

Abstract
YcaO enzymes catalyze several post-translational modifications on peptide substrates, including thioamidation, which substitutes an amide oxygen with sulfur. The majority of predicted thioamide-forming YcaO enzymes are encoded adjacent to TfuA, which when present, is required for thioamidation. While activation of the peptide amide backbone is well established for YcaO enzymes, the function of TfuA has remained enigmatic. Here we characterize the TfuA protein involved in methyl-coenzyme M reductase thioamidation and demonstrate that TfuA catalyzes the hydrolysis of thiocarboxylated ThiS (ThiS-COSH), a proteinaceous sulfur donor, and enhances the affinity of YcaO towards the thioamidation substrate. We also report the first crystal structure of a TfuA, which displays a new protein fold. Our structural and mutational analyses of TfuA have uncovered conserved binding interfaces with YcaO and ThiS in addition to revealing a hydrolase-like active site featuring a Ser/Lys catalytic pair.
Introduction
The YcaO superfamily (Protein Family PF02624)1 currently consists of 16,000 members (UniProt)2 and is prevalent in bacteria and archaea. The most widely studied YcaO function is the ATP-dependent cyclodehydration of serine, threonine, and cysteine to form azoline heterocycles on peptides3,4. The heterocyclized products fall into several classes of ribosomally synthesized and post-translationally modified peptides (RiPPs), including linear azol(in)e-containing peptides, cyanobactins, bottromycins, and thiopeptides4. Formation of peptide backbone azolines often requires a partner protein to engage the peptide substrate5,6. Known partner proteins leverage a RiPP precursor recognition element (RRE) that is either fused to or encoded next to the YcaO7,8.
Recent work expanded the known transformations catalyzed by YcaO, including macrolactamidination (e.g., bottromycin) and amidination (klebsazolicin)9. YcaO-dependent thioamidation is also established for the thiopeptides Sch 18640, thiopeptin, and saalfelduracin10, and methyl-coenzyme M reductase (MCR)11,12. YcaO has also been potentially implicated in thioamidation of ribosomal protein uL1613. Despite the variable outcomes, each YcaO-catalyzed reaction proceeds through hydroxyl, thiol, or amine nucleophilic attack on the peptide backbone to yield an amide oxyanion that is subsequently O-phosphorylated to give a presumptive hemiorthoamide intermediate4. During thioamidation, a sulfide/bisulfide equivalent acts as the nucleophile. The reaction ends with the formation of the thioamide and phosphate as a byproduct12. Replacement of oxygen with sulfur in the peptide backbone has profound implications on the bond rotation barrier, hydrogen bonding, propensity to adopt the cis conformation, and protease resistance14.
The biosynthetic gene clusters for nearly all thioamide-containing RiPPs encode a YcaO, and are referred to as thioamitides9. Characterized thioamitides include the thioviridamides15–17, certain thiopeptides10, and thiovarsolin18 (Extended Data Fig. 1). In each case, the YcaO is encoded directly adjacent to a “TfuA-like” protein (PF07812, ~2,000 UniProt entries), a functionally undefined protein with no sequence similarity to any characterized protein. As noted4, the original tfuA gene encodes a 650-residue protein (GenBank: AAB17513) implicated in trifolitoxin biosynthesis19. This “original” TfuA contains domains homologous to transcriptional activators, TolB, and tetratricopeptide repeats and unfortunately bears no sequence similarity to proteins now annotated as “TfuA-like” proteins. A gene neighboring the “original” tfuA (GenBank: AAB17515; 247-residue protein) is not involved to trifolitoxin biosynthesis but is homologous to the TfuA proteins pertinent to this study. Further, this TfuA is encoded adjacent to a YcaO. We believe an error was made when assigning the TfuA-like superfamily.
The co-occurrence of TfuA and YcaO suggested these proteins collaborate, prompting a study that demonstrated TfuA and YcaO are required to convert thiostrepton to a thioamidated analog10. TfuA-associated YcaOs also modify MCR11, a large protein ubiquitous to methanogenic and methanotrophic archaea that catalyzes the anaerobic production and consumption of methane20,21. In the alpha subunit (McrA), a Gly near the active site is converted to thioglycine20,22. Deletion and complementation studies in the genetically tractable methanogen Methanosarcina acetivorans demonstrated that ycaO and tfuA were necessary and sufficient for MCR thioamidation in vivo11. Mutants lacking thioglycine exhibited severe growth defects and were unable to grow at temperatures well-tolerated by the parental strain. These data and a recent study22 suggested that thioamidation and other modifications provide local structural stability to the MCR active site and fine-tune the enzymatic activity. Thioglycine is present in all MCR proteins characterized to date23, consistent with ycaO being universally present in methanogens and methanotrophs11. The majority of sequenced methanogens also encode TfuA directly adjacent to ycaO12.
The lack of TfuA in ~10% of sequenced methanogens prompted a detailed analysis of the thioamidation reaction12. The YcaO-catalyzed thioamidation on an MCR-derived peptide was reconstituted in vitro using proteins from three distinct methanogens. YcaOs from methanogens lacking TfuA (e.g., Methanocaldococcus jannaschii and Methanopyrus kandleri) installed thioglycine independent of any additional proteins. In contrast, thioamidation catalyzed by the M. acetivorans YcaO was greatly facilitated by the cognate TfuA. Despite the importance of TfuA in vivo and in vitro, the biochemical role remained elusive. Unlike the partner proteins of azoline-forming YcaOs, TfuA does not directly engage the peptide substrate. This leaves several possible functions for TfuA, including activating YcaO and/or mediating sulfur delivery.
Previous thioamidation studies employed millimolar sulfide concentrations, reflecting the ecological conditions of some methanogens24; however, other methanogens are isolated from low-sulfide environments25 and a carrier protein may be necessary for sulfur metabolism. In this study, we identified a genome-wide correlation between TfuA and ThiS homolog (PF02597), which uses a thiocarboxylated C-terminus (ThiS-COSH) to deliver sulfur for various biosynthetic pathways26. McrA peptide thioamidation was then reconstituted in vitro using TfuA, YcaO, and ThiS-COSH from Methanothermobacter sp. CaT2 at a much higher efficiency relative to free sulfide. The rate enhancement was quantified and two functional roles for TfuA were demonstrated: (i) ThiS-COSH hydrolysis and (ii) enhancement of YcaO affinity for the McrA peptide. We also report the first crystal structure of TfuA, revealing a novel didomain fold. Structure- and bioinformatics-guided mutagenesis revealed a conserved Ser/Lys catalytic pair for TfuA and other residues that interface with YcaO. Our results support a model where TfuA and YcaO function together as a thioamide synthetase: TfuA delivers sulfide to YcaO via ThiS-COSH hydrolysis while YcaO catalyzes thioamide formation via an ATP-dependent activation of the peptide backbone.
Results
Cooccurrence of TfuA and ThiS homologs
Given that a direct role for TfuA in peptide binding has been ruled out12, another possibility is mediating sulfur delivery to YcaO. ThiS homologs are occasionally found near tfuA-ycaO loci11,18, prompting the hypothesis that ThiS could provide the required sulfur. The defining feature of the ThiS superfamily is a conserved Gly-Gly C-terminal motif, which undergoes adenylation by an activating enzyme (ThiF, PF00899) and thionation to form a C-terminal thiocarboxylate26,27. The thiocarboxylate is a known sulfur donor for the biosynthesis of thiamine (ThiS-COSH)28, molybdopterin (MoaD-COSH)29, 2-thiouridine (Urm1/TtuB-COSH)30,31, free methionine (HcyS-COSH)32, and the natural products thioplatensimycin and thioplatencin (PtmS-COSH)33. Despite the shared carboxylate activation step, the enzymes that release sulfide equivalents from ThiS homologs vary substantially in sequence and mechanism. The occasional proximity to tfuA-ycaO led us to investigate a potential functional link between TfuA and methanogenic homologs of ThiS.
The genomic neighborhood was analyzed for all TfuA proteins in UniProt (n = 2,042)2,34. A large majority (89%) are encoded directly adjacent to ycaO, with 17 instances (~0.8%) of a TfuA-YcaO fusion product, supporting a related function. A locally encoded thiS was identified for 6% of TfuA proteins, including some methanogens (Extended Data Fig. 1). In comparison, thiS was absent from the genomic neighborhood of methanogenic ycaO lacking a companion tfuA. This observation led to a genome-wide correlation analysis between tfuA and thiS on all the completed methanogen genomes (n = 111). Hidden Markov Model (HMM)-based retrieval of TfuA and ThiS homologs revealed that all genomes encoding TfuA also encoded at least one ThiS homolog (n = 94, Extended Data Fig. 2, Supplementary Dataset). Among the 17 genomes lacking tfuA, 12 also lack thiS. The remaining five genomes belong to the divergent Methanomassiliicoccales methanogen class35. Collectively, TfuA and ThiS homologs are strongly correlated in methanogens (p-value = 1.6×10−12), suggesting a statistically plausible functional linkage.
ThiS as the sulfur donor for McrA thioamidation
To confirm or refute ThiS involvement in McrA thioamidation, proteins from Methanothermobacter sp. CaT2 (Mt) were heterologously expressed as maltose-binding protein (MBP)- and His6-tagged proteins and purified to homogeneity: MtThiS (WP_048176273), MtTfuA (WP_048175617) and MtYcaO (WP_048175616, Supplementary Fig. 1). We employed Mt proteins here owing to the superior solution stability of MtYcaO. Akin to previous reports12,36, a synthetic peptide derived from MtMcrA was used as the substrate (GG-440RLGFYGYDLQD450). To determine if thiocarboxylated MtThiS (hereafter, MtThiS-COSH) was a competent sulfur donor, we generated MtThiS-COSH by treating tag-free MtThiS with purified MtThiF (WP_048176081), ATP, and sodium sulfide. Formation of MtThiS-COSH was confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS, Extended Data Fig. 3). The site of thiocarboxylation was localized to the C-terminus of MtThiS after treatment with endoproteinase GluC, a result corroborated by high-resolution and tandem MS (HR-MS/MS: calc., 892.5074 Da; obsv., 892.5060 Da; error, 1.6 ppm; Extended Data Fig. 4). Additionally, the presence of a collision-induced [M-34 Da] daughter ion was consistent with release of H2S from the parent ion.
Reactions were then initiated using MtThiS-COSH, MtYcaO, MtTfuA, McrA peptide, and ATP. An endpoint MALDI-TOF-MS assay was used to monitor McrA modification. Under the conditions employed, McrA peptide was fully processed when all reaction components were present (Fig. 1a). Replacement of MtThiS-COSH with MtThiS-COOH or omission of MtYcaO gave no detectable thioamidation. Omission of MtTfuA from an otherwise identical reaction severely diminished reaction progress, as a very low intensity ion consistent with thioamidated McrA peptide was visible. The thioamidated McrA peptide from the full reaction was confirmed by HR-MS/MS (calc., 1476.6577 Da; obsv., 1476.6555 Da; error, 1.5 ppm) and localized to the central Gly (Extended Data Fig. 5).
Figure 1. MS and kinetic analysis of TfuA-mediated thioamidation.
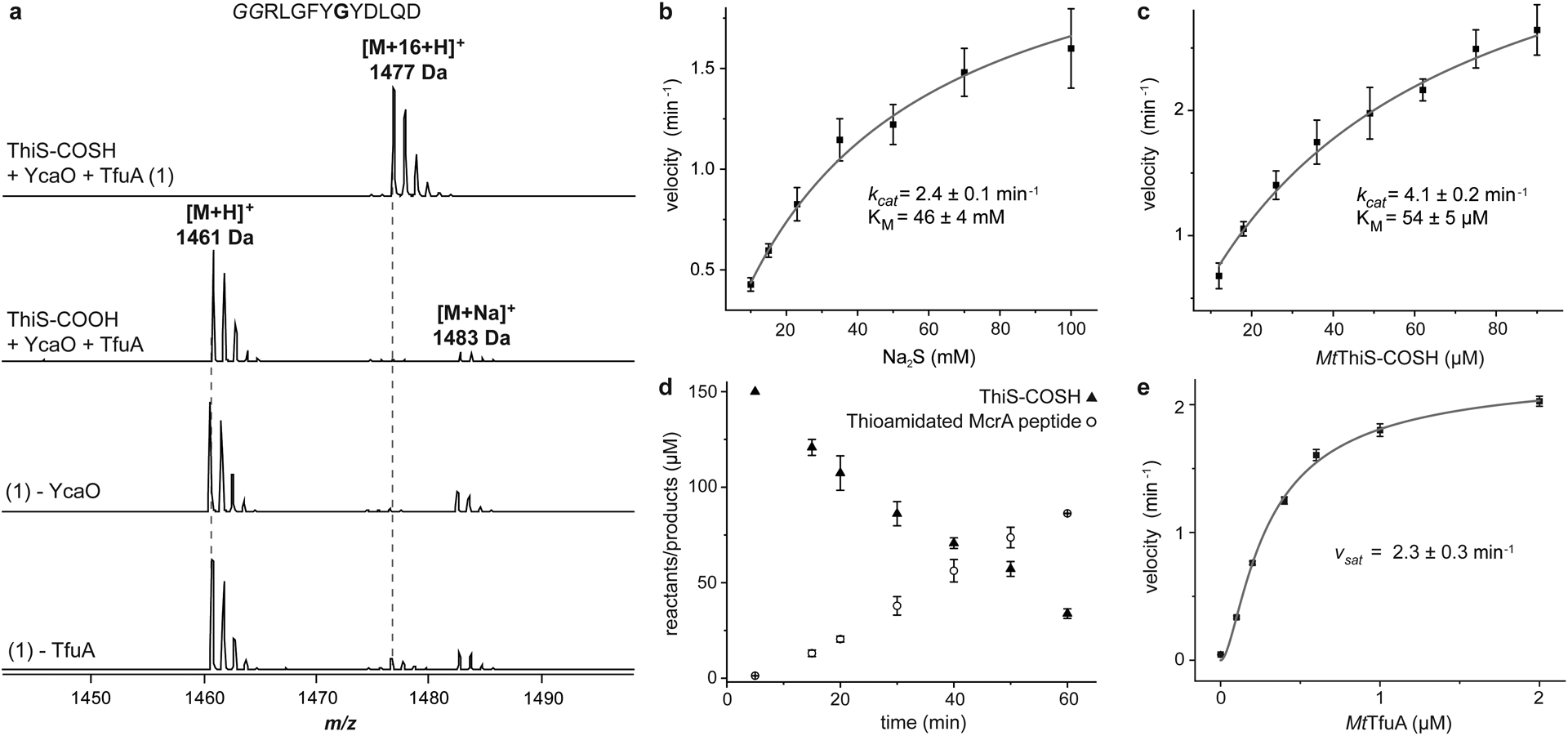
(a) MALDI-TOF mass spectra of the McrA-derived peptide substrate (m/z 1,461 Da, Gly to be modified is bold) under different reaction conditions. Top, McrA peptide after reaction with MtYcaO, MtTfuA, MtThiS-COSH, and ATP (full reaction) showing a single thioamidated product (m/z 1,477 Da). Controls replace MtThiS-COSH with MtThiS-COOH and omit either MtYcaO or MtTfuA (bottom). HPLC-MS kinetic analysis of McrA peptide thioamidation with Na2S (b) or MtThiS-COSH (c) as the sulfur donor. Initial velocities are presented as the estimated slope ± SE derived from linear regression analysis (n = 4 time points examined with duplicates). Initial velocities are fitted into a Michaelis-Menten model via non-linear regression analysis. The resultant kcat ± SE and KM ± SE are reported. (d) McrA thioamidation (open circles) and ThiS-COSH hydrolysis (filled triangles) were monitored by HPLC-MS and fluorescence quantification, respectively. Data are presented as mean values ± SD (n = 3 independent experiments). (e) Rate of McrA thioamidation as a function of TfuA concentration. Initial velocity was determined as in panels b and c (n = 4 time points examined with duplicates), plotted against TfuA concentration, and fitted into a substrate activation model to estimate the velocity at a saturating concentration of TfuA (vsat ± SE).
