

Abstract
Worldwide, each year over 30,000 patients undergo an allogeneic hematopoietic stem cell transplantation with the intent to cure high-risk hematologic malignancy, immunodeficiency, metabolic disease, or a life-threatening bone marrow failure syndrome. Despite substantial advances in donor selection and conditioning regimens and greater availability of allograft sources, transplant recipients still endure the morbidity and mortality of graft-versus-host disease (GVHD). Herein, we identify key aspects of acute and chronic GVHD pathophysiology, including host/donor cell effectors, gut dysbiosis, immune system and cytokine imbalance, and the interface between inflammation and tissue fibrosis. In particular, we also summarize the translational application of this heightened understanding of immune dysregulation in the design of novel therapies to prevent and treat GVHD.
Keywords: graft-versus-host disease, pathophysiology, prevention, therapy, regulation, tolerance
1. INTRODUCTION
Transplantation of allogeneic bone marrow or granulocyte-colony stimulating factor (G-CSF)-mobilized peripheral blood stem cells (G-PBSCs) [hereafter, allogeneic hematopoietic stem cell transplantation (allo-HSCT)] is a cornerstone curative therapy for patients with high-risk hematological malignancies or life-threatening lymphohematopoietic disorders. Cytoreductive and immunosuppressive chemotherapy minimizes malignancy and provides immune suppression to prevent rejection of donor grafts containing stem and progenitor cells, mature myeloid cells, and innate and adaptive lymphocytes. While most donor sources provide HLA-matched related or unrelated donor grafts, alternative sources, especially half-matched (haploidentical) family members and, in some instances, variably mismatched cord blood, are increasingly being used.
Despite immunoprophylaxis, donor responses to host HLA and/or minor histocompatibility (miH) antigen disparities result in graft-versus-host disease (GVHD) in 30–70% of allogeneic transplant recipients. Acute GVHD (aGVHD), a cytolytic, tissue-destructive process involving skin, gut, liver, and, recently demonstrated, lung and central nervous system, is most often diagnosed 3–12 weeks after HSCT but may occur later (1). aGVHD is initiated by innate immune cells activated during conditioning-regimen-induced inflammation and by tissue injury amplified by adaptive immune responses. Chronic GVHD (cGVHD), immunologically distinct from aGVHD, is characterized by T helper type 2 (Th2)- rather than Th1-skewed responses, immune dysregulation and autoimmunity, and/or fibrosis (2). Typically presenting ≥6 months after HSCT, cGVHD is heterogeneous, can affect most tissues, and often involves the skin with lichenoid plaques, eyes with sicca symptoms, joints, and skin and lung fibrosis.
Improved understanding of GVHD immunology in murine systems has facilitated translation of novel therapies such as kinase inhibitors, approved for GVHD treatment. Here we highlight cellular and molecular pathways exploitable for GVHD prevention and therapy while discussing the overarching need for regulation and eventual tolerance. Although GVHD and graft-versus-leukemia (GVL)/graft-versus-tumor responses are interrelated, due to the scope of that topic, we refer the reader to a recent publication that details this relationship more extensively than can be done here (reviewed in 3).
2. ACUTE GVHD INITIATION
Key aGVHD mechanisms, subject of intense study in albeit imperfect rodent models (4, 5), are predicated on recipient antigen-presenting cell (APC)-donor T cell interactions (6) (Figure 1). Subsequent T cell responses occur in a profoundly inflamed, lymphodepleted environment where alloantigen availability is ubiquitous and indefinite. Patients often have been pretreated with extensive chemotherapy, accompanied by broad-spectrum antibiotics disrupting the microbiota ecosystem within the gastrointestinal tract, especially the ileum, where microbiota and innate cells constantly interact (6, 7).
Figure 1.
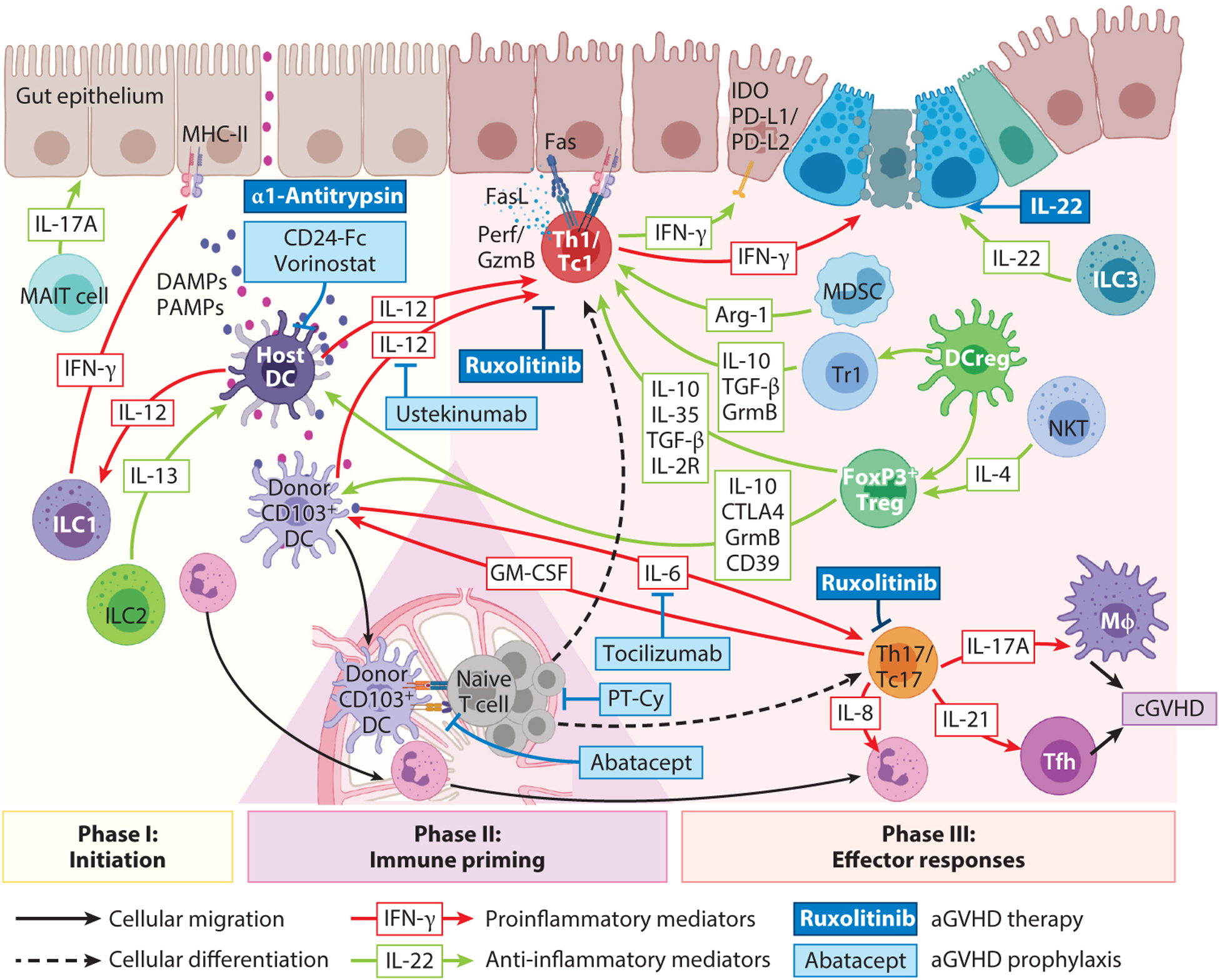
aGVHD (acute GVHD) pathogenesis and treatment. Phase I: GVHD initiation. Pretransplantation conditioning causes barrier integrity loss, gut lumen pathogen-associated molecular pattern (PAMP) translocation, and dying host cell release of damage-associated molecular patterns (DAMPs) that activate host antigen-presenting cells (APCs). Host mucosal-associated invariant T (MAIT) cells produce IL-17A, which prevents intestinal dysbiosis. Activated APCs secrete IL-12, inducing host type 1 innate lymphoid cells (ILC1s) and T cells to release IFN-γ, which upregulates MHC-II on nonprofessional APCs (epithelial and stromal cells). APC activation is suppressed by IL-13-producing ILC2s, or by regulatory T cells (Tregs) utilizing secreted (IL-10) or contact-dependent (CTLA4, granzyme B, CD39) mechanisms, and modified by Siglec G-CD24 pathway agonists (e.g., CD24-Fc) or IL-12 inhibition (ustekinumab). Histone deacetylase inhibitor vorinostat and α−1-antitrypsin inhibit PAMP/DAMP-induced APC proinflammatory cytokine production and costimulation. Phase II: Donor T cell priming. Donor T cell activation follows interaction with recipient hematopoietic APCs [e.g. dendritic cells (DCs)] or nonhematopoietic APCs activated during phase I. Reconstituting donor CD103+ DCs migrate to draining lymph nodes, where they further prime donor T cells. Abatacept (CTLA4-Ig) disrupts Tcell:APC costimulation to inhibit aGVHD. Alloreactive donor T cell expansion is curtailed by early posttransplantation cyclophosphamide (PT-Cy). Phase III: effector responses. Primed T cells differentiate to type 1 T helper (Th1)/type 1 CD8+ T (Tc1) or Th17/Tc17 cells, directed by IL-12 or IL-6, respectively, and are susceptible to the Jak1/2 inhibitor ruxolitinib, IL-12/23p40 (ustekinumab), or IL-6R (tocilizumab) blocking monoclonal antibodies. Th1/Tc1 cells secrete IFN-γ, which is intestinal stem cell cytotoxic, while augmenting the negative regulatory indoleamine-2,3-dioxygenase (IDO) and PD-L1/PD-L2 pathways to restrain aGVHD. Th17/Tc17 cells secrete proinflammatory cytokines that mediate tissue pathology both directly and by recruitment of secondary effectors [e.g., neutrophils, Mφs (macrophages)]. IL-22-secreting ILC3s or exogenous IL-22 supplementation support intestinal stem cell regeneration. Myeloid-derived suppressor cells (MDSCs) suppress T effector functions via an arginase-1-dependent mechanism. Tregs [supported by IL-4-producing natural killer T (NKT) cells and regulatory DC cells (DCregs)] and regulatory type 1 T cells exert multiple pathways to control T effector cells. Abbreviations: cGVHD, chronic GVHD; GM-CSF, granulocyte-macrophage colony-stimulating factor; Tfh, T follicular helper; Tr1, regulatory type 1 T cell. Figure adapted
Pretransplantation conditioning invokes inflammation, intestinal microbiota translocation of pathogen-associated molecular pattern (PAMP) molecules, and danger-associated molecular pattern (DAMP) molecule release from dying host cells (reviewed in 1). PAMPs, including bacterium-derived lipopolysaccharide and peptide-glycans, stimulate Toll-like receptors (TLRs) on innate cells, such as monocytes/macrophages and dendritic cells (DCs), initiating an inflammatory cytokine cascade of IL-12, IL-23, and IL-6 (6, 7). DAMPs (HMGB1, ATP, and uric acid) amplify inflammatory cytokine secretion, culminating in high local (gut) IL-12 levels (6). IL-12 promotes IFN-γ secretion by murine and human type 1 innate lymphoid cells (ILC1s) (8, 9) and recipient T conventional (Tcon) cells, markedly upregulating MHC expression on intestinal epithelial cells, enabling APC function and donor T cell priming to initiate lethal gut aGVHD (7). Conversely, ILC2s downregulate gut aGVHD via IL-13 secretion (10). Likewise, peritransplantation IL-22 supports gastrointestinal cytoprotective ILC3s (11).
Recipient APCs in lymphoid structures initiate naive donor T cell differentiation, which dominates the aGVHD effector phase (12). Profound lymphodepletion and extensive inflammation postconditioning drive rapid homeostatic expansion and differentiation, enabling T cell entry into target tissues, where they interact with professional and nonprofessional APCs (6, 13). Recipient DCs in primary lymphoid organs are responsible for dramatic clonal donor T cell deletion (13). Conversely, aGVHD is exacerbated in their absence, a finding attributed to uncontrolled donor T cell expansion (13) and an observed increase in the inflammatory chemokine CCL2, which recruits precursors to refill the mucosal DC compartment (14). In some (15) but not all (16) murine studies, aGVHD is exacerbated in B cell-deficient recipients, with regulation by IL-10; in macrophage-deficient recipients, aGVHD is exacerbated due to loss of regulation of CD47-mediated signals (17).
Hematopoietic recipient APCs appear critical for MHC-I-dependent aGVHD (18), but no single professional APC type has been identified as critical for MHC-II-dependent aGVHD, suggesting redundancy (6). Nonprofessional APCs, epithelial and mesenchymal cells, can initiate MHC-II-dependent aGVHD following conditioning and microbiota disruption (6, 13, 19). While immunological pathways involved in initiating MHC-I- versus MHC-II-dependent aGVHD differ, PAMP and DAMP signals are central to both. Lastly, donor DCs within the aGVHD-damaged colon respond to microbiota locally. In mice and humans undergoing allogeneic HSCT, disruption of microbiota homeostasis (dysbiosis) can modulate intestinal inflammation and injury, thereby accelerating aGVHD, with the patterns in mice and humans mirroring each other (20). In mice and humans, aGVHD caused expansion of loss of anaerobic bacterial order Clostridiales and expansion of the gram-positive lactic acid bacteria order Lactobacillales (20, 21). Intestinal dysbiosis recently has been reported in patients with cGVHD, and those with higher plasma butyrate and propionate levels appeared to be protected from this complication (22). Along with aGVHD, broad-spectrum antibiotic use after allogeneic HSCT (23) causes loss of commensals such as the anerobic Blautia (24) and overall diversity that is itself a predictor of mortality in patients undergoing allogeneic HSCT (25). Autologous fecal transplants can restore bacterial species diversity in allogeneic HSCT patients, but effects on GVHD are yet to be definitively demonstrated (26). For further information, the reader is referred to two recent reviews on this subject (27, 28).
In mice, upon donor DC activation, DCs expand and migrate to mesenteric lymph nodes (LNs), promoting T cell priming, differentiation, and gut homing integrin receptor imprinting in a feed-forward cascade for aGVHD that is driven by donor T cell granulocyte-macrophage colony-stimulating factor (GM-CSF) secretion (19, 29).
2.1. Acute GVHD T Effectors
Following priming, naive T cells differentiate and are licensed for tissue destruction. DAMPs and PAMPs (reviewed in 30) and strong alloantigen-driven T cell receptor (TCR) stimulation favor Th1 over Th17 skewing (31). T-bet-expressing Th1 cells arise under polarizing conditions such as where IL-12 is generated by macrophages and DCs, augmented by T cell, natural killer (NK) cell, and antigen-presenting cell production of IFN-γ and macrophage, mononuclear cell, and DC production of IL-18, leading to IFN-γ, IL-2, and TNF-α secretion by Th1 cells (32). Without immunoprophylaxis, autocrine and paracrine IL-2 signaling in Th1/type 1 CD8+ T (Tc1) cells supported high proliferation seen in nonhuman primates and patients receiving posttransplantation cyclophosphamide (PT-Cy), respectively (33, 34). Excessive Th1 cytokine production in mice led to the initial concept that aGVHD is primarily driven by Th1/Tc1- rather than Th2/Tc2-associated immunopathology (35). Donor T cell IFN-γ was also directly cytotoxic to intestinal stem cells during gastrointestinal murine aGVHD (36), yet it induced a host tissue-protective program by upregulating indoleamine-2,3-dioxygenase (IDO) (37) and PD-L1 (38). IFN-γ ablation or blockade shifted aGVHD from gastrointestinal damage to pulmonary pneumonitis (39). During murine aGVHD, Th/Tc1 cells exert multiple overlapping and redundant cytotoxicity pathways, of Fas/FasL, perforin/granzymes, and TRAIL (TNF-related apoptosis-inducing ligand) (40, 41). Perforin/granzyme-mediated cytotoxicity was more prominent in murine miH-disparate CD8+ T cell-driven aGVHD and likely central in clinical aGVHD (40). FasL was highly expressed by Th/Tc1 cells, whereas Fas, induced in murine aGVHD tissues, caused aGVHD damage (41).
