

Abstract
GPCR-regulated cAMP production from endosomes can specify signaling to the nucleus by moving the source of cAMP without changing its amount. How this is possible remains unknown because cAMP gradients dissipate over the nanoscale whereas endosomes typically localize microns from the nucleus. We show that the key location-dependent step for endosome-encoded transcriptional control is nuclear entry of cAMP-dependent protein kinase (PKA) catalytic subunits. These are sourced from punctate accumulations of PKA holoenzyme that are densely distributed in the cytoplasm and titrated by global cAMP into a discrete metastable state, in which catalytic subunits are bound but dynamically exchange. Mobile endosomes containing activated receptors collide with the metastable PKA puncta and pause in close contact. We propose that these properties enable cytoplasmic PKA to act collectively like a semiconductor, converting nanoscale cAMP gradients generated from endosomes into microscale elevations of free catalytic subunit to direct downstream signaling.
INTRODUCTION
G protein-coupled receptors (GPCRs) comprise nature’s largest family of signaling receptors, regulate essentially every physiological process and define an important class of therapeutic targets. Rhodopsin, a light-activated GPCR, was long-recognized to couple to heterotrimeric G proteins on intracellular membranes1. Until recently, ligand-activated GPCRs were widely believed to lack this ability and activate heterotrimeric G proteins exclusively from the plasma membrane. Evidence accumulating over the past decade indicates that ligand-activated GPCRs can also initiate G protein signaling from internal membranes, including endosomes, and that receptor activation from internal membranes can produce distinct downstream effects relative to activation from the plasma membrane2–7. Although this concept is now well established, much remains unknown about how intracellular GPCR-G protein activation achieves distinct downstream effects. How this occurs is particularly problematic for signal transduction that is communicated downstream by diffusible cytoplasmic mediators such as cAMP.
One way to distinguish signaling from endosomes is to change the kinetics of global cytoplasmic cAMP elevation, using endosomes or endosome-associated membranes to sustain cAMP production and the plasma membrane to generate cAMP transiently. Such a kinetic encoding scheme has been demonstrated for polypeptide hormone receptors that have a prolonged residence time in the endocytic network and are thought to form stable signaling complexes there8–11. Using endocytosis to sustain global cAMP elevation is advantageous in some physiological contexts, such as producing stable responses to pulsatile hormone release12 or driving cAMP diffusion across cell boundaries in tissues13, but it has severe limitations in other contexts because cells lose the ability to respond to temporal fluctuations of ligand.
Signaling by the human beta-2 adrenergic receptor (β2AR) provides an example. This GPCR responds physiologically to changes in extracellular catecholamine concentration that span a wide range of time scales, through sequential bouts of Gs-coupled stimulation of cAMP production from the plasma membrane and then endosomes after agonist-induced receptor internalization14. Unlike polypeptide hormone receptors which are stably sequestered after endocytosis, β2ARs recycle efficiently and cycle continuously in the prolonged presence of agonist15, thus activating Gs transiently from each location during iterative trafficking cycles16. Endosomal activation has only a small effect on global cAMP elevation but is essential for efficient downstream signaling to cAMP-dependent gene transcription. Further, signaling to the nucleus has been explicitly demonstrated to occur as a consequence of moving the subcellular location of cAMP production rather than changing the total amount of cAMP produced17,18.
The existence of such ‘location-encoded’ signaling has been clearly demonstrated experimentally but remains problematic from first principles. The reason is a profound discordance between distance scales. Diffusion inherently limits the range of local cAMP gradients around endosomes to a sub-micron distance scale, but endosomes that are known sites of internal β2AR-Gs activation14 and regulated synthesis of cAMP19 are typically located much farther from the nucleus (Supplementary Fig. 1). This raises the fundamental unanswered question of how endosomal cAMP is able to communicate location-encoded information over a distance that far exceeds the range of local gradients.
Here we propose a simple solution to this problem which emerged from systematically investigating the cellular basis for endocytosis-dependent β2AR signaling to the nucleus via the cAMP cascade, and in a cell model expressing all relevant proteins at endogenous levels.
RESULTS
A cell model for location-encoded cAMP signaling
We verified that endocytic blockade has little effect on the global cytoplasmic cAMP increase produced by endogenous β2AR activation in HEK293 cells using a previously described luminescence-based cAMP biosensor14,20, and added to this system an IRES-linked Renilla luciferase cassette to verify reliability of the small inhibition that was observed (see Methods). In contrast to its relatively weak effect on global cAMP elevation, endocytic blockade strongly suppressed induction of the major cAMP-dependent target gene PCK1 (Fig. 1a,b, Extended Data Fig. 1a). We confirmed this selectivity using three independent methods of endocytic blockade (Extended Data Fig. 1b,c and Supplementary Fig. 2a–d). We further verified, by agonist titration, that the small reduction of global cAMP elevation produced by endocytic blockade cannot account for this pronounced suppression of transcriptional signaling (Fig. 1c and Extended Data Fig. 1d–g).
Figure 1.
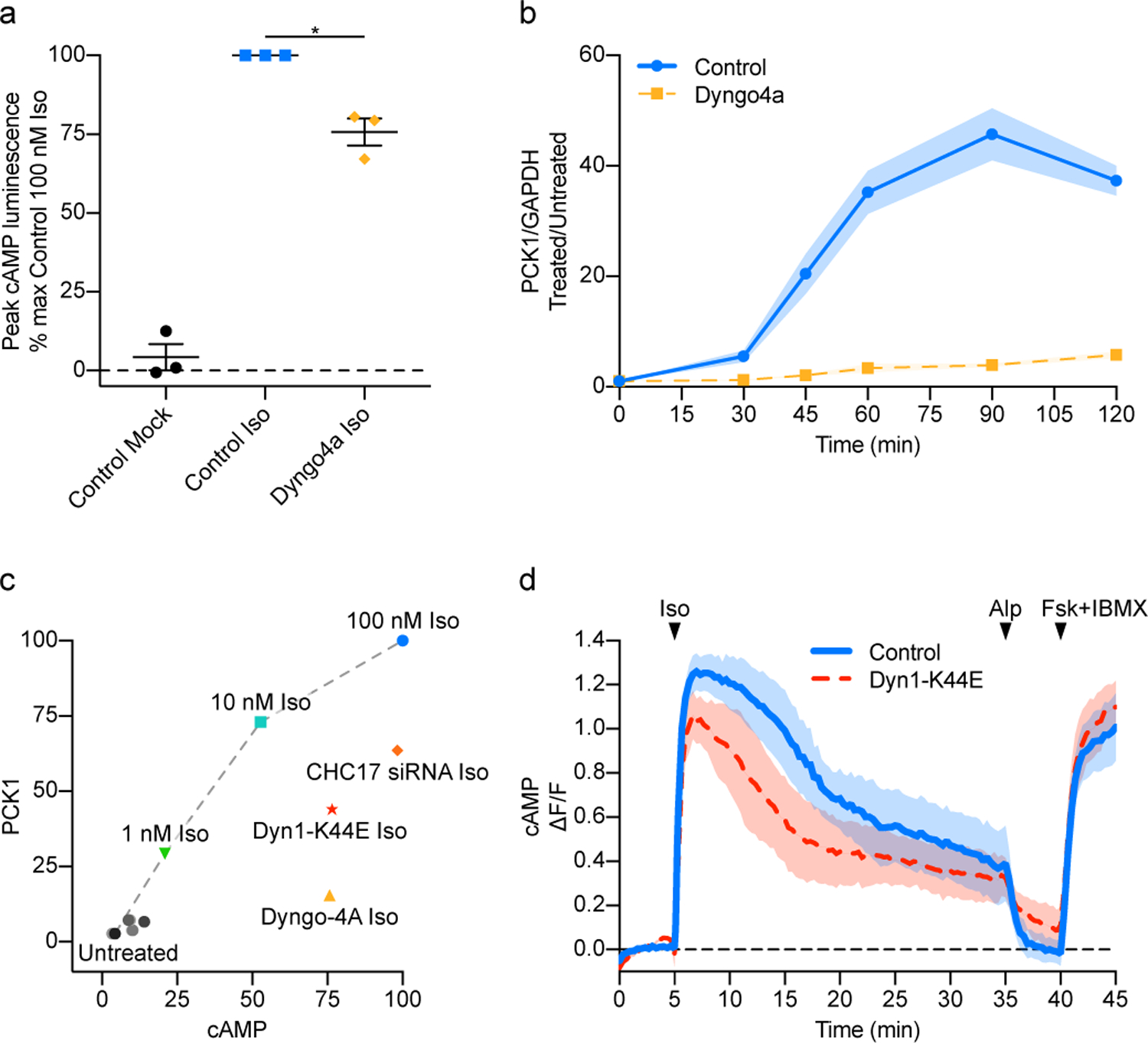
β2AR-induced transcriptional signaling is endocytosis-dependent and independent of magnitude and kinetics. a and b, HEK293 cells were pretreated with either DMSO (control) or chemical inhibitor of dynamin (30 μM Dyngo4a) and followed by either no treatment (mock) or 100 nM Iso treatment. a, Endocytic blockade by Dyngo4a reduces β2AR-stimulated cAMP. The maximum luminescence value measured in cells expressing the cAMP luciferase biosensor was determined and normalized to the control Iso condition. (n = 3 with ≥ two technical replicates per biological replicate, p = 0.0048, two-tailed unpaired t-test). b, Quantitative RT-PCR for PCK1 and GAPDH transcript levels was performed on samples treated with 100 nM Iso for two hours. (DMSO vs 30 μM Dyngo4a, n = 4 with 3 technical replicates per biological replicate, Interaction, Time and Pretreatment p < 0.0001; p < 0.0001 for times 45, 60, 90, and 120 minutes with Sidak’s multiple comparisons test, two-way ANOVA). c, Endocytic inhibition results in a smaller reduction in overall cAMP than PCK1 induction. Comparing overall cAMP and PCK1 induction among different endocytic inhibition methods (mutant dynamin1, siRNA depletion of clathrin, pharmacological inhibitor of dynamin) and different Iso doses. Respective untreated control conditions are shown in black or gray circles in the bottom left of the plot. Gray dotted line connects Iso dose conditions. d, Kinetics of cAMP production over time. Time course of HEK293 cells co-expressing the cAMP fluorescence biosensor GG4B and pcDNA3 (control) or mCherry-Dyn1-K44E (control, n = 4; Dyn1-K44E n = 3, cells ≥ 13 per biological replicate; Interaction p = 0.9934, p < 0.0001 for Time and Transfection, two-way ANOVA). All data are mean ± sem (shaded areas).
β2AR activation elicits a biphasic global cAMP response
We developed a cAMP biosensor capable of higher temporal resolution by fusing a high-affinity cAMP-binding domain derived from human Epac2 to a circularly permuted green fluorescent protein and screening for variants with favorable properties (Supplementary Fig. 2e). The selected biosensor (GG4B) detected a rapid cAMP increase after Iso application that peaked within several minutes after initial application and decayed over 5–10 minutes thereafter in the continued presence of agonist (Fig. 1d, blue line). Calibrated ∆F/F determination (Supplementary Fig. 2f) indicated that global cAMP during this ‘peak’ signaling phase briefly exceeds ~5 μM.
The cytoplasmic cAMP concentration did not return fully to baseline in the prolonged presence of agonist. Rather, it remained elevated at a lower level. This was not an artifact of incomplete sensor reversibility because alprenolol (Alp, 10 µM), a competitive β2AR antagonist, caused the fluorescence response to rapidly return to baseline. Calibrated ΔF/F determination estimated the cAMP concentration maintained during this ‘plateau’ phase in the range of 100–300 nM.
Neither phase of the endogenous β2AR-stimulated elevation of cytoplasmic cAMP required endocytosis. Endocytic inhibition imposed by Dyn1-K44E fused to mCherry (we avoided Dyngo4a due to interfering autofluorescence) slightly lowered the peak phase of global cAMP elevation and moderately shortened its duration, but we observed little change in the plateau phase (Fig. 1d, red line) or overall kinetics of cAMP fluctuations (Extended Data Fig. 1h).
Nuclear accumulation of PKAcat is endocytosis dependent
We next assessed phosphorylation of the cAMP response element binding protein at Ser133 (pCREB), a key intermediate step in the transcriptional transduction pathway downstream of cAMP-dependent protein kinase (PKA)21. In contrast to the relatively small effect of endocytic blockade on global cytoplasmic cAMP elevation, the CREB phosphorylation was markedly suppressed after endocytic inhibition imposed either by Dyn1-K44E (Fig. 2a) or clathrin knockdown (Fig. 2b). Multiple cellular mechanisms have been proposed to explain CREB phosphorylation by PKA, including nuclear entry of PKA catalytic subunits (PKAcat) after cAMP-driven dissociation from holoenzyme in the cytoplasm and direct activation of a nucleus-restricted pool of PKA holoenzyme. Of these, nuclear entry of PKAcat has a generally higher sensitivity for cAMP and slower kinetics22,23. Because endocytic blockade primarily suppressed endogenous CREB phosphorylation observed at later time points after agonist application, and clearly after the global cAMP concentration had fallen to its lower plateau (Fig. 1d), we hypothesized that endosomal cAMP production promotes nuclear entry of PKAcat.
Figure 2.
