

Abstract
Repetitive sequences throughout the genome are a major source of endogenous DNA damage, due to the propensity of many of them to form alternative non-B DNA structures that can interfere with replication, transcription, and DNA repair. These repetitive sequences are prone to breakage (fragility) and instability (changes in repeat number). Repeat fragility and expansions are linked to several diseases, including many cancers and neurodegenerative diseases, hence the importance of understanding the mechanisms that cause genome instability and contribute to these diseases. This review focuses on recent findings of mechanisms causing repeat fragility and instability, new associations between repeat expansions and genetic diseases, and potential therapeutic options to target repeat expansions.
Keywords: chromosome fragility, replication fork stalling and restart, microsatellite instability (MSI), R-loops, repeat expansion diseases
Introduction
Genome integrity is constantly under threat due to both endogenous and exogenous DNA damaging sources. One source of endogenous DNA damage is repetitive sequences that have the ability to form alternative secondary structures different from B-form DNA. These structure-forming repeats can interfere with various cellular processes including replication, transcription, and DNA repair. Structure-forming repeat sequences are prone to chromosome breakage and are enriched at breakpoints of genomic rearrangements in cancer cells [1]. Also, a still growing number of neurodegenerative diseases are caused by repeat expansions that occur in both coding and non-coding regions of the genome [2]. Many diseases linked to repeat instability do not have any successful treatment options, highlighting the importance of understanding mechanisms that cause these diseases and to develop potential therapeutic options in the future. In this review, we discuss mechanisms that contribute to repeat instability, focusing on the most recent advances, new associations between repeat expansions and genetic diseases, and potential therapies to contract expanded repeats.
Repeats Interfere with Replication to Cause Genome Instability
Repetitive sequences throughout the genome serve as potential barriers to replication that can result in fork stalling and collapse. Various regions throughout the genome termed fragile sites are prone to chromosome breakage, especially under replication stress. Several recent advances have confirmed that DNA structures play an important role in common fragile site (CFS) fragility. Sinai et al. inserted an AT-rich sequence predicted to form hairpin structures from CFS FRA16C into a normally non-fragile ectopic site and observed recurrent chromosome gaps, indicating that the inserted AT-rich sequences interfered with completion of replication [3]. Another common fragile site, FRA16D, contains a polymorphic AT repeat (Flex1) that stalls replication and causes fragility when inserted into a yeast chromosome [4]. A recent study showed that Flex1 causes a length-dependent increase in fragility that is strongly correlated with the lengths that caused fork stalling and form cruciform structures in vivo [5]. The AT repeat fragility was dependent on the Mus81-Mms4 nuclease complex working in the context of the Slx4 scaffold [5], which is the same nuclease complex shown to be required for breaks at FRA16D and other CFSs in human cells (reviewed in [6]) (Fig. 1a). It appears that AT repeat fragility may be a wide-spread phenomenon as a genome-wide study to identify sites of fork collapse upon inhibition of the ATR checkpoint kinase, detected through RPA-ChIP and BrITL (breaks identified by TdT labeling), identified AT-rich repeats as the most commonly represented sites in human cells [7]. Interestingly, the same study showed that (CAGAGG)n and (CACAG)n repeats, which form quadruplex structures, were most commonly identified as sites of fork collapse in mouse cells, indicating that the most problematic repeats may vary by organism [7]. Poly(dA:dT) tracts, that are unwinding elements but may also form triplex secondary structures, were demonstrated to be a causal factor of fork stalling and breakage under replication stress within early-replicating fragile sites in activated B cells isolated from mice [8]. Recent evidence also suggests that TG-repeats in stickleback fish contribute to recurrent deletions at the Pel locus due to their ability to form alternative secondary structures, driving evolution in these organisms [9]. Overall, these various findings indicate that replication fork stalling and subsequent breakage at structure-forming repeats are a significant source of genome instability across multiple organisms and conditions.
Figure 1. Fork stalling at structure forming repeats results in repeat fragility and instability.
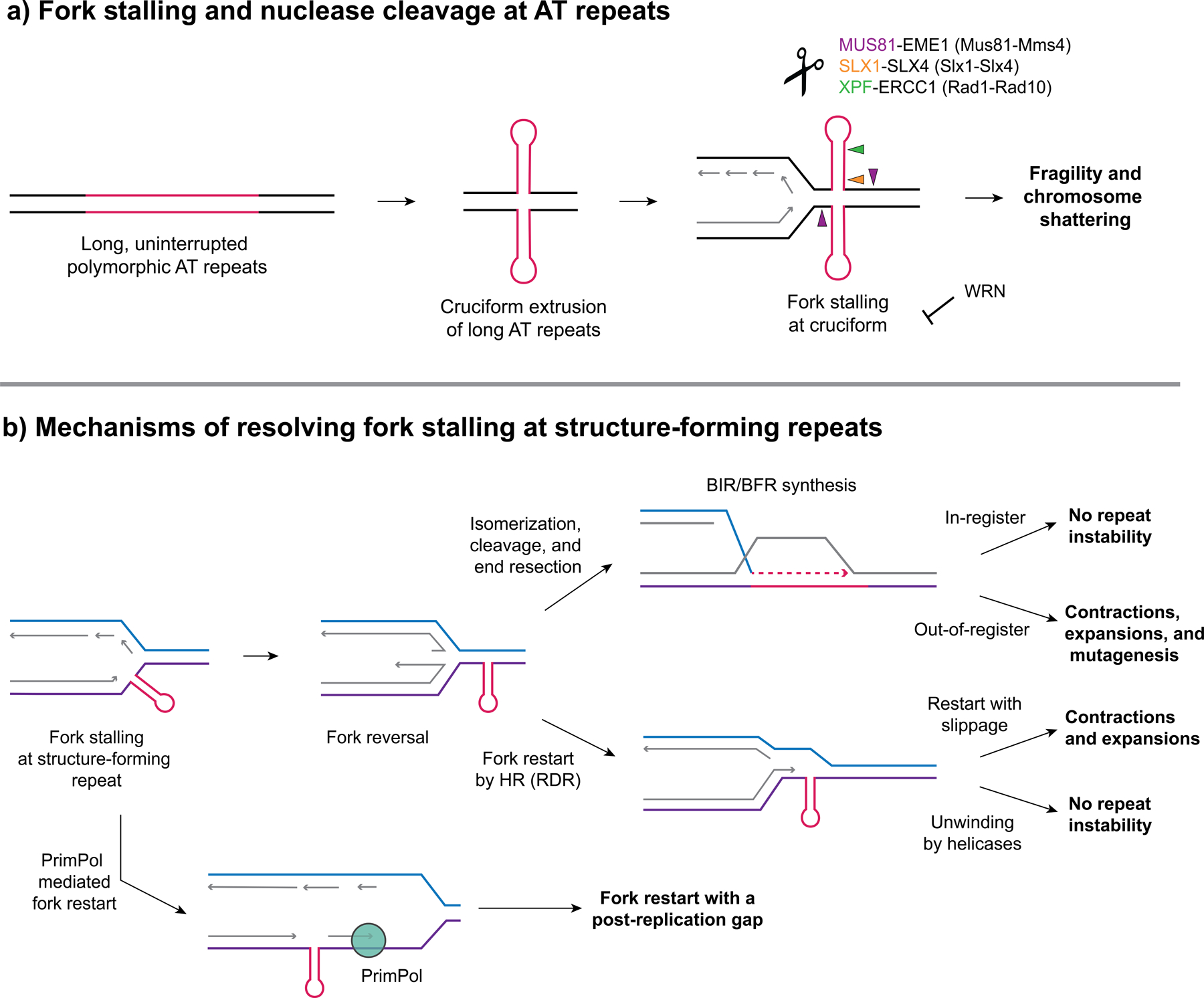
(a) Long, uninterrupted polymorphic AT repeats have the potential to form cruciform structures that serve as a barrier to replication and cause fork stalling and ATR activation. WRN (Werner Syndrome) helicase (a RecQ helicase) can be recruited to unwind the structure and prevent fork collapse. Loss of WRN results in chromosome shattering in MMR deficient MSI cancers with expanded AT repeats. (b) Pathways to resolve fork stalling at structure-forming repeats (a hairpin is shown but it could also be a G4 or triplex structure). Fork stalling can occur due to structure-forming repeats serving as a barrier to replication on either the leading or lagging strand and can result in fork reversal (a resected reversed fork is shown). Fork restart can occur through several pathways: (1) repriming past the structure, e.g. by PrimPol (2) through recombination-dependent replication (RDR), using the displaced 3’ end from a reversed fork and template strand invasion, and (3) through a BIR-like pathway after fork cleavage and end resection (referred to as broken fork repair, BFR). These pathways can result in expansions or contractions if slippage or out-of-register invasion or structure bypass occurs. Alternatively, unwinding of the structure by helicases during restart can avoid repeat instability. Exposed ssDNA accumulating during BIR can result in repeat-induced mutagenesis (RIM).