These results indicated that MtThiS-COSH was a robust sulfur donor for McrA thioamidation and that MtTfuA was critical for efficient McrA modification. We next employed a liquid chromatography mass spectrometry (LCMS)-based assay to enable a direct comparison between MtThiS-COSH and Na2S as sulfur donors and to quantify the TfuA-dependent rate enhancement. First, MtThiS-COSH or Na2S were employed as sulfur donors in reactions containing a saturating concentration of MtTfuA. The catalytic efficiencies were then compared under Michaelis-Menten conditions, demonstrating that MtThiS-COSH was ~1,500-fold more efficient than Na2S [kcat/KM (ThiS-COSH) = 77 min−1mM−1, kcat/KM (Na2S) = 0.053 min−1mM−1, Fig. 1]. To further evaluate MtThiS-COSH as a sulfur donor, the production of thioamidated McrA peptide and consumption of MtThiS-COSH were monitored by LCMS and a lissamine rhodamine sulfonyl azide (LRSA)-based assay that reacts selectively with the thiocarboxylate (Supplementary Fig. 2)37. The rate of MtThiS-COSH consumption was comparable to thioamide formation. Next, the reaction rate was monitored as a function of MtTfuA concentration while the concentration of all other components was fixed. In the absence of MtTfuA, thioamidation proceeded with an initial rate of 0.05 ± 0.01 min−1, whereas MtTfuA potentiated the rate ~50-fold (2.3 ± 0.3 min−1, Fig. 1) under saturating conditions. Given that MtYcaO activates the amide undergoing thioamidation, and MtThiS-COSH is the sulfur donor, we proposed that MtTfuA likely facilitates sulfur delivery from MtThiS-COSH to MtYcaO.
TfuA binds to and hydrolyzes ThiS-COSH
To probe sulfur delivery, reactions with ThiS-COSH in the presence of MtTfuA and/or MtYcaO in [18O]-H2O were performed. Significant [18O] incorporation into MtThiS-COOH was observed only in the presence of MtTfuA (m/z 878 Da indicates mono-[18O] incorporation, Fig. 2). Analysis by HR-MS/MS unambiguously localized the [18O] label to the C-terminus of MtThiS-COOH (Supplementary Fig. 3). Subsequently, sulfide production38 and consumption of MtThiS-COSH were monitored during the reaction with MtTfuA. A concurrent increase of sulfide and decrease of MtThiS-COSH was observed in an MtTfuA-dependent manner (Extended Data Fig. 6). In comparison, MtYcaO alone or in the presence of McrA peptide and ATP led to a basal level of MtThiS-COSH hydrolysis. These data indicated that TfuA catalyzed ThiS-COSH hydrolysis, which has not previously been observed as a sulfur delivery strategy. Upon replacement of MtThiS-COSH with a divergent member of the superfamily (WP_011023978, M. acetivorans ThiS), thioamidation was severely impaired (Supplementary Fig. 4), suggesting a specific interaction between MtThiS and MtTfuA.
Figure 2. TfuA catalyzes the hydrolysis of ThiS-COSH.
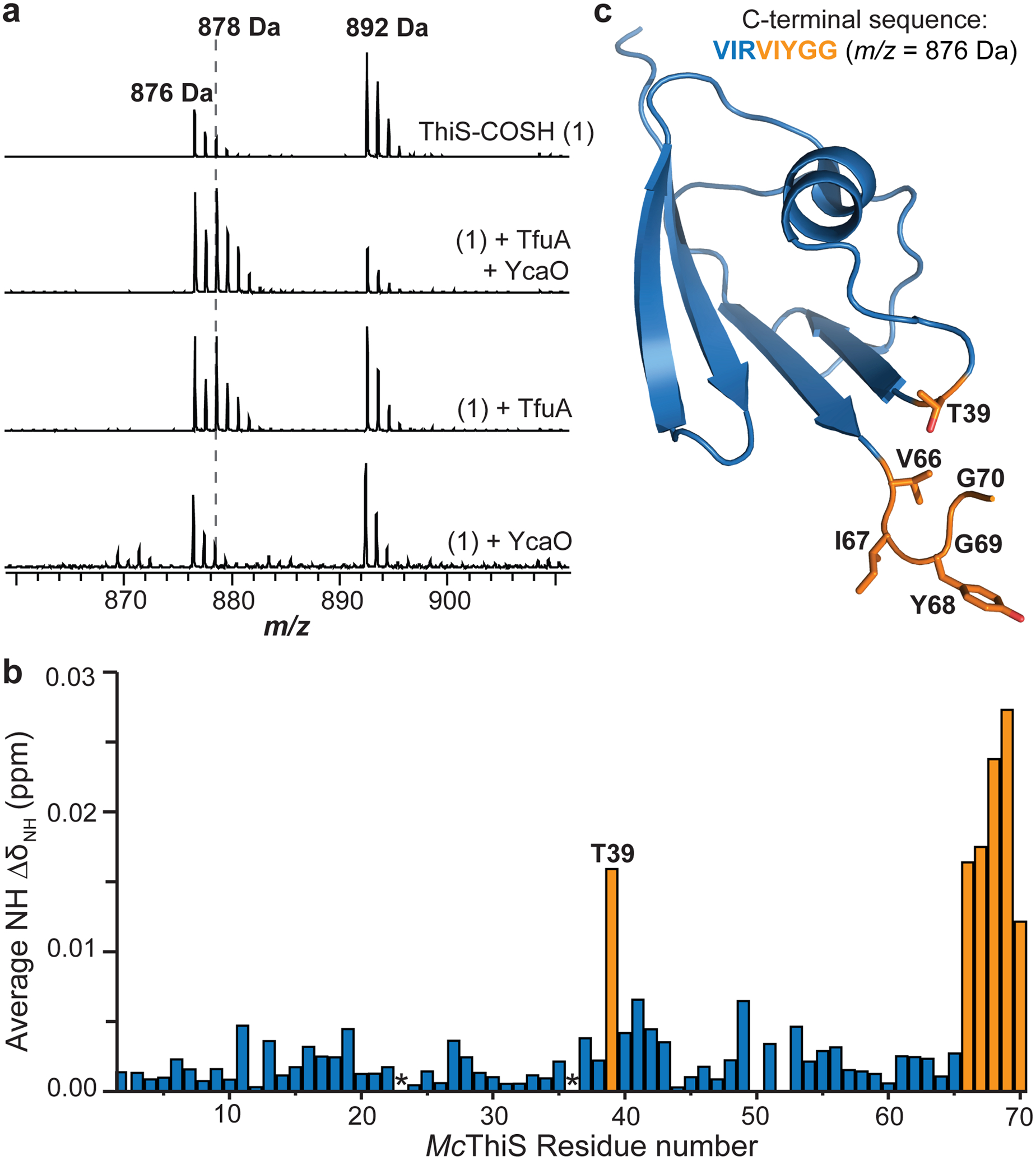
(a) Top, MALDI-TOF mass spectrum of the C-terminal GluC fragment of MtThiS-COSH (m/z 892 Da). Upon treatment with TfuA and YcaO in [18O]-H2O, MtThiS-COSH undergoes hydrolysis to MtThiS-COOH with incorporation of [18O] (m/z 878 Da). Reactions omitting either YcaO or TfuA show the hydrolysis is TfuA-dependent. (b) NMR spectroscopy was used to monitor amide NH chemical shift perturbation of McThiS upon titration with MtTfuA. Residues with an average chemical shift change >0.01 ppm are orange. Asterisks, Pro residues. (c) NMR solution structure of McThiS (PDB code: 1RJY), with residues showing the largest chemical shift perturbation upon titration with TfuA highlighted in orange.
To further examine the TfuA-ThiS interaction, chemical shift perturbation experiments were conducted using 1H, 15N-Heteronuclear Multiple-Quantum Correlation (HMQC) NMR spectroscopy. Owing to an available solution NMR structure39 for a MtThiS homolog differing only by two residues (Methanothermococcus thermolithotrophicus (Mc), PDB code: 1RYJ, Supplementary Fig. 5), McThiS was prepared using site-directed mutagenesis. McThiS-COSH was efficiently hydrolyzed by wild-type MtTfuA and served as a competent sulfur donor for McrA peptide thioamidation (Supplementary Fig. 5). 15N-labeled McThiS was prepared, and spectral changes upon titration with MtTfuA were monitored. Six amide signals shifted significantly as a function of MtTfuA concentration (Extended Data Fig. 7). The gradual change in chemical shift indicated the TfuA-ThiS interaction was in fast-exchange. The most significant chemical shift perturbations occurred between 0.5 mM and 1.2 mM TfuA, suggesting a high micromolar KD. Notably, the chemical shifts for all six residues were of approximately equivalent magnitudes, indicative of a single binding event. In congruence with TfuA catalyzing ThiS-COSH hydrolysis, the five C-terminal residues (Val66 to Gly70) were the most perturbed (Fig. 2). McThiS-Thr39 also experienced a substantial perturbation, which was attributed to C-terminal proximity. The formation of a MtTfuA and MtThiS-COSH complex was further confirmed via native MS (nMS, Supplementary Fig. 6). Under the conditions employed, MtThiS-COSH formed a homodimer, consistent with a homologous crystal structure (PDB code: 2CU3), while only monomeric TfuA was detected. Ions representing the 7+ to 15+ charge states for the MtTfuA:MtThiS-COSH heterodimer were detected (deconvoluted native mass: calc., 34,911.64 Da; obsv., 34,911.68 Da; error, 0.6 ppm).
TfuA enhances YcaO affinity for the McrA peptide
To efficiently transfer sulfide from ThiS-COSH to McrA, TfuA would presumably also interact with YcaO. To evaluate the putative protein-protein interaction, several experiments were conducted, including co-expression/purification, size-exclusion chromatography, isothermal titration calorimetry, microscale thermophoresis, and nMS. However, none definitively demonstrated a MtTfuA-MtYcaO interaction. A co-crystal structure of YcaO from M. jannaschii (MjYcaO) bound to its cognate peptide (PDB code: 6PEU) shows a mostly closed active site that requires a conformational change to allow entry of the peptide substrate and sulfur donor36. TfuA could theoretically fulfill this role, and such allostery could presumably also alter YcaO affinity for the McrA peptide. To test this hypothesis, an established fluorescent polarization (FP) binding assay was used to quantify binding between FITC (fluorescein isothiocyanate)-labeled McrA peptide and YcaO36. FITC-labeled McrA peptide bound to MtYcaO with a KD of 7 μM and exhibited no (or very weak) affinity towards MtTfuA (Fig. 3 and Supplementary Fig. 7). The MtYcaO-McrA peptide interaction was then monitored as a function of MtTfuA concentration. At a saturating concentration (~10 μM), an 8-fold increase in affinity between MtYcaO and McrA peptide was observed. Further, the polarization value with MtTfuA was significantly higher, suggesting the formation of a larger complex. As a control, MjYcaO, a TfuA-independent protein, bound McrA peptide at equal affinity irrespective of the non-cognate MtTfuA (Supplementary Fig. 7). These data are consistent with a specific TfuA-YcaO interaction. Notably, addition of MtThiS to the assay did not significantly alter the polarization maximum or apparent KD, indicating the YcaO:McrA affinity is TfuA-dependent and ThiS-independent (Supplementary Fig. 8). Although attempts to co-purify TfuA and YcaO proteins from methanogens were unsuccessful, likely due to a low-affinity interaction, YcaO from Azospirillum sp. B510 (BAI72909.1) co-purified with the cognate His-tagged TfuA protein (BAI72908.1, Extended Data Fig. 8). This result carries implications for thioamitide biosynthesis, as the YcaO:TfuA interaction is not restricted to methanogens.
Figure 3. TfuA and YcaO enhances the activity of each other.
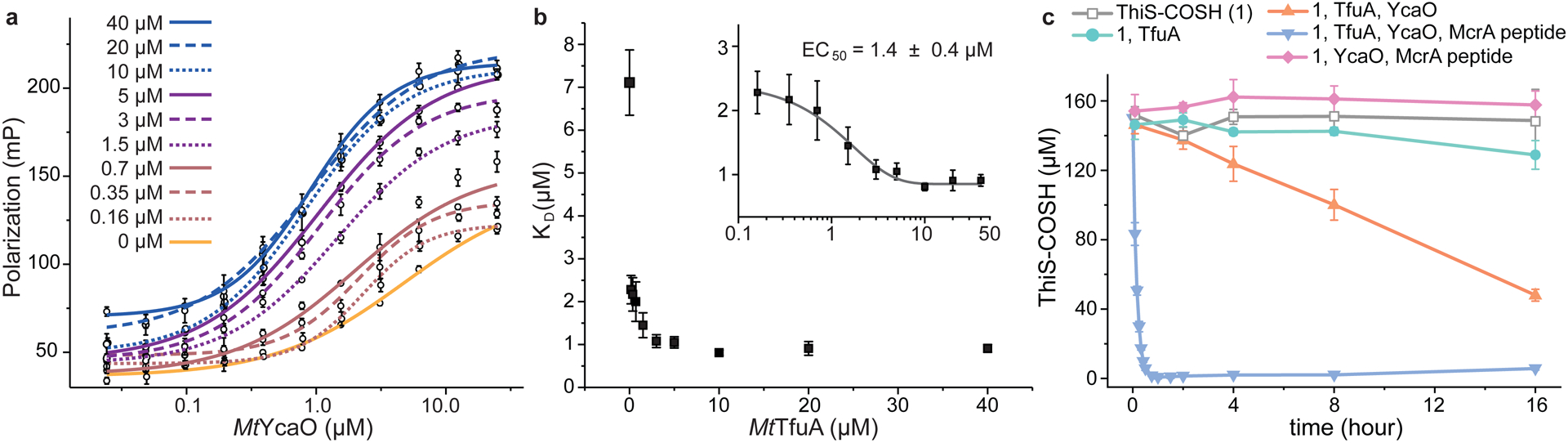
(a) Fluorescence polarization analysis of FITC-labeled McrA peptide binding to increasing concentrations of YcaO. Identical titrations were carried out with increasing concentration of TfuA (indicated by variable colors). Polarization values are reported as mean values ± SD (n = 3 independent experiments) and fitted into dose-response models. (b) The dissociation constants (KD ± SE) are derived from the non-linear regression analysis (dose-response model) using data in panel a. The inset replots the KD on a log-scale and fits them into a dose-response model. The resultant EC50 ± SE is reported. (c) Fluorescence quantification of ThiS-COSH over a 16-h period in the presence of varied reaction components, presented as mean values ± SD (n = 3 independent experiments).
YcaO and McrA peptide influence TfuA activity
Since TfuA enhances the YcaO-McrA interaction, it seemed plausible that YcaO and McrA could also affect the thiocarboxylate-hydrolyzing activity of TfuA. Therefore, MtThiS-COSH hydrolysis was monitored with and without MtYcaO and McrA peptide using the LRSA-based assay (Fig. 3). ATP was omitted to prevent sulfide incorporation into the pending thioamide. While the presence of MtYcaO enhanced hydrolysis by 5-fold (0.1 μM/min), the presence of MtYcaO and McrA peptide induced a 400-fold rate enhancement (7.9 μM/min) relative to MtTfuA alone (0.02 μM/min). Sulfide generation from identical reactions was also quantified. Congruent with the consumption of MtThiS-COSH, sulfide liberated from ThiS-COSH was potentiated in the presence of MtYcaO and McrA peptide (Supplementary Fig. 9). Upon omitting TfuA, the reaction of MtYcaO, MtThiS-COSH, and McrA peptide generated a background level of sulfide and a negligible amount of MtThiS-COOH. These data indicated that MtYcaO and McrA peptide significantly enhance MtTfuA-catalyzed ThiS-COSH hydrolysis.
ThiS-COSH hydrolysis with TfuA, YcaO, and McrA peptide present was next compared to reactions including ATP, allowing for McrA thioamidation. Without ATP, MtThiS-COSH was nearly quantitatively hydrolyzed and an equivalent amount of free sulfide was detected with no thioamide formation (Extended Data Fig. 9). The initial rate of MtThiS-COSH consumption was comparable with and without ATP; however, the reaction with ATP ceased to consume more ThiS-COSH than supplied McrA and resulted in a basal-level of sulfide, suggesting that ATP prevented the unproductive hydrolysis of ThiS-COSH. In the presence of all reaction components, sulfur transfer from ThiS-COSH to McrA was highly efficient.
Atomic-resolution structure of M. acetivorans TfuA
With an established biochemical role for TfuA, a structural characterization commenced. While efforts to crystallize MtTfuA were unsuccessful, the crystal structure of M. acetivorans TfuA (MaTfuA) was determined to 1.65 Å resolution using crystallographic phases derived from the selenomethionine-labeled protein. The structure shows a di-domain topology, with Ala7-Val111 and Asp203-Ala223 forming an α/β fold, containing a 4-helical bundle insertion formed by residues Ile130-Glu201 (Fig. 4). A DALI search40 against the PDB failed to identify any proteins sharing the same overall fold but did establish local similarities with various proteins mainly based on alignments with the α/β fold domain. The highest similarities occurred with the periplasmic solute-binding components of ABC transporters involved in binding sugars (PF13407). For example, the myo-inositol-binding protein from Caulobacter vibriodes41 (PDB code: 4IRX, Extended Data Fig. 10) gave a Z-score of 9 with the similarity restricted to the α/β fold domain (143 Cα atoms could be aligned, RMSD of 3.8 Å).