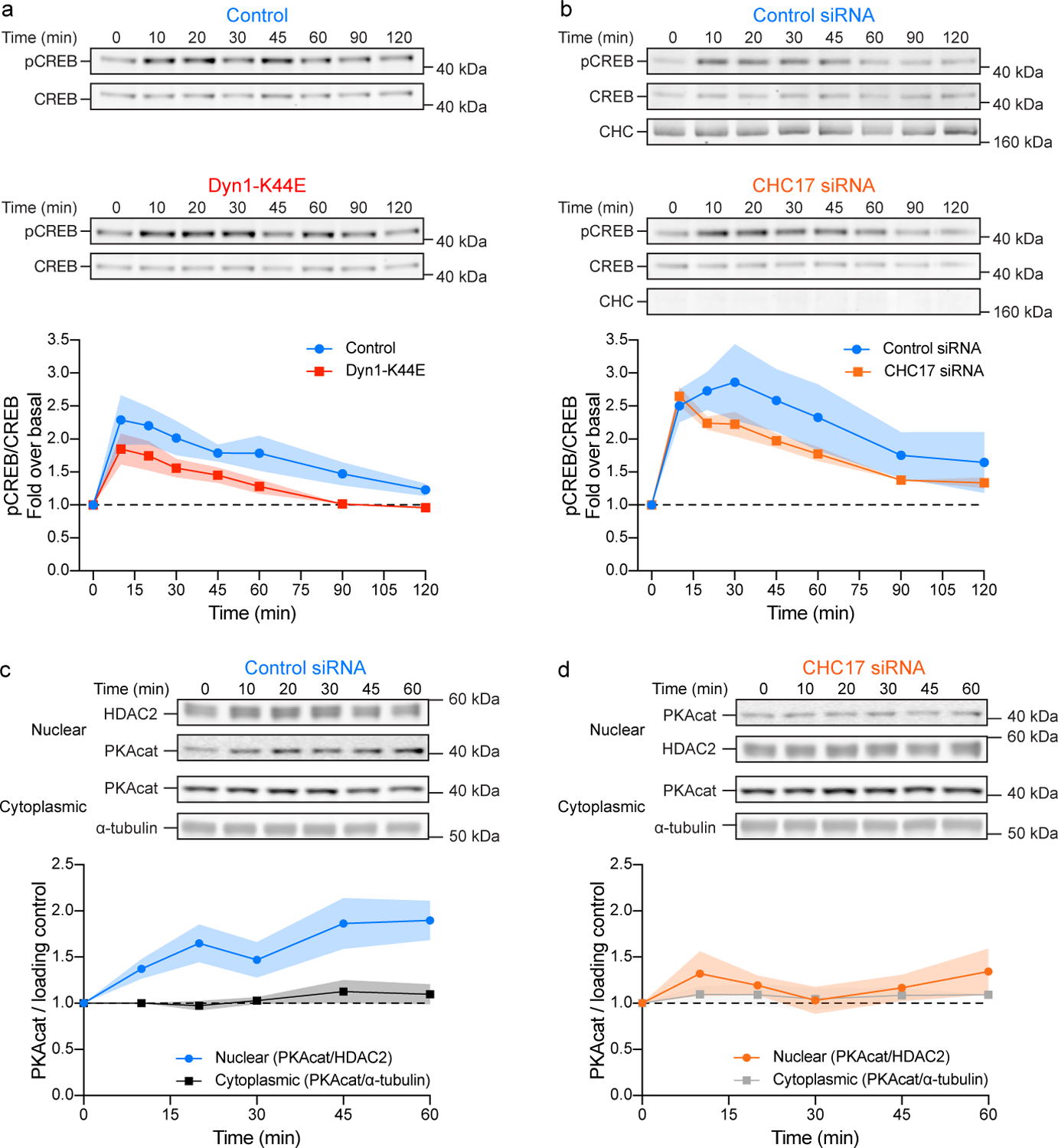
Iso-stimulated CREB phosphorylation and PKAcat nuclear entry are endocytosis dependent. a and b, Western blot analysis of CREB phosphorylation (Ser133) in HEK293 cells untreated and treated with 100 nM Iso. Cells expressing mCherry-Dyn1 (control) or mCherry-Dyn1-K44E (a) or scramble (control) or clathrin (CHC17) siRNA (b). Top, representative western blot probing CREB phosphorylation at Ser133 (pCREB) and CREB. Bottom, quantification of western blots in a (n = 3 biological replicates, Interaction p = 0.9041, Time p < 0.0001, Transfection p = 0.0005; two-way ANOVA) and b (n = 7 biological replicates, Interaction p = 0.8413, Time p < 0.0001, Transfection p = 0.0181; two-way ANOVA). c and d, Western blot analysis of PKAcat in nuclear and cytoplasmic fractions from cells untreated and treated with 100 nM Iso. Cells expressing scramble siRNA (c, control) or clathrin siRNA (d, CHC17). Top, representative western blots probing for PKA catalytic subunit α in nuclear and cytoplasmic fractions. HDAC2 and α-tubulin were used as nuclear and cytoplasmic loading controls, respectively. Bottom, quantification of western blots in c (nuclear n = 3, cytoplasmic n = 4 biological replicates; Interaction p = 0.0763, Time p = 0.0100 and Fraction p < 0.0001; two-way ANOVA) and d (n = 4 biological replicates, ns, Interaction p = 0.8805, Time p = 0.5095 and Fraction p = 0.1782; two-way ANOVA). All data are mean ± sem (shaded areas).
Supporting this hypothesis, subcellular fractionation revealed a consistent increase of PKAcat detected in isolated nuclei after endogenous β2AR activation (Fig. 2c). Further, the time course of nuclear PKAcat accumulation overlapped the later, endocytosis-dependent component of CREB phosphorylation (Fig. 2d and Extended Data Fig. 2a,b). Moreover, endocytic blockade strongly reduced this nuclear accumulation of PKAcat (Fig. 2d).
PKAcat accumulation in the nuclear fraction occurred without a detectable decrease in the amount in the cytoplasmic fraction (Fig. 2c,d, black and gray lines), suggesting that the net amount accumulated is small. We verified this by quantifying PKAcat in subcellular fractions, using HDAC2 as a nuclear marker, and estimated the net amount of nuclear accumulation to be in the range of 1–2% of total cellular PKAcat (Extended Data Fig. 2d). Despite its small overall amount, nuclear accumulation of PKAcat appeared to represent a key endocytosis-dependent step in the downstream transduction pathway because subsequent steps were found to be similarly endocytosis-dependent (Extended Data Fig. 2a–c).
Imaging nuclear entry of PKAcat
We next sought to independently verify the endocytosis-dependent nature of nuclear accumulation of PKAcat in intact cells. We pursued a fluorescence imaging approach, applying a recently developed strategy to label the endogenous protein using CRISPR-mediated gene editing and self-complementing split fluorescent protein fusion24,25. We targeted PRKACA, the predominant PKAcat isoform in most tissues26, and used split mNeonGreen2 (mNG2) due to its high brightness27 (Supplementary Fig. 3a–c and d, top row). Confocal microscopy detected endogenously labeled PKAcat primarily in the cytoplasm. In unstimulated cells PKAcat was accumulated in cytoplasmic puncta, colocalizing with native PKAcat as well as PKAreg (Supplementary Fig. 3e–g). Some of these puncta overlapped Golgi membranes (Supplementary Fig. 3h,i), consistent with Golgi localization of native22,28 and tagged25 PKAcat - PKAreg complexes shown previously. However, endogenously labeled PKAcat was not restricted to the Golgi apparatus because punctate accumulations were also observed elsewhere in the cytoplasm at lower density (Supplementary Fig. 3h,i).
We began by examining the effect of supraphysiological cAMP elevation produced by applying the direct adenylyl cyclase activator forskolin (Fsk) in the presence of the broad-spectrum phosphodiesterase (PDE) inhibitor IBMX. In this condition, PKAcat persistently delocalized to a diffuse cytoplasmic distribution and a small increase of PKAcat fluorescence was visible in the nucleus after 60 min (Supplementary Fig. 3j), consistent with nuclear entry of PKAcat described previously after supraphysiological pathway activation22,28. However, we did not detect a significant increase by this assay after endogenous β2AR activation using Iso (Fig. 3a, top row). We considered that imaging might be insufficiently sensitive to detect the small amount of net accumulation produced by physiological activation, particularly if the added bulk of mNG211 in complex with mNG21–10 impedes labeled protein entry through nuclear pores.
Figure 3.
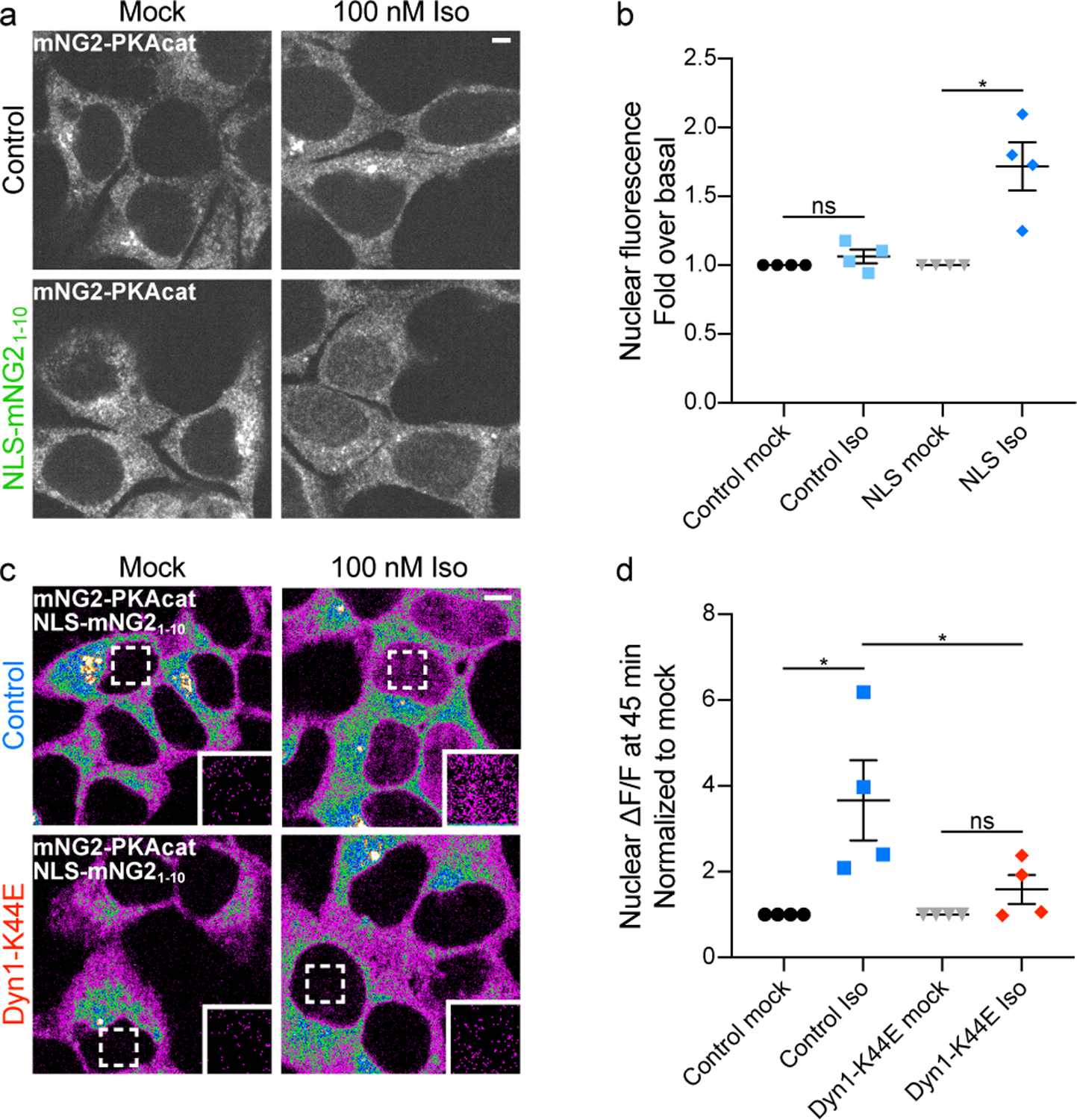
Independent verification of endocytosis-dependent nuclear entry of PKAcat. a, Representative spinning disk confocal images of fixed cells untransfected (control, top) and transfected with nuclear localized mNG21–10 (bottom). Cells were untreated (mock) or stimulated with 100 nM Iso. b, Quantification of fixed cells in a (n = 4, cells ≥ 74 per biological replicate; control 0 min vs 45 min p = 0.8625, NLS 0 min vs 45 min p = 0.0003; ordinary one-way ANOVA). c, Representative spinning disk confocal images of live cells expressing mNG211-PKAcat, mNG21–10 and nuclear localized NLS-mNG21–10. Cells were transfected with pcDNA3 (control, or mCherry-Dyn-K44E and cells were untreated (mock, top) or treated with 100 nM Iso (bottom) at 0 minutes. Inset shows nuclear ROI shown as dotted line square at 0 minutes. d, Quantification of cells expressing pcDNA3 (control) or mCherry-Dyn1-K44E from c, data normalized to the corresponding untreated 45 minute time point (n = 4; control mock vs Iso p < 0.0001, Dyn1-K44E mock vs Iso p = 0.7726, control Iso vs Dyn1-K44E Iso p = 0.0004 with Sidak’s multiple comparisons test; ordinary one-way ANOVA). All data are mean ± sem (shaded areas). Scale bars = 5 μm.
Accordingly, we modified the assay to include a nucleus-localized version of mNG21–10 (NLS-mNG21–10) together with cytoplasmic mNG21–10 (Supplementary Fig. 3d, bottom row). We reasoned that mNG211-fused PKAcat would complex either with mNG21–10 in the cytoplasm or with NLS-mNG21–10 after nuclear entry, creating a kinetic trap in the nucleus to detect uptake of mNG211-fused PKAcat into the nucleus while reducing potential mass effects before entry. Indeed, with this modification, an increase of nuclear fluorescence indicating entry of endogenously labeled PKAcat was evident visually (Fig. 3a, bottom row) and verified quantitatively (Fig. 3b) after native β2AR activation by Iso.
Nuclear entry of PKAcat is endocytosis-dependent
We adapted the nuclear entry assay for real time imaging of living cells at 37˚C. A time-dependent signal reporting PKAcat entry was observed (Extended Data Fig. 3a–c), consistent with the biochemical fractionation data. Further, nuclear entry of PKAcat was suppressed in the presence of endocytic blockade imposed by either Dyn1-K44E (Fig. 3c,d and Extended Data Fig. 3d,e) or clathrin knockdown (Extended Data Fig. 3f–i). Suppression of PKAcat nuclear entry was most pronounced after prolonged β2AR activation, whereas an earlier component of uptake appeared less sensitive to endocytic blockade (Extended Data Fig. 3d,g). This suggests that nuclear entry of PKAcat can be driven irrespective of endocytosis during the peak phase of endogenous β2AR signaling, when the global cAMP concentration is high, but requires cAMP production from endosomes during the plateau phase when global cAMP is lower.