A new study provides a very important link between fragility at AT repeats and cancers caused by microsatellite instability (MSI). Previous studies demonstrated that WRN (Werner Syndrome) helicase, a RecQ DNA helicase, is essential for survival in mismatch repair (MMR) deficient cells with MSI, and that loss of WRN resulted in increased chromatin bridges, chromosome fragmentation, and micronuclei (see [10] and references therein). These studies identified WRN as a synthetic lethal target for potential MSI cancer therapeutics. The Nussenzweig lab followed up on this evidence to determine a mechanism through which WRN helicase acts in MSI cancers and why is it necessary for viability. They used END-seq to determine the genome-wide sites of double-strand breaks (DSBs) formed in MSI cancer cell lines upon depletion of WRN and observed that these breaks accumulate primarily at AT repeats [10]. Interestingly, the repeats at the breakage sites were expanded compared to non-MSI control cell lines and were susceptible to MUS81-EME1-SLX4 nuclease cleavage, in agreement with the Kaushal et al. data described above [5,10]. Overall, the authors propose that in MSI cancers, MMR deficiencies contribute to AT repeat expansions which then can form cruciform-like secondary structures that stall replication forks [10]. Fork stalling causes ATR activation and recruitment of WRN to aid in completion of DNA replication [10]. Upon loss of WRN, MUS81-EME1 endonuclease cleaves at the expanded AT repeats, resulting in chromosome fragmentation and cell death [10] (Fig. 1a).
Replication fork stalling occurs at telomeres due to the G-quadruplex (G4) structures formed by telomeric sequences, and telomeres display features of fragile sites (reviewed in [11]). Internal G4 structures can also cause fork stalling, which can be overcome by repriming by PrimPol (reviewed in [12]) (Fig. 1b). Recent studies demonstrate that the Exo1 exonuclease is important for preventing telomere length instability, chromosomal aberrations, and cell death, especially when cells are treated with a G4-stabilizer [13,14]. Exo1 was proposed to process forks stalled within the telomere and mediate repair by a recombination-based repair mechanism [13]. The Warsaw Breakage Syndrome associated DDX11 helicase was also recently found to resolve G4 structures and protect cells from DNA damage during replication and improper sister chromatid cohesion [15].
In addition to fragility, replication problems at structure-forming repeats can also lead to repeat expansions and contractions (reviewed in [16,17]). One ended breaks, for example caused by fork collapse, can trigger break-induced replication (BIR). Large-scale expansions at CAG/CTG, CGG/CCG, and GAA/TTC repeats are dependent on proteins known be involved in BIR, such as Pol32, Pif1, and HR proteins Rad51 and Rad52 [18–20] (Fig. 1b). It is not yet clear if the BIR mechanism involves fork restart in S phase using the as yet unreplicated chromosome ahead of the stall as a template (broken fork repair, BFR), or whether it occurs in G2, using the replicated sister chromatid or another chromosome as the template (Fig. 1b). Recent results show that a late S-phase event that is likely fork restart happens after fork collapse at an expanded CAG/CTG repeat [21,22].
The lagging strand, which has single-stranded stretches exposed during replication, is particularly prone to allowing DNA structure formation. Khristich et al. found that large-scale contractions of GAA repeats occurred primarily during lagging strand replication and were dependent on their ability to form a H-DNA triplex structure [23]. Contractions are proposed to occur by bypass of the template structure by Pol δ, and are exacerbated by mutations that affect Pol δ processivity [23]. Another response to lagging strand hairpins can be a template switch to copy from the sister chromatid, which has been shown by multiple groups to cause repeat expansions (see [17] for review). A new appreciation is that histone modifications are required to facilitate efficient D-loop extension during sister-chromatid recombination and prevent CAG repeat expansions in a yeast model. These include histone H4 acetylation on lysines 12 and 16 and histone H2A.1 phosphorylation on threonine 126 [24,25]. In human cells the histone deacetylase HDAC3 stimulates CAG repeat expansions [26]. The Lahue group recently identified that, MSH3 previously identified to be required for CAG expansions, is the target of HDAC3 deacetylation, which is required for MutSβ nuclear localization [27]. HDAC2 also mildly enhances CAG expansions at the HD locus, perhaps by altering chromatin structure of the locus [28].
Transcription-Induced DNA Structures
Transcription can also pose a threat to genome integrity since it involves unwinding of the DNA double helix. One byproduct of transcription that is particularly relevant for structure-forming repeats is R-loops [29,30]. When the transcribed strand is engaged in an DNA:RNA hybrid, it can allow DNA structure formation on the exposed single-stranded non-transcribed strand, and this is particularly likely in G-rich sequences such as expandable CGG, CAG, or G4C2 repeats (see [2,31] for review). An R-loop with a G4 structure is referred to as a G-loop, and with a slipped-out hairpin an S-loop (Fig. 2). R-loops at the FMR1 and C9orf72 gene loci that can contain expanded CGG and G4C2 repeats, respectively, were recently mapped at the nucleotide level using bisulfite footprinting and deep sequencing, confirming that their presence allows structure formation on the non-transcribed strand in vivo and showing that even non-expanded alleles form unusually long, stable R-loops of ~500–800 bp at these loci [32]. These results are consistent with earlier models suggesting that G-loops can also form as a hybrid between the ssDNA and ssRNA structures [33] (Fig. 2). Su and Freudenreich investigated how R-loops at expanded CAG repeats cause repeat instability and fragility, and demonstrated that expanded CAG repeats engaged in R-loops are prone to cytosine deamination, which recruits the base excision repair (BER) pathway to cause repeat contractions [34]. In addition, cleavage by the MutLγ (Mlh1-Mlh3) nuclease was R-loop dependent, providing a possible mechanism for how this nuclease acts inappropriately on CAG or CTG hairpins to cause repeat expansions (see [31] for review).
Figure 2. Predicted models of R-loop formation at structure-forming repeats.