More recently, Th17/Tc17 cell expansion early after allo-HSCT, mediated by IL-6, has been shown to recruit neutrophils to inflammatory sites by secreted CXCL8; neutrophils are important in gastrointestinal aGVHD pathogenesis, and Th17/Tc17-specific ablation early after HSCT protected mice from aGVHD (42–44). In vitro differentiated murine or human Th17 cell transfer caused lethal aGVHD with skin and lung manifestations (45, 46), while neutralizing IL-17A (45) partially reversed aGVHD. In mice, nonhuman primates, and humans, Th17/Tc17 cells were increasingly important in orchestrating GVHD as time progressed, reflecting their resistance to pharmacological immune suppression relative to Th1 effectors (45, 47, 48). Tc17 cells post-transplantation were poorly cytotoxic but produced proinflammatory cytokines IL-17A, IL-22, GM-CSF, and IFN-γ, contributing to aGVHD (43). Allogeneic donor T cell GM-CSF licensed donor-derived phagocytes to produce inflammatory mediators and directly expanded donor DCs to increase indirect alloantigen presentation and secrete costimulatory cytokines such as IL-23, providing a pathway that amplified aGVHD (19, 49, 50). Thus, Th17 and Tc17 cells generate large amounts of inflammatory mediators distinct from those of Th1 and Tc1 cells that can amplify aGVHD (43). In contrast, host IL-22 deficiency accelerated gut aGVHD (11).
2.2. Peritransplantation Immunity Modulation by Innate and Nonconventional T Cells
Recipient innate-like cells modulate peritransplantation immunity. NK cells are relatively radioresistant and supported (e.g., IL-2, IL-15) and activated (e.g., IL-12, IL-18) by cytokines and activating and inhibitory receptor ligation (e.g., by killer inhibitor receptors) in the context of MHC disparity, classically missing self-MHC-I (51). Activated NK cells reject MHC-disparate allogeneic hematopoietic stem cell grafts and produce large amounts of IFN-γ, enhancing recipient APC activation and alloantigen presentation (51, 52).
Natural killer T (NKT) cells, innate-like T cells that express both NK and T cell markers, can be divided into type I and type II subsets by their semi-invariant or diverse TCR-restricted response to glycolipid or an alternative (e.g., sulfite ligands), respectively, when expressed by the MHC-I-like molecule CD1d (53). NKT cells rapidly produce high cytokine levels following TCR ligation. In mice, CD4+ type I NKTs attenuated GVHD via IL-4-dependent donor regulatory T cell (Treg) induction (54). Total lymphoid irradiation conditioning and antithymocyte globulin enriched recipient regulatory NKT cells, associated with low aGVHD rates in patients (55). Conversely, donor CD4− NKT cells increased recipient APC function and cytotoxic T lymphocyte priming, promoting antitumor immunity (56). The role of type II NKT cells in GVHD is less clear.
Mucosal-associated invariant T (MAIT) cells represent a large proportion of the innate-like T cell population, predominantly CD8+ with a semi-invariant TCR responsive to bacterial riboflavin metabolites presented in the MHC-I-like molecule MR1 (57). MAIT cells reside principally at mucosal surfaces to prevent dysbiosis before transplantation and secrete large amounts of cytokines important in mucosal barrier function such as IL-17A (58). Recipient mice lacking MAIT cells have compromised mucosal integrity, augmented donor DC alloantigen presentation, heightened donor T cell priming, and severe colonic aGVHD relative to wild-type mice (58). A similar but even more exaggerated phenotype was seen in recipients lacking IL-17A, reflecting extensive dysbiosis in these animals at steady state (59). Clinically, G-CSF mobilized MAIT cells into peripheral blood (60), and donor MAIT cell number reductions were associated with dysbiosis and transplant-related mortality (61).
Lastly, γδ T cells, a major skin and mucosal (e.g., gastrointestinal tract) T cell population, are characterized by innate-like cytokine (e.g., IFN-γ, TNF) and cytotoxic (e.g., perforin/granzyme) responses to stress ligands, are not MHC restricted, and are important in pathogen responses, tissue repair, and regeneration (62). Recipient γδ T cells appear activated during conditioning, augmenting the APC function of donor T cell priming to increase aGVHD (63). Donor γδ T cells did not invoke aGVHD directly and are of interest in T cell-depleted bone marrow transplantation (BMT) due to the possibility of mediating pathogen- and leukemia-specific immunity (64). Although clinical data are mixed for associations of robust γδ T cell reconstitution and improved transplantation outcomes, increasing αβ T cell-depleted graft use raised renewed interest in γδ T cells (reviewed in 65).
2.3. Myeloid Cell Acute GVHD Amplification
Neutrophils and CD103+ DCs participate in aGVHD initiation and are promising targets to prevent aGVHD. Neutrophil-mediated insult to aGVHD-target organs requires (a) transplantation-conditioning-regimen-mediated tissue damage, (b) leukotaxis orchestrated by tissue-resident donor T cells, and (c) commensal translocation from the intestine (44). This alignment occurs very early after transplantation and depends on surviving host neutrophils that traffic to and invade the terminal ileum by day +3 (44), coinciding with alloreactive donor T cell arrival. Selective neutrophil depletion by anti-Ly6G monoclonal antibody (mAb) reduced aGVHD lethality. Intralesional neutrophil burden appeared to correlate with patient aGVHD severity. During gastrointestinal aGVHD, ileal mucosal barrier damage allowed host neutrophils to exit and interact with donor T cells in mesenteric LNs, potentiating alloimmune inflammation (66) and contributing to very early stages of aGVHD pathogenesis. Donor-derived CD103+CD11b− DCs critically exacerbated alloimmune intestinal inflammation; presented exogenous alloantigen; migrated to mesenteric LNs; interacted with donor T cells, producing IL-6 and IL-12 that costimulated donor T cells; and ensured ongoing gut damage by directing α4β7+ donor T cell intestinal migration (29).
3. ACUTE GVHD PREVENTION
An increasingly accepted composite end point for allo-HSCT success is one-year severe aGVHD (grade III/IV)-free, cGVHD (requiring systemic therapy)-free, and relapse-free survival, termed modified GRFS (67). Traditional strategies to prevent aGVHD developed by Storb and coworkers (68, 69) in dogs centered on a calcineurin inhibitor (CNI) (cyclosporin or tacrolimus) with methotrexate after myeloablative conditioning or mycophenolate mofetil (MMF) after non-myeloablative conditioning, although significant aGVHD still occurred. Sirolimus (rapamycin) with CNI/MMF increased survival and reduced aGVHD in the nonmyeloablative setting and is now a standard combination (70). GVHD prophylaxis with steroids requires careful timing and dosing to avoid negative outcomes (71) and is not widely utilized. Pretransplantation antithymocyte globulin in patients depletes T cells, modulates leukocyte/endothelial interactions, induces B cell apoptosis, interferes with DC function and Treg and NKT cell induction, and reduces severe aGVHD and cGVHD, albeit without survival benefits (72, 73). Routine antithymocyte globulin use remains debated between continents (74). Specific naive T cell depletion from grafts demonstrated low cGVHD rates (75) (Figure 1).
More recently, high-dose PT-Cy on days 3 and 4 after HSCT, with or without CNI/MMF, has been shown to be well-tolerated and highly effective in haploidentical transplantation, permitting excellent clinical outcomes with relatively low severe aGVHD and cGVHD rates. PT-Cy is increasingly studied in HLA-matched transplantation (76, 77). In preclinical models, PT-Cy caused alloreactive T cell depletion, spared Tregs, and induced tolerance (78). Selective naive T cell depletion resulted in low incidence of cGVHD and severe aGVHD (75). Although CCR5 knockout donor T cells in nonirradiated mice lessened aGVHD by failing to access Peyer patches, aGVHD was accelerated in irradiated mice receiving MHC-disparate grafts; nonetheless, in a phase 2 clinical study, maraviroc, a small-molecule CCR5 antagonist, reduced aGVHD (79).
Approaches blocking PAMP or DAMP signaling preconditioning consist of mAbs to inhibit IL-12p40 (7) or agonists for the Siglec-G-CD24 axis (80), respectively. Pre-HSCT, postconditioning IL-6 inhibition has an early suggestion of efficacy (81). IL-6 or IL-12/IL-23p40 cytokines from CD103+CD11b− DCs can be neutralized with anti-IL-6R or anti-IL-12/IL-23 mAbs and are being investigated in aGVHD prevention trials (81, 82). Histone deacetylase inhibitors (HDACis) downregulated TLR-induced proinflammatory cytokine secretion and DC costimulatory molecules and induced IDO and Tregs (83). Recipients of reduced-intensity-conditioned, related donor transplants given CNIs/MMF and an HDACi (vorinostat) had low aGVHD rates (84). Short-chain fatty acids including butyrate from fermented resistant starch or microbiota manipulation including fecal microbiota transfer favored intestinal peripheral Treg (pTreg) induction and are being tested for aGVHD prevention (85–87). In a phase 2 trial (NCT01743131), T cell costimulation blockade with CTLA4-Ig, a fusion protein of IgG1-modified Fc and human CTLA4 extracellular domain that binds CD80 or CD86 with high affinity, improved aGVHD in HLA-mismatched and -matched unrelated donor HSCT (NCT01012492).
3.1. Steroid-Refractory Acute GVHD Therapy
Ruxolitinib, a small-molecule JAK1 and JAK2 inhibitor, regulated proinflammatory cytokine secretion and signaling and limited mesenteric LN neutrophil accumulation (66). JAK1/2 inhibitors that block STAT1 and STAT3 phosphorylation (i.e., downstream signaling of cytokines including IL-12 and IL-6) have been highly effective in treating steroid-refractory (SR) aGVHD (88). In a phase 3 randomized SR aGVHD trial, ruxolitinib significantly extended overall median survival (88), and it is now approved by the US Food and Drug Administration (FDA) for SR aGVHD therapy. In addition to targeting pathogenic cytokines, ruxolitinib has the potential to limit beneficial common γ chain receptor cytokines, such as IL-2, IL-7, and IL-15, by its inhibitory effects on JAK1 (reviewed in 89). Thus, data in mice and humans show that ruxolitinib reduces the number and function of Tregs, cytotoxic T lymphocytes, and NK cells (89). While ruxolitinib achieves high remission rates in SR GVHD, durable responses are less robust and possibly due to impaired Treg activity (88). Further, broad JAK inhibition by ruxolitinib is linked to an increased risk of opportunistic infections, which is an important consideration when treating transplant recipients (90). Other treatments under investigation have included α−1-antitrypsin, an anti-inflammatory protease inhibitor and urinary-derived human chorionic gonadotropin (HCG) that promoted regulatory responses; IL-22-Fc fusion protein, lithium, and epidermal growth factor (present in HCG) to increase intestinal epithelial cell regeneration; and anti-P-selectin glycoprotein ligand-1 mAb to impede leukocyte migration into tissues (91–93). Extracorporeal photopheresis (ECP), to treat aGVHD and cGVHD in some centers, is proposed to promote Treg activity and tolerance without immune suppression (94) but is expensive and labor intensive and requires central venous access. Regarding efficacy, a recent review of GVHD treatment revealed that studies were typically small, retrospective, and heterogeneous with respect to patient characteristics, treatment schedule, and outcome assessment (95). While in general a majority of patients achieved partial response or better, response rates varied by the organs affected and without direct treatment comparators, such that definitive efficacy conclusions are difficult to reach.
4. CHRONIC GVHD
cGVHD, a heterogeneous immunological complication, is the major responsible factor for nonrelapse mortality in patients surviving ≥2 years after allo-HSCT (2). Risk factors include G-PBSCs, mismatched or unrelated donor grafts, female-to-male transplantations, older recipient age, and aGVHD history. As a multi-organ system disorder affecting virtually every organ, cGVHD can be a highly debilitating disease, magnified by few effective therapies and often requiring team management that includes specialists of the organs. Poor therapeutic responses are ascribed to diverse pathogenesis, preclinical models inadequately simulating the clinical spectrum, nonspecific diagnostic criteria, and absence of validated biomarkers. Its chronic nature requires long observation and tissue fibrosis that may be irreversible. Despite these obstacles, considerable progress has been made through cGVHD National Institutes of Health consensus conferences that refined diagnostic criteria and improved standards for designing and performing clinical trials in conjunction with the FDA (96, 97). One composite metric for allo-HSCT success is GRFS, modified to include no cGVHD requiring systemic therapy (67). Here we focus on cGVHD immunology related to disease pathogenesis, prevention, and therapy.
4.1. Murine Chronic GVHD Pathogenesis
While mouse cGVHD models only recapitulate a part of the clinical spectrum (reviewed in 4, 5), biological insights leading to clinical studies and therapies can be placed on a solid foundation, especially if interrogations and testing are conducted in multiple models with distinct pathophysiology. The first described cGVHD models were induced by CD4+ T cells and were profibrotic (reviewed in 5). After lethal irradiation, recipients were given miH-disparate donor bone marrow plus splenocytes, and by one to two months they developed scleroderma and, in some strains, progressive fibrosis and increased immunoglobulin; mortality rates were high. CD4+ T cell clones isolated from cGVHD mice were noncytotoxic and proliferated in response to MHC-II (98). Other models consisted of parental grafts of modest- to high-dose splenocytes given to nonirradiated F1 recipients. These cGVHD models are CD4+ T cell driven, nonlethal (with one exception), and predicated on autoantibody production, seen in a majority of cGVHD patients but not associated with disease severity or activity (99). Disease manifestations include lupus-like disease, Sjögren syndrome (sialoadenitis), polyarthritis, sclerosing cholangitis, and scleroderma. These latter models are flawed in simulating clinical cGVHD due to lack of pre-HSCT conditioning and absence of donor bone marrow infusion (only high-dose splenocytes are given). Without conditioning-induced inflammation, host hematopoietic cells remain to produce autoantibody, leading to glomerulonephritis, a very infrequent cGVHD complication.
cGVHD was theorized to be attributable to T cell responses to autoantigens (98), as later supported by the finding of GVHD-induced dysplasia (100). Proof-of-concept could be found in lethally irradiated mice given T cell-depleted, fully disparate bone marrow from MHC-II-deficient donors to mimic the proposed failed thymic negative selection that might cause cGVHD (101). Mice developed skin (thickened dermis, fat loss, epidermis atrophy, follicle loss) and salivary gland involvement, pancytopenia, autoreactive T cell proliferation, and delayed but high lethality rates, preventable by pre-BMT thymectomy. Intriguingly, thymus and spleen Treg numbers were unaffected. In vivo activated FOXP3+CD103+ (αEβ7) Tregs isolated from irradiated recipients of miH-disparate donor grafts expressed CCR5 but not CD62L, migrated directly to cGVHD tissues, and were more potent than CD25highFOXP3+CD4+ Tregs in treating established cGVHD (102).
In irradiated MHC-disparate and parent-into-F1 graft recipients given a lower T cell dose, aGVHD was mild to moderate, cGVHD developed with scleroderma, and inflammation in liver and salivary glands was seen (103). Donor DCs defective in MHC-II antigen presentation led to Treg deficiency and development of cGVHD that was ameliorated by donor Treg infusion. In contrast to xenogeneic aGVHD models, cGVHD models using human hematopoietic cells that simulate human cGVHD have been lacking. Recently, a model of sublethally irradiated immunodeficient mice implanted with human fetal thymus and liver fragments and then given autologous fetal HSCs supported human T cells, B cells, and macrophages; scleroderma, lung and liver fibrosis typically occurred >100 days post-HSCT, and disease severity correlated inversely with FOXP3+ thymocytes (104).