Entry occurs when PKAcat is in cytoplasmic puncta
Because nuclear entry of cytoplasmic PKAcat during the plateau phase appears to be a key endocytosis-dependent step in the integrated signaling pathway, we next investigated the properties of the cytoplasmic PKAcat pool from which it is sourced. As described above, endogenously labeled PKAcat localized in unstimulated cells to a heterogeneous population of cytoplasmic puncta (Fig. 4a, top row). Activation of endogenous β2ARs produced a rapid delocalization of PKAcat into a diffuse cytoplasmic distribution, similar to that observed after supraphysiological activation by Fsk and IBMX. However, in contrast to supraphysiological activation which produced a sustained delocalization of PKAcat, endogenous β2AR activation delocalized PKAcat transiently and re-localization to a punctate distribution occurred within 10–20 minutes in the continued presence of agonist (Fig. 4a, bottom row and Supplementary Video 1). To our knowledge, such sequential de/re-localization of PKAcat has not been previously described but it is fully consistent with the kinetics of the endogenous cAMP response, with PKAcat delocalizing during the peak phase of high global cAMP concentration and re-localizing to discrete puncta in the plateau phase. Both PKAcat delocalization and re-localization were observed in the presence of endocytic blockade (Fig. 4b,c and Extended Data Fig. 4), indicating that this reversible redistribution process is driven by global cAMP concentration and does not require cAMP production from endosomes.
Figure 4.
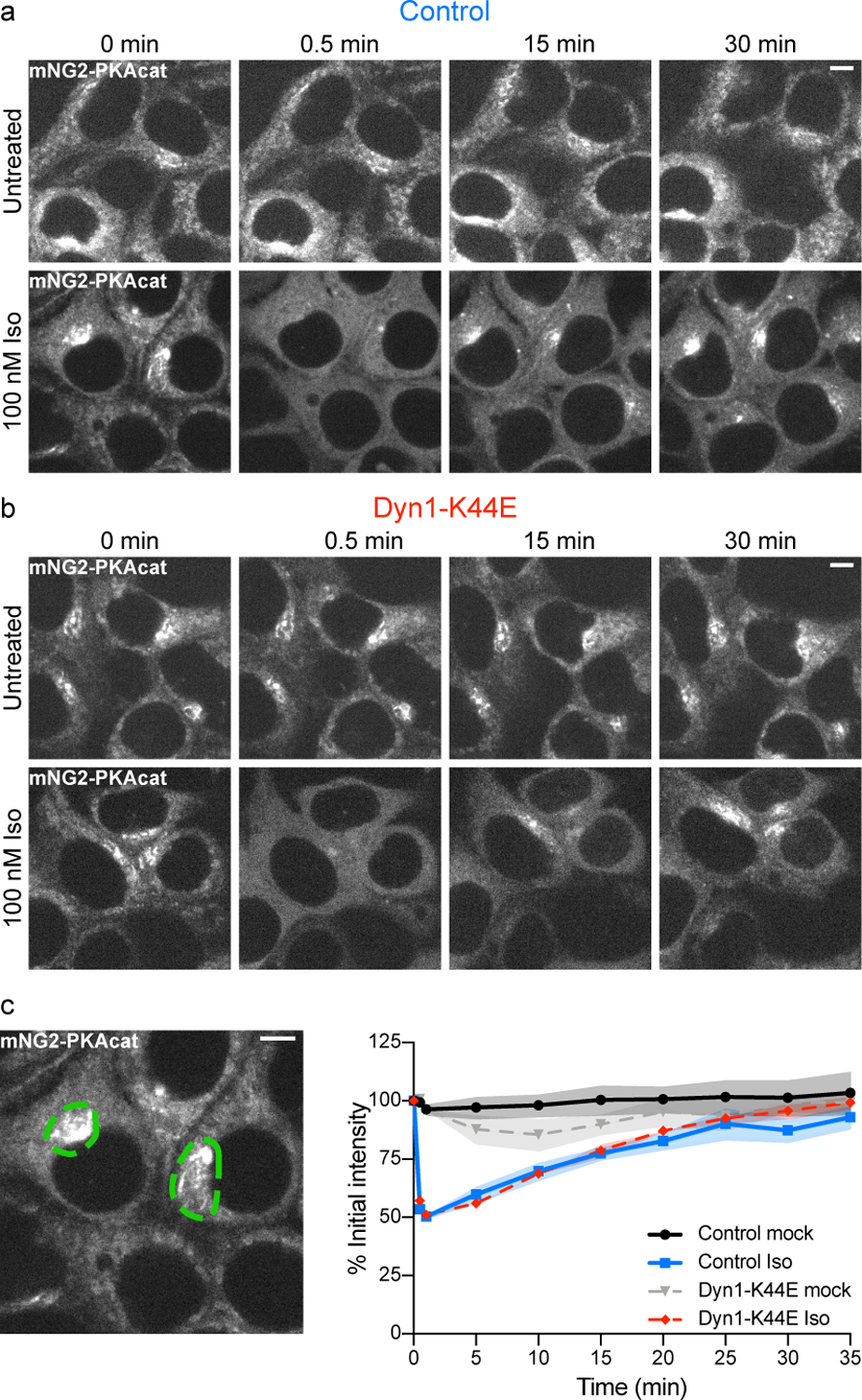
Cytoplasmic PKAcat sequentially disperses and relocalizes during prolonged β2AR activation. a and b, Representative live cell spinning disk confocal images of endogenously tagged mNG2-PKAcat HEK293T cells. Cells expressing pcDNA3 (a, control) or mCherry-Dyn1-K44E (b) were untreated (mock, top) or treated with 100 nM Iso (bottom) at 0 minutes. c, Quantification of experiments described in (a and b). Examples of ROIs drawn around the perinuclear region in green. (n = 3, cells ≥ 5 per biological replicate; Interaction, Time and Treatment p < 0.0001; control mock vs control Iso p < 0.0001, Dyn1-K44E mock vs Dyn1-K44E Iso p < 0.0001, control mock vs Dyn1-K44E mock p = 0.0437, control Iso vs Dyn1-K44E Iso p = 0.6556 with Tukey’s multiple comparisons test; two-way ANOVA). Data are mean ± sem (shaded areas). Scale bars = 5 μm.
PKAcat dynamically exchanges between puncta
Nuclear entry of PKAcat clearly continued after this apparently complete re-localization into cytoplasmic PKA puncta, raising the question of whether there exists any free PKAcat in the cytoplasm during the plateau phase to support the observed nuclear entry. To investigate this, we applied fluorescence recovery after photobleaching (FRAP) analysis, assessing return of endogenously labeled PKAcat fluorescence in sequential confocal images acquired at 10 s intervals beginning 5 s after photobleaching a selected cytoplasmic region. Under these conditions, we expected freely diffusible PKAcat to fully recover in the bleached region before the first post-bleach frame, as predicted based on previously measured rates of free PKAcat diffusion in the cytoplasm (Supplementary Table 1) and verified experimentally using EGFP as a reference with mass comparable to PKAcat (Fig. 5a, white box indicates bleached region).
Figure 5.
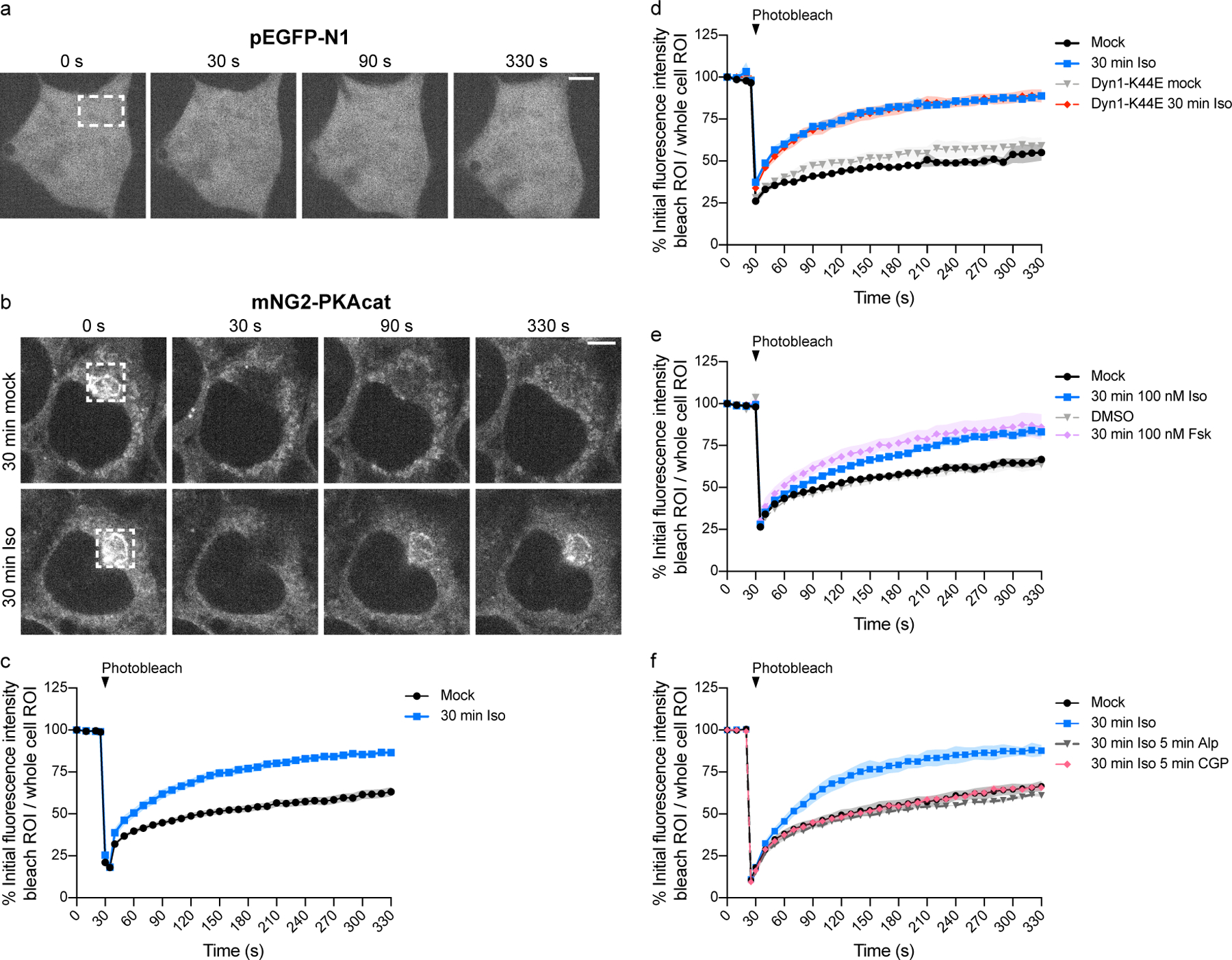
Cytoplasmic PKAcat dynamically exchanges during prolonged β2AR activation. a and b, Cells were photobleached (white box) at 30 seconds and fluorescence recovery was monitored after photobleaching. Representative live cell spinning disk confocal images of HEK293 cells transfected with pEGFP-N1 in a and HEK293T mNG2-PKAcat cells in b. b, Untransfected cells (control) were untreated (mock) or treated with 100 nM Iso for 30 minutes before live imaging. c-f, Quantification of photobleaching. c, Cells were untreated (mock) or treated with 100 nM Iso for 30 minutes before photobleaching. Data combined from d-f (n = 9, cells ≥ 3 per biological replicate). d, Cells were untransfected or transfected with mCherry-Dyn1-K44E and untreated (mock) or treated with 100 nM Iso for 30 minutes before photobleaching (n = 3, cells ≥ 3 per biological replicate; Interaction, Time and Treatment p < 0.0001; Iso vs Dyn1-K44E Iso p = 0.8710; mock vs Iso, Dyn1-K44E mock vs Dyn1-K44E and mock vs Dyn1-K44E mock p <0.0001). e, Cells were untreated (mock) or treated with 100 nM Iso, DMSO or 100 nM Fsk for 30 minutes before photobleaching (n = 3, cells ≥ 3 per biological replicate; Interaction, Time and Treatment p < 0.0001; mock vs DMSO p = 0.2619; mock vs Iso, DMSO vs Fsk and Iso vs Fsk p < 0.0001). f, Untransfected cells were untreated (mock) or treated with 100 nM Iso for 30 minutes followed by no treatment or treatment with 10 μM Alprenolol or 10 μM CGP-12177 for 5 minutes before photobleaching (n = 3, cells = 9 per biological replicate; Interaction, Time and Treatment p < 0.0001; mock vs Iso CGP p = 0.9425, mock vs Iso, Iso vs Iso Alp and Iso vs Iso CGP p < 0.0001). Data are mean ± sem (shaded area). Significance in d-f was determined by two-way ANOVA with Tukey’s multiple comparisons test. Scale bars = 5 μm.
Bleaching mNG2-PKAcat in unstimulated cells produced a dark spot that persisted for tens of seconds in the post-bleach image series (Fig. 5b, top row and Supplementary Video 2), indicating stable binding of PKAcat in the basal state. A small fluorescence increase was observed over an extended time scale, but intensity in the bleached region never reached a value greater than in surrounding (unbleached) regions despite PKAcat having been highly concentrated there in the pre-bleach image. This suggests that the slow fluorescence recovery observed primarily reflects overall redistribution of the cytoplasm during cell movement, rather than actual subunit exchange.
A different result was observed in cells driven into the plateau signaling phase by pre-exposing cells to Iso for 30 min and imaging them in its continued presence. Here, labeled PKAcat was similarly localized in cytoplasmic puncta. However, fluorescence recovery was observed in the bleached region to a level clearly exceeding that in surrounding regions (Fig. 5b, bottom row and Supplementary Video 3). This was verified by quantifying fluorescence recovery between the two conditions, indicating the presence of a significant exchangeable pool of PKAcat during the plateau phase relative to the unstimulated state (Fig. 5c and Extended Data Fig. 5a). The amount of this exchangeable pool, estimated by calculating the difference between respective fluorescence recovery curves (Extended Data Fig. 5b), was 20–30% of total. Fluorescence recovery indicative of dynamic subunit exchange was most obvious in the perinuclear region, likely due to a high density of scaffolded regulatory subunits in this region, but it was also evident elsewhere in the cytoplasm (Extended Data Fig. 5c).