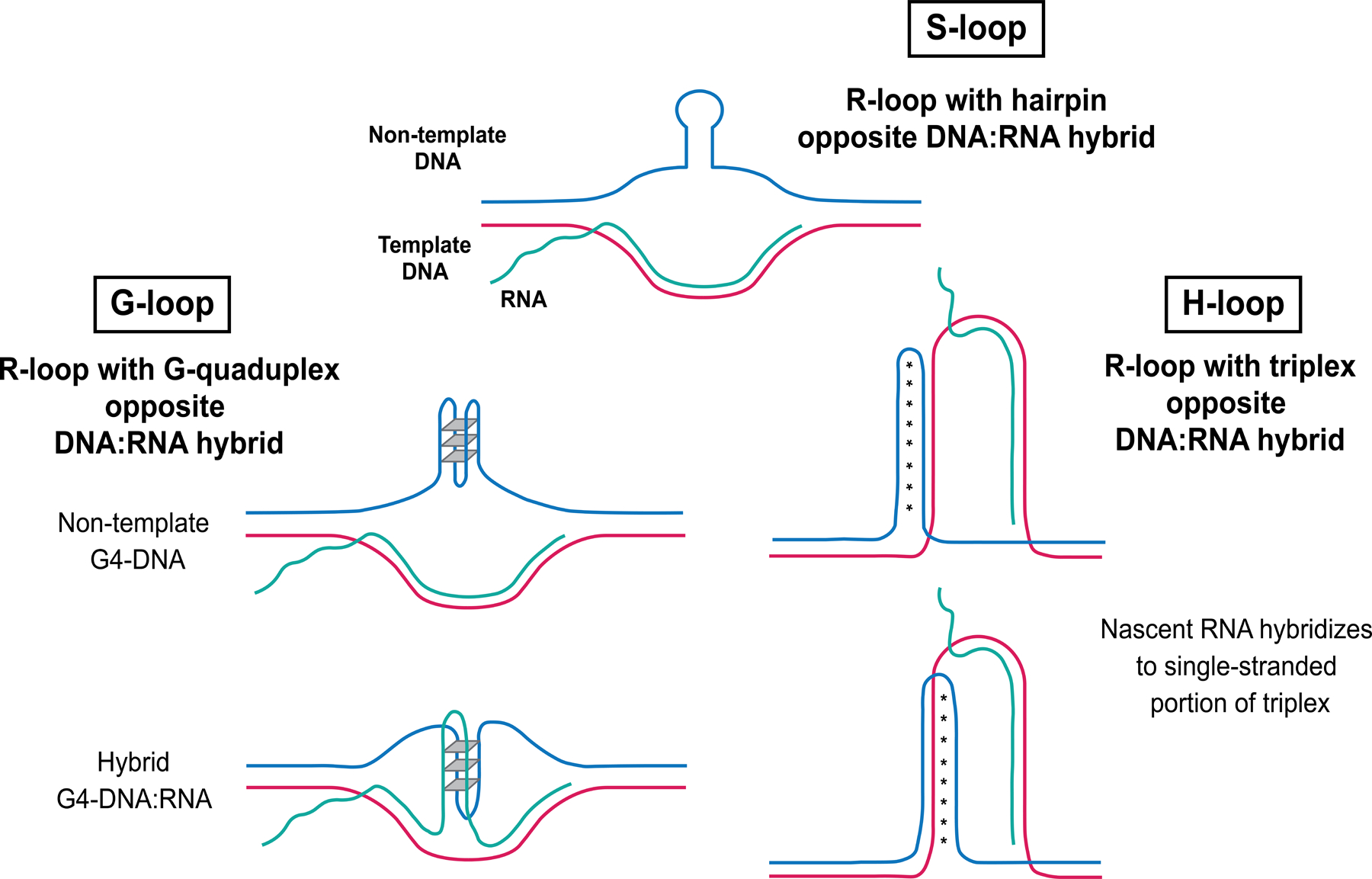
S-loops contain a hairpin structure formed on the non-template displaced single DNA strand opposite a DNA:RNA hybrid. G-loops contain a G-quadruplex opposite a DNA:RNA hybrid; either the displaced non-template single DNA strand can form a G-quadruplex (top) or a hybrid DNA:RNA G-quadruplex can form in the context of an R-loop (bottom). H-loops contain a triplex or H-DNA structure opposite a DNA:RNA hybrid. Nascent RNA can bind the single-stranded portion of the triplex structure and the triplex can form in two orientations, in which Hoogsteen bonding occurs either between two purine strands (top) or a purine and pyrimidine strand (bottom).
Even though they aren’t GC rich, expanded GAA repeats have also been shown to form R-loops. Neil et al. examined the role of DNA:RNA hybrids in GAA repeat stability and found that loss of RNase H (which cleaves the RNA of DNA:RNA hybrids) resulted in an increase in GAA expansions [20]. However, unlike canonical R-loops, GAA repeats may form a novel type of structure they termed an H-loop, which is a combination of an R-loop and triplex H-DNA [20] (Fig. 2). There is evidence that PrimPol repriming activity may prevent unscheduled R-loop formation at GAA repeats by helping to restart stalled replication and preventing ssDNA from accumulating [35].
H3K9 methylation has recently been shown to suppress DNA:RNA hybrid-induced instability of satellite repeats in Caenorhabditis elegans [36,37]. More specifically, C. elegans with loss of the MET-2 (SETDB1 homolog) H3K9 methyltransferase accumulate satellite repeat transcripts, sequences that are normally marked by H3K9me2 and are repressed. These transcripts can bind to simple and satellite repeat sequences, resulting in the formation of DNA:RNA hybrids [36,37]. Simple repetitive transcripts may be even more dangerous to genomic integrity than unique sequences as they lack signals for RNA processing and can form structures that may stall replication. A genome-wide screen examining synthetic lethality in met-2 mutants identified RNA processing, nuclear RNA degradation, and DNA repair and replication fork stability factors [37]. For example the BRCA1/BARD1 complex was found to be partially redundant with MET-2, as it prevents the accumulation of satellite repeat transcripts and DNA:RNA hybrid formation that contribute to genomic instability and germline lethality in C. elegans [37].
In addition to DNA:RNA hybrids, transcription itself can cause repeat instability through unwinding of DNA and introduction of negative supercoils behind RNA Polymerase II (RNAPII) (see [38] for review). Koch et al. demonstrated that defects in chromatin remodeling upon loss of Isw1 causes an increase in CAG repeat expansions and this was dependent on both transcription through the CAG repeat tract and nucleotide excision repair (NER) and BER proteins [39]. Isw1 is known to be important for nucleosome spacing, which was altered in the mutant cells following transcription. The authors proposed that improper establishment of nucleosomes after RNAPII passage in cells lacking Isw1 leads to CAG/CTG hairpin formation, which triggers BER and NER to cause CAG instability [39].
Interference with Repair causes Repeat Instability
DNA repair pathways are a double-edged sword. Though they are meant to protect the genome and maintain genome integrity, in the context of repetitive DNA they can lead to inappropriate repair, repeat instability, or genome rearrangements. Some recent advances have shed light on the players and what can go wrong.
Multiple NER nucleases have recently been shown to target structure-forming repetitive sequences. XPF-ERCC1 (Rad1-Rad10 in yeast) causes deletions and translocations due to cleavage of cruciform, inverted repeat, and H-DNA triplex structures, resulting in genome instability [5,40,41]. Additionally, the MutSβ (MSH2-MSH3) complex recognizes Z-DNA as damage, resulting in recruitment of and cleavage by XPF-ERCC1 to cause deletions and translocations [42]. These nucleases act in both replication-dependent and replication-independent pathways to cause repeat fragility.