In another model, sublethally irradiated miH-disparate transplant recipient strains developed cGVHD, driven by donor CD4+ T cells and B cells, that resulted in IgG autoantibodies, glomerulonephritis, and scleroderma (105). In new murine cGVHD models, the clinical and immunological features representing different aspects of the varied clinical cGVHD spectrum were reported (106, 107). Increasing irradiation from sublethal (105) to lethal in the same miH-disparate transplant recipient strains as above resulted in cGVHD of skin, lung, salivary, and lacrimal glands, associated with inflammation (106). This model simulates features of the aGVHD-to-cGVHD transition occurring in some patients and reminiscent of the overlap syndrome, wherein aGVHD and cGVHD coexist. Therefore, strategies that attack either component may have efficacy in reducing some or all of cGVHD. Donor CD4+ T cells and B cells but not an intact thymus are required, adding donor Tregs on day 0 prevented cGVHD, and passively transferred donor immunoglobulin from cGVHD mice resulted in its deposition in thymus and skin, perpetuating skin cGVHD (108). Donor Th22 and IL-22+ Th17 cells also have been implicated in murine and human sclerodermatous cGVHD (109). Additionally, Th17 cells infiltrated skin and lung, contributing to fibrosis; the role of cytokines in cGVHD has been reviewed (110).
In a nonscleroderma cGVHD model, recipients conditioned with a regimen extrapolated from the clinic, cyclophosphamide and heavy irradiation, and given fully MHC-disparate donor T cell-depleted bone marrow with a limited T cell number (14–40-fold less than for aGVHD) developed multi-organ system (lung, liver, tongue, thymus, spleen, and colon but not skin or lacrimal or salivary glands) cGVHD (107). Bronchiolitis obliterans (BO), small-airway injury leading to fibrosis, was documented by histopathology that also showed CD4+ T cell and B220+ B cell infiltration and pulmonary function indicative of a mixed obstructive-restrictive airflow pattern. BO occurs in 10–15% of cGVHD patients and is considered diagnostic of lung cGVHD; except lung transplantation, there are no curative therapies, and five-year survival is only 15%.
In cGVHD/BO, donor B cells secrete antihost reactive, isotype-switched IgG2c, deposited in organs such as lung, liver, and colon, resulting in collagen deposition with ensuing fibrosis (Figure 2). An increase in germinal center (GC) formation, T follicular helper (Tfh) cells, and GC B cells, along with low T follicular regulatory (Tfr) cells and Tfr:Tfh ratio, were seen (111). Tfh cells (CD4+FOXP3−ICOS+IL21R+IL6R+CXCR5+c-Maf+STAT3+BCL6+), Tfr cells (CD4+FOXP3+CD25+PD-1+CXCR5+ICOS+BCL6+Blimp-1+), and GC B cells accumulate in secondary lymphoid organs (SLOs) in the B cell-rich zone. Tfh and GC B cells cooperate to produce long-lived, IL-6-driven, antibody-secreting plasma cells and memory B cells. Somatic mutation of B cells encoding B cell receptor-encoding genes is followed by selection of high-affinity cells responsible for generating antihost-specific-antibody-secreting cells. GC disruption by infusing lymphotoxin-receptor immunoglobulin fusion protein and mAbs that block GC and extrafollicular (pre-GC) Tfh cell:B cell interactions (e.g., CD40L, ICOS/ICOSL, IL-21/IL21R, or OX40/OX40L pathways) prevents GC initiation and maintenance; however, the requirement for GCs is not universal (107, 111, 112). Th2 cells secrete IL-4 to promote B cell isotype switching, and Th17 cells secrete IL-17 and IL-21, which differentiate Tcon cells into pathogenic Tfh cells that are linked to cGVHD lichenoid skin lesions and lung fibrosis (113, 114). As in patients, G-CSF-mobilized donor grafts have a predilection to cause murine cGVHD with prominent scleroderma and high levels of Th17 cells, which recruit macrophages, a rich source of profibrotic TGF-β, essential for lung and skin fibrosis (114, 115).
Figure 2.

Chronic GVHD (cGVHD) pathogenesis and treatment. Phase I: Inflammation. Pretransplantation conditioning and acute GVHD (aGVHD) cause thymus injury. Phase II: T and B cell dysfunction. The resulting thymic dysfunction impairs donor regulatory T cell (Treg) generation and the negative selection of autoreactive donor T cell clones that escape into the periphery. Balanced Treg/T effector cell reconstitution can be restored by low-dose IL-2 treatment. Donor T follicular helper (Tfh) cells expand in secondary lymphoid organs and promote germinal center (GC) reactions and survival, expansion, and differentiation of donor B cells into antihost-immunoglobulin-producing plasma cells via IL-21 secretion. T follicular regulatory (Tfr) cells suppress Tfh cell:B cell interactions and antibody production. The Rho-associated coiled-coil kinase 2 (ROCK2) inhibitor, KD025, blocks Th17 differentiation and GC reactions by inhibiting Tfh cell generation. Donor B cells receive activating signals via multiple pathways during cGVHD. B cell receptor signaling activates the tyrosine kinase Syk, inhibited by fostamatinib and Entospletinib, and Bruton tyrosine kinase (BTK), inhibited by ibrutinib. IL-21 receptor (IL-21R) signals via the Jak1/2-STAT3 pathway and can be targeted by Jak1/2 inhibitor ruxolitinib or KD025. B cell-activating factor (BAFF) provides crucial prosurvival and mitogenic signals that can be blocked by anti-BAFF antibodies (e.g., belimumab). Donor B cell subsets can be depleted by the anti-CD20 antibody rituximab. Differentiated plasma cells secrete affinity-matured, antihost reactive immunoglobulin. Phase III: Fibrosis. Plasma cell-derived immunoglobulin is deposited in host tissues and stimulates donor CSF1-R+ macrophages, putatively via Fc receptor (FcR) ligation, to secrete profibrotic mediators. These include TGF-β that stimulates fibroblast proliferation and extracellular matrix production via the tyrosine-kinase receptor TGF-βR and is inhibitable by nilotinib and imatinib. The Hedgehog pathway inhibitor glasdegib and ROCK2 inhibitor KD025 each can directly ameliorate fibrosis. Abbreviations: Ab, antibody; CSF1, colony-stimulating factor 1; DC, dendritic cell; HSCT, hematopoietic stem cell transplantation; Mφ, macrophage; Th17, type 17 T helper. Figure adapted from images created with BioRender.com.
Collectively, these cGVHD preclinical data suggested three cGVHD pathophysiological processes: (a) early inflammation due to tissue injury, (b) thymic injury and T cell and B cell dysregulation, and (c) fibrosis (116) (Figure 2). These can exist as distinct phases or coexist with other phases. Importantly, all phases do not need to occur, nor do they need to occur in sequence; each associated pathophysiological mechanism in cGVHD points to potential therapeutic options for preclinical and ultimately clinical testing.
4.2. Human Chronic GVHD Pathogenesis
There are similarities between murine and human cGVHD pathogenesis, taking into account that the cGVHD spectrum is, in aggregate, represented in part by the summation of findings in multiple murine models. The importance of a T cell:B cell response observed in many rodent cGVHD models has parallelisms in patients. Human cGVHD pathogenesis has been recently reviewed (2). Early patient observations documented combined cellular and humoral immune deficiency, attributed to decreased CD4+ T cells, altered B cell function, and increased CD8+ T cells and nonspecific suppressors. Patients had naive B cell lymphopenia, B cell homeostasis was altered, and regulatory B cells (Bregs) were deficient. Elevated BAFF levels promote survival of activated autoreactive and alloreactive B cells by increased B cell receptor responses and intracellular Syk and BLNK phosphorylation (117). Immature and transitional peripheral B cells were associated with hypogammaglobulinemia and active cGVHD with CD21− B cells; the latter have an exhaustion phenotype (CD10−, CD27−, CD20high, and high expression of multiple inhibitor receptors) and serve as a cGVHD severity biomarker (118).
Antibodies to H-Y antigens were seen in male recipients of female grafts (119). Activating antibodies to nonpolymorphic autoantigens such as PDGFR induce tyrosine phosphorylation, ERK1/2-reactive oxygen species, and fibrosis (120). Antibodies directed to membrane antigens have been found in cGVHD patients (121). Although isotype-switched IgG deposition in murine cGVHD target tissues and high levels of Tfh cells in mice with inflammatory cGVHD (122, 123) and cGVHD/BO have been reported (111), active cGVHD patients had lower levels of circulating Tfh cells (124) with a highly activated profile, dominated by Th2/Th17 subsets, and augmented capacity to promote B cell immunoglobulin secretion and maturation. Activated B cells and short-lived plasmablasts were increased, providing a source of IgG. The observed higher level of CXCL13, a chemokine that recruits T and B cells into GCs, is consistent with the interpretation that Tfh cells were retained in or drawn back to SLOs and GCs (124). As in murine sclerodermatous skin that has T cells coexpressing STAT3-dependent IFN-γ and IL-17, increased IL-17-producing CD4+ and CD8+ Th17 cells and mixed Th1/Th17 cells were demonstrated in lichenoid skin in many but not all studies (125, 126). In patients with skin cGVHD, Tfh cells were found to be increased locally in fibrotic skin lesions, where they drive inflammation and myofibroblast differentiation in an IL-21-dependent manner (127). As in aGVHD, microbe-derived short-chain fatty acids appear to be associated with higher cGVHD rates in patients.
One of the most striking findings in cGVHD patients is low Treg frequency, as seen in mouse cGVHD models (128). Decreased thymic naive Treg output is compensated for by increased memory Tregs in patients with CD4+ T cell lymphopenia. Treg deficiency is compounded by increased mitophagy, Fas-mediated apoptosis of pTregs, and decreased BCL-2 (129). A low level of circulating naive Tregs that would otherwise migrate to SLOs and GCs to become Tfr cells could lead to an unrestrained GC response.
4.3. Chronic GVHD Prevention
Therapy for moderate to severe cGVHD has been reviewed (125, 130). We focus on selected therapies based on murine models that exploit cGVHD immunobiology and use agents with cGVHD activity undergoing clinical testing (Figure 2). Systemic steroids are the accepted first-line therapy for moderate to severe cGVHD. Unfortunately, only 50–60% of patients respond, responses are often partial and not durable, and ~80% need second-line therapy that includes primarily T cell-directed agents such as CNIs, rapamycin, MMF, methotrexate, or cyclophosphamide. Therefore, a focus was placed on B cell targeting. With the flexibility of murine models in interrogating pathogenesis and treatment, new therapeutic agents were tested in preclinical mouse cGVHD models and human clinical-laboratory correlates.
4.3.1. Chronic GVHD therapy.
Taking lessons from B cell malignant diseases, researchers tested several clinically approved reagents, the first of which was anti-CD20 mAb (e.g., rituximab). Anti-CD20 mAb was effective in murine cGVHD prophylaxis but not therapy (111, 131). In SR cGVHD patients, Rituxan conferred some clinical benefit (132), with modest success likely due to low or absent CD20 levels of plasma cells and plasmablasts. Drugs for multiple myeloma, a malignant plasma cell disease, have a basis in murine cGVHD models and are in early phases of clinical cGVHD testing. These include the proteasome inhibitor bortezomib (133), which also targets proliferating T cells and DCs. Other agents being tested include immunoproteosomal inhibitors (e.g., carfilzomib, ixazomib) and IL-6/IL-6R mAbs.
Ibrutinib, a reversible Bruton tyrosine kinase (BTK) and IL-2-inducible T cell kinase (ITK) inhibitor, was efficacious in treating cGVHD/BO and scleroderma models. BTK was constitutively activated in cGVHD B cells, bone marrow BTK or T cell ITK deficiency was sufficient to prevent cGVHD/BO, and ibrutinib became the first FDA-approved GVHD drug for its efficacy in SR cGVHD therapy (134, 135). B cells from cGVHD mice and patients have high phosphorylated spleen tyrosine kinase (pSyk) levels; pSyk inhibitor (fostamatinib) was able to reverse ongoing cGVHD/BO, but only in some of the scleroderma models tested (136, 137). The pSyk inhibitor entospletinib was efficacious in preventing scleroderma and ocular cGVHD (137) but did not improve efficacy over placebo when combined with steroids as first-line therapy in the clinic (NCT02701634).
Ruxolitinib, recently FDA approved for SR aGVHD therapy (88), was efficacious in reversing cGVHD/BO; retrospective studies showed considerable efficacy in treating SR cGVHD patients, including those with inflammatory scleroderma (138). Results of a prospective phase 3 trial of ruxolitinib against best available therapy for SR cGVHD are expected soon (NCT03112603). A Rho-associated coiled-coil kinase 2 (ROCK2) inhibitor, KD025, decreased pSTAT3, required for generating inflammatory Th17 cells; increased pStat5, which supports Tregs; and had direct antifibrotic properties. KD025 treatment decreased Th17 cells (scleroderma model), GC B cells, and Tfh cells while increasing Tfr cells (cGVHD/BO model) (139). In human volunteers and in vitro cultures, KD025 inhibited IL-17 and IL-21 production and Tfh generation (140). Phase 2 clinical trials are promising, and results are forthcoming (NCT03640481). In a nonirradiated parent-into-F1 autoantibody-mediated cGVHD model, CTLA4-Ig blockade of CD28/B7 ligand costimulation (see below) reversed active cGVHD (141). A phase 1 trial of abatacept (CTLA4-Ig) given to treat SR cGVHD resulted in a 44% clinical partial response and an ~50% reduction in steroid dose (142). Other studies of SR cGVHD are ongoing (NCT01954979).
To increase the low Treg/Tcon cell levels in cGVHD patients, low-dose IL-2 was administered over eight weeks to selectively increase endogenous Tregs and restore homeostasis including in inflammatory scleroderma (143). In mice with ongoing cGVHD/BO, IL-2/anti-IL2 mAb complexes were effective, but they caused lethality when initiated, even in an abbreviated schedule early post-BMT, likely due to stimulating CD25high Tcon cells that caused aGVHD, whereas Tregs given to treat established cGVHD/BO proved effective (144). Clinical trials are exploring IL-2 with or without Treg infusion or ECP. An IL-2 mutein with a higher selectivity for Tregs is under investigation. ECP has been used for over two decades to treat GVHD, with evidence of responses, especially for skin cGVHD (reviewed in 145). Mechanisms of action include anti-inflammatory cytokines and antigen-specific Treg induction.
In the fibrotic phase, therapies are geared to repair and regenerate tissues and prevent further fibrosis. KD025 and each agent below reduced collagen deposition and lessened or reversed active cGVHD. The receptor tyrosine kinase inhibitor imatinib has selective dual inhibition of profibrotic TGF-β and PDGFR-β, as does nilotinib, a potent dual PDGFR-α and -β and TGF-β inhibitor. Imatinib and nilotinib each were effective in treating murine sclerodermatous cGVHD and have shown some clinical cGVHD responses (146). Studies in scleroderma suggested that macrophages, a rich TGF-β source, were critical for cGVHD. Macrophage depletion using an anti-CSF1R mAb was effective in cGVHD/BO and full MHC-disparate recipients of G-PBSCs that developed cGVHD/scleroderma (115). An ongoing clinical trial is exploring a high-affinity anti-CSF1R mAb for treatment-refractory cGVHD patients (NCT03604692). In a lethally irradiated miH-disparate cGVHD/scleroderma model, treatment with LDE223, a selective inhibitor of the Hedgehog coreceptor, Smoothened, ameliorated murine cGVHD (147), leading to an ongoing cGVHD therapeutic clinical trial of the Smoothened inhibitor, Glasdegib (NCT04111497). Due to the refractory nature of BO and organs that have extensive fibrosis or cross-linked collagen, antifibrotic therapies should continue to be explored. Since several cGVHD pathophysiological mechanisms can coexist, combinatorial therapy with agents described here is on the near horizon.