Figure 4. Crystal structure of MaTfuA.
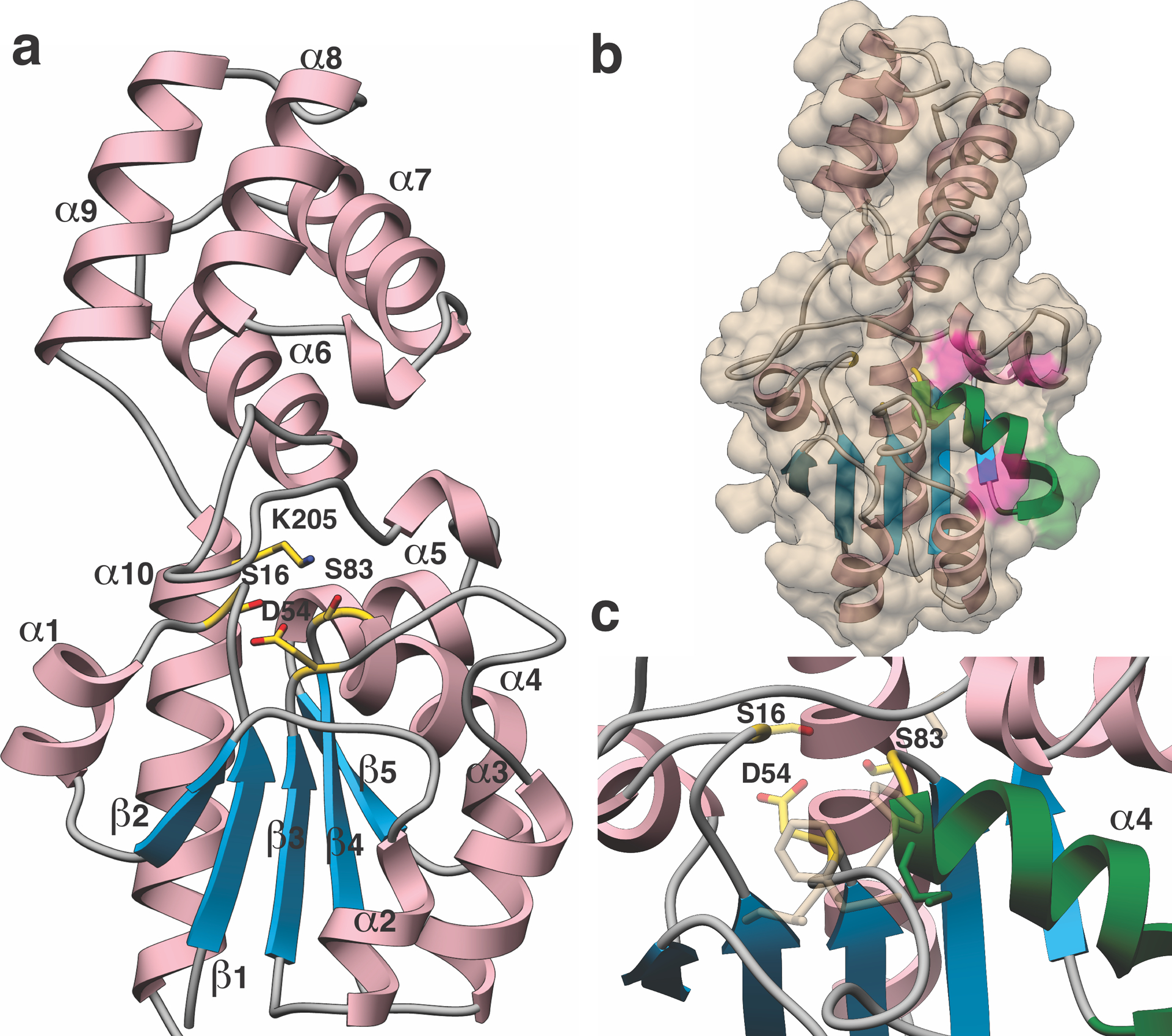
(a) Overall structure of MaTfuA showing a unique di-domain fold. Secondary structural elements are marked, and presumptive active site residues are shown in yellow sticks. (b) Surface representation of MaTfuA with the ribbon diagram superimposed. The α-helix that is implicated to mediate interactions with YcaO is colored green (α4). Additional residues involved in interactions with YcaO are colored in pink. (c) Close-up view of the putative active site of MaTfuA. Residues predicted to form the ThiS-binding pocket based on a superimposition with subtilisin BPN’ (3CNQ) are shown as tan colored sticks.
Structural similarities were also identified with a number of hydrolytic enzymes that contain a canonical α/β hydrolase fold. For example, the co-crystal structure of Bacillus amyloliquefaciens subtilisin BPN’ in complex with its prodomain42 (PDB code: 3CNQ, Extended Data Fig. 10) aligns with the MaTfuA structure with an RMSD of 2.0 Å over 100 Cα atoms. A superposition of the two structures revealed two notable features. First, a conserved hydrophobic loop composed of MaTfuA-Ile56-Phe58, Met84, and Val118 appear to form the substrate-binding pocket (Fig. 4 and Extended Data Fig. 10). Second, the Ser/His/Asp catalytic residues of subtilisin BPN’ are in the vicinity of MaTfuA-Ser16 (moderately conserved) and MaTfuA-Ser83 (strictly conserved), which in turn are adjacent to highly conserved Asp54 and Lys205 (Supplementary Fig. 10–11). These structural homology arguments support a hydrolytic role for TfuA, a common function for α/β fold-containing proteins43. HHpred analysis44 of MaTfuA also revealed extremely low-level similarity (E-value 27) towards the region surrounding the catalytic Ser of patatin-17 (PDB code: 4PK9, eukaryotic lipid acyl hydrolase)45. Notably, the catalytic Ser of patatin and MaTfuA-Ser83 align in the HHpred analysis and both reside in a conserved Gly-(Xaa)1–2-Ser-Xaa-Gly motif (Supplementary Fig. 12)45. Collectively, these analyses suggested TfuA might employ a Ser/Lys/Asp hydrolase-like active site.
Mutational and mechanistic analysis of TfuA
A metal ion-mediated hydrolytic mechanism for TfuA was first ruled out based on the lack of metals in the reaction buffer, no visible metals in the crystal structure, and no evidence of bound metal by nMS. TfuA-catalyzed ThiS-COSH hydrolysis also remained unchanged upon addition of EDTA (Supplementary Fig. 13). Conserved residues of TfuA (n = 18) were then targeted for mutational analysis. Although Mt proteins were examined in the following experiments, the amino acid similarity between MtTfuA and MaTfuA (49% identity) allows a direct structural interpretation of the biochemical data (residues are numbered based on MaTfuA hereafter). Single amino acid variants of MtTfuA were generated and purified with yields and purities equivalent to the wild-type protein (Supplementary Fig. 1). The MtTfuA variants were then evaluated in four distinct assays: (i) thioamidation of McrA peptide, (ii) ThiS-COSH hydrolysis with TfuA by [18O] incorporation, (iii) enhancement of ThiS-COSH hydrolysis with YcaO and McrA peptide via sulfide detection, and (iv) enhancement of YcaO-McrA affinity by TfuA (Table 1 and Supplementary Figs. 14–17).
Table 1. Activity of TfuA variants.
The hydrolysis of ThiS-COSH catalyzed by TfuA alone was assayed in [18O]-H2O and qualitatively (+/−) assessed by endpoint MALDI-TOF-MS. The enhancement of TfuA-catalyzed ThiS-COSH hydrolysis by YcaO and McrA peptide (ATP omitted) was assessed by endpoint sulfide quantification. ++, wild-type-like activity; +, reduced activity; −, activity equivalent to TfuA alone. The approximated affinity towards FITC-labeled McrA peptide (EC50 ± SE) in the presence of 1.5 μM YcaO and varied concentration of TfuA was determined by FP and subsequent regression analysis. McrA peptide thioamidation was qualitatively assessed with TfuA, YcaO, and ThiS-COSH by endpoint MALDI-TOF-MS. ++, wild-type-like activity; +, reduced activity; −, activity equivalent to omission of TfuA; (− −), inhibitory to thioamidation.
| TfuA variant (Ma numbering) | ThiS hydrolysis (TfuA alone) | ThiS hydrolysis enhancement (+YcaO/McrA) |
EC50 of McrA (μM) | McrA thioamidation |
|---|---|---|---|---|
| Wild-type | + | ++ | 1.9 ± 0.1 | ++ |
| S16A | + | ++ | 5.4 ± 2.1 | ++ |
| D54A | + | ++ | > 30 | + |
| F57A | + | − | 3.8 ± 1.3 | − |
| H65A | + | + | > 30 | + |
| E67A | + | ++ | 4.9 ± 0.7 | ++ |
| S83A | − | − | 4.1 ± 0.3 | (− −) |
| M84A | + | + | 3.2 ± 0.6 | + |
| L87A | + | − | > 30 | − |
| R88A | + | − | > 30 | − |
| E91D | + | ++ | 2.1 ± 0.3 | ++ |
| E91Q | + | − | > 30 | − |
| Y107A | + | ++ | > 30 | ++ |
| D115A | + | + | > 30 | − |
| V118A | − | − | > 30 | + |
| R170A | + | + | > 30 | + |
| D203A | + | ++ | 2.9 ± 1.2 | ++ |
| K205M | − | − | 1.1 ± 0.3 | − |
| R206A | + | ++ | 3.1 ± 0.5 | ++ |
| D208N | + | ++ | 3.9 ± 0.3 | ++ |
A Ser hydrolase mechanism was next evaluated. The top candidates from the crystal structure were individually replaced with Ala. MtTfuA-S16A retained ThiS-COSH hydrolysis while MtTfuA-S83A was devoid of ThiS-COSH hydrolytic activity (Table 1). In the absence of TfuA, thioamidation occurred very slowly and was detectable after long reaction times (Fig. 1 and Supplementary Fig. 18), possibly from non-enzymatic hydrolysis of ThiS-COSH. However, upon comparison of MtTfuA-S83A to a reaction lacking TfuA, no McrA thioamidation was detected, even after prolonged reaction times, demonstrating an inhibitory effect (Supplementary Fig. 18). This outcome was logical for a catalytically inactive variant that maintained interactions with MtThiS and MtYcaO, preventing sulfide from accessing the active site. Indeed, HMQC analysis of McThiS upon titration with MtTfuA-S83A revealed chemical shift perturbations near the McThiS C-terminus similar to wild-type TfuA (Supplementary Fig. 19). An FP-based assay demonstrated that MtTfuA-S83A also retained the ability to promote the MtYcaO-McrA substrate interaction (Table 1). Collectively, these data support a catalytic role for MtTfuA-Ser83. Two charged residues near Ser83 were next evaluated. MtTfuA-D54A was decreased in thioamidation activity while MtTfuA-K205M was entirely inactive, consistent with a Ser83/Lys205 catalytic pair. Likewise, MtTfuA-V118A was inactive as a ThiS-COSH hydrolase and showed significantly impaired thioamidation (Table 1). The superimposition of MaTfuA structure with that of subtilisin BPN’ revealed that Val118 could be involved in ThiS binding (Extended Data Fig. 10). To evaluate this hypothesis, a HMQC-based chemical shift perturbation experiment was performed on McThiS titrated with MtTfuA-V118A. The McThiS spectra remained unchanged, consistent with a loss of contact with MtTfuA-V118A (Supplementary Fig. 20).
TfuA residues that failed to potentiate the YcaO:McrA peptide interaction were next examined (Table 1). The majority of such variants retained ThiS-COSH hydrolytic activity but displayed diminished or abolished thioamide formation (i.e., H65A, L87A, R88A, E91Q, D115A, and R170A), highlighting the interaction with YcaO as an essential functional role of TfuA. The ThiS-COSH hydrolytic activity of these TfuA variants also were not enhanced by the additional of YcaO-McrA. From the crystal structure, these residues are solvent-exposed and located on the same face of the protein, with Leu87, Arg88, and Glu91 located on α4, forming a highly probable interface with YcaO (Fig. 4). Notably, TfuA variant E91D restored the deficiencies observed with E91Q, indicative of participation in an ionic interaction with YcaO.
During the course of the TfuA mutational analysis, we noted that MaTfuA contains only a single, Cys residue and MtTfuA contains no Cys (Supplementary Fig. 10). Further, there are no conserved Cys among methanogenic YcaO (Supplementary Fig. 21). However, with a Cys-dependent persulfide (R-S-SH) based sulfur transfer mechanism prevalent in biosynthetic pathways46, all Cys in MtYcaO were substituted with Ala. Each variant exhibited wild-type-like thioamidation activity (Supplementary Fig. 22), refuting a persulfide-mediated sulfur transfer mechanism.
Discussion
We have characterized peptide backbone thioamidation catalyzed by YcaO, TfuA, and the sulfur donor ThiS-COSH. Two biochemical roles were assigned to TfuA, which had no previously known function. TfuA enhances the affinity of YcaO for the McrA peptide and catalyzes ThiS-COSH hydrolysis. Our analyses suggest that TfuA employs a Ser83/Lys205 catalytic pair47, which was supported by three lines of evidence: (i) remote structural and sequence similarity with the active site of known serine hydrolases, (ii) deficient catalysis upon substituting Ser83/Lys205, and (iii) intact affinity of the substituted variants towards YcaO and ThiS. Based on the available data, a reaction scheme was proposed featuring the Ser/Lys pair and a hydrophobic ThiS binding pocket (Fig. 5).
Figure 5. Reaction scheme of YcaO-TfuA thioamide synthetase.
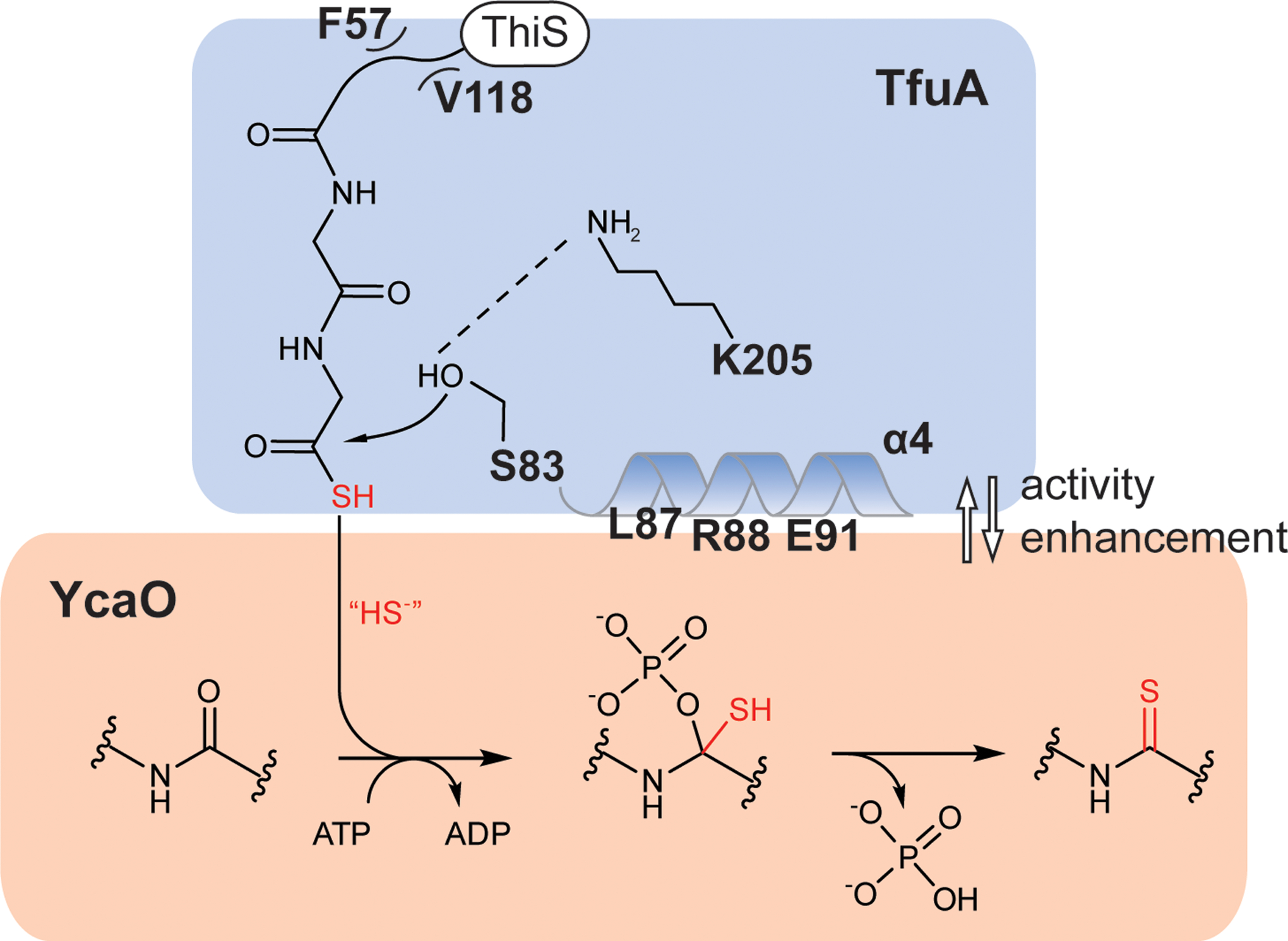
TfuA utilizes a Ser/Lys catalytic pair to hydrolyze ThiS-COSH. TfuA residues that mediate the hydrophobic interaction with ThiS are labeled; α-helix four, which mediates the YcaO interaction is highlighted. The resulting sulfide equivalent is delivered to the YcaO active site, providing the nucleophile for forming the thioamide via an O-phosphorylated hemiorthoamide intermediate.