Despite clear evidence for PKAcat exchange in the cytoplasm during the plateau signaling phase, the kinetics of fluorescence recovery in the bleached region had an overall half-time on the order of 50–60 seconds (Extended Data Fig. 5c,d). This is ~50-fold longer than would be expected for free protein diffusion into the bleached region (≲1 s). In principle, such slowed fluorescence recovery could arise in two ways29. First, it could result from equilibration of free PKAcat with the bleached protein occupying a limiting number of immobilized binding sites, such that fluorescence recovery rate is limited by the rate of bleached protein dissociation. Second, recovery rate could be limited by a low concentration of free cytoplasmic PKAcat, with most PKAcat bound due to excess binding sites and exchanging by occasional diffusional ‘hops’ from one site to another. We favor the latter possibility because PKAreg is known to be in significant (~10-fold) molar excess relative to PKAcat30 (Supplementary Table 1), excluding the first model. We also note that the rate of PKAcat binding to PKAreg approaches the diffusion limit in vitro31–33 (Supplementary Table 1), also compatible with a hop regime. Slowed recovery of fluorescent PKAcat during the plateau signaling phase thus appears to reflect a continuous process of free PKAcat binding to, and unbinding from, PKA regulatory subunits immobilized in the cytoplasm. Because the fluorescence recovery data indicate ~50-fold slowing relative to free protein diffusion, we estimate that ~2% of cytoplasmic PKAcat is free at steady state in this buffered ‘hop’ regime. Because a much larger fraction of cytoplasmic PKAcat is exchangeable under this condition (20–30% estimated from determination of the mobile fraction), our data further indicate that the small fraction of free PKAcat which is present in the cytoplasm during the plateau signaling phase is heavily (≳10-fold) buffered.
PKAcat exchange depends on global cAMP
PKAcat FRAP kinetics measured during the plateau signaling phase were not significantly affected by endocytic blockade (Fig. 5d and Extended Data Fig. 5d,e), indicating that dynamic exchange of PKAcat is determined primarily by global cAMP elevation and does not require local cAMP from endosomes. To independently verify this, we used a low concentration of Fsk (100 nM) in the absence of IBMX, which drives only a slight elevation of global cAMP concentration (Supplementary Fig. 2e) and no detectable net dissociation of PKAcat (Extended Data Fig. 5f,g). This condition also produced comparable PKAcat exchange as assessed by FRAP (Fig. 5e and Extended Data Fig. 5d,e). Moreover, the Iso-induced increase in PKAcat exchange was reversed not only by excess Alp, a β2AR antagonist that accesses both surface and internal receptors, but also by CGP-12177 which is cell-impermeant and selectively blocks surface receptors (Fig. 5f and Extended Data Fig. 5d,e). This indicates that the global cAMP elevation which is responsible for determining PKAcat buffering in the cytoplasm originates primarily from residual cAMP production stimulated by receptor activation at the plasma membrane.
Endosomes contact dynamic PKA puncta
A clue to how the location of cAMP production might impact downstream signaling emerged from the observation that endosomes containing internalized β2ARs, and verified to be in an activated conformation using a previously described nanobody-derived biosensor (Nb80-mApple, Extended Data Fig. 6a)34, achieve close proximity to individual PKA puncta in the cytoplasm (Fig. 6a). Line scan analysis indicated that, whereas β2ARs present in the plasma membrane are largely separate from the cytoplasmic PKA puncta, Iso-induced internalization results in activated receptor-containing endosomes moving into very close proximity (Fig. 6b).
Figure 6.
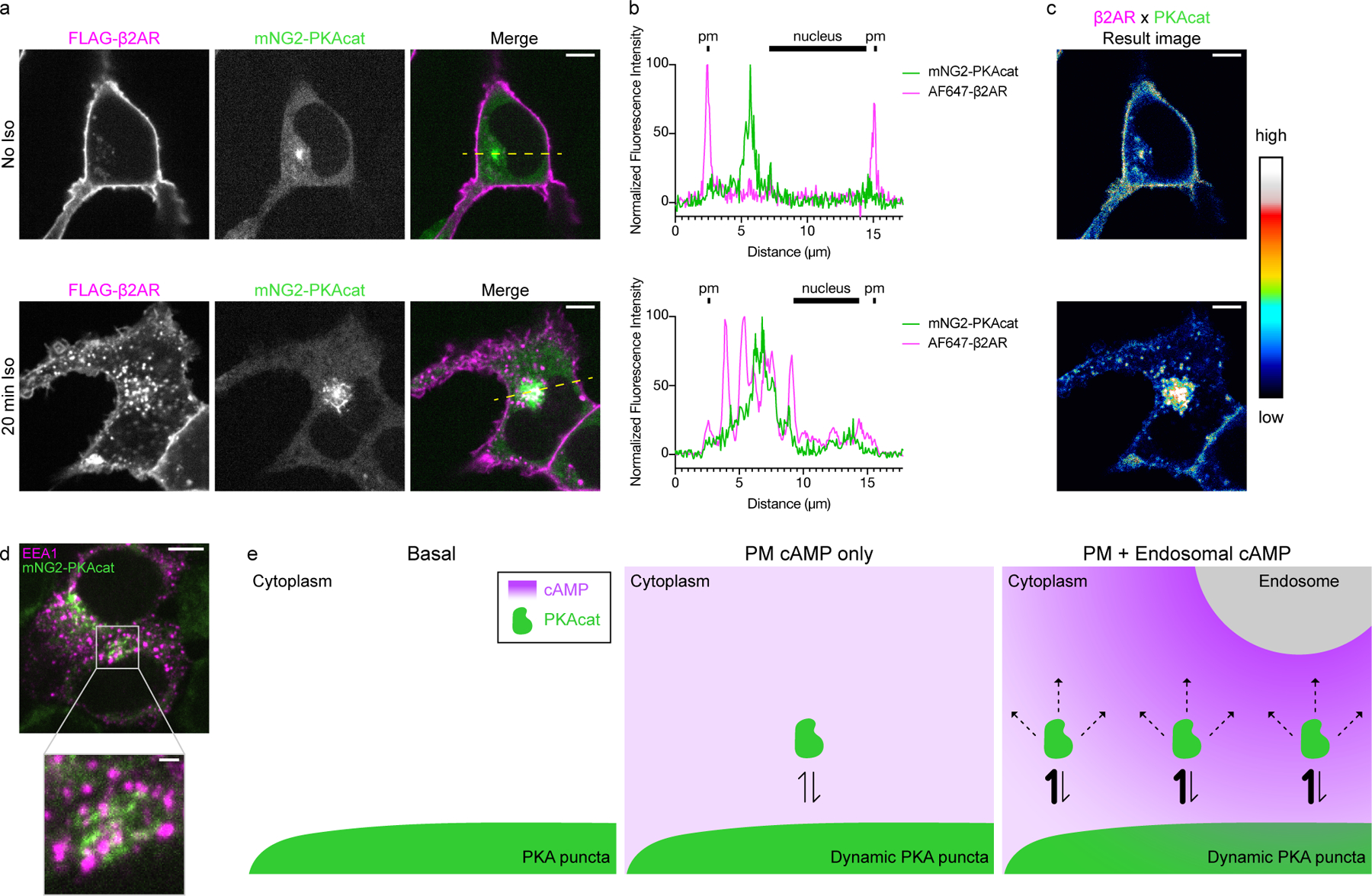
Endocytosis moves activated β2ARs into close proximity with dynamic PKAcat. a, HEK293T mNG2-PKAcat cells transfected with FLAG-β2AR and labeled with M1-FLAG-647. Cells were treated with 100 nM Iso and images were taken at 20 minutes post Iso addition. b, Normalized fluorescence intensity profiles of lines shown in a for mNG2-PKAcat and FLAG-β2AR. c, Potential sites of signaling in cells. Result image from image multiplication of normalized images of mNG2-PKAcat x FLAG-β2AR in a. d, Representative fixed cell spinning disk confocal images of HEK293T mNG2-PKAcat cells stained for endogenous EEA1. Scale bars = 5 μm, except in the inset of d where the scale bar = 1 μm. e, Model proposed for selective signaling by β2AR. Cartoon model based on the proximity of endosomes and dynamic PKA puncta show in the inset in d. At basal state, little cAMP is produced in the cytoplasm. PKAcat exists at stable puncta in the cytoplasm. After prolonged agonist, low levels of cAMP are produced at the plasma membrane and fill the cell allowing for dynamic exchange of PKAcat. Endosomes add local production of cAMP at a local high concentration, increasing the amount of dynamic PKAcat exchange.
Because the resolution limit of our optical imaging method (~250 nm) is comparable to the estimated distance scale of local cAMP gradients (Supplementary Fig. 1), we mapped the overlap between endosomes and PKAcat puncta within this relevant ‘diffusion range’ by calculating a product image between the corresponding fluorescence channels (Fig. 6c). Such regions of close approach were observed throughout the cytoplasm, and most frequently in the perinuclear region where the PKAcat puncta are most highly concentrated. In addition, when examined in sequential image series acquired from live cells, individual endosomes and PKAcat puncta appeared to collide. Close appositions were also observed in cells not over-expressing β2AR, using endogenous EEA1 to mark the relevant signaling endosomes in fixed specimens (Fig. 6d and Extended Data Fig. 6b), and collisions with individual PKA puncta were verified in live cell image series using an EEA1-derived FYVE domain to mark these endosomes35. Moreover, we noted that close appositions between endosomes and concentrated PKAcat puncta vary in dwell time, with some encounters lasting less than 5 s (the temporal resolution limit of our imaging analysis) and others persisting as longer-lasting endosome-to-PKA ‘contact sites’ (Extended Data Fig. 6c and Supplementary Video 4).
DISCUSSION
The present study addressed the fundamental unanswered question of how endocytosis is able to specify long-range cellular cAMP signaling by moving the location of cAMP synthesis rather than changing its overall amount or duration. We identify nuclear entry of PKAcat as a key endocytosis-dependent step in downstream transduction. We then identify several properties of the cytoplasmic PKA pool that simply solve the problem of location-encoded cAMP signaling. Specifically, we show that PKA is concentrated in discrete puncta which are titrated by global cAMP into a dynamic buffering regime, as defined by PKAcat being largely bound but able to dynamically exchange. Such metastable PKA puncta are densely distributed in the cytoplasm, and endosomes containing activated receptors move rapidly among them. Activated endosomes frequently collide with individual PKA puncta, creating ‘contacts’ within the range of local cAMP gradients which last for a variable dwell time (Supplementary video 4).
Our results support a model with two key elements (Fig. 6e). First, a low global cAMP elevation during the plateau phase titrates cytoplasmic PKA puncta to act as reservoirs of concentrated PKAcat, poised for sensitive local release by cAMP. In this way, PKA puncta ‘detect’ nanoscale cAMP gradients around colliding endosomes through local dissociation. Second, PKAcat exchange between puncta ‘transmits’ the elevations over a longer range. We estimate, from previously measured values (Supplementary Table 2), that free PKAcat can diffuse several microns in bulk cytoplasm before rebinding. This exceeds the distance between individual PKA puncta, and the high density of puncta inherently favors local capture36. Thus, PKAcat is in a dynamic equilibrium between adjacent puncta, enabling the puncta to act collectively as a cytoplasmic transmission network. While only a small fraction of PKAcat (~2%) is free at steady state during the plateau signaling phase, this amount is sufficient to account for the nuclear accumulation of PKAcat that is observed (1–2%) during the endocytosis-dependent downstream transcriptional response. Accordingly, the location-encoded cAMP signaling scheme that we delineate here appears to be capable of achieving high overall sensitivity.
Much remains to be learned about specific biochemical and biophysical properties of the cytoplasmic PKA network, and about its integrated signaling functions. Toward the latter goal, we suggest that it is useful to consider the cytoplasmic PKA network in simpler terms, as a bulk material. In the unstimulated state, the network resembles an ‘insulator’, based on PKAcat being stably bound. In the peak cAMP phase, the network resembles a ‘conductor’ with PKAcat fully dissociated. During the plateau signaling phase, the network is titrated into a ‘semiconductor’ state, as defined by a small fraction of PKAcat being free and a large fraction bound but in dynamic exchange. An analogous semiconductor state has been described in solid state materials, in which electrical charge carriers are largely bound but a small fraction dynamically exchanges between binding sites. This is useful in electronic devices to enable sensitive detection of signals that are introduced locally at junctions in the material differing in net concentration of dynamically bound carriers. Here, regions of high concentration act as sensitive reservoirs for local release of carriers into the free pool, and the local increase is transmitted through the material by diffusional hops37,38. We propose that dynamic PKA puncta confer an analogous property on the cytoplasm by providing reservoirs for sensitive release of PKAcat in response to local cAMP gradients, and then transmitting local increases through the network by diffusional hops of PKAcat. Accordingly, the cytoplasmic PKA network appears to act as a kind of a semiconductor material to mediate spatial decoding of cAMP signals.
It is now clear that cAMP can produce highly localized effects under physiological conditions, with recent studies highlighting restricted movements of cAMP through binding PKA regulatory subunits39 and local hydrolysis40. The present results are fully compatible with these concepts, and with previous evidence that cAMP effects are highly buffered41,42. The present results extend this understanding to dynamic buffering of PKAcat. In addition, they reveal a discrete role of cytoplasmic PKA as an integrated protein network mediating the local detection, and longer-range transmission, of endomembrane-encoded cAMP information.
MATERIALS AND METHODS
DNA constructs
Dynamin1 plasmids
Both rat dynamin1 wildtype and dominant negative K44E mutant were N-terminally tagged with mCherry and cloned into pcDNA3 (Clontech/Takara Bio, Mountain View, CA).
cAMP luminescence biosensor with Renilla luciferase
pSF CMV GloSensor20F IRES Rluc was generated from pSF CMV EMCV (IRES) Rluc (Boca Scientific, Dedham, MA; OG296) and from pGloSensor-20F (Promega, Madison, WI; E1171). The full length GloSensor20F coding sequence including stop codon was amplified by PCR and inserted into pSF CMV EMCV (IRES) Rluc between the CMV promoter and IRES. This was accomplished by linearizing pSF CMV EMCV (IRES) Rluc with EcoRI and EcoRV digestion and InFusion cloning (Clontech/Takara Bio).
Nuclear localized mNG21–10
pcDNA3.1+ 2xNLS mNG21–10 IRES TagBFP was generated by a multiple insert ligation with Quick Ligase (New England BioLabs, Ipswich, MA; M2200) to clone all inserts into pcDNA3.1+ (Clontech/Takara Bio). A gBlock (IDT, Coralville, IA) encoding 2x NLS (SV40) mNG21–10 was obtained and PCR amplified with flanking 5’ BamHI and 3’ AgeI sites. An IRES fragment was PCR amplified from pSF CMV EMCV (IRES) Rluc (Boca Scientific) with flanking 5’ AgeI and 3’ XhoI sites. Lastly, TagBFP was PCR amplified with flanking 5’ XhoI and 3’ XbaI sites. The multiple insert ligation was performed with digested inserts 2x NLS mNG21–10 (cut BamHI and AgeI), IRES (cut AgeI and XhoI), TagBFP (cut XhoI and XbaI) and linearized pcDNA3.1+ (cut BamHI and XbaI).