In contrast, other nucleases have been found to be protective of repeat instability. A new player on the scene is FANCD2 and FANCI-associated nuclease 1 (FAN1), which was identified in a GWA study as a modifier of Huntington’s disease (HD) onset [43–45]. FAN1 protects against CAG repeat expansions in HD cell lines in a dose-dependent and nuclease-independent manner, and knockout of FAN1 increases CAG repeat expansion in HD induced pluripotent stem cells (iPSCs) [45,46]. FAN1 also protects against somatic CGG repeat expansions in a Fragile X mouse model [47]. Interestingly, it was recently shown that Fan1-dependent somatic CAG expansions in HD knock-in mice are dependent on the presence of MLH1, indicating that it acts downstream of a MutL-dependent process [48].
MMR proteins have long been known to play a role in repeat instability, and some new data sheds light on possible mechanisms. It was observed earlier that CAG expansions in an HD mouse model, as well as all germ line and somatic CGG expansions in a Fragile X mouse, were dependent on MutLγ (MLH1-MLH3) [49,50]. MutLα and MutLβ were also found to prevent CGG expansions in mouse embryonic stem cells, as PMS1 and PMS2 prevented expansions similarly to MLH1 and MLH3 [51]. MLH3 nuclease activity is required for CGG expansions in a mouse stem cell model, consistent with evidence from a yeast model that Mlh3 nuclease activity causes CAG repeat fragility and instability [34,52]. Therefore, MutLγ cleavage activity is implicated as a key component of its inappropriate action at repeats and its target is likely a conserved feature of hairpin-forming sequences. However EXO1, which normally acts downstream of MutLγ in meiosis, protects against somatic CGG repeat expansions in a Fragile X mouse model [50]. Therefore, EXO1 may process a MutLγ cleaved structure to prevent expansions. There is also evidence of crosstalk between the MMR and BER machinery: MSH2-MSH3 stimulates Pol β to copy through DNA structures on the template strand and displace a 5’ flap during BER, which promotes CAG and GAA trinucleotide expansion [53]. In the absence of MSH2-MSH3, Pol β bypasses the loop structure, resulting in repeat deletions [53]. Overall, the evidence shows that various repair pathways act inappropriately in the context of DNA structures to contribute to repeat instability.
Repeats Traveling to Specific Nuclear Domains for Repair
There is mounting evidence that several types of DNA damage relocate within the nucleus for repair, including persistent DSBs, DSBs within rDNA and heterochromatin, and collapsed forks due to severe replication stress or replication fork barriers (RFBs), including structure-forming repeats (see [54] for review). In yeast, persistent DSBs can relocate to either the NPC in all cell cycle phases or the SUN domain protein Mps3 in the inner nuclear membrane during S and G2 phases [55]. While relocation to the NPC can promote BIR or other events requiring strand invasion, association with Mps3 prevents aberrant recombination from occurring, indicating that different destinations can control repair outcome ([56] and reviewed in ([57,58]). However, when relocation fails, genome instability occurs. Recent evidence demonstrates that collapsed forks that encounter a CAG repeat replication barrier in yeast relocate to the nuclear pore complex (NPC) through a sumoylation-mediated mechanism that prevents chromosomal breaks and end loss events [22]. This relocation also suppresses Rad52-mediated repeat instability [21]. Interestingly, Rad51 was excluded from the CAG repeat when it was in the nuclear interior, only associating after movement to the NPC [22]. In a recent study of eroded telomeres, it was found that mutating the NPC basket protein Nup1 impairs relocalization of both telomeric and expanded CAG repeats [59]. In both cases, the Nup1 defect led to altered repair: either increased Rad52-dependent CAG repeat contractions or increased sister chromatid recombination at telomeres, providing evidence that relocation is important for suppressing inappropriate HR at repetitive DNA [59]. Related, Maestroni et al. showed that loss of telomerase and Bqt4, a protein involved in anchoring telomeres to the nuclear envelope, resulted in enhanced subtelomeric recombination [60].
In higher eukaryotes including Drosophila and mammalian cells repair within repetitive heterochromatic DNA is also controlled by nuclear position (see [61] for review). DSBs within pericentromeric heterochromatin of the Drosophila genome, mainly consisting of satellite repeats, relocalize to the nuclear periphery (see [62] for review). In a recent development, this was shown to occur by directed motion along nuclear actin filaments [63]. Impairment of relocalization resulted in genomic instability including chromosome fusions, aneuploidy, and abnormal satellite DNA copy number [63]. In mammalian cells, the Soutoglou lab showed that breaks within heterochromatic satellite DNA move to the periphery of the heterochromatin domain in S/G2 in a manner dependent on chromatin relaxation [64]. In both systems, Rad51 is excluded from the heterochromatin domain, only becoming associated after the movement, though interestingly this was not the case for mammalian centromeric repeats [64]. Though the rDNA is an actively transcribed area it contains many tandemly repeated genes and is prone to accumulating deletions. In both yeast and mammalian cells, DSBs within the rDNA move to the periphery of the nucleolus for repair and defects in this process lead to rDNA hyperrecombination and genome instability (see [58,61] for review). A recent study of DSB mobility in the rDNA of human cells showed that movement to the nucleolar periphery is an active process that involves actin as well as the nuclear envelope-associated LINC complex [65].
In addition to relocation for repair, 3D genome organization appears to play a more constitutive role in preventing repeat expansions. Disease-associated loci containing short tandem repeats, including FMR1, HTT, DMPK, FXN, C9orf72, and ATXN1, localize to TAD (topologically associated domain) boundaries [66]. The authors tested cells from Fragile X Syndrome (FXS) patients and healthy siblings and found that at the FMR1 locus, FXS patients with expanded repeats (>600 CGG repeats) exhibited disrupted TAD boundaries and CCCTC-binding factor (CTCF) binding that correlated to FMR1 silencing, compared to healthy siblings that did not exhibit these phenotypes [66]. However, another study found that CTCF binding and chromatin interactions were unchanged upon CAG/CTG repeat expansions at the DMPK and HTT loci [67]. These studies highlight that chromatin interactions may have different effects at different loci.
New Repeat Expansion Diseases and Potential Therapies
The Genetic Modifiers of Huntington’s Disease Consortium (GeM-HD) has been working to identify factors that alter age at onset of motor symptoms and progression of HD. The 2019 GeM-HD study clearly distinguished for the first time that uninterrupted CAG repeat length and not polyglutamine length is the driving factor in timing of age at onset of HD motor symptoms [44]. This highlights the extreme importance of repeat instability in driving HD and likely other neurodegenerative diseases. Previously, it had been demonstrated that somatic instability of CAG repeats is associated with age at onset of HD symptoms, specifically within tissues in the brain that are most affected (striatum and cortex), suggesting that factors that impact somatic instability may be modifiers of the disease [68]. Sure enough, genome-wide association (GWA) analysis revealed genetic modifiers of HD are genes involved in DNA repair: MLH1, FAN1, PMS1, MSH3, DHFR, PMS2, and LIG1, and polymorphisms that either increase or decrease expression of these factors can either delay or advance age at onset of HD [44].