A notable characteristic of the TfuA/YcaO pair is their interdependency: TfuA enhances YcaO affinity for the McrA peptide, while YcaO-McrA induces increased ThiS-COSH hydrolytic activity from TfuA. Additionally, the bidirectional enhancement was abolished when the same set of TfuA residues were substituted, underlining a critical role in mediating the protein-protein interactions. From the crystal structure of MaTfuA, most of these residues are posited on or near an α-helix directly preceding the Ser nucleophile, providing an explanation for how YcaO-interaction regulates TfuA activity (Fig. 4). Notably, ThiS-COSH hydrolysis and sulfide release were augmented when YcaO and the McrA peptide were present. This can be justified by a significant YcaO conformational change upon McrA binding, as evidenced by the available structures of MjYcaO36. With ATP, YcaO, and TfuA, sulfide delivery from ThiS-COSH to the McrA backbone is efficient, with very little sulfide being lost to bulk solvent (Extended Data Fig. 9). There are two probable mechanisms for this transfer: (i) ThiS-COSH is hydrolyzed to generate sulfide very near (or channeled directly to) the active site of YcaO, or (ii) ThiS-COSH attacks the activated McrA peptide to form a thioester-hemiorthoamide intermediate (Supplementary Fig. 23) that could undergo subsequent hydrolysis to the thiolate, followed by thioamide formation and phosphate elimination. While the second mechanism cannot be ruled out, we favor the first based on several observations: (i) Sulfide is considerably less hindered than ThiS-COSH and a more suitable candidate to enter the McrA- and ATP-bound active site of YcaO, which is already complexed with TfuA. (ii) Sulfide is rapidly generated from ThiS-COSH upon reaction with TfuA, YcaO, and McrA peptide. It is difficult to reconcile the robust sulfide production if ThiS-COSH were the direct nucleophilic species for thioamidation. Notably, the initial rate of ThiS-COSH hydrolysis to generate sulfide (-ATP) was similar to the initial rate of thioamidation (+ATP), suggesting that sulfide production is on-pathway. (iii) While there is ~900-fold difference in KM between ThiS-COSH and sulfide for the thioamidation reaction, the kcat values are comparable. This suggests that the nucleophile utilized by YcaO in the two reactions are the same chemical species (i.e., sulfide/bisulfide). (iv) There is no evidence of a direct interaction between YcaO and ThiS-COSH. If the unstable thioester-hemiorthoamide intermediate did form, it would presumably be readily hydrolyzed. However, only minute levels of ThiS-COSH hydrolysis were detected when TfuA was omitted, even with large amounts of ATP and McrA peptide supplied to the reaction.
To the best of our knowledge, this study provides the first in vitro characterization of an archaeal ThiS sulfur carrier protein and enforces an earlier in vivo study that associated a Haloferax volcanii ThiS homolog with molybdopterin biosynthesis48. Given the data presented, we anticipate that ThiS-COSH, TfuA, and YcaO will similarly collaborate in thioamitide biosynthesis. One notable difference between thioamitide and McrA thioamidation is the presence of a leader region in the thioamitide precursor peptide. Although the data show that YcaO, not TfuA, binds the McrA peptide, whether this remains true for thioamitides and how the leader region is involved warrants further exploration.
Additional remaining questions include the precise mechanism of sulfide containment and transfer between TfuA and YcaO. As a persulfide-mediated mechanism is not possible, we propose that there could be a sulfide conduit connecting the active sites of TfuA and YcaO if they are not already in close proximity. Further, the evolutionary history of TfuA could be further delineated. Methanogenesis is believed to have originated in sulfide-rich environments and it is known that the fitness consequences of non-thioamidated MCR are severe13. Therefore, as methanogens began to adapt to lower sulfide environments, a mechanism to maintain efficient thioamidation of MCR likely had to emerge.
Online Methods
General materials and methods.
Oligonucleotides were purchased from Integrated DNA Technologies. Restriction endonucleases, DNA polymerases, and T4 DNA ligase were obtained from New England Biolabs. Amicon Ultra-4 centrifugal filters were purchased from EMD Millipore. Chemical reagents were purchased from Sigma Aldrich. The synthetic peptide was obtained from GenScript. Escherichia coli DH5α and E. coli BL21(DE3) cells were used for plasmid maintenance and protein overproduction, respectively. The genomic DNA of Methanothermobacter sp. CaT2 was purchased from the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures (DSM 24414). The strain Azospirillum sp. B510 was obtained from the Japan Collection of Microorganisms (JCM 14679). DNA sequencing was performed by the DNA sequencing facility at the University of Illinois at Urbana-Champaign.
Molecular biology techniques.
The MBP-fusion proteins used in this study were cloned from as reported before12 into the pET28 vector with an N-terminal MBP-His tag. The MtTfuA was cloned into the pETDuet-1 vector with an N-terminal His tag. The AzoTfuA and AzoYcaO were cloned into the first and second multiple cloning sites (MCSs) in pETDuet-1 vector, respectively, with an N-terminal His tag preceding the first MCS. These constructs were used for protein expression and as the templates for site-directed mutagenesis. Mutagenesis was performed using the QuikChange method (Agilent) with Q5 Polymerase (NEB). The primers used in this study were listed in Supplementary Table 1. All mutagenesis was verified by DNA sequencing.
Bioinformatics.
For Sequence Similarity Network (SSN) generation and genomic neighborhood analysis, TfuA domain-containing protein sequences (2,042 as of December 2020) from the UniProt database2 were used as the input for Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST)34. The SSN was visualized with an alignment score of 60 using the organic layout within Cytoscape49; sequences with 100% identity were conflated into a single node. The genes encoded within ten open-reading frames of the TfuA sequence were examined using the Genome Neighborhood Network tool34. TfuA sequences with neighboring ThiS (PF02597) are noted on the SSN. The UniProt records of TfuA longer than 500 amino acids were examined manually for TfuA-YcaO fusions.
For the correlation analysis, all genome assemblies (111) from GenBank denoted as “complete” from the classes Methanobacteria (31), Methanococci (20), Methanopyri (1), Methanomicrobia (53), and the order Methanomassiliicoccales (6) were retrieved for the co-occurrence analysis (Supplementary dataset). Hidden Markov Models (HMMs) for the YcaO (protein family PF02624), TfuA (PF07812), and ThiS superfamilies were downloaded from the Pfam database1. Hmmsearch of each HMM was performed against all 111 genomes using HMMER 3.3. The absence or presence of each protein homolog was determined based on the hmmsearch result using the trusted cut-off from the Pfam HMMs. The data were visualized along with a maximum-likelihood phylogenetic tree of methanogenic YcaO (generated with FastTree 2.050 with default settings) using iTOL51. The correlation analysis was conducted in R studio with Fisher’s exact test.
Expression and purification of MBP-tagged proteins.
The plasmids with constructs of interest were transformed into E. coli BL21(DE3) RIPL cells for protein expression. Cells were grown for 24 h on Luria-Bertani (LB) agar plates containing appropriate antibiotics at 37 °C. Single colonies were picked to inoculate 10 mL of LB containing appropriate antibiotics and grown at 37 °C for 16–18 h. This culture was used to inoculate 1 L of LB with appropriate antibiotics and grown to an optical density at 600 nm (OD600) of 0.6–0.8 at 37 °C and then placed on ice for 10 min. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was then added to a final concentration of 0.3 mM, followed by an induction period at 18 °C for 14–18 h. Cells were harvested via centrifugation at 4,500 × g for 10 min at 4 °C and resuspended in 30 mL lysis buffer [25 mM Tris pH 8.0, 500 mM NaCl, 2.5% glycerol (v/v), 0.1% Triton X-100 (v/v)] containing 4 mg/mL lysozyme, 2 μM leupeptin, 2 μM benzamidine HCl, 2 μM E64, and 30 mM phenylmethylsulfonyl fluoride (PMSF). Cells were lysed by sonication for three cycles (50 s each) at 20% amplitude, with 10 min intervals of resting at 4 °C. Insoluble cell debris was removed by centrifugation at 17,000 × g for 90 min at 4 °C. The resulting supernatant was loaded onto a pre-equilibrated amylose resin (NEB; 5 mL of resin per L of cells). The column was washed with 30 mL of lysis buffer followed by 20 mL of wash buffer (lysis buffer without Triton X-100). The MBP-tagged proteins were eluted using 30 mL elution buffer [50 mM Tris-HCl pH 7.5, 300 mM NaCl, 2.5% glycerol (v/v), 10 mM maltose]. The eluent was concentrated using a 30 kDa molecular weight cut-off (MWCO) Amicon Ultra centrifugal filter (EMD Millipore). A buffer exchange with 10 × volume of protein storage buffer [50 mM HEPES pH 7.5, 300 mM NaCl, 2.5% glycerol (v/v), 0.5 mM TCEP] was performed prior to final concentration and storage. Protein concentrations were determined based on their absorbance at 280 nm (theoretical extinction coefficients were calculated using the ExPASy ProtParam tool). Purity was visually inspected by Coomassie-stained SDS-PAGE gel (Supplementary Fig. 1).
To obtain MaTfuA, MtTfuA, MtThiS, and McThiS protein without the MBP-His tag for crystallography, biochemical, and NMR studies, respectively, the aforementioned eluent was digested by addition of 1:100 (w/w) tobacco etch virus (TEV) protease and buffer exchanged to the wash buffer using Amicon Ultra centrifugal filters. The proteins were loaded onto a pre-equilibrated Ni-NTA column to remove the MBP-His tag. The flow-through was collected and concentrated using a 3 kDa MWCO Amicon Ultra centrifugal filter. The resulting proteins were subjected to a Superdex 200 (for TfuA) or 75 (for ThiS) size-exclusion column (GE Healthcare Lifesciences) and a single peak from the 280 nm absorbance trace corresponding to the desired protein was collected and used.
Expression and purification of the His-tagged MtTfuA and AzoTfuA.
The proteins are expressed the same way as MBP-tagged proteins. Cells were harvested similarly and resuspended in 30 mL Ni lysis buffer [25 mM Tris pH 8.0, 500 mM NaCl, 15 mM imidazole, 2.5% glycerol (v/v), 0.1% Triton X-100 (v/v)] containing 4 mg/mL lysozyme, 2 μM leupeptin, 2 μM benzamidine HCl, 2 μM E64, and 30 mM PMSF. Cells were lysed the same way as MBP-tagged proteins and the resulting supernatant was loaded onto a pre-equilibrated Ni-NTA resin (NEB; 5 mL of resin per L of cells). The column was washed with 30 mL of Ni lysis buffer followed by 20 mL of Ni wash buffer [25 mM Tris pH 8.0, 500 mM NaCl, 30 mM imidazole, 2.5% glycerol (v/v)]. The proteins were eluted using 30 mL elution buffer [50 mM Tris-HCl pH 7.5, 300 mM NaCl, 250 mM imidazole, 2.5% glycerol (v/v)]. The eluent was concentrated using a 10 kDa (MtTfuA) or 30 kDa (AzoTfuA) MWCO Amicon Ultra centrifugal filter and buffer exchanged to the storage buffer. Protein concentrations were determined based on their absorbance at 280 nm and the purity was visually inspected by Coomassie-stained SDS-PAGE gel (Supplementary Fig. 1).
Preparation of ThiS-COSH using MBP-ThiF and inorganic sulfide.
To form the thiocarboxylic acid group on Mt or MaThiS, 100 μM tag-free ThiS was allowed to react with 15 μM MBP-ThiF, 10 mM Na2S or Li2S in the synthase buffer [50 mM Tris-HCl pH 7.5, 80 mM NaCl, 20 mM MgCl2, 5 mM ATP] at room temperature for 16 h. Upon completion, the reaction was desalted with a PD-10 column (GE Healthcare) per manufacturer’s instructions using wash buffer and subsequently loaded onto a pre-equilibrated Ni-NTA column (1 mL of resin per mL of reaction). The column was washed with 2 mL of wash buffer and 3 mL of Ni wash buffer. The flow-through from all steps were collected, pooled, and buffer-exchanged to storage buffer using a 3 kDa MWCO Amicon Ultra centrifugal filter and dialysis. The resulting ThiS-COSH was analyzed by MALDI-TOF-MS (desalted using a ZipTip and eluted into 70% aq. MeCN) with sinapic acid as the matrix. Reflector positive mode was used on a Bruker UltrafleXtreme instrument (Bruker Daltonics) at the University of Illinois School of Chemical Sciences Mass Spectrometry Laboratory to acquire the spectra. Data were analyzed using the Bruker FlexAnalysis software. The concentration of MtThiS-COSH was also determined through reaction with LRSA as described below, buffer exchanged (10,000-fold) to remove the free dye, absorbance measurement (568 nm), and comparison to a standard curve.
Reconstitution and MS-based analysis of thioamide formation.
The synthetic McrA peptide substrate as reported previously36 (Genescript, 100 μM), MBP-MtYcaO (1 μM), MBP-MtTfuA (1 μM), TEV protease (0.5 μM), and purified Mt, Ma, or McThiS-COSH (150 μM) were let react in the synthase buffer at room temperature. The reactions were initiated by adding the MtThiS-COSH and part of the reaction mixture was quenched at different time points by adding 67% MeOH. The precipitate was removed from the quenched reaction mixture by centrifugation (15,000 × g for 1 min) and the supernatant was analyzed by MALDI-TOF-MS using α-Cyano-4-hydroxycinnamic acid (CHCA) as the matrix or LC-MS (Shimadzu LCMS-2020). To be specific, 10 μL the supernatants were mixed with 90 μL water and injected onto the LC-MS system equipped with a C18 column (Macherey-Nagel, 4.6 × 250 mm, 5 μm particle size). Acetonitrile and 0.01% (v/v) formic acid were used as the mobile phases. A linear gradient of 20 to 56% acetonitrile over 9 min at 1 mL/min was used to separate the thioamide-containing peptide from the starting material. The concentration of the thioamidated product was determined by the ratio of the area under the peaks corresponding to the reactant and product (retention times are ~1 min apart).
For testing the MtTfuA variants, a slightly modified reaction condition was used: McrA peptide substrate (40 μM), MBP-MtYcaO (2 μM), MBP-MtTfuA (2 μM), TEV protease (1 μM), MtThiS-COSH (~50 μM). The same batch of MtThiS-COSH was used to test each variant, wild-type MtTfuA, and a control with no MtTfuA simultaneously. The reactions were quenched at 60 min and examined by MALDI-TOF-MS.
Quantification of ThiS-COSH by reacting with LRSA.
To quantify MtThiS-COSH as shown in Fig. 1d, the aforementioned reactions were quenched the same way and the resulting protein precipitate containing MtThiS-COSH was re-dissolved in 5 μL LRSA buffer [50 mM K2HPO4 pH 6, 6 M urea]. Once dissolved, 3 equivalents of LRSA were added and mixed by vortex. The labeling reaction was let proceed for 40 min in the dark and quenched by adding 1 μL of 250 mM TCEP. This was incubated for 1 h in the dark prior to the addition of 1 μL water and 1.5 μL SDS loading dye [250 mM Tris-HCl pH 6.8, 5% β-mercaptoethanol (v/v), 0.02% bromophenol blue (w/v), 30% glycerol (v/v), 10% SDS (w/v)], and heated to 95 °C for 5 min. Samples were loaded onto a 12% PAGE gel and ran in the dark using tricine buffer as described previously37. A low voltage (30V to 50V) was used through the stacking gel to ensure sufficient stacking. The resulting gel was analyzed using a Typhoon 9410 imager and quantified using the Array & Microplate Analysis application of the ImageQuant software. All the gels were subsequently stained with Coomassie blue and inspected visually. For ThiS-COSH quantification in Fig. 3c and Extended Data Fig. 9, 7 μL of reaction was quenched by adding LRSA buffer (5 μL). The resulting solution was allowed to react with LRSA and analyzed as described above.