GG4B cAMP fluorescence biosensor
The cAMP fluorescence biosensor was derived using a cpGFP variant derived from Green GECO and the high affinity cAMP-binding domain (CNBD-B domain) derived from the human Epac2 protein RAPGEF4, hence its name ‘GG4B’. Human RAPGEF4 (B isoform) in pDONR221 (DNASU, HsCD00039697) and CMV-G-GECO1.043 (Addgene #32447) were PCR-amplified, and InFusion (Clontech) cloning was used to create a series of constructs varying in the sequence of linker regions according to a systematic linker variation strategy used previously to generate other cpGFP-based biosensors44. Constructs were selected by cell-based assay for high sensitivity and a rapid fluorescence increase upon cAMP elevation. The GG4B variant used in the present study corresponds to the following engineered protein construct cloned into the pCAGG-SE expression vector45: MLYK - [residues K149-R458 from RAPGEF4 (this includes the hinge region of high-affinity cAMP binding site)] - LVSH - [residues N27-N267 from CMV-G-GECO1.0 (this corresponds to cpGFP)] - FNNP - [residues D459-Q662 from RAPGEF4 (this includes the lid region of the cAMP binding site)] - EF (EcoRI site) - stop.
FLAG-β2AR pcDNA3.0
N-terminally FLAG-tagged human β2AR was previously described15. This construct was used to validate endocytic blockade effects and visualize receptor localization in cells. All signaling experiments were carried out using endogenous β2AR activation.
pmApple-N1-Nb80
Nb80 C-terminally tagged with mApple was previously described34.
DsRed-EEA1 (2xFYVE)
A construct containing tandem FYVE domains derived from EEA1 was N-terminally tagged with DsRed was previously described14,35.
AC9-EGFP pcDNA3.1+
AC9 C-terminally tagged with EGFP was generated by a three-part ligation with Quick Ligase. AC9 was obtained from FLAG-AC919 by PCR amplification with flanking 5’ KpnI and 3’ NotI sites. Next, EGFP was amplified from pEGFP-N1 with flanking 5’ NotI and 3’ XbaI sites. The multiple insert ligation was performed with digested inserts AC9 (cut KpnI and NotI) and EGFP (cut NotI and XbaI) and linearized pcDNA3.1+ (cut KpnI and XbaI).
Cell culture and treatments
Human HEK293 and HEK293T cells (ATCC, CRL-1573 and 3216) were grown in DMEM (Gibco/Thermo Fisher, Waltham, MA J; 11965) supplemented with 10% fetal bovine serum (UCSF Cell Culture Facility, San Francisco, CA), and maintained at 37°C and 5% CO2 in a humidified incubator. Cells were treated with the following ligands and inhibitor: isoproterenol (Iso; Sigma, St. Louis MO; 6504), alprenolol, (Alp; Tocris, Minneapolis, MN; 2806), CGP-12177 (CGP; Tocris,1134), forskolin (Fsk; Sigma, F6886), isobutylmethylxanthine (IBMX; Sigma, I5879), and Dyngo4a (abcam, Cambridge, MA; ab120689). Prior to Dyngo4a treatment, cells were changed to serum-free media (DMEM without added FBS).
DNA transfections
Cells were seeded at 70% confluency in a well of a 6 well plate or a 35 mm imaging dish (MatTek, Ashland, MA; P35G-1.5–14-C or CellVis, Mountain View, CA; D35–20-1.5-N). DNA was transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA; 11668) according to the manufacturer’s protocol, with medium changed the next day and used for experimentation within 24–48 hours after transfection.
siRNA transfections
Cells were seeded at 30% confluency in a 10 cm dish and transfected overnight with 200 pmol either AllStars Negative Control (Qiagen, Germantown, MD; 1027281) or CHC17 (5’- AAGCAATGAGCTGTTTGAAGA-3’) siRNA with RNAiMax Lipofectamine (Invitrogen) according to the manufacturer’s protocol. Media was changed the next day and cells recovered for another 48 hours before experimentation. Unlabeled CHC17 siRNA and CHC17 siRNA labeled with AlexaFluor555 (3’ end of sense strand) were obtained from Qiagen with the above target sequence. If DNA transfection was also required, this was accomplished using Lipofectamine 2000 24 hours before experimentation.
In vitro calibration of cAMP fluorescent biosensor
HEK293 cells were transiently transfected with pCAGG-GG4B and harvested 48 hours after transfection by dissociation in ice-cold PBS supplemented with 1 mM EDTA. Cell pellets (1,000 g for 10 min) were resuspended in cold 100 mM KCl, 30 mM HEPES pH7.4 freshly supplemented with PMSF and lysed using a Dounce homogenizer followed by centrifugation at 40,000 g for 10 minutes. The supernatant was maintained on ice, diluted to 1 mg/mL total protein concentration and then supplemented with 100 μM IBMX before assays. Fluorescence was monitored using 100 μl aliquots in a 96-well plate, at room temperature and in the presence of added cAMP as indicated, using a Gemini XPS fluorimeter (Molecular Devices, San Jose, CA) set to 488 nm excitation and 525 nm emission. Mean values derived from duplicate fluorescence determinations were analyzed according to a single-site binding model (F / F0 = max * [cAMP] / {[cAMP] + KD}) fit by a non-linear regression method using Prism 8 (GraphPad, San Diego, CA).
Generation of gene-edited PKAcat cells
HEK293T cells were grown as previously described, additionally supplemented with penicillin/streptomycin (100 µg/ml; UCSF Cell Culture Facility). Synthetic nucleic acid reagents were obtained from Integrated DNA Technologies (IDT). sgRNAs and Cas9/sgRNA ribonucleoprotein (RNP) complexes were prepared as described previously46.
For the knock-in of mNG211 and GFP11, 200-nt homology-directed recombination (HDR) templates were ordered as single-stranded DNA ultramer oligos (Supplementary Table 3). Cas9 protein (pMJ915 construct, containing two nuclear localization sequences) was purified following protocols described previously47 and obtained from the University of California, Berkeley Macrolab. To increase HDR efficiency, HEK293T cells stably expressing GFP1–10 or mNG21–10 were treated with nocodazole (200 ng/mL; Sigma) for 15 hours before electroporation46. Cas9/sgRNA RNP complexes were assembled with Cas9 protein (100 pmol) and sgRNA (130 pmol) just before electroporation and combined with the corresponding HDR template in a final volume of 10 μl. Nocodazole-treated HEK293T cells stably expressing FP1–10 were resuspended to 104 cells/μl in SF solution immediately before electroporation. For each sample, 20 μl of cells was added to the 10 μl RNP/template mixture. Cells were quickly electroporated on an Amaxa 96-well shuttle Nucleofector device (Lonza, Morristown, NJ) using the CM-130 program and transferred to 12-well plate with pre-warmed media.
Electroporated cells were cultured normally for 5–10 days before FACS selection of positive cells. Both mNG2 and GFP cells were sorted in the 488-GFP channel on a FACSAria II (BD Biosciences, San Jose, CA) in the Laboratory for Cell Analysis at UCSF.
DNA sequence validation of gene-edited PKAcat cells
Genomic DNA from each sorted cell type was isolated using the PureLink Genomic DNA mini kit (K1820, Invitrogen) according to the manufacturer’s protocol. Three sets of primer pairs were designed for each cell line: forward and reverse primers outside of the insert, forward outside with reverse in the insert, forward inside the insert and reverse outside the insert (Supplementary Table 4). PCR amplification was carried out with 200 ng isolated genomic DNA, 0.5 μM forward and reverse primers, Q5 Hot Start Master Mix, 1M Betaine, and water to a total volume of 50 μl using the following program: 98°C 5 min, [98°C 20 s, gradient 15 s, 72°C 30 s] x 30 cycles, 72°C 3 min and then 4°C. PCR reaction products were purified and sequenced (Quintara Biosciences, South San Francisco, CA) as indicated in Supplementary Fig. 3b.
Quantitative real time PCR
RNA was isolated from HEK293 cells using the QIAshredder and RNeasy Mini kit (Qiagen, 79654 and 74104, respectively). After ligand treatment, cells were placed on ice and washed once with cold PBS. Then cells were lysed, and RNA was extracted according to the manufacturer’s protocol. RNA was eluted in 30–50 μl of DEPC-treated water, and the concentration of each sample was determined.
cDNA was generated from RNA using the SuperScript III First-strand synthesis system (Invitrogen, 18080–051). Briefly, 2 μg of RNA was used per reaction primed with oligo dT. The reverse transcription reaction was performed according to the manufacturer’s protocol.
qRT-PCR was performed using a StepOnePlus (Applied Biosystems, Foster City, CA) instrument. cDNA generated from extracted RNA was used as the input for the qRT-PCR, with amplification by Power SYBR Green PCR master mix (Applied Biosystems, 4367659). Levels of transcripts were normalized to GAPDH. The primers used are listed in Supplementary Table 5.
cAMP luminescence assay
HEK293 cells stably transfected with GloSensor20F-IRES-Rluc were lifted and resuspended in imaging media (DMEM without phenol red supplemented with 30 mM HEPES; Gibco, 31053 and 15640 respectively). These cells were then incubated with 1.6 mM D-luciferin (Gold Biotechnology, St. Louis, MO; LUCNA-1g) for one hour at 37°C and 5% CO2 in a humidified incubator in a 24 well plate. Immediately before imaging, cells were treated with imaging media only or imaging media with Iso and placed into a 37°C heated light-proof chamber, then imaged every 10 seconds for 20 minutes. When the cAMP luminescence image series finished, stop buffer (to eliminate firefly luciferase luminescence; 25 mM HEPES pH 7.4, 1 mM EDTA, 0.4 mM DTT, 0.2% TritonX-100, 2% glycerol) with 10 μM coelenterazine (Gold Biotechnology, CZ2.5) was added to each well. Renilla luciferase luminescence was immediately imaged at 10 second intervals for one minute.
Images were acquired using a 512 × 512 pixel electron multiplying CCD camera (Hamamatsu Photonics, Japan; C9100–13) using μManager 1.448,49. Analysis was carried using the multiple ROI analysis tool in the Fiji implementation of ImageJ50–52. ROIs were drawn around each well, and corresponding background ROIs were placed in an area without cells. Intensity over time was measured and corrected for background luminescence for both firefly and renilla luciferase values. The renilla luminescence values over the time series was averaged (renillaavg). A luminescence intensity ratio (if/r) of firefly to renillaavg luminescence was calculated per well over time. Next, each time series was initialized to the average of the first 5 time points (inew = if/r - i5avg). The max (average of the top 5 proximal values) of each Iso treated condition was determined, and each condition was normalized to the max of the control Iso treated sample.
Spinning Disk Confocal Imaging
Live and fixed cell imaging was primarily performed on a Nikon Ti inverted spinning disk confocal microscope (Yokogawa CSU-W1) with 405-, 488-, 561- and 640-solid-state lasers (Andor, United Kingdom), fitted with Plan Apo λ 20× 0.75 NA, Plan Apo VC 60× 1.4 NA, and Plan Apo VC 100× 1.4 NA objectives (Nikon), and an Andor Zyla 4.2 sCMOS camera controlled by μManager 2.1 beta software (https://www.micro-manager.org). Cells were kept at 37°C in a temperature- and humidity-controlled chamber (Okolab, San Bruno, CA) during live imaging.
Spinning disk confocal images were also acquired on two other systems. Live and fixed cell imaging was performed on a Nikon Ti inverted spinning disk confocal microscope (Yokogawa CSU-22) with a custom laser launch (100 mW at 405, 488, 561, and 640 nm; Coherent OBIS, Santa Clara, CA) fitted with an Apo TIRF 100x/1.49 NA Oil objective (Nikon), and a Photometrics Evolve Delta electron multiplying CCD camera (Tuscon, AZ) controlled by Nikon Elements 5. For live cell imaging, cells were kept in a temperature- and humidity-controlled chamber (Okolab) during imaging. Live and fixed cell imaging was also performed using a Nikon TE2000 inverted microscope fitted with an Apo TIRF 100x/1.49 NA Oil objective (Nikon), spinning disk confocal scan unit (Yokogawa CSU-10), 488 and 561 nm laser illumination (Vortran), and custom-built chamber and objective heater for imaging at 37˚C. Images were detected using an EMCCD camera (Andor iXon Ultra 897) operated in the linear range and controlled by μManager.
Imaging analysis
Images analysis was primarily performed by drawing whole cell or subcellular ROIs with background ROIs placed in a nearby area without cells to determine the background signal. The fluorescence intensity was normally background subtracted and each time point was either normalized to the initial intensity for each condition or the change in fluorescence relative to the initial intensity was calculated at each time point and divided by the basal value to determine ΔF/F. In cells transfected with 2x NLS mNG21–10 IRES TagBFP and mCherry-Dyn1-K44E or AlexaFluor555-CHC17 siRNA, transfected cells for analysis were identified by the 405 nm and 647 nm channels.
Fixed imaging
Cells were plated on poly-D-lysine (Sigma, P8920) coated coverslips (Fisher, 12–545-100) in 12 well plates. Cells untreated or treated with 100 nM Iso for 45 minutes at 37°C. Cells fixed in 3.7% formaldehyde (Fisher, F79) in modified BRB80 (80 mM PIPES pH 6.8, 1 mM MgCl2, 1 mM CaCl2) for 20 minutes. For cells to be stained, cells were blocked in 3% BSA (Sigma, A7030), 0.1% Triton X-100 (Sigma, T8787) in PBS. Next, cells were stained using antibodies (Supplementary Table 6) and with 1 µg/ml DAPI (20 mg/ml stock in water, Sigma, D9542) or 1 μM DRAQ5 (Invitrogen, 62251) in PBS. Then the coverslips were mounted on glass slides with ProLong Gold Antifade mounting media (Invitrogen, P10144). Slides were stored for a minimum of 24 hours to allow mounting media to dry before imaging.
Fluorescence recovery after photobleaching
A Rapp Optoelectronic UGA-40 photobleaching system was used to photobleach mNG2-PKAcat ROIs in the perinuclear or general cytoplasmic regions using a 473 nm laser (Vortran, Sacramento, CA). Cells were imaged at 10 second intervals for 330 seconds. Photobleaching occurred approximately at 30 seconds.