Since uninterrupted CAG repeat length is a major determinant of HD motor symptoms, it has become a central target in therapeutics for HD. As use of endonucleases, such as ZFNs (zinc-finger nucleases), TALENs (transcription-activator like effector nucleases), and CRISPR-Cas9, have become popular gene editing tools, these have been used to target trinucleotide repeats to shorten their length as a potential therapy for repeat expansion diseases [69]. Mosbach et al. showed that a TALEN targeting expanded CAG repeats can contract them below pathological length and the induced DSB is repaired through single-strand annealing (SSA) [70] (Fig. 3a). Cinesi et al. demonstrated that use of CRISPR-Cas9 D10A nickase promotes CAG contractions by activating nick repair within the repeat tract [71] (Fig. 3b). The use of Cas9 nickases is a potentially promising therapeutic, as inducing nicks rather than DSBs likely helps to avoid expansions from occurring [71]. A follow up study excised the CAG tract from the HTT gene in HD patient-derived fibroblasts and observed a reduction in huntingtin protein level upon excision by Cas9 [72] (Fig. 3b). However, use of CRISPR-Cas9 to induce a DSB within an expanded CAG/CTG tract led to frequent chromosomal deletions in yeast, demonstrating some downsides of using this system to promote repeat contractions [73]. Studies excising other expanded repeats, such as CGG from FMR1 and GAA from FXN, resulted in reactivation of expression of genes that are silenced upon repeat expansion (reviewed in [69]).
Figure 3. Recent advances in contracting or removing expanded CAG/CTG repeat tracts.
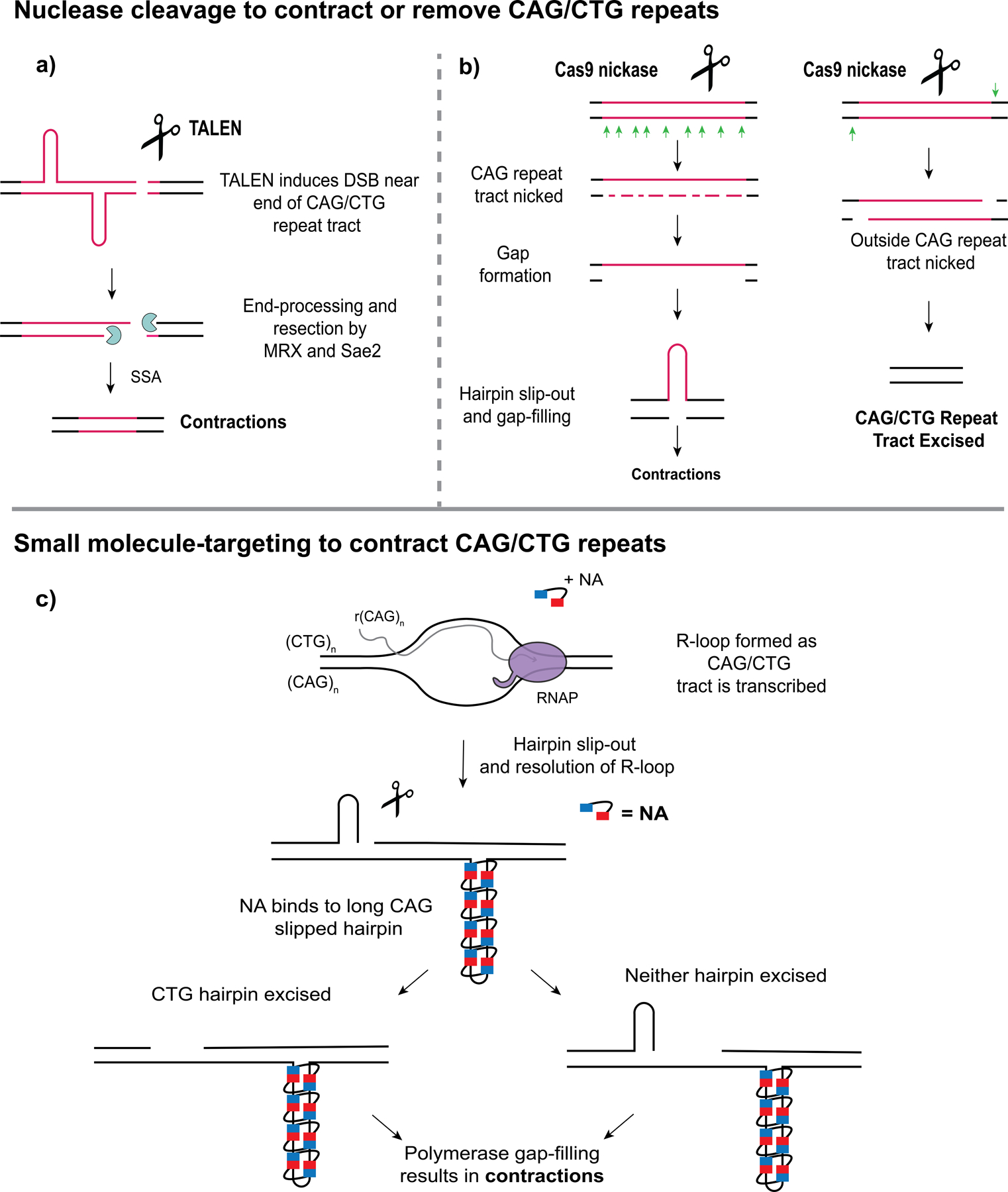
(a) A TALEN (transcription-activator like effector nuclease) targeting expanded CAG/CTG repeats induces a DSB near the end of the repeat tract. The DSB is processed and resected by the MRX (Mre11-Rad50-Xrs2) endonuclease complex stimulated by Sae2. Repair of the gap created by end-processing occurs through single-strand annealing (SSA), resulting in contractions [70]. (b) Use of the Cas9 D10A nickase to contract or remove an expanded CAG repeat tract. Left, Cinesi et al. (2016) introduced nicks throughout the CAG repeat tract, resulting in contractions [71]. Right, Dabrowska et al. (2018) excised the CAG repeat tract by nicking outside of the tract [72]. (c) Nakamori et al. (2020) used a small molecule, NA (naphthyridine-azaquinolone) to target slipped-out CAG repeats to promote contractions [74]. NA binds only to long CAG slip-outs, for example that could form upon resolution of R-loops. Though the NA-bound CAG tract is resistant to repair, CTG hairpins on the opposite strand promote nicking or hairpin excision, and polymerase fill-in of the resulting gap will result in a bias towards contractions.
A recent study tested a small molecule, called NA (naphthyridine-azaquinolone), that specifically binds slipped CAG repeats as a potential therapeutic for promoting contractions of expanded CAG repeats [74] (Fig. 3c). This molecule was found to be highly specific, only binding to long slip-outs, and blocked their repair in a manner dependent on transcription [74]. Additionally, NA was able to contract expanded CAG repeats in striatum tissue when injected into the brain of R6/2 HD mice [74]. This is a promising approach and the hope is that this molecule can be optimized to be used as a potential therapy in the future. In another promising approach, treatment of an HD mouse model with the HDAC3-selective inhibitor RGFP966 suppressed CAG expansions in the striatum and prevented cognitive decline [26].
In the last 2 years several new repeat expansions linked to various genetic diseases have been identified through repeat-primed PCR and long-read sequencing. A biallelic pentanucleotide repeat expansion was identified in CANVAS (cerebellar ataxia, neuropathy, and vestibular areflexia syndrome) patients within intron 2 of the RFC1 (replication factor C subunit 1) gene (reviewed in [2]). There is a range of potential genotypes present at this locus in healthy and CANVAS individuals, including (AAAAG)11 in healthy individuals, and (AAAAG)exp, (AAAGG)exp, and (AAGGG)exp in the disease state. The sequence from normal to disease-causing appears not only to expand, but the nucleotide content of the repeat unit changes, gaining more guanines and losing adenines. Additionally, intronic ATTTT repeat expansions containing an (ATTTC)exp interruption were identified in several loci, including STARD7, MARCH6, SAMD12, TNRC6A, and RAPGEF2, linked to various types of familial adult myoclonic epilepsy (FAME) (reviewed in [2]). A study examining the evolution of a similar repeat expansion in the DAB1 locus that is linked to spinocerebellar ataxia type 37 (SCA37) revealed a potential mechanism in which the ATTTT allele underwent a T-to-C mutation to create the ATTTC interruption that is present in affected individuals [75]. Also through repeat-primed PCR and long-read sequencing, a (GGC)n expansion was identified in patients with NIID (neuronal intranuclear inclusion disease) within the NOTCH2NLC gene [76]. Most recently, through genome-wide interrogation, gene-associated rare tandem repeat expansions were linked to autism [77]. Future studies are expected to determine mechanisms of repeat expansions at these loci to elucidate potential genetic causes of these diseases.
