bernard F, et al. (2006) Incidence of adrenal insufficiency after severe traumatic brain injury varies according to definition used: clinical implications. Br J Anaesth 96, 72–76 [DOI] [PubMed] [Google Scholar]
- 80.Dong T, et al. (2016) Cortisol-induced immune suppression by a blockade of lymphocyte egress in traumatic brain injury. J Neuroinflammation 13, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mracsko E, et al. (2014) Differential effects of sympathetic nervous system and hypothalamic-pituitary-adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav Immun 41, 200–209 [DOI] [PubMed] [Google Scholar]
- 82.Catania A, et al. (2009) Detrimental consequences of brain injury on peripheral cells. Brain Behav Immun 23, 877–884 [DOI] [PubMed] [Google Scholar]
- 83.Alali AS, et al. (2017) Beta-blockers and Traumatic Brain Injury: A Systematic Review, Meta-analysis, and Eastern Association for the Surgery of Trauma Guideline. Ann Surg 266, 952–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Khalili H, et al. (2020) Beta-Blocker Therapy in Severe Traumatic Brain Injury: A Prospective Randomized Controlled Trial. World J Surg 44, 1844–1853 [DOI] [PubMed] [Google Scholar]
- 85.Ko A, et al. (2016) Early propranolol after traumatic brain injury is associated with lower mortality. J Trauma Acute Care Surg 80, 637–642 [DOI] [PubMed] [Google Scholar]
- 86.Bellinger DL and Lorton D (2018) Sympathetic Nerve Hyperactivity in the Spleen: Causal for Nonpathogenic-Driven Chronic Immune-Mediated Inflammatory Diseases (IMIDs)? Int J Mol Sci 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Korner A, et al. (2019) Sympathetic nervous system controls resolution of inflammation via regulation of repulsive guidance molecule A. Nat Commun 10, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mirakaj V, et al. (2014) Vagus nerve controls resolution and pro-resolving mediators of inflammation. J Exp Med 211, 1037–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Braun M, et al. (2017) Activation of Myeloid TLR4 Mediates T Lymphocyte Polarization after Traumatic Brain Injury. J Immunol 198, 3615–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hsieh CL, et al. (2014) CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma 31, 1677–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Makinde HM, et al. (2017) Nonclassical Monocytes Mediate Secondary Injury, Neurocognitive Outcome, and Neutrophil Infiltration after Traumatic Brain Injury. J Immunol 199, 3583–3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Morganti JM, et al. (2016) Age exacerbates the CCR2/5-mediated neuroinflammatory response to traumatic brain injury. J Neuroinflammation 13, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chaban V, et al. (2020) Systemic Inflammation Persists the First Year after Mild Traumatic Brain Injury: Results from the Prospective Trondheim Mild Traumatic Brain Injury Study. J Neurotrauma [DOI] [PMC free article] [PubMed]
- 94.Henry RJ, et al. (2020) Microglial depletion with CSF1R inhibitor during chronic phase of experimental traumatic brain injury reduces neurodegeneration and neurological deficits. J Neurosci [DOI] [PMC free article] [PubMed]
- 95.Doran SJ, et al. (2019) Sex Differences in Acute Neuroinflammation after Experimental Traumatic Brain Injury Are Mediated by Infiltrating Myeloid Cells. J Neurotrauma 36, 1040–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Villapol S, et al. (2017) Sexual dimorphism in the inflammatory response to traumatic brain injury. Glia 65, 1423–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kumar A, et al. (2013) Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging 34, 1397–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Johnson VE, et al. (2013) Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136, 28–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ramlackhansingh AF, et al. (2011) Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 70, 374–383 [DOI] [PubMed] [Google Scholar]
- 100.Kerr NA, et al. (2019) Human Lung Cell Pyroptosis Following Traumatic Brain Injury. Cells 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kumar A, et al. (2017) Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J Neuroinflammation 14, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mondello S, et al. (2020) Circulating Brain Injury Exosomal Proteins following Moderate-To-Severe Traumatic Brain Injury: Temporal Profile, Outcome Prediction and Therapy Implications. Cells 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Devoto C, et al. (2020) Exosomal MicroRNAs in Military Personnel with Mild Traumatic Brain Injury: Preliminary Results from the Chronic Effects of Neurotrauma Consortium Biomarker Discovery Project. J Neurotrauma [DOI] [PMC free article] [PubMed]
- 104.Kumar A, et al. (2019) Neutral Sphingomyelinase Inhibition Alleviates LPS-Induced Microglia Activation and Neuroinflammation after Experimental Traumatic Brain Injury. J Pharmacol Exp Ther 368, 338–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ni H, et al. (2019) Exosomes Derived From Bone Mesenchymal Stem Cells Ameliorate Early Inflammatory Responses Following Traumatic Brain Injury. Front Neurosci 13, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]