Kinetic analysis of McrA peptide thioamidation.
For the kinetic analysis of Na2S, the McrA peptide substrate (100 μM), MBP-MtYcaO (1 μM), MBP-MtTfuA (4 μM), and various concentrations of Na2S (10–100 mM) were let react in the high-tris synthase buffer [400 mM Tris-HCl pH 7.5, 80 mM NaCl, 20 mM MgCl2, 5 mM ATP] at 22 °C. The reaction mixtures excluding Na2S were equilibrated with TEV protease (0.5 μM) for 30 min to remove the MBP tag prior to reaction initiation by the addition of Na2S. The reactions were then quenched at four different time points by adding 67% MeOH. The resulting quenched reactions (in duplicates) were analyzed by LC-MS as described above to determine the product concentration. The product concentrations were plotted against time and fitted to a standard linear model (OriginPro9.1) to estimate the initial velocities (vo ± SE). The vo /[MtYcaO] were plotted against the Na2S concentrations and fitted to a Michaelis-Menten kinetic model.
For the kinetic analysis of MtThiS-COSH, the McrA peptide substrate (100 μM), MBP-MtYcaO (0.2 or 1 μM), MBP-MtTfuA (4 μM), and various concentrations of MtThiS-COSH (12–90 μM) were let react in the synthase buffer at 22 °C. The reaction mixtures excluding MtThiS-COSH were equilibrated with TEV protease (0.5 μM) for 30 min to remove the MBP tag prior to reaction initiation by adding MtThiS-COSH. The reactions were quenched, analyzed, and fitted into a Michaelis-Menten kinetics model the same way as for Na2S.
For the kinetic analysis of MtTfuA, the McrA peptide substrate (100 μM), MBP-MtYcaO (1 μM), MtThiS-COSH (200 μM), and various concentrations of MBP-MtTfuA (0–2 μM) were let react in the synthase buffer at 22 °C. The reaction mixtures excluding ThiS-COSH were treated with TEV protease as described above prior to reaction initiation by adding MtThiS-COSH. The reactions were quenched and analyzed as described above, and then plotted against the MtTfuA concentrations and fitted to the substrate activation kinetics model (Eq. 1) using OriginPro9.1.
| Eq. 1 |
Tandem MS characterization of the products.
The reactions were desalted using a ZipTip and eluted into 70% aq. MeCN. The eluents were directly infused into a ThermoFisher Scientific Orbitrap Fusion ESI-MS using an Advion TriVersa Nanomate 100. The MS was calibrated and tuned with Pierce LTQ Velos ESI Positive Ion Calibration Solution (ThermoFisher). The MS was operated using the following parameters: resolution, 100,000; isolation width (MS/MS), 1 m/z; normalized collision energy (MS/MS), 70; activation q value (MS/MS), 0.4; activation time (MS/MS), 30 ms. Fragmentation was performed using collision-induced dissociation (CID) at 35 to 70%. The resulting data were averaged and analyzed using the Qualbrowser application of Xcalibur software.
MtThiS-COSH hydrolysis in [18O]-H2O and MALDI-TOF analysis.
MtThiS-COSH was lyophilized to remove H2O and resuspended in [18O]-H2O. For the hydrolysis reaction, 5 μM of MtTfuA (1 μL) was incubated with 45 μM MtThiS-COSH in [18O]-H2O (9 μL) at room temperature for 1 h. The reaction was stopped by the addition of endoproteinase GluC (NEB) at the final concentration of 5 ng/μL and incubation at 37 °C for 16 h. The resulting fragment of MtThiS-COSH was analyzed by MALDI-TOF-MS (desalted using a ZipTip and eluted into 70% aq. MeCN) with CHCA as the matrix. The variants were tested the same way as wild-type MtTfuA using the same batch of ThiS-COSH.
Quantification of sulfide using 7-amino-4-methylcoumarin.
The reaction buffer [50 mM Tris-HCl pH 7.5, 500 mM NaCl, 2% Glycerol (v/v)] used for this assay was first degassed with N2 for 30 min. 7-amino-4-methylcoumarin38 (7-AMC, Sigma-Aldrich) was dissolved in dimethylformamide (DMF) at the concentration of 20 mM and subsequently diluted to 2 mM with the reaction buffer. Samples for the sulfide standard curve were prepared by letting Na2S standard solutions (15 μL, 0–500 μM) react with 2 mM 7-AMC (15 μL) in dark at room temperature for 60 min. Fluorescence intensity (λEx/Em = 340/450 nm) of the reactions was measured in triplicates using a Tecan infinite M200PRO plate reader.
For the various analysis of sulfide generation from MtThiS-COSH hydrolysis, the reactions were allowed to proceed at room temperature and quenched via flash freezing. The resulting solutions were stored in −80 °C for less than 24 h and then incubated with an equal volume (15 μL) of 7-AMC (2 mM) for 60 min in the dark at room temperature. The fluorescence quantification was performed as described above, and the sulfide concentration was calculated based on the standard curve. The TfuA variants were tested simultaneously with the same batch of MtThiS-COSH.
Preparation of 15N-labeled McThiS.
The pET28-MBP-McThiS construct was transformed into E. coli BL21(DE3) RIPL cells for protein expression. Cells were grown the same way as for other MBP-tagged proteins (in 1 L LB) to an OD600 of 0.6–0.8 and then harvested via centrifugation at 4,500 × g for 10 min. The cells were resuspended in 45 mL of 22 mM KH2PO4, 43 mM Na2HPO4, 8.5 mM NaCl and pelleted again via centrifugation (4,500 × g for 10 min). M9 media (0.5 L) containing 15N-labeled NH4Cl [22 mM KH2PO4, 43 mM Na2HPO4, 8.5 mM NaCl, 18 mM 15NH4Cl (Cambridge Isotope Laboratories), 0.4% Glucose, 2 mM MgSO4, 0.1 mM CaCl2, BME vitamin mix (Sigma-Aldrich), trace mineral supplement (ATCC)] with appropriate antibiotics was used to resuspend and culture the cells for 1 h at 30 °C. IPTG was then added to a final concentration of 0.3 mM, followed by an induction period at 18 °C for 18 h. The MBP-McThiS was harvested and purified the same way as for other MBP-tagged proteins. Affinity-purified proteins were then buffer exchanged to the NMR buffer [25 mM Na2HPO4 pH 6.5, 100 mM NaCl, 10 mM DTT, 20 μM ZnCl2] using the Superdex 75 size-exclusion column. Protein concentrations were determined by Bradford colorimetric assay and the sample concentration was adjusted to 330 μM.
NMR spectroscopy.
The sample was prepared by adding 30 μL D2O (99.96 atom % D; Sigma-Aldrich) into 270 μL of purified 15N-labeled McThiS in NMR buffer to achieve a final concentration of 300 μM. The sample was loaded into a D2O-lock Shigemi tube and NMR spectra were recorded on an Agilent VNMRS 750 MHz narrow bore magnet spectrometer equipped with a 5 mm triple resonance (1H-13C-15N) triaxial gradient probe and pulse-shaping capabilities. Samples were held at 25°C during acquisition. Standard Varian pulse sequences were used for the 1H, 15N-HMBC with 1,024 points in t2 over the spectrum width of 16 ppm (0.085 sec acquisition time), 512 points in t1 over the spectrum width of 36 ppm, and 16 scans. Solvent suppression by presaturation (PRESAT) was employed for all the experiments. Spectra were recorded with VNMRJ 3.2A. Spectra were imported into MestReNova 10.0.2, phased and analyzed. Changes in average amide chemical shifts are calculated based on Eq. 2, in which ΔδN stands for the change in the amide nitrogen’s chemical shift, and the ΔδH stands for the change in the amide proton’s chemical shift.
| Eq. 2 |
Native mass spectrometry (nMS) and data analysis.
Purified MtThiS-COSH and His-tagged MtTfuA were buffer exchanged extensively (10,000 folds) with 3 kDa or 10 kDa MWCO Amicon Ultra centrifugal filters to 20 mM ammonium acetate, respectively. MtThiS-COSH and His-tagged MtTfuA were mixed to final concentrations of 10 μM and 5 μM, respectively, and immediately infused into a ThermoFisher Q Exactive Ultra High Mass Range (UHMR) mass spectrometer using an Advion TriVersa Nanomate. The MS was calibrated and tuned with cesium iodide. The data were acquired in positive mode using the following parameters: ionization voltage, 1.4 kV; capillary temperature, 175 °C; desolvation voltage, 125V with in-source trapping; trapping gas pressure, 2; resolution, 25,000; scan range, 2,025–5,000 m/z. The resulting data were averaged and analyzed using the Qualbrowser application of Xcalibur software and deconvoluted with BioPharma Finder v3.1 and ReSpect deconvolution algorithm (mass range: 7,000–40,000 Da; charge state range: 4–20; minimum adjacent charges: 4–8; deconvolution mass tolerance: 20 ppm).
SeMet-labeled MaTfuA expression and purification.
SeMet labeled samples of MaTfuA was produced using the methionine suppression method. A small starter culture (~10 mL) in LB media of E. coli BL21(DE3) cells containing the MBP-MaTfuA plasmid was inoculated into 1 L of M9 medium supplemented with 4 g of glucose, 120 mg of MgSO4, 4.2 mg of FeSO4, 0.5 mg of thiamine, and appropriate amount of selective antibiotic. The M9 culture was grown at 37 °C to OD600 of ~0.4 and then placed on ice for 10 min before adding six amino acids (100 mg of L-Lys, L-Phe, and L-Thr, as well as 50 mg of L-Leu, L-Ile, and L-Val). The culture was then grown at 37 °C for 15 min with shaking. 50 mg of SeMet was subsequently added, followed by additional 15-min shaking. IPTG was then added to a final concentration of 0.3 mM, followed by an induction period at 18 °C for 18 h. The SeMet-labeled MaTfuA was harvested and purified as described above. Incorporation of SeMet was confirmed by mass spectrometry.
Crystallization of MaTfuA.
The purified and concentrated tag-free MaTfuA proteins (10 mg/mL) were used for crystallization in 1:1 ratio (v/v) mixing with the precipitant solution by the hanging drop vapor diffusion. The precipitant solution consists of 100 mM sodium citrate tribasic/citric acid pH 5.5 and 40% (v/v) PEG 600. Crystals of both SeMet and native MaTfuA reached their maximum size after 3–4 days. Crystals were vitrified by direct immersion into liquid nitrogen without the addition of any cryoprotectants. All crystallographic data were collected at LS-CAT (Sector 21, Advanced Photon Source, Argonne National Labs, IL) using either an MX-300 or Eiger 9M detector. Data were integrated and scaled using autoProc52.
Phasing, structure determination and refinement of MaTfuA.
Crystallographic phases were determined using single wavelength anomalous diffraction data collected from crystals of SeMet labeled MaTfuA collected at the Se absorption edge (λ=0.97856 Å). A total of 6 Se atom sites were located using SHELLX53 and subsequent maximum likelihood refinement of heavy atom parameters was carried out using SHARP54. The resultant map (mean figure of merit of 0.322) was further improved using solvent flattening using SOLOMON and allowed for the near complete automated model building using wARP55. The preliminary model was used as a probe to determine the structure of native MaTfuA using the molecular replacement method as implemented in PHASER56. Additional cycles of manual rebuilding using COOT57, interspersed with crystallographic refinement using REFMAC558, resulted in the final model. Anisotropic B-factor refinement using Phenix was used during the final cycles of refinement and resulted in a 2% decrease in free R value. Relevant data collection, phasing, and refinement statistics may be found in Supplementary Table 2.
Fluorescence polarization (FP) binding assay.
FITC-labeled McrA peptide with an N-terminal Gly-Gly linker (GG-RLGFYGYDLQD; for clarity, these peptides do not contain any thioamides) were generated and purified as described previously36. Peptide binding to the MtYcaO and MtTfuA or its variants was assessed by equilibrium FP at 25 °C in non-binding-surface, 384-black-well polystyrene microplates (Corning) using a Synergy H4 Hybrid plate reader (BioTek) with λex/em = 485/538 nm. The data were recorded using Gen5 software. For each titration, the MBP-MtYcaO was serially diluted into the binding buffer [50 mM HEPES, pH 7.5, 300 mM NaCl, 1 mM ATP, 2.5% glycerol (v/v)], mixed with various concentration (0 to 40 μM) of MBP-MtTfuA and TEV protease (5 μM) at room temperature with shaking for 1 h. FITC-labeled McrA peptide (25 nM) was added into the protein mixture and equilibrated for 1 h with shaking at room temperature before data acquisition. Data from three independent titrations were background subtracted and fitted using a standard dose-response curve in OriginPro9.1 to estimate the dissociation constants (KD).
For testing the effect of MtTfuA variants on the peptide binding to the MtYcaO, each MBP-MtTfuA variant was serially diluted into the binding buffer, mixed with MBP-MtYcaO (1.5 μM) and TEV protease (5 μM) at room temperature with shaking for 1 h. FITC-labeled McrA peptide (25 nM) was added, equilibrated for 1 h, and the data were acquired as described above.
Data Availability Statement
We declare that all the data supporting the findings of this study are available within the manuscript, Supplementary Dataset, or Supplementary Information. Plasmids are available upon request. X-ray crystallographic coordinates were deposited at Protein Data Bank under PDB code 6XP8. Source data is provided for Fig. 1–3 and Extended Data Fig. 6, 8, 9. Public databases used in the study include Pfam (PF02624, PF07812, PF02597, PF00899, PF13407), UniProt, GenBank (AAB17515, AAB17513, WP_048176273, WP_048175617, WP_048175616, WP_048176081, WP_011023978), and Protein Data Bank (1RYJ, 2CU3, 6PEU, 4IRX, 3CNQ, 4PK9). The accession codes are also provided in the manuscript upon the first occurrence.
Extended Data
Extended Data Fig. 1. TfuA-associated RiPP structures and TfuA sequence analysis.