Quantitative image analysis was performed on unprocessed images using Fiji. ROIs were drawn in the bleached region to measure average fluorescence in each sequential frame. Corresponding ROIs in areas without cells were used for background subtraction. To examine kinetics, the fluorescence intensity measured immediately after photobleaching was set to zero. To correct for overall bleaching of the cell, background-subtracted whole cell ROIs were measured at each time point and used to normalize the fluorescence measured in the bleach region. Data were analyzed using a single site exponential fit (one-phase association) using Prism 8 (GraphPad) to determine half-time of fluorescence recovery and using the maximum (plateau) value from the curve fit to estimate mobile fraction.
Proximity analysis of mNG2-PKAcat to β2AR endosomes
Line scan analysis of β2AR and PKAcat fluorescence from the shown line was carried out in Fiji using the plot profile function to measure pixel values along the line.
Result product image from the normalized β2AR and normalized PKAcat was generated with MATLAB 2018b (MathWorks, Natick MA). Briefly, the normalized image from each channel was obtained by first drawing a background ROI. The mean fluorescence in the background ROI was subtracted from the entire image. The average of the top 1% intensities was used to determine the average max intensity. The background subtracted image was divided by the average max intensity to generate the normalized image. The normalized images were multiplied on a pixel-by-pixel basis to calculate the product image (MATLAB immultiply function).
Protein collection and western blot analysis
Cells were typically lysed with RIPA buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100, 0.5% Sodium Deoxycholate, 0.1% Sodium Dodecyl Sulfate) containing Complete Mini, EDTA-free (Roche, Mannheim, Germany; 11836170001) and PhosSTOP (Roche, 04906837001) for CREB phosphorylation experiments. Lysates were then transferred to microcentrifuge tubes and spun for 10 minutes at 14,000 rpm at 4°C. The supernatant was then saved and used for western blot experiments. Protein concentration was determined by BCA Protein Assay (Thermo Fisher, 23225).
Nuclear-cytoplasmic separation was performed using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, 78833) according to the manufacturer’s protocol, fractions were then prepared for western blot analysis.
Protein samples were prepared for western blot analysis by adding NuPage LDS Sample Buffer (Invitrogen, NP0007) and 1 mM dithiothreitol (DTT; Sigma, D0632). Samples were loaded into a NuPAGE 4–12% Bis-Tris Protein gel (Invitrogen, NP0335BOX) and transferred to nitrocellulose.
After transfer onto nitrocellulose, membranes were stained with Ponceau S solution (Sigma, P7170) to check transfer quality. Membranes were washed with PBS to remove the Ponceau S solution before blocking with Odyssey blocking buffer. For blots where phosphorylated CREB was to be detected, Odyssey TBS blocking buffer (LI-COR Biosciences, Lincoln, NE; 927–50000) was used, otherwise all other blots used Odyssey PBS blocking buffer (LI-COR Biosciences, 927–40000). Primary antibodies were used to probe for proteins of interest with the corresponding secondary antibodies (Supplementary Table 7). The blots were imaged on an Odyssey Infrared Imaging System (LI-COR Biosciences) and bands were analyzed by ROI analysis with background subtraction in Fiji. Scans of uncropped and unprocessed blots are provided in Source Data files.
Flow cytometry
Cell surface fluorescence of HEK293 cells stably expressing FLAG-β2AR was measured to assess receptor internalization, following exposure to 100 nM Iso for 30 minutes at 37°C to internalize receptors to steady state. After treatment, cells were washed with cold PBS three times, then incubated with 1 μg/ml AlexaFluor647-conjugated (Invitrogen, A20173) M1 mouse anti Flag monoclonal (Sigma, F-3040) antibody at 4°C for one hour on a shaker. Cells were mechanically lifted and mean fluorescence intensity of 10,000 cells was read by flow cytometry for each sample on a FACSCalibur (BD Biosciences) using CellQuest Pro (BD Biosciences). Internalization was calculated as the mean fluorescence intensity from the (untreated sample - agonist treated sample) / untreated sample. Each condition had three technical replicates per biological replicate.
Statistical analysis and reproducibility
All data are shown as mean ± standard error of the mean (sem) from at least 3 biologically independent experiments, unless otherwise indicated. Images are representative of at least 3 biologically independent experiments. Statistical analyses (included in source data files where applicable) to determine significance were performed using Prism 8 (GraphPad) for unpaired t-test, one- and two-way analysis of variance (ANOVA, alpha 0.05).
Extended Data
Extended Data Fig. 1. Characterization of endocytic blockade on β2AR and the effects of endocytic blockade on the cAMP signaling pathway.
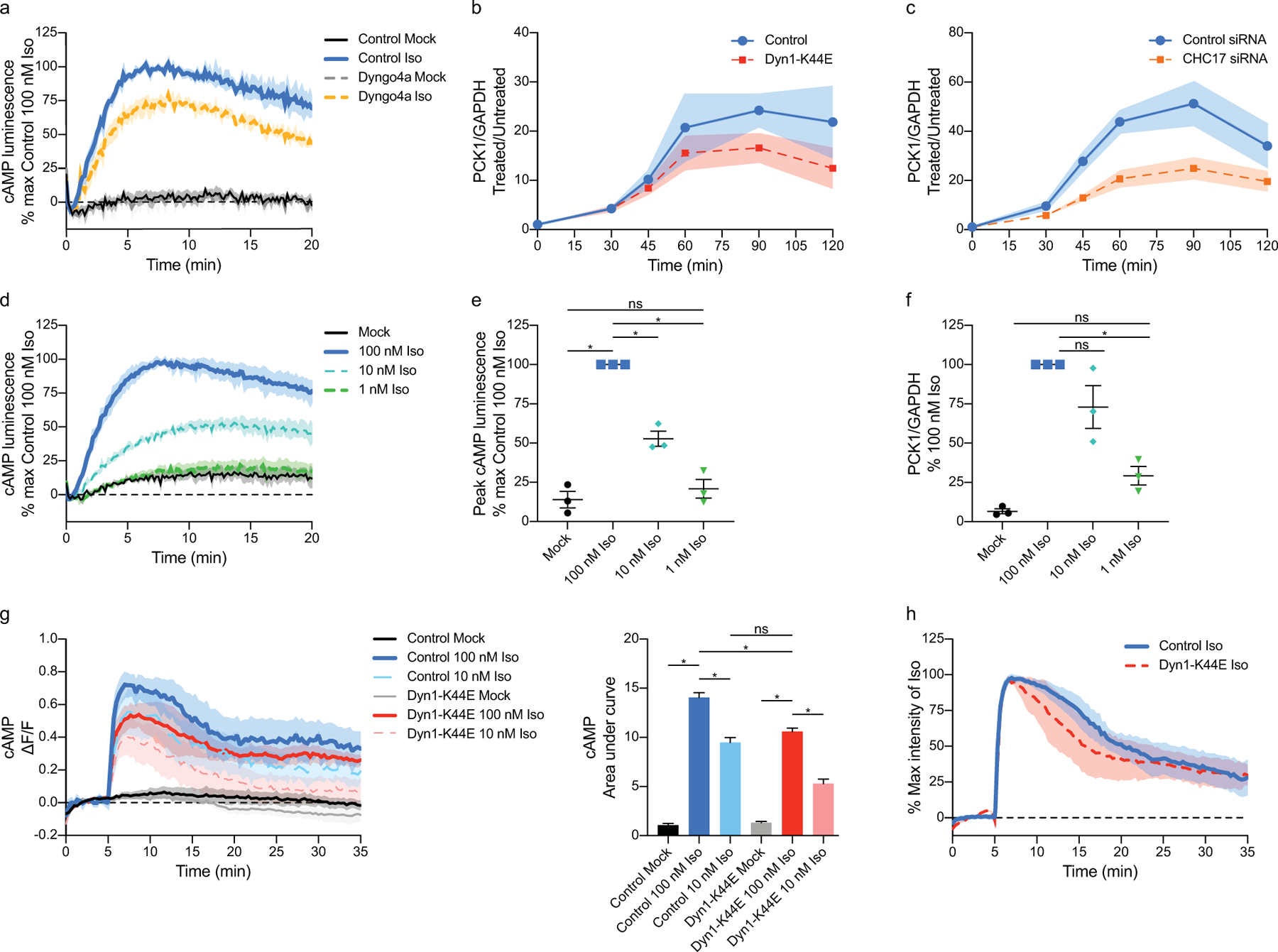
a, cAMP luminescence time course from Fig. 1a (n = 3 biological replicates; control vs Dyngo4a mock p = 0.0469; control mock vs Iso, Dyngo4a mock vs Iso and control vs Dyngo4a Iso, p < 0.0001). b and c, Quantitative RT-PCR was performed on cells transfected with (b) mCherry-Dyn1 (control) or mCherry-Dyn1-K44E, or (c) Control siRNA or CHC17 siRNA, then untreated or treated with 100 nM Iso. PCK1 and GAPDH transcript levels determined by qRT-PCR. Dyn1 vs Dyn1K44E (n = 6 biological replicates, Interaction = 0.6313, Time p < 0.0001, Transfection p = 0.0476). Control siRNA vs CHC17 siRNA (n = 6 biological replicates, Interaction p = 0.0260, Time p < 0.0001, Transfection p < 0.0001). d, Iso stimulates cAMP luminescence over time. Cells were untreated (mock) or treated with 100 nM, 10 nM, or 1 nM Iso at 0 minutes. (n = 3 biological replicates; Interaction, Time and Treatment p < 0.0001; mock vs 1 nM Iso, mock vs 10 nM Iso and mock vs 100 nM Iso p < 0.0001). e, Maximum cAMP luminescence from d (n = 3 biological replicates; mock vs 1 nM Iso p = 0.7889; 10 nM vs 100 nM Iso, p = 0.0004; mock vs 100 nM Iso and 1 nM vs 100 nM Iso p < 0.0001). f, Induction of PCK1 increases with concentration of Iso. qRT-PCR was performed on cells untreated (mock) and treated with 100 nM, 10 nM, and 1 nM Iso (n = 3 biological replicates; mock vs 1 nM Iso, p = 0.2323; mock vs 100 nM Iso, p < 0.0001; 1 nM vs 100 nM Iso, p = 0.0006; 10 nM vs 100 nM Iso, p = 0.1270). g, Comparison of cAMP production using different doses of Iso and endocytic blockade with mutant dynamin. GG4B and pcDNA3 (control) or mCherry-Dyn1-K44E expressing cells were untreated (mock) or treated with 100 or 10 nM Iso. Area under the curve (right) shown for the quantification of the time course (left) (n = 4, cells ≥ 9 per biological replicate; control 10 nM Iso vs. Dyn1-K44E 100 nM Iso p = 0.3350; all other comparisons p < 0.0001). h, Kinetics of cAMP response. Each cAMP response from Fig. 1d was normalized to the peak. (control, n = 4, Dyn1-K44E n = 3, Interaction p = 0.9776, Time p < 0.0001, Transfection p = 0.0634). All data are mean ± sem (shaded areas). Significance determined by ordinary one-way ANOVA (e-g), or two-way ANOVA (a-d, h) with Tukey’s (a, d) or Sidak’s (c, e-g) multiple comparisons tests.
Extended Data Fig. 2. Endocytosis blockade reduces β2AR-stimulated cAMP downstream signaling.
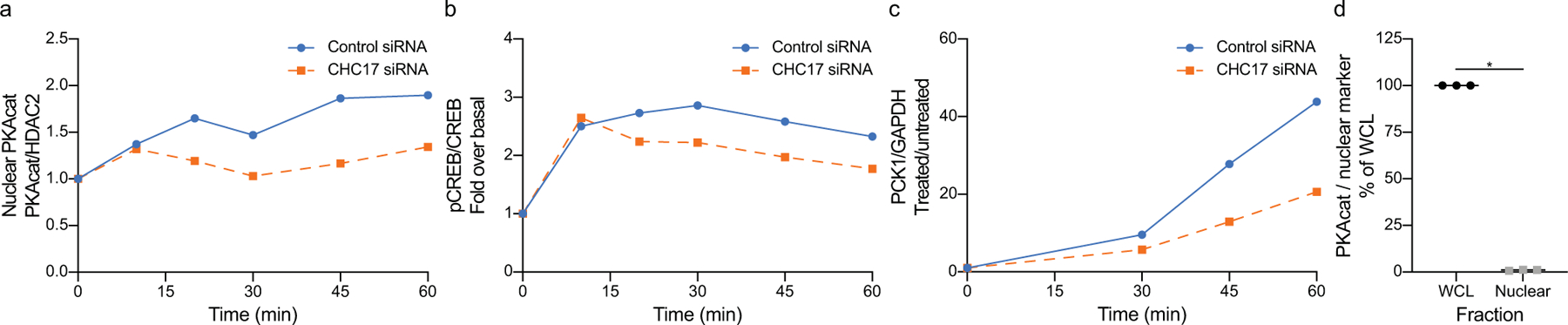
a-c, Nuclear cAMP signaling cascade responses are shown in the same time scale (up to 60 minutes) in cells transfected with control siRNA or CHC17 siRNA. a, Western blot analysis of PKAcat from nuclear samples treated with 100 nM Iso (adapted from Fig. 2c and d). b, Western blot analysis of CREB phosphorylation from whole cell lysates after 100 nM Iso treatment (adapted from Fig. 2b). c, qRT-PCR quantitation of cells treated with 100 nM Iso (adapted from Extended Data Fig. 1c). d, Western blot quantification of PKAcat from whole cell lysate and nuclear samples (n = 3 biological replicates, p < 0.0001, two-tailed unpaired t-test). All data are mean ± sem.
Extended Data Fig. 3. Detection of PKAcat by live microscopy gene-edited mNG2-PKAcat HEK293T cells.