5. IMMUNE DEFICIENCY AFTER ALLO-HSCT
Allo-HSCT results in immune deficiency of varying severity and duration, depending on donor source and graft manipulation. It is most profound with cord blood and T cell-depleted grafts. In older recipients, allo-HSCT results in lower thymic function and both aGVHD and cGVHD (148). Even after T cell-replete allogeneic BMT, reconstituted T cells, initially derived from transplanted mature donor T cells, were oligoclonal (149). Full reconstitution of a broad T cell repertoire requires T cell precursors in the bone marrow to undergo thymic positive selection of Tcon cells and Tregs with TCRs responding to low- and intermediate-affinity peptide-self-MHC, respectively. This process can take 24 months or more in older adults or those with aGVHD-induced thymic injury (148, 149), during which time T cell lymphopenia and Treg deficiency predispose the recipient to infections and disease recurrence as well as autoimmunity, respectively.
Donor NK cells were reduced in preclinical models and patients in the presence of aGVHD (150, 151). Their expansion and maturation were dependent on IL-15 signaling, and consumption by CD4+ and CD8+ Tcon cells during aGVHD starved NK cells of this critical growth signal. In mice, acquired NK cell deficiency in aGVHD impaired NK cell-dependent pathogen-specific [e.g., cytomegalovirus (CMV)] and leukemia-specific immunity (150). Similarly, donor B cell reconstitution and antibody secretion also were impaired during aGVHD, due to profound Th1/Th17 inflammatory conditions (152). GVHD-targeted clearance of recipient plasma cells resulted in diminished host pathogen-specific IgG permissive of CMV reactivation (153). Conventional and plasmacytoid DC subsets were sensitive to aGVHD and numerically and functionally deficient during aGVHD (154). Exogenous antigen uptake and presentation within MHC-II were grossly impaired in conventional DCs during aGVHD, leading to defective pathogen-specific T cell priming (155). This MHC-II defect caused failure of pTreg homeostasis and cGVHD (103). Chronic inflammation during aGVHD created profound T effector cell proliferative defects (35). Thus, aGVHD had striking multifaceted innate and adaptive immune system effects even without factoring in pharmacological immune suppression administered to control the disease itself. Thus prevention of aGVHD and cGVHD is associated with dramatic amelioration of immune defects typically seen after allo-HSCT.
6. IMMUNE TOLERANCE AND GVHD
Tolerance is the active mechanism of maintaining unresponsiveness to self-antigens and certain foreign antigens by T cell deletion in the thymus (central tolerance) and clonal deletion, anergy, immunological ignorance, and immune regulation in the periphery (peripheral tolerance) (156) (Figure 3). Failed thymic negative selection of high-affinity peptide-self-MHC-reactive T cells may occur as a result of lack of peripheral antigen expression in Aire+ DCs or medullary thymic epithelial cell (mTEC) injury or dysfunction due to older age, conditioning injury, CNIs, or direct donor T cell cytotoxicity during aGVHD (157–159). GVHD induced thymic ILC3 depletion with loss of IL-22 and impaired thymic regeneration (160). Diminished output due to thymus injury restricted TCR repertoire and impaired thymic Treg (tTreg) generation in mouse models and patients (128). Inflammatory aGVHD evolution to autoimmunity-driven cGVHD also can be facilitated by thymus injury allowing autoreactive T cell clones to escape negative selection (101, 158). Keratinocyte growth factor, IL-7, IL-22, sex steroid hormone ablation, IGF-1, growth hormone, stem cell-derived mTECs, thymic organoids, and an injectable bone marrow-like scaffold each have been investigated for accelerating thymic recovery and T cell output without autoreactivity (reviewed in 159).
Figure 3.
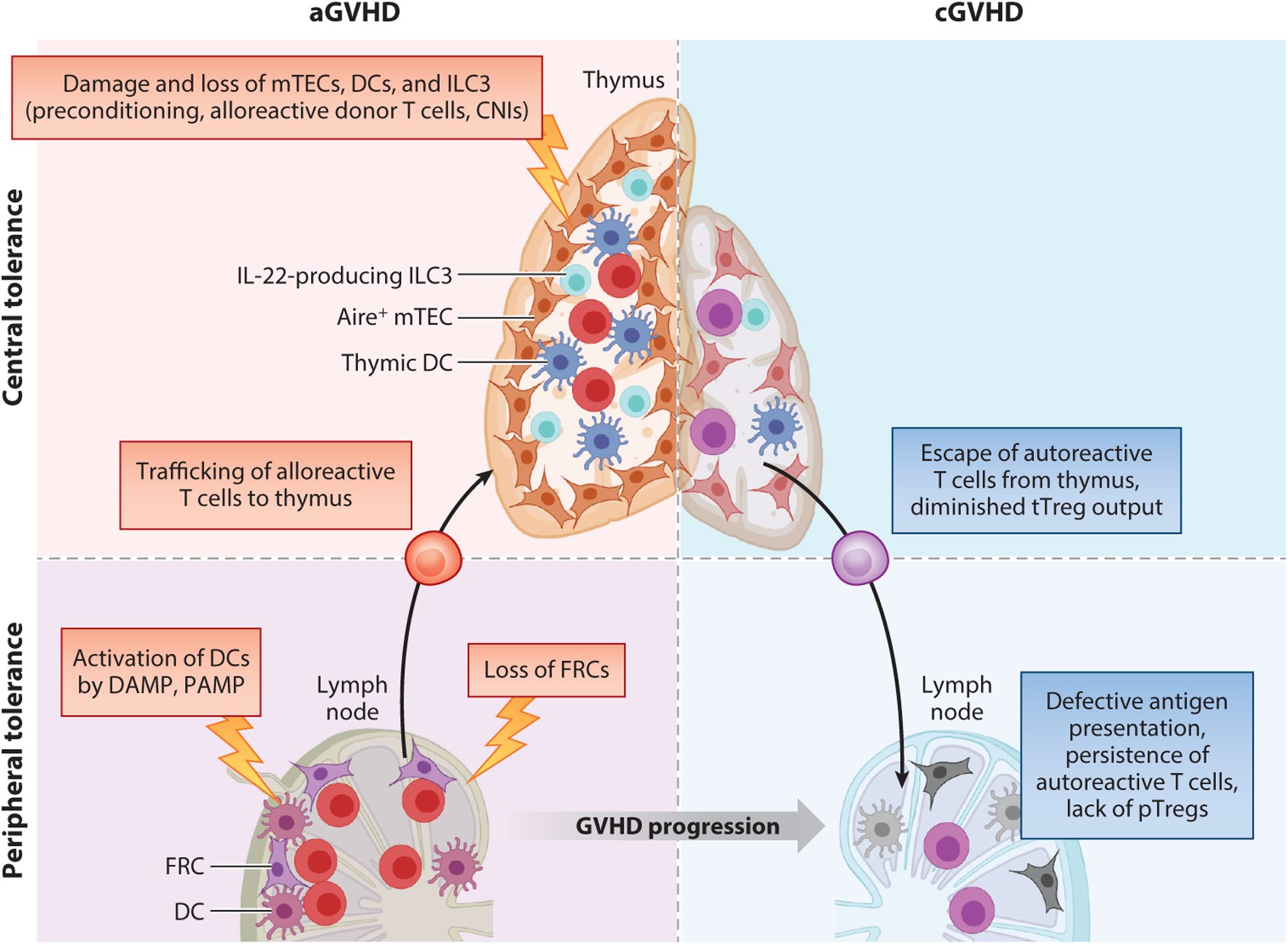
Failure of immune tolerance during GVHD. During acute GVHD (aGVHD) damage-associated molecular pattern (DAMP)/pathogen-associated molecular pattern (PAMP)-induced activation of host antigen-presenting cells (APCs) in secondary lymphoid organs (SLOs) results in switch from donor T cell anergy to activation and dysfunction or deletion of fibroblastic reticular cells (FRCs) and donor dendritic cells (DCs). Donor alloreactive T cells infiltrate the thymus, exerting cytotoxicity of thymic DCs, Aire-expressing medullar thymic epithelial cells (mTECs), and thymus type 3 innate lymphoid cells (ILC3s) that are already damaged by both the conditioning regimen and use of calcineurin inhibitors (CNIs). Thymic damage results in impaired negative selection, allowing escape of self-reactive T cell clones, and diminished thymic regulatory T cell (Treg) generation. Potentially self-reactive T cell clones escaping to the periphery fail to undergo peripheral tolerance in SLOs because of impaired antigen presentation on DCs and FRCs damaged during aGVHD. The outcome of impaired thymus function is the persistence of self-reactive lymphocytes. These defects in APCs within SLOs also impede peripheral Treg (pTreg) generation, causing profound Treg defects. Altogether, these mechanisms contribute to the failure of central and peripheral immune tolerance. Abbreviations: cGVHD, chronic GVHD; tTreg, thymic Treg. Figure adapted from images created with BioRender.com.
In the periphery, DAMPs and PAMPs release primed host DCs, increase costimulatory molecules, and shift the immune system from anergy induction to alloimmunity (161). As aGVHD progresses, impaired antigen presentation by engrafted donor DCs fails to support the conversion of self-reactive T cells to Tregs, i.e., pTreg differentiation; failed pTreg generation further disrupts peripheral tolerance mechanisms and promotes cGVHD (103). aGVHD damages SLO fibroblastic reticular cells blocking regeneration. With the loss of peripheral tissue-associated antigen presentation, especially of intestinal antigens, autoaggressive donor T cell clones accumulated and cGVHD ensued (162). Breakdown in tolerance during aGVHD does not proceed unchallenged: aGVHD-induced tissue inflammation upregulates inhibitory pathways, including PD-L1 and IDO pathways, to restrain donor T cell activation. While their absence exacerbates immunopathology, these pathways are seldom sufficient to offset the aggressive inflammatory and tissue-destructive aGVHD process. Instead, tTregs or pTregs dampen alloreactivity and favor tolerance. Moreover, tolerance-inducing T cells can pass on their regulatory properties to other T cells to propagate peripheral regulation, known as linked suppression, unless it is broken by viruses or UV-induced tissue injury.
6.1. Costimulatory and Coinhibitory Molecules
T cell activation requires at least two signals. The first is delivered by intracellular signaling domains triggered by antigen-specific TCR engagement of antigen-MHC. The second is provided by CD28 binding to B7 ligands (CD80 or CD86) or TNF/TNFR superfamily members such as OX40 and CD40L engaging their ligands on APCs. T cells receiving the first but not second costimulatory signal acquire antigen-specific nonresponsiveness, termed anergy. In contrast to constitutive CD28 expression, the CD28 superfamily member ICOS is upregulated within one to two days after T cell activation, amplifying initial CD28/B7 ligand responses to produce IL-4, IL-6, and IL-10. OX40 and CD40L provide later T cell costimulation, proliferation, and apoptosis resistance. Blockade of any of these pathways diminishes aGVHD and cGVHD.
Combined anti-CD80 and anti-CD86 mAb infusion is needed to reduce murine aGVHD mortality; either mAb alone confers no or partial protection from aGVHD lethality mediated by CD8+ T cells and CD4+ T cells, respectively (163). Blocking CD28 mAbs prevented aGVHD by permitting CTLA-4/B7 ligand coinhibitory function to be unopposed by costimulatory CD28/B7 ligand engagement. CD28 costimulation requires mammalian target of rapamycin (mTOR) signaling and is inhibited by sirolimus (rapamycin). Tcon cells are highly rapamycin sensitive, and mTOR blockade increases Treg:Tcon cell ratio early after allo-HSCT, potentially contributing to the observed aGVHD reduction (164). Preclinical studies show sirolimus and CD28 blockade synergistically prevent aGVHD (165). Transcriptomics identified aberrant donor T cell Aurora kinase A activity as associated with sirolimus failure and breakthrough aGVHD; the role of Aurora kinase A activity in driving aGVHD was validated in a murine aGVHD lethality model (33).
CTLA4-Ig infusion reduced murine aGVHD lethality without causing primary graft failure, despite inhibiting engraftment-promoting donor T cell function (166). Abatacept (CTLA4-Ig) appears highly promising for aGVHD prevention (167). In a multi-arm, phase 2 study (NCT01743131), CNI, methotrexate, and abatacept significantly decreased moderate to severe aGVHD among 7/8 and 8/8 HLA-matched, unrelated allo-HSCT recipients, resulting in abatacept receiving FDA Breakthrough Therapy designation for prevention of moderate to severe aGVHD in unrelated donor recipients. Future trials are warranted to test abatacept for cGVHD prevention. Overall, aGVHD prophylaxis, excluding CNIs, will likely assist with durable immune tolerance by supporting Treg recovery at least in part by lowering proinflammatory cytokines and limiting suppression of antileukemia T effector function.
Other coinhibitory molecules such as PD-1 counterbalance costimulation (168). PD-1, an immunoglobulin superfamily member, is coinhibitory. Administering anti-PD-1-blocking mAb or PD-1-deficient donor T cells caused rapid aGVHD lethality associated with an IFN-γ burst (168). PD-1 ligands (PD-L1 and PD-L2) were upregulated in aGVHD target organs; host deficiency of either markedly accelerated aGVHD lethality (38). While blocking anti-PD-1 mAbs have a high therapeutic impact in treating nontransplantation patients with refractory or relapsed hematologic malignancies, peritransplantation or late post-HSCT administration increased aGVHD risk associated with a deficiency in PD-1+ T cells implicated in tolerance induction (168). Increased aGVHD was not observed when the blocking anti-CTLA-4 mAb, ipilimumab, was given as relapse therapy after allo-HSCT (168). The apparent greater aGVHD-inducing properties of anti-PD-1 mAb compared to anti-CTLA-4 mAb could be due to higher PD-1/PD-1 ligand levels or favorable expression kinetics (later versus earlier), distribution (peripheral tissues versus LNs), cell surface stability versus recycling, or higher mAb efficacy (168). The impact of these and other costimulatory and coinhibitory molecules on aGVHD has been recently reviewed (168).
6.2. CD4+ Tregs
Strategies augmenting regulatory mechanisms are being applied as GVHD prevention and therapy in patients (169). CD4+CD25high Tregs express the master regulator transcription factor FOXP3, critical for their development and fate and suppressor functions (170). Thymic Tregs have a self-antigen-specific TCR repertoire (170). In contrast, pTregs are differentiated from mature CD4+ T cells extrathymically in response to self-antigens and foreign antigens in the context of immunoregulatory molecules such as retinoic acid and TGF-β (170). Tregs suppress T cell responses by a variety of mechanisms relevant to GVHD control. These include cytolytic killing of T effector cells and APCs by granzyme B, perforin, and FasL; modifying DCs to be less conducive to alloreactive T cell priming; releasing immune-suppressive cytokines; and inhibiting T effector cell function by Treg CTLA-4 surface molecule expression that causes B7 ligand transendocytosis. Additional mechanisms include depletion of IL-2 and amino acids as well as reduced ATP production.
Both tTregs and pTregs are referred to as natural Tregs (nTregs), in contrast to induced Tregs (iTregs), which are generated in vitro by exposure of Tcon cells upon TCR engagement in the presence of IL-2, TGF-β, rapamycin, and/or retinoic acid (170). Unlike the case of nTregs that have a fully demethylated FOXP3 promoter and stable FOXP3 expression, iTreg demethylation is partial. Further, iTregs lack the locked-in transcription factor signature of tTregs (171), rendering iTregs inherently unstable and giving rise to ex-Tregs that have converted to T effector cells under proinflammatory conditions (reviewed in 172). FOXP3 stabilization in iTregs (CD4+ and CD8+) is bolstered by rapamycin exposure, Jak2 inhibitors, all-trans retinoic acid, vitamin C, transcription factors id2 and Blimp-1, decreased Dnmt3a, and high Dnmt1 and EZH2 chromatin-modifying enzyme expression (169). In vitro polyclonal alloantigen-specific CD4+ iTregs generated from CD4+ Tcon cells are potently suppressive in vitro and in vivo in alloantigen-bearing mice (173). Despite a propensity for destabilization, CD4+ iTregs can efficiently prevent murine allogeneic and human xenogeneic GVHD in preclinical models (174, 175), and they have been tested in a completed phase 1 trial (NCT01634217).