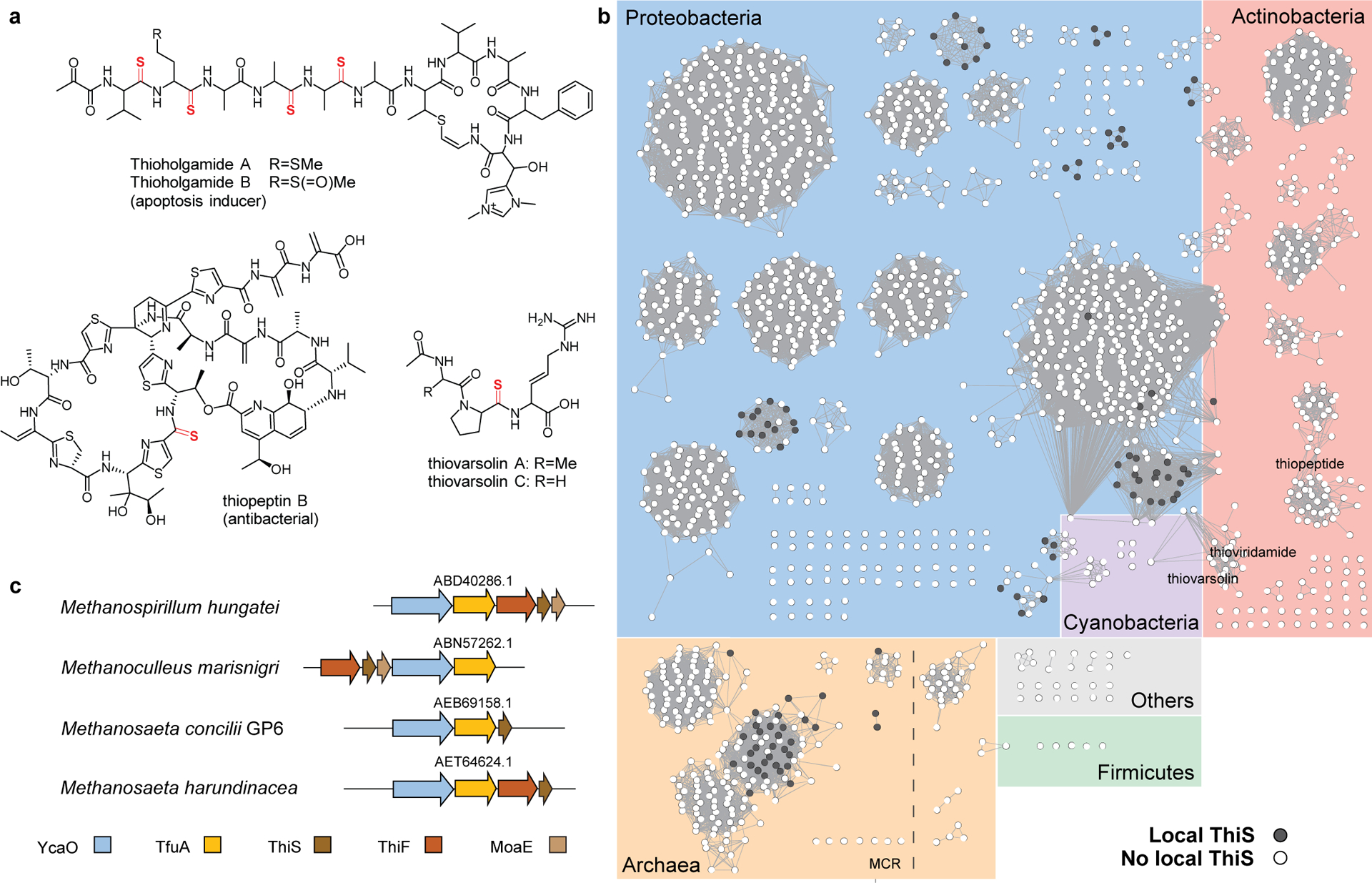
(a) Representative thioamitide structures with thioamide moiety shown in red. (b) A sequence similarity network (SSN) of the TfuA protein family PF07812 (n = 2,042 sequences) was generated with protein sequences 100% identical are conflated to a single node, and an alignment score of 60 (BLAST-P expectation value of 10−60) was used as the edge cut-off. Background is shaded based on taxonomy: archaea (orange), Proteobacteria (blue), Actinobacteria (pink), Cyanobacteria (purple), Firmicutes (green), and others (grey). Nodes representing TfuA proteins encoded within ten open-reading frames of a ThiS homolog are black. (c) Gene neighborhood diagrams for selected methanogens encoding TfuA near ThiS. NCBI accession identifiers are given for the TfuA proteins.
Extended Data Fig. 2. Correlation analysis of TfuA and ThiS from methanogens with complete genomes.
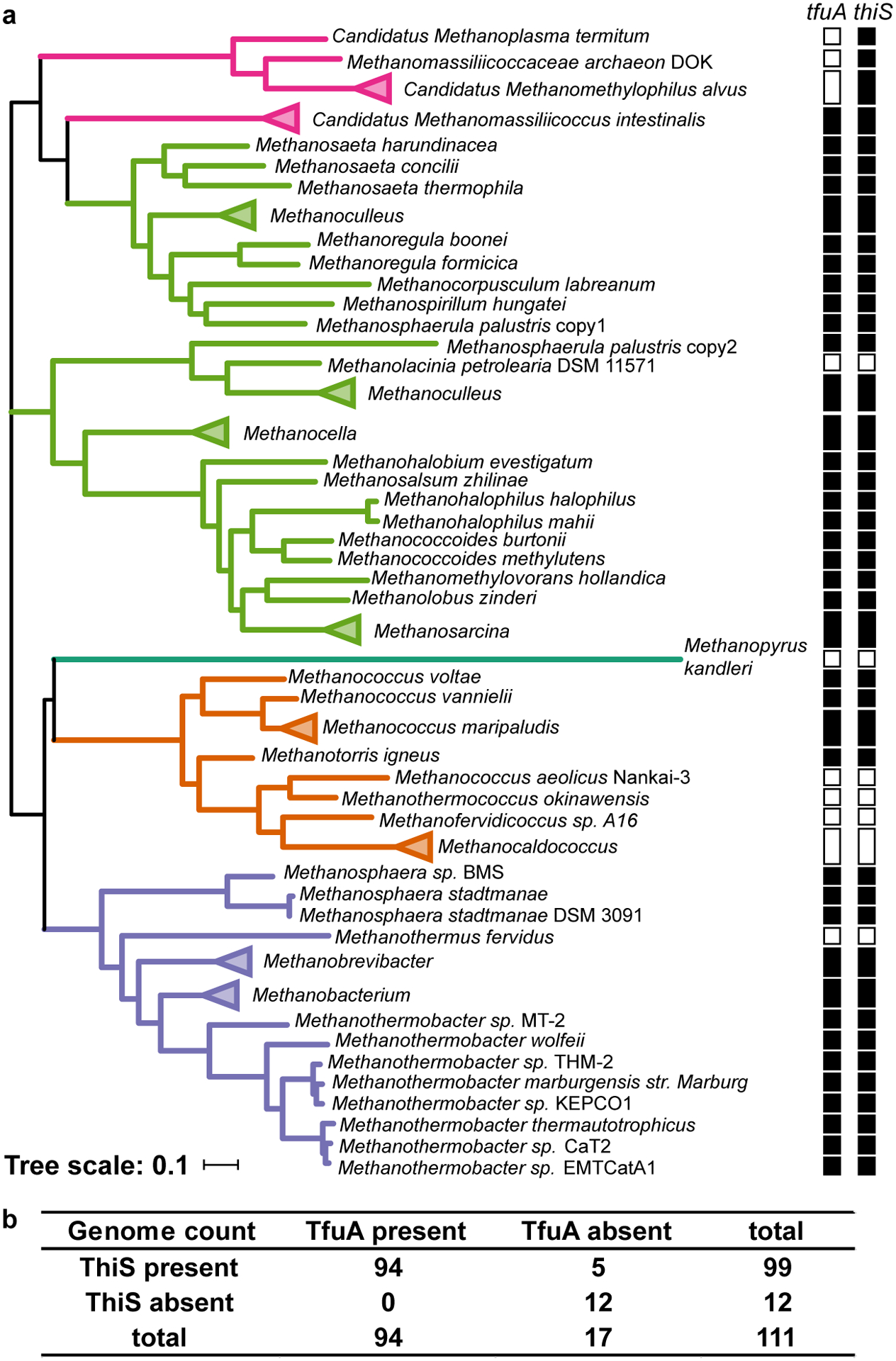
(a) Maximum-likelihood tree of YcaO proteins from methanogens (n = 111 sequences) with clades colored based on taxonomic group: Methanobacteria (purple), Methanococci (orange), Methanopyri (teal), Methanomicrobia (green), and Methanomassiliicoccales (magenta). The absence (open square) or presence (filled square) of tfuA and thiS within each genome is denoted. (b) The co-occurrence table calculated for the correlation analysis between TfuA and ThiS. The P-value calculated using Fisher’s exact test is 1.6 × 10−12.
Extended Data Fig. 3. MALDI-TOF-MS analysis of MtThiS-COSH.
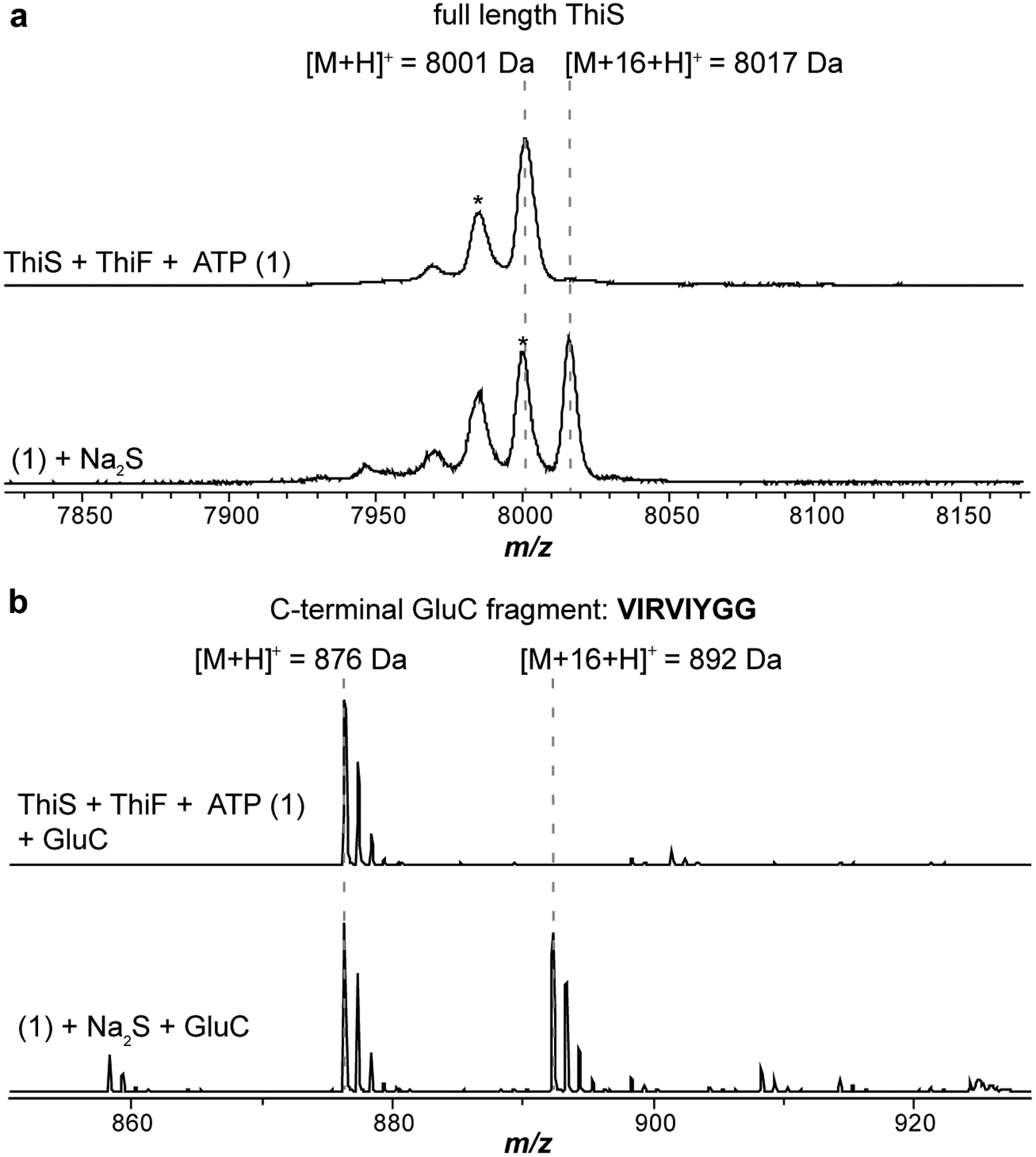
(a) Top, MS spectrum of MtThiS (m/z 8,001 Da). Bottom, MS spectrum of MtThiS-COSH (thiocarboxylated C-terminus, m/z 8,017 Da). (b) The same sample as above was digested with endoproteinase GluC. Shown is the spectral window surrounding the C-terminal peptide: VIRVIYGG.
Extended Data Fig. 4. High-resolution and tandem MS of the MtThiS-COSH C-terminal fragment.
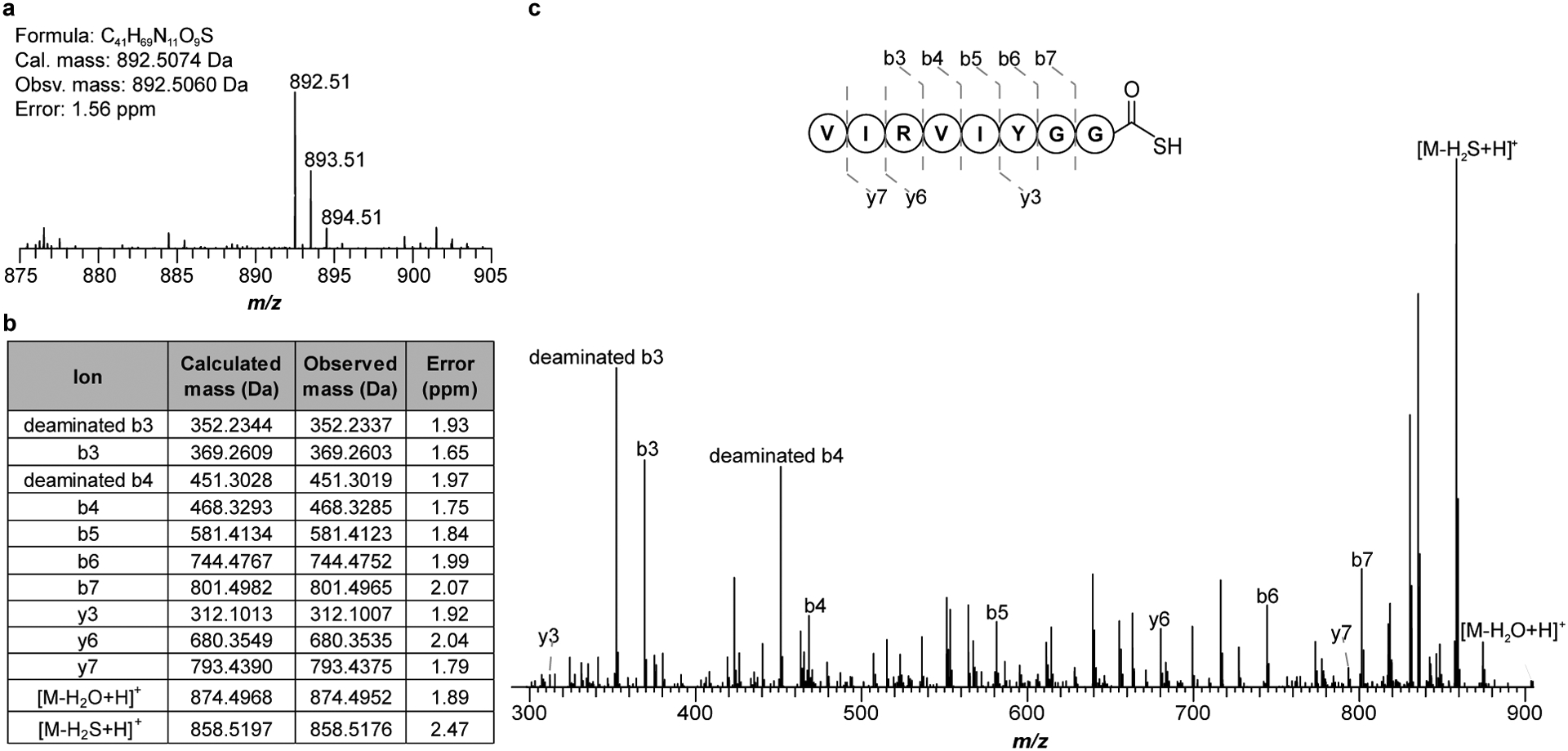
(a) High-resolution broadband spectrum of the C-terminal GluC peptide fragment of MtThiS-COSH. (b) m/z 892.51 was subjected to CID (collision-induced dissociation) with assigned ions indicated in tabular form. (c) Tandem mass spectrum (MS/MS) confirming the location of the +16 Da mass change to the C-terminus. CID also promotes the formal loss of H2S (obsv. Δm/z 33.9884 Da; calc. Δm/z 33.9877 Da) from the parent ion.
Extended Data Fig. 5. High-resolution and tandem MS of the thioamidated McrA peptide.