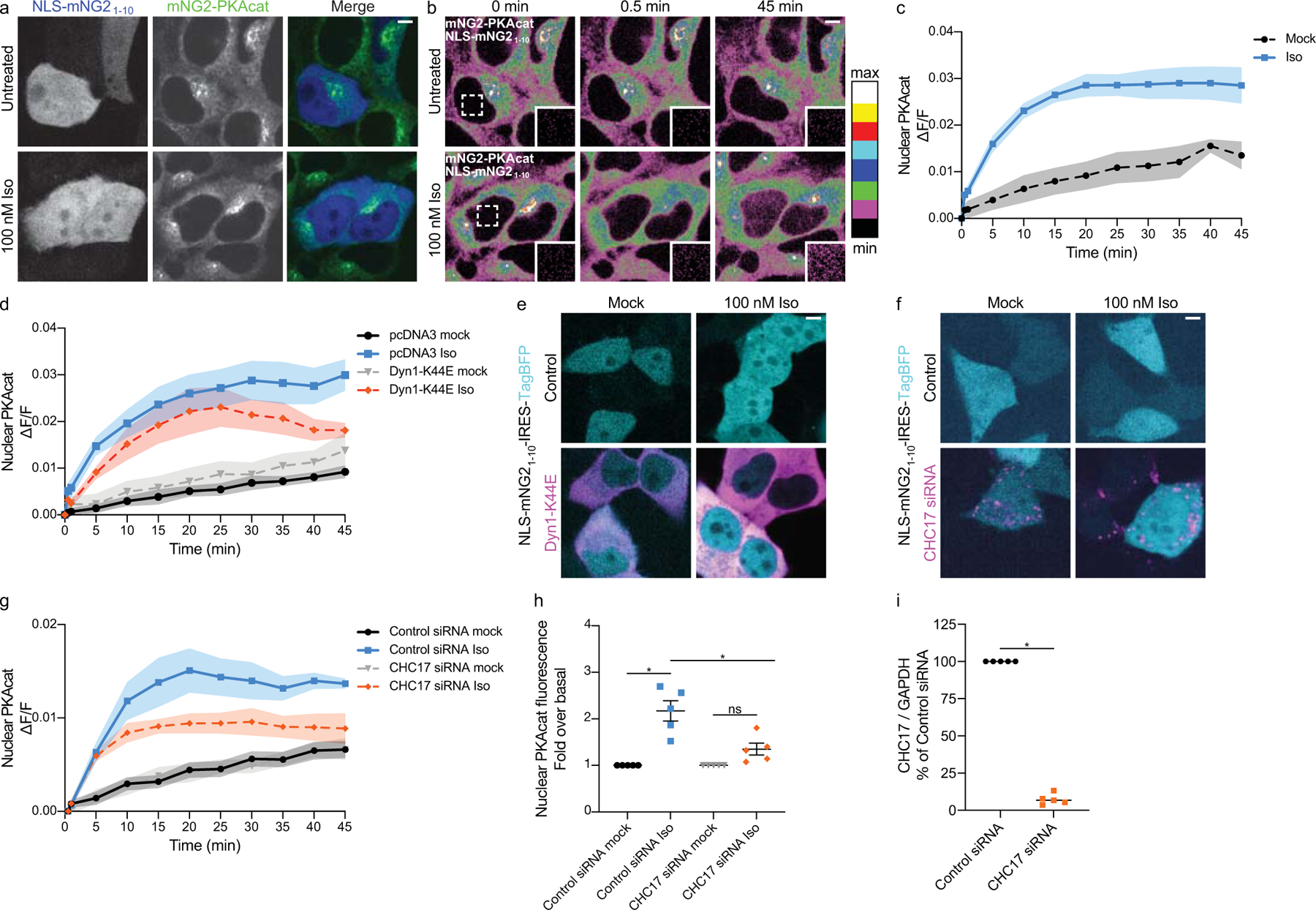
a, Identifying cells expressing NLS-mNG21–10 IRES TagBFP for analysis. Images correspond with cells in b. b, Representative spinning disk confocal images of live cells expressing mNG211-PKAcat, mNG21–10 and nuclear localized NLS-mNG21–10. Cells were untreated (mock) or treated with 100 nM Iso at 0 minutes. Inset shows nuclear ROI shown as dotted line square. c and d, Quantification of PKAcat nuclear accumulation. c, Cells were untreated (mock) or stimulated with 100 nM Iso at 0 minutes in c (n = 3, cells ≥ 21 per biological replicate; Interaction and Time p = 0.0346, Treatment p < 0.0001; p < 0.05 mock vs Iso time = 5–35 and 45 minutes with Sidak’s multiple comparisons test; two-way ANOVA). d, Cells expressing pcDNA3 (control) or mCherry-Dyn1-K44E were untreated (mock) or treated with 100 nM Iso at 0 minutes. (n = 4, cells ≥ 7 per biological replicate; Interaction p = 0.7938, Time and Transfection p < 0.0001, two-way ANOVA). e and f, Identifying cells for analysis. mNG2-PKAcat cells expressing NLS-mNG21–10 IRES TagBFP were co-transfected with NLS-mNG21–10 IRES TagBFP and control or Dyn1-K44E DNA (e) or CHC17 siRNA (f). e, Images correspond to cells in Fig. 3c. g, Quantification of PKAcat nuclear accumulation in mNG2-PKAcat cells transfected with ASN siRNA (control) or AF555-CHC17 siRNA (n = 5, cells ≥ 9 per biological replicate; Interaction, Time and Transfection p < 0.0001; Control siRNA vs CHC17 siRNA mock p = 0.8237, all other comparisons p < 0.0001; Sidak’s multiple comparisons test, two-way ANOVA). h, Quantification of cells transiently expressing ASN siRNA (control) or AF555-CHC17 siRNA from g, data normalized to the corresponding untreated 45 minute time point (n = 5 biological replicates; p = 0.0017 Control siRNA untreated vs Iso, p = 0.5632 CHC17 siRNA untreated vs Iso, p = 0.03 Control siRNA Iso vs CHC17 siRNA Iso; Sidak’s multiple comparisons test, ordinary one-way ANOVA). i, Validation of CHC17 knockdown in imaging experiments quantified in g and h. qRT-PCR for CHC17 and GAPDH transcript levels (n = 5 biological replicates, p < 0.0001, two-tailed unpaired t-test). All data are mean ± sem (shaded areas). Scale bars = 5 μm.
Extended Data Fig. 4. Identification of cells expressing mCherry-Dyn1-K44Ein HEK293T mNG2-PKAcat cells.
Cells expressing mCherry-Dyn1-K44E were identified to determine which cells would be used for analysis. Images correspond with image of cells from Fig. 4b. Scale bar = 5 μm.
Extended Data Fig. 5. Fluorescence recovery after photobleaching analysis.
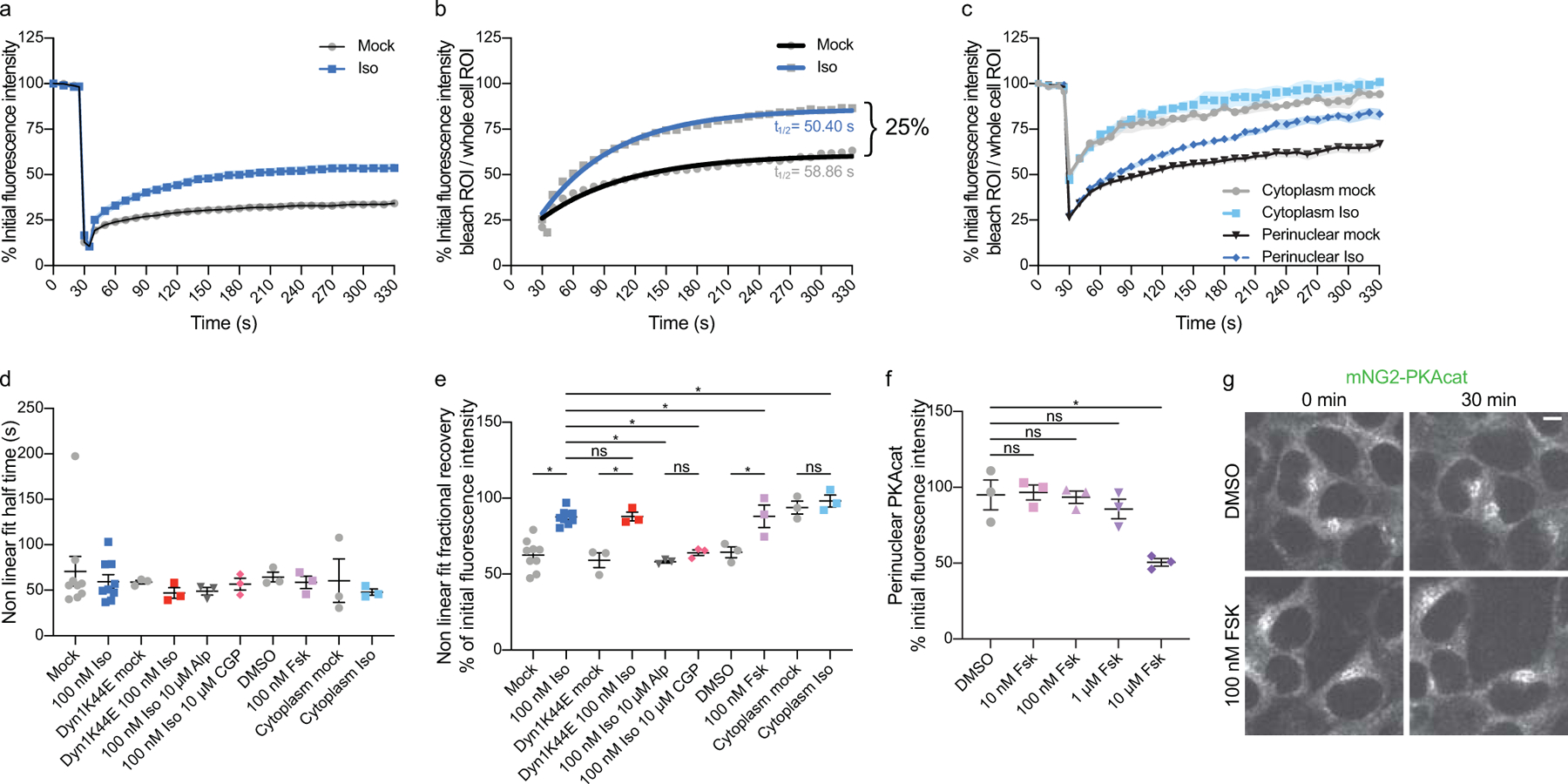
a, Quantification from Fig. 5c without normalizing to whole cell fluorescence. b, Photobleaching recovery curves with non-linear fit. Recovery curves from Fig. 5c are replotted without pre-photobleaching. Each recovery curve was fit using non-linear regression and an exponential one-phase association model. The Iso condition (blue) has a half-time of 50.40 s and fractional recovery of 86.04%. The untreated condition (gray) has a half-time of 58.86 s and a fractional recovery of 61.08% (n = 3, cells ≥ 3 per biological replicate; Interaction, Time and Transfection p < 0.0001; p < 0.05 for untreated vs Iso for time = 30–300 s with Sidak’s multiple comparisons test, two-way ANOVA). c, Photobleaching in different regions of the cell. Cells untreated or treated with 100 nM Iso for 30 minutes are photobleached in a perinuclear or cytoplasmic region (n = 3, cells ≥ 3 per biological replicate; Interaction, Time and Transfection p < 0.0001; all comparisons p < 0.0001 with Sidak’s multiple comparisons test, two-way ANOVA). d and e, Half times and percent maximal recovery of all photobleaching conditions determined by a non-linear regression and an exponential one-phase association model. d, Half times (n ≥ 3, cells ≥ 3 per biological replicate; p = 0.9645, ordinary one-way ANOVA). e, Percent maximal recovery (n ≥ 3, cells ≥ 3 per biological replicate; mock vs Iso and Iso vs Iso Alp p < 0.0001; Dyn1-K44E mock vs Iso and Iso vs Iso Alp p = 0.0005; DMSO vs Fsk p = 0.0057; Perinuclear ROI vs Cytoplasmic ROI p = 0.3736; Iso Alp vs Iso CGP p = 0.9847; Cyto mock vs Iso p = 0.9988; control vs Dyn1-K44E Iso and Iso vs Fsk p > 0.9999; Sidak’s multiple compariosons test, ordinary one-way ANOVA). f and g, Validation of PKAcat return to the perinuclear region after 100 nM forskolin (Fsk). f, Quantification of perinuclear PKAcat 30 minutes after DMSO, 100 nM, 10 μM and 10 μM Fsk treatment (n = 3, cells ≥ 20 per biological replicate; DMSO vs 10 nM Fsk 0.9995; DMSO vs 100 nM Fsk 0.9997; DMSO vs 1 μM Fsk 0.7704; DMSO vs 10 μM Fsk 0.0017; Sidak’s multiple comparisons test, ordinary one-way ANOVA). g, Representative live cell spinning disk confocal images of mNG2-PKAcat before and after 30 minute treatment with DMSO and 100 nM Fsk. Scale bar = 5 μm. Data are mean ± sem.
Extended Data Fig. 6. Endocytosis moves activated β2ARs into close proximity with dynamic PKAcat.