Both aGVHD and cGVHD are associated with abnormal Treg reconstitution. aGVHD induced nTreg instability and suppressed pTreg differentiation by IL-6, IL-21, or IL-27 activation of STAT3, skewing Tcon cells toward T effector cells (176, 177). Early after allo-HSCT, tTreg output was compromised and pTregs were prone to apoptosis and reduced proliferative capacity (128). Corruption of peripheral host antigen presentation resulted in defective pTreg generation (103). Depleting donor graft Tregs accelerated aGVHD; conversely, the adoptive transfer of ex vivo expanded or freshly isolated donor or third-party but not host Tregs reduced aGVHD (178). Most studies currently isolate human Tregs from peripheral blood, although third-party umbilical cord blood-derived Tregs have been successfully used in clinical trials (179). Adoptively transferred polyclonal Tregs are most effective if used in nonphysiologically high Treg:Tcon cell ratios (1:1). In rodent cells, compared to polyclonal Tregs, in vitro antigen-specific Treg adoptive transfer required lower Treg numbers to abrogate murine aGVHD (180). Achieving high ratios requires vigorous polyclonal or alloantigen-specific ex vivo or in vivo expansion. In vivo Treg expansion is facilitated by transfer of donor Tregs into lymphopenic recipients two days before T effector cells, thereby allowing in vivo lymphopenia-induced homeostatic proliferation to occur (181). nTregs and iTregs can be supported by donor IL-27 pretreatment and TNFR2 agonists. Alloantigen-specific Treg expansion or engineered chimeric antigen receptor (CAR)-Tregs directed against alloantigens (144) are appealing, although manufacturing logistics and costs are challenges at present.
Tregs have their highest efficacy in the early aGVHD priming phase, wherein Treg homing to SLOs via CD62L and GVHD-target organs via CCR5 allows Treg colocalization with and suppression of T effector cells (181). Phase 2 trials of freshly isolated donor Tregs transferred before Tcon cells in haploidentical HSCT recipients without immunosuppression (182) or HLA-matched HSCT with single agent (CNI or rapamycin) (183) prevented aGVHD and cGVHD in high-risk patients without increasing relapse rates. Rapamycin, unlike CNIs, mitigated aGVHD, preferentially expanding graft-derived pTregs due to the differential sensitivity of T effector cells versus Tregs to mTOR inhibition (184). Despite reversal of ongoing aGVHD (185) and cGVHD (144) in mice by ex vivo expanded Tregs, treatment of advanced GVHD in patients has not been sufficiently tested. While Treg transfer alleviated clinical symptoms and permitted reduction in immunosuppressive drugs in SR cGVHD patients (186, 187), only transient improvement in stage IV aGVHD was seen (187).
CNIs blunt T cell IL-2 production, which is essential for Treg survival, and exogenous IL-2 can reverse CNI-mediated Treg dysfunction (188). In contrast to the case of CNIs, IL-2 production is unaffected by rapamycin, which improved Treg persistence in nonhuman primates, further increased by IL-2 coadministration (189). Low-dose IL-2 improved cGVHD by supporting high-affinity IL-2R+ Tregs in the periphery and promoting the emergence of new Treg clones, resulting in durable cGVHD remission without significant toxicities (143, 190). IL-2 increased endogenous tTregs (190), particularly notable given their severely impaired reconstitution in cGVHD patients (128). Treg reconstitution was seen when OX40/OX40L pathway blockade was combined with rapamycin; together they remarkably enable long-term aGVHD-free survival to be achieved in nonhuman primates (191).
6.3. CD8+ Tregs
Compared to CD4+ Tregs, thymic CD8+ T cells express low or negligible FOXP3. CD8+ Tregs can be generated in vitro by TGF-β and IL-2 (192) or in vivo in the periphery of aGVHD mice from donor CD8+FOXP3− T cells in response to alloantigen exposure (193). CD4+ Treg-associated surface marker expression is shared with CD8+ Tregs, albeit at lower levels, with differential IL-10 and IFN-γ production, respectively (193). In vivo, CD8+ Tregs were generated early after allo-HSCT, wherein their expansion dominated over CD4+ iTregs, were massively induced by rapamycin plus IL-2, and were highly effective in suppressing aGVHD (193). As compared to CD4+ Tregs, under inflammatory conditions, CD8+ Tregs more readily lost FOXP3 expression and were less responsive to rapamycin- and IL-2-supported FOXP3 stabilization (184). Interestingly, CD4+ iTregs were more potent than CD8+ iTregs in suppressing aGVHD, but they compromised GVL, whereas the opposite was true for CD8+ iTregs. Importantly, coinfusing CD4+ iTregs and CD8+ iTregs maximally inhibited aGVHD while preserving GVL (194).
6.4. Regulatory Type 1 T Cells
Regulatory type 1 T (Tr1) cells, first described in tolerant HLA-disparate fetal liver allo-HSCT patients, are CD4+CD25−FOXP3− T cells that coexpress CD49b and Lag3 in some settings and have shared surface antigens with CD4+ Tregs (PD-1, GITR, TIGIT, CD39/73, Tim-3, OX40, ICOS) (195). Tr1 cells produce IL-10, IL-5, GM-CSF, TGF-β, IFN-γ, and granzyme B. Tr1 cells can be generated in vitro from CD4+ Tcon cells using tolerogenic IL-10-producing DCs or via IL-10 expression in Tcon cells (195). During aGVHD, Tr1 cells become a more dominant population as a result of donor mononuclear cell IL-27, and Tr1 depletion results in aGVHD exacerbation (196). Tr1 differentiation is driven by a transcriptional network that includes T-bet and Blimp-1, stabilized in commitment by Eomesodermin and dependent upon unique metabolic profiles (196). A pilot study utilizing an IL-10-anergized donor lymphocyte product enriched for alloantigen-specific Tr1 cells in patients with high-risk hematologic malignancies demonstrated feasibility and safety (197).
6.5. Bregs
Bregs are a phenotypically diverse subset of B lymphocytes in mice and humans that exert their immunosuppressive and immunomodulatory functions via IL-10 secretion (reviewed in 198). Abnormal B cell reconstitution after allo-HSCT results in Breg deficiency and functional impairment during cGVHD, whereas early reconstitution of IL-10-producing Bregs after allo-HSCT may protect from cGVHD (199–202). In mouse models, endogenous host cells and donor Bregs protect from GVHD, and cotransfer of CD5+ Bregs also protects from this disease (203, 204). The therapeutic potential of this cell population was suggested by the fact that a higher dose of human CD24+CD38bright Bregs in the donor graft was associated with lower-grade aGVHD rates (204).
6.6. Myeloid Cells with Tolerogenic or Suppressive Function
Mouse CD49+CD200R3+ regulatory DCs (DCregs) are present in low frequencies in normal mice and can be generated in vitro from bone marrow precursors by IL-10, TGF-β, and low-dose GM-CSF. DCregs induce T cell anergy, promote CD4+ Treg differentiation, and prevent murine aGVHD and cGVHD (205, 206). In vitro low-dose GM-CSF, vitamin D3, HDACi, rapamycin, or steroids (reviewed in 207) and bone marrow precursors cultured with flt3L and TLR agonists generate tolerogenic DCs (207). In vivo recipient flt3L treatment or donor flt3L or G-CSF treatment pretransplantation expanded tolerogenic immature CCR9+ DCs, CD8α+ DCs, or CD34+ regulatory monocytes, and therefore each was capable of aGVHD amelioration (208, 209). Mesenchymal stromal cells cocultured with human peripheral blood monocytes generated M2-like macrophages, which, upon adoptive transfer, prevented lethal xenogeneic aGVHD (210). Macrophage colony-stimulating factor (M-CSF; CSF1) administered pre-HSCT expanded recipient macrophages that engulfed activated donor T cells via CD47-dependent mechanisms and ameliorated aGVHD (17). Human monocytes cultured with low M-CSF doses generated regulatory macrophages that induced tolerance to transplanted kidneys (211), indicative of their therapeutic potential.
Bone marrow myeloid-derived suppressor cells (MDSCs) are characterized as polymorpho-nuclear (PMN-MDSCs) (CD11b+Ly6G+Ly6Clow) or monocytic (M-MDSCs) (CD11b+Ly6G− Ly6Chigh) (212). In peripheral blood, PMN-MDSCs express CD15, while M-MDSCs do not (212). MDSCs are immune suppressive and readily derived from murine bone marrow with the use of G-CSF and GM-CSF (213). A distinct IL-13-producing MDSC subset exhibited substantial potency in preventing aGVHD, primarily via release of immune-suppressive arginase-1 (213). MDSC-induced donor Th2 polarization and Th2 cells were found to be required for maximal MDSC benefits in suppressing aGVHD (214). Increased MDSC frequency within G-PBSC grafts was associated with a significantly lower aGVHD risk (215). However, the murine aGVHD proinflammatory milieu impaired adoptively transferred MDSCs due to inflammasome activation with subsequent MDSC maturation into APCs and loss of suppressor function (216). Overcoming this obstacle, if needed for human MDSCs, will speed MDSC translation for aGVHD prophylaxis (216, 217).
7. PERSPECTIVE
The GVHD continuum is associated with the evolution of alloantigen presentation, costimulatory environments, and subsequent T cell effector functionality. The early phase of aGVHD pathogenesis is predominantly mediated by Th1/Tc1 cells, with hyperproliferation and high cytotoxicity driving disease. These cells arise in response to the transplant-conditioning-induced cytokine storm and resultant release of DAMPs and PAMPs, and they can often be targeted by standard immunosuppression regimens, which focus on the inhibition of proliferation and NFAT-driven T cell signaling.
Central to the efficacy of these agents is attenuation of effector function rather than the prevention of T cell priming and clonal expansion, which predisposes to GVHD following drug withdrawal in the absence of adequate regulation. Looking forward, the recently developed strategies that deplete alloreactive T cells and/or promote regulatory pathways appear better for the induction of long-term tolerance. As time progresses after transplantation, Th17/Tc17 cells may become a major driving force of GVHD, secreting proinflammatory cytokines, providing a cellular reservoir for effector alloimmune cells, and supporting the Tfh-driven immune response that characterizes cGVHD. Because of this evolution, different approaches are likely needed to effectively control later-onset GVHD, focusing on inhibition of aberrant Th17/Tc17 and GC B cell differentiation.
Given late-onset GVHD relative resistance to traditional broad immune-suppressant drugs (and the associated toxicity), targeted tyrosine kinase inhibitors and mAbs with acceptable safety profiles are likely to be more frequently utilized in this setting. A challenge in cGVHD management is the presence of fibrosis that can be debilitating and slow to reverse or irreversible. Antifibrotic drugs that speed up collagen resolution and tissue remodeling are few in number and are an urgent need in the field. Thus we are in an era when the use of broad immune-suppressive drugs developed in the last century is likely to give way to more targeted biological agents, based on a better understanding of disease pathophysiology.
ACKNOWLEDGMENTS
This work was supported by research grants from the National Institutes of Health (NIH): National Heart, Lung, and Blood Institute grants R01 HL148164 (G.R.H.), 2R01 56067 and R01 HL11879 (B.R.B.), HL095791 (L.S.K.), and R01 HL133823 (B.C.B.); National Institute of Allergy and Infectious Diseases grants R37 AI 34495 and P01 AI 056299 (B.R.B.), U19 AI051731 (L.S.K.); National Cancer Institute grants P01 CA142106 and 2P01 065493 (B.R.B.) and U01 CA244291 (G.R.H); and NIH/FDA grant FD004099 (L.S.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. G.R.H. is an Andy Hill CARE Distinguished Researcher. V.T. received an American Society of Transplantation and Cellular Therapy New Investigator Award Grant and Be the Match Foundation Amy Strelzer Manasevit Research Program Grant.
DISCLOSURE STATEMENT
G.R.H. has received funding from Roche for a clinical study of tocilizumab in acute GVHD prophylaxis and consulted for Generon and NapaJen. B.C.B. reports pacritinib is supplied by CTI BioPharma for the conduct of clinical trial NCT 02891603 and a pending patent, WO2017058950A1, for methods of treating transplant rejection. B.C.B. holds patents related to CD4+ T cell pSTAT3 as a marker and therapeutic target of acute GVHD (WO2015120436A2); for the use of JAK inhibitors for rejection and GVHD prevention (WO2017058950A1); and for the use of CD83-targeted chimeric antigen receptor T cells in GVHD prevention, immune tolerance, autoimmunity, and acute myeloid leukemia therapy (WO2019165156). At this time, neither B.C.B. nor the University of Minnesota (or Moffitt Cancer Center) have received payment related to claims described in the patent. B.C.B. has received honoraria for participating in advisory board discussions for Incyte Corp and CTI BioPharma within the past 5 years.
B.R.B. has received funding from Kadmon Pharmaceuticals for preclinical studies, is an Advisory Board member for a KD025 cGVHD trial, has received funding from BlueRock Therapeutics, serves as a consultant for BlueRock Therapeutics and Magenta Therapeutics, and is a cofounder of Tmunity Therapeutics. L.S.K. is on the Scientific Advisory Board for HiFiBio. She has received funding from Kymab Limited, Magenta Therapeutics, BlueBird Bio, and Regeneron Pharmaceuticals. She reports consulting fees from Equillium; FortySeven, Inc.; Novartis, Inc.; EMD Serono; Gilead Sciences; and Takeda Pharmaceuticals. L.S.K. reports grants and personal fees from Bristol Myers Squibb and has a patent “Method to prevent relapse after transplant,” which is pending, and a patent “Method to prevent GVHD after transplant,” with royalties paid.