Funding acknowledgment:
C.H.F. is funded by NIGMS (GM122880) and NSF (MCB1817499).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement
The authors declare no conflict of interest.
References:
- 1.Bacolla A, Tainer JA, Vasquez KM, Cooper DN: Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res 2016, 44:5673–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khristich AN, Mirkin SM: On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J Biol Chem 2020, 295:4134–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irony-Tur Sinai M, Salamon A, Stanleigh N, Goldberg T, Weiss A, Wang YH, Kerem B: AT-dinucleotide rich sequences drive fragile site formation. Nucleic Acids Res 2019, 47:9685–9695.* The authors determined that AT-rich sequences are a driving factor of fragile site formation by inserting an AT-rich sequence from the FRA16C CFS into a non-fragile ectopic location in the genome. This ectopic location then exhibited recurrent chromosome gaps.
- 4.Zhang H, Freudenreich CH: An AT-Rich Sequence in Human Common Fragile Site FRA16D Causes Fork Stalling and Chromosome Breakage in S. cerevisiae. Mol Cell 2007, 27:367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaushal S, Wollmuth CE, Das K, Hile SE, Regan SB, Barnes RP, Haouzi A, Lee SM, House NCM, Guyumdzhyan M, et al. : Sequence and Nuclease Requirements for Breakage and Healing of a Structure-Forming (AT)n Sequence within Fragile Site FRA16D. Cell Rep 2019, 27:1151–1164.e5.* This study determined that a polymorphic AT repeat from the common fragile site FRA16D exhibited AT-length dependent fragility when inserted into a yeast chromosome. This fragility was dependent on Mus81-Mms4, Slx1-Slx4, and Rad1-Rad10 nucleases and breaks required the Sae2/MRX complex for processing and healing.
- 6.Minocherhomji S, Hickson ID: Structure-specific endonucleases: Guardians of fragile site stability. Trends Cell Biol 2014, 24:321–327. [DOI] [PubMed] [Google Scholar]
- 7.Shastri N, Tsai YC, Hile S, Jordan D, Powell B, Chen J, Maloney D, Dose M, Lo Y, Anastassiadis T, et al. : Genome-wide Identification of Structure-Forming Repeats as Principal Sites of Fork Collapse upon ATR Inhibition. Mol Cell 2018, 72:222–238.e11.* This study detected sites of fork collapse genome-wide upon ATR inhibition, using RPA-ChIP and BrITL (breaks identified by TdT labeling). In human cells, AT repeats were most commonly identified, whereas (CAGAGG)n and (CACAG)n repeats, which form quadruplex structures, were most commonly identified in mouse cells. Tubbs et al., 2018 did a similar study using HU and mouse B cells and the END-seq method, and identified poly(dA:dT) tracts as the most common repeat at early-replicating fragile sites.
- 8.Tubbs A, Sridharan S, van Wietmarschen N, Maman Y, Callen E, Stanlie A, Wu W, Wu X, Day A, Wong N, et al. : Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 2018, 174:1127–1142.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie KT, Wang G, Thompson AC, Wucherpfennig JI, Reimchen TE, Maccoll ADC, Schluter D, Bell MA, Vasquez KM, Kingsley DM: DNA fragility in the parallel evolution of pelvic reduction in stickleback fish. Science (80- ) 2019, 84:81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Wietmarschen N, Sridharan S, Nathan WJ, Tubbs A, Chan EM, Callen E, Wu W, Belinky F, Tripathi V, Wong N, et al. : Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 2020, 586:1–7.** The authors followed up on previous studies identifying WRN (Werner Syndrome) helicase as a synthetic lethal target in MSI (microsatellite instability) cancers. Interestingly, many AT repeat loci were found to be expanded in the MMR-deficient MSI cancer cell lines, and these were the sites susceptible to breakage in the absence of WRN. They proposed a mechanism in which expanded AT repeats form a DNA structure which stalls replication forks but can be resolved by ATR-phosphorylated WRN. In the absence of WRN, cleavage of the cruciform-like structures at sites of stalled replication by MUS81-EME1 results in fragility and chromosome shattering.
- 11.Higa M, Fujita M, Yoshida K: DNA replication origins and fork progression at mammalian telomeres. Genes (Basel) 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lerner LK, Sale JE: Replication of G quadruplex DNA. Genes (Basel) 2019, 10:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stroik S, Kurtz K, Lin K, Karachenets S, Myers CL, Bielinsky A-K, Hendrickson EA: EXO1 resection at G-quadruplex structures facilitates resolution and replication. Nucleic Acids Res 2020, 48:4960–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matmati S, Lambert S, Géli V, Coulon S: Telomerase Repairs Collapsed Replication Forks at Telomeres. Cell Rep 2020, 30:3312–3322.e3. [DOI] [PubMed] [Google Scholar]
- 15.van Schie JJM, Faramarz A, Balk JA, Stewart GS, Cantelli E, Oostra AB, Rooimans MA, Parish JL, de Almeida Estéves C, Dumic K, et al. : Warsaw Breakage Syndrome associated DDX11 helicase resolves G-quadruplex structures to support sister chromatid cohesion. Nat Commun 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neil AJ, Kim JC, Mirkin SM: Precarious maintenance of simple DNA repeats in eukaryotes. BioEssays 2017, 39:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polleys EJ, House NCM, Freudenreich CH: Role of recombination and replication fork restart in repeat instability. DNA Repair (Amst) 2017, 56:156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JC, Harris ST, Dinter T, Shah KA, Mirkin SM: The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat Struct Mol Biol 2017, 24:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kononenko AV, Ebersole T, Vasquez KM, Mirkin SM: Mechanisms of genetic instability caused by (CGG)n repeats in an experimental mammalian system. Nat Struct Mol Biol 2018, 25:669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neil AJ, Liang MU, Khristich AN, Shah KA, Mirkin SM: RNA-DNA hybrids promote the expansion of Friedreich’s ataxia (GAA) n repeats via break-induced replication. Nucleic Acids Res 2018, 46:3487–3497.* The authors of this study examined the role of DNA:RNA hybrids in GAA repeat stability and found that that accumulation of DNA:RNA hybrids at GAA repeats results in GAA expansions. They termed the proposed structure, in which a triplex structure forms opposite an DNA:RNA hybrid, an H-loop.
- 21.Su XA, Dion V, Gasser SM, Freudenreich CH: Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes Dev 2015, 29:1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whalen JM, Dhingra N, Wei L, Zhao X, Freudenreich CH: Relocation of Collapsed Forks to the Nuclear Pore Complex Depends on Sumoylation of DNA Repair Proteins and Permits Rad51 Association. Cell Rep 2020, 31:107635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khristich AN, Armenia JF, Matera RM, Kolchinski AA, Mirkin SM: Large-scale contractions of Friedreich’s ataxia GAA repeats in yeast occur during DNA replication due to their triplex-forming ability. Proc Natl Acad Sci U S A 2020, 117:1628–1637.* This study identified lagging-strand replication as a mechanism for large-scale GAA contractions. Contractions are proposed to occur as Pol δ bypasses the triplex structure formed by the GAA repeats.