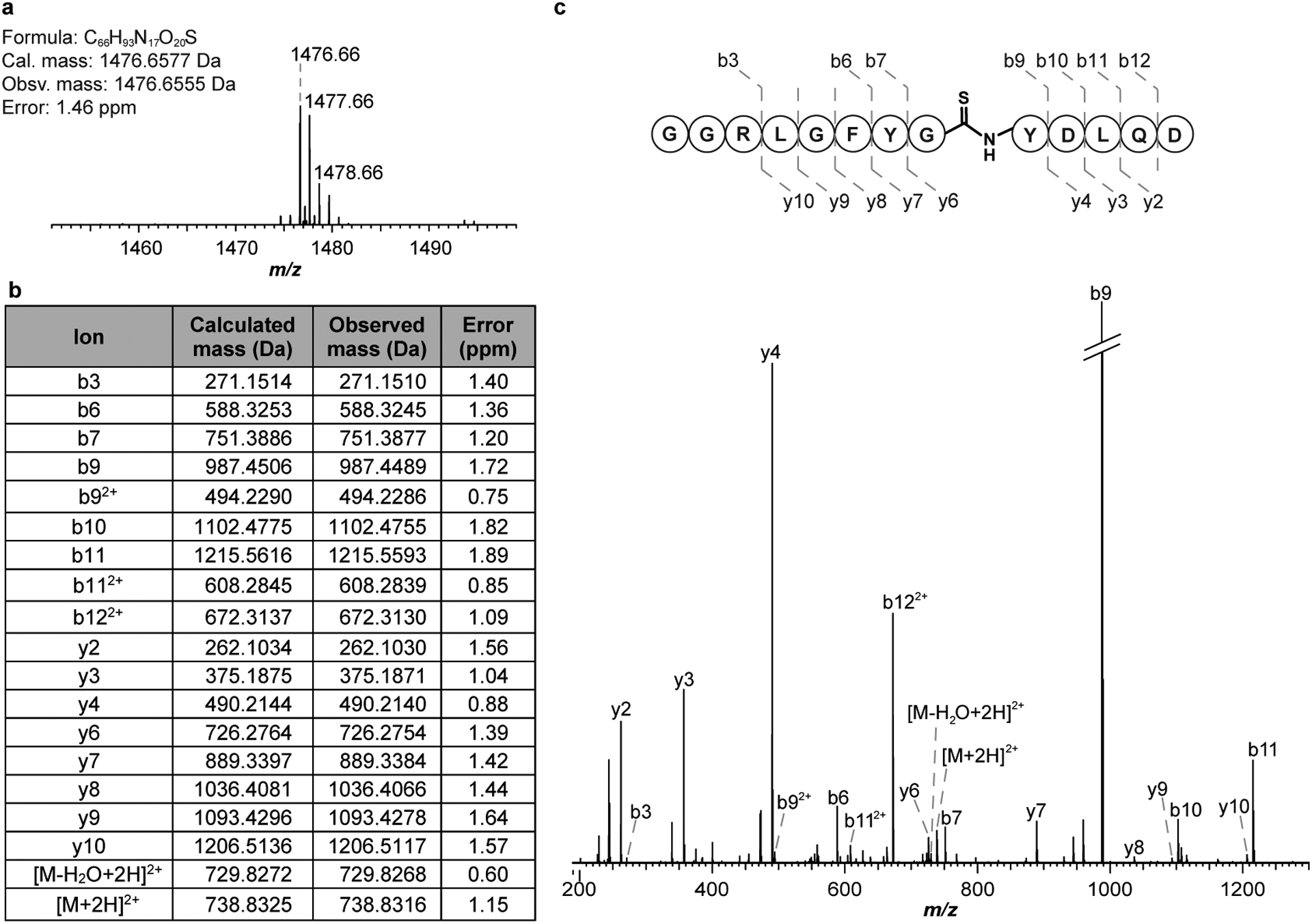
(a) The thioamidated McrA peptide (GG-RLGFYGYDLQD) was characterized by HRMS. (b) m/z 1,476.66 was subjected to CID with assigned ions indicated in tabular form. (c) MS/MS spectrum confirming the location of the +16 Da mass change to the central Gly residue. Thioamide bond cleavage was not observed under the applied CID conditions.
Extended Data Fig. 6. TfuA catalyzes ThiS-COSH hydrolysis to generate sulfide.
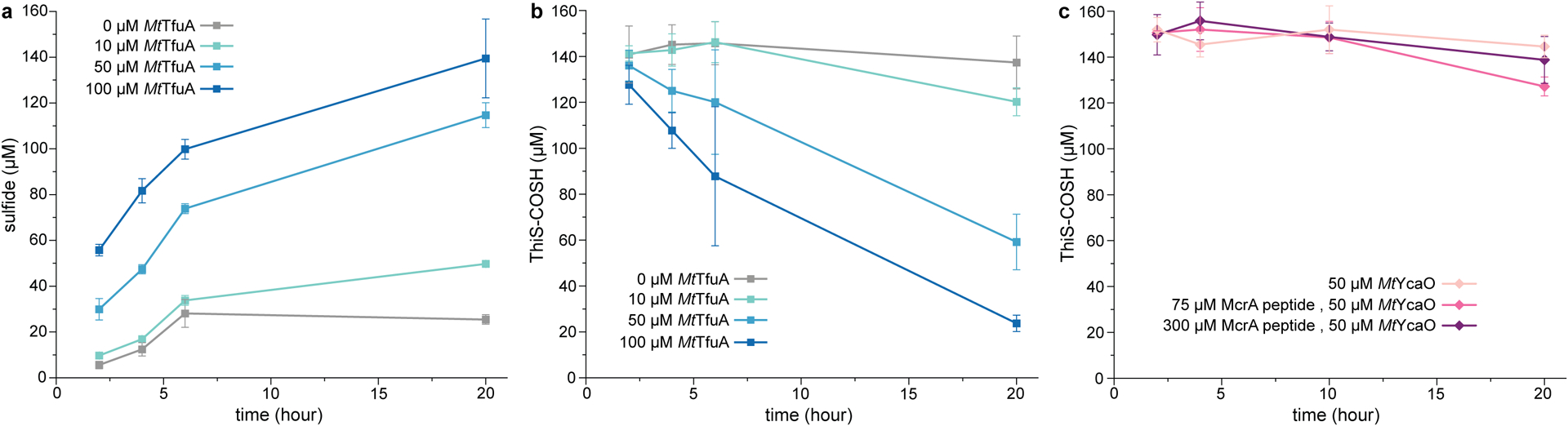
Quantification of sulfide (a) and MtThiS-COSH (b) via fluorescence detection at different time points of the reaction between MtThiS-COSH (150 μM) and MtTfuA of various concentrations. Sulfide concentrations were measured via reaction with 7-amino-4-methylcoumarin followed by fluorescence quantification. ThiS-COSH concentrations were determined using the LRSA-based assay. Data are presented as mean values ± SD (n = 3 independent experiments) (c) Fluorescence quantification of MtThiS-COSH at different time points after reaction with 50 μM MtYcaO, 5 mM ATP, and McrA peptide of various concentrations. Data are presented as mean values ± SD (n = 3 independent experiments)
Extended Data Fig. 7. McThiS 1H,15N-HMQC spectra upon MtTfuA titration.
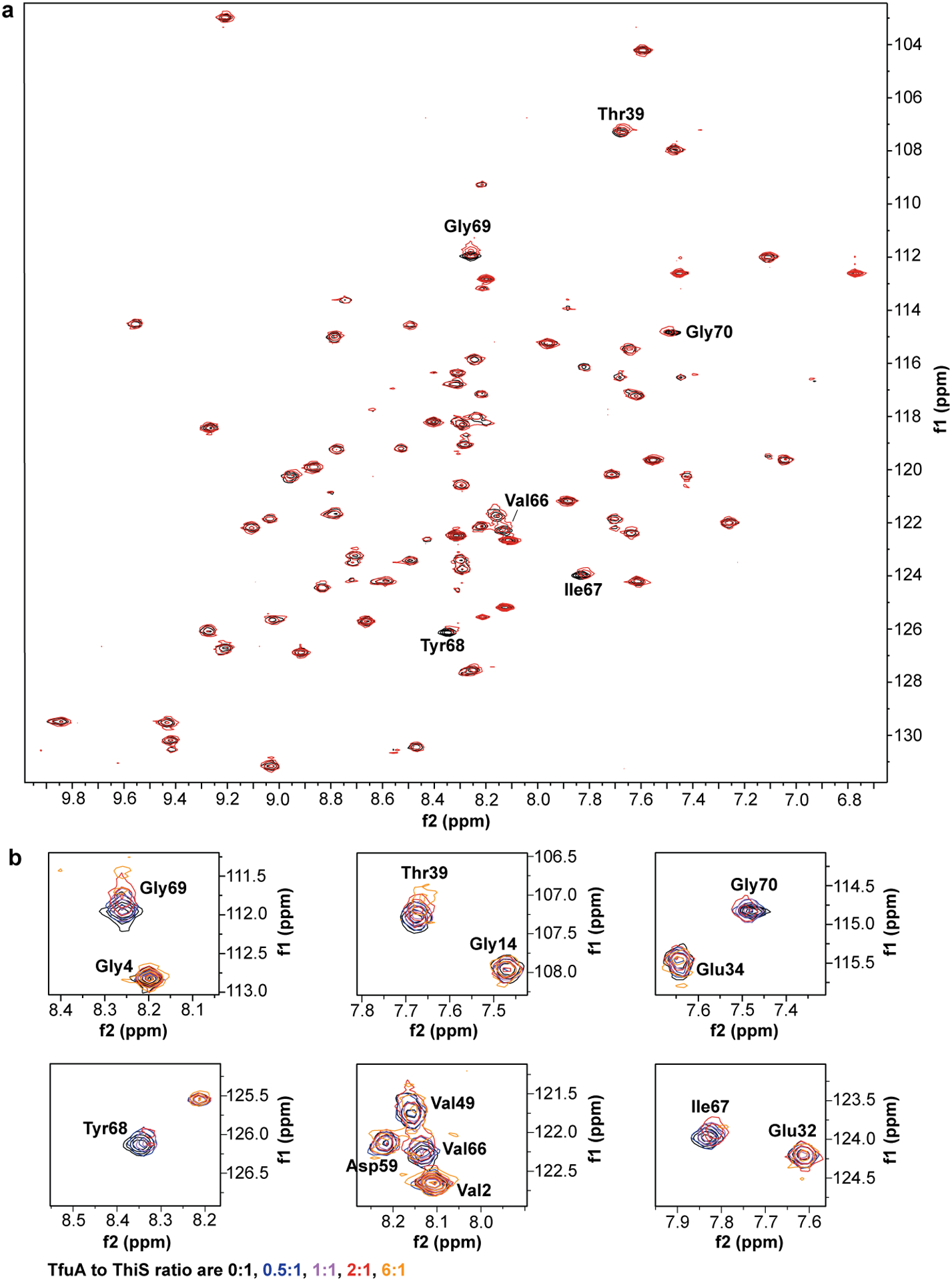
(a) An overlay of McThiS 1H,15N-HMQC spectra in the presence (red) and absence (black) of MtTfuA (2 equiv.). Residues with significant chemical shift perturbations are labeled. (b) Zoomed regions of the spectrum showing McThiS residues with the largest chemical shift changes. The MtTfuA: McThiS ratio varies from 0:1 to 6:1 equiv. (MtTfuA concentrations vary from 110 μM to 1.2 mM).
Extended Data Fig. 8. SDS-PAGE analysis of AzoYcaO co-purifying with His-AzoTfuA.
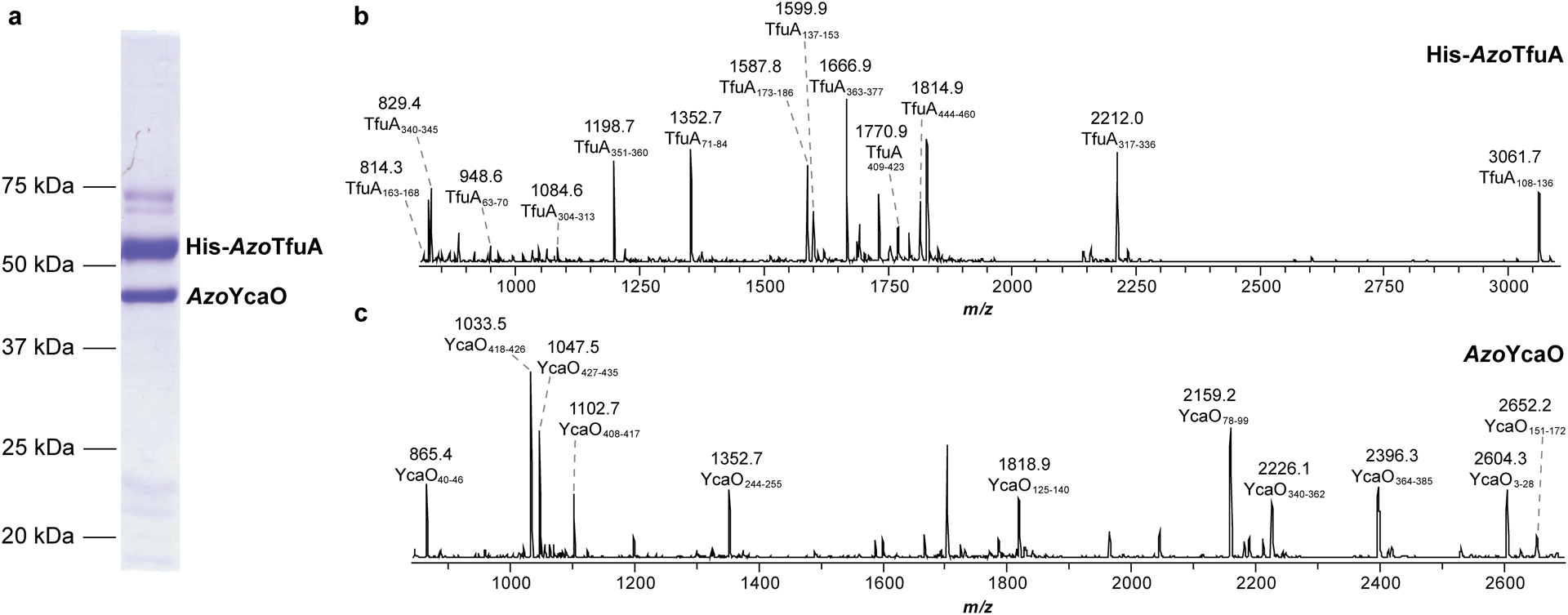
(a) A Coomassie-stained SDS-PAGE gel showing untagged AzoYcaO (BAI72909.1) and His6-AzoTfuA (BAI72908.1) from Azospirillum sp. B510 co-expressed heterologously and co-purified using Ni-NTA affinity chromatography. In-gel trypsin digestion and subsequent MS analysis was performed to confirm the identity of the suspected His6-AzoTfuA (b) and AzoYcaO (c) bands. MALDI-TOF mass spectra of the tryptic peptides are labeled with subscripts corresponding native amino acid sequence covered. The identified tryptic fragments cover ~40% of the total protein sequence for both proteins.
Extended Data Fig. 9. The presence of ATP alters the outcome of ThiS-COSH hydrolysis.
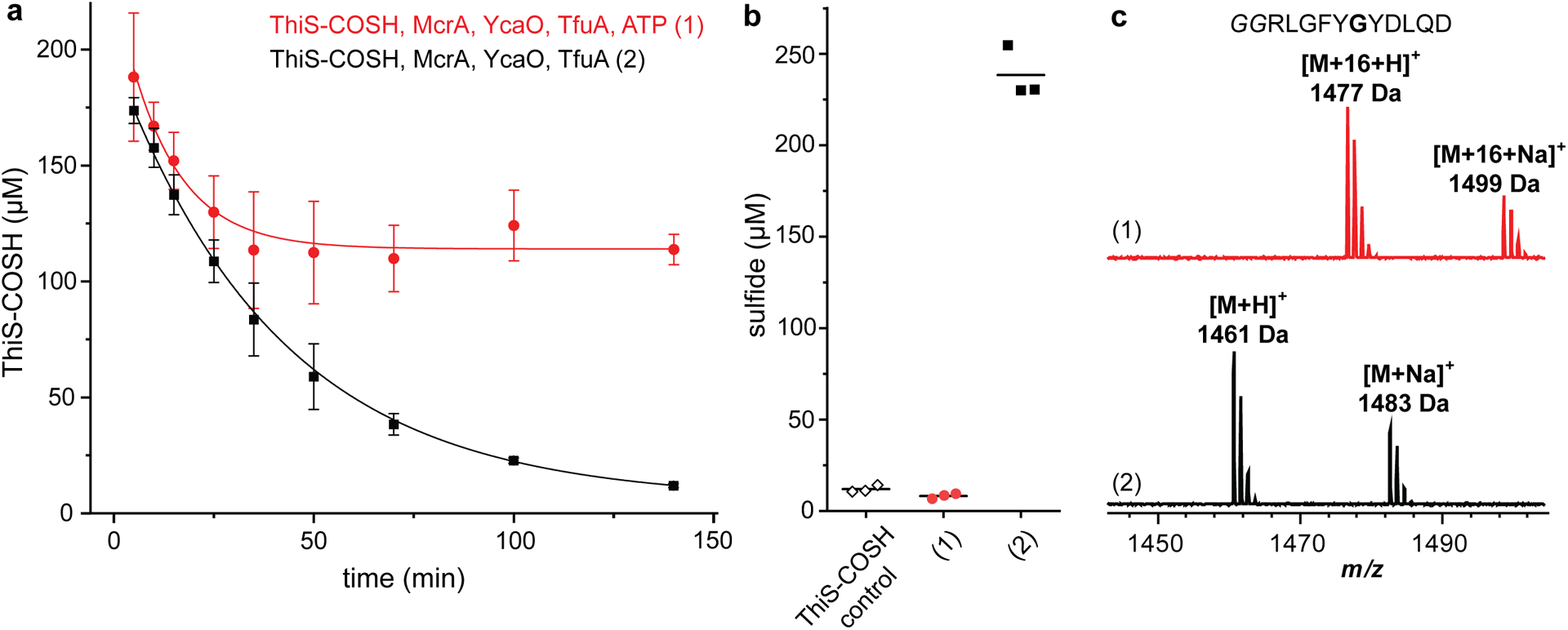
(a) Fluorescence quantification of MtThiS-COSH (initially 200 μM) upon reaction with MtTfuA (3 μM), MtYcaO (3 μM), and McrA peptide (100 μM), in the presence (red circles) or absence (black squares) of ATP (5 mM). Data are presented as mean values ± SD (n = 3 independent experiments). An exponential decay model was used to fit the data. (b) Fluorescence quantification of sulfide production for MtThiS-COSH control and reactions in panel a at 140 min. Individual data points (n = 3 independent experiments) and mean values (lines) are presented. (c) MALDI-TOF mass spectra of the McrA peptide from reactions in panel a at 140 min.