Glossary
- Allo-HSCT
allogeneic hematopoietic stem cell transplantation
- G-PBSC
G-CSF-mobilized peripheral blood stem cells
- miH
minor histocompatibility antigen
- aGVHD
acute graft-versus-host disease
- cGVHD
chronic graft-versus-host disease
- APC
antigen-presenting cell
- PAMP
pathogen-associated molecular pattern
- DAMP
danger-associated molecular pattern
- LN
lymph node
- Treg
regulatory T cell
- BMT
bone marrow transplantation
- pTreg
peripheral Treg
- BO
bronchiolitis obliterans
- GC
germinal center
- Tfr cell
T follicular regulatory cell
- Breg
regulatory B cell
- tTreg
thymus-derived Treg
LITERATURE CITED
- 1.Zeiser R, Blazar BR. 2017. Acute graft-versus-host disease—biologic process, prevention, and therapy. N. Engl. J. Med 377:2167–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeiser R, Blazar BR. 2017. Pathophysiology of chronic graft-versus-host disease and therapeutic targets. N. Engl. J. Med 377:2565–79 [DOI] [PubMed] [Google Scholar]
- 3.Blazar BR, Hill GR, Murphy WJ. 2020. Dissecting the biology of allogeneic HSCT to enhance the GvT effect whilst minimizing GvHD. Nat. Rev. Clin. Oncol In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeiser R, Blazar BR. 2016. Preclinical models of acute and chronic graft-versus-host disease: How predictive are they for a successful clinical translation? Blood 127:3117–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schroeder MA, DiPersio JF. 2011. Mouse models of graft-versus-host disease: advances and limitations. Dis. Model. Mech 4:318–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koyama M, Hill GR. 2019. The primacy of gastrointestinal tract antigen-presenting cells in lethal graft-versus-host disease. Blood 134:2139–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koyama M, Mukhopadhyay P, Schuster IS, Henden AS, Hülsdünker J, et al. 2019. MHC class II antigen presentation by the intestinal epithelium initiates graft-versus-host disease and is influenced by the microbiota. Immunity 51(5):885–98.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, et al. 2017. ILC1 confer early host protection at initial sites of viral infection. Cell 171:795–808.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, et al. 2013. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 38:769–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruce DW, Stefanski HE, Vincent BG, Dant TA, Reisdorf S, et al. 2017. Type 2 innate lymphoid cells treat and prevent acute gastrointestinal graft-versus-host disease. J. Clin. Investig 127:1813–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanash AM, Dudakov JA, Hua G, O’Connor MH, Young LF, et al. 2012. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity 37:339–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson BE, McNiff J, Yan J, Doyle H, Mamula M, et al. 2003. Memory CD4+ T cells do not induce graft-versus-host disease. J. Clin. Investig 112:101–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koyama M, Kuns RD, Olver SD, Raffelt NC, Wilson YA, et al. 2012. Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat. Med 18:135–42 [DOI] [PubMed] [Google Scholar]
- 14.Jordan-Garrote A, Brede C, Riedel SS, Bauerlein CA, Ritz M, et al. 2012. Depletion of host dendritic cells during the effector phase of GVHD enhances acute GVHD and mortality. Biol. Blood Marrow Transplant 18:S329 [Google Scholar]
- 15.Rowe V, Banovic T, Macdonald KP, Kuns R, Don AL, et al. 2006. Host B cells produce IL-10 following TBI and attenuate acute GVHD after allogeneic bone marrow transplantation. Blood 107:2485–92 [DOI] [PubMed] [Google Scholar]
- 16.Li H, Demetris AJ, McNiff J, Matte-Martone C, Tan HS, et al. 2012. Profound depletion of host conventional dendritic cells, plasmacytoid dendritic cells, and B cells does not prevent graft-versus-host disease induction. J. Immunol 188:3804–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashimoto D, Chow A, Greter M, Saenger Y, Kwan WH, et al. 2011. Pretransplant CSF-1 therapy expands recipient macrophages and ameliorates GVHD after allogeneic hematopoietic cell transplantation. J. Exp. Med 208:1069–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, et al. 1999. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science 285:412–15 [DOI] [PubMed] [Google Scholar]
- 19.Gartlan KH, Koyama M, Lineburg KE, Chang K, Ensbey KS, et al. 2019. Donor T-cell-derived GM-CSF drives alloantigen presentation by dendritic cells in the gastrointestinal tract. Blood Adv. 3:2859–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenq RR, Ubeda C, Taur Y, Menezes CC, Khanin R, et al. 2012. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J. Exp. Med 209:903–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stein-Thoeringer CK, Nichols KB, Lazrak A, Docampo MD, Slingerland AE, et al. 2019. Lactose drives Enterococcus expansion to promote graft-versus-host disease. Science 366:1143–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Markey KA, Schluter J, Gomes ALC, Littmann ER, Pickard AJ, et al. 2020. The microbe-derived short-chain fatty acids butyrate and propionate are associated with protection from chronic GVHD. Blood 136:130–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shono Y, Docampo MD, Peled JU, Perobelli SM, Velardi E, et al. 2016. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci. Transl. Med 8:339ra71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jenq RR, Taur Y, Devlin SM, Ponce DM, Goldberg JD, et al. 2015. Intestinal Blautia is associated with reduced death from graft-versus-host disease. Biol. Blood Marrow Transplant 21:1373–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peled JU, Gomes ALC, Devlin SM, Littmann ER, Taur Y, et al. 2020. Microbiota as predictor of mortality in allogeneic hematopoietic-cell transplantation. N. Engl. J. Med 382:822–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taur Y, Coyte K, Schluter J, Robilotti E, Figueroa C, et al. 2018. Reconstitution of the gut microbiota of antibiotic-treated patients by autologous fecal microbiota transplant. Sci. Transl. Med 10:eaap9489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shono Y, van den Brink MRM. 2018. Gut microbiota injury in allogeneic haematopoietic stem cell transplantation. Nat. Rev. Cancer 18:283–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staffas A, Burgos da Silva M, van den Brink MR. 2017. The intestinal microbiota in allogeneic hematopoietic cell transplant and graft-versus-host disease. Blood 129:927–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koyama M, Cheong M, Markey KA, Gartlan KH, Kuns RD, et al. 2015. Donor colonic CD103+ dendritic cells determine the severity of acute graft-versus-host disease. J. Exp. Med 212:1303–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toubai T, Mathewson ND, Magenau J, Reddy P. 2016. Danger signals and graft-versus-host disease: current understanding and future perspectives. Front. Immunol 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purvis HA, Stoop JN, Mann J, Woods S, Kozijn AE, et al. 2010. Low-strength T-cell activation promotes Th17 responses. Blood 116:4829–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy KM, Reiner SL. 2002. The lineage decisions of helper T cells. Nat. Rev. Immunol 2:933–44 [DOI] [PubMed] [Google Scholar]
- 33.Furlan SN, Watkins B, Tkachev V, Flynn R, Cooley S, et al. 2015. Transcriptome analysis of GVHD reveals aurora kinase A as a targetable pathway for disease prevention. Sci. Transl. Med 7:315ra191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ross D, Jones M, Komanduri K, Levy RB. 2013. Antigen and lymphopenia-driven donor T cells are differentially diminished by post-transplantation administration of cyclophosphamide after hematopoietic cell transplantation. Biol. Blood Marrow Transplant 19:1430–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krenger W, Falzarano G, Delmonte J Jr., Snyder KM, Byon JC, Ferrara JL. 1996. Interferon-gamma suppresses T-cell proliferation to mitogen via the nitric oxide pathway during experimental acute graft-versus-host disease. Blood 88:1113–21 [PubMed] [Google Scholar]
- 36.Takashima S, Martin ML, Jansen SA, Fu Y, Bos J, et al. 2019. T cell-derived interferon-γ programs stem cell death in immune-mediated intestinal damage. Sci. Immunol 4:eaay8556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Taylor PA, Mellor AL, et al. 2008. Indoleamine 2,3-dioxygenase is a critical regulator of acute graft-versus-host disease lethality. Blood 111:3257–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saha A, Aoyama K, Taylor PA, Koehn BH, Veenstra RG, et al. 2013. Host programmed death ligand 1 is dominant over programmed death ligand 2 expression in regulating graft-versus-host disease lethality. Blood 122:3062–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burman AC, Banovic T, Kuns RD, Clouston AD, Stanley AC, et al. 2007. IFNγ differentially controls the development of idiopathic pneumonia syndrome and GVHD of the gastrointestinal tract. Blood 110:1064–72 [DOI] [PubMed] [Google Scholar]
- 40.Baker MB, Altman NH, Podack ER, Levy RB. 1996. The role of cell-mediated cytotoxicity in acute GVHD after MHC-matched allogeneic bone marrow transplantation in mice. J. Exp. Med 183:2645–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin T, Brunner T, Tietz B, Madsen J, Bonfoco E, et al. 1998. Fas ligand- mediated killing by intestinal intraepithelial lymphocytes: participation in intestinal graft-versus-host disease. J. Clin. Investig 101:570–779449689 [Google Scholar]
- 42.Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, et al. 2010. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 115:335–43 [DOI] [PubMed] [Google Scholar]
- 43.Gartlan KH, Markey KA, Varelias A, Bunting MD, Koyama M, et al. 2015. Tc17 cells are a proinflammatory, plastic lineage of pathogenic CD8+ T cells that induce GVHD without antileukemic effects. Blood 126:1609–20 [DOI] [PubMed] [Google Scholar]
- 44.Schwab L, Goroncy L, Palaniyandi S, Gautam S, Triantafyllopoulou A, et al. 2014. Neutrophil granulocytes recruited upon translocation of intestinal bacteria enhance graft-versus-host disease via tissue damage. Nat. Med 20:648–54 [DOI] [PubMed] [Google Scholar]
- 45.Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, Serody JS. 2009. In vitro-differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood 113:1365–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delens L, Ehx G, Somja J, Vrancken L, Belle L, et al. 2019. In vitro Th17-polarized human CD4+ T cells exacerbate xenogeneic graft-versus-host disease. Biol. Blood Marrow Transplant 25:204–15 [DOI] [PubMed] [Google Scholar]
- 47.Furlan SN, Watkins B, Tkachev V, Cooley S, Panoskaltsis-Mortari A, et al. 2016. Systems analysis uncovers inflammatory Th/Tc17-driven modules during acute GVHD in monkey and human T cells. Blood 128:2568–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varelias A, Gartlan KH, Kreijveld E, Olver S, Lor M, et al. 2015. Lung parenchyma-derived IL-6 promotes IL-17A-dependent acute lung injury after allogeneic stem cell transplantation. Blood 125(15):2435–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tugues S, Amorim A, Spath S, Martin-Blondel G, Schreiner B, et al. 2018. Graft-versus-host disease, but not graft-versus-leukemia immunity, is mediated by GM-CSF-licensed myeloid cells. Sci. Transl. Med 10(469):eaat8410 [DOI] [PubMed] [Google Scholar]
- 50.Piper C, Zhou V, Komorowski R, Szabo A, Vincent B, et al. 2020. Pathogenic Bhlhe40+ GM-CSF+ CD4+ T cells promote indirect alloantigen presentation in the GI tract during GVHD. Blood 135:568–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Degli-Esposti MA, Smyth MJ. 2005. Close encounters of different kinds: Dendritic cells and NK cells take centre stage. Nat. Rev. Immunol 5:112–24 [DOI] [PubMed] [Google Scholar]
- 52.Sungur CM, Tang-Feldman YJ, Zamora AE, Alvarez M, Pomeroy C, Murphy WJ. 2013. Murine NK-cell licensing is reflective of donor MHC-I following allogeneic hematopoietic stem cell transplantation in murine cytomegalovirus responses. Blood 122:1518–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rossjohn J, Pellicci DG, Patel O, Gapin L, Godfrey DI. 2012. Recognition of CD1d-restricted antigens by natural killer T cells. Nat. Rev. Immunol 12:845–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pillai AB, George TI, Dutt S, Strober S. 2009. Host natural killer T cells induce an interleukin-4-dependent expansion of donor CD4+CD25+Foxp3+ T regulatory cells that protects against graft-versus-host disease. Blood 113:4458–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lowsky R, Takahashi T, Liu YP, Dejbakhsh-Jones S, Grumet FC, et al. 2005. Protective conditioning for acute graft-versus-host disease. N. Engl. J. Med 353:1321–31 [DOI] [PubMed] [Google Scholar]
- 56.Morris ES, Macdonald KP, Rowe V, Banovic T, Kuns RD, et al. 2005. NKT cell-dependent leukemia eradication following stem cell mobilization with potent G-CSF analogs. J. Clin. Investig 115:3093–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, et al. 2012. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491:717–23 [DOI] [PubMed] [Google Scholar]
- 58.Varelias A, Bunting MD, Ormerod KL, Koyama M, Olver SD, et al. 2018. Recipient mucosal-associated invariant T cells control GVHD within the colon. J. Clin. Investig 128:1919–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Varelias A, Ormerod KL, Bunting MD, Koyama M, Gartlan KH, et al. 2017. Acute graft-versus-host disease is regulated by an IL-17-sensitive microbiome. Blood 129:2172–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Varelias A, Gartlan KH, Wilkinson AN, Olver SD, Samson LD, et al. 2019. Expansion of IL-17A-secreting MAIT cells in peripheral blood following stem cell mobilization. Blood Adv. 3(5):718–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhattacharyya A, Hanafi LA, Sheih A, Golob JL, Srinivasan S, et al. 2018. Graft-derived reconstitution of mucosal-associated invariant T cells after allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant 24:242–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nielsen MM, Witherden DA, Havran WL. 2017. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat. Rev. Immunol 17:733–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maeda Y, Reddy P, Lowler KP, Liu C, Bishop DK, Ferrara JL. 2005. Critical role of host γδ T cells in experimental acute graft-versus-host disease. Blood 106:749–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Minculescu L, Marquart HV, Ryder LP, Andersen NS, Schjoedt I, et al. 2019. Improved overall survival, relapse-free-survival, and less graft-versus-host-disease in patients with high immune reconstitution of TCR gamma delta cells 2 months after allogeneic stem cell transplantation. Front. Immunol 10:1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Minculescu L, Sengelov H. 2015. The role of gamma delta T cells in haematopoietic stem cell transplantation. Scand. J. Immunol 81:459–68 [DOI] [PubMed] [Google Scholar]
- 66.Hulsdunker J, Ottmuller KJ, Neeff HP, Koyama M, Gao Z, et al. 2018. Neutrophils provide cellular communication between ileum and mesenteric lymph nodes at graft-versus-host disease onset. Blood 131:1858–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Magenau J, Braun T, Gatza E, Churay T, Mazzoli A, et al. 2019. Assessment of individual versus composite endpoints of acute graft-versus-host disease in determining long-term survival after allogeneic transplantation. Biol. Blood Marrow Transplant 25:1682–88 [DOI] [PubMed] [Google Scholar]
- 68.Storb R, Deeg HJ, Whitehead J, Appelbaum F, Beatty P, et al. 1986. Methotrexate and cyclosporine compared with cyclosporine alone for prophylaxis of acute graft versus host disease after marrow transplantation for leukemia. N. Engl. J. Med 314:729–35 [DOI] [PubMed] [Google Scholar]
- 69.Niederwieser D, Maris M, Shizuru JA, Petersdorf E, Hegenbart U, et al. 2003. Low-dose total body irradiation (TBI) and fludarabine followed by hematopoietic cell transplantation (HCT) from HLA-matched or mismatched unrelated donors and postgrafting immunosuppression with cyclosporine and mycophenolate mofetil (MMF) can induce durable complete chimerism and sustained remissions in patients with hematological diseases. Blood 101:1620–29 [DOI] [PubMed] [Google Scholar]
- 70.Sandmaier BM, Kornblit B, Storer BE, Olesen G, Maris MB, et al. 2019. Addition of sirolimus to standard cyclosporine plus mycophenolate mofetil-based graft-versus-host disease prophylaxis for patients after unrelated non-myeloablative haemopoietic stem cell transplantation: a multicentre, randomised, phase 3 trial. Lancet Haematol. 6:e409–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ruutu T, Volin L, Parkkali T, Juvonen E, Elonen E. 2000. Cyclosporine, methotrexate, and methylprednisolone compared with cyclosporine and methotrexate for the prevention of graft-versus-host disease in bone marrow transplantation from HLA-identical sibling donor: a prospective randomized study. Blood 96:2391–98 [PubMed] [Google Scholar]