- 24.House NCM, Polleys EJ, Quasem I, Mejia MD la R, Joyce CE, Takacsi-Nagy O, Krebs JE, Fuchs SM, Freudenreich CH: Distinct roles for S. Cerevisiae H2A copies in recombination and repeat stability, with a role for H2A.1 threonine 126. Elife 2019, 8:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.House NCM, Yang JH, Walsh SC, Moy JM, Freudenreich CH: NuA4 Initiates Dynamic Histone H4 Acetylation to Promote High-Fidelity Sister Chromatid Recombination at Postreplication Gaps. Mol Cell 2014, 55:818–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suelves N, Kirkham-McCarthy L, Lahue RS, Ginés S: A selective inhibitor of histone deacetylase 3 prevents cognitive deficits and suppresses striatal AG repeat expansions in Huntington’s disease mice. Sci Rep 2017, 7:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams GM, Paschalis V, Ortega J, Muskett FW, Hodgkinson JT, Li GM, Schwabe JWR, Lahue RS: HDAC3 deacetylates the DNA mismatch repair factor MutSβ to stimulate triplet repeat expansions. Proc Natl Acad Sci U S A 2020, 117:23597–23605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kovalenko M, Erdin S, Andrew MA, Claire J St., Shaughnessey M, Hubert L, Neto JL, Stortchevoi A, Fass DM, Mouro Pinto R, et al. : Histone deacetylase knockouts modify transcription, CAG instability and nuclear pathology in Huntington disease mice. Elife 2020, [DOI] [PMC free article] [PubMed]
- 29.Lin Y, Dent SYR, Wilson JH, Wells RD, Napierala M: R loops stimulate genetic instability of CTG·CAG repeats. Proc Natl Acad Sci U S A 2010, 107:692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy K, Tam M, Bowater RP, Barber M, Tomlinson M, Nichol Edamura K, Wang YH, Pearson CE: Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res 2011, 39:1749–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freudenreich CH: R-loops: targets for nuclease cleavage and repeat instability. Curr Genet 2018, 64:789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diab MA, Mor-Shaked H, Cohen E, Cohen-Hadad Y, Ram O, Epsztejn-Litman S, Eiges R: The g-rich repeats in FMR1 and C9orf72 loci are hotspots for local unpairing of DNA. Genetics 2018, 210:1239–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng KW, Xiao S, Liu JQ, Zhang JY, Hao YH, Tan Z: Co-transcriptional formation of DNA: RNA hybrid G-quadruplex and potential function as constitutional cis element for transcription control. Nucleic Acids Res 2013, 41:5533–5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su XA, Freudenreich CH: Cytosine deamination and base excision repair cause R-loop–induced CAG repeat fragility and instability in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 2017, 114:E8392–E8401.* The authors determined that R-loop-mediated instability of CAG repeats occurs through cytosine deamination of ssDNA that becomes exposed upon R-loop formation, followed by repair of deaminated cytosines through base excision repair (BER). R-loop-mediated fragility of expanded CAG repeats was found to occur through a mechanism requiring the Mlh1-Mlh3 (MutLγ) nuclease.
- 35.Šviković S, Crisp A, Tan‐Wong SM, Guilliam TA, Doherty AJ, Proudfoot NJ, Guilbaud G, Sale JE: R‐loop formation during S phase is restricted by PrimPol‐mediated repriming. EMBO J 2019, 38:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeller P, Padeken J, Van Schendel R, Kalck V, Tijsterman M, Gasser SM: Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat Genet 2016, 48:1385–1395. [DOI] [PubMed] [Google Scholar]
- 37.Padeken J, Zeller P, Towbin B, Katic I, Kalck V, Methot SP, Gasser SM: Synergistic lethality between BRCA1 and H3K9me2 loss reflects satellite derepression. Genes Dev 2019, 33:436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin Y, Hubert L, Wilson JH: Transcription destabilizes triplet repeats. Mol Carcinog 2009, 48:350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koch MR, House NC, Cosetta CM, Jong RM, Salomon CG, Joyce CE, Philips EA, Su XA, Freudenreich CH: The Chromatin Remodeler Isw1 Prevents CAG Repeat Expansions During Transcription in Saccharomyces cerevisiae. Genetics 2018, 208:963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu S, Wang G, Bacolla A, Zhao J, Spitser S, Vasquez KM: Short inverted repeats are hotspots for genetic instability: Relevance to cancer genomes. Cell Rep 2015, 10:1674–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao J, Wang G, del Mundo IM, McKinney JA, Lu X, Bacolla A, Boulware SB, Zhang C, Zhang H, Ren P, et al. : Distinct Mechanisms of Nuclease-Directed DNA-Structure-Induced Genetic Instability in Cancer Genomes. Cell Rep 2018, 22:1200–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKinney JA, Wang G, Mukherjee A, Christensen L, Subramanian SHS, Zhao J, Vasquez KM: Distinct DNA repair pathways cause genomic instability at alternative DNA structures. Nat Commun 2020, 11:1–12.* The authors proposed a mechanism in which Z-DNA is recognized by the MutSβ (MSH2-MSH3) complex as DNA damage, resulting in recruitment of nucleotide excision repair (NER) nuclease XPF-ERCC1 and subsequently deletions and translocations. This is consistent with previous findings from the Vasquez lab identifying NER nucleases as a cause of fragility of H-DNA and short inverted repeats (IRs).
- 43.Ciosi M, Maxwell A, Cumming SA, Hensman Moss DJ, Alshammari AM, Flower MD, Durr A, Leavitt BR, Roos RAC, Holmans P, et al. : A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine 2019, 48:568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee J-M, Correia K, Loupe J, Kim K-H, Barker D, Hong EP, Chao MJ, Long JD, Lucente D, Vonsattel JPG, et al. : CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell 2019, 178:887–900.e14.** The GeM-HD (Genetic Modifiers of Huntington’s Disease) consortium clearly determined for the first time that uninterrupted CAG repeat length and not polyglutamine length is linked to age of onset of HD motor symptoms. The authors also identified polymorphisms within many DNA repair genes as genetic modifiers of age of at onset, including MSH3, FAN1, and LIG1.
- 45.Kim KH, Hong EP, Shin JW, Chao MJ, Loupe J, Gillis T, Mysore JS, Holmans P, Jones L, Orth M, et al. : Genetic and Functional Analyses Point to FAN1 as the Source of Multiple Huntington Disease Modifier Effects. Am J Hum Genet 2020, 107:96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goold R, Flower M, Moss DH, Medway C, Wood-Kaczmar A, Andre R, Farshim P, Bates GP, Holmans P, Jones L, et al. : FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet 2019, 28:650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao XN, Usdin K: FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair (Amst) 2018, 69:1–5.* Following up on the identification of FAN1 as a genetic modifier of HD, this study was the first to show that FAN1 protects against CGG expansions in a Fragile X mouse model. Similarly, Goold et al. (2019) found that loss of FAN1 resulted in an increase in CAG repeat length at the HTT locus in both U2OS cells and HD-derived iPSCs, confirming a general role for this nuclease in maintaining structure-forming repeats.