Extended Data Fig. 10. Structural comparison of MaTfuA and proteins with partial structural similarity.
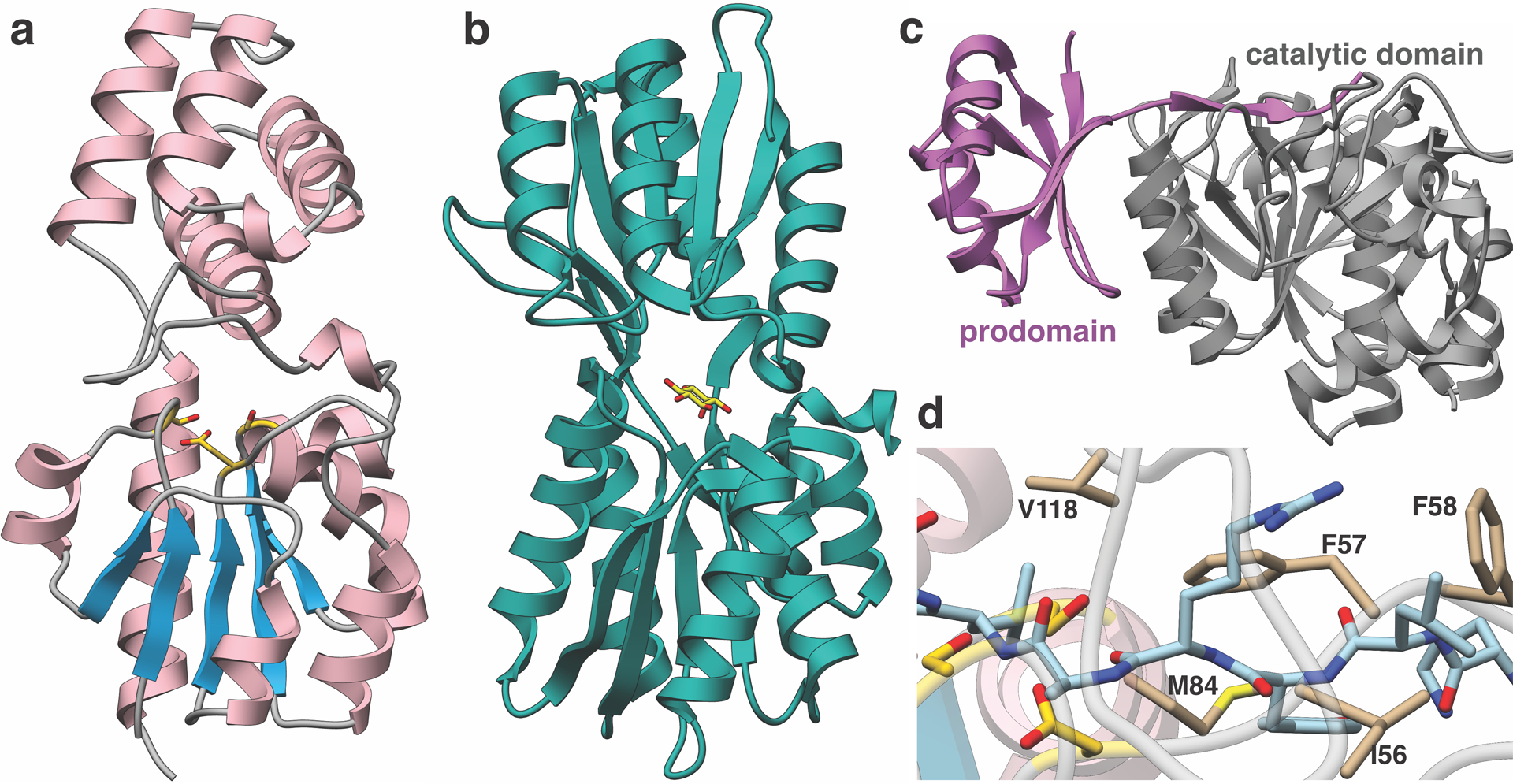
(a) Overall structure of MaTfuA. (b) Structure of myo-inositol-binding protein (PDB code: 4IRX) bound to its substrate (yellow stick) shows similarities with the α/β fold of MaTfuA (c) Structure of subtilisin BPN’ in complex with its prodomain (PDB code: 3CNQ) shows similarities with the α/β fold of MaTfuA. (d) Close-up view of the superimposition between MaTfuA and 3CNQ shows the putative binding pocket for a peptide substrate. Residues that form the presumptive binding pocket are shown as tan (stick); the prodomain bound by subtilisin BPN’ is shown in blue (stick).
Supplementary Material
Acknowledgements
We thank Drs. Lingyang Zhu and Justin Arrington for assistance with the NMR and nMS experiments, respectively. We thank Xiao Rui Guo from the Mitchell group for acquiring the high-resolution mass spectral data. This work was supported in part by National Institutes of Health Grant GM097142 (to D.A.M.), GM131347 (to S.K.N.) and the Alice Helm Graduate Research Excellence Fellowship in Microbiology (to A.L.). The Bruker UltrafleXtreme MALDI TOF/TOF mass spectrometer was purchased in part with a grant from the National Institutes of Health (S10 RR027109 A).
Footnotes
Competing Financial Interests Statement
The authors declare no competing financial interest.
References
- 1.El-Gebali S et al. The Pfam protein families database in 2019. Nucleic Acids Res 47, D427–D432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.UniProt Consortium, T. UniProt: the universal protein knowledgebase. Nucleic Acids Res 46, 2699 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunbar KL, Melby JO & Mitchell DA YcaO domains use ATP to activate amide backbones during peptide cyclodehydrations. Nat. Chem. Biol 8, 569–575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burkhart BJ, Schwalen CJ, Mann G, Naismith JH & Mitchell DA YcaO-Dependent Posttranslational Amide Activation: Biosynthesis, Structure, and Function. Chem. Rev 117, 5389–5456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunbar KL et al. Discovery of a new ATP-binding motif involved in peptidic azoline biosynthesis. Nat. Chem. Biol 10, 823–829 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunbar KL, Tietz JI, Cox CL, Burkhart BJ & Mitchell DA Identification of an Auxiliary Leader Peptide-Binding Protein Required for Azoline Formation in Ribosomal Natural Products. J. Am. Chem. Soc 137, 7672–7677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burkhart BJ, Hudson GA, Dunbar KL & Mitchell DA A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nat. Chem. Biol 11, 564–570 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koehnke J et al. Structural analysis of leader peptide binding enables leader-free cyanobactin processing. Nat. Chem. Biol 11, 558–563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montalbán-López M et al. New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep (2020) doi: 10.1039/D0NP00027B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwalen CJ, Hudson GA, Kille B & Mitchell DA Bioinformatic Expansion and Discovery of Thiopeptide Antibiotics. J. Am. Chem. Soc 140, 9494–9501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nayak DD, Mahanta N, Mitchell DA & Metcalf WW Post-translational thioamidation of methyl-coenzyme M reductase, a key enzyme in methanogenic and methanotrophic Archaea. eLife 6, e29218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahanta N, Liu A, Dong S, Nair SK & Mitchell DA Enzymatic reconstitution of ribosomal peptide backbone thioamidation. Proc. Natl. Acad. Sci 115, 3030–3035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson ZL et al. Structure of the bacterial ribosome at 2 Å resolution. eLife 9, e60482 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahanta N, Szantai-Kis DM, Petersson EJ & Mitchell DA Biosynthesis and Chemical Applications of Thioamides. ACS Chem. Biol 14, 142–163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Izawa M, Kawasaki T & Hayakawa Y Cloning and Heterologous Expression of the Thioviridamide Biosynthesis Gene Cluster from Streptomyces olivoviridis. Appl. Environ. Microbiol 79, 7110–7113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frattaruolo L, Lacret R, Cappello AR & Truman AW A Genomics-Based Approach Identifies a Thioviridamide-Like Compound with Selective Anticancer Activity. ACS Chem. Biol 12, 2815–2822 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Kjaerulff L et al. Thioholgamides: Thioamide-Containing Cytotoxic RiPP Natural Products. ACS Chem. Biol 12, 2837–2841 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Santos-Aberturas J et al. Uncovering the unexplored diversity of thioamidated ribosomal peptides in Actinobacteria using the RiPPER genome mining tool. Nucleic Acids Res 47, 4624–4637 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breil B, Borneman J & Triplett EW A newly discovered gene, tfuA, involved in the production of the ribosomally synthesized peptide antibiotic trifolitoxin. J. Bacteriol 178, 4150–4156 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ermler U, Grabarse W, Shima S, Goubeaud M & Thauer RK Crystal Structure of Methyl-Coenzyme M Reductase: The Key Enzyme of Biological Methane Formation. Science 278, 1457–1462 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Scheller S, Goenrich M, Boecher R, Thauer RK & Jaun B The key nickel enzyme of methanogenesis catalyses the anaerobic oxidation of methane. Nature 465, 606–608 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Nayak DD et al. Functional interactions between posttranslationally modified amino acids of methyl-coenzyme M reductase in Methanosarcina acetivorans. PLOS Biol 18, e3000507 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kahnt J et al. Post-translational modifications in the active site region of methyl-coenzyme M reductase from methanogenic and methanotrophic archaea. FEBS J 274, 4913–4921 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Sieprawska-Lupa M, Whitman WB & White RH Cysteine Is Not the Sulfur Source for Iron-Sulfur Cluster and Methionine Biosynthesis in the Methanogenic Archaeon Methanococcus maripaludis. J. Biol. Chem 285, 31923–31929 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lomans BP et al. Isolation and Characterization of Methanomethylovorans hollandica gen. nov., sp. nov., Isolated from Freshwater Sediment, a Methylotrophic Methanogen Able To Grow on Dimethyl Sulfide and Methanethiol. Appl. Environ. Microbiol 65, 3641–3650 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maupin-Furlow JA Ubiquitin-like proteins and their roles in archaea. Trends Microbiol 21, 31–38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehmann C, Begley TP & Ealick SE Structure of the Escherichia coli ThiS-ThiF Complex, a Key Component of the Sulfur Transfer System in Thiamin Biosynthesis,. Biochemistry 45, 11–19 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorrestein PC, Zhai H, McLafferty FW & Begley TP The Biosynthesis of the Thiazole Phosphate Moiety of Thiamin: The Sulfur Transfer Mediated by the Sulfur Carrier Protein ThiS. Chem. Biol 11, 1373–1381 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Rudolph MJ, Wuebbens MM, Rajagopalan KV & Schindelin H Crystal structure of molybdopterin synthase and its evolutionary relationship to ubiquitin activation. Nat. Struct. Biol 8, 42–46 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Leidel S et al. Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature 458, 228–232 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Chen M et al. The [4Fe-4S] cluster of sulfurtransferase TtuA desulfurizes TtuB during tRNA modification in Thermus thermophilus. Commun. Biol 3, 1–9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krishnamoorthy K & Begley TP Protein Thiocarboxylate-Dependent Methionine Biosynthesis in Wolinella succinogenes. J. Am. Chem. Soc 133, 379–386 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong L-B et al. Biosynthesis of thiocarboxylic acid-containing natural products. Nat. Commun 9, 2362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zallot R, Oberg N & Gerlt JA The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 58, 4169–4182 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borrel G et al. Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genomics 15, 679 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong S-H, Liu A, Mahanta N, Mitchell DA & Nair SK Mechanistic Basis for Ribosomal Peptide Backbone Modifications. ACS Cent. Sci 5, 842–851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krishnamoorthy K & Begley TP Reagent for the Detection of Protein Thiocarboxylates in the Bacterial Proteome: Lissamine Rhodamine B Sulfonyl Azide. J. Am. Chem. Soc 132, 11608–11612 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen B et al. Fluorescent probe for highly selective and sensitive detection of hydrogen sulfide in living cells and cardiac tissues. The Analyst 138, 946–951 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Yee A et al. An NMR approach to structural proteomics. Proc. Natl. Acad. Sci 99, 1825–1830 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holm L DALI and the persistence of protein shape. Protein Sci 29, 128–140 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herrou J & Crosson S myo-inositol and D-ribose ligand discrimination in an ABC periplasmic binding protein. J. Bacteriol 195, 2379–2388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruan B, London V, Fisher KE, Gallagher DT & Bryan PN Engineering Substrate Preference in Subtilisin: Structural and Kinetic Analysis of a Specificity Mutant. Biochemistry 47, 6628–6636 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Ollis DL et al. The α/β hydrolase fold. Protein Eng. Des. Sel 5, 197–211 (1992). [Google Scholar]
- 44.Zimmermann L et al. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol 430, 2237–2243 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Rydel TJ et al. The Crystal Structure, Mutagenesis, and Activity Studies Reveal that Patatin Is a Lipid Acyl Hydrolase with a Ser-Asp Catalytic Dyad ‡. Biochemistry 42, 6696–6708 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Mueller EG Trafficking in persulfides: delivering sulfur in biosynthetic pathways. Nat. Chem. Biol 2, 185–194 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Ekici ÖD, Paetzel M & Dalbey RE Unconventional serine proteases: Variations on the catalytic Ser/His/Asp triad configuration. Protein Sci 17, 2023–2037 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miranda HV et al. E1- and ubiquitin-like proteins provide a direct link between protein conjugation and sulfur transfer in archaea. Proc. Natl. Acad. Sci 108, 4417–4422 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shannon P et al. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price MN, Dehal PS & Arkin AP FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLOS ONE 5, e9490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Letunic I & Bork P Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47, W256–W259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vonrhein C et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr 67, 293–302 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Usón I & Sheldrick GM An introduction to experimental phasing of macromolecules illustrated by SHELX; new autotracing features. Acta Crystallogr. Sect. Struct. Biol 74, 106–116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vonrhein C, Blanc E, Roversi P & Bricogne G Automated structure solution with autoSHARP. Methods Mol. Biol. Clifton NJ 364, 215–230 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Alharbi E, Bond PS, Calinescu R & Cowtan K Comparison of automated crystallographic model-building pipelines. Acta Crystallogr. Sect. Struct. Biol 75, 1119–1128 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Oeffner RD et al. On the application of the expected log-likelihood gain to decision making in molecular replacement. Acta Crystallogr. Sect. Struct. Biol 74, 245–255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Casañal A, Lohkamp B & Emsley P Current developments in Coot for macromolecular model building of Electron Cryo-microscopy and Crystallographic Data. Protein Sci. Publ. Protein Soc 29, 1069–1078 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kovalevskiy O, Nicholls RA, Long F, Carlon A & Murshudov GN Overview of refinement procedures within REFMAC5: utilizing data from different sources. Acta Crystallogr. Sect. Struct. Biol 74, 215–227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We declare that all the data supporting the findings of this study are available within the manuscript, Supplementary Dataset, or Supplementary Information. Plasmids are available upon request. X-ray crystallographic coordinates were deposited at Protein Data Bank under PDB code 6XP8. Source data is provided for Fig. 1–3 and Extended Data Fig. 6, 8, 9. Public databases used in the study include Pfam (PF02624, PF07812, PF02597, PF00899, PF13407), UniProt, GenBank (AAB17515, AAB17513, WP_048176273, WP_048175617, WP_048175616, WP_048176081, WP_011023978), and Protein Data Bank (1RYJ, 2CU3, 6PEU, 4IRX, 3CNQ, 4PK9). The accession codes are also provided in the manuscript upon the first occurrence.