- 72.Mohty M 2007. Mechanisms of action of antithymocyte globulin: T-cell depletion and beyond. Leukemia 21:1387–94 [DOI] [PubMed] [Google Scholar]
- 73.Kroger N, Solano C, Wolschke C, Bandini G, Patriarca F, et al. 2016. Antilymphocyte globulin for prevention of chronic graft-versus-host disease. N. Engl. J. Med 374:43–53 [DOI] [PubMed] [Google Scholar]
- 74.Soiffer RJ, Kim HT, McGuirk J, Horwitz ME, Johnston L, et al. 2017. Prospective, randomized, double-blind, phase III clinical trial of anti-T-lymphocyte globulin to assess impact on chronic graft-versus-host disease-free survival in patients undergoing HLA-matched unrelated myeloablative hematopoietic cell transplantation. J. Clin. Oncol 35:4003–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C, et al. 2015. Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J. Clin. Investig 125:2677–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luznik L, O’Donnell PV, Fuchs EJ. 2012. Post-transplantation cyclophosphamide for tolerance induction in HLA-haploidentical bone marrow transplantation. Semin. Oncol 39:683–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McCurdy SR, Kasamon YL, Kanakry CG, Bolanos-Meade J, Tsai HL, et al. 2017. Comparable composite endpoints after HLA-matched and HLA-haploidentical transplantation with post-transplantation cyclophosphamide. Haematologica 102:391–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wachsmuth LP, Patterson MT, Eckhaus MA, Venzon DJ, Gress RE, Kanakry CG. 2019. Post-transplantation cyclophosphamide prevents graft-versus-host disease by inducing alloreactive T cell dysfunction and suppression. J. Clin. Investig 129:2357–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reshef R, Ganetsky A, Acosta EP, Blauser R, Crisalli L, et al. 2019. Extended CCR5 blockade for graft-versus-host disease prophylaxis improves outcomes of reduced-intensity unrelated donor hematopoietic cell transplantation: a phase II clinical trial. Biol. Blood Marrow Transplant 25:515–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Toubai T, Hou G, Mathewson N, Liu C, Wang Y, et al. 2014. Siglec-G-CD24 axis controls the severity of graft-versus-host disease in mice. Blood 123:3512–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kennedy GA, Varelias A, Vuckovic S, Le Texier L, Gartlan KH, et al. 2014. Addition of interleukin-6 inhibition with tocilizumab to standard graft-versus-host disease prophylaxis after allogeneic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol. 15:1451–59 [DOI] [PubMed] [Google Scholar]
- 82.Pidala J, Beato F, Kim J, Betts B, Jim H, et al. 2018. In vivo IL-12/IL-23p40 neutralization blocks Th1/Th17 response after allogeneic hematopoietic cell transplantation. Haematologica 103:531–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reddy P, Sun Y, Toubai T, Duran-Struuck R, Clouthier SG, et al. 2008. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J. Clin. Investig 118:2562–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi SW, Braun T, Chang L, Ferrara JL, Pawarode A, et al. 2014. Vorinostat plus tacrolimus and mycophenolate to prevent graft-versus-host disease after related-donor reduced-intensity conditioning allogeneic haemopoietic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol. 15:87–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, et al. 2016. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat. Immunol 17:505–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shono Y, van den Brink MRM. 2018. Gut microbiota injury in allogeneic haematopoietic stem cell transplantation. Nat. Rev. Cancer 18:283–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kohler N, Zeiser R. 2018. Intestinal microbiota influence immune tolerance post allogeneic hematopoietic cell transplantation and intestinal GVHD. Front. Immunol 9:3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zeiser R, von Bubnoff N, Butler J, Mohty M, Niederwieser D, et al. 2020. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N. Engl. J. Med 382(19):1800–10 [DOI] [PubMed] [Google Scholar]
- 89.MacDonald KPA, Betts BC, Couriel D. 2018. Emerging therapeutics for the control of chronic graft-versus-host disease. Biol. Blood Marrow Transplant 24:19–26 [DOI] [PubMed] [Google Scholar]
- 90.Heine A, Brossart P, Wolf D. 2013. Ruxolitinib is a potent immunosuppressive compound: Is it time for anti-infective prophylaxis? Blood 122:3843–44 [DOI] [PubMed] [Google Scholar]
- 91.Magenau JM, Goldstein SC, Peltier D, Soiffer RJ, Braun T, et al. 2018. α1-Antitrypsin infusion for treatment of steroid-resistant acute graft-versus-host disease. Blood 131:1372–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, et al. 2015. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528:560–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martin PJ. 2020. How I treat steroid-refractory acute graft-versus-host disease. Blood 135:1630–38 [DOI] [PubMed] [Google Scholar]
- 94.Rafei H, Kharfan-Dabaja MA, Nishihori T. 2017. A critical appraisal of extracorporeal photopheresis as a treatment modality for acute and chronic graft-versus-host disease. Biomedicines 5(4):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nygaard M, Wichert S, Berlin G, Toss F. 2020. Extracorporeal photopheresis for graft-vs-host disease: a literature review and treatment guidelines proposed by the Nordic ECP Quality Group. Eur. J. Haematol 104:361–75 [DOI] [PubMed] [Google Scholar]
- 96.Jagasia MH, Greinix HT, Arora M, Williams KM, Wolff D, et al. 2015. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-Versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol. Blood Marrow Transplant 21:389–401.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee SJ, Wolff D, Kitko C, Koreth J, Inamoto Y, et al. 2015. Measuring therapeutic response in chronic graft-versus-host disease:. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-Versus-Host Disease: IV. The 2014 Response Criteria Working Group report. Biol. Blood Marrow Transplant 21:984–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Parkman R 1989. Graft-versus-host disease: an alternative hypothesis. Immunol. Today 10:362–64 [DOI] [PubMed] [Google Scholar]
- 99.Kuzmina Z, Gounden V, Curtis L, Avila D, Taylor T, et al. 2015. Clinical significance of autoantibodies in a large cohort of patients with chronic graft-versus-host disease defined by NIH criteria. Am. J. Hematol 90:114–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fukushi N, Arase H, Wang B, Ogasawara K, Gotohda T, et al. 1990. Thymus: a direct target tissue in graft-versus-host reaction after allogeneic bone marrow transplantation that results in abrogation of induction of self-tolerance. PNAS 87:6301–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sakoda Y, Hashimoto D, Asakura S, Takeuchi K, Harada M, et al. 2007. Donor-derived thymic-dependent T cells cause chronic graft-versus-host disease. Blood 109:1756–64 [DOI] [PubMed] [Google Scholar]
- 102.Zhao D, Zhang C, Yi T, Lin CL, Todorov I, et al. 2008. In vivo-activated CD103+CD4+ regulatory T cells ameliorate ongoing chronic graft-versus-host disease. Blood 112:2129–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Leveque-El mouttie L, Koyama M, Le Texier L, Markey KA, Cheong M, et al. 2016. Corruption of dendritic cell antigen presentation during acute GVHD leads to regulatory T-cell failure and chronic GVHD. Blood 128:794–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lockridge JL, Zhou Y, Becker YA, Ma S, Kenney SC, et al. 2013. Mice engrafted with human fetal thymic tissue and hematopoietic stem cells develop pathology resembling chronic graft-versus-host disease. Biol. Blood Marrow Transplant 19:1310–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang C, Todorov I, Zhang Z, Liu Y, Kandeel F, et al. 2006. Donor CD4+ T and B cells in transplants induce chronic graft-versus-host disease with autoimmune manifestations. Blood 107:2993–3001 [DOI] [PubMed] [Google Scholar]
- 106.Young JS, Wu T, Chen Y, Zhao D, Liu H, et al. 2012. Donor B cells in transplants augment clonal expansion and survival of pathogenic CD4+ T cells that mediate autoimmune-like chronic graft-versus-host disease. J. Immunol 189:222–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Srinivasan M, Flynn R, Price A, Ranger A, Browning JL, et al. 2012. Donor B-cell alloantibody deposition and germinal center formation are required for the development of murine chronic GVHD and bronchiolitis obliterans. Blood 119:1570–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jin H, Ni X, Deng R, Song Q, Young J, et al. 2016. Antibodies from donor B cells perpetuate cutaneous chronic graft-versus-host disease in mice. Blood 127:2249–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gartlan KH, Bommiasamy H, Paz K, Wilkinson AN, Owen M, et al. 2018. A critical role for donor-derived IL-22 in cutaneous chronic GVHD. Am. J. Transplant 18:810–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.MacDonald KP, Blazar BR, Hill GR. 2017. Cytokine mediators of chronic graft-versus-host disease. J. Clin. Investig 127:2452–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Flynn R, Du J, Veenstra RG, Reichenbach DK, Panoskaltsis-Mortari A, et al. 2014. Increased T follicular helper cells and germinal center B cells are required for cGVHD and bronchiolitis obliterans. Blood 123:3988–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Deng R, Hurtz C, Song Q, Yue C, Xiao G, et al. 2017. Extrafollicular CD4+ T-B interactions are sufficient for inducing autoimmune-like chronic graft-versus-host disease. Nat. Commun 8:978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Forcade E, Paz K, Flynn R, Griesenauer B, Amet T, et al. 2017. An activated Th17-prone T cell subset involved in chronic graft-versus-host disease sensitive to pharmacological inhibition. JCI Insight 2(12):e92111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hill GR, Olver SD, Kuns RD, Varelias A, Raffelt NC, et al. 2010. Stem cell mobilization with G-CSF induces type 17 differentiation and promotes scleroderma. Blood 116:819–28 [DOI] [PubMed] [Google Scholar]
- 115.Alexander KA, Flynn R, Lineburg KE, Kuns RD, Teal BE, et al. 2014. CSF-1-dependant donor-derived macrophages mediate chronic graft-versus-host disease. J. Clin. Investig 124:4266–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cooke KR, Luznik L, Sarantopoulos S, Hakim FT, Jagasia M, et al. 2017. The biology of chronic graft-versus-host disease: a task force report from the National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-Versus-Host Disease. Biol. Blood Marrow Transplant 23:211–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sarantopoulos S, Stevenson KE, Kim HT, Bhuiya NS, Cutler CS, et al. 2007. High levels of B-cell activating factor in patients with active chronic graft-versus-host disease. Clin. Cancer Res 13:6107–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rozmus J, Kariminia A, Abdossamadi S, Storer BE, Martin PJ, et al. 2019. Comprehensive B cell pheno-typing profile for chronic graft-versus-host disease diagnosis. Biol. Blood Marrow Transplant 25:451–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zorn E, Miklos DB, Floyd BH, Mattes-Ritz A, Guo L, et al. 2004. Minor histocompatibility antigen DBY elicits a coordinated B and T cell response after allogeneic stem cell transplantation. J. Exp. Med 199:1133–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Svegliati S, Olivieri A, Campelli N, Luchetti M, Poloni A, et al. 2007. Stimulatory autoantibodies to PDGF receptor in patients with extensive chronic graft-versus-host disease. Blood 110:237–41 [DOI] [PubMed] [Google Scholar]
- 121.Wang KS, Kim HT, Nikiforow S, Heubeck AT, Ho VT, et al. 2017. Antibodies targeting surface membrane antigens in patients with chronic graft-versus-host disease. Blood 130:2889–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang M, Wu Y, Bastian D, Iamsawat S, Chang J, et al. 2018. Inducible T-cell co-stimulator impacts chronic graft-versus-host disease by regulating both pathogenic and regulatory T cells. Front. Immunol 9:1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wu Y, Schutt S, Paz K, Zhang M, Flynn RP, et al. 2018. MicroRNA-17–92 is required for T-cell and B-cell pathogenicity in chronic graft-versus-host disease in mice. Blood 131:1974–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Forcade E, Kim HT, Cutler C, Wang K, Alho AC, et al. 2016. Circulating T follicular helper cells with increased function during chronic graft-versus-host disease. Blood 127:2489–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Socie G, Ritz J. 2014. Current issues in chronic graft-versus-host disease. Blood 124:374–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Radojcic V, Pletneva MA, Yen HR, Ivcevic S, Panoskaltsis-Mortari A, et al. 2010. STAT3 signaling in CD4+ T cells is critical for the pathogenesis of chronic sclerodermatous graft-versus-host disease in a murine model. J. Immunol 184:764–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Taylor DK, Mittereder N, Kuta E, Delaney T, Burwell T, et al. 2018. T follicular helper-like cells contribute to skin fibrosis. Sci. Transl. Med 10(431):eaaf5307 [DOI] [PubMed] [Google Scholar]
- 128.Matsuoka K, Kim HT, McDonough S, Bascug G, Warshauer B, et al. 2010. Altered regulatory T cell homeostasis in patients with CD4+ lymphopenia following allogeneic hematopoietic stem cell transplantation. J. Clin. Investig 120:1479–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Murase K, Kim HT, Bascug OR, Kawano Y, Ryan J, et al. 2014. Increased mitochondrial apoptotic priming of human regulatory T cells after allogeneic hematopoietic stem cell transplantation. Haematologica 99:1499–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zeiser R, Sarantopoulos S, Blazar BR. 2018. B-cell targeting in chronic graft-versus-host disease. Blood 131:1399–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Johnston HF, Xu Y, Racine JJ, Cassady K, Ni X, et al. 2014. Administration of anti-CD20 mAb is highly effective in preventing but ineffective in treating chronic graft-versus-host disease while preserving strong graft-versus-leukemia effects. Biol. Blood Marrow Transplant 20:1089–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cutler C, Kim HT, Bindra B, Sarantopoulos S, Ho VT, et al. 2013. Rituximab prophylaxis prevents corticosteroid-requiring chronic GVHD after allogeneic peripheral blood stem cell transplantation: results of a phase 2 trial. Blood 122:1510–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pai CC, Chen M, Mirsoian A, Grossenbacher SK, Tellez J, et al. 2014. Treatment of chronic graft-versus-host disease with bortezomib. Blood 124:1677–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dubovsky JA, Flynn R, Du J, Harrington BK, Zhong Y, et al. 2014. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J. Clin. Investig 124:4867–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Miklos D, Cutler CS, Arora M, Waller EK, Jagasia M, et al. 2017. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood 130:2243–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Flynn R, Allen JL, Luznik L, MacDonald KP, Paz K, et al. 2015. Targeting Syk-activated B cells in murine and human chronic graft-versus-host disease. Blood 125:4085–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Poe JC, Jia W, Di Paolo JA, Reyes NJ, Kim JY, et al. 2018. SYK inhibitor entospletinib prevents ocular and skin GVHD in mice. JCI Insight 3(19):e122430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zeiser R, Burchert A, Lengerke C, Verbeek M, Maas-Bauer K, et al. 2015. Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: a multicenter survey. Leukemia 29:2062–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, et al. 2016. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood 127:2144–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Weiss JM, Chen W, Nyuydzefe MS, Trzeciak A, Flynn R, et al. 2016. ROCK2 signaling is required to induce a subset of T follicular helper cells through opposing effects on STATs in autoimmune settings. Sci. Signal 9:ra73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Via CS, Rus V, Nguyen P, Linsley P, Gause WC. 1996. Differential effect of CTLA4Ig on murine graft-versus-host disease (GVHD) development: CTLA4Ig prevents both acute and chronic GVHD development but reverses only chronic GVHD. J. Immunol 157:4258–67 [PubMed] [Google Scholar]
- 142.Nahas MR, Soiffer RJ, Kim HT, Alyea EP 3rd, Arnason J, et al. 2018. Phase 1 clinical trial evaluating abatacept in patients with steroid-refractory chronic graft-versus-host disease. Blood 131(25):2836–45 [DOI] [PubMed] [Google Scholar]