- 48.Loupe JM, Pinto RM, Kim K-H, Gillis T, Mysore JS, Andrew MA, Kovalenko M, Murtha R, Seong I, Gusella JF, et al. : Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington’s disease knock-in mice is blocked by Mlh1 knock-out. Hum Mol Genet 2020, 00:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinto RM, Dragileva E, Kirby A, Lloret A, Lopez E, Claire St., Panigrahi GB, Hou C, Holloway K, Gillis T, et al. : Mismatch Repair Genes Mlh1 and Mlh3 Modify CAG Instability in Huntington’s Disease Mice: Genome-Wide and Candidate Approaches. PLoS Genet 2013, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao X, Zhang Y, Wilkins K, Edelmann W, Usdin K: MutLγ promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet 2018, 14:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller CJ, Kim GY, Zhao X, Usdin K: All three mammalian MutL complexes are required for repeat expansion in a mouse cell model of the Fragile X-related disorders. PLoS Genet 2020, 16:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayward BE, Steinbach PJ, Usdin K: A point mutation in the nuclease domain of MLH3 eliminates repeat expansions in a mouse stem cell model of the Fragile X-related disorders. Nucleic Acids Res 2020, doi: 10.1093/nar/gkaa573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lai Y, Budworth H, Beaver JM, Chan NLS, Zhang Z, McMurray CT, Liu Y: Crosstalk between MSH2-MSH3 and polβ promotes trinucleotide repeat expansion during base excision repair. Nat Commun 2016, 7:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whalen JM, Freudenreich CH: Location, location, location: The role of nuclear positioning in the repair of collapsed forks and protection of genome stability. Genes (Basel) 2020, 11:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, Gasser SM: SWR1 and INO80 chromatin remodelers contribute to dna double-strand break perinuclear anchorage site choice. Mol Cell 2014, 55:626–639. [DOI] [PubMed] [Google Scholar]
- 56.Horigome C, Bustard DE, Marcomini I, Delgoshaie N, Tsai-Pflugfelder M, Cobb JA, Gasser SM: PolySUMOylation by Siz2 and Mms21 triggers relocation of DNA breaks to nuclear pores through the Slx5/Slx8 STUbL. Genes Dev 2016, 30:931–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Freudenreich CH, Su XA: Relocalization of DNA lesions to the nuclear pore complex. FEMS Yeast Res 2016, 16:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hauer MH, Gasser SM: Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev 2017, 31:2204–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aguilera P, Whalen J, Minguet C, Churikov D, Freudenreich C, Simon MN, Géli V: The nuclear pore complex prevents sister chromatid recombination during replicative senescence. Nat Commun 2020, 11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maestroni L, Reyes C, Vaurs M, Gachet Y, Tournier S, Géli V, Coulon S: Nuclear envelope attachment of telomeres limits TERRA and telomeric rearrangements in quiescent fission yeast cells. Nucleic Acids Res 2020, 48:3029–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mitrentsi I, Yilmaz D, Soutoglou E: How to maintain the genome in nuclear space. Curr Opin Cell Biol 2020, 64:58–66. [DOI] [PubMed] [Google Scholar]
- 62.Amaral N, Ryu T, Li X, Chiolo I: Nuclear Dynamics of Heterochromatin Repair. Trends Genet 2017, 33:86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caridi CP, D’agostino C, Ryu T, Zapotoczny G, Delabaere L, Li X, Khodaverdian VY, Amaral N, Lin E, Rau AR, et al. : Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 2018, 559:54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsouroula K, Furst A, Rogier M, Heyer V, Maglott-Roth A, Ferrand A, Reina-San-Martin B, Soutoglou E: Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol Cell 2016, 63:293–305. [DOI] [PubMed] [Google Scholar]
- 65.Marnef A, Finoux AL, Arnould C, Guillou E, Daburon V, Rocher V, Mangeat T, Mangeot PE, Ricci EP, Legube G: A cohesin/HUSH- And LINC-dependent pathway controls ribosomal DNA double-strand break repair. Genes Dev 2019, 33:1175–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun JH, Zhou L, Emerson DJ, Phyo SA, Titus KR, Gong W, Gilgenast TG, Beagan JA, Davidson BL, Tassone F, et al. : Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 2018, 175:224–238.e15.* Through 5C experiments to examine 3D genome and chromatin interactions, the authors found that disease-associated short tandem repeats (daSTRs) at FMR1, HTT, DMPK, FXN, C9orf72, and ATXN1 loci localized to TAD boundaries. Additionally, at the FMR1 locus, expansion of CGG repeats ablated chromatin interactions and TAD organization. This indicates that overall genome organization may contribute to disease phenotypes of repeat expansion diseases.
- 67.Ruiz Buendía G, Leleu M, Marzetta F, Vanzan L, Tan J, Ythier V, Randall E, Marques A, Baubec T, Murr R, et al. : Three-dimensional chromatin interactions remain stable upon CAG/CTG repeat expansion. Sci Adv 2020, doi: 10.1126/sciadv.aaz4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Swami M, Hendricks AE, Gillis T, Massood T, Mysore J, Myers RH, Wheeler VC: Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet 2009, 18:3039–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mosbach V, Poggi L, Richard GF: Trinucleotide repeat instability during double-strand break repair: from mechanisms to gene therapy. Curr Genet 2019, 65:17–28. [DOI] [PubMed] [Google Scholar]
- 70.Mosbach V, Poggi L, Viterbo D, Charpentier M, Richard GF: TALEN-Induced Double-Strand Break Repair of CTG Trinucleotide Repeats. Cell Rep 2018, 22:2146–2159. [DOI] [PubMed] [Google Scholar]
- 71.Cinesi C, Aeschbach L, Yang B, Dion V: Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase. Nat Commun 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dabrowska M, Juzwa W, Krzyzosiak WJ, Olejniczak M: Precise Excision of the CAG Tract from the Huntingtin Gene by Cas9 Nickases. Front Neurosci 2018, 12:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mosbach V, Viterbo D, Descorps-Declère S, Poggi L, Vaysse-Zinkhöfer W, Richard GF: Resection and repair of a Cas9 double-strand break at CTG trinucleotide repeats induces local and extensive chromosomal deletions. PLoS Genet 2020, 16:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakamori M, Panigrahi GB, Lanni S, Gall-Duncan T, Hayakawa H, Tanaka H, Luo J, Otabe T, Li J, Sakata A, et al. : A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat Genet 2020, 52:146–159.** The authors used the small molecule NA (naphthyridine-azaquinolone) to stabilize long-slipped CAG repeats and promote CAG contractions. They show that the compound works in vivo in an HD mouse model to cause contractions. This is a promising potential future therapeutic option for HD and other genetic diseases caused by CAG repeat expansions.
- 75.Loureiro JR, Oliveira CL, Mota C, Castro AF, Costa C, Loureiro JL, Coutinho P, Martins S, Sequeiros J, Silveira I: Mutational mechanism for DAB1 (ATTTC) n insertion in SCA37: ATTTT repeat lengthening and nucleotide substitution. Hum Mutat 2019, 40:404–412. [DOI] [PubMed] [Google Scholar]
- 76.Sone J, Mitsuhashi S, Fujita A, Mizuguchi T, Hamanaka K, Mori K, Koike H, Hashiguchi A, Takashima H, Sugiyama H, et al. : Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 2019, 51:1215–1221. [DOI] [PubMed] [Google Scholar]
- 77.Trost B, Engchuan W, Nguyen CM, Thiruvahindrapuram B, Dolzhenko E, Backstrom I, Mirceta M, Mojarad BA, Yin Y, Dov A, et al. : Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature 2020, doi: 10.1038/s41586-020-2579-z.* Through genome-wide interrogation, the authors identified tandem repeat expansions that are linked to autism within both known and novel loci. Tandem repeat motifs were identified at different genetic loci, indicating that multiple expansion-prone motifs may contribute. Although the effect is small (2.6% contribution), these repeat expansions are a potential cause of autism in some children.