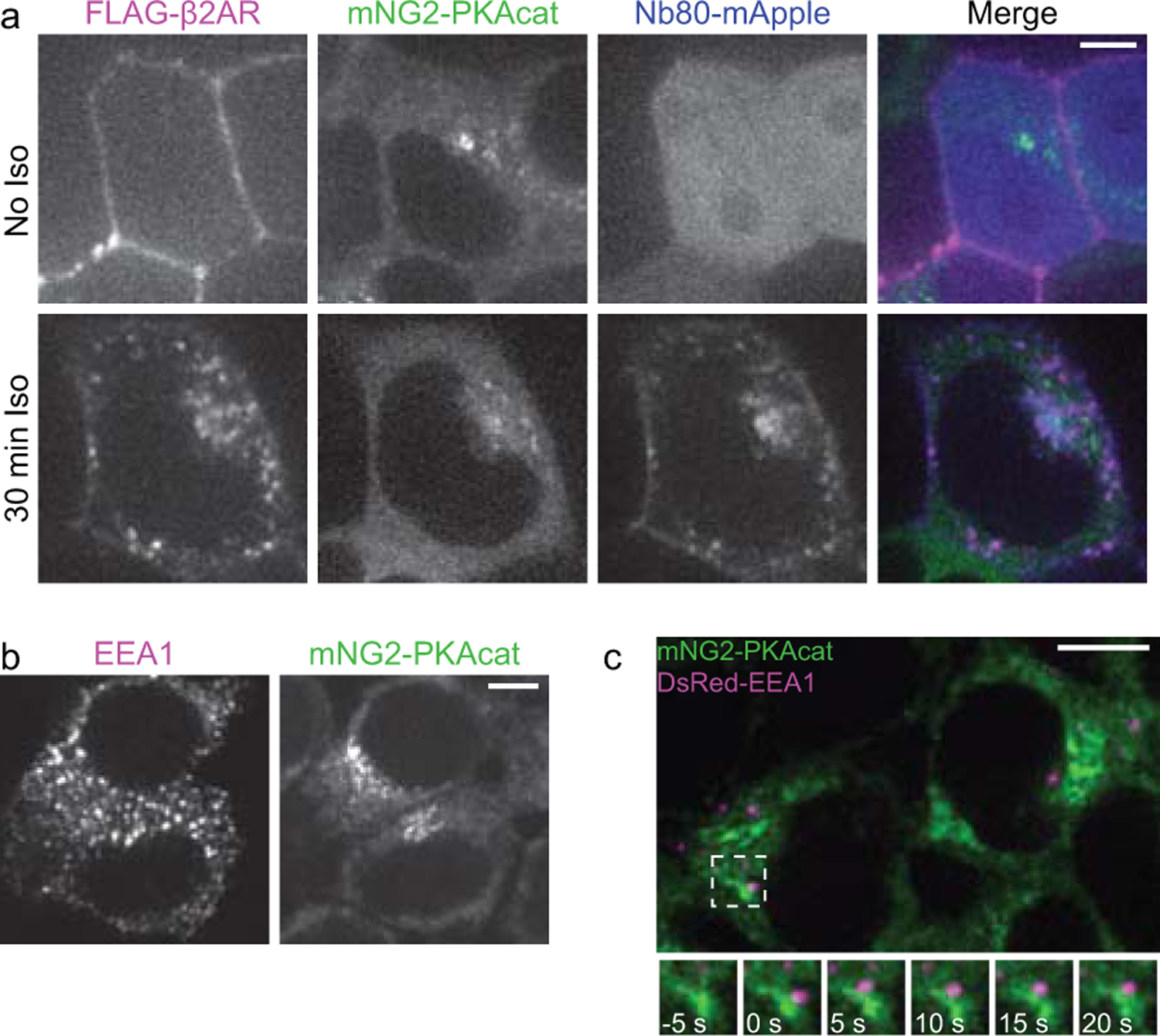
a, Cells were transfected with FLAG-β2AR and Nb80-mApple. Surface FLAG-β2ARs were labeled with M1-FLAG-647 for 15 minutes prior to imaging. Representative spinning disk confocal images of live cells were taken before and 30 minutes after 100 nM Iso addition. b, Spinning disk confocal fixed images of mNG2-PKAcat cells stained for EEA1. Individual channels from merged image in Fig. 6d. c, Spinning disk confocal live images of mNG2-PKAcat cells transiently expressing DsRed-EEA1. Cells were pretreated with 100 nM Iso for 25 minutes and imaged for 5 minutes at 5 second intervals. Scale bar = 5 μm.
Supplementary Material
Statistical source data file for Supplementary Figure 2.
Movie of a live confocal image series of endogenously labeled mNG2-PKAcat (green) and endosomes marked by DsRed-EEA1(2x FYVE) (magenta), providing a representative example of sequential dispersal and punctate relocalization of PKAcat during the biphasic β2AR response. A 25 min series is shown, with image capture every 20 s. Elapsed time is indicated at the upper left (mm:ss). 100 nM Iso was bath applied at 2:40. Scale bar = 5 µm.
Movie of fluorescence recovery after photobleaching of an untreated endogenously labeled mNG2-PKAcat cell. Cells were untreated for 30 minutes prior to imaging and photobleached at 30 seconds. Recovery was observed for 300 seconds after photobleaching. Corresponds to images in Fig 5b, top row. Scale bar = 5 µm.
Movie of a live confocal image series (corresponding with Extended Data Fig. 6c) of endogenously labeled mNG2-PKAcat (green and endosomes marked by DsRed-EEA1(2x FYVE) (magenta). A 50 s series is shown, with image capture every 5 s. The green channel displays endogenously labeled mNG2-PKAcat. The magenta channel displays endosomes marked by DsRed-EEA1(2x FYVE). A calculated image corresponding to the product of the green and magenta channels is overlaid and displayed using the built-in ‘fire’ lookup table of ImageJ. Scale bar = 5 µm.
Movie of fluorescence recovery after photobleaching of an Iso treated endogenously labeled mNG2-PKAcat cell. Cells were treated for 30 minutes with 100 nM Iso prior to imaging, and photobleached at 30 seconds. Recovery was observed for 300 seconds after photobleaching. Corresponds to images in Fig 5b, bottom row. Scale bar = 5 µm.
ACKNOWLEDGMENTS
We thank A. Marley for developing the cAMP fluorescence biosensor and S. Kotowski for generating the DsRed-tagged dynamin1 constructs. We thank B. Lobingier for help with the development of the cAMP luminescence assay. We also thank N. Tsvetanova, R. Irannejad, D. Jullié and members of the von Zastrow lab for valuable advice and discussion, as well as K. Thorn, D. Larsen, and K. Herrington (Nikon Imaging Center, UCSF, National Institute of Health 1S10OD017993-01A1) for technical support and expertise. We also thank M. Chevalier, R. Dror, N. L’Etoile, R. D. Mullins, S. Taylor, O. Weiner and J. Zhang for valuable discussion. These studies were supported by the National Institute on Drug Abuse (DA012864 and DA010711 to M.v.Z.), the National Institute of Mental Health (MH120212 to M.v.Z.), the National Heart, Lung and Blood Institute (HL129689 to G.E.P.), the National Institute of Biomedical Imaging and Bioengineering (EB022798 to B.H.), and the American Heart Association (15PRE21770003 to V.P., 16PRE26420057 to G.E.P.).
Footnotes
COMPETING FINANCIAL INTERESTS STATEMENT
The authors declare no competing interests.
DATA AVAILABILITY
The data that support the findings of this study are available from the authors upon reasonable request. Source data and uncropped blots for Fig. 1–5, Extended Data Fig. 1, 2, 3, 5 and Supplementary Fig. 3 are provided with this paper.
REFERENCES
- 1.Kwok-Keung Fung B & Stryer L Photolyzed rhodopsin catalyzes the exchange of GTP for bound GDP in retinal rod outer segments. Proc. Natl. Acad. Sci 77, 2500–2504 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsvetanova NG, Irannejad R & von Zastrow M G protein-coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes. J. Biol. Chem 290, 6689–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pavlos NJ & Friedman PA GPCR Signaling and Trafficking: The Long and Short of It. Trends Endocrinol. Metab 28, 213–226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calebiro D & Godbole A Internalization of G-protein-coupled receptors: Implication in receptor function, physiology and diseases. Best Pract. Res. Clin. Endocrinol. Metab 32, 83–91 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Vilardaga JP, Jean-Alphonse FG & Gardella TJ Endosomal generation of cAMP in GPCR signaling. Nature Chemical Biology 10, 700–706 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohse MJ & Hofmann KP Spatial and temporal aspects of signaling by G-protein-coupled receptors. Mol. Pharmacol 88, 572–578 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Lobingier BT & von Zastrow M When trafficking and signaling mix: How subcellular location shapes G protein-coupled receptor activation of heterotrimeric G proteins. Traffic 20, 130–136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calebiro D et al. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol 7, e1000172 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrandon S et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat. Chem. Biol 5, 734–742 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wehbi VL et al. Noncanonical GPCR signaling arising from a PTH receptor-arrestin- Gβγ complex. Proc. Natl. Acad. Sci 110, 1530–1535 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomsen ARB et al. GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 166, 907–919 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keenan DM & Veldhuis JD Pulsatility of hypothalamo-pituitary hormones: A challenge in quantification. Physiology 31, 34–50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyga S et al. Persistent cAMP Signaling by Internalized LH Receptors in Ovarian Follicles. Endocrinology 157, 1613–1621 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Irannejad R et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao TT, Deacon HW, Reczek D, Bretscher A & von Zastrow M A kinase-regulated PDZ-domain interaction controls endocytic sorting of the β2-adrenergic receptor. Nature 401, 286–290 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Uchida Y, Rutaganira FU, Jullié D, Shokat KM & Von Zastrow M Endosomal phosphatidylinositol 3-kinase is essential for canonical GPCR signaling. Mol. Pharmacol 91, 65–73 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsvetanova NG & von Zastrow M Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol 10, 1061–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Banion CP, Vickerman BM, Haar L & Lawrence DS Compartmentalized cAMP Generation by Engineered Photoactivated Adenylyl Cyclases. Cell Chem. Biol 26, 1393–1406.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lazar AM et al. G protein-regulated endocytic trafficking of adenylyl cyclase type 9. Elife 9, e58039 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Binkowski BF et al. A luminescent biosensor with increased dynamic range for intracellular cAMP. ACS Chem. Biol 6, 1193–1197 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez GA & Montminy MR Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59, 675–680 (1989). [DOI] [PubMed] [Google Scholar]
- 22.Nigg EAA, Hilz H, Eppenberger HMM & Dutly F Rapid and reversible translocation of the catalytic subunit of cAMP-dependent protein kinase type II from the Golgi complex to the nucleus. EMBO J 4, 2801–2806 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clister T et al. AKAP95 Organizes a Nuclear Microdomain to Control Local cAMP for Regulating Nuclear PKA. Cell Chemical Biology 26, 885–891.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamiyama D et al. Versatile protein tagging in cells with split fluorescent protein. Nat. Commun 7, 11046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leonetti MD et al. A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc. Natl. Acad. Sci 113, E3501–E3508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uhler MD, Chrivia JC & McKnight GS Evidence for a second isoform of the catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem 261, 15360–3 (1986). [PubMed] [Google Scholar]
- 27.Feng S et al. Improved split fluorescent proteins for endogenous protein labeling. Nat. Commun 8, 370 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mavillard F, Hidalgo J, Megias D, Levitsky KL & Velasco A PKA-Mediated Golgi remodeling during cAMP signal transmission. Traffic 11, 90–109 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Sprague BL & McNally JG FRAP analysis of binding: Proper and fitting. Trends Cell Biol 15, 84–91 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Walker-Gray R, Stengel F & Gold MG Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and cross-linking approaches. Proc. Natl. Acad. Sci 114, 10414–10419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng X, Phelps C & Taylor SS Differential Binding of cAMP-dependent Protein Kinase Regulatory Subunit Isoforms Iα and IIβ to the Catalytic Subunit. J. Biol. Chem 276, 4102–4108 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Zawadzki KM & Taylor SS cAMP-dependent protein kinase regulatory subunit type IIbeta: active site mutations define an isoform-specific network for allosteric signaling by cAMP. J. Biol. Chem 279, 7029–36 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Schlosshauer M & Baker D Realistic protein-protein association rates from a simple diffusional model neglecting long-range interactions, free energy barriers, and landscape ruggedness. Protein Sci 13, 1660–1669 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irannejad R et al. Functional selectivity of GPCR-directed drug action through location bias. Nat. Chem. Biol 13, 799–806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gillooly DJ et al. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J 19, 4577–4588 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berg HC & Purcell EM Physics of Chemoreception. Biophys. J 20, 193–219 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ashcroft NW & Mermin ND Solid State Physics (Harcourt, Inc., 1976). [Google Scholar]
- 38.Sze SM Physics of Semiconductor Devices (John Wiley & Sons, Inc., 1981). [Google Scholar]
- 39.Zhang JZ et al. Phase separation of a PKA regulatory subunit controls cAMP compartmentation and oncogenic signaling. Cell 182, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bock A et al. Optical mapping of cAMP signaling at the nanometer scale. Cell 182, 1–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beavo JA, Bechtel PJ & Krebs EG Activation of protein kinase by physiological concentrations of cyclic AMP. Proc. Natl. Acad. Sci 71, 3580–3 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koschinski A & Zaccolo M Activation of PKA in cell requires higher concentration of cAMP than in vitro: implications for compartmentalization of cAMP signalling. Sci. Rep 7, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS REFERENCES
- 43.Zhao Y et al. An Expanded Palette of Genetically Encoded Ca2+ Indicators. Science (80-. ) 333, 1888–1891 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patriarchi T et al. Imaging neuromodulators with high spatiotemporal resolution using genetically encoded indicators. Nat. Protoc 14, 3471–3505 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Niwa H, Yamamura K & Miyazaki J Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199 (1991). [DOI] [PubMed] [Google Scholar]
- 46.Lin S, Staahl BT, Alla RK & Doudna JA Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3, e04766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (80-. ) 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edelstein A, Amodaj N, Hoover K, Vale R & Stuurman N Computer Control of Microscopes Using µManager. in Current Protocols in Molecular Biology 92, 14.20.1–14.20.17 (John Wiley & Sons, Inc., 2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edelstein AD et al. Advanced methods of microscope control using μManager software. J. Biol. Methods 1, 1–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abràmoff MD, Magalhães PJ & Ram SJ Image Processing with ImageJ. Biophotonics Int 11, 36–42 (2004). [Google Scholar]
- 51.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistical source data file for Supplementary Figure 2.
Movie of a live confocal image series of endogenously labeled mNG2-PKAcat (green) and endosomes marked by DsRed-EEA1(2x FYVE) (magenta), providing a representative example of sequential dispersal and punctate relocalization of PKAcat during the biphasic β2AR response. A 25 min series is shown, with image capture every 20 s. Elapsed time is indicated at the upper left (mm:ss). 100 nM Iso was bath applied at 2:40. Scale bar = 5 µm.
Movie of fluorescence recovery after photobleaching of an untreated endogenously labeled mNG2-PKAcat cell. Cells were untreated for 30 minutes prior to imaging and photobleached at 30 seconds. Recovery was observed for 300 seconds after photobleaching. Corresponds to images in Fig 5b, top row. Scale bar = 5 µm.
Movie of a live confocal image series (corresponding with Extended Data Fig. 6c) of endogenously labeled mNG2-PKAcat (green and endosomes marked by DsRed-EEA1(2x FYVE) (magenta). A 50 s series is shown, with image capture every 5 s. The green channel displays endogenously labeled mNG2-PKAcat. The magenta channel displays endosomes marked by DsRed-EEA1(2x FYVE). A calculated image corresponding to the product of the green and magenta channels is overlaid and displayed using the built-in ‘fire’ lookup table of ImageJ. Scale bar = 5 µm.
Movie of fluorescence recovery after photobleaching of an Iso treated endogenously labeled mNG2-PKAcat cell. Cells were treated for 30 minutes with 100 nM Iso prior to imaging, and photobleached at 30 seconds. Recovery was observed for 300 seconds after photobleaching. Corresponds to images in Fig 5b, bottom row. Scale bar = 5 µm.
Data Availability Statement
The data that support the findings of this study are available from the authors upon reasonable request. Source data and uncropped blots for Fig. 1–5, Extended Data Fig. 1, 2, 3, 5 and Supplementary Fig. 3 are provided with this paper.