- 143.Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, et al. 2011. Interleukin-2 and regulatory T cells in graft-versus-host disease. N. Engl. J. Med 365:2055–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.McDonald-Hyman C, Flynn R, Panoskaltsis-Mortari A, Peterson N, MacDonald KP, et al. 2016. Therapeutic regulatory T-cell adoptive transfer ameliorates established murine chronic GVHD in a CXCR5-dependent manner. Blood 128:1013–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hart JW, Shiue LH, Shpall EJ, Alousi AM. 2013. Extracorporeal photopheresis in the treatment of graft-versus-host disease: evidence and opinion. Ther. Adv. Hematol 4:320–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zerr P, Distler A, Palumbo-Zerr K, Tomcik M, Vollath S, et al. 2012. Combined inhibition of c-Abl and PDGF receptors for prevention and treatment of murine sclerodermatous chronic graft-versus-host disease. Am. J. Pathol 181:1672–80 [DOI] [PubMed] [Google Scholar]
- 147.Zerr P, Palumbo-Zerr K, Distler A, Tomcik M, Vollath S, et al. 2012. Inhibition of hedgehog signaling for the treatment of murine sclerodermatous chronic graft-versus-host disease. Blood 120:2909–17 [DOI] [PubMed] [Google Scholar]
- 148.Krenger W, Hollander GA. 2008. The immunopathology of thymic GVHD. Semin. Immunopathol 30:439–56 [DOI] [PubMed] [Google Scholar]
- 149.Simons L, Cavazzana M, Andre I. 2019. Concise Review: Boosting T-cell reconstitution following allogeneic transplantation—current concepts and future perspectives. Stem Cells Transl. Med 8:650–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bunting MD, Varelias A, Souza-Fonseca-Guimaraes F, Schuster IS, Lineburg KE, et al. 2017. GVHD prevents NK-cell-dependent leukemia and virus-specific innate immunity. Blood 129:630–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ullrich E, Salzmann-Manrique E, Bakhtiar S, Bremm M, Gerstner S, et al. 2016. Relation between acute GVHD and NK cell subset reconstitution following allogeneic stem cell transplantation. Front. Immunol 7:595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Storek J, Wells D, Dawson MA, Storer B, Maloney DG. 2001. Factors influencing B lymphopoiesis after allogeneic hematopoietic cell transplantation. Blood 98:489–91 [DOI] [PubMed] [Google Scholar]
- 153.Martins JP, Andoniou CE, Fleming P, Kuns RD, Schuster IS, et al. 2019. Strain-specific antibody therapy prevents cytomegalovirus reactivation after transplantation. Science 363:288–93 [DOI] [PubMed] [Google Scholar]
- 154.Reddy V, Iturraspe JA, Tzolas AC, Meier-Kriesche HU, Schold J, Wingard JR. 2004. Low dendritic cell count after allogeneic hematopoietic stem cell transplantation predicts relapse, death, and acute graft-versus-host disease. Blood 103:4330–35 [DOI] [PubMed] [Google Scholar]
- 155.Markey KA, Koyama M, Kuns RD, Lineburg KE, Wilson YA, et al. 2012. Immune insufficiency during GVHD is due to defective antigen presentation within dendritic cell subsets. Blood 119:5918–30 [DOI] [PubMed] [Google Scholar]
- 156.Thangavelu G, Blazar BR. 2019. Achievement of tolerance induction to prevent acute graft-versus-host disease. Front. Immunol 10:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Na IK, Lu SX, Yim NL, Goldberg GL, Tsai J, et al. 2010. The cytolytic molecules Fas ligand and TRAIL are required for murine thymic graft-versus-host disease. J. Clin. Investig 120:343–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dertschnig S, Hauri-Hohl MM, Vollmer M, Hollander GA, Krenger W. 2015. Impaired thymic expression of tissue-restricted antigens licenses the de novo generation of autoreactive CD4+ T cells in acute GVHD. Blood 125:2720–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chaudhry MS, Velardi E, Dudakov JA, van den Brink MR. 2016. Thymus: the next (re)generation. Immunol. Rev 271:56–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dudakov JA, Mertelsmann AM, O’Connor MH, Jenq RR, Velardi E, et al. 2017. Loss of thymic innate lymphoid cells leads to impaired thymopoiesis in experimental graft-versus-host disease. Blood 130:933–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhang Y, Louboutin JP, Zhu J, Rivera AJ, Emerson SG. 2002. Preterminal host dendritic cells in irradiated mice prime CD8+ T cell-mediated acute graft-versus-host disease. J. Clin. Investig 109:1335–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dertschnig S, Evans P, Santos ESP, Manzo T, Ferrer IR, et al. 2020. Graft-versus-host disease reduces lymph node display of tissue-restricted self-antigens and promotes autoimmunity. J. Clin. Investig 130:1896–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Blazar BR, Sharpe AH, Taylor PA, Panoskaltsis-Mortari A, Gray GS, et al. 1996. Infusion of anti-B7.1 (CD80) and anti-B7.2 (CD86) monoclonal antibodies inhibits murine graft-versus-host disease lethality in part via direct effects on CD4+ and CD8+ T cells. J. Immunol 157:3250–59 [PubMed] [Google Scholar]
- 164.Pidala J, Hamadani M, Dawson P, Martens M, Alousi AM, et al. 2020. Randomized multicenter trial of sirolimus versus prednisone as initial therapy for standard-risk acute GVHD: the BMT CTN 1501 trial. Blood 135:97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Watkins BK, Tkachev V, Furlan SN, Hunt DJ, Betz K, et al. 2018. CD28 blockade controls T cell activation to prevent graft-versus-host disease in primates. J. Clin. Investig 128:3991–4007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Blazar BR, Taylor PA, Linsley PS, Vallera DA. 1994. In vivo blockade of CD28/CTLA4: B7/BB1 interaction with CTLA4-Ig reduces lethal murine graft-versus-host disease across the major histocompatibility complex barrier in mice. Blood 83:3815–25 [PubMed] [Google Scholar]
- 167.Koura DT, Horan JT, Langston AA, Qayed M, Mehta A, et al. 2013. In vivo T cell costimulation blockade with abatacept for acute graft-versus-host disease prevention: a first-in-disease trial. Biol. Blood Marrow Transplant 19:1638–49 [DOI] [PubMed] [Google Scholar]
- 168.Kean LS, Turka LA, Blazar BR. 2017. Advances in targeting co-inhibitory and co-stimulatory pathways in transplantation settings: the Yin to the Yang of cancer immunotherapy. Immunol. Rev 276:192–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Blazar BR, MacDonald KPA, Hill GR. 2018. Immune regulatory cell infusion for graft-versus-host disease prevention and therapy. Blood 131:2651–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. 2020. Regulatory T cells and human disease. Annu. Rev. Immunol 38:541–66 [DOI] [PubMed] [Google Scholar]
- 171.Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, et al. 2012. A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat. Immunol 13:972–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sawant DV, Vignali DA. 2014. Once a Treg, always a Treg? Immunol. Rev 259:173–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu Y, Wu Y, Wang Y, Cai Y, Hu B, et al. 2015. IL-35 mitigates murine acute graft-versus-host disease with retention of graft-versus-leukemia effects. Leukemia 29:939–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Semple K, Yu Y, Wang D, Anasetti C, Yu XZ. 2011. Efficient and selective prevention of GVHD by antigen-specific induced Tregs via linked-suppression in mice. Biol. Blood Marrow Transplant 17:309–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hippen KL, Merkel SC, Schirm DK, Nelson C, Tennis NC, et al. 2011. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am. J. Transplant 11:1148–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Laurence A, Amarnath S, Mariotti J, Kim YC, Foley J, et al. 2012. STAT3 transcription factor promotes instability of nTreg cells and limits generation of iTreg cells during acute murine graft-versus-host disease. Immunity 37:209–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bucher C, Koch L, Vogtenhuber C, Goren E, Munger M, et al. 2009. IL-21 blockade reduces graft-versus-host disease mortality by supporting inducible T regulatory cell generation. Blood 114:5375–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Taylor PA, Lees CJ, Blazar BR. 2002. The infusion of ex vivo activated and expanded CD4+CD25+ immune regulatory cells inhibits graft-versus-host disease lethality. Blood 99:3493–99 [DOI] [PubMed] [Google Scholar]
- 179.Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, et al. 2016. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood 127:1044–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Martin GH, Gregoire S, Landau DA, Pilon C, Grinberg-Bleyer Y, et al. 2013. In vivo activation of transferred regulatory T cells specific for third-party exogenous antigen controls GVH disease in mice. Eur. J. Immunol 43:2263–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nguyen VH, Zeiser R, Dasilva DL, Chang DS, Beilhack A, et al. 2007. In vivo dynamics of regulatory T-cell trafficking and survival predict effective strategies to control graft-versus-host disease following allogeneic transplantation. Blood 109:2649–56 [DOI] [PubMed] [Google Scholar]
- 182.Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, et al. 2014. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood 124:638–44 [DOI] [PubMed] [Google Scholar]
- 183.Meyer EH, Laport G, Xie BJ, MacDonald K, Heydari K, et al. 2019. Transplantation of donor grafts with defined ratio of conventional and regulatory T cells in HLA-matched recipients. JCI Insight 4(10):e127244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhang P, Tey SK, Koyama M, Kuns RD, Olver SD, et al. 2013. Induced regulatory T cells promote tolerance when stabilized by rapamycin and IL-2 in vivo. J. Immunol 191:5291–303 [DOI] [PubMed] [Google Scholar]
- 185.Riegel C, Boeld TJ, Doser K, Huber E, Hoffmann P, Edinger M. 2020. Efficient treatment of murine acute GvHD by in vitro expanded donor regulatory T cells. Leukemia 34:895–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Theil A, Tuve S, Oelschlagel U, Maiwald A, Dohler D, et al. 2015. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy 17:473–86 [DOI] [PubMed] [Google Scholar]
- 187.Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, et al. 2009. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin. Immunol 133:22–26 [DOI] [PubMed] [Google Scholar]
- 188.Whitehouse G, Gray E, Mastoridis S, Merritt E, Kodela E, et al. 2017. IL-2 therapy restores regulatory T-cell dysfunction induced by calcineurin inhibitors. PNAS 114:7083–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Furlan SN, Singh K, Lopez C, Tkachev V, Hunt DJ, et al. 2020. IL-2 enhances ex vivo-expanded regulatory T-cell persistence after adoptive transfer. Blood Adv. 4:1594–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, et al. 2013. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci. Transl. Med 5:179ra43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tkachev V, Furlan SN, Watkins B, Hunt DJ, Zheng HB, et al. 2017. Combined OX40L and mTOR blockade controls effector T cell activation while preserving Treg reconstitution after transplant. Sci. Transl. Med 9(408):eaan3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zheng SG, Wang JH, Koss MN, Quismorio F Jr., Gray JD, Horwitz DA. 2004. CD4+ and CD8+ regulatory T cells generated ex vivo with IL-2 and TGF-β suppress a stimulatory graft-versus-host disease with a lupus-like syndrome. J. Immunol 172:1531–39 [DOI] [PubMed] [Google Scholar]
- 193.Robb RJ, Lineburg KE, Kuns RD, Wilson YA, Raffelt NC, et al. 2012. Identification and expansion of highly suppressive CD8+FoxP3+ regulatory T cells after experimental allogeneic bone marrow transplantation. Blood 119:5898–908 [DOI] [PubMed] [Google Scholar]
- 194.Heinrichs J, Li J, Nguyen H, Wu Y, Bastian D, et al. 2016. CD8+ Tregs promote GVHD prevention and overcome the impaired GVL effect mediated by CD4+ Tregs in mice. Oncoimmunology 5:e1146842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gregori S, Roncarolo MG. 2018. Engineered T regulatory type 1 cells for clinical application. Front. Immunol 9:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhang P, Lee JS, Gartlan KH, Schuster IS, Comerford I, et al. 2017. Eomesodermin promotes the development of type 1 regulatory T (TR1) cells. Sci. Immunol 2(10):eaah7152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bacchetta R, Lucarelli B, Sartirana C, Gregori S, Lupo Stanghellini MT, et al. 2014. Immunological outcome in haploidentical-HSC transplanted patients treated with IL-10-anergized donor T cells. Front. Immunol 5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Alhabbab RY, Nova-Lamperti E, Aravena O, Burton HM, Lechler RI, et al. 2019. Regulatory B cells: development, phenotypes, functions, and role in transplantation. Immunol. Rev 292:164–79 [DOI] [PubMed] [Google Scholar]
- 199.Shimabukuro-Vornhagen A, Hallek MJ, Storb RF, von Bergwelt-Baildon MS. 2009. The role of B cells in the pathogenesis of graft-versus-host disease. Blood 114:4919–27 [DOI] [PubMed] [Google Scholar]
- 200.de Masson A, Bouaziz JD, Le Buanec H, Robin M, O’Meara A, et al. 2015. CD24hiCD27+ and plasmablast-like regulatory B cells in human chronic graft-versus-host disease. Blood 125:1830–39 [DOI] [PubMed] [Google Scholar]
- 201.Khoder A, Sarvaria A, Alsuliman A, Chew C, Sekine T, et al. 2014. Regulatory B cells are enriched within the IgM memory and transitional subsets in healthy donors but are deficient in chronic GVHD. Blood 124:2034–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sarvaria A, Basar R, Mehta RS, Shaim H, Muftuoglu M, et al. 2016. IL-10+ regulatory B cells are enriched in cord blood and may protect against cGVHD after cord blood transplantation. Blood 128:1346–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Weber M, Stein P, Prufer S, Rudolph B, Kreft A, et al. 2014. Donor and host B cell-derived IL-10 contributes to suppression of graft-versus-host disease. Eur. J. Immunol 44:1857–65 [DOI] [PubMed] [Google Scholar]
- 204.Hu Y, He GL, Zhao XY, Zhao XS, Wang Y, et al. 2017. Regulatory B cells promote graft-versus-host disease prevention and maintain graft-versus-leukemia activity following allogeneic bone marrow transplantation. Oncoimmunology 6:e1284721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sato K, Yamashita N, Yamashita N, Baba M, Matsuyama T. 2003. Regulatory dendritic cells protect mice from murine acute graft-versus-host disease and leukemia relapse. Immunity 18:367–79 [DOI] [PubMed] [Google Scholar]
- 206.Fujita S, Sato Y, Sato K, Eizumi K, Fukaya T, et al. 2007. Regulatory dendritic cells protect against cutaneous chronic graft-versus-host disease mediated through CD4+CD25+Foxp3+ regulatory T cells. Blood 110:3793–803 [DOI] [PubMed] [Google Scholar]
- 207.Yu H, Tian Y, Wang Y, Mineishi S, Zhang Y. 2019. Dendritic cell regulation of graft-vs.-host disease: immunostimulation and tolerance. Front. Immunol 10:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Teshima T, Reddy P, Lowler KP, KuKuruga MA, Liu C, et al. 2002. Flt3 ligand therapy for recipients of allogeneic bone marrow transplants expands host CD8α+ dendritic cells and reduces experimental acute graft-versus-host disease. Blood 99:1825–32 [DOI] [PubMed] [Google Scholar]
- 209.Hadeiba H, Sato T, Habtezion A, Oderup C, Pan J, Butcher EC. 2008. CCR9 expression defines tolerogenic plasmacytoid dendritic cells able to suppress acute graft-versus-host disease. Nat. Immunol 9:1253–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bouchlaka MN, Moffitt AB, Kim J, Kink JA, Bloom DD, et al. 2017. Human mesenchymal stem cell-educated macrophages are a distinct high IL-6-producing subset that confer protection in graft-versus-host-disease and radiation injury models. Biol. Blood Marrow Transplant 23:897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hutchinson JA, Riquelme P, Sawitzki B, Tomiuk S, Miqueu P, et al. 2011. Cutting edge: immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J. Immunol 187:2072–78 [DOI] [PubMed] [Google Scholar]
- 212.Gabrilovich DI. 2017. Myeloid-derived suppressor cells. Cancer Immunol. Res 5:3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, et al. 2010. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood 116:5738–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Messmann JJ, Reisser T, Leithauser F, Lutz MB, Debatin KM, Strauss G. 2015. In vitro-generated MDSCs prevent murine GVHD by inducing type 2 T cells without disabling antitumor cytotoxicity. Blood 126:1138–48 [DOI] [PubMed] [Google Scholar]
- 215.Vendramin A, Gimondi S, Bermema A, Longoni P, Rizzitano S, et al. 2014. Graft monocytic myeloid-derived suppressor cell content predicts the risk of acute graft-versus-host disease after allogeneic transplantation of granulocyte colony-stimulating factor-mobilized peripheral blood stem cells. Biol. Blood Marrow Transplant 20:2049–55 [DOI] [PubMed] [Google Scholar]
- 216.Koehn BH, Apostolova P, Haverkamp JM, Miller JS, McCullar V, et al. 2015. GVHD-associated, inflammasome-mediated loss of function in adoptively transferred myeloid-derived suppressor cells. Blood 126:1621–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Koehn BH, Saha A, McDonald-Hyman C, Loschi M, Thangavelu G, et al. 2019. Danger-associated extracellular ATP counters MDSC therapeutic efficacy in acute GVHD. Blood 134:1670–82 [DOI] [PMC free article] [PubMed] [Google Scholar]